Contents
- Primary biliary cirrhosis
- Biliary tract
- Primary biliary cirrhosis causes
- Primary biliary cirrhosis stages
- Primary biliary cirrhosis Pathophysiology
- Primary biliary cirrhosis prevention
- Primary biliary cirrhosis signs and symptoms
- Primary biliary cirrhosis complications
- Primary biliary cirrhosis diagnosis
- Primary biliary cirrhosis differential diagnosis
- Primary biliary cirrhosis treatment
- Table 1. Current and emerging medical therapies in primary biliary cholangitis
- Table 2. Current international guidelines in the management of primary biliary cholangitis
- Treating the symptoms
- Treatment for fatigue
- Treatment for itching
- Treatment for dry eyes and mouth
- Primary biliary cirrhosis complications treatment
- Primary biliary cirrhosis liver transplantation
- What can I do to help prevent further liver damage?
- Home remedies
- Primary biliary cirrhosis diet
- Primary biliary cirrhosis prognosis
- Primary biliary cirrhosis life expectancy
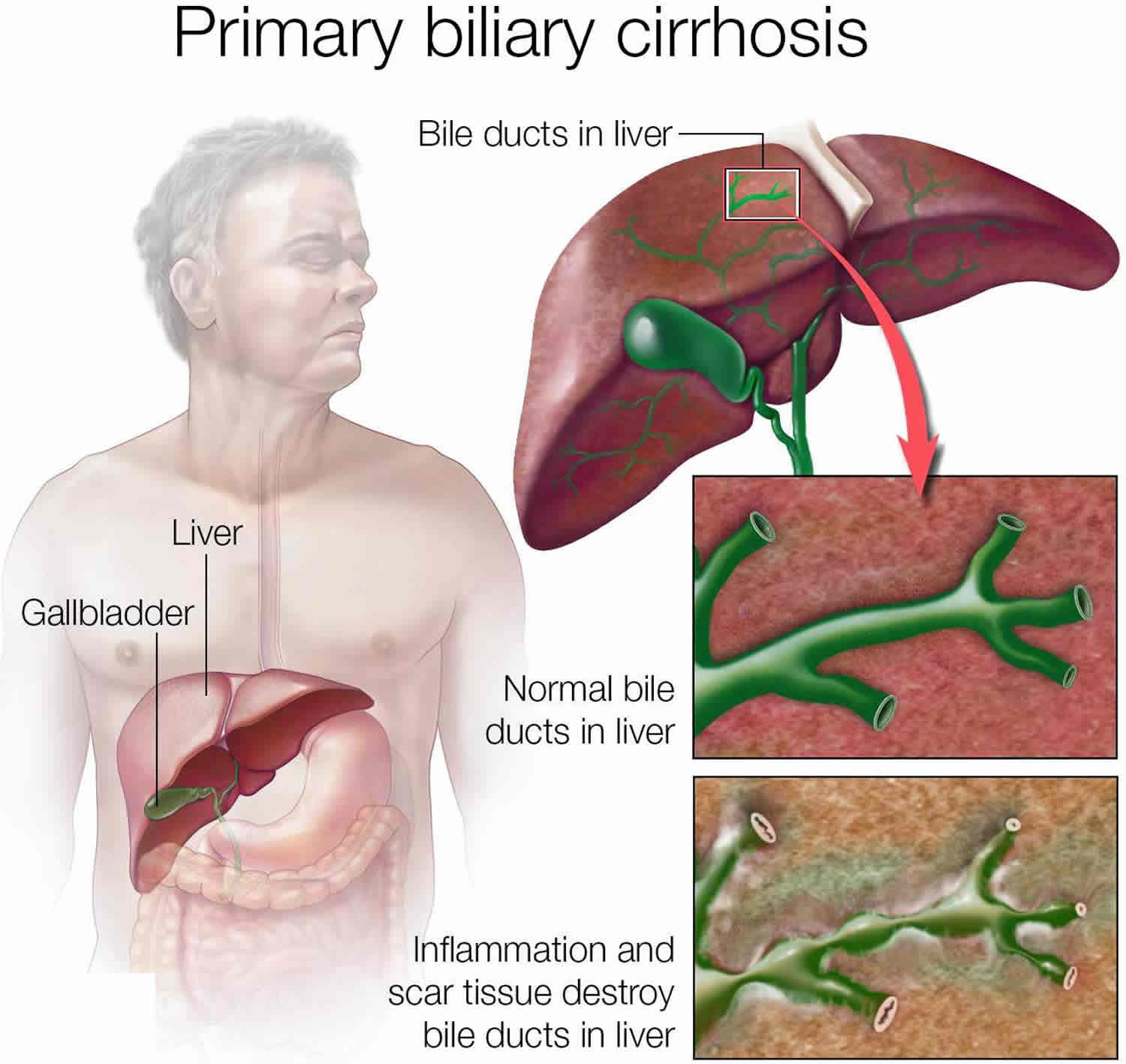
Primary biliary cirrhosis
Primary biliary cirrhosis is now known as primary biliary cholangitis or PBC is a chronic progressive autoimmune liver disease in which the small bile ducts in your liver (small intrahepatic bile ductules with diameter < 100 µm) become inflamed (cholangitis) and are slowly being destroyed 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15. Bile is a fluid made in your liver. Bile aids with digestion and absorbing certain vitamins and fats. Bile also helps your body get rid of cholesterol, toxins and worn-out red blood cells. Primary biliary cirrhosis is believed to be an autoimmune disease in which a person’s own immune system becomes overactive and attacks normal, healthy bile duct cells. Researchers think a combination of genetic and environmental factors triggers the disease. When the bile ducts are damaged, bile can back up in your liver and over time can lead to liver inflammation (hepatitis), poor liver function and irreversible scarring of liver tissue (cirrhosis) and eventually liver failure (end-stage liver disease) 16.
Common early symptoms of primary biliary cirrhosis may include:
- Fatigue (feeling tired).
- Itchy skin, called pruritus.
Other common early symptoms may include:
- Abdominal pain or pain in the upper-right side of your abdomen
- Nausea
- Poor appetite
- Weight loss
- Arthritis or joint pain
- Dry eyes and dry mouth due to Sjögren’s syndrome
As primary biliary cirrhosis gets worse, your symptoms may include:
- Yellowing of the skin and eyes, called jaundice.
- Pain in the upper right abdomen.
- Swelling of the spleen, called splenomegaly.
- Bone, muscle or joint pain, called musculoskeletal pain.
- Swollen feet and ankles (edema).
- Buildup of fluid in the abdomen due to liver failure, called ascites.
- Fatty deposits, called xanthomas, on the skin around the eyes, eyelids or in the creases of the palms, soles, elbows or knees.
- Darkening of the skin that’s not related to sun exposure, called hyperpigmentation.
- Weak and brittle bones, called osteoporosis, which can lead to fractures.
- High cholesterol.
- Diarrhea that may include greasy stools, called steatorrhea.
- Underactive thyroid, called hypothyroidism.
- Weight loss.
- Weakness.
- Dark urine.
Primary biliary cirrhosis affects both sexes, but is more common in women 17. Primary biliary cirrhosis disease ratio among females to males is 9:1 11. Researchers estimated that in 2014 about 58 out of every 100,000 U.S. women and about 15 out of every 100,000 U.S. men had primary biliary cirrhosis 18. Many people do not have symptoms when they are first diagnosed and may not develop symptoms for several years 19, 20, 21. Early symptoms may include fatigue (the most common symptom), itchy skin (pruritus), and abdominal pain. As the disease progresses, people with primary biliary cirrhosis may develop weakness, nausea, diarrhea, swelling in the legs and feet (edema), bone and joint pain, jaundice (your skin and the whites of your eyes turn yellow caused by the build-up of bilirubin in your blood), dark urine, and xanthomas (yellowish, fatty skin deposits that can appear anywhere on the body, often on joints, tendons, and eyelids). The symptoms of primary biliary cirrhosis can significantly impair the quality of your life 19.
The diagnosis of primary biliary cirrhosis may involve blood tests, imaging studies such as X-ray or ultrasound of your liver, and sometimes, a liver biopsy. Blood tests may include tests for anti-mitochondrial antibodies (which may confirm the diagnosis), liver function tests, and cholesterol tests. Abnormal blood test results commonly lead to the diagnosis in people with primary biliary cirrhosis who do not have symptoms.
Asymptomatic patients with abnormal liver chemistry, especially abnormal alkaline phosphatase (ALP), should be evaluated for primary biliary cirrhosis (PBC). Patients who present with vague right upper quadrant pain, unexplained pruritus, fatigue, jaundice, skin hyperpigmentation, and unexplained weight loss should also be evaluated for primary biliary cirrhosis (PBC). Patients who are suspected of having primary biliary cirrhosis should undergo a right abdominal ultrasound, magnetic resonance cholangiopancreatogram (MRCP), or endoscopic retrograde cholangiopancreatogram (ERCP) to rule out the extrahepatic biliary obstruction 11. Once the extrahepatic obstruction is ruled out, antimitochondrial antibody (AMA) should be obtained. In some instances, IgM is elevated.
In cases of atypical disease presentation with elevated alkaline phosphatase (ALP) but normal antimitochondrial antibody (AMA), alternative diagnosis and liver biopsy should be considered for diagnosis. However, with typical clinical features of primary biliary cirrhosis and positive antimitochondrial antibody (AMA) and a normal ALP, liver biopsy is not required. Patients with primary biliary cirrhosis also have a deranged lipid profile due to cholestasis. Fifty percent of patients have elevated cholesterol, which clinically manifests as xanthomas and xanthelasmas 11. Primary biliary cirrhosis patients also have iron deficiency anemia due to chronic blood loss secondary to portal hypertensive gastropathy. Primary biliary cirrhosis patients who have already developed cirrhosis may have coagulopathy (elevated prothrombin time), thrombocytopenia, and leukopenia in addition to anemia.
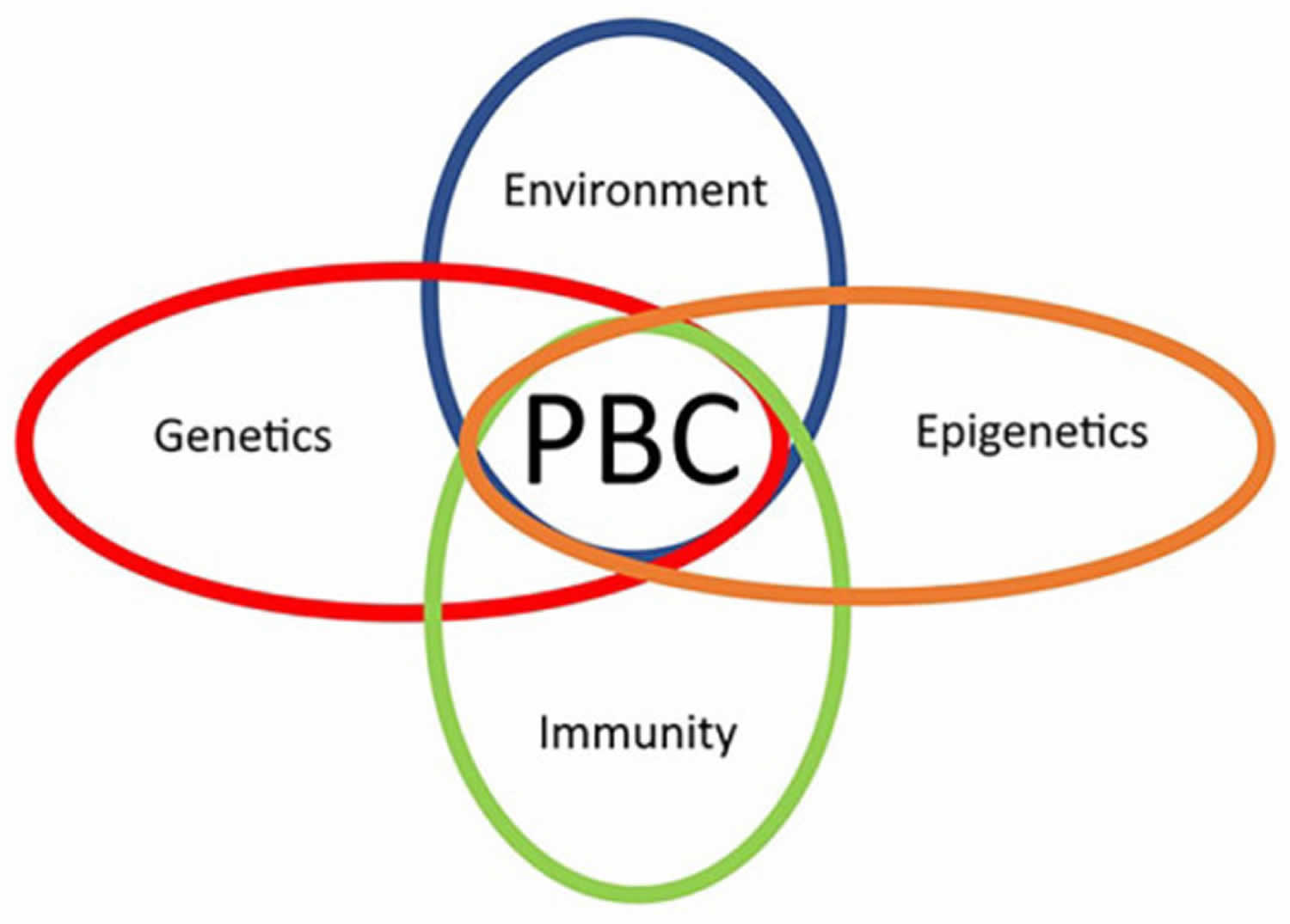
Primary biliary cirrhosis is considered an autoimmune disease in which the immune system malfunctions and mistakenly attacks a person’s healthy bile duct cells, causing the inflammation and damage. It is thought to be caused by a combination of genetic susceptibility and environmental triggers (multifactorial inheritance) 19.
Medication can slow liver damage, especially if treatment begins early. The first treatment recommended for people with primary biliary cirrhosis is ursodiol, also called ursodeoxycholic acid (UDCA), which has been shown to slow disease progression and reduce the need for a liver transplant 19. Obeticholic acid (OCA) is available as a second-line treatment either in combination with UDCA (in those with an inadequate response to UDCA), or by itself (in those who are not able to tolerate UDCA) 19. The symptoms of primary biliary cirrhosis typically do not improve with UDCA or OCA, so individual symptoms are treated separately 19. A liver transplant may be needed when primary biliary cirrhosis leads to liver failure 16.
The rate of progression varies greatly among people with primary biliary cirrhosis, and the disease may progress over many decades before resulting in end-stage liver disease and its complications 19.
Figure 1. Location of the human liver
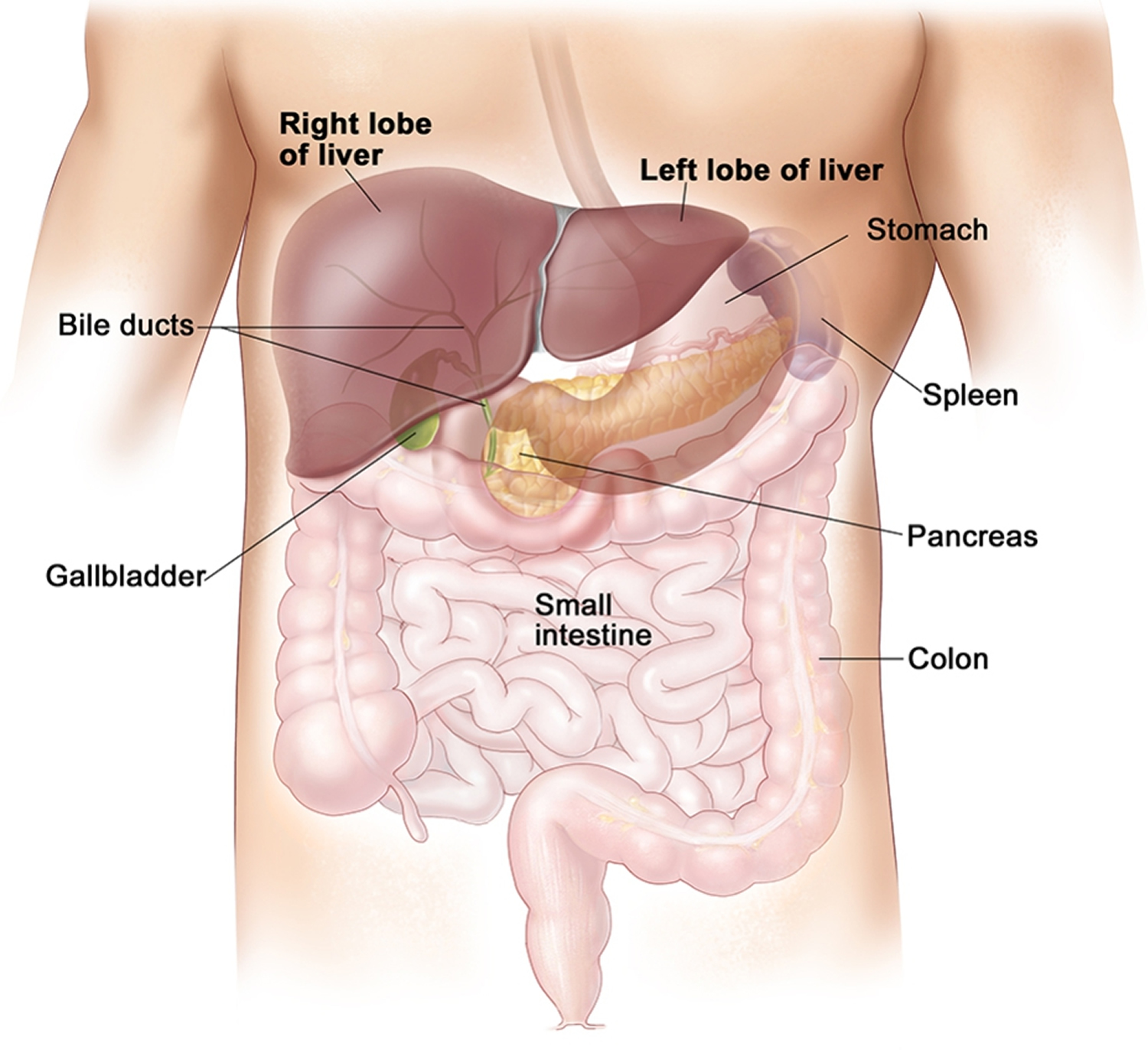
Figure 2. Liver anatomy
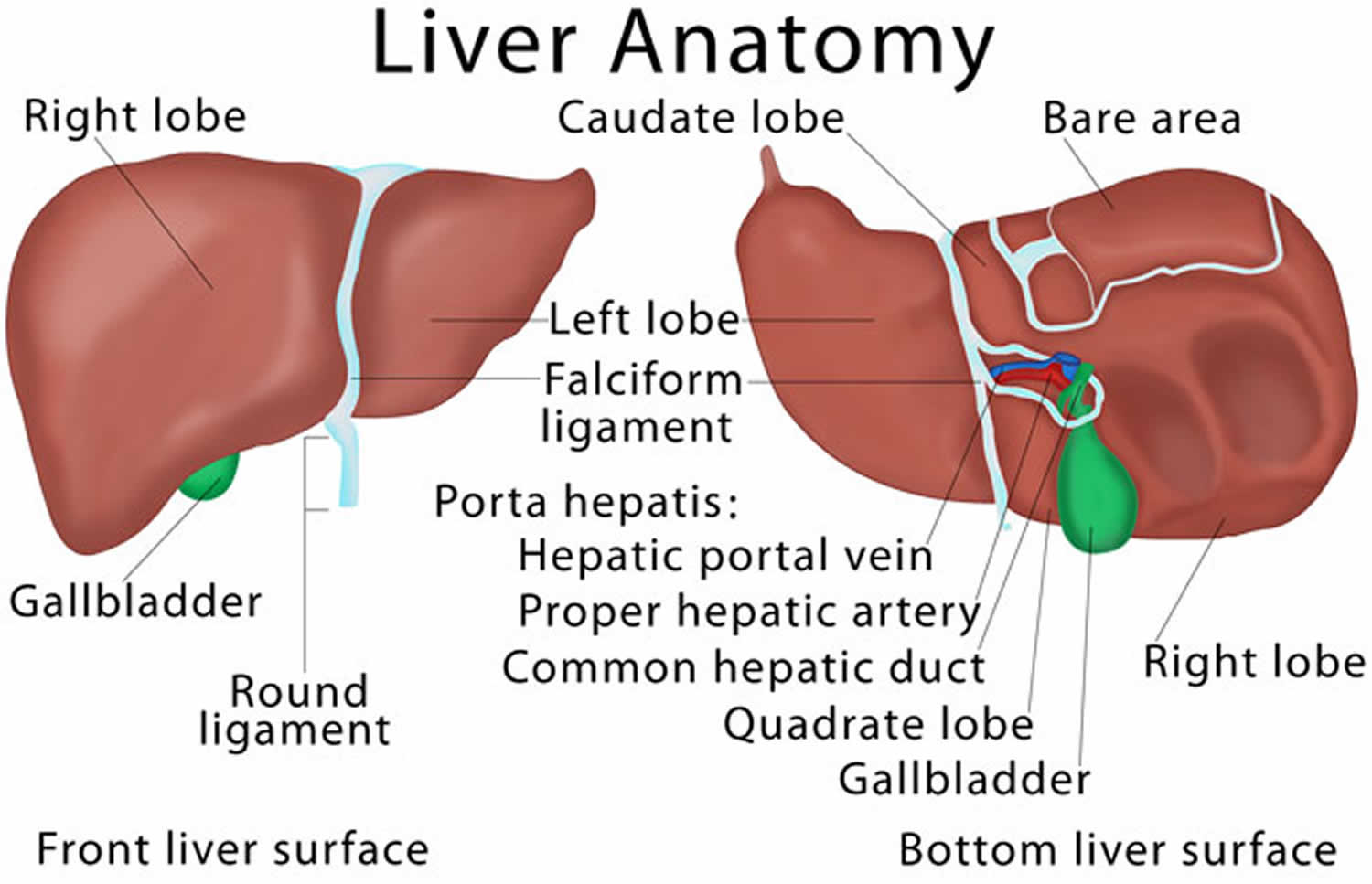
Figure 3. Liver cellular architecture
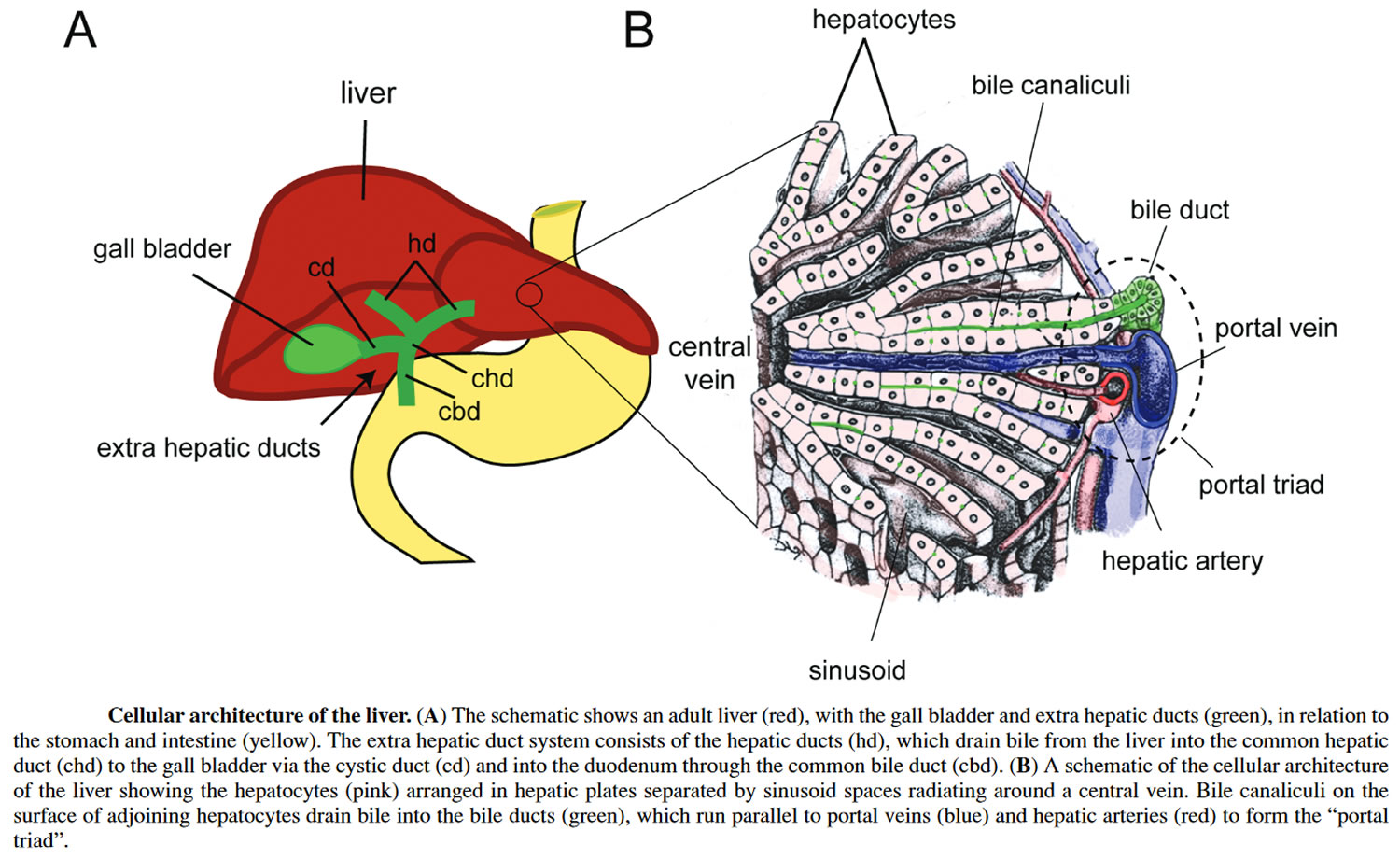
Figure 4. Bile duct anatomy
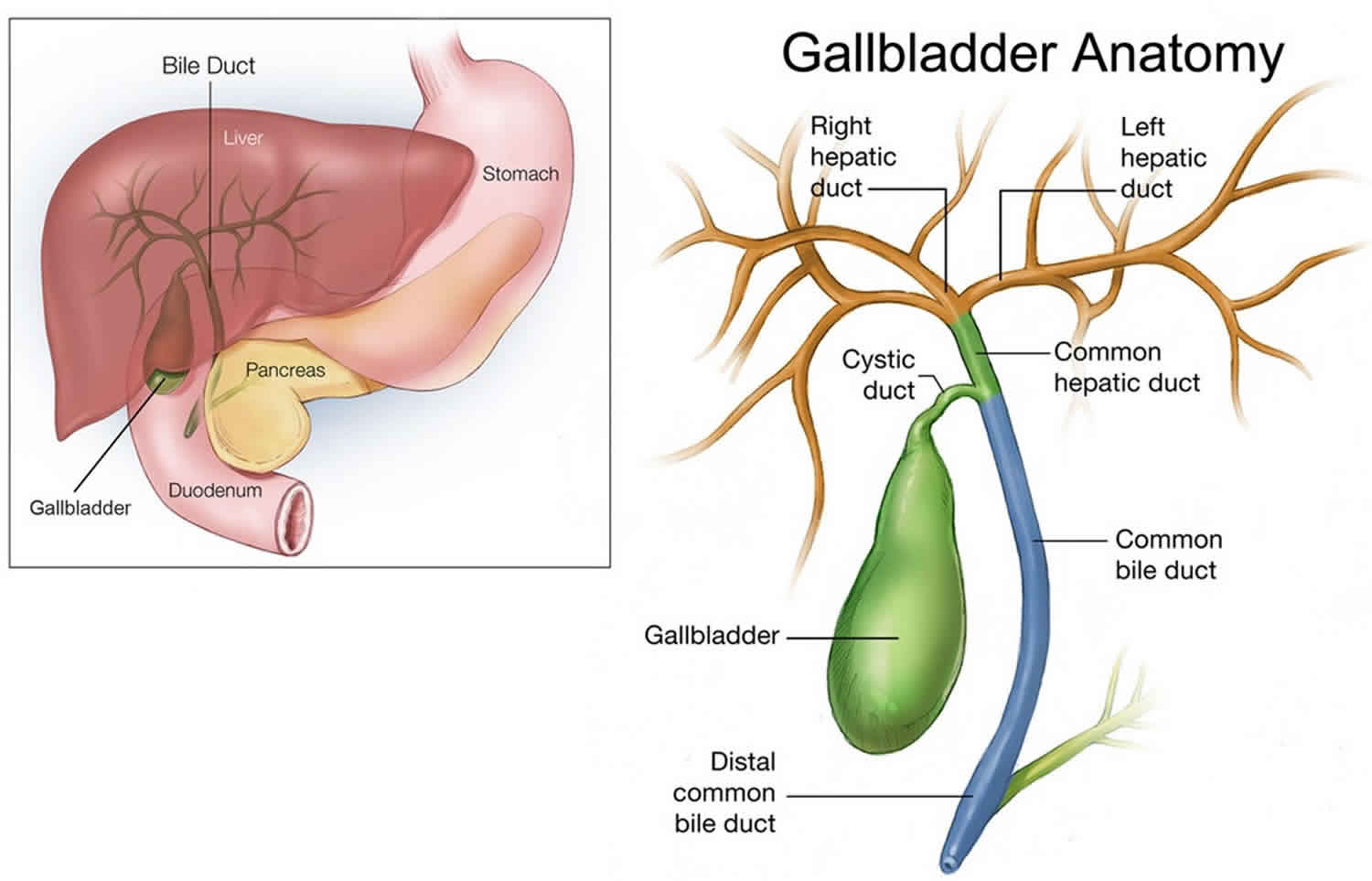
Biliary tract
Biliary tract also called biliary tree or biliary system, is the whole network of various sized bile ducts branching through the liver (hepatic duct), gall bladder and bile ducts, and how they work together to make, store and secrete bile. The name biliary tract is used to refer to all of the ducts, structures and organs involved in the production, storage and secretion of bile. Bile, required for the digestion of food, is excreted by the liver into passages that carry bile toward the hepatic duct, which joins with the cystic duct (carrying bile to and from the gallbladder) to form the common bile duct, which opens into the intestine. The path is as follows: bile canaliculi → canals of Hering → interlobular bile ducts → intrahepatic bile ducts → left and right hepatic ducts merge to form → common hepatic duct exits liver and joins → cystic duct (from gall bladder) forming → common bile duct → joins with pancreatic duct → forming ampulla of Vater → enters duodenum.
Bile consists of water, electrolytes, bile acids, cholesterol, phospholipids and conjugated bilirubin. Some components are synthesised by hepatocytes (liver cells), the rest are extracted from the blood by the liver.
Bile is secreted by the liver into small ducts that join to form the common hepatic duct. Between meals, secreted bile is stored in the gall bladder. During a meal, the bile is secreted into the duodenum to rid the body of waste stored in the bile as well as aid in the absorption of dietary fats and oils.
Pressure inside in the biliary tree can give rise to gallstones and lead to cirrhosis of the liver.
The biliary tract can also serve as a reservoir for intestinal tract infections. Since the biliary tract is an internal organ, it has no somatic nerve supply, and biliary colic due to infection and inflammation of the biliary tract is not a somatic pain. Rather, pain may be caused by luminal distension, which causes stretching of the wall.
An obstruction of the biliary tract can result in jaundice, a yellowing of the skin and whites of the eyes.
Figure 3. Biliary system of the liver
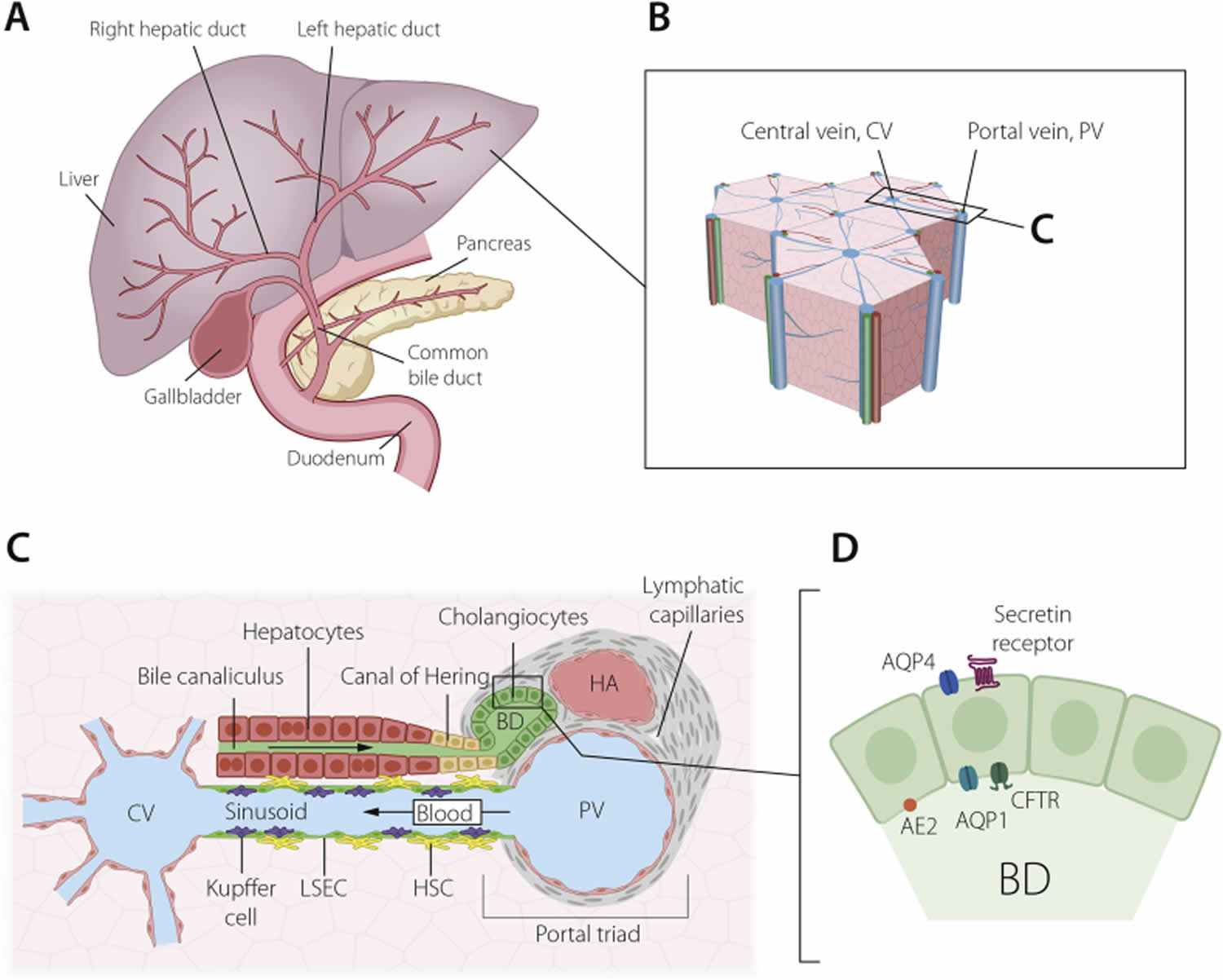
Footnotes: The biliary system of the liver. (A) Schematic depiction of the extra- and intra-hepatic bile duct systems and links to the gall bladder. (B) The hexagonal lobular structure of the liver, with a central vein (CV) surrounded by six portal veins (PV), each paired with a bile duct and hepatic artery, a trio known as the portal triad, enlarged in (C). These three structures are embedded in portal vein mesenchyme, which also contains a lymphatic system.Blood flows centripetally from the portal veins and hepatic arteries to the central vein, along sinusoids lined by liver sinusoidal endothelial cells (LSECs), Kupffer cells and hepatic stellate cells (HSCs).Bile flows instead along bile canaliculi formed by hepatocytes, towards the canals of Hering and into the bile ducts. (D) Bile ducts are highly polarized structures, with an apical cilium (not pictured) and apicobasal distribution of channels and receptors, including anion exchange protein 2 (AE2), aquaporin 1 and 4 (AQP1, AQP4), the cystic fibrosis transmembrane receptor (CFTR) and the secretin receptor.
Abbreviations: CV = central vein; PV = portal vein; LSECs = liver sinusoidal endothelial cells; HSCs = hepatic stellate cells; BD = bile duct; AE2 = anion exchange protein 2; AQP1 = aquaporin 1; AQP4 = aquaporin 4; CFTR = cystic fibrosis transmembrane receptor
[Source 14 ]Figure 4. Biliary tree
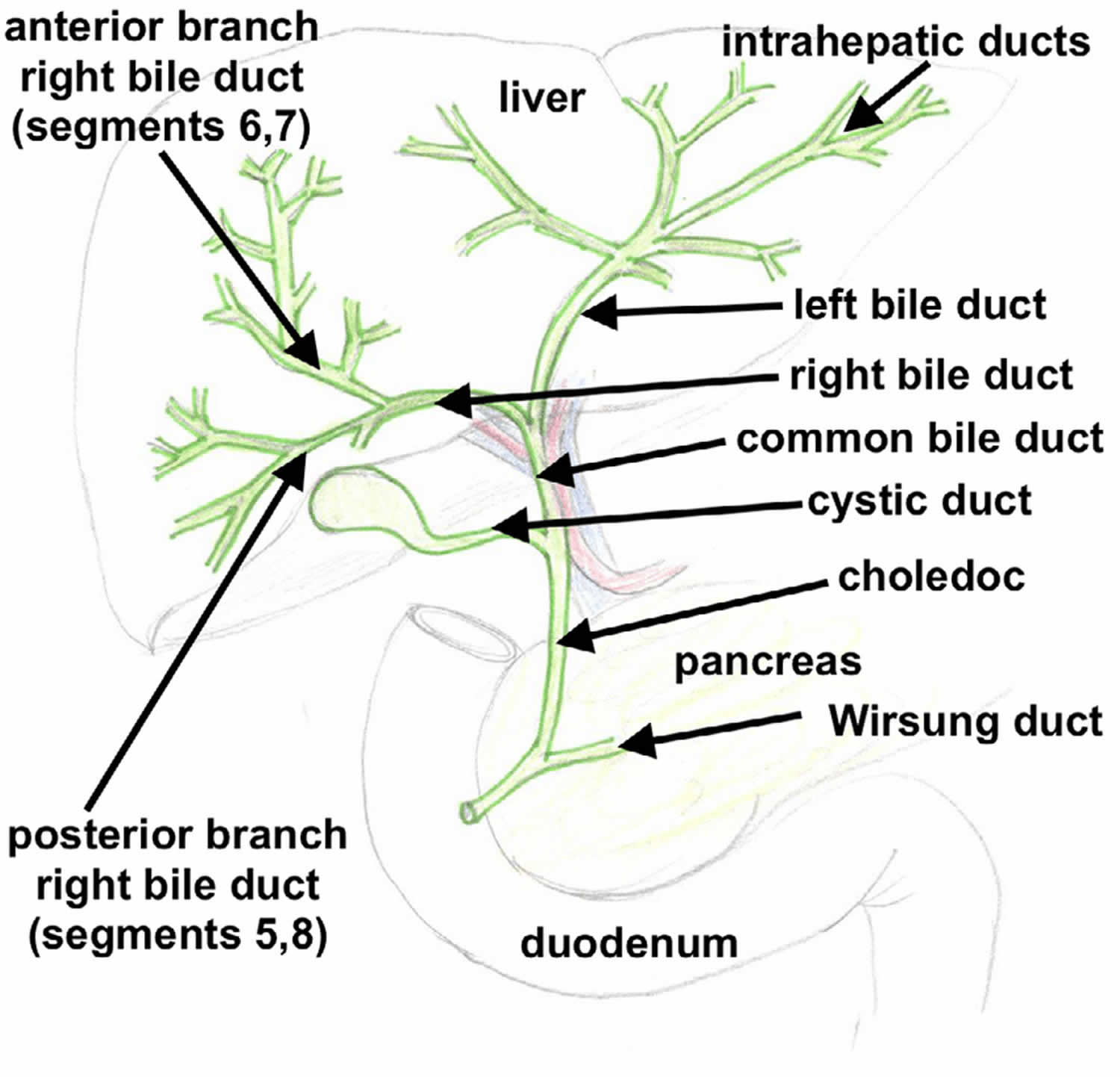
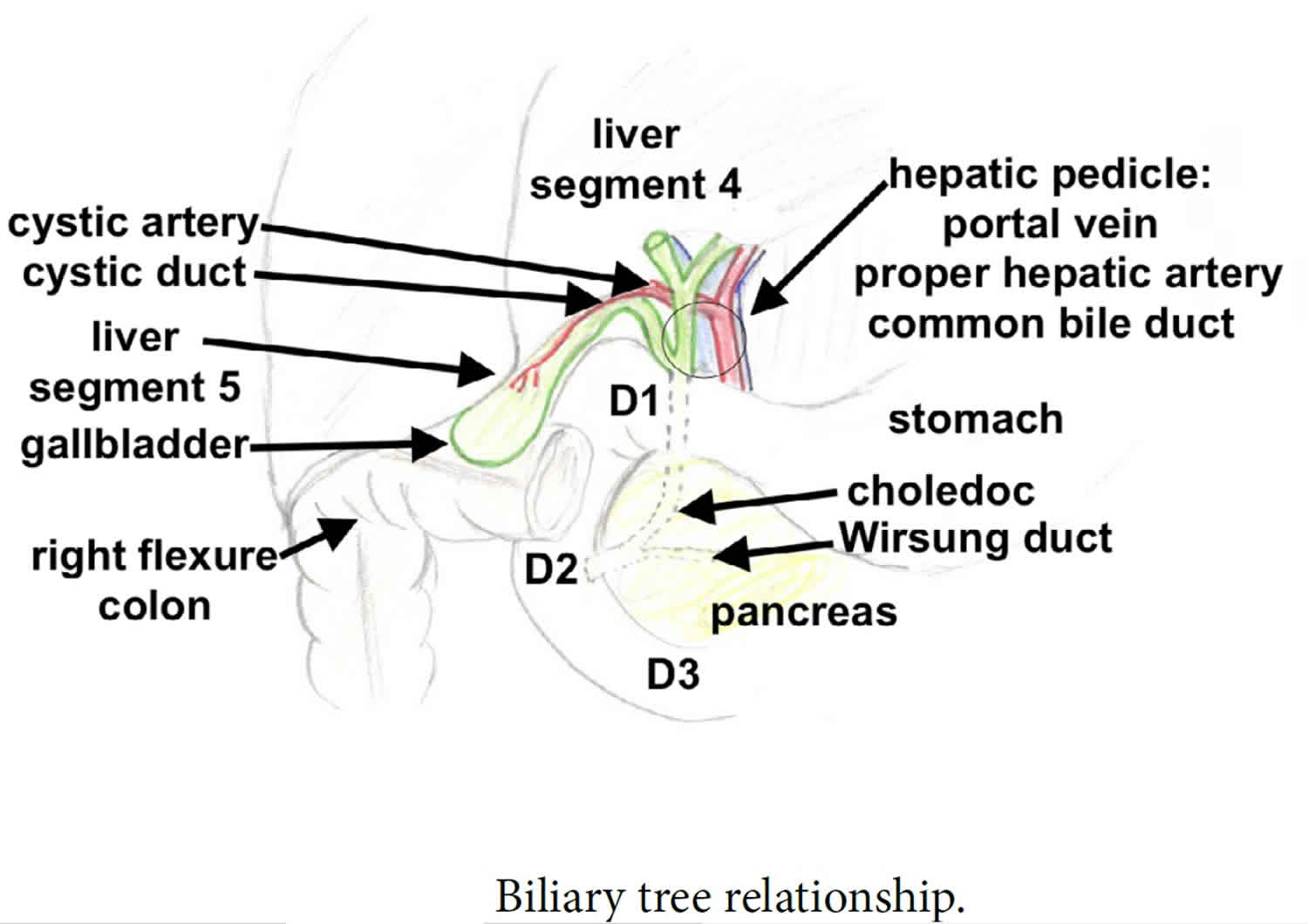
Biliary tract system
The biliary tract refers to the path by which bile is secreted by the liver then transported to the duodenum, the first part of the small intestine. A structure common to most members of the mammal family, the biliary tract is often referred to as a tree because it begins with many small branches which end in the common bile duct, sometimes referred to as the trunk of the biliary tree. The duct, the branches of the hepatic artery, and the portal vein form the central axis of the portal triad. Bile flows in the direction opposite to that of the blood present in the other two channels.
The biliary tract system is usually referred to as the biliary tract or system, and can include the use of the term “hepatobiliary” when used to refer just to the liver and bile ducts. The name biliary tract is used to refer to all of the ducts, structures and organs involved in the production, storage and secretion of bile.
The biliary tract is as follows:
- Bile canaliculi → canals of Hering → intrahepatic bile ductule (in portal tracts / triads) → interlobular bile ducts → left and right hepatic ducts → these merge to form the common hepatic duct, this exits the liver and joins with the cystic duct from gall bladder → together these form the common bile duct which joins the pancreatic duct → these pass through the ampulla of Vater and enter the duodenum.
Primary biliary cirrhosis causes
The exact cause of primary biliary cirrhosis is unknown. Many experts think that a person’s tendency to have an overactive immune system (an autoimmune disease) in which the body turns against its own cells, which may be genetic and unknown environmental triggers play a role in causing primary biliary cirrhosis 22, 20, 23, 24, 25, 26. Possible environmental triggers include:
- Infections
- Cigarette smoking
- Exposure to certain chemicals
The liver inflammation seen in primary biliary cirrhosis starts when certain types of white blood cells called T cells (T lymphocytes) start to collect in the liver. Normally, these immune cells detect and help defend against germs, such as bacteria and viruses. But in primary biliary cirrhosis, they mistakenly destroy the healthy cells lining the small bile ducts in the liver.
Inflammation in the smallest bile ducts spreads and eventually damages other cells in the liver. As the cells die, they’re replaced by scar tissue, also known as fibrosis, that can lead to cirrhosis. Cirrhosis is scarring of liver tissue that makes it difficult for your liver to work properly.
Figure 5. Factors involved in primary biliary cirrhosis pathophysiology
Risk factors for primary biliary cirrhosis
The following factors may increase your risk of primary biliary cirrhosis:
- Sex. Most people with primary biliary cirrhosis are women.
- Age. It’s most likely to occur in people 30 to 60 years old.
- Genetic factors. You’re more likely to get primary biliary cirrhosis if you have a family member who has or had it 27, 28, 29.
- Geography. Primary biliary cirrhosis is most common in people of northern European descent, but primary biliary cirrhosis can affect all ethnicities and races.
Researchers think that genetic factors combined with certain environmental factors trigger primary biliary cirrhosis. These environmental factors may include 30, 31, 32:
- Infections caused by bacteria, fungi or parasites (e.g., recurrent urinary tract infections)
- Smoking
- Exposure to toxic chemicals.
Who is more likely to develop primary biliary cirrhosis?
Primary biliary cirrhosis is more common in:
- Women than in men.
- People who are middle aged. The average age at diagnosis is 60 33.
- People who are white compared with other racial or ethnic groups.
- People who have a parent or sibling particularly an identical twin with primary biliary cirrhosis.
Primary biliary cirrhosis stages
Primary biliary cirrhosis staging system devised by Scheuer is as follows 34, 35:
- Stage 1 Portal stage. Portal stage is associated with bile duct changes or portal inflammation
- Stage 2 Periportal fibrosis or inflammation with dilated but intact portal tracts
- Stage 3 Septal stage: Septal fibrosis with active inflammation or paucicellular septa
- Stage 4 Cirrhotic stage: Variously sized nodules and varying degrees of inflammation.
Primary biliary cirrhosis Pathophysiology
The pathogenesis of primary biliary cirrhosis is thought to be related to the interaction between genetic predisposition and environmental triggers. The genetic predisposition is suggested by a strong prevalence of the disease in first-degree relatives, with an odds ratio (OR) of 11 11. An odds ratio (OR) of 11 means the odds of the outcome are 11 times higher in the exposed group, or there is a 1000% increase in the odds of the outcome with the exposure. There is also a high degree of concordance in monozygotic twins. Daughters of index women (first identified case in a family) have the highest relative risk for the development of primary biliary cirrhosis. Several human leukocyte antigen (HLA) allele associations have been reported with primary biliary cirrhosis, which includes DRB1, DR3, DPB1, DQA1, and DQB1. HLA-DRB1*08 is common in European and Asian descent, whereas HLA-DRB1*11 is protective 15, 36, 37.
The environmental triggers include toxic waste, cigarette smoking, nail polish, hair dye, and various xenobiotics (eg, Escherichia coli, Mycobacterium gordonae, Novosphingobium aromaticivorans) 11. These environmental triggers induce the autoimmune reaction in genetically susceptible patients, which is evident by the presence of a humoral (B-cell-mediated) and cellular response (T-cell-mediated) to an intracytoplasmic antigen, the presence of anti-mitochondrial antibody (AMA), and the involvement of T lymphocytes in the destruction of bile ducts 11. In addition, bacteria containing lipoylated proteins lead to immune response targeting their lipoylated proteins via molecular mimicry. When apoptosis occurs in somatic cells, the exposed epitope is blocked by the attachment of a glutathione residue.
Primary biliary cirrhosis is associated with highly specific autoantibodies. The anti-mitochondrial antibody (AMA) is found in 95% of the cases. The anti-mitochondrial antibody (AMA) binds to lipoic acid containing the E2 component of the pyruvate dehydrogenase complex located on the mitochondrial inner membrane 11. Other antibodies associated with primary biliary cirrhosis are antinuclear antibodies (ANA), anti-multiple nuclear dot antibodies (anti-MND), anticentromere antibodies, and antinuclear envelop antibodies 11. In particular, antinuclear antibodies (ANA) and anti-multiple nuclear dot antibodies (anti-MND) are considered to be surrogate markers in anti-mitochondrial antibody (AMA)-negative primary biliary cirrhosis 11.
Primary biliary cirrhosis prevention
Working together, you and your doctor can help prevent these specific complications:
- Increased pressure in the portal vein (portal hypertension). Your doctor is likely to screen and monitor you for portal hypertension and enlarged veins if you have liver disease.
- Weak bones (osteoporosis). Exercise most days of the week can help increase your bone density. If you have osteoporosis, your treatment may include calcium and vitamin D supplements.
- Vitamin deficiencies. Your doctor may recommend supplements of vitamins A, D, E and K to improve vitamin levels. Avoid taking herbs or nutritional supplements without talking to your doctor first.
Primary biliary cirrhosis signs and symptoms
More than half of people with primary biliary cirrhosis do not have any noticeable symptoms when first diagnosed with primary biliary cirrhosis. The disease may be diagnosed when blood tests are done for other reasons, such as routine testing. Symptoms eventually develop over the next 5 to 20 years. Those who do have symptoms at diagnosis typically have poorer outcomes.
Doctors diagnose up to 6 in 10 people with primary biliary cirrhosis before symptoms begin 38, 39. People with primary biliary cirrhosis and no symptoms are identified through blood tests. Some people do not have symptoms for years after they have been diagnosed with primary biliary cirrhosis.
Common early symptoms of primary biliary cirrhosis may include 40, 41, 42:
- Fatigue (feeling tired).
- Itchy skin, called pruritus.
Other common early symptoms may include:
- Abdominal pain or pain in the upper-right side of your abdomen
- Nausea
- Poor appetite
- Weight loss
- Arthritis or joint pain
- Dry eyes and dry mouth due to Sjögren’s syndrome
As primary biliary cirrhosis gets worse, your symptoms may include 40, 41, 42, 43, 44, 45:
- Yellowing of the skin and eyes, called jaundice.
- Pain in the upper right abdomen.
- Swelling of the spleen, called splenomegaly.
- Bone, muscle or joint pain, called musculoskeletal pain.
- Swollen feet and ankles (edema).
- Buildup of fluid in the abdomen due to liver failure, called ascites.
- Fatty deposits, called xanthomas, on the skin around the eyes, eyelids or in the creases of the palms, soles, elbows or knees.
- Darkening of the skin that’s not related to sun exposure, called hyperpigmentation.
- Weak and brittle bones, called osteoporosis, which can lead to fractures.
- High cholesterol.
- Diarrhea that may include greasy stools, called steatorrhea.
- Underactive thyroid, called hypothyroidism.
- Weight loss.
- Weakness.
- Dark urine.
Primary biliary cirrhosis associated conditions
People with primary biliary cirrhosis may have other autoimmune diseases, including:
- Autoimmune thyroid diseases, conditions in which the immune system attacks the thyroid gland
- Raynaud’s disease
- Sjögren’s syndrome
- Scleroderma
- Autoimmune hepatitis
Women with primary biliary cirrhosis may also have frequent urinary tract infections (UTIs).
Primary biliary cirrhosis complications
As liver damage worsens, primary biliary cirrhosis can cause serious health problems, including:
- Liver scarring or cirrhosis. Cirrhosis makes it difficult for your liver to work and may lead to liver failure. It means the later stage of primary biliary cholangitis. People with primary biliary cholangitis and cirrhosis have a poor medical outlook. They also have a higher risk of other complications.
- Increased pressure in the portal vein, called portal hypertension. Blood from your intestine, spleen and pancreas enters your liver through a large blood vessel called the portal vein. When scar tissue from cirrhosis blocks normal blood flow through your liver, blood backs up. This causes increased pressure inside the vein. Also, because blood doesn’t flow correctly through your liver, drugs and other toxins aren’t filtered properly from your bloodstream.
- Enlarged veins (varices). When blood flow through the portal vein is slowed or blocked, blood may back up into other veins. It usually backs up into those in your stomach and esophagus. Increased pressure may cause delicate veins to break open and bleed. Bleeding in the upper stomach or esophagus is a life-threatening emergency. It requires immediate medical care.
- Enlarged spleen (splenomegaly). Your spleen may become swollen with white blood cells and platelets. This is because your body no longer filters toxins out of the bloodstream as it should.
- Gallstones and bile duct stones. If bile cannot flow through the bile ducts, it may harden into stones in the ducts. These stones can cause pain and infection.
- Liver cancer. Liver scarring increases your risk of liver cancer. If you have liver scarring, you’ll need regular cancer screening.
- Weak bones or osteoporosis. People with primary biliary cirrhosis have an increased risk of weak, brittle bones that may break more easily.
- Vitamin deficiencies. Not having enough bile affects your digestive system’s ability to absorb fats and the fat-soluble vitamins (vitamin A, vitamin D, vitamin E and vitamin K). Because of this, some people with advanced primary biliary cirrhosis may have low levels of these vitamins. Low vitamin levels can result in a variety of health problems, including night blindness and bleeding disorders.
- High cholesterol. Up to 80% of people with primary biliary cirrhosis have high cholesterol.
- Decreased mental function, called hepatic encephalopathy. Some people with advanced primary biliary cirrhosis and cirrhosis have personality changes. They also may have problems with memory and concentration.
- Increased risk of other disease. Primary biliary cirrhosis is associated with other metabolic or immune system disorders, including thyroid problems, dry eyes and mouth disorder called Sjogren’s syndrome, limited scleroderma (CREST syndrome) and rheumatoid arthritis.
Liver complications
Primary biliary cirrhosis can also damage the liver, leading to liver complications.
Cirrhosis
In cirrhosis, scar tissue replaces healthy liver tissue and prevents your liver from working normally. As cirrhosis gets worse, the liver begins to fail.
Portal hypertension
- swelling in your legs, ankles, or feet, called edema
- buildup of fluid in your abdomen, called ascites
- enlarged veins—called varices—in your esophagus, stomach, or intestines, which can lead to internal bleeding if the veins burst
- confusion or difficulties thinking caused by the buildup of toxins in your brain, called hepatic encephalopathy
Liver failure
Cirrhosis may eventually lead to liver failure, also called end-stage liver disease. With liver failure, your liver is badly damaged and stops working. People with liver failure may require a liver transplant.
Liver cancer
Research suggests that people with cirrhosis caused by primary biliary cirrhosis, as well as men with primary biliary cirrhosis have an increased risk for liver cancer.
Primary biliary cirrhosis diagnosis
Your doctor can diagnose primary biliary cirrhosis based on your medical and family history, a physical exam, and the results of medical tests.
Your doctor will ask you about your symptoms. He or she will also ask:
- whether you have a history of certain autoimmune diseases
- whether one of your parents or siblings has been diagnosed with primary biliary cirrhosis
- about your history of infections and exposure to certain chemicals
Your doctor will examine your body, use a stethoscope to listen to sounds in your abdomen, and tap or press on specific areas of your abdomen. He or she will:
- look for yellowing of the whites of your eyes and your skin
- check to see if your liver and spleen are larger than they should be
- check for abdominal tenderness or pain, particularly in the upper right side of your abdomen
The following tests and procedures may be used to diagnose primary biliary cirrhosis.
Blood tests:
- Cholesterol test. More than half the people with primary biliary cirrhosis have extreme increases in blood fats (lipids), including total cholesterol level, which may be a sign that their liver is not working properly.
- Liver tests. These blood tests check the levels of enzymes that may signal liver disease and bile duct injury. Higher-than-normal levels of the liver enzyme alkaline phosphatase (ALP) occur in people with diseases that destroy or block the bile ducts, such as primary biliary cirrhosis.
- Antibody tests for signs of autoimmune disease. Blood tests may be done to check for anti-mitochondrial antibodies (AMAs). Anti-mitochondrial antibodies (AMA) are found in the blood of about 95 percent of people with primary biliary cirrhosis 46, 43. Anti-mitochondrial antibodies (AMA) almost never occur in people without the disease, even if they have other liver disorders. Therefore, a positive AMA test is considered a very reliable sign of primary biliary cirrhosis. However, a small number of people with primary biliary cirrhosis (5–10% of primary biliary cirrhosis cases) don’t have anti-mitochondrial antibodies (AMAs) 47, 48. Although anti-mitochondrial antibody (AMA) has diagnostic value, it has no prognostic value. In other words, AMA titer and subtypes are not associated with disease severity and outcome 49, 50, 51. Although there was a report that treatment with ursodeoxycholic acid (UDCA) may decrease the AMA titer, it is controversial whether the AMA titer is associated with treatment response 52, 53.
- Primary biliary cirrhosis-specific anti-nuclear antibodies (ANA) (anti-GP210 and/or anti-SP100) might help to diagnose AMA-negative subjects. Interestingly, anti-SP100 and anti-GP210 are specific for primary biliary cirrhosis and correlate with disease severity 54.
Your doctor may diagnose primary biliary cirrhosis if you have anti-mitochondrial antibodies (AMAs) and higher-than-normal levels of alkaline phosphatase (ALP) in your blood, even if you have no other signs or symptoms of the disease.
Imaging tests may not be needed. However, they may help your doctor confirm a diagnosis or rule out other conditions with similar signs and symptoms. Imaging tests looking at the liver and bile ducts may include:
- Ultrasound. Ultrasound uses high-frequency sound waves to produce images of structures inside your body.
- FibroScan. Using an ultrasound-like probe, this test can detect scarring of the liver.
- Magnetic resonance cholangiopancreatography (MRCP). This special magnetic resonance imaging (MRI) exam creates detailed images of your organs and bile ducts.
- Magnetic resonance elastography (MRE). MRI is combined with sound waves to create a visual map (elastogram) of internal organs. The test is used to detect hardening of your liver that might be a sign of cirrhosis.
- Endoscopic retrograde cholangiopancreatography (ERCP). The doctor passes a thin, flexible tube down your throat and injects dye into the area of your small intestine where your bile ducts empty. A tiny camera attached to the end of the tube provides a picture of your bile ducts. This test may be done with or instead of an MRCP. But, it’s invasive and may cause complications. With advances in MRI, it’s usually not needed for diagnosis.
If the diagnosis is still uncertain, your doctor may perform a liver biopsy. A small sample of liver tissue is removed through a small incision using a thin needle. It’s examined in a laboratory, either to confirm the diagnosis or to determine the extent (stage) of the disease.
Your doctor may perform a liver biopsy to 37, 55:
- rule out other diseases that may be causing your symptoms
- confirm the diagnosis of primary biliary cirrhosis
- determine whether the disease is advanced—as shown by the amount of liver scarring or cirrhosis—or very active.
Primary biliary cirrhosis diagnostic criteria
The diagnostic criteria for primary biliary cirrhosis include an absence of any other liver disease, no evidence of extrahepatic biliary obstruction on imaging, and at least 2 out of 3 of the following 40, 41, 42, 56, 11:
- Elevation of alkaline phosphatase (ALP) at least 1.5 times the upper limit of normal
- Presence of antimitochondrial antibody (AMA) with a titer of 1:40 or higher
- Histopathological evidence of primary biliary cirrhosis (nonsuppurative destructive cholangitis or “florid duct lesion” and destruction of interlobular bile ducts with a predominance of lymphocytic infiltration).
Primary biliary cirrhosis differential diagnosis
The differential diagnoses of primary biliary cirrhosis comprise all the diseases resulting in cholestasis, pruritis, and a deranged liver profile. All middle-aged women presenting with pruritis and jaundice must be evaluated in the context of primary biliary cirrhosis. Some of the more significant differentials are shown below:
Primary biliary cirrhosis treatment
There’s no cure for primary biliary cirrhosis, but medications are available to help slow the progression of the disease and prevent complications. Options include:
- Ursodeoxycholic acid (UDCA). This medicine, also known as ursodiol (Actigall, Urso), is commonly used first 57. Ursodeoxycholic acid (UDCA) helps move bile through your liver 58. UDCA (ursodiol) at a dose of 13–15 mg/kg/day doesn’t cure primary biliary cirrhosis, but it can slow the progression of liver damage, it seems to improve liver function and reduce liver scarring 57, 59, 60:1–15. doi: 10.3109/00365529409103618)). Most studies have indicated that alkaline phosphatase (ALP) and total bilirubin are the two most meaningful variables in evaluating the UDCA response. It’s less likely to help with itching and fatigue. Side effects may include weight gain, hair loss and diarrhea. People who respond to ursodiol early in the course of primary biliary cirrhosis can live longer without needing a liver transplant. Up to 40% of patients with PBC will denote a suboptimal biochemical response to UDCA 61; therefore, other treatment options are necessary. If you do not respond to ursodiol, your doctor may prescribe obeticholic acid (Ocaliva).
- Obeticholic acid (Ocaliva). This is the newest medication approved by the Food and Drug Administration for primary biliary cirrhosis 62. Studies show that when given alone or combined with ursodiol (UDCA) for 12 months it can help improve liver function. However, its use is often limited because it can cause increased itching.
- Fibrates (Tricor). Researchers aren’t exactly sure how this medicine works to help ease primary biliary cirrhosis symptoms. But, when taken with UDCA, it has reduced liver inflammation and itching in some people. More studies are needed to determine long-term benefits.
- Budesonide. When combined with UDCA, the corticosteroid budesonide may be of potential benefit for primary biliary cirrhosis. However, this medicine is associated with steroid-related side effects for people with more advanced disease. More long-term trials are necessary before budesonide can be recommended for treating primary biliary cirrhosis.
- Other medications. Many other drugs have been used or studied for treating primary biliary cirrhosis with mixed results. They include methotrexate (Trexall) and colchicine (Colcrys).
- Liver transplant. When medications no longer control primary biliary cirrhosis and the liver begins to fail, a liver transplant may help prolong life. A liver transplant replaces your diseased liver with a healthy one from a donor. Liver transplantation is associated with very good long-term outcomes for people with primary biliary cirrhosis. However, sometimes the disease comes back several years later in the transplanted liver.
Figure 6. Primary biliary cirrhosis treatment algorithm
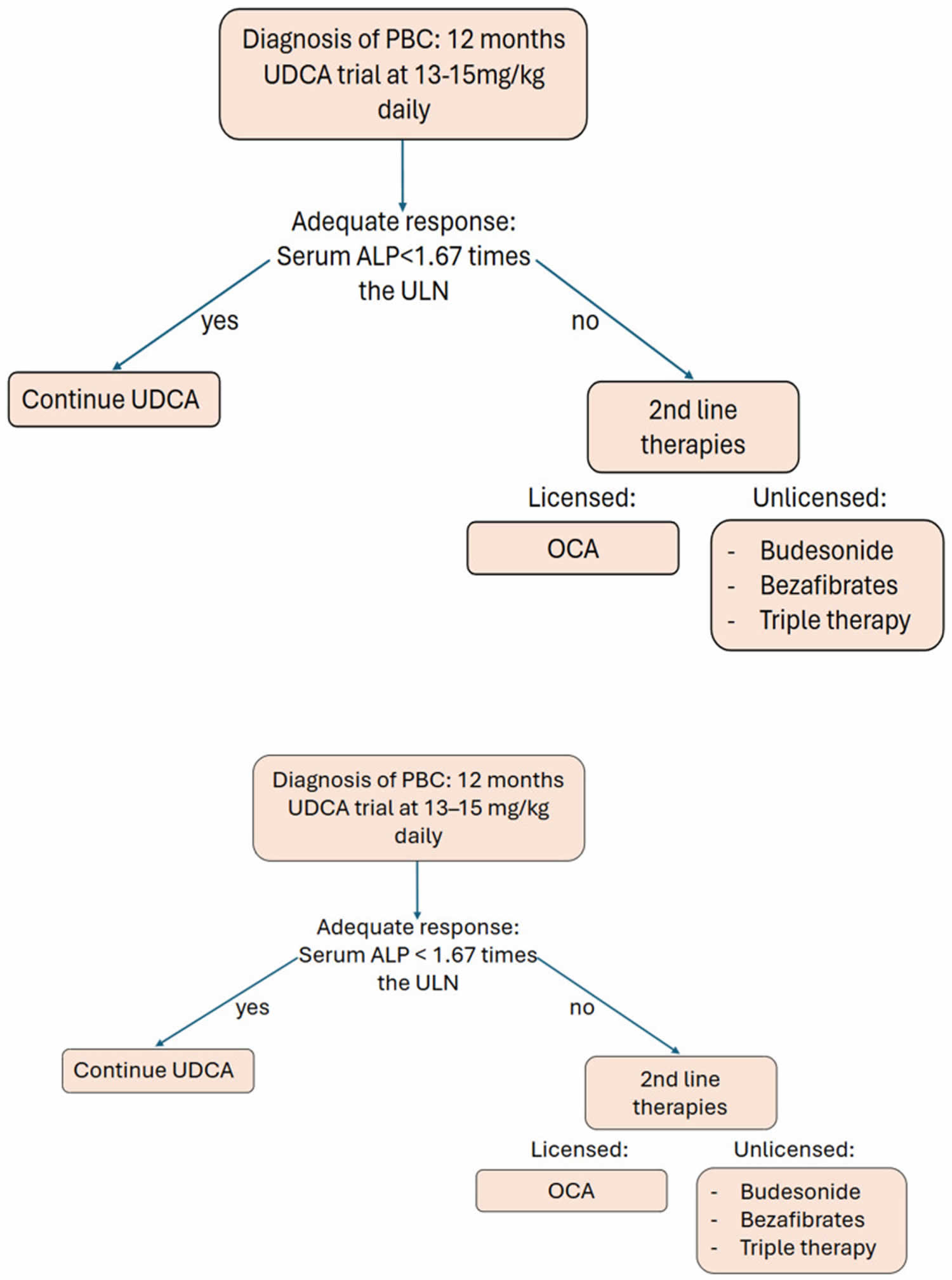
Footnotes: Pathway for patient care in regard to disease-modifying management of primary biliary cirrhosis (PBC).
Abbreviations: PBC = Primary Biliary Cholangitis; UDCA = ursodeoxycholic acid; ALP = alkaline phosphatase; ULN = upper limit of normal; OCA = Obetacholic Acid.
[Source 3 ]Table 1. Current and emerging medical therapies in primary biliary cholangitis
| Current Licenced Therapies | |
|---|---|
| 1. Ursodeoxycholic acid (UDCA) | The first-line therapy for PBC has three proposed mechanisms of action: firstly, conjugates of UDCA; secondly, UDCA stimulates the biliary secretion of bile acids, and lastly, the inhibition of hepatocyte apoptosis. |
| 2. Obeticholic acid (OCA) | The second-line therapy for PBC treatment is derived from the primary human bile acid chenodeoxycholic acid, and it agonises the farnesoid X receptor. |
| Alternate Therapies | |
| 1. Bezafibrate | It treats PBC through its action as a bile acid-sequestrant. Although it is not yet recommended by the British Society of Gastroenterology guidelines, clinical data suggest it could perform well as a second-line option. |
| 2. Fenofibrate | It treats PBC by acting as a bile acid-sequestrant. It is currently being explored as an adjunct first-line therapy alongside UDCA in treating naive patients against UDCA alone. |
| 3. Budesonide | As an immunosuppressive steroid, budesonide could have a role in PBC therapy through control of autoimmune hepatocellular inflammation. However, the evidence for the use of budesonide as a second-line therapy for PBC is limited. Budesonide’s future in PBC treatment is potentially as a component of PBC triple therapy. |
| 4. Triple therapy | As PBC is an autoimmune disorder, the addition of immunosuppressive therapies to UCDA could be of a therapeutic value. However, further research is needed in this area. |
| Emerging novel therapies | |
| Elafibranor and Seladelpar | Seladelpar and elafibranor are novel peroxisome proliferator-activated receptor (PPAR) agonist being explored for the treatment of PBC. PPAR-delta is a hormone receptor that modulates lipid metabolism. |
| Linierxibiat | Linerixibat, a minimally absorbed, oral, small-molecule ileal bile acid transporter (IBAT) inhibitor has been reported to attenuate cholestatic pruritus associated with PBC. |
| Setaxinib | Setanaxib is a NOX1/4 inhibitor that has shown anti-fibrotic effects in in vitro and animal studies. However, it is currently being investigated for its efficacy and safety in patients with PBC. |
Abbreviations: UDCA = Ursodeoxycholic acid; ALP = Alkaline phosphatase; ULN = upper limit of normal; OCA = Obetacholic Acid
[Source 3 ]Table 2. Current international guidelines in the management of primary biliary cholangitis
| Clinical Guidelines | European Association for the Study of the Liver | British Society of Gastroenterology | American college of gastroenterology |
|---|---|---|---|
| First-Line management | Ursodeoxycholic acid (UDCA) 13–15 mg/kg/day | UDCA 13–15 mg/kg/day | UDCA 13–15 mg/kg/day |
| Criteria to consider before starting second-line therapy | Biochemical response post 1 year of UDCA treatment:
| Biochemical response post 1 year of UDCA treatment:
| Biochemical response post 6–12 months of UDCA treatment:
|
| Second-line management | Obetacholic Acid (OCA)
| Obetacholic Acid (OCA)
| Obetacholic Acid (OCA)
|
| Alternative therapies | Unlicensed therapies are not recommended by the European Association for the Study of the Liver | Unlicensed therapies are not recommended by the British Society of Gastroenterology | If the above are not successful, patients should be considered for clinical trials. |
Abbreviations: UDCA = Ursodeoxycholic acid; ALP = Alkaline phosphatase; ULN = upper limit of normal; OCA = Obetacholic Acid
[Source 3 ]Treating the symptoms
- Your doctor may recommend treatments to control the signs and symptoms of primary biliary cirrhosis and make you more comfortable.
Treatment for fatigue
- Primary biliary cirrhosis causes fatigue. But, your daily habits, proper diet and exercise, and other health conditions can affect how tired you feel. Researchers are investigating whether a medicine called modafinil (Provigil) may help reduce fatigue in people with primary biliary cirrhosis. More research is needed. It is important to also be tested to exclude thyroid disease since it is more common in people with primary biliary cirrhosis.
Treatment for itching
- Antihistamines such as diphenhydramine (Benadryl, others) and loratadine (Claritin, others) are commonly used to reduce itching. They may help with sleep if itching keeps you awake. For mild itchy skin, your doctor may prescribe hydroxyzine (Vistaril). For more severe itchy skin, your doctor may prescribe cholestyramine (Locholest, Questran).
- Cholestyramine (Questran) is a powder that must be mixed with food or liquids. Though cholestyramine works for most people, the taste is unpleasant.
- Rifampin (Rifadin, others) is an antibiotic that may stop itching. Exactly how it does this is unknown. Researchers think it may block the brain’s response to itch-inducing chemicals in the blood.
- Opioid antagonists such as those containing naloxone (Bunavail, Evzio) and naltrexone (Vivitrol) may help itching related to liver disease. Like rifampin, these drugs seem to reduce the itching sensation by acting on your brain.
- Sertraline is a selective serotonin reuptake inhibitor (SSRI) medicine that increases serotonin in the brain. It can help reduce itching.
Treatment for dry eyes and mouth
- Artificial tears and saliva substitutes, available over-the-counter or by prescription, can help ease dry eyes and mouth. Chewing gum or sucking on hard candy also can help you make more saliva and relieve dry mouth.
Primary biliary cirrhosis complications treatment
Doctors treat the complications of primary biliary cirrhosis with medicines and dietary supplements. Your doctor may recommend changes in your diet and lifestyle.
- High blood cholesterol levels. If you have higher-than-normal blood cholesterol levels, your doctor may prescribe medicines called statins and recommend lifestyle changes.
- Osteoporosis. For osteoporosis, your doctor may prescribe medicines that slow or stop bone loss and improve bone density. Your doctor may recommend dietary supplements of calcium and vitamin D.
- Fat-soluble vitamin deficiencies. For fat-soluble vitamin deficiencies, your doctor may recommend dietary supplements of vitamins A, D, E, and K. Follow your doctor’s instructions on the type and amount of vitamins you should take.
- Cirrhosis. If primary biliary cirrhosis leads to cirrhosis, doctors can treat the health problems related to cirrhosis with medicines, surgery, and other medical procedures. If cirrhosis leads to liver failure, you may need a liver transplant.
- Treatment for increased pressure in the portal vein or portal hypertension. Your doctor is likely to screen and monitor you for portal hypertension and enlarged veins if you have more advanced scarring from liver disease. Fluid in your abdomen is a common side effect of portal hypertension. For mild fluid in the abdomen, your doctor may only recommend limiting salt in your diet. More-severe cases may require medicines known as diuretics or a procedure to drain the fluid called paracentesis.
Primary biliary cirrhosis liver transplantation
A liver transplant is surgery to replace a diseased liver with a healthy liver. Liver transplant is the standard gold treatment for primary biliary cirrhosis. Your doctor will consider a liver transplant when your primary biliary cirrhosis leads to liver failure. Patients with primary biliary cirrhosis develop complications related to cirrhosis (hepatic encephalopathy, recurrent ascites, severe portal hypertensive gastropathy, bleeding, or hemorrhage secondary to gastric or esophageal varices). They also have disabling symptoms such as fatigue, intractable pruritus, and severely deranged bilirubin levels in the absence of liver cancer. These patients should be evaluated for a liver transplant. Doctors consider liver transplants only after they have ruled out all other treatment options. Talk with your doctor to find out whether a liver transplant is right for you.
Your donated liver may be from:
- A donor who has recently died and has not had liver injury. This type of donor is called a cadaver donor.
- Sometimes, a healthy person will donate part of their liver to a person with a diseased liver. For example, a parent may donate to a child. This kind of donor is called a living donor. The liver can regrow itself in a living donor. Both people most often end up with fully working livers after a successful transplant.
The donor liver is transported in a cooled salt-water (saline) solution that preserves the organ for up to 8 hours. The necessary tests can then be done to match the donor with the recipient.
The new liver is removed from the donor through a surgical cut in the upper abdomen. It is placed into the person who needs the liver (called the recipient) and attached to the blood vessels and bile ducts. The operation may take up to 12 hours. The recipient will often need a large amount of blood through a transfusion.
Liver transplant surgery is often not recommended for people who have:
- Certain infections, such as tuberculosis or osteomyelitis.
- Difficulty taking medicines several times each day for the rest of their lives.
- Heart or lung disease or other life-threatening diseases.
- A history of cancer.
- Infections, such as hepatitis, that are considered to be active. But now Hepatitis C can be cured and medicine for Hepatitis B started before transplant.
- Cigarette use or an alcohol or drug use disorder, or other risky lifestyle habits.
- A lack of a good support system.
People who receive a liver transplant may reject the new liver. This means that their immune system sees the new liver as a foreign substance and tries to destroy it. To avoid rejection, almost all transplant recipients must take medicines that suppress their immune response for the rest of their lives. This is called immunosuppressive medications. Although the immunosuppressive medication helps prevent organ rejection, it also puts people at a higher risk for infection and cancer.
If you take immunosuppressive medicine, you need to be regularly screened for cancer. The medicines may also cause high blood pressure and high cholesterol, and increase the risks for diabetes.
A successful transplant requires close follow-up with your provider and your transplant team. You must always take your medicine as directed.
Liver transplantation risks
Liver transplantation carries significant risks, both during and after the surgery, including complications from the procedure itself, rejection of the new liver, infections, and long-term side effects from immunosuppressant medications. Understanding these risks is crucial for patients considering or undergoing a liver transplant.
Risks of any anesthesia include:
- Problems breathing or with blood pressure
- Reactions to medicines
Risks of any surgery include:
- Bleeding
- Heart attack or stroke
- Infection
Liver transplant surgery and management after surgery carry major risks. There is an increased risk for infection because you must take medicines that suppress the immune system to prevent transplant rejection. Signs of infection include:
- Diarrhea
- Drainage from your surgical wound
- Fever
- Jaundice
- Redness
- Swelling
- Tenderness.
Before the liver transplantation procedure
Your doctor will refer you to a transplant center. The transplant team will want to make sure that you are a good candidate for a liver transplant. You will make a few visits over several weeks or months. You will need to have blood drawn and imaging tests done.
If you are the person getting the new liver, the following tests will be done before the procedure:
- Tissue and blood typing to make sure your body will not reject the donated liver
- Blood tests or skin tests to check for infection
- Heart tests such as an electrocardiogram (ECG), echocardiogram, or cardiac catheterization
- Tests to look for early cancer
- Tests to look at your liver, gallbladder, pancreas, small intestine, and the blood vessels around the liver
- Colonoscopy, depending on your age
You may choose to look at one or more transplant centers to determine which is best for you.
- Ask the center how many transplants they perform every year, and their survival rates. Compare these numbers to those of other transplant centers.
- Ask what support groups they have available, and what travel and housing arrangements they offer.
- Ask what is the average waiting time for a liver transplant.
If your liver transplant team thinks you are a good candidate for a liver transplant, you will be put on a national waiting list. Your place on the waiting list is based on a number of factors. Key factors include the type of liver problems you have, how severe your disease is, and the likelihood that a transplant will be successful. The amount of time you spend on a waiting list is most often not a factor in how soon you get a liver, with the possible exception of children.
While you are waiting for a liver, follow these steps:
- Follow any diet your liver transplant team recommends.
- Do not drink alcohol or use drugs.
- Do not smoke.
- Keep your weight in the appropriate range. Follow the exercise program your doctor recommends.
- Take all medicines prescribed for you. Report changes in your medicines and any new or worsening medical problems to the transplant team.
- Follow-up with your regular doctor and transplant team at any appointments that have been made.
- Make sure your transplant team has your correct phone numbers, so they can contact you immediately if a liver becomes available. Make sure that, no matter where you are going, you can be contacted quickly and easily.
- Have everything ready ahead of time to go to the hospital.
After the liver transplantation procedure
If you received a donated liver, you will likely need to stay in the hospital for a week or longer. After that, you will need to be closely followed up by a doctor for the rest of your life. You will have regular blood tests after the transplant.
The recovery period is about 6 to 12 months. Your transplant team may ask you to stay close to the hospital for the first 3 months. You will need to have regular check-ups, with blood tests and x-rays for many years.
What can I do to help prevent further liver damage?
To help prevent further liver damage, you can do the following:
- Carefully follow your doctor’s instructions, and take your medicines and dietary supplements as directed.
- Quit smoking.
- Do not drink any alcohol or use illegal drugs.
- Have regular checkups, as recommended by your doctor.
- Talk with your doctor before taking
- prescription medicines
- over-the-counter medicines
- dietary supplements
- complementary and alternative medicines
- Try to keep a healthy body weight.
Home remedies
You may feel better if you take good care of your overall health. Here are some things you can do to improve some primary biliary cirrhosis symptoms and, possibly, help prevent certain complications:
- Choose reduced-sodium foods. Opt for low-sodium foods or naturally sodium-free foods, since sodium contributes to tissue swelling and to the buildup of fluid in your abdominal cavity (ascites).
- Never eat oysters or other raw shellfish. Such seafood can carry infection-causing bacteria, which can be dangerous for people with liver disease.
- Exercise most days of the week. Exercise may reduce your risk of bone loss.
- Avoid alcohol. Your liver processes the alcohol you drink, and the added stress can cause liver damage. Generally, people with primary biliary cirrhosis should abstain from alcohol.
- Check with your doctor before starting new medications or dietary supplements. Because your liver isn’t working normally, you’ll likely be more sensitive to the effects of over-the-counter and prescription medications, as well as some dietary supplements, so check with your doctor before taking anything new.
Coping and support
Living with a chronic liver disease with no cure can be frustrating. Fatigue alone can have a profound impact on your quality of life. Each person finds ways to cope with the stress of a chronic disease. In time, you’ll find what works for you. Here are some ways to get started:
- Learn about your condition. The more you understand about primary biliary cirrhosis, the more active you can be in your own care. In addition to talking with your doctor, look for information at your local library and on websites affiliated with reputable organizations such as the American Liver Foundation.
- Take time for yourself. Eating well, exercising and getting enough rest can help you feel better. Try to plan ahead for times when you may need more rest.
- Get help. If friends or family want to help, let them. Primary biliary cirrhosis can be exhausting, so accept the help if someone wants to do your grocery shopping, wash a load of laundry or cook your dinner. Tell those who offer to help what you need.
- Seek support. Strong relationships can help you maintain a positive attitude. If friends or family have a hard time understanding your illness, you may find that a support group can be helpful.
Primary biliary cirrhosis diet
You should eat a well-balanced and nutritious diet. Good nutrition is important in all stages of primary biliary cirrhosis to help your liver work properly and manage complications. When primary biliary cirrhosis leads to cirrhosis, you may develop malnutrition because cirrhosis can cause:
- loss of appetite, which will cause you to eat less
- changes in your metabolism
- reduced absorption of nutrients
Your doctor can recommend a healthy eating plan that is well balanced and provides enough calories and nutrients. If you have vitamin deficiencies, your doctor may recommend foods that are high in vitamins A, D, E, and K. Your doctor may recommend that you eat foods high in calcium and vitamin D to help prevent osteoporosis.
What foods should I avoid eating if I have primary biliary cirrhosis?
You should avoid eating raw or undercooked shellfish such as oysters, fish, meat, and unpasteurized milk. Bacteria or viruses from these foods may cause severe infections in people with liver disease. Your doctor may recommend that you make healthy food choices and avoid high-salt foods and foods that are high in fat and carbohydrates, especially those with added sugars.
If you have primary biliary cirrhosis, your doctor will recommend that you quit smoking and stop drinking alcohol or, at least, limit your intake to no more than one or two drinks per week. If you have primary biliary cholangitis and cirrhosis, you should completely stop drinking alcohol.
Primary biliary cirrhosis prognosis
The most reliable indicators of a patient’s prognosis (outlook) from primary biliary cirrhosis are the rise in serum bilirubin level and the Mayo risk score. Based on bilirubin levels, the prognosis can be determined as follows 11:
- Bilirubin constantly above 2 – mean survival is 4 years
- Bilirubin constantly above 6 – mean survival is 2 years
- Bilirubin constantly above 10 – mean survival is 1.4 years
A study determined that fatigue levels may also be a prognostic indicator of primary biliary cirrhosis 63.
Primary biliary cirrhosis life expectancy
Primary biliary cirrhosis outcome can vary. If primary biliary cirrhosis is not treated, most people will die without a liver transplant. About one quarter of people who have had primary biliary cirrhosis for 10 years will have liver failure. Doctors can now use a statistical model to predict the best time to do the liver transplant. Other diseases, such as hypothyroidism and anemia, can also develop.
In patients with low serum albumin, survival can range from 3 to 6 years 11. However, if the bilirubin levels are consistently high, survival is significantly reduced, with an average survival rate of 1.7 years 11. Almost all patients with primary biliary cirrhosis develop moderate-to-severe fatigue, which persists even after a liver transplant. Universally, once patients develop symptoms, the outcome is grim 64.
- Komori A, Kugiyama Y. Hard-to-treat autoimmune hepatitis and primary biliary cholangitis: The dawn of a new era of pharmacological treatment. Clin Mol Hepatol. 2025 Jan;31(1):90-104. doi: 10.3350/cmh.2024.0821[↩]
- Jallouli I, Doulberis M, Kountouras J. Primary biliary cholangitis: a summary of pathogenesis and therapies. Ann Gastroenterol. 2025 Mar-Apr;38(2):121-132. doi: 10.20524/aog.2025.0953[↩]
- Warsop Z, Anand N, Al Maliki H, De Souza S, Kamyab A, Al Hadad A, Alrubaiy L. Up-to-Date Snapshot of Current and Emerging Medical Therapies in Primary Biliary Cholangitis. J Pers Med. 2024 Nov 30;14(12):1133. doi: 10.3390/jpm14121133[↩][↩][↩][↩]
- Sylvia D, Tomas K, Marian M, Martin J, Dagmar S, Peter J. The treatment of primary biliary cholangitis: from shadow to light. Therap Adv Gastroenterol. 2024 Jul 29;17:17562848241265782. doi: 10.1177/17562848241265782. Erratum in: Therap Adv Gastroenterol. 2024 Aug 31;17:17562848241277276. doi: 10.1177/17562848241277276[↩][↩]
- Yang Y, He X, Rojas M, Leung PSC, Gao L. Mechanism-based target therapy in primary biliary cholangitis: opportunities before liver cirrhosis? Front Immunol. 2023 May 30;14:1184252. doi: 10.3389/fimmu.2023.1184252[↩]
- Trivella J, John BV, Levy C. Primary biliary cholangitis: Epidemiology, prognosis, and treatment. Hepatol Commun. 2023 Jun 2;7(6):e0179. doi: 10.1097/HC9.0000000000000179[↩]
- Khor SS, Ueno K, Nishida N, Kawashima M, Kawai Y, Aiba Y, Hitomi Y, Nagasaki M, Nakamura M, Tokunaga K. Novel HLA allele associations with susceptibility, staging, symptomatic state, autoimmune hepatitis and hepatocellular carcinoma events for primary biliary cholangitis in the Japanese population. Front Immunol. 2023 May 31;14:1151502. doi: 10.3389/fimmu.2023.1151502[↩]
- Martini F, Balducci D, Mancinelli M, Buzzanca V, Fracchia E, Tarantino G, Benedetti A, Marzioni M, Maroni L. Risk Stratification in Primary Biliary Cholangitis. J Clin Med. 2023 Sep 1;12(17):5713. doi: 10.3390/jcm12175713[↩]
- Park JW, Kim JH, Kim SE, Jung JH, Jang MK, Park SH, Lee MS, Kim HS, Suk KT, Kim DJ. Primary Biliary Cholangitis and Primary Sclerosing Cholangitis: Current Knowledge of Pathogenesis and Therapeutics. Biomedicines. 2022 May 31;10(6):1288. doi: 10.3390/biomedicines10061288[↩]
- Primary Biliary Cholangitis (Primary Biliary Cirrhosis). https://www.niddk.nih.gov/health-information/liver-disease/primary-biliary-cholangitis/definition-facts[↩]
- Pandit S, Samant H. Primary Biliary Cholangitis. [Updated 2023 Feb 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459209[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Gunaydin M, Bozkurter Cil AT. Progressive familial intrahepatic cholestasis: diagnosis, management, and treatment. Hepat Med. 2018 Sep 10;10:95-104. doi: 10.2147/HMER.S137209[↩]
- Adam L, Zoldan K, Hofmann M, Schultheiss M, Bettinger D, Neumann-Haefelin C, Thimme R, Böettler T. Follicular T Helper Cell Signatures in Primary Biliary Cholangitis and Primary Sclerosing Cholangitis. Hepatol Commun. 2018 Sep 7;2(9):1051-1063. doi: 10.1002/hep4.1226[↩]
- Tam PKH, Yiu RS, Lendahl U, Andersson ER. Cholangiopathies – Towards a molecular understanding. EBioMedicine. 2018 Sep;35:381-393. doi: 10.1016/j.ebiom.2018.08.024. Epub 2018 Sep 17. Erratum in: EBioMedicine. 2018 Oct;36:564. doi: 10.1016/j.ebiom.2018.09.044[↩][↩]
- Isayama H, Tazuma S, Kokudo N, et al. PSC guideline committee Members: Ministry of Health, Labour and Welfare (Japan) Research Project, The Intractable Hepatobiliary Disease Study Group. Clinical guidelines for primary sclerosing cholangitis 2017. J Gastroenterol. 2018 Sep;53(9):1006-1034. doi: 10.1007/s00535-018-1484-9 Erratum in: J Gastroenterol. 2022 Jun;57(6):453-454. doi: 10.1007/s00535-022-01867-7[↩][↩]
- Primary Biliary Cholangitis (Primary Biliary Cirrhosis). https://www.niddk.nih.gov/health-information/liver-disease/primary-biliary-cholangitis/all-content[↩][↩]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J Hepatol. July, 2017; 67(1):145-172. https://www.ncbi.nlm.nih.gov/pubmed/28427765[↩]
- Lu M, Zhou Y, Haller IV, et al. Increasing prevalence of primary biliary cholangitis and reduced mortality with treatment. Clinical Gastroenterology and Hepatology. 2018;16(8):1342–1350.e1. doi: 10.1016/j.cgh.2017.12.033[↩]
- Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology. January, 2019; 69(1):394-419. https://www.aasld.org/sites/default/files/guideline_documents/PracticeGuidelines-PBC-November2018.pdf[↩][↩][↩][↩][↩][↩][↩]
- McGee EE, Castro FA, Engels EA, Freedman ND, Pfeiffer RM, Nogueira L, Stolzenberg-Solomon R, McGlynn KA, Hemminki K, Koshiol J. Associations between autoimmune conditions and hepatobiliary cancer risk among elderly US adults. Int J Cancer. 2019 Feb 15;144(4):707-717. doi: 10.1002/ijc.31835[↩][↩]
- Parés A, Albillos A, Andrade RJ, Berenguer M, Crespo J, Romero-Gómez M, Vergara M, Vendrell B, Gil A. Primary biliary cholangitis in Spain. Results of a Delphi study of epidemiology, diagnosis, follow-up and treatment. Rev Esp Enferm Dig. 2018 Oct;110(10):641-649. doi: 10.17235/reed.2018.5665/2018[↩]
- Ebrahimi H, Naderian M, Sohrabpour AA. New Concepts on Reversibility and Targeting of Liver Fibrosis; A Review Article. Middle East J Dig Dis. 2018 Jul;10(3):133-148. doi: 10.15171/mejdd.2018.103[↩]
- Tanaka A, Leung PSC, Gershwin ME. Pathogen infections and primary biliary cholangitis. Clin Exp Immunol. 2019 Jan;195(1):25-34. doi: 10.1111/cei.13198[↩]
- Juran B.D., Lazaridis K.N. Environmental factors in primary biliary cirrhosis. Semin. Liver Dis. 2014;34:265–272. doi: 10.1055/s-0034-1383726[↩]
- Webb G.J., Hirschfield G.M. Using GWAS to identify genetic predisposition in hepatic autoimmunity. J. Autoimmun. 2016;66:25–39. doi: 10.1016/j.jaut.2015.08.016[↩]
- Bianchi I., Carbone M., Lleo A., Invernizzi P. Genetics and epigenetics of primary biliary cirrhosis. Semin. Liver Dis. 2014;34:255–264. doi: 10.1055/s-0034-1383725[↩]
- Selmi C., Mayo M.J., Bach N., Ishibashi H., Invernizzi P., Gish R.G., Gordon S.C., Wright H.I., Zweiban B., Podda M., et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: Genetics, epigenetics, and environment. Gastroenterology. 2004;127:485–492. doi: 10.1053/j.gastro.2004.05.005[↩]
- Lazaridis K.N., Juran B.D., Boe G.M., Slusser J.P., de Andrade M., Homburger H.A., Ghosh K., Dickson E.R., Lindor K.D., Petersen G.M. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrhosis. Hepatology. 2007;46:785–792. doi: 10.1002/hep.21749[↩]
- Gulamhusein A.F., Lazaridis K.N. Primary biliary cholangitis, DNA, and beyond: The Relative contribution of genes. Hepatology. 2018;68:19–21. doi: 10.1002/hep.29783[↩]
- Gershwin M.E., Selmi C., Worman H.J., Gold E.B., Watnik M., Utts J., Lindor K.D., Kaplan M.M., Vierling J.M., USA PBC Epidemiology Group Risk factors and comorbidities in primary biliary cirrhosis: A controlled interview-based study of 1032 patients. Hepatology. 2005;42:1194–1202. doi: 10.1002/hep.20907[↩]
- Prince M.I., Ducker S.J., James O.F. Case-control studies of risk factors for primary biliary cirrhosis in two United Kingdom populations. Gut. 2010;59:508–512. doi: 10.1136/gut.2009.184218[↩]
- Liang Y., Yang Z., Zhong R. Smoking, family history and urinary tract infection are associated with primary biliary cirrhosis: A meta-analysis. Hepatol. Res. 2011;41:572–578. doi: 10.1111/j.1872-034X.2011.00806.x[↩]
- Lu M, Li J, Haller IV, et al. Factors associated with prevalence and treatment of primary biliary cholangitis in united states health systems. Clinical Gastroenterology and Hepatology. 2018;16(8):1333–1341.e6. doi: 10.1016/j.cgh.2017.10.018[↩]
- Scheuer P. Primary biliary cirrhosis. Proc R Soc Med. 1967 Dec;60(12):1257-60. https://pmc.ncbi.nlm.nih.gov/articles/instance/1901478/pdf/procrsmed00161-0027.pdf[↩]
- Namisaki T, Moriya K, Kitade M, et al. Clinical significance of the Scheuer histological staging system for primary biliary cholangitis in Japanese patients. Eur J Gastroenterol Hepatol. 2017 Jan;29(1):23-30. doi: 10.1097/MEG.0000000000000765[↩]
- Ehrlich L, Scrushy M, Meng F, Lairmore TC, Alpini G, Glaser S. Biliary epithelium: A neuroendocrine compartment in cholestatic liver disease. Clin Res Hepatol Gastroenterol. 2018 Sep;42(4):296-305. doi: 10.1016/j.clinre.2018.03.009[↩]
- Rodríguez Lugo DA, Coronado Tovar JJ, Solano Villamarin GA, Otero Regino W. Colangitis biliar primaria. Parte 1. Actualización: generalidades,epidemiología, factores involucrados, fisiopatología y manifestaciones clínicas [Primary biliary cholangitis. Part 1. State of the art, epidemiology, physiopathology and clinical manifestations]. Rev Gastroenterol Peru. 2017 Oct-Dec;37(4):357-364. Spanish.[↩][↩]
- Carrion AF, Rosen JD, Levy C. Understanding and treating pruritus in primary biliary cholangitis. Clinical Liver Disease. 2018;22(3):517–532. doi: 10.1016/j.cld.2018.03.005[↩]
- Poupon R. Clinical manifestations, diagnosis, and prognosis of primary biliary cholangitis (primary biliary cirrhosis). http://www.uptodate.com/contents/clinical-manifestations-diagnosis-and-prognosis-of-primary-biliary-cholangitis-primary-biliary-cirrhosis[↩]
- Hirschfield G.M., Dyson J.K., Alexander G.J.M., Chapman M.H., Collier J., Hübscher S., Patanwala I., Pereira S.P., Thain C., Thorburn D., et al. The British Society of Gastroenterology/UK-PBC primary biliary cholangitis treatment and management guidelines. Gut. 2018;67:1568–1594. doi: 10.1136/gutjnl-2017-315259[↩][↩][↩]
- European Association for the Study of the Liver EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017;67:145–172. doi: 10.1016/j.jhep.2017.03.022[↩][↩][↩]
- Younossi Z.M., Bernstein D., Shiffman M.L., Kwo P., Kim W.R., Kowdley K.V., Jacobson I.M. Diagnosis and Management of Primary Biliary Cholangitis. Am. J. Gastroenterol. 2019;114:48–63. doi: 10.1038/s41395-018-0390-3[↩][↩][↩]
- Prince MI, Chetwynd A, Craig WL, Metcalf JV, James OF. Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut. 2004 Jun;53(6):865-70. doi: 10.1136/gut.2003.023937. Erratum in: Gut. 2004 Aug;53(8):1216.[↩][↩]
- Guañabens N., Cerdá D., Monegal A., Pons F., Caballería L., Peris P., Parés A. Low bone mass and severity of cholestasis affect fracture risk in patients with primary biliary cirrhosis. Gastroenterology. 2010;138:2348–2356. doi: 10.1053/j.gastro.2010.02.016[↩]
- Menon K.V., Angulo P., Weston S., Dickson E.R., Lindor K.D. Bone disease in primary biliary cirrhosis: Independent indicators and rate of progression. J. Hepatol. 2001;35:316–323. doi: 10.1016/S0168-8278(01)00144-1[↩]
- Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69(1):394–419. doi: 10.1002/hep.30145[↩]
- Selmi C., Gershwin M.E. Chronic Autoimmune Epithelitis in Sjogren’s Syndrome and Primary Biliary Cholangitis: A Comprehensive Review. Rheumatol. Ther. 2017;4:263–279. doi: 10.1007/s40744-017-0074-2[↩]
- Muratori P., Muratori L., Gershwin M.E., Czaja A.J., Pappas G., MacCariello S., Granito A., Cassani F., Loria P., Lenzi M., et al. ‘True’ antimitochondrial antibody-negative primary biliary cirrhosis, low sensitivity of the routine assays, or both? Clin. Exp. Immunol. 2004;135:154–158. doi: 10.1111/j.1365-2249.2004.02332.x[↩]
- Dahlqvist G., Gaouar F., Carrat F., Meurisse S., Chazouilleres O., Poupon R., Johanet C., Corpechot C., French Network of Immunology Laboratories Large-scale characterization study of patients with antimitochondrial antibodies but nonestablished primary biliary cholangitis. Hepatology. 2017;65:152–163. doi: 10.1002/hep.28859[↩]
- Yamagiwa S., Kamimura H., Takamura M., Aoyagi Y. Autoantibodies in primary biliary cirrhosis: Recent progress in research on the pathogenetic and clinical significance. World J. Gastroenterol. 2014;20:2606–2612. doi: 10.3748/wjg.v20.i10.2606[↩]
- Joshi S., Cauch-Dudek K., Heathcote E.J., Lindor K., Jorgensen R., Klein R. Antimitochondrial antibody profiles: Are they valid prognostic indicators in primary biliary cirrhosis? Am. J. Gastroenterol. 2002;97:999–1002. doi: 10.1111/j.1572-0241.2002.05620.x[↩]
- Tang L., Zhong R., He X., Wang W., Liu J., Zhu Y., Li Y., Hou J. Evidence for the association between IgG-antimitochondrial antibody and biochemical response to ursodeoxycholic acid treatment in primary biliary cholangitis. J. Gastroenterol. Hepatol. 2017;32:659–666. doi: 10.1111/jgh.13534[↩]
- Zandanell S., Strasser M., Feldman A., Strebinger G., Aigner G., Niederseer D., Laimer M., Mussnig B., Paulweber B., Datz C., et al. Similar clinical outcome of AMA immunoblot-M2-negative compared to immunoblot-positive subjects over six years of follow-up. Postgrad. Med. 2021;133:291–298. doi: 10.1080/00325481.2021.1885945[↩]
- Granito A., Yang W.H., Muratori L., Lim M.J., Nakajima A., Ferri S., Pappas G., Quarneti C., Bianchi F.B., Bloch D.B., et al. PML nuclear body component Sp140 is a novel autoantigen in primary biliary cirrhosis. Am. J. Gastroenterol. 2010;105:125–131. doi: 10.1038/ajg.2009.596[↩]
- Khanna A, Jopson L, Howel D, Bryant A, Blamire A, Newton JL, Jones DE. Rituximab Is Ineffective for Treatment of Fatigue in Primary Biliary Cholangitis: A Phase 2 Randomized Controlled Trial. Hepatology. 2019 Nov;70(5):1646-1657. doi: 10.1002/hep.30099. Epub 2018 Sep 22. Erratum in: Hepatology. 2020 Jan;71(1):404. doi: 10.1002/hep.31083[↩]
- Örnolfsson K.T., Lund S.H., Olafsson S., Bergmann O.M., Björnsson E.S. Biochemical response to ursodeoxycholic acid among PBC patients: A nationwide population-based study. Scand. J. Gastroenterol. 2019;54:609–616. doi: 10.1080/00365521.2019.1606931[↩]
- European Association for the Study of the Liver EASL Clinical Practice Guidelines: The Diagnosis and Management of Patients with Primary Biliary Cholangitis. J. Hepatol. 2017;67:145–172. doi: 10.1016/j.jhep.2017.03.022[↩][↩]
- Lazaridis K.N., Gores G.J., Lindor K.D. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J. Hepatol. 2001;35:134–146. doi: 10.1016/S0168-8278(01)00092-7[↩]
- You H., Ma X., Efe C., Wang G., Jeong S.H., Abe K., Duan W., Chen S., Kong Y., Zhang D., et al. APASL clinical practice guidance: The diagnosis and management of patients with primary biliary cholangitis. Hepatol. Int. 2022;16:1–23. doi: 10.1007/s12072-021-10276-6[↩]
- Hofmann A.F. Pharmacology of ursodeoxycholic acid, an enterohepatic drug. Scand. J. Gastroenterol. 1994;29((Suppl. S204[↩]
- Lammers W.J., van Buuren H.R., Hirschfield G.M., Janssen H.L., Invernizzi P., Mason A.L., Ponsioen C.Y., Floreani A., Corpechot C., Mayo M.J., et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: An international follow-up study. Gastroenterology. 2014;147:1338–1349.e5. doi: 10.1053/j.gastro.2014.08.029[↩]
- Sepe V., Distrutti E., Fiorucci S., Zampella A. Farnesoid X receptor modulators 2014–present: A patent review. Expert Opin. Ther. Pat. 2018;28:351–364. doi: 10.1080/13543776.2018.1459569[↩]
- Björnsson E, Kalaitzakis E, Neuhauser M, Enders F, Maetzel H, Chapman RW, Talwalkar J, Lindor K, Jorgensen R. Fatigue measurements in patients with primary biliary cirrhosis and the risk of mortality during follow-up. Liver Int. 2010 Feb;30(2):251-8. doi: 10.1111/j.1478-3231.2009.02160.x[↩]
- Vieira Barbosa J, Vionnet J, Sciarra A, Sempoux C, Aubert V, Moradpour D, Fraga Christinet M. Cholangite biliaire primitive : mise à jour [Primary biliary cholangitis : an update]. Rev Med Suisse. 2018 Aug 29;14(616):1489-1494. French.[↩]





{kind=link}