Contents
What is CHARGE syndrome,
CHARGE is an abbreviation for several of the features common in the disorder: Coloboma, Heart defects, Atresia choanae (also known as choanal atresia), Restricted growth and development, Genital abnormalities, and Ear abnormalities 1). CHARGE syndrome is a congenital condition (present from birth) that affects many areas of the body. The pattern of malformations varies among individuals with CHARGE syndrome, and the multiple health problems can be life-threatening in infancy. Neonates with CHARGE syndrome often have multiple life-threatening medical conditions. Feeding difficulties are a major cause of morbidity in all age groups. Affected individuals usually have several major characteristics or a combination of major and minor characteristics.
The major characteristics of CHARGE syndrome are common in this disorder and occur less frequently in other disorders. Most individuals with CHARGE syndrome have a gap or hole in one of the structures of the eye (coloboma), which forms during early development. A coloboma may be present in one or both eyes and may impair a person’s vision, depending on its size and location. Some affected individuals also have abnormally small or underdeveloped eyes (microphthalmia). In many people with CHARGE syndrome, one or both nasal passages are narrowed (choanal stenosis) or completely blocked (choanal atresia), which can cause difficulty breathing. Affected individuals frequently have cranial nerve abnormalities. The cranial nerves emerge directly from the brain and extend to various areas of the head and neck, controlling muscle movement and transmitting sensory information. Abnormal function of certain cranial nerves can cause swallowing problems, facial paralysis, a sense of smell that is diminished (hyposmia) or completely absent (anosmia), and mild to profound hearing loss. People with CHARGE syndrome also typically have middle and inner ear abnormalities, which can contribute to hearing problems, and unusually shaped external ears.
While the minor characteristics of CHARGE syndrome are common in this disorder, they are also frequently present in people without the disorder. The minor characteristics include heart defects; slow growth starting in late infancy; delayed development of motor skills, such as sitting unsupported and walking; and an opening in the lip (cleft lip) with or without an opening in the roof of the mouth (cleft palate). Affected individuals frequently have hypogonadotropic hypogonadism, which affects the production of hormones that direct sexual development. As a result, males with CHARGE syndrome are often born with an unusually small penis (micropenis) and undescended testes (cryptorchidism). Abnormalities of external genitalia are seen less often in affected females. Puberty can be incomplete or delayed in affected males and females. Another minor feature of CHARGE syndrome is tracheoesophageal fistula, which is an abnormal connection (fistula) between the esophagus and the trachea. Most people with CHARGE syndrome also have distinctive facial features, including a square-shaped face and differences in appearance between the right and left sides of the face (facial asymmetry). Affected individuals have a wide range of cognitive function, from normal intelligence to major learning disabilities with absent speech and poor communication.
Less common features of CHARGE syndrome include kidney abnormalities; immune system problems; abnormal curvature of the spine (scoliosis or kyphosis); and limb abnormalities, such as extra fingers or toes (polydactyly), missing fingers or toes (oligodactyly), an inward and upward turning foot (club foot), and abnormalities of the long bones of the arms and legs.
CHARGE syndrome is a rare disorder occuring in approximately 1 in 8,500 to 10,000 newborns. CHARGE syndrome affects males and females in equal numbers and has been seen in all races and on every continent. There are far more cases of CHARGE than those described in the medical literature. Many cases are misdiagnosed or undiagnosed, especially in children with fewer problems. Although many features of CHARGE are apparent at birth, some features will not become apparent for weeks, months, or perhaps years later.
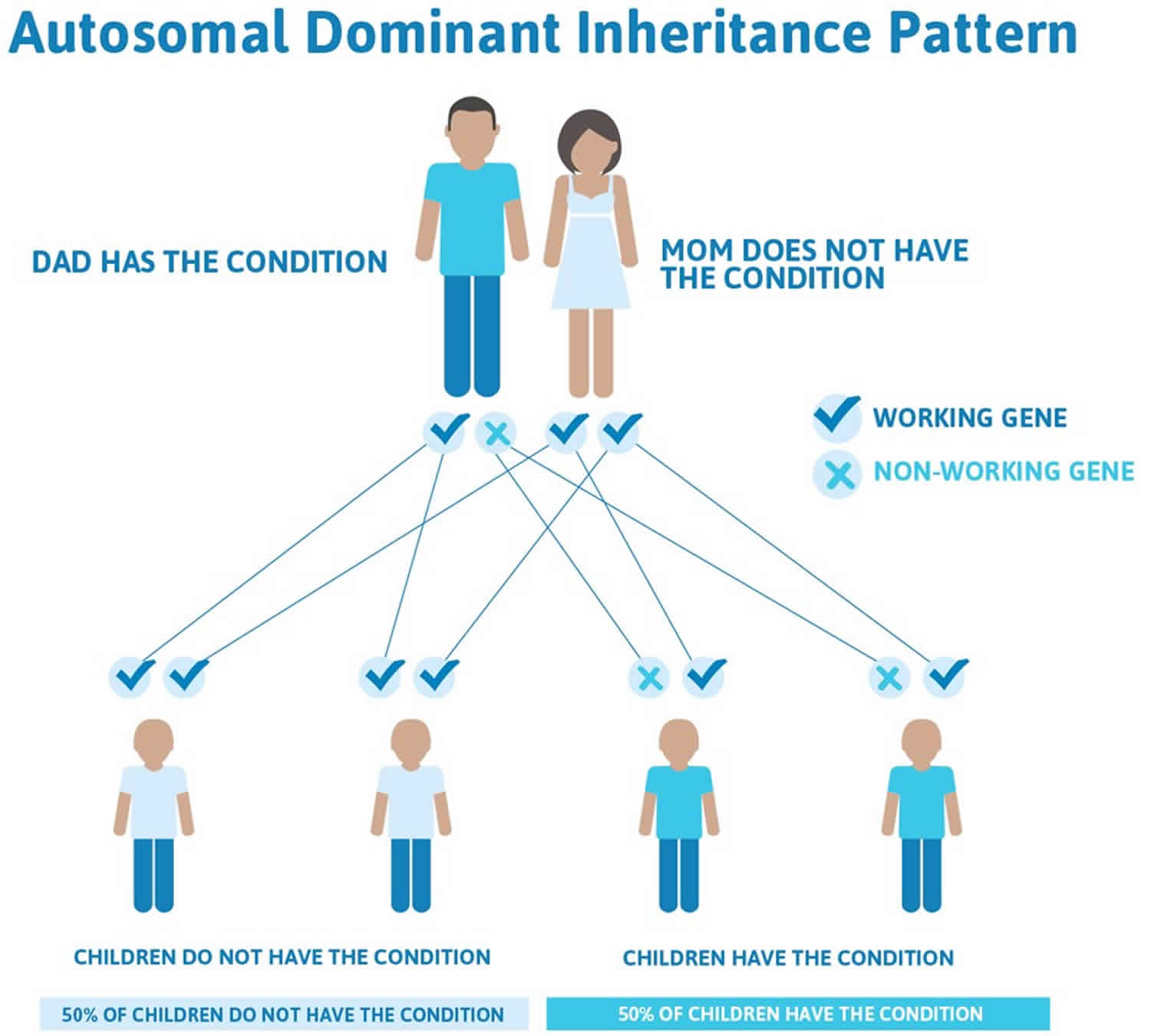
The CHD7 gene is the only gene in which mutations are known to cause CHARGE syndrome. CHARGE syndrome is usually not inherited, it typically occurrs due to a new (de novo) CHD7 gene mutation in the affected individual. Most cases of CHARGE syndrome represent simplex cases (i.e., a single occurrence in a family) from new mutations in the CHD7 gene and occur in people with no history of the disorder in their family 2). However, rare familial CHARGE syndrome cases are inherited in an autosomal dominant manner have been described. All individuals who have a CHD7 mutation have some features of CHARGE syndrome (i.e. penetrance is 100%). In rare instances, one parent may have mild features, and the family history may appear to be negative because of failure to recognize the mild features of the condition. The recurrence risk of CHARGE for parents with one affected child is low, around 2-3 percent 3). The recurrence risk for an adult with CHARGE syndrome to have an affected child may be as high as 50 percent (see Inheritance pattern below).
The risk to the siblings of an affected individual depends on the genetic status of the individual’s parents. If a parent of an affected child also has CHARGE syndrome, the risk for each sibling to inherit the condition is 50%. If neither parent is affected, the risk to each sibling of an affected child is estimated to be 2%-3%, most likely attributable to germline mosaicism. Prenatal diagnosis for pregnancies at increased risk is possible if the disease-causing CHD7 mutation has been identified in an affected family member 4).
The diagnosis of CHARGE syndrome is based on a combination of major and minor characteristics and temporal bone imaging 5). In more than half of all cases, mutations in the CHD7 gene cause CHARGE syndrome 6). When caused by a mutation in the CHD7 gene, it can be inherited in an autosomal dominant pattern; although most cases of CHARGE syndrome result from new (de novo) mutations in the gene and occur in people with no history of the condition in their family 7). CHD7 gene, encoding the chromodomain helicase DNA binding protein, is the only gene currently known to be associated with CHARGE syndrome. Sequence analysis of the CHD7 coding region detects pathogenic variants in most individuals with typical CHARGE syndrome (i.e., having the four major characteristics or three major and three minor characteristics). Overall, CHD7 gene analysis in individuals with either typical CHARGE syndrome or a milder phenotype (i.e., fewer major characteristics) detects pathogenic variants in about 65%-70% of cases.
Although there is no specific treatment or cure for CHARGE syndrome, there may be ways to manage the symptoms. A team of doctors is often needed to figure out the treatment options for each person 8).
Key points
- CHARGE syndrome remains a clinical diagnosis. Genetic confirmation can be made in the majority of patients by detection of heterozygous mutations in the CHD7 gene.
- Absent/hypoplastic semicircular canals are present in the majority of patients with CHARGE syndrome and are highly predictive of the presence of a CHD7 mutation.
- Early involvement of a cardiologist, ophthalmologist, endocrinologist, geneticist and ear, nose and throat surgeon is recommended.
- Complete thymic aplasia rarely occurs but leads to severe combined immune deficiency. Persistent lymphopenia in a patient with CHARGE syndrome must always be investigated. The prevalence of other immune defects in CHARGE syndrome remains unclear.
Is CHARGE syndrome inherited?
CHARGE syndrome is usually not inherited, typically occurring due to a new (de novo) gene mutation in the affected individual 9). However, rare familial cases inherited in an autosomal dominant manner have been described.
All individuals who have a CHD7 mutation have some features of CHARGE syndrome (i.e. penetrance is 100%). In rare instances, one parent may have mild features, and the family history may appear to be negative because of failure to recognize the mild features of the condition.
The risk to the siblings of an affected individual depends on the genetic status of the individual’s parents. If a parent of an affected child also has CHARGE syndrome, the risk for each sibling to inherit the condition is 50%. If neither parent is affected, the risk to each sibling of an affected child is estimated to be 2%-3% 10), most likely attributable to germline mosaicism. Prenatal diagnosis for pregnancies at increased risk is possible if the disease-causing CHD7 mutation has been identified in an affected family member 11).
Is genetic testing available for CHARGE syndrome?
Genetic testing is available for CHARGE syndrome. The CHD7 gene is the only gene in which mutations are known to cause CHARGE syndrome. The CHD7 mutation detection rate when sequence analysis is performed is estimated to be 65%-70% for all typical and suspected cases combined 12))).
GeneTests lists the names of laboratories that are performing clinical genetic testing for CHARGE syndrome. To view the contact information for these laboratories, click here (https://www.ncbi.nlm.nih.gov/gtr/). Please note that most of the laboratories listed through GeneTests do not accept direct contact from patients and their families. Therefore, if you are interested in learning more, you will need to work with a health care provider or a genetics professional.
How can I find a genetics professional in my area?
To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
What causes CHARGE syndrome
Mutations in the CHD7 13) gene cause most cases of CHARGE syndrome. The CHD7 gene provides instructions for making a protein that regulates gene activity (expression) by a process known as chromatin remodeling. Chromatin is the complex of DNA and protein that packages DNA into chromosomes. The structure of chromatin can be changed (remodeled) to alter how tightly DNA is packaged. When DNA is tightly packed, gene expression is lower than when DNA is loosely packed. Chromatin remodeling is one way gene expression is regulated during development.
Most mutations in the CHD7 gene lead to the production of an abnormal CHD7 protein that is broken down prematurely. Shortage of this protein is thought to disrupt chromatin remodeling and the regulation of gene expression. Changes in gene expression during embryonic development likely cause the signs and symptoms of CHARGE syndrome.
About one-third of individuals with CHARGE syndrome do not have an identified mutation in the CHD7 gene. The cause is unknown in these individuals, but researchers suspect that other genetic and/or environmental factors may be involved.
Inheritance pattern
The CHD7 gene is the only gene in which mutations are known to cause CHARGE syndrome. 97% of CHD7 mutations in CHARGE syndrome is usually not inherited, it typically occurrs due to a new (de novo) CHD7 gene mutation in the affected individual. Most cases of CHARGE syndrome represent simplex cases (i.e., a single occurrence in a family) from new mutations in the CHD7 gene and occur in people with no history of the disorder in their family 14). However, rare familial CHARGE syndrome cases are inherited in an autosomal dominant manner have been described. All individuals who have a CHD7 mutation have some features of CHARGE syndrome (i.e. penetrance is 100%). In rare instances, one parent may have mild features, and the family history may appear to be negative because of failure to recognize the mild features of the condition. The recurrence risk of CHARGE for parents with one affected child is low, around 2-3 percent 15). The recurrence risk for an adult with CHARGE syndrome to have an affected child may be as high as 50 percent (see Inheritance pattern below).
The risk to the siblings of an affected individual depends on the genetic status of the individual’s parents. If a parent of an affected child also has CHARGE syndrome, the risk for each sibling to inherit the condition is 50%. If neither parent is affected, the risk to each sibling of an affected child is estimated to be 2%-3% 16), most likely attributable to germline mosaicism. Prenatal diagnosis for pregnancies at increased risk is possible if the disease-causing CHD7 mutation has been identified in an affected family member 17).
However, autosomal dominant inheritance with transmission from parent to child has been reported in rare cases. In rare cases, an affected person inherits the mutation from an affected parent. The inheritance pattern of other cases of CHARGE syndrome is unknown. If neither parent is affected, the empiric risk to siblings of an index case (child with CHARGE syndrome) is approximately 2%-3%, most likely attributable to germline mosaicism. Prenatal diagnosis for pregnancies at increased risk is possible if the CHD7 pathogenic variant has been identified in an affected family member.
Figure 1. CHARGE syndrome mutations in the CHD7 gene – autosomal dominant inheritance pattern (in rare cases only)
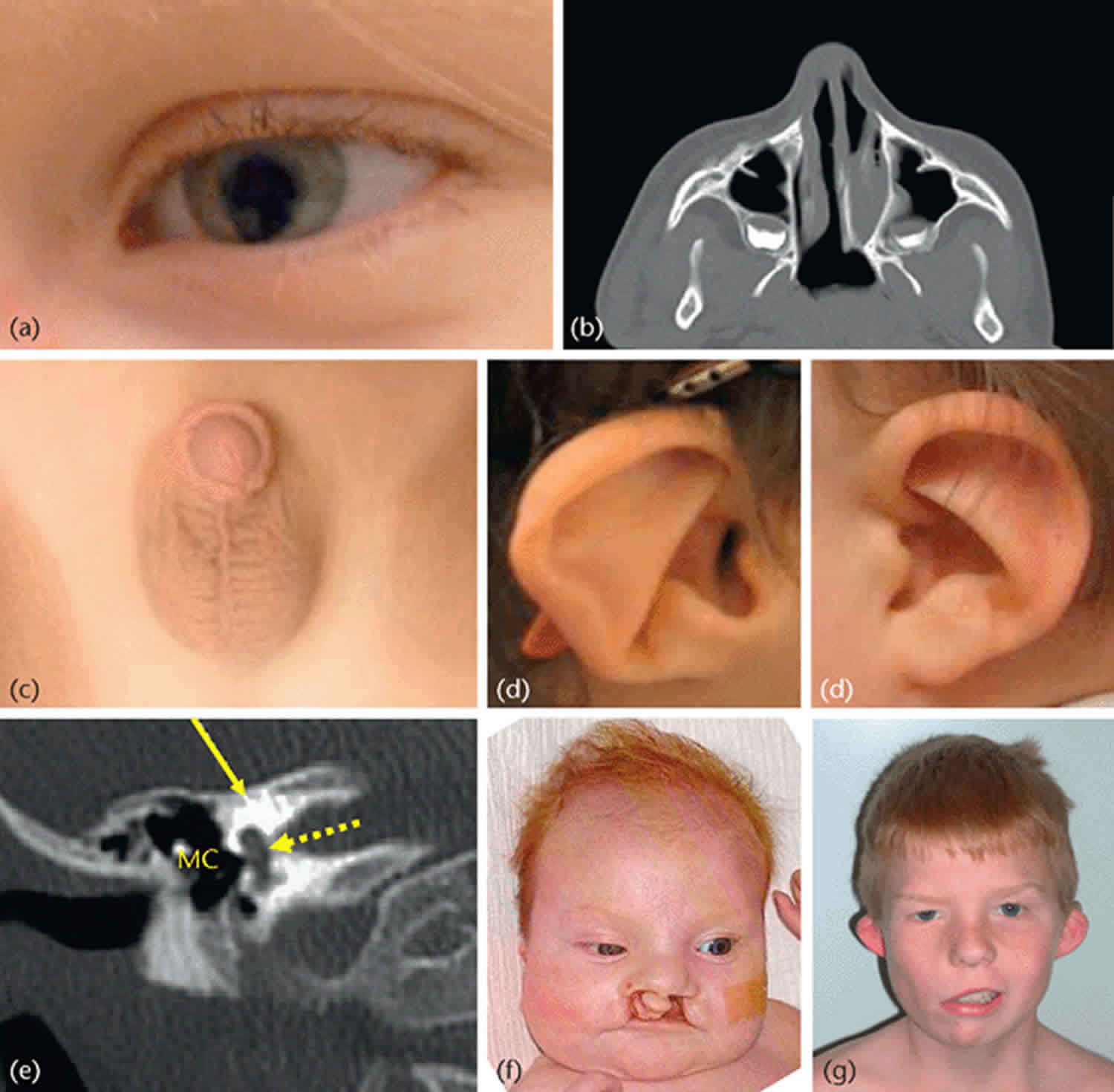
CHARGE syndrome symptoms
CHARGE syndrome affects multiple organ systems, resulting in multiple problems apparent at birth. Other characteristics of CHARGE syndrome may not become apparent until later in life. The diagnosis of CHARGE syndrome should be made by a medical geneticist based on the presence of at least one major criterion and several minor and/or occasional criteria of CHARGE syndrome (see below).
CHARGE syndrome is characterized by the following 18):
- Unilateral or bilateral coloboma of the iris, retina-choroid, and/or disc with or without microphthalmos (80%-90% of individuals)
- Unilateral or bilateral choanal atresia or stenosis (50%-60%)
- Cranial nerve dysfunction resulting in hyposmia or anosmia, unilateral or bilateral facial palsy (40%), impaired hearing, and/or swallowing problems (70%-90%)
- Abnormal outer ears, ossicular malformations, Mondini defect of the cochlea and absent or hypoplastic semicircular canals (>90%)
- Cryptorchidism in males and hypogonadotropic hypogonadism in both males and females
- Developmental delay
- Cardiovascular malformations (75%-85%)
- Growth deficiency (70%-80%)
- Orofacial clefts (15%-20%)
- Tracheoesophageal fistula (15%-20%)
Major Diagnostic Criteria
Features seen commonly in CHARGE syndrome (the 4 C’s), rarely in other conditions: Coloboma, Cranial nerve abnormalities, Choanal atresia, typical CHARGE Ear.
Table 1. Major Diagnostic Characteristics of CHARGE Syndrome
| Characteristics | Manifestations | Frequency |
|---|---|---|
| Ocular coloboma | Coloboma of the iris, retina, choroid, disc; microphthalmos | 80%-90% |
| Choanal atresia or stenosis 1, 2 | Unilateral/bilateral: bony or membranous atresia/stenosis | 50%-60% |
| Cranial nerve dysfunction or anomaly | I: hyposmia or anosmia | Frequent |
| VII: facial palsy (unilateral or bilateral) | >40% | |
| VIII: hypoplasia of auditory nerve | Frequent | |
| IX/X: swallowing problems with aspiration | 70%-90% | |
| Characteristic CHARGE syndrome ear | Outer ear: short, wide ear with little or no lobe, “snipped off” helix, prominent antihelix that is often discontinuous with tragus, triangular concha, decreased cartilage; often protruding and usually asymmetric (see Figure 2) 19) | 80%-100% |
| Middle ear: ossicular malformations 4 | ||
| Mondini defect of the cochlea 5 | ||
| Temporal bone abnormalities; absent or hypoplastic semicircular canals 5 |
Footnotes:
1. Cleft palate may substitute for this characteristic in some individuals.
2. The diagnosis is confirmed by non-enhanced CT scan in axial sections.
4. The combination of ossicular malformations and inner ear defects can result in a mixed (conductive and sensorineural) hearing loss with a wedge-shaped audiogram.
5. Most commonly determined by CT of the temporal bones
Coloboma
A coloboma is a cleft or failure to close of the eyeball during fetal development. This can result in a keyhole-shaped pupil (iris coloboma) and/or abnormalities in the retina, macula or optic nerve. Very small eyes (microphthalmia) or missing eyes (anophthalmia) can be severe forms of coloboma. Colobomas of the retina or optic nerve may result in significant vision loss, including blind spots, problems with depth perception or legal blindness. Colobomas occur most frequently in the retina and are present in at least 70-90% of patients with CHARGE syndrome. Examination of 38 eyes in 19 patients with CHARGE syndrome and confirmed CHD7 mutations revealed colobomata affected the posterior segment of 35 eyes in 18 patients. Both retinochoroidal and optic disk colobomata were observed bilaterally in 15 patients and unilaterally in 3 patients. The coloboma involved the macula totally or partially in 21 eyes of 13 patients. Bilateral large retinochoroidal colobomata are the typical ophthalmic feature of CHARGE syndrome in patients with confirmed CHD7 mutations; however, even eyes with large colobomata can form maculas. Many children with colobomas (even just an iris coloboma) may be sensitive to bright light (photophobia). Surgery cannot correct ocular colobomas. Near-sightedness or far-sightedness can be helped with glasses. Sunglasses and a hat with a protective bill can help the photophobia.
Cranial nerve abnormalities
Sensorineural (nerve) hearing loss in CHARGE syndrome is due to abnormalities in cranial nerve 8. Cranial CT scan often reveals a hypoplastic cochlea (81%) with absent semicircular canals in most cases. Hearing loss and difficulty with balance are the most common features associated with cochlear hypoplasia and absent semicircular canals. CHARGE syndrome is associated with characteristic external ears that tend to protrude and lack lobes. The hearing loss can range from a mild hearing loss to profound deafness. Hearing loss can be very difficult to measure in young children. Many children with CHARGE syndrome receive cochlear implants to aid their sensorineural hearing loss. Most also have balance problems (vestibular abnormalities) associated with absent semicircular canals, which is a key finding in making the diagnosis of CHARGE syndrome.
Most children with CHARGE syndrome have swallowing problems (cranial nerves 9/10). These swallowing problems include the inability to coordinate suck and swallow, leading to gagging and aspiration of food into the lungs (which can cause pneumonia). Many children require feeding via a gastrostomy tube (tube directly into the stomach through the abdominal wall) until they are able to swallow safely.
Many children with CHARGE syndrome have asymmetric facial palsy resulting in paralysis of one side of the face (cranial nerve 7). This results in a lack of facial expression, which is important when a child is working with teachers or therapists.
Most children with CHARGE syndrome have an absent or reduced sense of smell (cranial nerve 1), which complicates learning to eat normally. Most patients with CHARGE syndrome have absent or abnormal olfactory bulbs in MRI, leading to a diminished sense of smell. Smell-testing can predict the presence of hypogonadotropic hypogonadism. The combination of defective olfaction (anosmia or hyposmia) with hypogonadotropic hypogonadism (termed Kallman syndrome) results in small external genitalia. This is very common in CHARGE syndrome and warrants consultation with an endocrinologist.
Choanal atresia
Choanae are the passages from the back of the nose to the throat that make it possible to breathe through the nose. In about half of all children with CHARGE syndrome, these passages may be blocked (atresia) or narrowed (stenosis). Among 12 patients with bilateral choanal atresia, 10 had related malformations, 3 of which had CHARGE syndrome. Surgery can often correct these defects. Patients with unilateral atresia can usually be corrected with 1 surgical procedure at a later age (median 6 years, range 6 months to 18 years), while patients with the bilateral form need a median of 2.85 interventions at an early age (median 25 days, range 6 days-6 years). If both sides are affected, immediate measures must be taken to allow the newborn to breathe properly and prevent respiratory failure.
CHARGE ear
Most children with CHARGE syndrome have unusual external ears. The “typical CHARGE ear” is short and wide with little or no earlobe. The helix (outer fold) may end abruptly in mid-ear. The center of the ear (concha) is often very triangular in shape. The ears are often floppy and may stick out due to weak cartilage. The two ears often look different from each other. There are also typical findings in the middle ear in CHARGE, including malformed bones of the middle ear (93%) and incomplete cochlea (Mondini defect), which is diagnosed with an MRI scan. In many cases, the external ear can be unique enough to suspect the diagnosis of CHARGE before examining other features, and a temporal bone CT scan to look for absent semicircular canals and evaluate the choanae for atresia or stenosis should prompt mutation analysis of CHD7 to confirm the diagnosis.
Figure 2. CHARGE syndrome ear
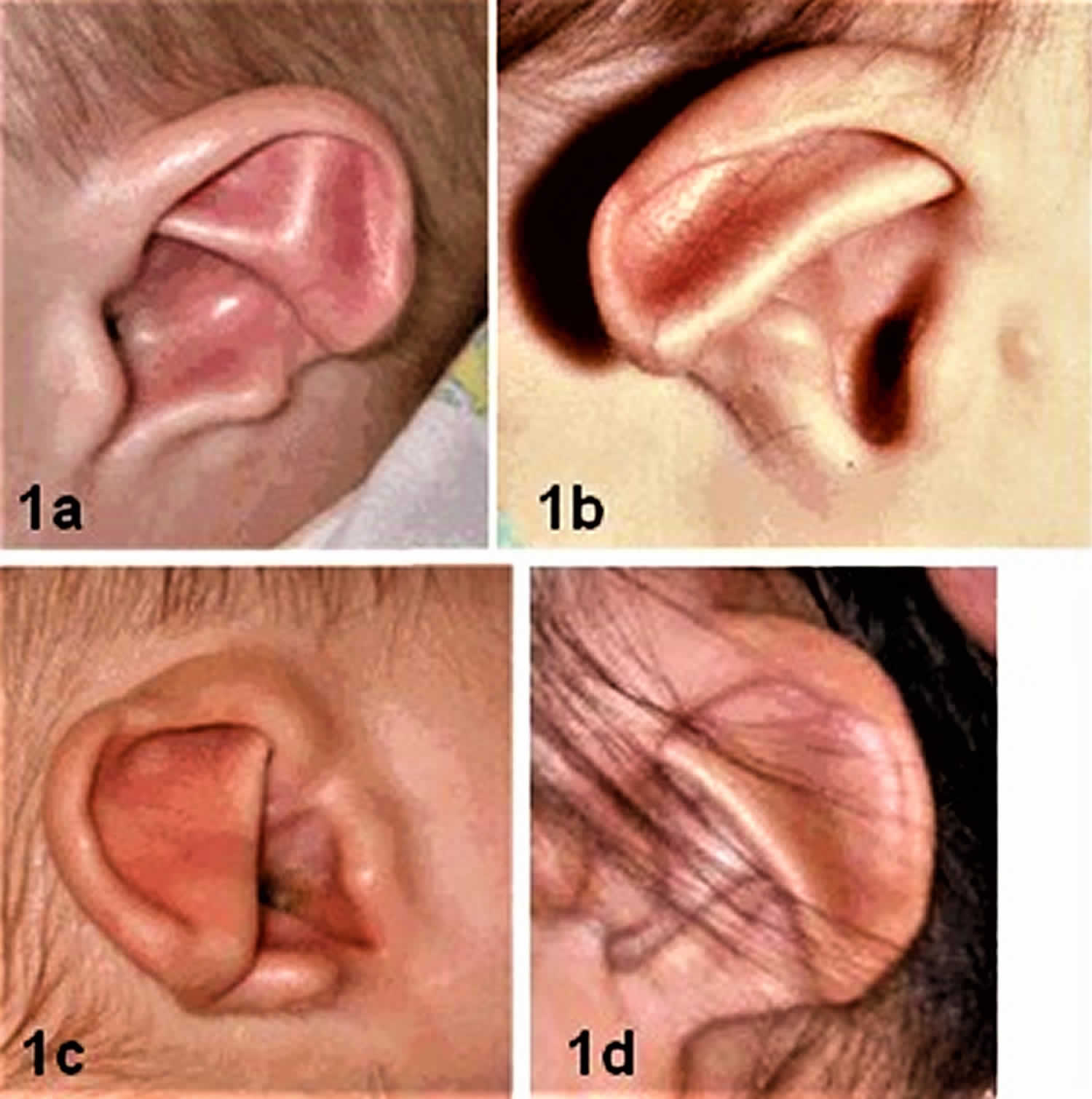
Footnote:
1a. Clipped-off helix, prominent antihelix that extends to the outer helical rim, antihelix discontinuous with the antitragus; absent lobe
1b. Antihelix discontinuous with the antitragus; very small lobe. Preauricular tag occurs occasionally.
1c. Clipped off helix, prominent antihelix that extends to helical margin and does not connect with antitragus, triangular concha and absent lobe
1d. Thin, unfolded helix, prominent inferior antihelix with notch between it and antitragus, rudimentary lobe
[Source 21) ]Hearing loss
Hearing loss is one of the most common features of CHARGE syndrome. Hearing loss can vary from mild to profound. The hearing loss can be difficult to quantify, requiring multiple brain stem audio evoked response tests over several months. Thelin et al 22) reported a characteristic wedge-shaped audiometric pattern of mixed hearing loss and verified that hearing loss is progressive in some individuals 23). The presence of facial paralysis was found to predict reliably the presence of sensorineural hearing loss 24).
The sensorineural component of the hearing loss is often associated with a Mondini malformation of the cochlea. Hypoplasia of the auditory nerve has also been described.
The conductive component of the hearing loss may result from malformed or absent ossicles, fixation of the ossicular chain to the wall of the tympanic cavity, absence of the stapedius muscle, absence of the oval window, and obliteration of the round window 25). The conductive component may fluctuate with middle ear disease.
Chronic recurrent otitis media is common.
Vestibular abnormalities
With appropriate imaging, abnormalities of the semicircular canals are found in as many as 95% of affected individuals 26).
Absence or hypoplasia of the semicircular canals impairs balance, especially when combined with visual loss. The resulting poor balance contributes to delays in motor development.
Minor Diagnostic Criteria
Features less specific to CHARGE syndrome and/or not consistent enough to be considered major: heart defects, genital abnormalities, kidney abnormalities, cleft lip or palate, tracheoesophageal fistula or esophageal atresia, poor growth, hypotonia, typical CHARGE syndrome face, and typical CHARGE syndrome hand.
Table 2. Minor Diagnostic Characteristics of CHARGE Syndrome
| Characteristics | Manifestations | Frequency |
|---|---|---|
| Genital hypoplasia | Males: micropenis, cryptorchidism Females: hypoplastic labia | 50%-60% |
| Males and females: delayed puberty secondary to hypogonadotropic hypogonadism | Frequent | |
| Developmental delay 1 | Delayed milestones, hypotonia | ≤100% |
| Cardiovascular malformation | Including conotruncal defects (e.g., tetralogy of Fallot), AV canal defects, and aortic arch anomalies | 75%-85% |
| Growth deficiency | Short stature, usually postnatal with or without growth hormone deficiency | 70%-80% |
| Orofacial cleft | Cleft lip and/or palate | 15%-20% |
| Tracheoesophageal (TE) fistula | TE defects of all types | 15%-20% |
| Distinctive facial features | Square face with broad prominent forehead, prominent nasal bridge and columella, flat midface (see Figure 3) 27) | 70%-80% |
Footnote:
1. May be primarily the result of illness, dual sensory impairment, and vestibular dysfunction
[Source 28) ]Heart defects
About 75-80% of children with CHARGE syndrome have congenital heart defects. Although all types of heart defects have been seen in children with CHARGE syndrome, the most common are tetralogy of Fallot (33%), VSD (ventricular septal defect), AV (atriventricular) canal defect, and aortic arch anomalies. The heart defects can range from an innocent murmur to life-threatening heart defects involving the outflow tracts of the heart. Most require medication and/or surgery. Severe heart defects are a major cause of death in children with CHARGE syndrome. The heart defects in CHARGE syndrome are similar to those seen in Deletion 22q11.2 syndrome.
Genital abnormalities
Most boys with CHARGE syndrome have a small penis, often with undescended testes (cryptorchidism). The urethral opening may not be at the end of the penis (hypospadias). Girls may have small labia. Among 46 boys with hypogonadotropic hypogonadism, 14 (30.4%) had Kallmann syndrome, 4 (8.7%) had CHARGE syndrome and 28 (60.9%) had hypogonadotropic hypogonadism without an olfaction deficit or olfactory bulb hypoplasia. Most children with CHARGE syndrome require hormone therapy to achieve puberty due to hypogonadotropic hypogonadism 29), and a pediatric endocrinologist should evaluate their pituitary gonadal axis.
Kidney abnormalities
About 40% of children with CHARGE syndrome have kidney abnormalities 30). These can include hydronephrosis (extra fluid in the kidneys) or reflux (backflow into the kidneys); horseshoe kidney; small or absent kidney; or multicystic dysplastic kidneys. All children with CHARGE should have a kidney ultrasound.
Cleft lip and/or cleft palate
About 25% of children with CHARGE have a cleft lip or cleft palate. The cleft lip can be one-sided or two-sided and may or not include the palate. A positive family history of any individual with an apparently isolated unilateral major CHARGE anomaly, or someone with a few of the minor features, should precipitate testing the affected child and both parents for CHD7. Some have cleft palate without cleft lip. Submucous cleft palate (just the muscle, not the bone in the roof of the mouth) may be hard to diagnose.
Tracheoesophageal Fistula/Esophageal atresia
About 15-20% of children with CHARGE syndrome are born with an esophageal atresia, where the food pipe is not connected to the stomach or with tracheoesophageal fistula, where there is a connection between the windpipe (trachea) and the food pipe (esophagus). Both of these conditions require surgery. In addition, the trachea may be weak or floppy due to weak cartilage. This can complicate surgery to treat these conditions.
Swallowing problems
Feeding can be associated with coughing, choking, nasal regurgitation, aspiration, and/or gastroesophageal reflux 31). Aspiration and swallowing dysfunction are common in children with CHARGE syndrome and are primarily the result of cranial nerve 9/10 abnormalities often complicated by choanal atresia or cleft palate.
Flexible endoscopic evaluation of swallowing and/or video swallow study often show pooling, premature spillage, poor hypopharyngeal motility, or laryngeal penetration 32). A large number of children require nasogastric or gastric (G-tube) feeding, often for several years. Swallowing may eventually improve spontaneously; however, some adults continue to avoid foods that are difficult to swallow.
Gastroesophageal reflux is common.
Poor growth
Although birth weight is usually normal, many children with CHARGE syndrome are small after birth. Sometimes this is due to nutrition problems, heart problems or multiple illnesses. Some children with CHARGE syndrome have growth hormone insufficiency, which can be evaluated with a growth hormone stimulation test 33).
Hypotonia of the trunk
Most children with CHARGE syndrome have upper body hypotonia (weakness). They are weak, especially in the trunk, and may have sloping shoulders. This weakness, especially combined with balance problems and/or vision problems, will delay walking. The average age of walking is about 3 or 4 years in children with CHARGE syndrome, and this results from the combination of hypotonia and diminished balance due to their underdeveloped semicircular canals.
Typical CHARGE face
Children with CHARGE syndrome often look similar to one another. The typical child has a square face, with broad prominent forehead, arched eyebrows, large eyes, occasional droopy eyelids, a prominent nasal bridge with square root, small nostrils, prominent nasal columella, flat midface, small mouth, occasional small chin, which improves with age. The face is often very asymmetric.
Figure 3. CHARGE syndrome face

Footnote:
Face
2a. Female age 2 1/2 years; square face, round eye, straight nose with broad nasal root, unilateral facial palsy
2b. Female age five years; mild expression of CHARGE facies; relatively square face, prominent columella of the nose. Note sloping shoulders.
2c. Male age seven years; square face, somewhat broad nasal root. Note prominent ear with unfolded helix and wide neck.
2d. Female age nine years; square face, round eyes, wide neck, sloping shoulders. Note lack of facial expression as a result of bilateral facial palsy.
2e. Male age 15 years. Note longer but still somewhat square face, wide neck with sloping shoulders.
2f. Female age 18 years; square, asymmetric face, prominent ears, head tilted back, wide neck, and sloping shoulders
[Source 34) ]Typical CHARGE hand
Many children with CHARGE syndrome have a small thumb, broad palm with “hockey-stick” palmar crease, and short fingers.
Figure 4. CHARGE syndrome hand
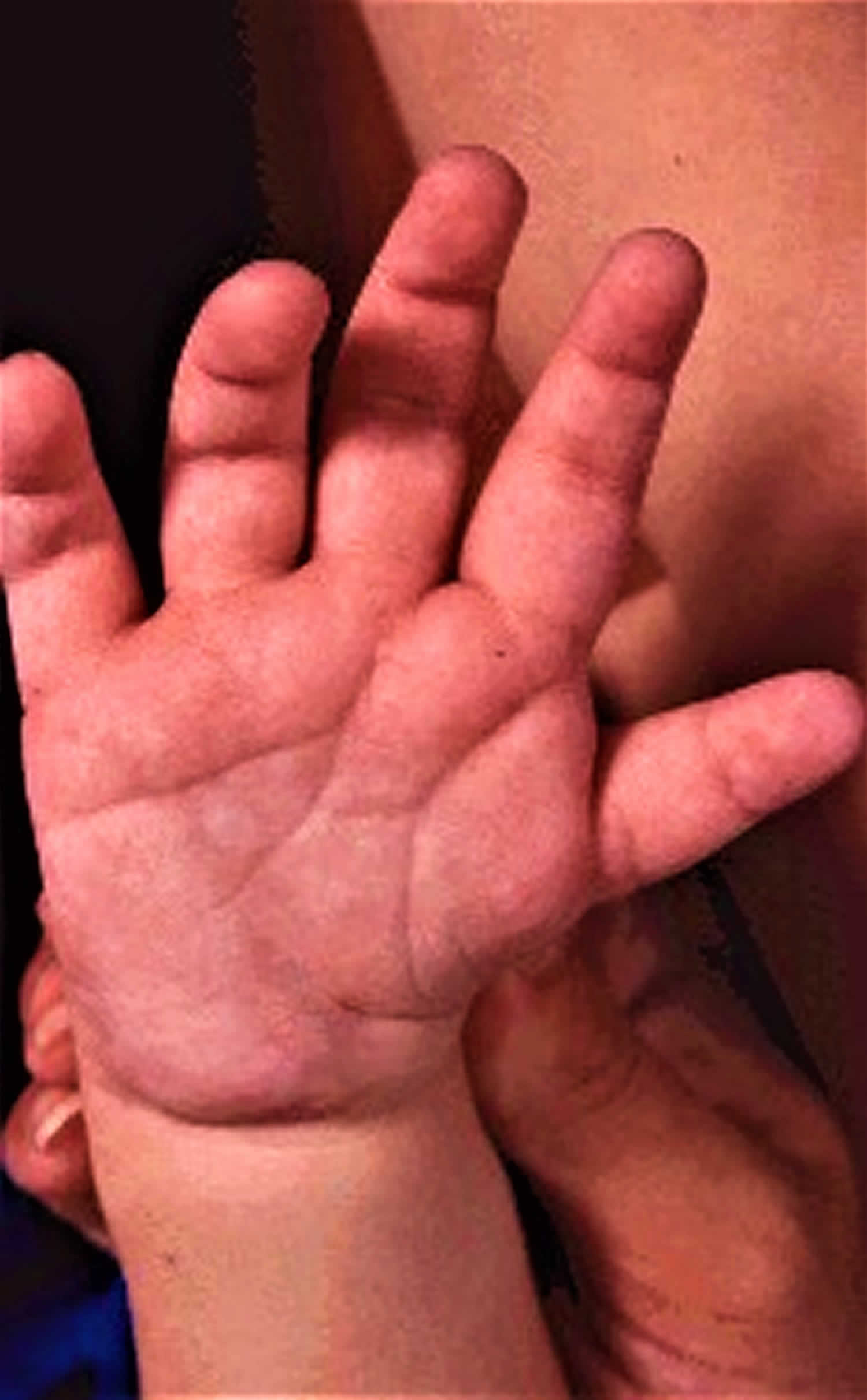
Footnote: Typical CHARGE hand: square hand, short fingers, finger-like thumb, hockey-stick palmar crease
[Source 35) ]Other Common Findings
These features may be important for management, but not very helpful in making the diagnosis. Brain abnormalities, including small head (microcephaly), enlarged cerebral ventricles or other abnormalities identified by brain imaging such as MRI or CT scan are occasionally seen 36). Apnea and seizures are rarely seen in children with CHARGE syndrome. Weak cartilage (as seen in the ears) can also affect the trachea (windpipe) making it weak. Sometimes the baby has a very weak cry due to laryngomalacia (weak vocal cords). A few children with CHARGE syndrome have missing or extra nipples. Some have a relatively wide neck, with occasional cervical vertebral abnormalities. Rarely, children with CHARGE syndrome have an umbilical hernia, omphalocele or limb abnormalities, such as abnormal thumbs or extra fingers.
A few children with CHARGE syndrome have DiGeorge sequence, consisting of a complex heart defect, immune deficiency, and abnormalities of the thyroid and parathyroid glands 37). Because these features are also seen in velocardiofacial syndrome, children with possible CHARGE syndrome and no mutation in CHD7 should have array comparative genomic hybridization testing done. Some children with CHARGE syndrome appear to have a poor immune response even, and the presence of hypocalcemia should prompt an immunologic evaluation. Most children with CHARGE syndrome and immunodeficiency have T-cell deficiency.
Many children with CHARGE syndrome develop scoliosis 38), even as relatively young children. This may be due in part to their weak upper body, but a skeletal survey should be carried out to exclude skeletal anomalies, particularly those of the cervical spine.
Dental anomalies include protruding jaw, overbite, hypodontia of permanent dentition, and poor mineralization of the enamel 39).
Sinusitis may be a major problem in childhood and in older children is often caused by “silent” gastroesophageal reflux. Sinusitis is a frequent cause of severe pain, often manifested by sudden changes in behavior, including apparently aggressive behaviors.
Developmental Delay
Most young children with CHARGE syndrome are developmentally delayed. Often, this is primarily due to sensory deficits (vision and/or hearing loss) and frequent illnesses and hospitalizations as infants and young children. In one report, mean age for head holding was five months, sitting independently 14.8 months, and walking unaided 33 months 40). Although developmentally delayed, many children with CHARGE syndrome will show significant catch up in later childhood, manifesting normal intellectual abilities, and ending up as independent adults. It is not possible to predict eventual development for any one child, and early intervention with a deaf-blind specialist is essential to remediate their sensory deficits and prevent behavioral problems. Regardless of the extent of inner ear anomalies and intellectual faculties, cochlear implantation with careful treatment planning can be a highly effective option for hearing rehabilitation in children with sensorineural hearing loss and CHARGE syndrome.
As children with CHARGE syndrome grow older, challenging behaviors can emerge. Some children display autistic-like behaviors such as hand waving or head banging. Often, these are attempts at communication by a child who has not yet developed language or other communication skills due to hearing and vision problems. These behaviors occur less often when a communication system (speech, signs, or a combination of both) is established. Older individuals with CHARGE syndrome may show signs of obsessive-compulsive disorder. Many children with CHARGE syndrome begin communication using sign language or some form of gestures and communication boards. Those with cochlear implants or hearing aids that bring hearing into the normal range will usually switch over to oral speech at some point. Learning signs first does not keep children from speaking later.
Speech/language delay. Language development is often delayed because of hearing loss and further exacerbated by reduced vision that impairs lip reading and perception of body language cues.
Cognitive development and psychological assessment. Delayed motor and/or language development cannot be used to predict cognitive potential of affected individuals 41).
Assessment of cognitive abilities is difficult because of lack of standardized tools to evaluate individuals with both visual and hearing impairment. Raqbi et al 42) showed that the intellectual performance of individuals with CHARGE syndrome ranged from major learning disability with no speech and poor communication to almost normal. They demonstrated that despite marked delay in motor milestones in children ages birth to three years, intellectual outcome in 50% was satisfactory. Only 25% of the studied group had a poor intellectual outcome. Raqbi et al 43) also showed that microcephaly, brain malformation, and extensive bilateral coloboma resulting in reduced vision were the only findings predictive of poor intellectual outcome. The results suggest that for approximately half of children with CHARGE syndrome, motor and speech/language delay is mainly secondary to multiple sensory deficits and not to CNS dysfunction.
Salem-Hartshorne & Jacob 44) showed that the range of adaptive behavior scores in individuals with CHARGE syndrome is broader and higher than previously reported. Those children with better walking skills and fewer medical problems scored higher on this scale than children with poorer walking skills and more medical problems. In this study, one half of the individuals obtained a standard score higher than 70 on the adaptive behavior scores at follow up; 13% scored above a standard score of 90.
Many adults with CHARGE syndrome are known to live independently and many are currently attending college, or are college graduates with advanced degrees 45).
Behavioral profile includes repetitive, obsessive-compulsive, aggressive, and self-abusive behaviors 46). Attention-deficit hyperactivity disorder (ADHD) is also seen in many individuals with CHARGE syndrome 47). Many behaviors regarded as aberrant or disruptive are attempts at communication about pain, unease, or frustration 48).
CHARGE syndrome life expectancy
Neonates with CHARGE syndrome often have multiple life-threatening medical conditions. Blake et al 49) reported poor survival if one or more of the following were present: cyanotic cardiac lesions, bilateral posterior choanal atresia, and tracheoesophageal fistula. In another study, poor life expectancy correlated with male gender, central nervous system (CNS) malformation, bilateral choanal atresia, and tracheoesophageal fistula 50). Issekutz et al 51) reported high mortality in infants with atrioventricular septal defects and in infants with a combination of ventriculomegaly and brain stem/cerebellar anomalies (13%). Feeding difficulties were also found to be a major cause of morbidity at all ages.
CHARGE syndrome diagnosis
Diagnostic criteria for CHARGE syndrome, a multiple malformation syndrome, are based on a combination of major and minor diagnostic characteristics. (CHARGE stands for coloboma, heart defects, choanal atresia, retarded growth and development, genital abnormalities, and ear anomalies.)
As described by Blake et al 52), and modified by Amiel et al 53) and Verloes 54), the major diagnostic characteristics of CHARGE syndrome are the following:
- Definite CHARGE syndrome. Individuals with all four major characteristics (see Table 1) or three major and three minor characteristics (see Table 2)
- Probable/possible CHARGE syndrome. Individuals with one or two major characteristics and several minor characteristics
Major characteristics are those that are common in CHARGE syndrome and relatively uncommon in other syndromes (see Table 1).
CHARGE syndrome treatment
The typical child with CHARGE syndrome is followed by an average of 17 different medical specialists and will have more than a dozen surgical procedures before he or she is 10 years old. Ideally, individuals with CHARGE would have a care coordinator to help coordinate and manage medical care. If this is available, take advantage of it. In reality, it is a parent (most often the mother) who takes on this role.
Below is a list of some of the medical issues that are often overlooked when physicians are concentrating on the more obvious medical issues.
Treatment of manifestations: Neonates require immediate evaluation of the airway, feeding, heart, and hearing. Management involves: tracheostomy and surgical correction of choanal atresia as needed; a multidisciplinary approach to feeding therapy including specialists in speech-language pathology, occupational therapy, and nutrition and gastrostomy as needed; routine care for heart defects; and hearing aids and hearing habilitation as soon as hearing loss is documented. Psychological/school evaluations should be performed by a team that includes specialists in deaf/blindness when dual sensory loss is present.
Prevention of secondary complications: Special attention to potential airway problems associated with anesthesia.
Surveillance: Regular ophthalmologic and audiologic evaluations; testing for hypogonadotropic hypogonadism if puberty has not occurred by age 13-14 years.
Management of children with CHARGE syndrome requires coordinated multidisciplinary care:
- Airway can be compromised from choanal atresia, TE fistula, aspiration pneumonias, tracheomalacia, or an aberrant subclavian vessel impinging on the trachea. Studies have shown that 15%-60% of individuals with CHARGE syndrome require tracheostomy 55).
- Heart defects are managed as in any individual with a congenital heart defect.
- Choanal atresia. Surgical correction by means of transnasal, transpalatal, or sublabial routes or airway bypass by tracheotomy or endotracheal intubation is usually necessary early in life. Multiple surgeries are often required to maintain the nasal airway.
- Feeding/swallowing dysfunction. In infancy, feeding can be compromised by oral-motor and/or sensory deficits. A multidisciplinary approach to therapy with specialists in speech-language pathology, occupational therapy, and nutrition can help promote early oral exploration and prevent or minimize oral defensiveness. For children with a G-tube, oral stimulation needs to be maintained to reduce future oral sensitivity/aversion.
- Gastroesophageal reflux can be significant enough to cause aspiration, often requiring Nissen fundoplication and G-tube insertion. Silent reflux should be considered in the evaluation of recurrent sinusitis.
- Renal evaluation. Renal ultrasound examination is recommended in all children. Evaluation for urinary tract infection is recommended in cases of unexplained fever or irritability in children unable to communicate.
- Endocrine evaluation. Early referral for endocrinology consultation is appropriate, especially if linear growth is falling from the normal curve in spite of adequate nutrition and normalized cardiac status. Some of these children may have growth hormone deficiency, which requires growth hormone replacement therapy. Individuals with hypogonadotropic hypogonadism may be considered for hormone replacement therapy for induction of puberty and for general health reasons including prevention of osteoporosis.
- Coloboma. Tinted glasses or a dark visor can be helpful for the photophobia that often accompanies iris colobomas. Parents, therapists, and teachers need to take into account visual field defects resulting from retinal coloboma and central visual defects resulting from optic nerve involvement and macular coloboma. For example, visual stimuli and sign language may need to be presented in child’s lower visual field. For eyes with visual potential, cycloplegic refraction and spectacle correction may be necessary, since substantive refractive errors of mildly or even moderately micro-ophthalmic eyes have been observed. Retinal detachment, a potential complication of retinal coloboma, can result in total blindness; therefore, any change in vision status should be treated as a medical emergency.
- Hearing loss. Hearing loss should be assumed until proven otherwise. Hearing aids and hearing habilitation (which may include sign language in addition to the auditory and speech training) should be started as soon as hearing loss is documented. Many children benefit from bone conduction aids or (especially at school) a frequency modulation system. Headbands can be used to help keep the hearing aids in place if the ear cartilage is floppy or if the tape that secures the aid to the scalp is ineffective. Pressure equalizing tube placement for chronic serous otitis is common and often needs to continue until adolescence. Cochlear implants have been successful in providing sound awareness and even speech recognition in the presence of cochlear abnormalities. Bauer et al 56) reported successful completion of cochlear implantation and measurable benefit in five individuals with CHARGE syndrome. Of note, variations in the temporal bone anatomy can lead to technical challenges and risk to the facial nerve during implantation. In some individuals, an aberrant course of the facial nerve may be a contraindication for cochlear implant 57).
- Communication. Establishing an appropriate system of communication is more difficult in the presence of both hearing loss and vision loss than in the presence of hearing loss alone. Depending on the degrees of hearing and vision loss, communication may start with touch cues, followed by object cues and proceeding to auditory/oral and/or sign language. Communication training initiated by age three years is critical to the eventual development of symbolic communication 58).
- Deaf-blind service referral. Children with CHARGE syndrome who have combined vision and hearing loss can be considered “deafblind,” an important designation used for qualifying for educational resources in many states. Of note, “deafblind” does not imply total hearing loss or total vision loss: most children with CHARGE syndrome have some residual vision and/or hearing but are still classified as “deafblind.” Referral to deafblind education services (e.g., the Deafblind Project within the state of residence) should be made as early as possible after birth so that the parents and project personnel can begin to plan together. A growing body of evidence indicates that normal language development can occur if hearing habilitation is started prior to age six months for hearing-impaired children, whether or not they are visually impaired. Assistance of the Deafblind team to provide consultation to the early childhood education team is highly recommended since many educators and speech therapists have little or no experience with dual sensory loss.
- Psychological/school evaluations should be performed by a team that includes specialists in deafblindness when dual sensory loss is present. If a deafblind specialist is not available when a psychologist does an evaluation, a vision educator can show the tester where to place materials and whether the lighting and contrast of the printed materials is adequate. A hearing educator can help place the child for optimal hearing and/or do sign language interpretation.
- Dental procedures, when necessary, may be performed under general anesthesia.
- Low muscle tone and poor balance predispose children to rapid exhaustion. Many children need adjustments to the classroom or therapy setting to allow for better truncal support. Frequent rest breaks may be needed. Many children can work for longer periods if allowed to do so in a supine position 59). Unpublished data suggest that hippotherapy (horseback riding) can be a helpful adjunct to physical therapy to prevent scoliosis as it requires frequent shifts in truncal muscular control — as do karate and other programs that promote good balance. Myofacial release can improve posture and flexibility.
- Sleep cycles are frequently disturbed even in those without significant visual impairments. Occasionally, sleep studies show obstructive apnea. If the cause is unknown, melatonin has been helpful for some children while others with severe visual impairment may need other medications to regulate sleep.
- Chronic constipation usually does not respond to simple measures such as increased fluid intake. Gastrointestinal consultation is often indicated.
- Obsessive-compulsive disorder (OCD). Behavioral therapy combined with stress reduction is sometimes helpful alone, but treatment with medications can be a useful adjunct.
- Pervasive developmental disorder. While the behaviors may mimic autism, there are differences 60). Sensory processing issues are likely implicated. Management by behavior therapy, stress reduction, and sometimes medication is indicated.
- Attention-deficit hyperactivity disorder (ADHD). In many instances, establishing an appropriate method of communication and providing adequate stimulation for exploration in a safe environment are more helpful than medication.
- Increased pain threshold may predispose children to behaviors that are incorrectly interpreted by others as aggressive. Understanding this is critical to devising appropriate interventional strategies.
Prevention of Secondary Complications
Facial palsy. Because the facial nerves are often ectopic, an MRI to determine the location of the facial nerves is appropriate before craniofacial surgery or cochlear implantation is considered.
Anesthesia. The airway problems associated with anesthesia in individuals with CHARGE syndrome can be attributed to choanal atresia, cleft lip and palate, and other upper-airway problems observed in approximately half of individuals with CHARGE syndrome. The soft cartilage and resultant floppy trachea add to potential anesthesia risk. Neurogenic incoordination of swallow and closure of the epiglottis may complicate the postoperative course, especially with repeated general anesthetics. If possible, procedures should be combined to reduce the overall use of anesthesia.
Surveillance
Regular ophthalmologic evaluations are appropriate to follow changes in acuity and risks for retinal detachment and/or cataract. Monitoring nonverbal infants and children who are unable to report subjective loss of vision can permit timely detection of retinal detachment and appropriate surgical repair where necessary.
Frequent retesting of hearing by a pediatric audiologist may be necessary to determine the exact type and extent of hearing loss and to assess the success of hearing habilitation.
Frequent clinical and radiologic dental evaluations should be performed.
Wheeler et al 61) recommended that LH and FSH be obtained between age two and three months, or by age 13-14 years if puberty has not occurred. If there is reason to suspect hypogonadotropic hypogonadism, a gonadotropin-releasing hormone (GnRH) stimulation test may be helpful.
References [ + ]


{kind=link}