Contents
What is brugada syndrome
Brugada syndrome is a rare but serious disorder characterized by sudden death associated with one of several ECG patterns characterized by incomplete right bundle-branch block and ST-segment elevations in the anterior precordial leads. Brugada syndrome affects the way electrical signals pass through the heart. It can cause the heart to beat dangerously fast. These unusually fast heartbeats – known as an arrhythmia – can be life threatening in some cases.
Brugada syndrome is usually caused by a faulty gene that’s inherited by a child from a parent. A simple heart test can be done to see if you have it.
Many people with Brugada syndrome don’t have any symptoms and don’t realize they have it.
Some people experience:
- Blackouts
- Fits (seizures)
- Occasional noticeable heartbeats (palpitations), chest pain, breathlessness, or dizziness.
Blackouts or syncope (temporary loss of consciousness usually related to insufficient blood flow to the brain) and cardiac arrest are the most common clinical manifestations leading to the diagnosis of Brugada syndrome. Nightmares or thrashing at night may occur. However, some patients remain asymptomatic and the diagnosis of Brugada syndrome is suggested by a routine electrocardiogram (ECG) showing ST-segment elevation in leads V1 through V3 1.
These symptoms can occur at any time, but are sometimes triggered by something such as a high temperature (fever), drinking lots of alcohol, or dehydration.
Symptoms typically first appear at around 30-40 years of age, but they can occur at any age. The exact prevalence of Brugada syndrome is unknown, although it is estimated to affect 5 in 10,000 people worldwide. This condition occurs much more frequently in people of Asian ancestry, particularly in Japanese and Southeast Asian populations.
Although Brugada syndrome affects both men and women, the condition appears to be 8 to 10 times more common in men. Researchers suspect that testosterone, a sex hormone present at much higher levels in men, may account for this difference.
Brugada syndrome usually is diagnosed in adults and, sometimes, in adolescents. It’s rarely diagnosed in young children.
A family history of sudden cardiac death is common, though not universal, as Brugada syndrome can occur sporadically. In about 20% of patients, atrial fibrillation is an associated arrhythmia 2.
The context of the cardiac event is important. In many cases, cardiac arrest occurs during sleep or rest. Cases occurring during physical activity are rare. Fever is often reported to trigger or exacerbate the clinical manifestations of Brugada syndrome.
Sudden death typically occurs around age 40. This condition may explain some cases of sudden infant death syndrome (SIDS), which is a major cause of death in babies younger than 1 year. Sudden infant death syndrome (SIDS) is characterized by sudden and unexplained death, usually during sleep.
Sudden unexplained nocturnal death syndrome is a condition characterized by unexpected cardiac arrest in young adults, usually at night during sleep. This condition was originally described in Southeast Asian populations, where it is a major cause of death. Researchers have determined that sudden unexplained nocturnal death syndrome and Brugada syndrome are the same disorder 3.
See your doctor if:
- you have unexplained blackouts or seizures
- one of your parents, siblings or children has been diagnosed with Brugada syndrome – this may mean you’re also at risk
- a close family member has died suddenly with no explanation – this can sometimes be the result of an undiagnosed heart problem like Brugada syndrome
They can refer you to a specialist heart doctor for some simple tests to check if you have Brugada syndrome or any other heart problem.
If you’ve already been diagnosed with Brugada syndrome, contact your specialist as soon as possible if you experience any symptoms.
A 2012 study suggested that the quality of symptoms prior to syncope can predict a benign or malignant cause in patients with Brugada syndrome. Specifically, the lack of a prodrome (early symptom indicating the onset of a disease or illness) was more common in patients with ventricular fibrillation documented as the cause of syncope 4.
The physical examination is usually normal in patients with Brugada syndrome. Nevertheless, physical examination is required to rule out other possible cardiac causes of syncope or cardiac arrest in an otherwise healthy patient (e.g, heart murmurs from hypertrophic cardiomyopathy or from a valvular or septal defect).
Three ECG patterns have been described in Brugada syndrome 5 (see Figure 1 and Table 1 below). Placing the right precordial leads in the second intercostal space has been proposed to add sensitivity to the ECG diagnosis of Brugada syndrome 6. Exercise stress testing may suppress ECG changes and arrhythmias.
Figure 1. Brugada syndrome ECG
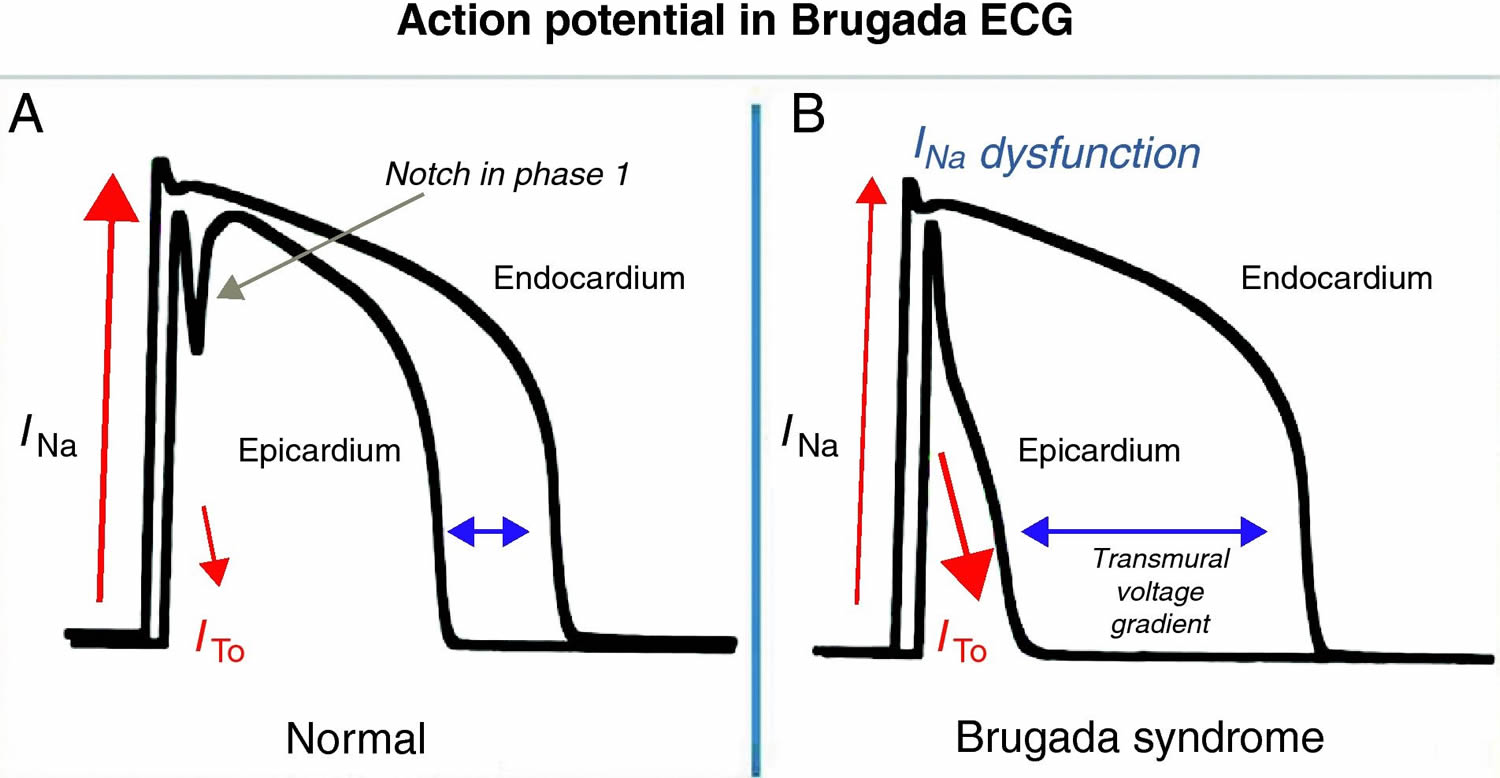
Figure 2. Brugada syndrome ECG
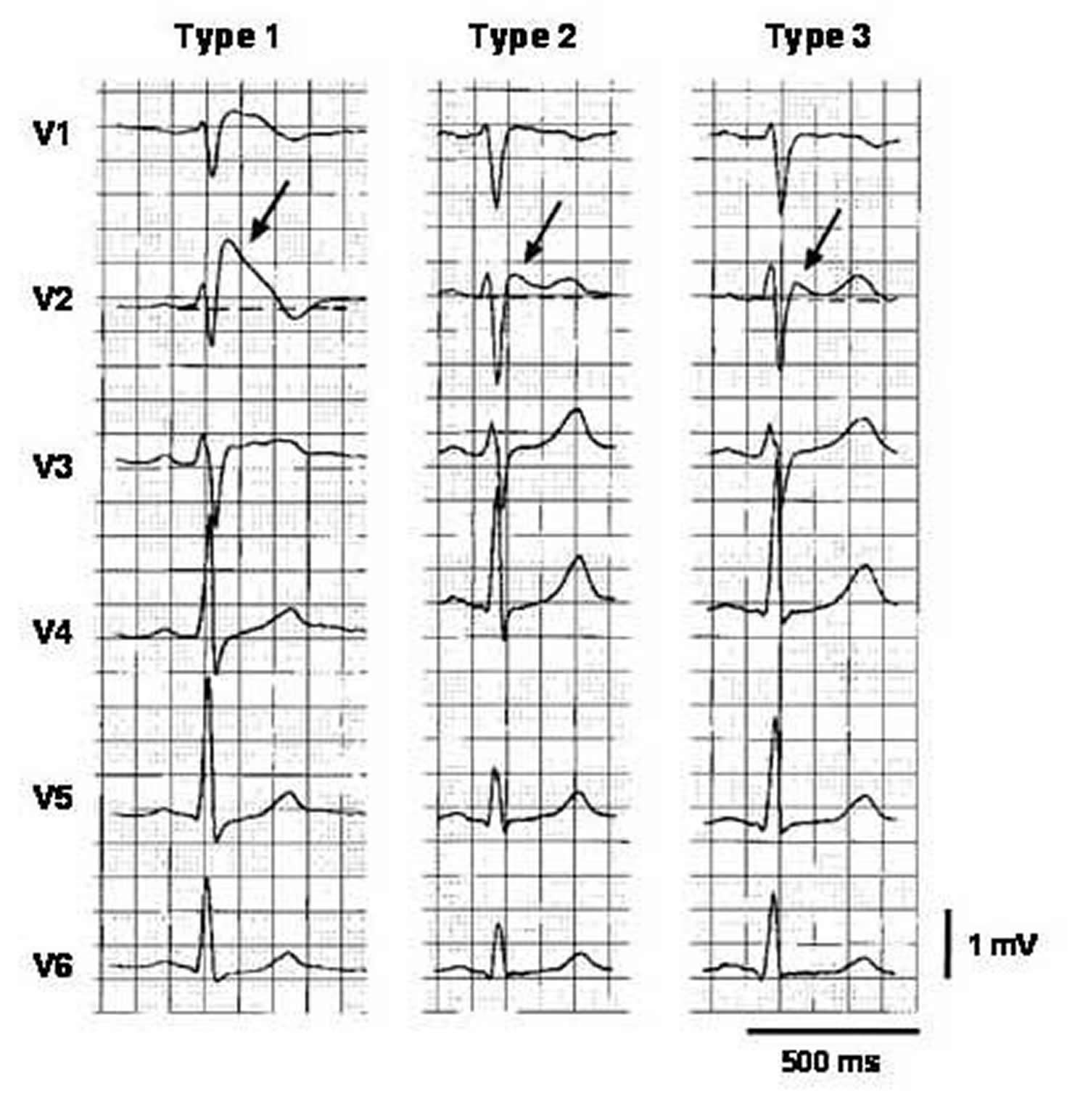 Note: Three types of ST-segment elevation in Brugada syndrome, as shown in the precordial leads on ECG in the same patient at different times. Left panel shows a type 1 ECG pattern with pronounced elevation of the J point (arrow), a coved-type ST segment, and an inverted T wave in V1 and V2. The middle panel illustrates a type 2 pattern with a saddleback ST-segment elevated by >1 mm. The right panel shows a type 3 pattern in which the ST segment is elevated <1 mm. According to a consensus report 7, the type 1 ECG pattern is diagnostic of Brugada syndrome.
Note: Three types of ST-segment elevation in Brugada syndrome, as shown in the precordial leads on ECG in the same patient at different times. Left panel shows a type 1 ECG pattern with pronounced elevation of the J point (arrow), a coved-type ST segment, and an inverted T wave in V1 and V2. The middle panel illustrates a type 2 pattern with a saddleback ST-segment elevated by >1 mm. The right panel shows a type 3 pattern in which the ST segment is elevated <1 mm. According to a consensus report 7, the type 1 ECG pattern is diagnostic of Brugada syndrome.Figure 3. Right Precordial ECG Traces From Blood Relatives of Brugada syndrome During Ajmaline Provocation
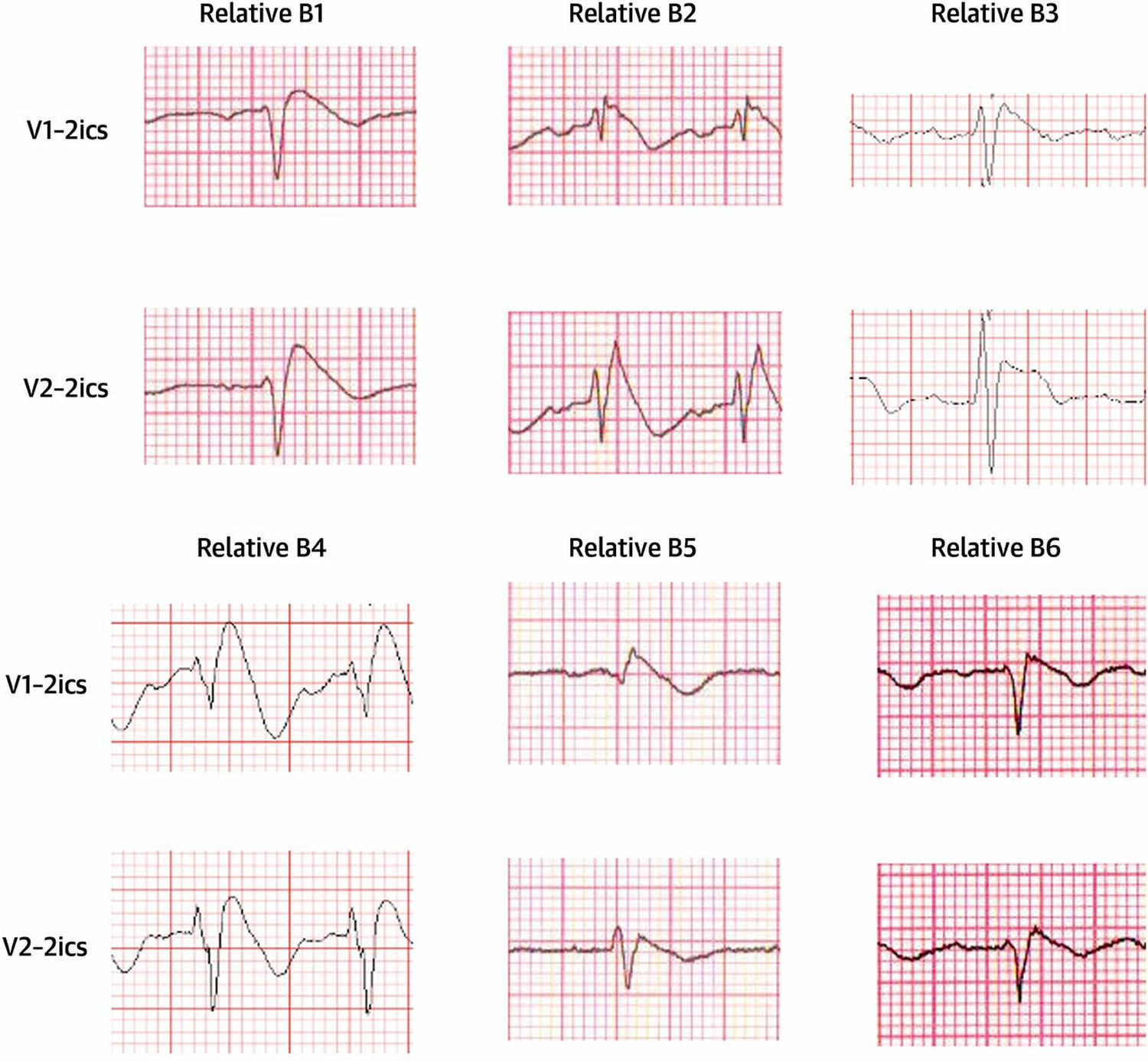
Note: ECG traces acquired following cranial displacement of electrode positions V1 and V2 into 2ics. 2ics = second intercostal space
Table 1. ECG Patterns in Brugada Syndrome
| Characteristic | Type 1 | Type 2 | Type 3 |
| J wave amplitude | ≥2 mm | ≥2 mm | ≥2 mm |
| T wave | Negative | Positive or biphasic | Positive |
| ST-T configuration | Cove-type | Saddleback | Saddleback |
| ST segment, terminal portion | Gradually descending | Elevated by ≥1 mm | Elevated by < 1 mm |
Figure 4. Brugada syndrome types ECG
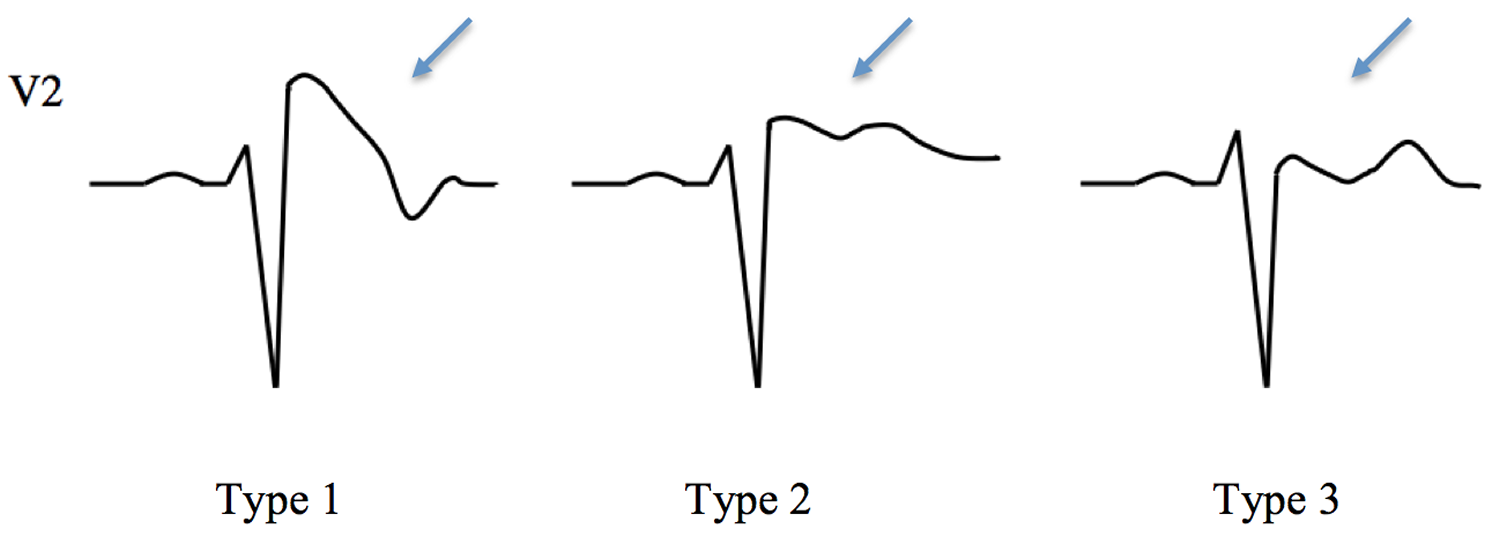
Recently, the QRS duration on 12-lead ECG has been suggested as a risk marker for vulnerability to dangerous arrhythmias 8. Inferolateral repolarization abnormalities have also been proposed to be a marker of risk 9.
Asymptomatic patients with a type 1 ECG pattern on routine ECG represent a difficult case. According to the latest consensus guidelines, a clinical electrophysiologist should evaluate patients in this situation 7. Patients should be risk-stratified using the techniques described below, and a decision on implantable cardioverter-defibrillator implantation should be made accordingly.
Brugada syndrome complications
Complications of Brugada syndrome require emergency medical care. They include:
- Sudden cardiac arrest. If not treated immediately, this sudden loss of heart function, breathing and consciousness, which often occurs while sleeping, is fatal. With fast, appropriate medical care, survival is possible. Administering cardiopulmonary resuscitation (CPR) — rapid compressions to the chest — and an external shock from an automatic external defibrillator can improve the chances of survival until emergency personnel arrive.
- Fainting (syncope). If you have Brugada syndrome and you faint, seek emergency medical attention.
Brugada syndrome causes
Brugada syndrome is a heart rhythm disorder. Each beat of your heart is triggered by an electrical impulse generated by special cells in the right upper chamber of your heart. Tiny pores, called channels, on each of these cells direct this electrical activity, which makes your heart beat.
Brugada syndrome can be caused by mutations in one of several genes. The most commonly mutated gene in this condition is SCN5A, which is altered in approximately 30 percent of affected individuals. This gene provides instructions for making a sodium channel, which normally transports positively charged sodium atoms (ions) into heart muscle cells. This type of ion channel plays a critical role in maintaining the heart’s normal rhythm. Mutations in the SCN5A gene alter the structure or function of the channel, which reduces the flow of sodium ions into cells. A disruption in ion transport alters the way the heart beats, leading to the abnormal heart rhythm and and spin electrically out of control in an abnormally fast and dangerous rhythm (ventricular fibrillation), which is characteristic of Brugada syndrome.
As a result, your heart doesn’t pump effectively and not enough blood travels to the rest of your body. This will cause fainting if that rhythm lasts for only a short time or sudden cardiac death if the heart remains in that bad rhythm.
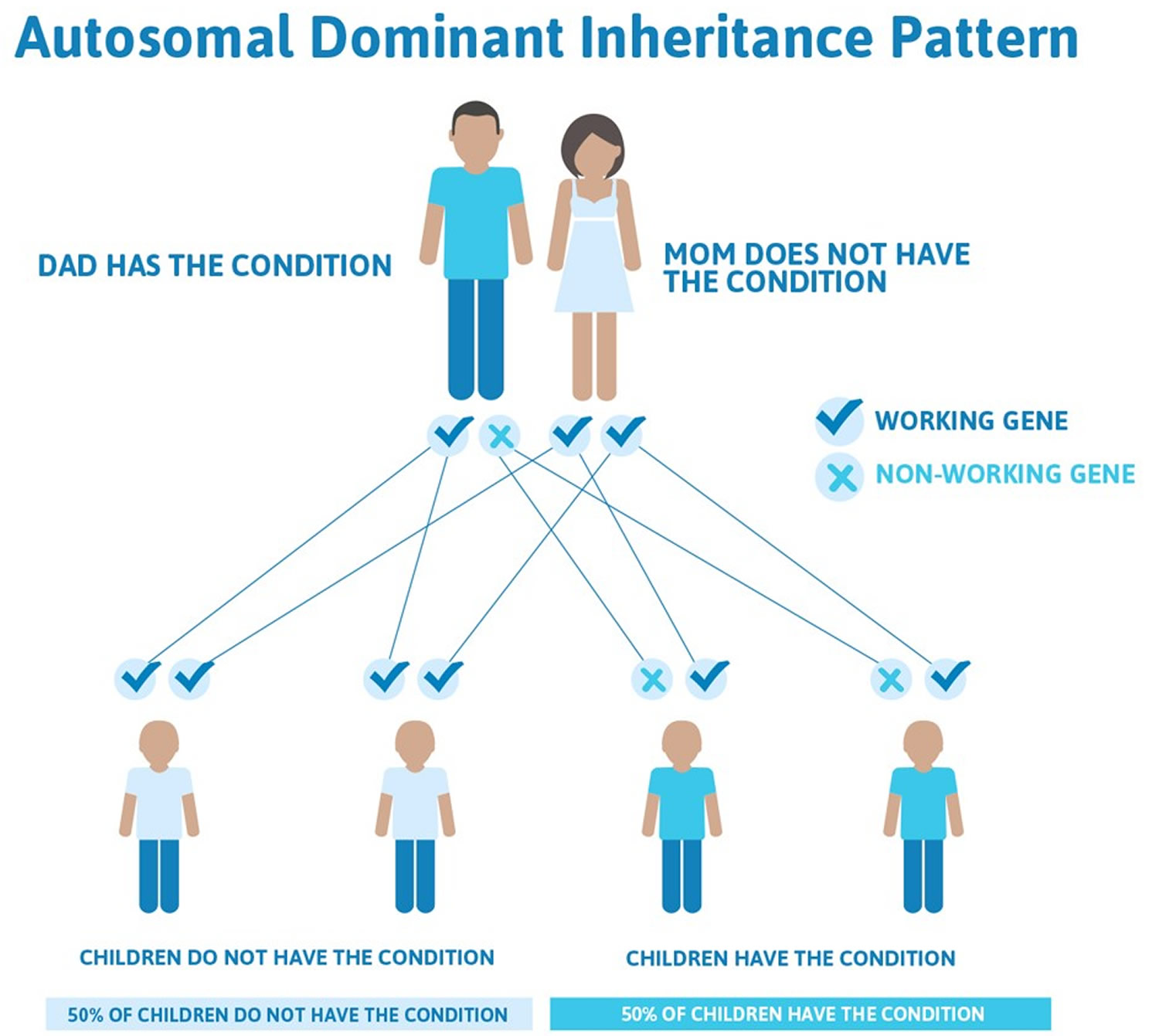
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition. Other cases may result from new mutations in the gene. These cases occur in people with no history of the disorder in their family.
Mutations in other genes can also cause Brugada syndrome. Together, these other genetic changes account for less than two percent of cases of the condition. Some of the additional genes involved in Brugada syndrome provide instructions for making proteins that ensure the correct location or function of sodium channels in heart muscle cells. Proteins produced by other genes involved in the condition form or help regulate ion channels that transport calcium or potassium into or out of heart muscle cells. As with sodium channels, proper flow of ions through calcium and potassium channels in the heart muscle helps maintain a regular heartbeat. Mutations in these genes disrupt the flow of ions, impairing the heart’s normal rhythm.
In affected people without an identified gene mutation, the cause of Brugada syndrome is often unknown, but it may also result from a hard-to-detect structural abnormality in your heart, imbalances in chemicals that help transmit electrical signals through your body (electrolytes). In some cases, certain drugs may cause a nongenetic (acquired) form of the disorder. Drugs that can induce an altered heart rhythm include cocaine and certain prescription medications used to treat some forms of arrhythmia, a condition called angina (which causes chest pain), high blood pressure, depression, and other mental illnesses. Abnormally high blood levels of calcium (hypercalcemia) or potassium (hyperkalemia), as well as unusually low potassium levels (hypokalemia), also have been associated with acquired Brugada syndrome. In addition to causing a nongenetic form of this disorder, these factors may trigger symptoms in people with an underlying mutation in SCN5A or another gene.
Figure 5. Brugada syndrome – autosomal dominant inheritance pattern
Risk factors for Brugada syndrome
Risk factors for Brugada syndrome include:
- Family history of Brugada syndrome. If other family members have had Brugada syndrome, you’re at an increased risk of having the condition.
- Being male. Adult men are more frequently diagnosed than are women. In young children and adolescents, however, boys and girls are diagnosed at about the same rate.
- Race. Brugada syndrome occurs more frequently in Asians than in other races.
- Fever. While having a fever doesn’t cause Brugada syndrome by itself, fever can irritate the heart and stimulate a Brugada-triggered faint or sudden cardiac arrest, especially in children.
Brugada syndrome signs and symptoms
Signs and symptoms in patients with Brugada syndrome may include the following:
- Syncope and cardiac arrest: Most common clinical manifestations; in many cases, cardiac arrest occurs during sleep or rest
- Nightmares or thrashing at night
- Asymptomatic, but routine ECG shows ST-segment elevation in leads V1-V3
- Associated atrial fibrillation (20%) 2
- Fever: Often reported to trigger or exacerbate clinical manifestations
The lack of a prodrome has been reported to be more common in patients with ventricular fibrillation documented as the cause of syncope in patients with Brugada syndrome 4.
Brugada syndrome diagnosis
A board-certified cardiologist who specializes in cardiac arrhythmic disorders (i.e, a clinical electrophysiologist) should evaluate patients with suspected Brugada syndrome. Consultation with a genetic counselor is indicated for genetic screening and counseling of patients and their relatives.
Many patients with Brugada syndrome are young and otherwise healthy and may present with syncope. Patients with syncope should not be assumed to have a benign condition, and a 12-lead ECG should be performed.
Most patients with Brugada syndrome have a normal physical examination. However, such an examination is necessary to exclude other potential cardiac causes of syncope or cardiac arrest in an otherwise healthy patient (e.g, heart murmurs from hypertrophic cardiomyopathy or from a valvular or septal defect).
A drug challenge with a sodium channel blocker should be considered in patients with syncope in whom no obvious cause is found. An experienced physician should interpret the ECGs, and an electrophysiologist should review them if possible.
Further testing may be indicated to exclude other diagnostic possibilities.
Additional tests
In patients with suspected Brugada syndrome, consider the following studies:
- 12-lead ECG in all patients with syncope
- Drug challenge with a sodium channel blocker in patients with syncope without an obvious cause
- Electrophysiologic study to determine the inducibility of arrhythmias for risk stratification
Laboratory tests that may aid in the diagnosis of Brugada syndrome include the following:
- Serum potassium and calcium levels: In patients presenting with ST-segment elevation in the right precordial leads. Both hypercalcemia and hyperkalemia may generate an ECG pattern similar to that of Brugada syndrome.
- Potassium and calcium levels: ECG patterns in patients with hypercalcemia and hyperkalemia similar to that of Brugada syndrome
- Creatine kinase-MB (CK-MB) and troponin levels: In patients with symptoms compatible with an acute coronary syndrome. Elevations indicate cardiac injury.
- Genetic testing for a mutation in SCN5A. Patients with high likelihood of Brugada syndrome may be genetically tested for a mutation in SCN5A, which codes for the alpha subunit Nav 1.5 of the cardiac sodium channel. The results of this test support the clinical diagnosis and are important for the early identification of family members at potential risk. However, the yield of genetic testing remains relatively low at this time, with mutations in SCN5A found in only 11-28% of index cases 10.
Further testing may be indicated to exclude other diagnostic possibilities.
Imaging studies
Echocardiography and/or MRI, primarily are needed to exclude arrhythmogenic right ventricular cardiomyopathy, as well as to assess for other potential causes of arrhythmias, such as hypertrophic cardiomyopathy, unsuspected myocardial injury, myocarditis, or aberrant coronary origins.
Brugada Syndrome Guidelines Summary
In its 2013 expert consensus statement on inherited primary arrhythmia syndromes, the Heart Rhythm Society/European Heart Rhythm Association/Asia Pacific Heart Rhythm Society 11 recommended a diagnosis of Brugada syndrome when the following criteria are met:
- ST-segment elevation with type I morphology ≥2 mm in ≥1 lead among the right precordial leads V1,V2 positioned in the 2nd, 3rd, or 4th intercostal space occurring either spontaneously or after provocative drug test with intravenous administration of Class I antiarrhythmic drugs
- Type 2 or type 3 ST-segment elevation in ≥1 lead among the right precordial leads V1,V2 positioned in the 2nd, 3rd, or 4th intercostal space when a provocative drug test with intravenous administration of class I antiarrhythmic drugs induces a type I ECG morphology
In 2015, the European Society of Cardiology 12 released guidelines for the management of ventricular arrhythmias and the prevention of sudden cardiac death that included the following specific recommendation for diagnosis of Brugada syndrome.
- Brugada syndrome is diagnosed in patients with ST-segment elevation with type 1 morphology ≥2 mm in one or more leads among the right precordial leads V1 and/or V2 positioned in the second, third, or fourth intercostal space, occurring either spontaneously or after provocative drug test with intravenous administration of sodium channel blockers (such as ajmaline, flecainide, procainamide or pilsicainide).
Genetic testing
In 2011, the Heart Rhythm Society and the European Heart Rhythm Association issued a joint expert consensus statement on genetic testing for channelopathies and cardiomyopathies with the following recommendations for Brugada syndrome testing 13:
- Consider comprehensive or BrS1 (SCN5A) targeted genetic testing for individuals with strong clinical index of suspicion for Brugada syndrome based on clinical history, family history, and expressed electrocardiographic phenotype.
- Mutation specific genetic testing for family members following identification of BrS mutation in an index case.
- Genetic testing is not indicated in the setting of an isolated type 2 or type 3 Brugada ECG pattern.
Brugada syndrome treatment
To date, the only treatment that has proven effective in treating ventricular tachycardia and fibrillation and preventing sudden death in patients with Brugada syndrome is implantation of an automatic implantable cardiac defibrillator 7.
Indications for implantable cardiac defibrillator implantation were published in the report of the Second Consensus Conference on Brugada syndrome 14. For patients at the two extremes of risk stratification, the decision to implant or not to implant an implantable cardiac defibrillator is relatively straightforward.
Patients with Brugada syndrome and a history of cardiac arrest must be treated with an implantable cardiac defibrillator. In contrast, asymptomatic patients with no family history of sudden cardiac death can be managed conservatively with close follow-up, and implantable cardiac defibrillator implantation is not recommended. Patients with intermediate clinical characteristics present the greatest challenge. For details about risk stratification and indications for implantable cardiac defibrillator implantation, readers are referred to the Second Consensus Conference report 14.
Patients with syncope or cardiac arrest and suspected or diagnosed Brugada syndrome must be hospitalized. Continuous cardiac monitoring is necessary until definitive treatment (i.e, implantable cardiac defibrillator placement) can be provided.
No pharmacologic therapy has been proven to reduce the occurrence of ventricular arrhythmias or sudden death; however, theoretically, drugs that counteract the ionic current imbalance in Brugada syndrome could be used to treat it. For example, quinidine, which blocks the calcium-independent transient outward potassium current, has been shown to normalize the ECG pattern in patients with Brugada syndrome 15. However, quinidine also blocks sodium (Na) currents, which could have contrary effects.
Management and prevention of sudden cardiac death
The following is a summary of recommendation included in the 2015 European Society of Cardiology guidelines 12 for management of of long QT syndrome and preventions of sudden cardiac death.
Lifestyle changes:
- Avoidance of drugs that may induce ST-segment elevation in right precordial leads
- Avoidance of excessive alcohol intake and large meals
- Prompt treatment of any fever with antipyretic drugs.
Implantable cardiac defibrillator implantation in patients who are survivors of an aborted cardiac arrest and/or have documented spontaneous sustained ventricular tachycardia (VT).
- Consider implantable cardiac defibrillator in patients with a spontaneous diagnostic type I ECG pattern and history of syncope.
- Consider quinidine or isoproterenol to treat electrical storms.
- Consider quinidine for patients who qualify for an implantable cardiac defibrillator but present a contraindication or refuse it and in patients who require treatment for supraventricular arrhythmias.
- Consider implantable cardiac defibrillator in patients who develop ventricular fibrillation during programmed ventricular stimulation with two or three extra stimuli at two sites.
- Consider catheter ablation in patients with a history of electrical storms or repeated appropriate implantable cardiac defibrillator shocks.
The 2013 Heart Rhythm Society/European Heart Rhythm Association/Asia Pacific Heart Rhythm Society guidelines 11 offer consistent recommendations. In addition, the guidelines find implantable cardiac defibrillator implantation is not indicated in asymptomatic Brugada syndrome patients with a drug-induced type I ECG and on the basis of a family history of sudden cardiac death alone.
A scientific statement published in 2015 by the American Heart Association and the American College of Cardiology on athletic competition by persons with known or suspected cardiac channelopathies includes the following recommendations related to Brugada syndrome 16:
- A heart rhythm specialist or genetic cardiologist experienced in cardiac channelopathies should conduct a thorough evaluation of an athlete in whom such a disorder has been diagnosed or is suspected.
- Symptomatic athletes with any suspected or diagnosed cardiac channelopathy should be restricted from all competitive sports until a comprehensive evaluation has been completed, the athlete and his or her family are well informed, a treatment program has been implemented, and the athlete has been asymptomatic on therapy for 3 months.
- Asymptomatic athletes who are genotype-positive/phenotype-negative for Brugada syndrome can be allowed to take part in all competitive sports if precautionary measures are taken, such as avoidance of drugs that exacerbate Brugada syndrome, electrolyte/hydration replenishment and avoidance of dehydration, avoidance or treatment of hyperthermia from febrile illnesses or training-related heat exhaustion or heat stroke, possession of a personal automatic external defibrillator as part of the athlete’s personal sports safety gear and establishment of an emergency action plan with the appropriate school or team officials.
- Competitive sports participation may be considered for an athlete with either previously symptomatic or electrocardiographically evident Brugada syndrome, assuming appropriate precautionary measures and disease-specific treatments are in place and the athlete has been asymptomatic on treatment for at least 3 months (although treatment involving an implantable cardioverter-defibrillator is subject to additional recommendations).
Physical activity
Because regular physical activity may increase vagal tone, sport may eventually enhance the likelihood of athletes with Brugada syndrome to have ventricular fibrillation and sudden cardiac death at rest or during recovery after exercise. Accordingly, Pelliccia et al 17 recommended that patients with a definite diagnosis of Brugada syndrome be restricted from participation competitive sports. However, no direct evidence supported this recommendation, and it was unclear whether asymptomatic carriers of SCN5A mutations should also be so restricted.
A scientific statement published in 2015 by the American Heart Association and the American College of Cardiology on athletic competition by persons with known or suspected cardiac channelopathies included the following recommendations related to Brugada syndrome 16:
- That a heart rhythm specialist or genetic cardiologist experienced in cardiac channelopathies conduct a thorough evaluation of an athlete in whom such a disorder has been diagnosed or is suspected
- That asymptomatic athletes who are genotype-positive/phenotype-negative for long-QT syndrome, catecholaminergic polymorphic ventricular tachycardia, Brugada syndrome, early repolarization syndrome, short-QT syndrome, or idiopathic ventricular fibrillation be allowed to take part in all competitive sports if precautionary measures, such as the avoidance of drugs that exacerbate Brugada syndrome, are undertaken
- That in an athlete with previously symptomatic or electrocardiographically evident Brugada syndrome, early repolarization syndrome, or short-QT syndrome, participation in competitive sports be considered if precautionary measures have been taken and disease-specific treatments are being administered and if the athlete has been asymptomatic on therapy for at least 3 months.
Medication
Theoretically, drugs that counteract the ionic current imbalance in Brugada syndrome could be used to treat it. For example, quinidine, which blocks the calcium-independent transient outward potassium current (Ito), has been shown to normalize the ECG pattern in patients with Brugada syndrome 15. However, quinidine also blocks sodium (Na) currents, which could have contrary effects.
Tedisamil, a potent independent transient outward potassium current (Ito) blocker without strong Na channel effects, may be more effective than quinidine 18. Isoproterenol, which boosts the L-type calcium current, can also counteract the ionic current imbalance 18.
Thus far, no drug therapy for Brugada syndrome is recommended because clinical trials have failed to convincingly prove effectiveness 14, 19, 20, 21. A number of medications can unmask the Brugada pattern on ECG and potentially exacerbate the clinical manifestations of Brugada syndrome. The Web site BrugadaDrugs.org has been established to educate patients and professionals about these potentially dangerous medications 22.
Long-Term Monitoring
A board-certified electrophysiologist should closely follow patients with Brugada syndrome. Taking a careful history is important, as not all syncope is necessarily arrhythmic in Brugada syndrome. For example, a clear prodrome suggesting vasovagal syncope does not suggest an adverse prognosis in an otherwise asymptomatic patient with a Brugada ECG pattern.
Planning a pregnancy if you have Brugada syndrome
Brugada syndrome is linked to genes you inherit from your parents. If you have the condition or have a family history of it, there’s a risk that any children you have could get it, too.
Speak to your heart specialist if you’re planning a pregnancy and:
- you or your partner have been diagnosed with Brugada syndrome
- you have a family history of the condition
You may be referred for genetic counseling to discuss the risk to your baby, and decide whether to have a blood test to look for a faulty gene linked to Brugada syndrome.
Coping and support
Finding out you have Brugada syndrome can be difficult. You may worry if your treatment will work or if other family members could be at risk. There are ways to cope with your feelings about your condition, including:
- Support groups. Finding out that you or a loved one has heart disease can be unnerving. Turning to friends and family for support is essential, but if you find you need more help, talk to your doctor about joining a support group. You may find that talking about your concerns with others who are experiencing the same difficulties can help.
- Continued medical checkups. If you have Brugada syndrome, it’s a good idea to regularly check in with your doctor to make sure you’re properly managing your heart condition. Regular checkups can help your doctor decide if you need to change your treatment and may help catch any new problems early.
- Brugada syndrome. https://emedicine.medscape.com/article/163751-clinical[↩]
- Bordachar P, Reuter S, Garrigue S, et al. Incidence, clinical implications and prognosis of atrial arrhythmias in Brugada syndrome. Eur Heart J. 2004 May. 25(10):879-84.[↩][↩]
- Brugada syndrome. https://ghr.nlm.nih.gov/condition/brugada-syndrome[↩]
- Take Y, Morita H, Toh N, Nishii N, Nagase S, Nakamura K, et al. Identification of high-risk syncope related to ventricular fibrillation in patients with Brugada syndrome. Heart Rhythm. 2012 May. 9(5):752-9.[↩][↩]
- Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, et al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation. 2002 Nov 5. 106(19):2514-9.[↩]
- Sangwatanaroj S, Prechawat S, Sunsaneewitayakul B, Sitthisook S, Tosukhowong P, Tungsanga K. New electrocardiographic leads and the procainamide test for the detection of the Brugada sign in sudden unexplained death syndrome survivors and their relatives. Eur Heart J. 2001 Dec. 22(24):2290-6.[↩]
- Antzelevitch C, Brugada P, Brugada J, Brugada R. Brugada syndrome: from cell to bedside. Curr Probl Cardiol. 2005 Jan. 30(1):9-54.[↩][↩][↩]
- Junttila MJ, Brugada P, Hong K, Lizotte E, DE Zutter M, Sarkozy A, et al. Differences in 12-lead electrocardiogram between symptomatic and asymptomatic Brugada syndrome patients. J Cardiovasc Electrophysiol. 2008 Apr. 19(4):380-3.[↩]
- Kamakura S, Ohe T, Nakazawa K, Aizawa Y, Shimizu A, Horie M, et al. Long-term prognosis of probands with Brugada-pattern ST-elevation in leads V1-V3. Circ Arrhythm Electrophysiol. 2009 Oct. 2(5):495-503.[↩]
- Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010 Jan. 7(1):33-46.[↩]
- [Guideline] Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013 Dec. 10 (12):1932-63.[↩][↩]
- [Guideline] Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015 Nov 1. 36 (41):2793-867.[↩][↩]
- [Guideline] Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011 Aug. 13 (8):1077-109.[↩]
- [Guideline] Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005 Feb 8. 111(5):659-70.[↩][↩][↩]
- Antzelevitch C, Brugada P, Brugada J, Brugada R, Nademanee K, Towbin J. The Brugada syndrome. Clinical approaches to tachyarrhythmias. Armonk NY: Futura Publishing Company; 1999.[↩][↩]
- [Guideline] Ackerman MJ, Zipes DP, Kovacs RJ, et al. Eligibility and Disqualification Recommendations for Competitive Athletes With Cardiovascular Abnormalities: Task Force 10: The Cardiac Channelopathies: A Scientific Statement From the American Heart Association and American College of Cardiology. Circulation. 2015 Dec 1. 132 (22):e326-9.[↩][↩]
- Pelliccia A, Fagard R, Bjørnstad HH, Anastassakis A, Arbustini E, Assanelli D, et al. Recommendations for competitive sports participation in athletes with cardiovascular disease: a consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J. 2005 Jul. 26(14):1422-45.[↩]
- Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol. 2001 Feb. 12(2):268-72.[↩][↩]
- Márquez MF, Salica G, Hermosillo AG, Pastelín G, Gómez-Flores J, Nava S, et al. Ionic basis of pharmacological therapy in Brugada syndrome. J Cardiovasc Electrophysiol. 2007 Feb. 18(2):234-40.[↩]
- Márquez MF, Salica G, Hermosillo AG, Pastelín G, Cárdenas M. Drug therapy in Brugada syndrome. Curr Drug Targets Cardiovasc Haematol Disord. 2005 Oct. 5(5):409-17.[↩]
- Yang F, Hanon S, Lam P, Schweitzer P. Quinidine revisited. Am J Med. 2009 Apr. 122(4):317-21.[↩]
- Postema PG, Wolpert C, Amin AS, Probst V, Borggrefe M, Roden DM, et al. Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up-to-date website (https://www.brugadadrugs.org/). Heart Rhythm. 2009 Sep. 6(9):1335-41.[↩]





{kind=link}