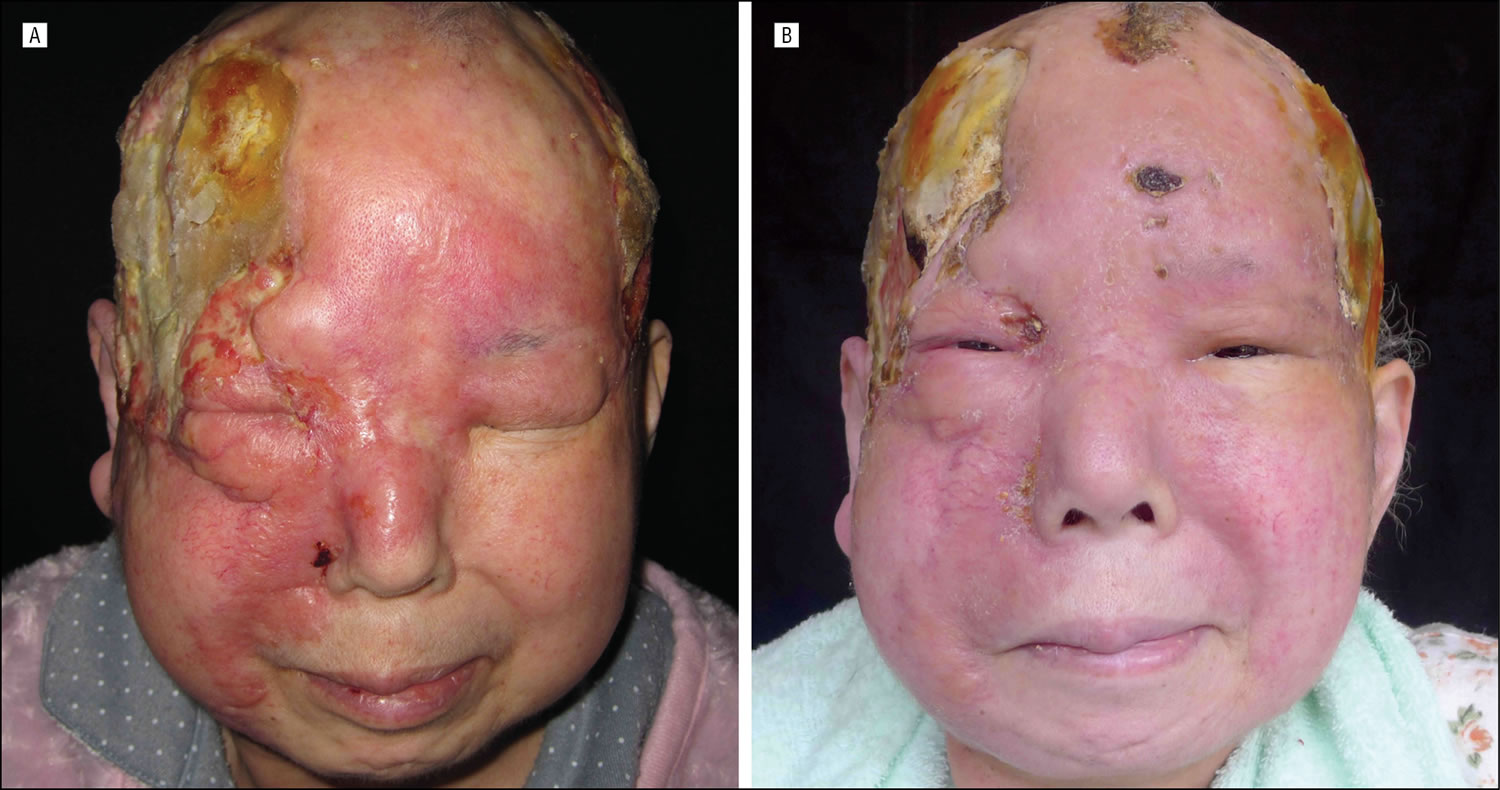
What is angiosarcoma
Angiosarcoma is an uncommon form of tissue cancer (sarcoma) that arises in the lining (vascular endothelial cells) of blood vessels or lymph vessels. Hemangiosarcomas start in blood vessel walls and lymphangiosarcomas start in lymph vessel walls. Angiosarcomas tend to be aggressive, recur locally, and spread widely. Angiosarcoma is derived from vascular tissues and accounts for only 2% to 4% of all soft tissue sarcomas 1, 2. Based on analyses of Surveillance, Epidemiology, and End Results program, 4.1% of angiosarcomas were diagnosed in the 26,758 cases of soft-tissue sarcoma available for analysis from 1978-2001. Angiosarcoma cancer could develop in any sites, including the skin, soft tissues, breast, liver, heart and spleen, and represent only 1% of primary malignant neoplasms in bone or marrow, but are more frequent in skin and soft tissue 3, 4. Cutaneous angiosarcomas are more common in the head and neck region and account for more than 60% of cases 5. The diagnosis of cutaneous angiosarcoma can be challenging because it often presents insidiously as a bruiselike lesion or a purplish papule that may be mistaken for a benign lesion such as hemangioma.
The mechanism of angiosarcoma is still unknown, while the related risk factors include previous exposure to radiation or some special chemotherapy for lymphoma 6. Predisposing factors include chronic lymphedema (Stewart-Treves syndrome) as from a radical mastectomy, radiotherapy, foreign materials (such as steel and plastic) in the body, and environmental agents (such as arsenic solutions used to spray grapevines and polyvinyl chloride in the plastic industry, thorium dioxide, arsenic, and radium). Angiosarcoma is not easy to be diagnosed just by clinical and radiological features. Thus, postoperative pathological examination remains the gold standard 7, 2. Comparing with the similar diseases such as hemangioma or hemangioendothelioma, which are benign or borderline malignant neoplasms, angiosarcoma is much more malignant 8. Angiosarcomas are aggressive and tend to recur locally, spread widely, and have a high rate of lymph node and systemic metastases. The rate of tumor-related death is high. The prognosis of angiosarcoma is very poor with a 1-year survival rate less than 50%, and the elder age, larger tumor size, and retroperitoneal location are poorer prognostic factors 9.
Angiosarcomas are rare neoplasms. Approximately 50% of angiosarcomas occur in the head and neck, but they account for less than 0.1% of head and neck malignancies 10.
Angiosarcomas demographic variation includes the following:
- African Americans in the United States are rarely affected by cutaneous angiosarcoma 10.
- Cutaneous angiosarcoma is more frequent in males than in females, with a male-to-female ratio of 2:1
- Bone and soft tissue angiosarcoma are also reported to be more frequent in males 11
- Bone angiosarcoma appears most often in adults (second to seventh decades of life)
- Cutaneous angiosarcoma of the head and neck tends to occur in the elderly populatio.
Epithelioid angiosarcoma
Epithelioid angiosarcoma is an extremely rare malignant disease, which accounts no more than 2% of all soft tissue sarcomas 12. Epithelioid angiosarcoma could lead to many different lesions, such as thyroid gland, skin, ureter, adrenal gland, kidney, small bowel, and so on. Epithelioid angiosarcoma is very hard to be diagnosed by clinical manifestation or radiological examinations 12.
In terms of pathology, epithelioid angiosarcoma could coexpress both vascular and epithelial markers, which make it named and hard to be distinguished from metastatic carcinomas. The first case about epithelioid angiosarcoma of bone was reported by Dr. Balicki, etc., in 1996 8, but there were no more than 30 cases reported all over the world until now 13. Epithelioid angiosarcoma of bone could involve the femur, tibia, talus, pelvis, spine, scapula, or multiple bones, but there were too few cases to do significant analysis 14.
The clinical manifestations of epithelioid angiosarcoma of bone were different according to the various locations of the primary lesions, while ostealgia and activity limitations were most commonly seen 14. However, it was hard to say which lesion was the primary one when epithelioid angiosarcoma had already made multiple bone destruction at the time of diagnosis. Since the extremely malignant features of epithelioid angiosarcoma, there were no absolutely standard treatments. Surgical treatment for local lesion with postoperative radiotherapy and chemotherapy was suggested to be effective, but sometimes it was just palliative treatment 15.
Under the microscope, epithelioid angiosarcoma is described to have large, rounded malignant cells with epithelioid features, as well as plentiful amphophilic or eosinophilic cytoplasm, and round to irregular vesicular nuclei with variably accentuated nucleoli. Epithelioid angiosarcoma cells are high-grade tumor cells and appear to be arranged in sheets, nests, cords, or rudimentary vascular channels 16.
Figure 1. Epithelioid angiosarcoma malignant cells (microscopy)
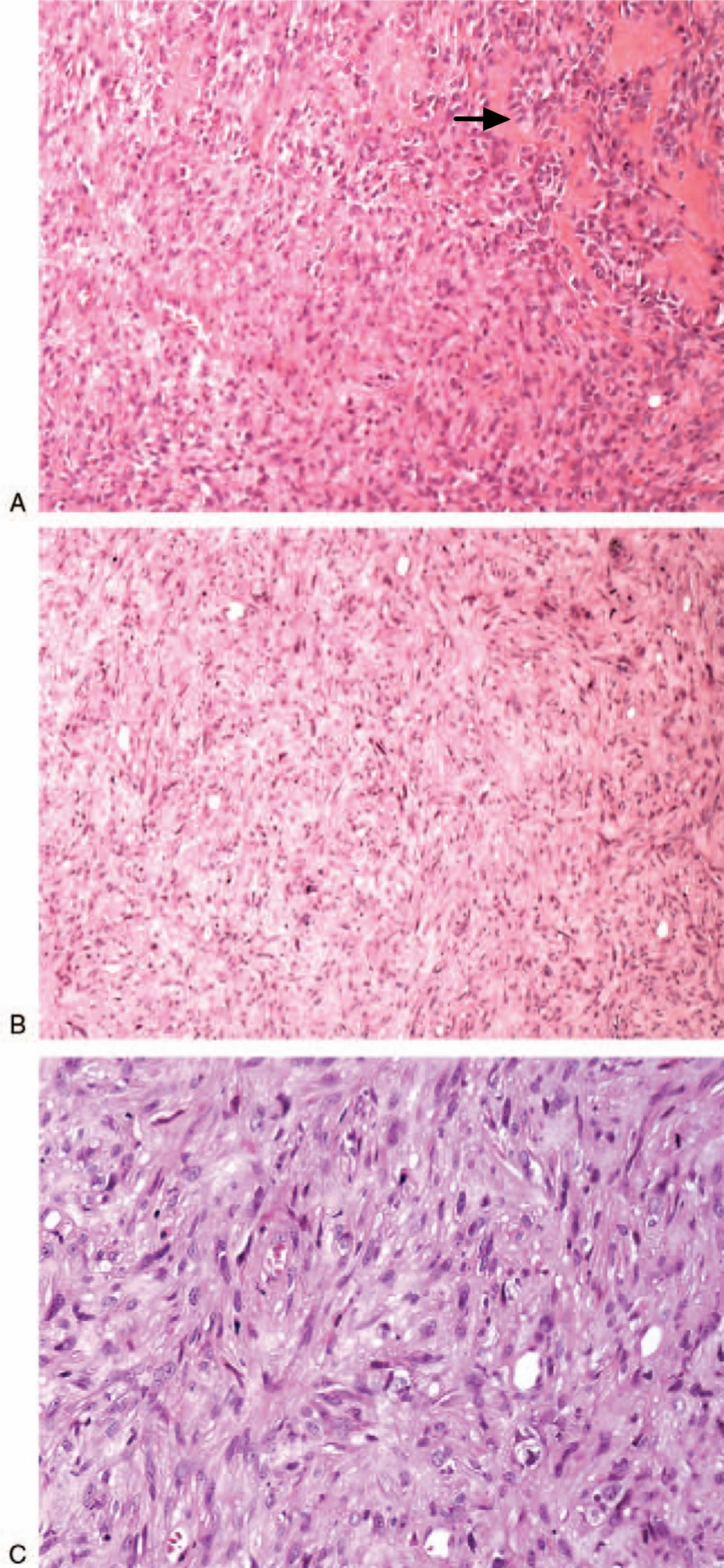
Note: (A) Hematoxylin-eosin staining of the mixture of tumor cells (the arrow) and fractured broken bone, 150×; (B) Hematoxylin-eosin staining of tumor cells, 60×; (C) Hematoxylin-eosin staining of those irregular tumor cells, 200×.
Immunohistochemistry is very significant in diagnosing epithelioid angiosarcoma. CD31 and CD34 are some usual vascular antigens and would be expressed by epithelioid angiosarcoma in different degrees 2. CD31 is considered to be a high specific and sensitive endothelial marker, and it is reported to be positive in 90% of all types of angiosarcoma. At the same time, the expression of CD34 in angiosarcoma remains controversial 7. As for the epithelial markers, cytokeratin (CK) could be positive at a rate of 30% to 50% in epithelioid angiosarcoma cells, while epithelial membrane antigen seems to be negative all the time 17. In our case, cytokeratin-7 was positive, while cytokeratin-19, cytokeratin-20, and epithelial membrane antigen were all negative.
One important differentiated diagnosis of epithelioid angiosarcoma is metastatic carcinoma. A higher proliferation index, some relatively typical morphological features, as well as the expression of both vascular and epithelial markers are very useful in distinguishing epithelioid angiosarcoma from some other carcinomas, such as epithelioid malignant mesothelioma, epithelioid haemangioma, epithelioid haemangioendothelioma, and so on 18. A focal positive expression of anion exchanger 1/ anion exchanger 3 might indicate a metastatic malignancy, but the negative expression of thyroid transcription factor 1 and anaplastic lymphoma kinase—sub-protein 8 denied the possibility of lung cancer, colorectal cancer, and lymphomas.
The prognosis of epithelioid angiosarcoma is very poor with a high rate of early metastasis and tumor-related death 18. An adequate surgery combined with the use of neoadjuvant and adjuvant chemoradiotherapy seems to improve the survival rate 18. It was extremely rare that the primary manifestation of epithelioid angiosarcoma was progressive ostealgia and multiple bone destruction, instead of a localized lesion.
Angiosarcoma scalp
Angiosarcoma of the scalp and neck is an often fatal tumor primarily of adult men. Furthermore, this angiosarcoma scalp primarily affects fair-skinned elderly men. Prior radiation may predispose. Angiosarcoma of the scalp is characterized by a bruise-like patches which may develop vascular papules, nodules, or plaques in an elderly patient, but variable clinical features are demonstrated (Figure 2). On progression, the lesions may ulcerate and bleed. This highly aggressive vascular tumor may eventuate in death from local invasion or distant metastases. Other less common presentations include a rosacea-like eruption, angioedema, a scarring alopecia, and skin-colored papules.
Angiosarcoma scalp prognosis is often poor. Some have suggested the following as poor prognostic signs:
- Diameter greater than 5 cm
- Satellite lesions
Cutaneous angiosarcoma of the head and neck region may involve the skin extensively with multifocal lesions, making it difficult to obtain a wide negative margin despite the attempt for radical resection. It also has a tendency to spread hematogenously. The 5-year survival rate for patients with cutaneous angiosarcoma of the head and neck region ranges from 10% to 54% 19. In the early experience at Mayo Clinic, of 13 patients treated between 1920 and 1970, only 2 patients lived longer than 5 years 20. In 2011, Albores-Saavedra et al 5 reported on a SEER (Surveillance, Epidemiology, and End Results) analysis of cutaneous angiosarcoma that included 135 cases of the scalp and neck, showing a 5-year and 10-year overall survival of 33.6% and 13.8%, respectively. Because of the rarity of the disease, most published studies are single-institution series with a relatively small number of patients.
Overexpression of vascular endothelial growth factor receptor (VEGFR) was observed in 80% of angiosarcomas 21 and VEGFR expression was also reported in angiosarcoma. Vascular endothelial growth factor receptor (VEGFR) expression was more frequent in better differentiated tumors and may play a role in predicting a particular patient’s clinical course 22.
Treatment is often difficult. Mortality is high. Whenever possible, surgical resection is a critical component of treatment. Wide surgical excision at an early stage is the best hope for cure. Mohs surgery has been used. Chemotherapy combined with radiotherapy has also been used. Adjuvant radiation therapy after maximal surgical resection is commonly administered to improve local control. Chemotherapeutic agents with activity against angiosarcoma include anthracyclines and taxanes. However, the role of adjuvant chemotherapy is not well defined. No prospective randomized studies have been conducted to provide treatment guidelines.
In an intriguing report 23, a patient with a β-adrenergic-positive multifocal stage T2 cutaneous angiosarcoma (≥20 cm) involving 80% of the scalp, left forehead, and left cheek, with no evidence of metastasis responded to propranolol 40 mg twice a day alone initially. Subsequently, propranolol 40 mg 3 times a day was combined with paclitaxel poliglumex, 2 mg/m2 infused weekly, and radiotherapy. During the subsequent 8 months, the tumor regressed with no detectable metastases. The authors suggest that β-blockade alone substantially reduced angiosarcoma proliferation and, in combination with standard therapy, is effective for reducing the size of the tumor and preventing metastases. Pazopanib treatment was helpful in one patient 24. Protracted intralesional interleukin-2 therapy induced complete remission in another 25.
Cutaneous angiosarcoma of the head and neck continues to be a difficult tumor to control and is therefore associated with poor outcomes 26. Further investigation is needed of novel treatment strategies, particularly effective systemic agents.
Figure 2. Angiosarcoma scalp – This older woman’s upper face and scalp was infiltrated with angiosarcoma. Note the contrast between the normal-colored skin on the right cheek and nose with the left cheek and forehead.
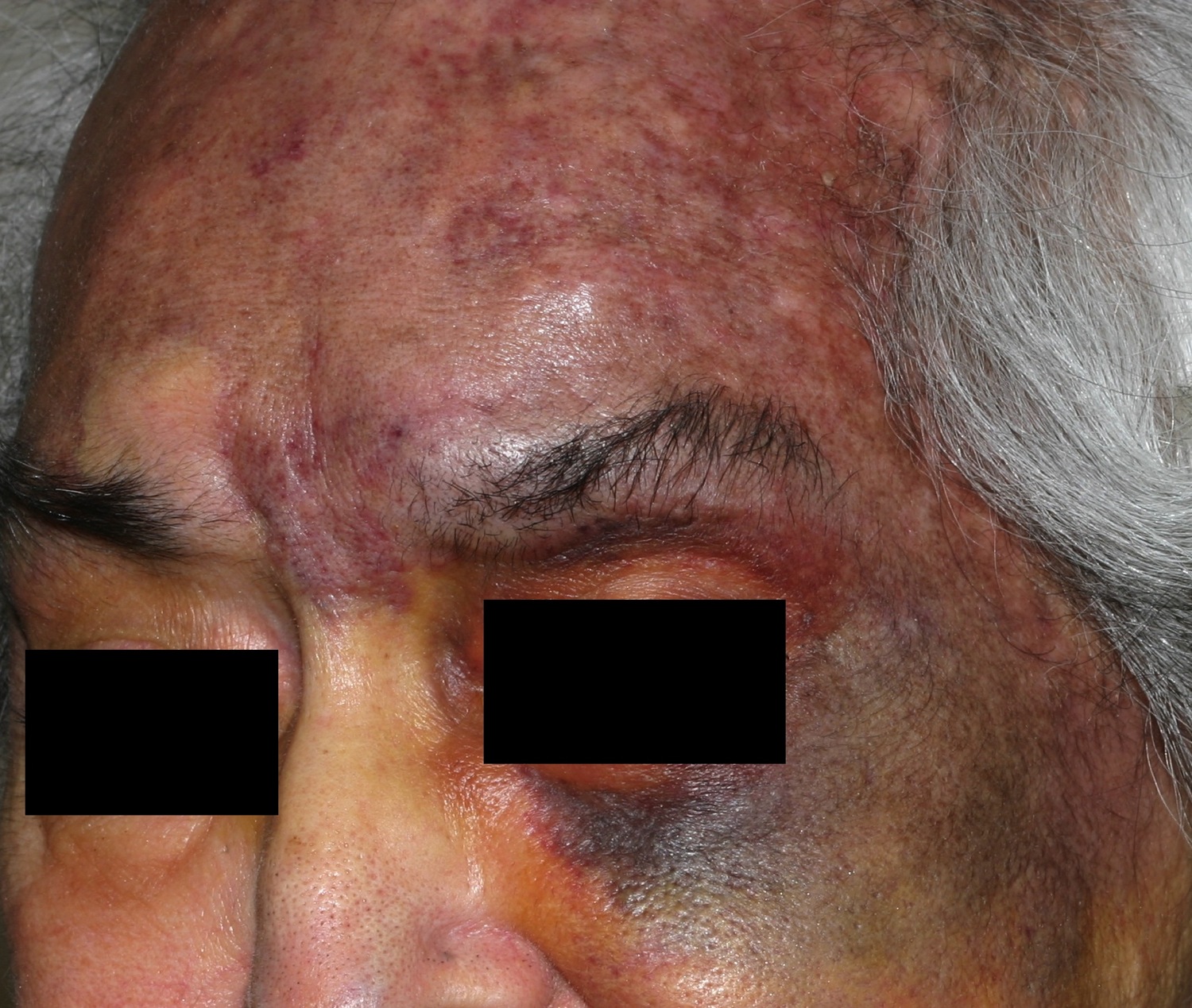


Angiosarcoma breast
Angiosarcomas of breast are rare neoplasms, accounting for about 0.05% of all primary malignancies of the breast. Angiosarcomas of breast occur sporadically in young women and usually present as palpable masses. They are aggressive tumors of endovascular origin and are often immunopositive for CD31, CD34 and Factor-VIII 27. Angiosarcoma breast is sometimes a complication of previous radiation treatment to the breast. Angiosarcoma breast can also occur in the affected arms of women with lymphedema, but this is not common. (Lymphedema is swelling that can develop after surgery or radiation therapy to treat breast cancer.) It can cause skin changes and/or a lump in the breast.
Angiosarcoma breast symptoms:
- Rapidly enlarging palpable mass without tenderness
- Often there is no pain
- Tumours often grow deep within breast tissue and cause diffuse breast enlargement with associated bluish skin discolouration
Angiosarcomas breast tend to grow and spread quickly. Treatment usually includes surgery to remove the breast (mastectomy), and is generally the same as for other sarcomas. Typically the axillary lymph nodes are not removed.
Angiosarcoma liver
Primary liver angiosarcoma is a rare and aggressive solid tumor, with high rates of local recurrence and distant metastasis, and poor prognosis 28. Primary liver angiosarcoma originates from liver vascular endothelial cells and lymphatic vessels cells. An estimated 200 cases are diagnosed annually in the world 29, with a male-to-female ratio of 3:1. Primary liver angiosarcoma has a predisposition to affect those between 60 and 80 years of age 30. No specific etiologic agent is identified in majority of patients with primary liver angiosarcoma. Some important risk factors for this disease include the exposure to vinyl chloride, radiocontrast material, the use of androgenic steroid and arsenic, and other chemicals 31.
The symptoms of primary liver angiosarcoma are nonspecific. Many tumors are found incidentally by diagnostic imaging, others are found as patients present with abdominal pain, anorexia, fatigue, weight loss, fever, and low back pain. Other reported symptoms/findings include jaundice, hemoperitoneum and acute hepatic failure. Primary hepatic angiosarcoma can metastasize to the lungs and hilar lymph nodes 32. Tumor markers such as AFP, CEA, CA19–9, and CA125 are usually not elevated. This makes primary liver angiosarcoma difficult to diagnose in its early stage.
Morphologically, primary liver angiosarcoma appear sole or multiple nodules or a dominant mass, or a mixed pattern. Pathologic diagnosis is golden standard for primary liver angiosarcoma. The optimal way to obtain tumor tissue is surgery. Percutaneous liver biopsy is considered to be dangerous and unreliable because of the necrosis and hemorrhage inside the tumor. Thus samples taken from these areas may produce false-negative results. Histopathology shows a variety of patterns of vascular channels, dilated sinusoidal or cavernous spaces. CD34, CD31 and factor VIII-related antigen can be positive. Other markers such ERG, Ki-67 and vimentin can be positive 33.
Most effective treatment modality is liver resection, however, there are no established chemotherapy regimens and moreover, chemotherapy is only palliative 34. Hepatic resection is rarely feasible but should be considered if the liver angiosarcoma is limited and the remainder of the liver is relatively normal. The prognosis of patients with primary liver angiosarcoma is poor with a median survival of 5-6 months. The causes of death of primary liver angiosarcoma patients include hepatic failure, disseminated intravascular coagulation, tumor rupture hemorrhagic shock and metastasis. Due to its low incidence, no standard treatment guideline exists for primary liver angiosarcoma. Some authors reported success with complete resection which can prolong the median survival to 17 months 35. However, surgical treatment is difficult to carry out in most patients, due to presence of multiple liver metastases or extrahepatic metastasis at the time of diagnosis. Liver transplant is unproved and is not considered to be a viable option due to high recurrence rate. One study reported that the overall median survival of 22 patients who underwent liver transplant was 6 months 36. If a complete surgical resection was possible and it was associated with a prolonged (10-year) postoperative survival 34.
Radiofrequency ablation of liver tumors has proven to be an effective way to control local lesions. E. Berber 37 reported that radiofrequency ablation for 18 patients with single liver metastases of sarcoma, local tumor recurrence is about 17% after 24 months of follow up. Therefore, radiofrequency ablation offers an alternative to surgery for inoperable patients. radiofrequency ablation was performed twice in our patient in order to achieve local tumor control. However, the emergence of new lesions rapidly after radiofrequency ablation treatment indicates the aggressive behavior of the cancer, thus making it urgent to start appropriate systemic treatment.
Chemotherapy has been reported to show clinical efficacy in unresectable primary liver angiosarcoma 35. Conventional chemotherapeutic agents for treatment of sarcoma include albumin paclitaxel, 5-fluorouracil, carboplatin, doxorubicin, ifosfamide, gemcitabine, dacarbazine, and newer agent trabectedin is approved for soft sarcoma in 2015 38. In recent years targeted therapies such as antiangiogenic drugs bevacizumab, sorafenib, and pazopanib showed shown efficacy in treatment of advanced soft tissue sarcoma. The small molecule vascular endothelial growth factor inhibitor pazopanib is a multiple target tyrosine kinase inhibitor, with activity against vascular endothelial growth factors 1, 2, and 3, and platelet derived growth factors. Pazopanib has received approval for the treatment of certain soft tissue sarcomas. In the phase III trial for metastatic soft tissue sarcoma, 372 patients with advanced soft tissue sarcoma whose tumors had progressed despite at least one line of chemotherapy, were randomly assigned either pazopanib or placebo. The difference in progression free survival in the pazopanib arm was statistically significant with a median of 4.6 months for pazopanib compared with 1.6 months for placebo 39.
In recent years, tumor immunotherapy has become an important therapeutic option for treatment of advanced cancer. Immune checkpoint inhibitors such as anti-PD-1 monoclonal antibodies have shown considerable efficacy in advanced tumors, especially for melanoma, renal cancer, and lung cancer. A retrospective analysis has shown a cohort of patients with relapsed metastatic sarcomas treated with nivolumab, the clinical benefit was observed in 50% of the evaluable patients 40. Currently, a clinical trial of PD-1 inhibitor nivolumab for sarcoma being conducted in the National Cancer Institute 41. Previous study suggested that targeting the VEGF axis may attenuate tumor-induced immunosuppression, allowing the tumor to become more responsive to immunotherapy when used in combination 42. It is in this context that Amin et al. designed a phase I clinical trial of nivolumab in combination with sunitinib or pazopanib in metastatic renal clear cell carcinoma (mRCC) National Cancer Institute 43. Nivolumab combined with TKI showed encouraging antitumor activity in this study with 53 patients. Objective response rate was higher with combination therapy up to about 50% than seen previously with nivolumab or TKI monotherapy in renal clear cell carcinoma, with a manageable toxicity profile, although renal and hepatic adverse effects were higher compared to monotherapy.
Angiosarcoma survival rates
All angiosarcomas tend to be aggressive and are often multicentric. These angiosarcoma cancers have a high local recurrence rate and metastasis because of their intrinsic biologic properties and because they are often misdiagnosed, leading to a poor prognosis and a high mortality rate. Malignant vascular tumors are clinically aggressive, difficult to treat, and have a reported 5-year survival rate around 20% 44. Advanced stage at presentation and lack of extensive excision are associated with higher recurrence, higher distant metastasis rates, and worsened survival.
The 5-year survival rate (or observed survival rate) refers to the percentage of patients who live at least 5 years after their cancer is diagnosed. Of course, many people live much longer than 5 years (and many are cured).
Five-year relative survival rates assume that some people will die of other causes and compare the observed survival with that expected for people without the cancer. This is a better way to see the effect of the cancer on survival.
Survival rates are often based on previous outcomes of large numbers of people who had the disease, but they cannot predict what will happen in any individual’s case. Many other factors might affect a person’s outlook, like the type of sarcoma, the location of the tumor, the treatment received, and the age of the patient. For example, sarcomas of the arms or legs have a better outcome than those found in other places. Also, older patients tend to have worse outcomes than younger people. Your doctor can tell you how the numbers below may apply to you, as he or she is familiar with your particular situation.
The rates below are based on the stage of the cancer at the time of diagnosis. When looking at survival rates, it’s important to understand that the stage of a cancer does not change over time, even if the cancer progresses. A cancer that comes back or spreads is still referred to by the stage it was given when it was first found and diagnosed, but more information is added to explain the current extent of the cancer. (And the treatment plan is adjusted based on the change in cancer status.)
The overall relative 5-year survival rate of people with soft tissue sarcomas is around 50% according to statistics from the National Cancer Institute (NCI). These statistics include people with Kaposi sarcoma, which has a poorer outlook than many sarcomas. The NCI doesn’t use the AJCC staging system. Instead, they group sarcomas only by whether they are still confined to the primary site (called localized) have spread to nearby lymph nodes or tissues (called regional); or have spread (metastasized) to sites away from the main tumor (called distant). The 5-year survival rates for soft tissue sarcomas have not changed much for many years. The corresponding 5-year relative survival rates were:
- 83% for localized sarcomas (56% of soft tissue sarcomas were localized when they were diagnosed)
- 54% for regional stage sarcomas; (19% were in this stage)
- 16% for sarcomas with distant spread (16% were in this stage)
The 10-year relative survival rate is only slightly worse for these stages, meaning that most people who survive 5 years are probably cured.
For sarcomas of the arms and legs, Memorial Sloan-Kettering Cancer Center has survival rates broken down by American Joint Committee on Cancer stage (these are for observed, not relative survival):
Table 1. Survival by Stage of Soft Tissue Sarcoma 45
| Stage | 5-year observed survival rate |
| I | 90% |
| II | 81% |
| III | 56% |
| IV | Not available |
Survival is worse when the sarcoma has developed somewhere other than the arms or legs. For example, the 5-year survival for retroperitoneal sarcomas is around 40% to 60%.
Angiosarcoma causes
It’s not clear what causes most angiosarcomas, though doctors have identified factors that may increase your risk of the disease.
Doctors know that something happens that causes a cell in the lining of a blood vessel or lymph vessel to develop an error (mutation) in its genetic code. The mutation tells the cell to grow quickly, making more abnormal cells. The abnormal cells continue living when other cells would die.
The result is a buildup of abnormal cells that grows from the affected blood vessel or lymph vessel. With time, cells may break off and spread (metastasize) to other areas of the body.
Risk factors for angiosarcoma cancer
Factors that may increase your risk of angiosarcoma include 46:
- Radiation therapy. Treatment with radiation for cancer or other conditions may increase your risk of angiosarcoma. A rare complication of radiation therapy, angiosarcoma typically occurs five to 10 years after treatment.
- Swelling caused by lymph vessel damage (lymphedema). Lymphedema is swelling caused by a backup of lymph fluid that occurs when the lymphatic system is blocked or damaged. Lymphedema is a risk whenever lymph nodes are removed during surgery — a technique that’s often used to treat cancer. Lymphedema can also occur in response to infection or other conditions.
- Chemicals. Liver angiosarcoma has been linked to exposure to several chemicals, including polyvinyl chloride in the plastic industry, thorium dioxide, radium and arsenic.
- Other carcinogens (e.g., bone wax, Dacron, metal bodies)
- Foreign materials (e.g., shrapnel, steel, surgical sponges)
- Environmental carcinogens
- AIDS
- Preexisting benign lesions (e.g., bone infarct, pagetoid bone, chronic osteomyelitis)
Angiosarcoma symptoms
Angiosarcomas are insidious, and they may not produce symptoms until the disease is well advanced 47. History should focus on identifying risk factors; however, most patients do not have these factors.
Angiosarcomas arising at different sites and in different organs have some distinct features, but the clinical manifestations they cause are associated with the amount of organ tissue replaced, as follows:
- Pathologic fractures, anemia, or hepatic dysfunction
- Other intrinsic characteristics of a malignant vascular proliferation (e.g.,, bleeding, thrombocytopenia, or intravascular disseminated coagulation)
- Compression of adjacent neurovascular structures that causes pain
Angiosarcoma that affects the skin
Most often, angiosarcoma occurs in the skin on the head and neck, particularly the scalp. Signs and symptoms of this form of angiosarcoma include:
- A raised, purplish area of skin that looks like a bruise
- A bruise-like lesion that grows larger over time
- A lesion that may bleed when scratched or bumped
- Swelling in the surrounding skin
Angiosarcoma that affects organs
When angiosarcoma affects organs, such as the liver or the heart, it often causes pain. Other symptoms depend on the location of the angiosarcoma.
Angiosarcoma diagnosis
Tests and procedures used in angiosarcoma diagnosis include:
- Physical exam. Your doctor will thoroughly examine you to understand your condition.
- Removing a sample of tissue for testing (biopsy). Your doctor will remove a sample of suspicious tissue for laboratory testing. Analysis in the lab can detect cancer cells and determine certain characteristics of your cancer cells that may help guide your treatment.
- Imaging tests. Imaging tests can give your doctor an idea of the extent of your cancer. Tests may include MRI, CT and positron emission tomography (PET). Which tests you undergo will depend on your particular situation.
Soft Tissue Sarcoma Stages
After someone is diagnosed with a soft tissue sarcoma, doctors will try to figure out if it has spread, and if so, how far. This process is called staging. The stage of a cancer describes how much cancer is in the body. It helps determine how serious the cancer is and how best to treat it. Doctors also use a cancer’s stage when talking about survival statistics.
The stages of soft tissue sarcomas range from stages I (1) through IV (4). As a rule, the lower the number, the less the cancer has spread. A higher number, such as stage IV, means cancer has spread more. And within a stage, an earlier letter means a lower stage. Although each person’s cancer experience is unique, cancers with similar stages tend to have a similar outlook and are often treated in much the same way.
The staging system most often used for soft tissue sarcomas is the American Joint Committee on Cancer (AJCC) TNM system, which is based on 4 key pieces of information:
- The extent of the tumor (T): How large is the cancer?
- The spread to nearby lymph nodes (N): Has the cancer spread to nearby lymph nodes?
- The spread (metastasis) to distant sites (M): Has the cancer spread to distant organs such as the lungs?
- The grade (G) of the cancer: How much do the sarcoma cells look like normal cells?
The grade of a sarcoma is determined using a system known as the French or FNCLCC system, and is based on 3 factors:
- Differentiation: Cancer cells are given a score of 1 to 3, with 1 being assigned when they look similar to normal cells and 3 being used when the cancer cells look very abnormal. Certain types of sarcoma are given a higher score automatically.
- Mitotic count: How many cancer cells are seen dividing under the microscope; given a score from 1 to 3 (a lower score means fewer cells were seen dividing)
- Tumor necrosis: How much of the tumor is made up of dying tissue; given a score from 0 to 2 (a lower score means there was less dying tissue present).
The scores for each factor are added to determine the grade for the cancer. Higher-grade cancers tend to grow and spread faster than lower-grade cancers.
- GX: The grade cannot be assessed (because of incomplete information).
- Grade 1 (G1): Total score of 2 or 3
- Grade 2 (G2): Total score of 4 or 5
- Grade 3 (G3): Total score of 6, 7 or 8.
There are different staging systems for soft tissue sarcomas depending on where the cancer is in the body.
- Head and neck
- Trunk and extremities (arms and legs)
- Abdomen and thoracic (chest) visceral organs
- Retroperitoneum
Numbers or letters after T, N, and M provide more details about each of these factors. Higher numbers mean the cancer is more advanced. Once a person’s T, N, and M categories have been determined, this information is combined in a process called stage grouping to assign an overall stage. Of the 4 main locations, only 2 (Trunk and Extremities and Retroperitoneum) have stage groupings.
The staging system in the table below uses the pathologic stage (also called the surgical stage). It is determined by examining tissue removed during an operation. Sometimes, if surgery is not possible right away or at all, the cancer will be given a clinical stage instead. This is based on the results of a physical exam, biopsy, and imaging tests. The clinical stage will be used to help plan treatment. Sometimes, though, the cancer has spread further than the clinical stage estimates, and may not predict the patient’s outlook as accurately as a pathologic stage.
The system described below is the most recent American Joint Committee on Cancer (AJCC) system, effective January 2018. Cancer staging can be complex, so ask your doctor to explain it to you in a way you understand.
Table 2. Trunk and Extremities Sarcoma Stages
| AJCC stage | Stage grouping | Trunk and Extremities Sarcoma Stage description* |
| IA | T1 N0 M0 G1 or GX | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| IB | T2, T3, T4 N0 M0 G1 or GX | The cancer is:
It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| II | T1 N0 M0 G2 or G3 | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIA
| T2 N0 M0 G2 or G3 | The cancer is larger than 5 cm (2 inches) but not more than 10 cm (T2). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIB | T3 or T4 N0 M0 G2 or G3 | The cancer is:
It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IV | Any T N1 M0 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has not spread to distant sites (M0). It can be any grade. |
| OR | ||
| Any T Any N M1 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has spread to distant sites such as the lungs (M1). It can be any grade. | |
*The following additional categories are not listed in the table above:
- TX: Main tumor cannot be assessed due to lack of information.
- T0: No evidence of a primary tumor.
- NX: Regional lymph nodes cannot be assessed due to lack of information.
Table 3. Retroperitoneum Sarcoma Stages
| AJCC stage | Stage grouping | Retroperitoneum Sarcoma Stage description* |
| IA | T1 N0 M0 G1 or GX | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| IB | T2, T3, T4 N0 M0 G1 or GX | The cancer is:
It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| II | T1 N0 M0 G2 or G3 | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIA
| T2 N0 M0 G2 or G3 | The cancer is larger than 5 cm (2 inches) but not more than 10 cm (T2). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIB | T3 or T4 N0 M0 G2 or G3 | The cancer is:
It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| OR | ||
| Any T N1 M0 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has not spread to distant sites (M0). It can be any grade. | |
| IV | Any T Any N M1 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has spread to distant sites such as the lungs (M1). It can be any grade. |
*The following additional categories are not listed in the table above:
- TX: Main tumor cannot be assessed due to lack of information.
- T0: No evidence of a primary tumor.
- NX: Regional lymph nodes cannot be assessed due to lack of information.
Angiosarcoma treatment
Which angiosarcoma treatment is best for you depends on your cancer’s location, its size and whether it has spread to other areas of your body.
Treatment options may include:
- Surgery. The goal of surgery is to remove the angiosarcoma entirely. Your surgeon will remove the cancer and some of the healthy tissue that surrounds it. In some cases surgery may not be an option, for example, if the cancer is very large or has spread to other areas of the body.
- Radiation therapy. Radiation therapy uses high-energy beams, such as X-rays and protons, to kill cancer cells. Radiation therapy is sometimes used after surgery to kill any cancer cells that remain. Radiation therapy may also be an option if you can’t undergo surgery.
- Chemotherapy. Chemotherapy is a treatment that uses drugs or chemicals to kill cancer cells. Chemotherapy may be an option if your angiosarcoma has spread to other areas of your body. In certain situations, it may be combined with radiation therapy if you can’t undergo surgery.
Treatment of soft-tissue angiosarcoma
For stage I angiosarcomas, the National Comprehensive Cancer Network (NCCN) recommends surgery to obtain adequate oncologic margins. For stage II-III disease that can be resected with acceptable functional outcomes, the NCCN also recommends preoperative radiation therapy (category 1) or preoperative chemoradiation or chemotherapy (category 2B) 48. The NCCN lists the following agents as having activity against angiosarcoma:
- Paclitaxel
- Docetaxel
- Vinorelbine
- Sorafenib
- Sunitinib
- Bevacizumab
Ravi et al 49 report exceptional response to treatment with pazopanib in a patient with angiosarcoma that harbored amplification of vascular endothelial growth factor receptor (VEGFR) and that had not responded to sorafenib. These authors suggest that a subset of patients with angiosarcoma with genomic alterations in vascular signaling genes may respond well to pazopanib.
Multiple randomized studies using doxorubicin-based chemotherapy fail to show a survival benefit from neoadjuvant chemotherapy. However, a meta-analysis suggests improved local control and disease-free survival with chemotherapy, but no survival advantage 50.
Response to preoperative chemotherapy is only 40-50%, with the most active regimens, and toxicity is significant. Consequently, specialists reserve preoperative chemotherapy for patients with high-grade lesions. The regimen is continued in those patients who respond with tumor shrinkage after two to three courses of multiagent chemotherapy after tumor resection.
Pasquier et al 23 reported effective treatment in seven patients with advanced angiosarcoma using the combination of twice-daily propranolol (40 mg) and weekly metronomic vinblastine (6 mg/m2) and methotrexate (35 mg/m2). All patients responded; one patient showed a complete response and three showed very good partial responses. Median progression-free and overall survival was 11 months (range 5–24) and 16 months (range 10–30), respectively. In January 2017, propranolol was granted orphan drug status in Europe for the treatment of soft-tissue sarcoma 51.
Response to neoadjuvant chemotherapy can be observed, but it does not always correlate with radiographic response.
Radiotherapy
The use of irradiation in conjunction with surgery continues to evolve and results in 80% of local control and excellent functional and cosmetic outcome 52. However, consider that 50% of angiosarcomas have distant metastases, and irradiation does not improve survival. Better definition of the extent of the disease with the use of MRI helps to further delineate the radiotherapy fields and decrease long-term morbidity. Intraoperative radiation, brachytherapy, or more external beam therapy can complement preoperative external beam radiotherapy.
The disadvantage of preoperative radiation is that a higher wound complication rate may delay surgery (1 wk of healing per 10 Gy of radiation delivered). The advantages of preoperative radiation are as follows:
- Optimization for surgery
- Smaller volume of external beam fields
- Less hypoxic tissue
- Potential to reduce the chance of intraoperative implantation
- Potential improvement in local control in advanced tumors
Bone angiosarcoma
Evidence of multicentricity must be sought before making any decision regarding therapy. Patients have presented with lesions affecting as many as 45 different bones. In such cases, consider neoadjuvant chemotherapy.
A chemotherapeutic regimen common for sarcomatous tumors can be administered (ifosfamide and doxorubicin used together or sequentially). If clinical or radiographic improvement is not observed, consider a second regimen with cyclophosphamide, etoposide, and cisplatin. Gemcitabine may be effective as second line or third-line therapy.
- Arachchi A, Van SL, Wang L, et al. Rare case of spontaneous splenic rupture. ANZ J Surg 2015;doi: 10.1111/ans.13192. https://www.ncbi.nlm.nih.gov/pubmed/26074015[↩]
- Collini P, Barisella M, Renne SL, et al. Epithelioid angiosarcoma of the thyroid gland without distant metastases at diagnosis: report of six cases with a long follow-up. Virchows Arch 2016;469:223–32. https://www.ncbi.nlm.nih.gov/pubmed/27229516[↩][↩][↩]
- Young RJ, Brown NJ, Reed MW, Hughes D, Woll PJ. Angiosarcoma. Lancet Oncol. 2010 Oct. 11(10):983-91.[↩]
- Lea & Febiger, Collins Andrew. Bone Tumors: Clinical, Radiologic, and Pathologic Correlations. 1991.[↩]
- Albores-Saavedra J, Schwartz AM, Henson DE, et al. Cutaneous angiosarcoma: analysis of 434 cases from the Surveillance, Epidemiology, and End Results Program, 1973-2007. Ann Diagn Pathol. 2011;15(2):93-97. https://www.ncbi.nlm.nih.gov/pubmed/21190880[↩][↩]
- Zwi LJ, Evans DJ, Wechsler AL, et al. Splenic angiosarcoma following chemotherapy for follicular lymphoma. Hum Pathol 1986;17:528–30. https://www.ncbi.nlm.nih.gov/pubmed/3516861[↩]
- Hasegawa T, Fujii Y, Seki K, et al. Epithelioid angiosarcoma of bone. Hum Pathol 1997;28:985–9. https://www.ncbi.nlm.nih.gov/pubmed/9269837[↩][↩]
- Danuta B, Ralf B, James M, et al. Multicentric epithelioid angiosarcoma of the bone. Blood Cells Mol Dis 1996;22:205–13. https://www.ncbi.nlm.nih.gov/pubmed/9075571[↩][↩]
- Milite D, Pilon F, Ferrari A, et al. Aortic epithelioid angiosarcoma after endovascular aneurysm repair. Ann Vasc Surg 2016;35:207.e217–21. https://www.ncbi.nlm.nih.gov/pubmed/27238982[↩]
- Sturgis EM, Potter BO. Sarcomas of the head and neck region. Curr Opin Oncol. 2003 May. 15(3):239-52.[↩][↩]
- Meis-Kindblom JM, Kindblom LG. Angiosarcoma of soft tissue: a study of 80 cases. Am J Surg Pathol. 1998 Jun. 22(6):683-97.[↩]
- Li Y, Zou X, Chang X, Chang X, Sun S, Zhang B. Right femoral pathological fracture caused by primary bone epithelioid angiosarcoma: Case report. Ling. S, ed. Medicine. 2017;96(27):e6951. doi:10.1097/MD.0000000000006951. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5502134/[↩][↩]
- Lang J, Chen L, Chen B, et al. Epithelioid angiosarcoma of the spine: A case report of a rare bone tumor. Oncol Lett 2014;7:2170–4. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4049740/[↩]
- Akio S, Yusuke T, Yoshinao O, et al. Aggressive clinical course of epithelioid angiosarcoma in the femur a case report. World J Surg Oncol 2014;12:281. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4167269/[↩][↩]
- Jesse H, Srinivas M. Epithelioid angiosarcoma a brief diagnostic review and differential diagnosis-review. Arch Pathol Lab Med 2011;135:268–72. https://www.ncbi.nlm.nih.gov/pubmed/21284449[↩]
- Jinbo Wu, Xiaojing Li, Xiuping Liu. Epithelioid angiosarcoma a clinicopathological study of 16 Chinese cases-pathology. Int J Clin Exp Pathol 2015;8:3901–9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4466961/[↩]
- Bone and soft tissue pathology. Modern Pathol 2015;28:12–30.[↩]
- Shi H, Zhen T, Zhang F, et al. Multifocal epithelioid angiosarcoma of the jejunum: a case report. Pathology 2016;48:91–3.https://www.ncbi.nlm.nih.gov/pubmed/27020220[↩][↩][↩]
- Dettenborn T, Wermker K, Schulze HJ, Klein M, Schwipper V, Hallermann C. Prognostic features in angiosarcoma of the head and neck: a retrospective monocenter study [published online May 15, 2014]. J Craniomaxillofac Surg. doi:10.1016/j.jcms.2014.05.002. https://www.ncbi.nlm.nih.gov/pubmed/24962043[↩]
- Hodgkinson DJ, Soule EH, Woods JE. Cutaneous angiosarcoma of the head and neck. Cancer. 1979;44(3):1106-1113. https://www.ncbi.nlm.nih.gov/pubmed/573173[↩]
- Zietz C, Rossle M, Haas C, Sendelhofert A, Hirschmann A, Sturzl M, et al. MDM-2 oncoprotein overexpression, p53 gene mutation, and vascular endothelial growth factor receptor (VEGFR) up-regulation in angiosarcomas. Am J Pathol 1998; 153: 1425–1433.[↩]
- Gennaro M, Valeri B, Casalini P, Carcangiu ML, Gronchi A, Conti AR, et al. Angiosarcoma of the breast and vascular endothelial growth factor receptor. Tumori 2010; 96: 930–935.[↩]
- Pasquier E, André N, Street J, Chougule A, Rekhi B, Ghosh J, et al. Effective Management of Advanced Angiosarcoma by the Synergistic Combination of Propranolol and Vinblastine-based Metronomic Chemotherapy: A Bench to Bedside Study. EBioMedicine. 2016 Apr. 6:87-95. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4856748/[↩][↩]
- JAAD 2014 Jan[↩]
- BJD 2015;172;156[↩]
- Patel SH, Hayden RE, Hinni ML, Wong WW, Foote RL, Milani S, Wu Q, Ko SJ, Halyard MY. Angiosarcoma of the Scalp and FaceThe Mayo Clinic Experience. JAMA Otolaryngol Head Neck Surg. 2015;141(4):335–340. doi:10.1001/jamaoto.2014.3584 https://jamanetwork.com/journals/jamaotolaryngology/fullarticle/2089814[↩]
- NATCON IASO Abstract 2015. Indian Journal of Surgical Oncology. 2016;7(3):273-294. doi:10.1007/s13193-016-0529-x. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5016334/[↩]
- Qiao Y, Yang J, Liu L, et al. Successful treatment with pazopanib plus PD-1 inhibitor and RAK cells for advanced primary hepatic angiosarcoma: a case report. BMC Cancer. 2018;18:212. doi:10.1186/s12885-018-3996-3. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5822655/[↩]
- Mani H, Van Thiel DH. Mesenchymal tumors of the liver. Clin Liver Dis. 2001;5(1):219–257. doi: 10.1016/S1089-3261(05)70162-8. https://www.ncbi.nlm.nih.gov/pubmed/11218917[↩]
- Awan S, Davenport M, Portmann B, Howard ER. Angiosarcoma of the liver in children. J Pediatr Surg. 1996;31(12):1729–1732. doi: 10.1016/S0022-3468(96)90065-2. https://www.ncbi.nlm.nih.gov/pubmed/8987004[↩]
- Makk L, Delmore F, Creech JL, Ogden LL, Fadell EH, Songster CL, Clanton J, Johnson M, Christopherson WM. Clinical and morphologic features of hepatic angiosarcoma in vinyl chloride workers. Cancer. 1976;37(1):149–163. doi: 10.1002/1097-0142(197601)37:1<149::AID-CNCR2820370122>3.0.CO;2-7. https://www.ncbi.nlm.nih.gov/pubmed/942881[↩]
- Kim HR, Rha SY, Cheon SH, Roh JK, Park YN, Yoo NC. Clinical features and treatment outcomes of advanced stage primary hepatic angiosarcoma. Ann Oncol. 2009;20(4):780–787. doi: 10.1093/annonc/mdn702. https://www.ncbi.nlm.nih.gov/pubmed/19179547[↩]
- Wang ZB, Yuan J, Chen W, Wei LX. Transcription factor ERG is a specific and sensitive diagnostic marker for hepatic angiosarcoma. World J Gastroenterol. 2014;20(13):3672–3679. doi: 10.3748/wjg.v20.i13.3672. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3974537/[↩]
- Timaran CH, Grandas OH, Bell JL. Hepatic angiosarcoma: long-term survival after complete surgical removal. Am Surg. 2000;66(12):1153–1157. https://www.ncbi.nlm.nih.gov/pubmed/11149588[↩][↩]
- Zheng YW, Zhang XW, Zhang JL, Hui ZZ, Du WJ LRM, Ren XB. Primary hepatic angiosarcoma and potential treatment options. J Gastroenterol Hepatol. 2014;29(5):906–911. doi: 10.1111/jgh.12506. https://www.ncbi.nlm.nih.gov/pubmed/24372769[↩][↩]
- Orlando G, Adam R, Mirza D, Soderdahl G, Porte RJ, Paul A, Burroughs AK, Seiler CA, Colledan M, Graziadei I, et al. Hepatic hemangiosarcoma: an absolute contraindication to liver transplantation–the European liver transplant registry experience. Transplantation. 2013;95(6):872–877. doi: 10.1097/TP.0b013e318281b902. https://www.ncbi.nlm.nih.gov/pubmed/23354302[↩]
- Berber E, Ari E, Herceg N, Siperstein A. Laparoscopic radiofrequency thermal ablation for unusual hepatic tumors: operative indications and outcomes. Surg Endosc. 2005;19(12):1613–1617. doi: 10.1007/s00464-005-0236-0. https://www.ncbi.nlm.nih.gov/pubmed/16247574[↩]
- Gordon EM, Sankhala KK, Chawla N, Chawla SP. Trabectedin for soft tissue sarcoma: current status and future perspectives. Adv Ther. 2016;33(7):1055–1071. doi: 10.1007/s12325-016-0344-3. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4939148/[↩]
- van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, Schoffski P, Aglietta M, Staddon AP, Beppu Y, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–1886. doi: 10.1016/S0140-6736(12)60651-5. https://www.ncbi.nlm.nih.gov/pubmed/22595799[↩]
- Paoluzzi L, Cacavio A, Ghesani M, Karambelkar A, Rapkiewicz A, Weber J, Rosen G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin Sarcoma Res. 2016;6:24. doi: 10.1186/s13569-016-0064-0. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5200964/[↩]
- https://clinicaltrials.gov/ct2/show/NCT02500797[↩]
- Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–251. doi: 10.1038/nrc3237. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3967236/[↩]
- https://clinicaltrials.gov/ct2/show/NCT01472081[↩]
- Lezama-del Valle P, Gerald WL, Tsai J, et al. Malignant vascular tumors in young patients. Cancer. 1998 Oct 15. 83(8):1634-9.[↩]
- Survival by Stage of Soft Tissue Sarcoma. https://www.cancer.org/cancer/soft-tissue-sarcoma/detection-diagnosis-staging/survival-rates.html[↩]
- DeVita VT Jr, Hellman S, Rosenberg SA, eds. Cancer: Principles & Practice of Oncology. 8th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2008.[↩]
- Lahat G, Dhuka AR, Hallevi H, Xiao L, Zou C, Smith KD, et al. Angiosarcoma: clinical and molecular insights. Ann Surg. 2010 Jun. 251(6):1098-106.[↩]
- [Guideline] NCCN. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Soft Tissue Sarcoma. https://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdf[↩]
- Ravi V, Sanford EM, Wang WL, Ross JS, Ramesh N, Futreal A, et al. Antitumor Response of VEGFR2- and VEGFR3-Amplified Angiosarcoma to Pazopanib. J Natl Compr Canc Netw. 2016 May. 14 (5):499-502.[↩]
- Budd GT. Management of Angiosarcoma. Curr Oncol Rep. 2002. 4(6):515-519.[↩]
- http://www.medscape.com/viewarticle/875136[↩]
- Mark RJ, Poen JC, Tran LM, Fu YS, Juillard GF. Angiosarcoma. A report of 67 patients and a review of the literature. Cancer. 1996 Jun 1. 77(11):2400-6.[↩]





{kind=link}