Contents
What is anca vasculitis
Antineutrophil cytoplasmic autoantibody (ANCA) causes vascular injury that leads to small-vessel vasculitis – also called ANCA vasculitis. ANCA (antineutrophil cytoplasmic autoantibody) vasculitis is characterized by microvascular inflammation, tissue necrosis, and circulating antineutrophil cytoplasmic autoantibodies (ANCAs) 1.
Antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis is the most common primary systemic small-vessel vasculitis to occur in adults. Although the cause is not always known, the incidence of ANCA vasculitis is increasing, and the diagnosis and management of patients may be challenging because of its relative infrequency, changing nomenclature, and variability of clinical expression.
Three systemic autoimmune small vessel vasculitis syndromes are associated with antineutrophil cytoplasmic autoantibodies (ANCAs); collectively, they’re called ANCA-associated vasculitis. ANCA associated vasculitis include:
- Microscopic polyangiitis (MPA)
- Granulomatosis with polyangiitis (GPA), formerly known as Wegener’s granulomatosis
- Eosinophilic granulomatosis with polyangiitis (EGPA), formerly known as Churg-Strauss syndrome
ANCA–associated small-vessel vasculitis includes microscopic polyangiitis, Wegener’s granulomatosis, Churg-Strauss syndrome, and drug-induced vasculitis 2. Better definition criteria and advancement in the technologies make these diagnoses increasingly common. Features that may aid in defining the specific type of vasculitic disorder include the type of organ involvement, presence and type of ANCA (myeloperoxidase–ANCA or proteinase 3–ANCA), presence of serum cryoglobulins, and the presence of evidence for granulomatous inflammation. Family physicians should be familiar with this group of vasculitic disorders to reach a prompt diagnosis and initiate treatment to prevent end-organ damage. Treatment usually includes corticosteroid and immunosuppressive therapy.
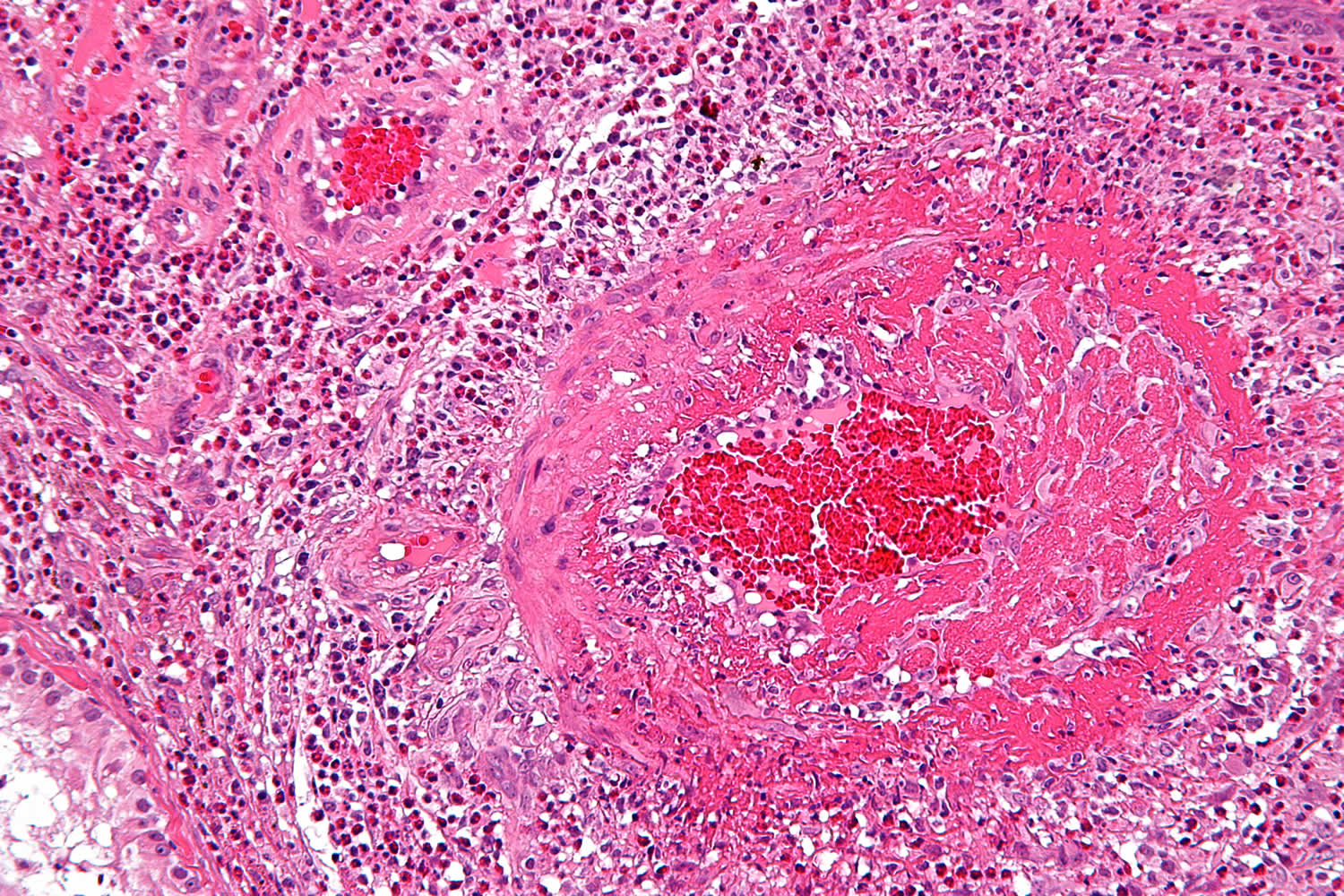
Figure 1. Clinical Manifestations of ANCA-Associated Small-Vessel Vasculitis
Note: GPA = Granulomatosis with polyangiitis (Wegener’s granulomatosis); MPA = Microscopic polyangiitis; EGPA = Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome)
The respiratory system is commonly involved in all three syndromes, which share certain histopathologic and clinical features. Other features are unique and disease-defining.
Diffuse alveolar hemorrhage is a potentially life-threatening complication of each of these syndromes. Patients with diffuse alveolar hemorrhage often require ICU care. Caused by pulmonary capillaritis, diffuse alveolar hemorrhage affects 20 to 30 percent of patients with microscopic polyangiitis and granulomatosis with polyangiitis, but less than 5 percent of patients with eosinophilic granulomatosis with polyangiitis. Other clinical manifestations caused by necrotizing small vessel vasculitis and capillaritis, such as glomerulonephritis, palpable purpura, scleritis, or sensory and motor mononeuropathies, also can occur in all three syndromes.
Granulomatous inflammation, which predominantly affects the respiratory tract, causes characteristic clinical features setting granulomatosis with polyangiitis and eosinophilic granulomatosis with polyangiitis apart from microscopic polyangiitis. The granulomatous inflammation is necrotizing and neutrophilic in granulomatosis with polyangiitis, but eosinophilic in eosinophilic granulomatosis with polyangiitis. The necrotizing granulomatous inflammation of granulomatosis with polyangiitis can cause pulmonary nodules or mass lesions and affect the large airways leading to subglottic and endobronchial stenoses. Asthma and peripheral blood eosinophilia, the defining characteristics of eosinophilic granulomatosis with polyangiitis, are not features of granulomatosis with polyangiitis or microscopic polyangiitis.
The type of ANCA also seems to affect the disease phenotype of ANCA-associated vasculitis. Two types of ANCAs are of clinical significance in patients with vasculitis:
- ANCAs causing a cytoplasmic immunofluorescence pattern (C-ANCAs) on ethanol-fixed neutrophils that react with proteinase 3 (PR3-ANCAs)
- ANCAs causing a perinuclear immunofluorescence pattern (P-ANCAs) on ethanol-fixed neutrophils that react with myeloperoxidase (MPO-ANCAs)
PR3-ANCAs occur in the vast majority of patients with GPA, while MPO-ANCAs occur far less frequently. In contrast, MPO-ANCAs are the predominant type of ANCAs in patients with both MPA and EGPA.
What are ANCA?
ANCA are specific antibodies for antigens in cytoplasmic granules of neutrophils and monocyte lysosomes, first reported in 1982 3. These antibodies can be detected with indirect immunofluorescence microscopy. Two major patterns of staining are present: cytoplasmic ANCA (c–ANCA) and peri-nuclear ANCA (p–ANCA). Specific immunochemical assays demonstrate that c–ANCA (cytoplasmic ANCA) is mainly antibodies to proteinase 3, and p–ANCA (peri-nuclear ANCA) is antibodies to myeloperoxidase. Using antigen-specific immunochemical assay to characterize ANCA (rather than the pattern of immunofluorescence microscopy) is more specific and more clinically relevant; therefore, the terms proteinase 3-ANCA (PR3-ANCA) and myeloperoxidase–ANCA (MPO–ANCA) are now in use 3.
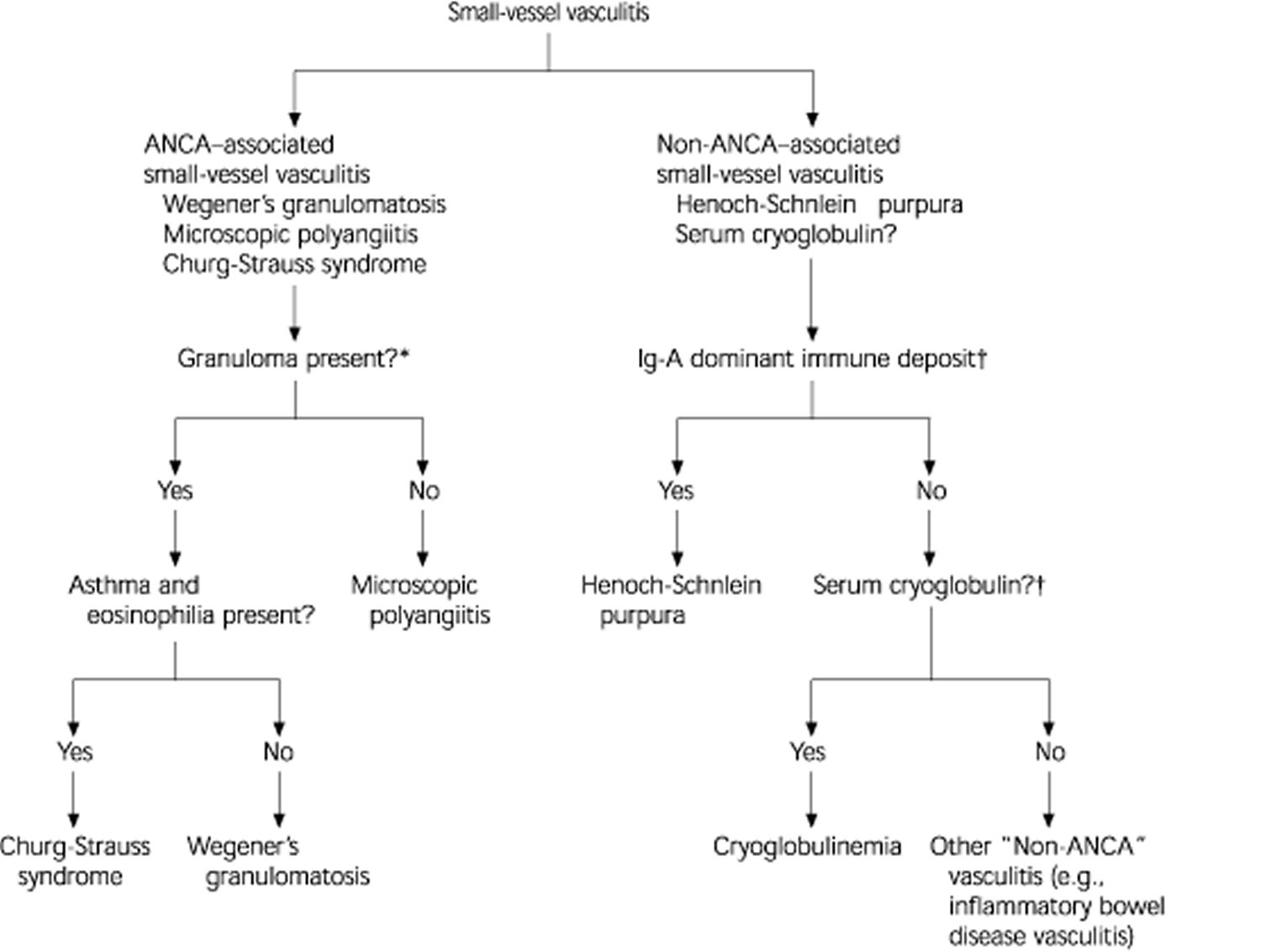
Figure 2 illustrates the differential diagnosis of ANCA and non–ANCA-associated vasculitis. About 10 percent of patients with microscopic polyangiitis (the most common type of ANCA–associated small vessel vasculitis) and Wegener’s granulomatosis have negative assays for ANCA; however, this finding does not completely rule out these diseases and ANCA titers do not always correlate with disease activity.3 On the other hand, a positive ANCA assay result is not solely diagnostic of ANCA–associated vasculitis.
Figure 2. Differential Diagnosis of Small-Vessel Vasculitis
Note:
Differential diagnosis of ANCA- and non-ANCA-associated small-vessel vasculitis. (ANCA = anti-neutrophilic cytoplasmic antibodies; Ig-A = immunoglobulin A)
*—In ANCA-associated vasculitis, the absence ofgranulomas differentiates microscopic polyangiitis from other types. Whereas in the presence ofgranuloma, the symptoms, the organ involved, and type of ANCA differentiate Granulomatosis with polyangiitis (Wegener’s granulomatosis) from Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome).
†—Testing for ANCA, vascular immunoglobulin A deposits, and serum cryoglobulins facilitates the diagnostic characterization of small vessel vasculitis.
[Source 4]ANCA associated vasculitis causes
Significant progress has been made over the last two decades in understanding the pathogenesis of ANCA associated vasculitis.
Clinical and experimental evidence supports the concept that a genetic predisposition for autoimmunity, epigenetic factors and environmental triggers are necessary for the loss of tolerance and development of an inflammatory milieu that supports the production of ANCAs. In the context of an inflammatory milieu, ANCAs can cause specific tissue inflammation and vascular injury by a variety of different mechanisms that involve direct interactions with the respective ANCAs’ target antigens PR3 or MPO.
Granulomatosis with polyangiitis (Wegener’s granulomatosis) and microscopic polyangiitis are idiopathic systemic vasculitides strongly associated with antineutrophil cytoplasmic autoantibodies (ANCA). In Granulomatosis with polyangiitis, ANCA are mostly directed against proteinase 3 (PR3), whereas in microscopic polyangiitis ANCA are directed against myeloperoxidase; increases in levels of these autoantibodies precede or coincide with clinical relapses in many cases. In vitro, ANCA can further activate primed neutrophils to release reactive oxygen species and lytic enzymes, and, in conjunction with neutrophils, can damage and lyse endothelial cells. Patients with Wegener’s granulomatosis or microscopic polyangiitis have an increased percentage of neutrophils that constitutively express PR3 on their membrane. These neutrophils can be stimulated by ANCA, without priming. In vivo, transfer of splenocytes from myeloperoxidase-deficient mice immunized with mouse myeloperoxidase into wild-type mice resulted in pauci-immune systemic vasculitis. A similar experiment in PR3-deficient mice did not cause significant vasculitic lesions. Together, clinical, in vitro and in vivo experimental data support a pathogenic role for ANCA in Wegener’s granulomatosis and microscopic polyangiitis, although this role is more evident for myeloperoxidase-specific ANCA than for PR3-specific ANCA. Several controlled trials have led to an evidence-based approach for the treatment of ANCA-associated vasculitis, and further studies, based on new insights into pathogenesis, are in progress.
Clinical and experimental evidence indicates that antineutrophil cytoplasmic autoantibodies (ANCAs) cause vascular injury by activating neutrophils 5. Neutrophils are the primary mediators of inflammation in ANCA vasculitis, because depletion of neutrophils protects against vascular lesions 6. Activated neutrophils have increased adherence and transmigration to the vascular endothelium, where they produce reactive oxygen species and release granule constituents, including proteolytic enzymes 7. These oxygen radicals and proteases activate the alternative complement pathway, in an animal and in vitro model, which amplifies neutrophil mediated inflammation 8.
A genome-wide association study found major histocompatibility complex (MHC) and non-MHC associations with ANCA associated vasculitis and genetic distinctions between granulomatosis with polyangiitis and MPO, and even more clearly between PR3- and MPO-ANCA-associated diseases, providing support for the concept that they are genetically distinct autoimmune disorders. The documented higher relapse rate of patients with PR3-ANCAs compared with MPO-ANCAs may have a genetic basis.
A large body of experimental work supports that B lymphocytes are essential for the development of ANCAs and disease activity, whereas T lymphocyte abnormalities seem to persist, particularly in patients with granulomatosis with polyangiitis, even during remission. The presence of ANCAs alone does not inevitably cause disease, but ANCAs seem necessary for the development of disease manifestations caused by capillaritis, such as alveolar hemorrhage, glomerulonephritis, scleritis or mononeuritis multiplex.
For all of these reasons, interventions aimed at B lymphocytes, T lymphocytes and ANCA have made inroads into the therapeutic arsenal for ANCA associated vasculitis.
ANCA vasculitis symptoms
The most common cutaneous lesion is palpable purpura—a slightly raised, non blanching eruption that usually begins in the lower extremities 2. Occasionally, the rash is vesicular or slightly ulcerated. Urticaria can also be a manifestation of small-vessel vasculitis. Unlike nonvasculitic allergic urticaria, vasculitic urticaria lasts more than one day and may evolve into purpuric lesions. The presence of hypocomplementemia may indicate that the vasculitis is immune complex-mediated rather than ANCA-associated primary vasculitis 2.
Table 1 summarizes the potential clinical manifestations of ANCA-associated small-vessel vasculitis that is generally shared by most types of small-vessel vasculitis. Small-vessel vasculitis should be suspected in any patient who presents with a multisystem disease that is not caused by an infectious or malignant process (e.g., renal dysfunction, skin rashes, pulmonary manifestations, or neurologic manifestation) 9. Constitutional symptoms are common. The frequency and combination of various system involvements vary among individual disease entities.
Table 1. Clinical Manifestations of ANCA-Associated Vasculitis
| System | Manifestations |
|---|---|
Constitutional | Fever, weight loss, anorexia, general malaise |
Musculoskeletal | Myalgia, arthralgia |
Skin | Palpable purpura, urticaria |
Kidneys | Proteinuria, hematuria, renal insufficiency, renal failure, necrotizing glomerulonephritis |
Respiratory tract | Dyspnea, cough, hemoptysis; lung infiltrate, interstitial lung disease, pulmonary hemorrhage |
Nervous system | Peripheral neuropathy, especially mononeuritis |
Gastrointestinal tract | Fecal blood, elevated liver enzymes; diarrhea, nausea, vomiting, abdominal pain |
Renal involvement in vasculitis may progress to renal failure. Results of biopsy of the kidney commonly reveals glomerulonephritis. Focal necrosis, crescentic formation and the absence or paucity of immunoglobulin deposits characterize glomerulonephritis in patients with ANCA-associated vasculitis 10.
Lung involvement ranges from fleeting focal infiltrates or interstitial disease to massive pulmonary hemorrhagic alveolar capillaritis. The latter is the most life-threatening feature of small-vessel vasculitis 3.
It is important, however, to differentiate small-vessel vasculitis from other diseases that result in multisystem manifestations. Diseases with widespread embolization to different organs (e.g., atheroembolic disease, endocarditis, antiphospholipid syndrome, and atrial myxoma) can produce similar clinical presentations 11. Persons with sepsis can also present with multisystem involvement. It is also important to realize that small-vessel vasculitis may be secondary to infections or malignancy. Some viral, bacterial, and fungal infections may be complicated by vasculitis, which is predominantly a dermal vasculitis 11. The diagnosis is suggested by the clinical history. Malignancy, such as lymphomas, leukemia, myeloprolifera-tive, and myelodysplastic syndromes may be associated with vasculitis; however, solid tumors are less commonly associated with vasculitis 11. It must be emphasized that underlying infectious or malignant causes should be thoroughly evaluated before the diagnosis of primary vasculitis is made—even if the ANCA assay result is positive.
Laboratory assessment should include a complete blood cell count and routine chemistry profile, urinalysis, fecal occult blood test, and chest radiography 12. There may be normocytic anemia, thrombo-cytosis, elevated erythrocyte sedimentation rate, increased liver function, or evidence of renal involvement. ANCA serum levels should also be measured. Other laboratory tests that should be performed to exclude ANCA-associated small-vessel vasculitis include antinu-clear antibody, rheumatoid factor, cryoglobulins, complement, antibodies to hepatitis B and C, and human immunodeficiency virus (HIV) testing 12. Chest and sinus computed tomographic scans may also be performed, if appropriate. Angiography may reveal evidence of medium- or large-vessel vasculitis. Pathologic examination of the involved tissue (e.g., skin, nerve, lung, or kidney) may aid in documenting the type of small-vessel vasculitis. Biopsy should be obtained from symptomatic and accessible sites. Biopsies from asymptomatic sites have a low yield of positive results 12.
ANCA-associated vasculitis is the most common primary type of vasculitis in older adults, while Henoch-Schönlein purpura is most common in children 2.
Table 2. Clinical Features That Favor Diagnosis of a Specific Type of Vasculitic Syndrome
| Clinical features | Probable type of small-vessel vasculitis |
|---|---|
Pulmonary and renal symptoms | Granulomatosis with polyangiitis |
Microscopic polyangiitis | |
Pulmonary-dermal symptoms | Cryoglobulinemia |
Henoch-Schönlein purpura | |
Asthma and eosinophilia | Eosinophilic granulomatosis with polyangiitis |
Upper respiratory tract involvement (e.g., sinusitis and otitis media) | Granulomatosis with polyangiitis |
Granulomatosis with polyangiitis (Wegener’s granulomatosis)
Granulomatosis with polyangiitis commonly has the classic triad of involvement of the upper respiratory tract, lungs, and kidneys 11. Upper respiratory tract signs and symptoms include sinusitis, nasal ulcers, otitis media, or hearing loss. Upper respiratory tract signs and symptoms are seen in 70 percent of patients and pulmonary infiltrates or nodules that may cavitate develop in 85 percent of patients 13. Serum antiprotease 3–ANCA (c–ANCA) is positive in 75 to 90 percent, although 20 percent may have positive p–ANCA Open lung biopsy is the most definitive diagnostic test. Sinus biopsy is diagnostic in only 30 percent of cases because inflammatory findings are often nonspecific and renal biopsy is also relatively nonspecific 11. Wegener’s granulomatosis can affect patients at any age, with the peak incidence during the fourth decade of life and is slightly more common in men 13.
Microscopic Polyangiitis
Microscopic polyangiitis is the most common ANCA–associated small-vessel vasculitis, and is characterized by the presence of ANCA and few or no immune deposits in the involved vessels 14. The kidneys are the most commonly affected organs in 90 percent of patients who have this type of vasculitis 2. Patients present with variable combinations of renal manifestations, palpable purpura, abdominal pain, cough, and hemoptysis 15. Most patients have positive MPO–ANCA (p–ANCA), although PR3–ANCA (c–ANCA) may be also present in 40 percent of patients 11. The most common age of onset is 40 to 60 years and is more common in men 11.
Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome)
Eosinophilic granulomatosis with polyangiitis is a rare disease and has three phases: allergic rhinitis and asthma, eosinophilic infiltrative disease resembling pneumonia, and systemic small vessel vasculitis with granulomatous inflammation 16. The vasculitic phase usually develops within three years of the onset of asthma. Almost all patients have more than 10 percent eosinophils in the blood. Coronary arteritis and myocarditis are the principal causes of morbidity and mortality 16. The age of onset varies from 15 to 70 years and is more common in men 16.
Drug-Induced Vasculitis
Drug-induced vasculitis usually develops within seven to 21 days after a drug is started and may be confined to the skin 2. Skin lesions are identical to those seen in systemic small vessel vasculitis. Drugs cause approximately 10 percent of vasculitic skin lesions. Drugs that have been implicated include penicillin, aminopenicillins, sulfonamides, allopurinol, thiazides, quinolones, hydantoins, and propylthiouracil 2. Some drugs, such aspropylthiouracil and hydralazine (Apresoline), appear to cause vasculitis by inducing ANCA 2.
ANCA vasculitis treatment
Treatment of patients with microscopic polyangiitis and granulomatosis with polyangiitis has three phases: (1) induction of remission, (2) maintenance of remission, and (3) treatment of relapse 17. Current induction therapy often consists of cyclophosphamide (Cytoxan) and corticosteroids. For aggressive disease, use of high-dose intravenous methylprednisolone for three days is recommended, combined with intravenous or oral cyclophosphamide 17. Tapering doses of prednisone should follow, along with cyclophosphamide maintenance for 12 to 18 months. The lowest dosage of steroids that controls the disease should be used, and infection should be considered if the symptoms appear to exacerbate. For patients in sustained remission at 12 months, the use of all medications may be gradually discontinued. Patients whose symptoms are under good control must, nevertheless, be closely followed at six-month intervals for signs and symptoms of relapse. During treatment with these agents, complete blood counts and liver function tests should be performed periodically 17.
Other treatment regimens that may be of benefit include methotrexate, azathioprine (Imuran), trimethoprim-sulfamethoxazole (Bactrim, Septra), plasma exchange, cyclosporine (Sandimmune), intravenous im-munoglobulin, and monoclonal antibodies 17.
During the use of potentially ulcerogenic immunosuppressive therapy, patients may be given H2-blockers or proton-pump inhibitors. Prophylactic treatment with fluconazole (Diflucan) orally for fungal infection may be considered, as well as trimethoprim-sulfamethoxazole (480 mg) three times weekly for prophylactic treatment of patients with pneumocystis carinii prophylaxis 17.
Patients with eosinophilic granulomatosis with polyangiitis usually respond to high-dose corticosteroid therapy alone, although some cases may require the addition of cytotoxic drugs 17.
Comorbid conditions that accelerate vascular damage, such as hypertension, diabetes, hypercholesterolemia, and smoking should be appropriately controlled.
In drug-induced vasculitis, the offending agent should be stopped. Antihistamines and nonsteroidal anti-inflammatory drugs help alleviate skin discomfort and reduce associated arthralgias and myalgias. Severe cutaneous disease may warrant oral corticosteroid therapy 14.
Randomized controlled trials and prospective observational cohort studies first showed that methotrexate could replace cyclophosphamide in patients with limited or nonsevere disease granulomatosis with polyangiitis (Wegener’s granulomatosis). For patients with microscopic polyangiitis and mild renal disease, mycophenolate mofetil (CellCept) might be the alternative.
Biologic response modifiers allowing mechanism-based treatment approaches have become available over the last decade. Targeting specific molecules, these agents can block immune pathways thought to cause maladaptive inflammation in autoimmune diseases by inhibiting pro-inflammatory cytokines, eliminating cells of defined lineage (B lymphocytes) or inhibiting their activation or recruitment (T lymphocytes, eosinophils).
The ability to specifically target B lymphocytes with rituximab (Rituxan), a chimeric monoclonal antibody against the B lymphocyte specific cell surface receptor CD20, has fundamentally changed the therapy of severe AAV. Following the first report of its use in ANCA associated vasculitis in 2001, experience with rituximab in ANCA associated vasculitis has rapidly expanded. The Rituximab Versus Cyclophosphamide for ANCA-Associated Vasculitis (RAVE) trial showed that rituximab was not inferior to cyclophosphamide for remission induction in severe GPA and MPA. In fact, for patients with relapsing disease, rituximab was found to be superior to cyclophosphamide. Based on the primary endpoint results of RAVE, the Food and Drug Administration (FDA) approved rituximab in combination with glucocorticoids for remission induction in newly diagnosed and relapsing severe granulomatosis with polyangiitis and microscopic polyangiitis.
The 18-month follow-up study of RAVE, showed that a single course of four once-weekly infusions of rituximab is as effective for remission induction and maintenance as 18 months of continuous immunosuppressant therapy with cyclophosphamide followed by azathioprine (Imuran). Long-term single-center cohort studies indicated that rituximab is also effective and safe for remission maintenance, particularly in chronically relapsing granulomatosis with polyangiitis. In 2013, most patients with granulomatosis with polyangiitis and microscopic polyangiitis can be managed without exposure to cyclophosphamide and its dreaded long-term toxicities.
A role for biologic response modifiers is also emerging for eosinophilic granulomatosis with polyangiitis. Glucocorticoids have long been the mainstay of treatment, and cyclophosphamide is used for disease activity that threatens the function of vital organs. Side effects of glucocorticoids represent the biggest challenge in the long-term management of this disease. The exact mechanisms of tissue injury in eosinophilic granulomatosis with polyangiitis remain unclear, but blood and tissue eosinophilia appear to be responsible for tissue damage.
Interleukin-5 (IL-5) mediates bone marrow release, tissue survival, maturation and activation of eosinophils. Furthermore, IL-5 levels are increased in patients with eosinophilic granulomatosis with polyangiitis and are associated with disease activity. Consequently, reducing the number of eosinophils and preventing their activation by inhibiting IL-5 appears to be a rational novel approach for eosinophilic granulomatosis with polyangiitis. Mepolizumab, a monoclonal antibody targeting IL-5, has shown promise in eosinophilic granulomatosis with polyangiitis. Small pilot trials demonstrated prompt and prolonged reduction of peripheral eosinophils, clinical improvement and reduction in glucocorticoid use. A large multicenter randomized controlled trial of this agent in eosinophilic granulomatosis with polyangiitis is underway. Small case series and a pilot trial suggest that rituximab may represent an alternative to cyclophosphamide in severe eosinophilic granulomatosis with polyangiitis, particularly if it’s MPO-ANCA-associated.
- Ciavatta DJ, Yang J, Preston GA, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. The Journal of Clinical Investigation. 2010;120(9):3209-3219. doi:10.1172/JCI40034. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2929711/[↩]
- Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337:1512–23.[↩][↩][↩][↩][↩][↩][↩][↩]
- Niles JL, Bottinger EP, Saurina GR, Kelly KJ, Pan G, Collins AB, et al. The syndrome of lung hemorrhage and nephritis is usually an ANCA-associated condition. Arch Intern Med. 1996;156:440–5.[↩][↩][↩]
- Jennette JC, Falk RJ. Small vessel vasculitis. N Engl J Med 1997;337:1512–23.[↩]
- Bansal PJ, Tobin MC. Neonatal microscopic polyangiitis secondary to transfer of maternal myeloperoxidase-antineutrophil cytoplasmic antibody resulting in neonatal pulmonary hemorrhage and renal involvement. Ann Allergy Asthma Immunol. 2004;93(4):398–401. doi: 10.1016/S1081-1206(10)61400-7. https://www.ncbi.nlm.nih.gov/pubmed/15521377[↩]
- Xiao H, et al. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol. 2005;167(1):39–45. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1603451/[↩]
- Kallenberg CG, Heeringa P, Stegeman CA. Mechanisms of Disease: pathogenesis and treatment of ANCA-associated vasculitides. Nat Clin Pract Rheumatol. 2006;2(12):661–670. doi: 10.1038/ncprheum0355. https://www.ncbi.nlm.nih.gov/pubmed/17133251[↩]
- Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170(1):52–64. doi: 10.2353/ajpath.2007.060573. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1762697/[↩]
- Guillevin L, Durand-Gasselin B, Cevallos R, Gayraud M, Lhote F, Callard P, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum. 1999;42:421–30.[↩]
- Pettersson EE, Sundelin B, Heigl Z. Incidence and outcome of pauci-immune necrotizing and crescentic glomerulonephritis in adults. Clin Nephrol. 1995;43:141–9.[↩]
- Kelley WN. Vasculitis and related disorders. In: Textbook of rheumatology. 5th ed. Philadelphia: Saunders, 1997:1079–1101[↩][↩][↩][↩][↩][↩][↩]
- Kelley WN. Vasculitis and related disorders. In: Textbook of rheumatology. 5th ed. Philadelphia: Saunders, 1997:1079–1101.[↩][↩][↩]
- Duna GF, Galperin C, Hoffman GS. Wegener’s granulomatosis. Rheum Dis Clin North Am. 1995;21:949–86.[↩][↩]
- Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–92.[↩][↩]
- Savage CO, Harper L, Adu D. Primary systemic vasculitis. Lancet. 1997;349:553–8.[↩]
- Guillevin L, Lhote F, Amouroux J, Gherardi R, Callard P, Casassus P. Antineutrophil cytoplasmic antibodies, abnormal angiograms and pathological findings in polyarteritis nodosa and Churg-Strauss syndrome: indications for the classification of vasculitides of the polyarteritis Nodosa Group. Br J Rheumatol. 1996;35:958–64.[↩][↩][↩]
- Jayne DR, Rasmussen N. Treatment of antineutrophil cytoplasmic autoantibody-associated systemic vasculitis: initiatives of the European Community Systemic Vasculitis Clinical Trials Study Group. Mayo Clin Proc. 1997;72:737–47.[↩][↩][↩][↩][↩][↩]





{kind=link}