Contents
Dubin Johnson syndrome
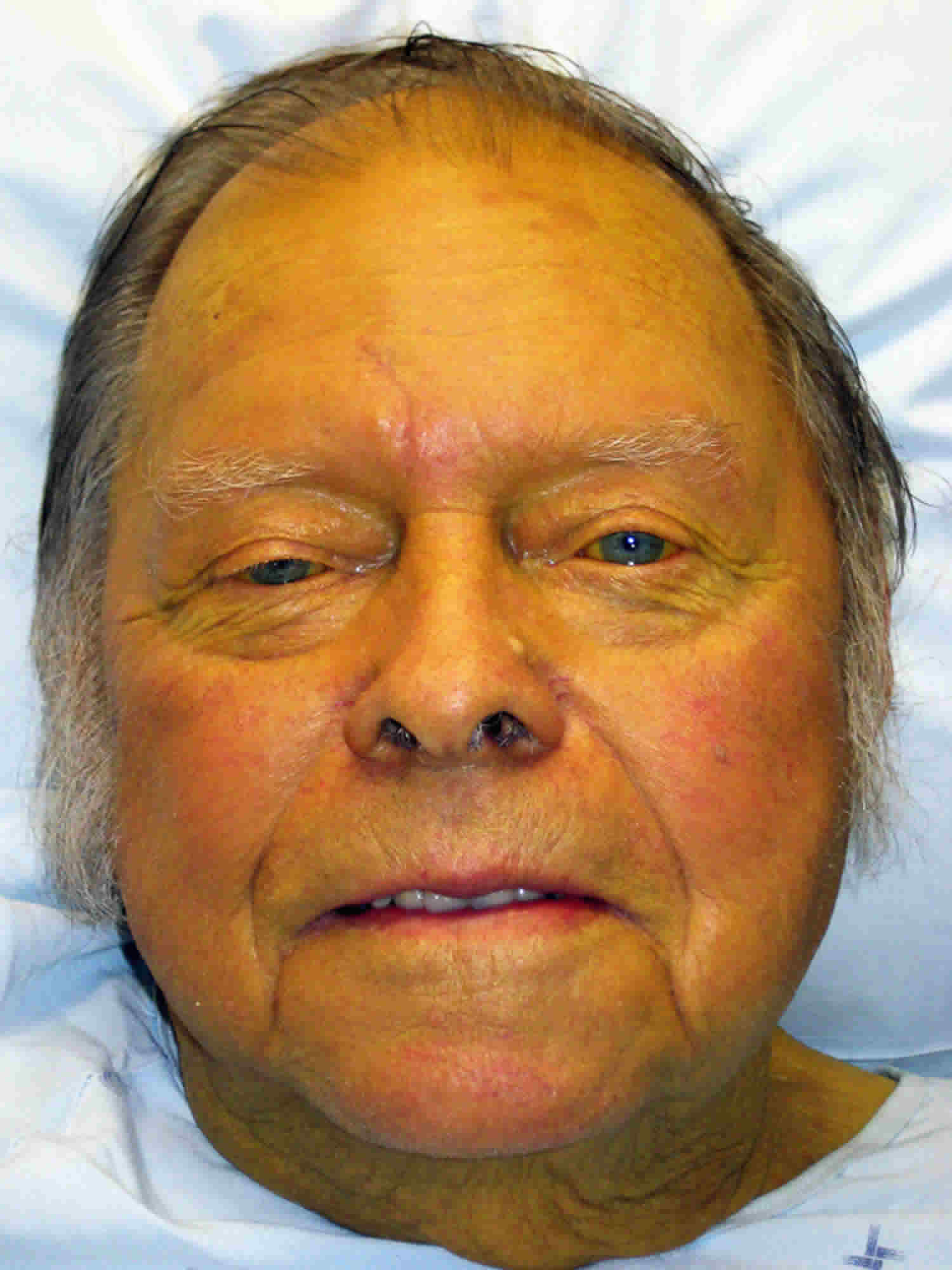
Dubin-Johnson syndrome is a rare autosomal recessive inherited disorder of bilirubin transport that is characterized by buildup of bilirubin in the bloodstream (conjugated hyperbilirubinemia), jaundice, which is a yellowing of the skin and whites of the eyes. In most affected people jaundice appears during adolescence or early adulthood 1, 2, 3, 4, 5, 6, 7, 8, 9. Jaundice is typically the only feature of Dubin-Johnson syndrome, but some people can experience weakness, mild abdominal pain, enlarged and tender liver, nausea, or vomiting. In most people with Dubin-Johnson syndrome, bilirubin pigment deposits build up in the liver but do not seem to impair liver function 10. The bilirubin pigment deposits make the liver appear black when viewed with medical imaging.
However, recent case reports have described an alternative presentation characterized by an intense, brilliant blue liver, expanding the phenotypic spectrum of Dubin-Johnson syndrome 11. This blue liver variant, although rare, should be considered in the differential diagnosis of blue liver syndrome, a term traditionally associated with oxaliplatin-induced sinusoidal obstruction syndrome 12.
Dubin-Johnson syndrome is caused by changes in a gene known as ABCC2 (ATP binding cassette subfamily C member 2) 10, 13. The ABCC2 gene provides instructions for producing a protein called multidrug resistance protein 2 (MRP2). This protein is one of a family of multidrug resistance proteins involved in the transport of substances out of cells 14. For example, MRP2 clears certain drugs from organs and tissues, playing a part in drug metabolism. Drug metabolism involves the breakdown of drugs into different chemical components allowing the drugs to have their intended effects and eventually be eliminated from the body. MRP2 also transports a substance called bilirubin out of liver cells and into bile (a digestive fluid produced by the liver). Bilirubin is produced during the breakdown of old red blood cells and has an orange-yellow tint. MRP2 is primarily found within the outer membrane that surrounds cells in the liver, with smaller amounts in the kidneys, intestine, and placenta.
Rarely, jaundice develops soon after birth in individuals with Dubin-Johnson syndrome. Affected infants typically also have enlarged livers (hepatomegaly) and a severely reduced ability to produce and release a digestive fluid called bile (cholestasis). As these children get older, their liver problems go away and they usually do not have any related health problems later in life. Factors that may worsen symptoms include: alcohol use, birth control pills, infection, and pregnancy. In most cases, treatment is not required.
The prevalence of Dubin-Johnson syndrome is estimated to be 1 in 100,000 people 10, 13. Dubin-Johnson syndrome appears to be most common in Iranian and Moroccan Jews living in Israel, with 1 in 1,300 individuals affected 15, 16, 17. Additionally, several people in the Japanese population have been diagnosed with Dubin-Johnson syndrome 2. Dubin-Johnson syndrome appears to be less common in other populations.
The diagnosis of Dubin-Johnson syndrome should be considered in all individuals with elevated conjugated bilirubin levels with otherwise normal liver function test findings.
Patients with Dubin-Johnson syndrome have a diagnostic abnormality in urinary coproporphyrin excretion, and abnormal distribution of the coproporphyrin isomers 1 and 3 in the urine is a characteristic feature of Dubin-Johnson syndrome. There are two naturally occurring coproporphyrin isomers, 1 and 3. Normally about 75% of the coproporphyrin in urine is coproporphyrin 3. In healthy people, the ratio of coproporphyrin 3 to coproporphyrin 1 is about 3.5:1. In patients with Dubin-Johnson syndrome, the total coproporphyrin content of the urine is normal, but >80% is coproporphyrin 1. Dubin-Johnson syndrome diagnosis can be confirmed by demonstrating an increase in the ratio of urinary coproporphyrin 1 to coproporphyrin 3. The levels of urinary coproporphyrin 1 levels are greater than coproporphyrin 3.
A combination of intense and prolonged visualization of the liver following intravenous administration of the radiopharmaceutical dye, with delayed to no visualization of the gallbladder, is unique to Dubin-Johnson syndrome.
The following tests can help diagnose Dubin Johnson syndrome:
- Liver enzyme levels (blood test)
- Serum bilirubin
- Urinary coproporphyrin levels
Although a liver biopsy is not necessary for the diagnosis of Dubin-Johnson syndrome, patients may be noted to have a dark liver during routine surgery (eg, cholecystectomy), prompting biopsy.
On liver biopsy in Dubin-Johnson syndrome, one can see the accumulation of dark, coarsely granular melanin-like pigment in centrilobular hepatocytes in a liver that otherwise looks normal. However, liver biopsy is not a recommendation for making a diagnosis.
Diagnosis of Dubin-Johnson syndrome can be made based on the presence of conjugated hyperbilirubinemia with no other abnormality of liver function tests and elevated urine coproporphyrin I fraction.
Invasive diagnostic tests should be avoided. Genetic testing for the ABCC2 gene is possible but used for scientific studies and not the clinical purpose 18, 19, 20.
Despite the marked liver discoloration, Dubin-Johnson syndrome is generally considered a benign condition, as liver function tests (aside from conjugated bilirubin elevation) remain within normal limits, and affected individuals do not develop progressive liver disease 21.
Can Dubin-Johnsons syndrome be passed along to another individual through sexual intercourse?
No, Dubin Johnson syndrome is caused by a gene mutation that is passed down (inherited) through families. Dubin Johnson syndrome is not a contagious condition and cannot be passed along to another individual through sexual intercourse.
Dubin Johnson syndrome causes
Dubin-Johnson syndrome is caused by changes in a gene known as ABCC2 (ATP binding cassette subfamily C member 2) 1, 2, 3, 4, 5, 6, 7, 8, 9. The ABCC2 gene provides instructions for making a protein called multidrug resistance protein 2 (MRP2) that transports certain substances out of the liver cells into the bile ducts. For example, MRP2 transports a substance called bilirubin out of liver cells and into bile (a digestive fluid produced by the liver). Bilirubin is produced during the breakdown of old red blood cells and has an orange-yellow tint 22, 11.
The MRP2 protein also clears certain drugs from organs and tissues, playing a part in drug metabolism 14. Drug metabolism involves the breakdown of drugs into different chemical components allowing the drugs to have their intended effects and eventually be eliminated from the body.
The multidrug resistance protein 2 (MRP2) is primarily found within the outer membrane that surrounds cells in your liver, with smaller amounts in your kidneys, intestine, and placenta.
The normal functioning multidrug resistance protein 2 (MRP2) works to secrete bilirubin into the bile, which is then transported to the gallbladder where it is stored. When the gall bladder is contracted during digestion, the bile is secreted into the intestine and then passes into the feces. Several different gene mutations have been identified that alter the function of the carrier protein.
ABCC2 gene mutations result in the production of multidrug resistance protein 2 (MRP2) with reduced or absent activity that cannot effectively transport substances out of cells. These mutations particularly affect moving bilirubin into bile. As a result, bilirubin accumulates in the body, causing a condition called hyperbilirubinemia. The buildup of bilirubin in the body causes the yellowing of the skin and whites of the eyes in people with Dubin-Johnson syndrome. The black liver in affected individuals is due to a buildup of different substance normally transported out of the liver by the multidrug resistance protein 2 (MRP2) produced from the ABCC2 gene.
Pregnancy or use of oral contraceptives may cause Dubin-Johnson syndrome to become apparent in females when no symptoms appeared previously 19.
Dubin-Johnson syndrome inheritance pattern
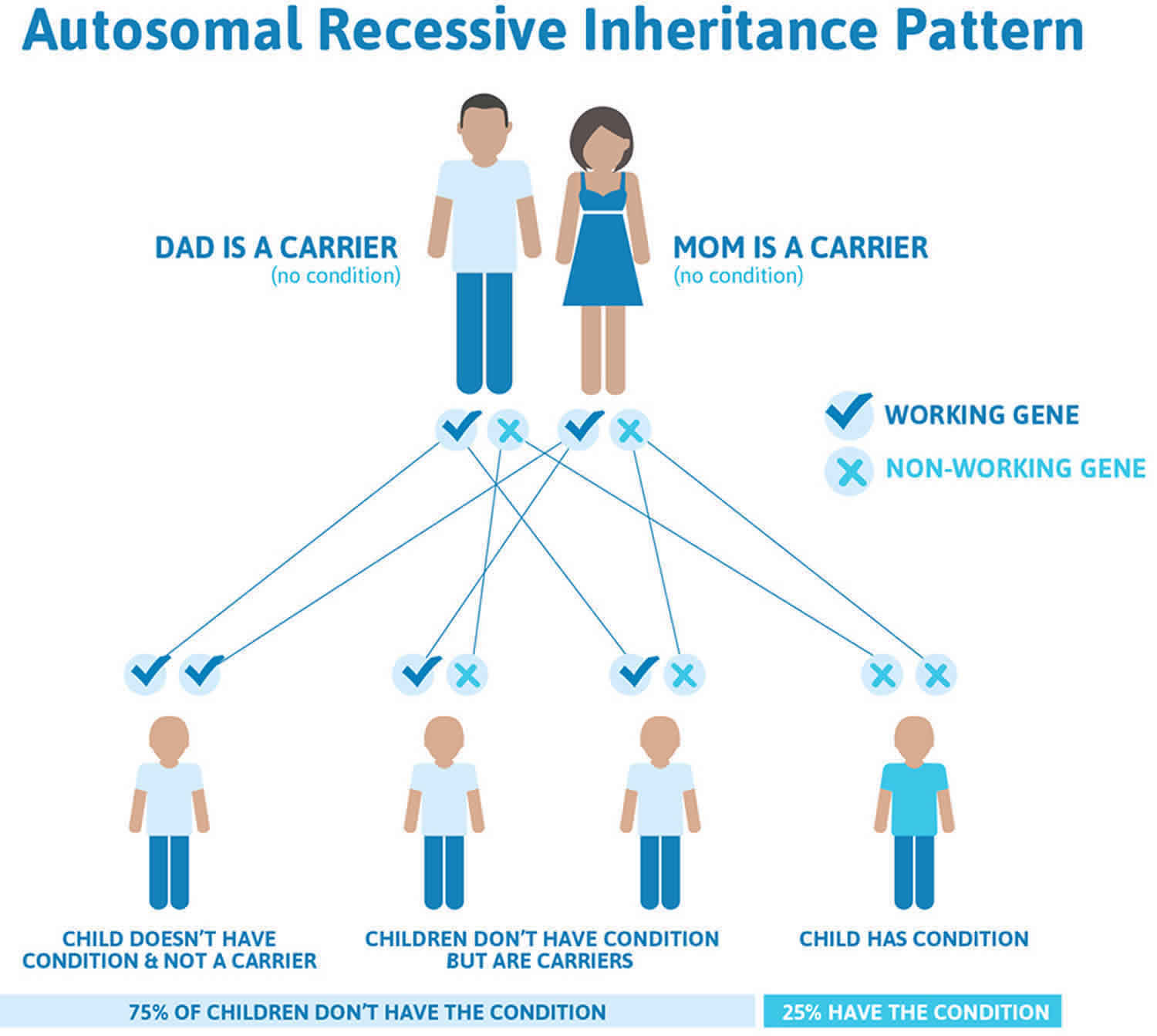
Dubin-Johnson syndrome is inherited in an autosomal recessive pattern, which means both copies of the ABCC2 gene in each cell have mutations. Recessive genetic disorders occur when an individual inherits a disease-causing gene mutation from each parent. If an individual receives one normal gene and one disease-causing gene mutation, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the gene variant and have an affected child is 25% with each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 1. Dubin-Johnson syndrome autosomal recessive inheritance pattern
Dubin Johnson syndrome symptoms
Jaundice, which is caused by excess bilirubin (bile pigment), is usually the only symptom of Dubin Johnson syndrome. Otherwise a physical examination is normal. Dubin Johnson syndrome rarely appears before puberty. Occasionally individual with Dubin Johnson syndrome may have an enlarged and tender liver (hepatomegaly) and complain of weakness and a painful abdomen, but the liver will function normally. There may sometimes be a mild recurrence of the jaundice. Pregnancy or use of oral contraceptives may cause the disease to become apparent in women when no symptoms appeared previously.
People with Dubin-Johnson syndrome have lifelong mild jaundice that may be made worse by:
- Alcohol
- Birth control pills
- Environmental factors that affect the liver
- Infection
- Pregnancy
- Fasting or dehydration
- Fatigue.
Dubin Johnson syndrome complications
Complications of Dubin-Johnson syndrome include jaundice (the most consistent finding) and enlarged and tender liver (hepatomegaly). Oral contraceptives, pregnancy, and intercurrent illness may exacerbate jaundice. Reduced prothrombin activity, resulting from lower levels of clotting factor VII, is found in 60% of patients.
Some neonates present with cholestasis, which may be severe. Increased fetal wastage was reported in one study. In a case report, gallstones in the gallbladder (cholecystolithiasis) and gallstones in the common bile duct (choledocholithiasis) developed in the presence of Dubin-Johnson syndrome 23.
Dubin Johnson syndrome diagnosis
The diagnosis of Dubin-Johnson syndrome should be considered in all individuals with elevated conjugated bilirubin levels with otherwise normal liver function test findings.
Patients with Dubin-Johnson syndrome have a diagnostic abnormality in urinary coproporphyrin excretion, and abnormal distribution of the coproporphyrin isomers 1 and 3 in the urine is a characteristic feature of Dubin-Johnson syndrome. There are two naturally occurring coproporphyrin isomers, 1 and 3. Normally about 75% of the coproporphyrin in urine is coproporphyrin 3. In healthy people, the ratio of coproporphyrin 3 to coproporphyrin 1 is about 3.5:1. In patients with Dubin-Johnson syndrome, the total coproporphyrin content of the urine is normal, but >80% is coproporphyrin 1. Dubin-Johnson syndrome diagnosis can be confirmed by demonstrating an increase in the ratio of urinary coproporphyrin 1 to coproporphyrin 3. The levels of urinary coproporphyrin 1 levels are greater than coproporphyrin 3.
A combination of intense and prolonged visualization of the liver following intravenous administration of the radiopharmaceutical dye, with delayed to no visualization of the gallbladder, is unique to Dubin-Johnson syndrome.
The following tests can help diagnose Dubin Johnson syndrome:
- Liver enzyme levels (blood test)
- Serum bilirubin
- Urinary coproporphyrin levels
Although a liver biopsy is not necessary for the diagnosis of Dubin-Johnson syndrome, patients may be noted to have a dark liver during routine surgery (eg, cholecystectomy), prompting biopsy.
On liver biopsy in Dubin-Johnson syndrome, one can see the accumulation of dark, coarsely granular melanin-like pigment in centrilobular hepatocytes in a liver that otherwise looks normal. However, liver biopsy is not a recommendation for making a diagnosis.
Diagnosis of Dubin-Johnson syndrome can be made based on the presence of conjugated hyperbilirubinemia with no other abnormality of liver function tests and elevated urine coproporphyrin I fraction.
Invasive diagnostic tests should be avoided. Histologically, the liver of Dubin-Johnson syndrome patients exhibits a distinctive brown-black pigment deposition within centrilobular hepatocytes, particularly around the central veins. Under light microscopy, the liver architecture appears normal, but the pigment accumulates as dark, granular deposits, a feature not observed in Rotor syndrome. Electron microscopy reveals that the pigment is localized within lysosomes. Histochemical staining and physicochemical analysis suggest that this pigment is a melanin-like polymer derived from defective bilirubin metabolism 24, 25.
Genetic testing for the ABCC2 gene is possible but used for scientific studies and not the clinical purpose 18, 19, 20.
Laboratory studies
Laboratory studies reveal conjugated hyperbilirubinemia, with total bilirubin serum levels usually in the 2- to 5-mg/dL range (but potentially as high as 25 mg/dL).
Results of other laboratory tests, including liver enzymes (aspartate aminotransferase [AST], alanine aminotransferase [ALT], and alkaline phosphatase [ALP]), serum albumin, and hematologic studies (eg, complete blood count [CBC], reticulocyte count), tend to be within reference ranges. Urine dipstick analysis may reveal bilirubinuria.
Prothrombin time is usually within normal limits, but it can be prolonged in Iranian Jewish patients with associated factor VII deficiency 26.
Reduced prothrombin activity resulting from lower levels of clotting factor VII is observed in 60% of patients with Dubin-Johnson syndrome.
Because MRP2 also transports leukotrienes into the bile, patients with Dubin-Johnson syndrome have defective biliary secretion and increased urinary excretion of leukotriene metabolites. This may become a noninvasive diagnostic assay for this condition 27.
Coproporphyrins
Coproporphyrins are byproducts of heme biosynthesis. Normally, coproporphyrin I is preferentially excreted in bile, whereas coproporphyrin III is preferentially excreted in urine.
The urinary excretion of coproporphyrin isomers, however, has a fairly unique pattern in patients with Dubin-Johnson syndrome and can be used as a pathognomonic feature of the condition when congenital erythropoietic porphyria and arsenic poisoning have been excluded.
An increase in the urinary excretion of coproporphyrin I and a decrease in the excretion of coproporphyrin III are observed in Dubin-Johnson syndrome 28. This results in total urinary coproporphyrin excretion (I+III) that is nearly normal when compared with unaffected individuals. The unique feature in Dubin-Johnson syndrome, however, is that 80% of the urinary coproporphyrin is type I in patients with Dubin-Johnson syndrome, compared with only 25% in other persons 29. Fecal coproporphyrin levels remain normal.
In persons who are heterozygous for Dubin-Johnson syndrome, an intermediate ratio of urinary coproporphyrin I to coproporphyrin III is observed; these levels have been used to create family trees and to establish the recessive nature of the condition.
How a defect in an apical transporter creates this variance in urinary isomers remains unexplained, with several possible pathogenic mechanisms.
Interestingly, for the first 2 days of life, healthy neonates have ratios of urinary coproporphyrin similar to those seen in patients with Dubin-Johnson syndrome; by 10 days of life, however, these levels convert to the normal adult ratio 30.
Imaging Studies
Studies such as the hepatobiliary iminodiacetic acid (HIDA) scan and computerized tomography (CT) of the abdomen are performed in patients with hyperbilirubinemia. Computed tomography (CT) scan findings in patients with Dubin-Johnson syndrome reportedly show a significantly higher attenuation than that seen in control subjects 31. The hepatobiliary iminodiacetic acid (HIDA) scan shows a characteristic pattern of delayed visualization or no visualization of the gallbladder and bile ducts in the presence of intense, homogenous, and prolonged visualization of the liver in the patients with Dubin-Johnson syndrome. Similarly, oral cholecystography will fail to visualize the gallbladder- subjecting many patients to unnecessary laparoscopic surgery because of a false positive result suggesting the presence of gallstones. Therefore, these studies generally are not needed if Dubin-Johnson syndrome is suspected 32.
Dubin Johnson syndrome differential diagnosis
Some features of the following disorders can be similar to those of Dubin Johnson syndrome and need to be considered in a patient prior to determining the diagnosis of Dubin Johnson syndrome.
Rotor syndrome is very similar to Dubin-Johnson syndrome in that the main symptom is also jaundice and both have increases in conjugated or direct bilirubin and otherwise normal liver characteristics (these are all easily measured in the chemistry lab). However, the liver maintains a normal color unlike the liver in patients with Dubin-Johnson syndrome, which appears black. Rotor syndrome is much less common than Dubin-Johnson syndrome.
Two other genetic diseases of bilirubin metabolism, Gilbert syndrome and Crigler-Najjar syndrome, also present with jaundice, but patients with these conditions have elevated unconjugated or indirect bilirubin.
Most diseases of the liver will have jaundice and can be confused with Dubin-Johnson syndrome. However, in almost all patients, the other liver tests, specifically the transaminases, will also be abnormal along with the bilirubin. In Dubin-Johnson syndrome the bilirubin elevation is usually mild, 2-3 mg/dL (normal is less than 1 mg/dL). Other causes of liver disease include drugs, infections, gallstones, tumors, ischemia or congestion (abnormal blood flow to the liver) other congenital diseases and inflammatory conditions, including autoimmune hepatitis. Liver abnormalities including jaundice can also be seen as part of several systemic illnesses that also affect the liver.
Primary biliary cholangitis (PBC), sclerosing cholangitis and autoimmune hepatitis are chronic progressive diseases of the liver and biliary system (the ducts that are within the liver and secrete the bile into the intestines). These three autoimmune disorders are caused by abnormalities in the immune system where the body literally attacks itself. Inflammation, obstruction or injury involving the bile ducts leads to jaundice. Excessive amounts of copper, usually excreted in the bile, also accumulate in the liver. The associated inflammation leads to scarring of the liver and decrease in function. Primary biliary cholangitis occurs mainly in females during the fourth to the seventh decade of life and has four progressive stages.
Among the liver infections, viral hepatitis particularly from hepatitis A (HAV), hepatitis B virus (HBV), and hepatitis C (HV) are worldwide the most common causes of liver disease. Hepatitis A is transmitted from water or food that has been exposed to an individual with an active hepatitis A infection. The virus is passed in the classic fecal oral pathway. Hepatitis B and hepatitis C are transmitted by parenteral means (IV, needle exposure, sexual contacts, tattoos, etc.). Hepatitis B begins with a prodrome including anorexia (loss of appetite), fever, nausea, lethargy and vomiting and usually results in jaundice. Hepatitis B is a common cause of chronic hepatitis leading to liver failure, cirrhosis and / or liver cancer. A mother with hepatitis B has a high likelihood of passing the virus to her baby unless the newborn is immunized and treated immediately after delivery. Hepatitis B can also be easily passed through bodily fluids such as blood, semen and possibly saliva. Hepatitis C is more insidious, meaning less clinically apparent and many people who have it do not know it. Like hepatitis B it is often spread from person to person through intravenous drug use, sexual contact and other injection treatments. Patients with Dubin-Johnson syndrome are often tested for these infections. Vaccination for hepatitis B is recommended universally and for hepatitis A if you are going into an endemic area and do not have natural immunity.
Table 1. Genetic differential diagnosis
| Disease | Mutation |
|---|---|
| Dubin-Johnson syndrome | Genetic mutation in the canalicular multispecific organic anion transporter (MRP2), encoded by the ABCC2 gene |
| Rotor syndrome | Autosomal recessive disorder caused by homozygous mutations in the SLCO1B1 and SLCO1B3 genes on chromosome 12; these genes encode the organic anion-transporting polypeptides OATP1B1 and OATP1B3, respectively |
| Progressive familial intrahepatic cholestasis (PFIC) 1 | ATP8B1 gene |
| Progressive familial intrahepatic cholestasis (PFIC) 2 | ABCB11 gene (located on chromosome 2) |
| Progressive familial intrahepatic cholestasis (PFIC) 3 | ABCB4 gene (located on chromosome 7) |
| Crigler-Najjar syndrome | Mutations in the UGT1A1 gene, located on chromosome 2, which encodes bilirubin uridine diphosphate-glucuronosyltransferase (B-UGT) |
| Gilbert-Meulengracht syndrome | Mutation in the promoter region of the UGT1A1 gene (2q37), leading to reduced activity of the uridine diphosphate-glucuronosyltransferase (UGT) enzyme |
| Wilson’s disease | Mutations in the ATP7B gene, located on the long arm (q) of chromosome 13 (13q14.3); this gene encodes the ATPase 2 protein |
Dubin Johnson syndrome treatment
Treatment of Dubin Johnson syndrome is symptomatic and supportive. Rarely, a severely reduced ability to produce and release bile from the liver (cholestasis) that occurs in neonatal Dubin-Johnson syndrome may benefit from phenobarbital and ursodeoxycholic acid 19. Phenobarbital increases bilirubin excretion, and the dose varies based on symptom severity and patient response. A starting dose of 15 to 30 mg/kg/day is typically given and then gradually increased until the desired effect is achieved 9. Ursodeoxycholic acid improves bile flow and has a hepatoprotective effect, and the recommended dose for Dubin-Johnson syndrome is 15 mg/kg/day in divided doses 9. Rifampicin is also one of the drugs that have shown improvement in hyperbilirubinemia in patients with Dubin-Johnson syndrome. Typically, the recommended dose of rifampicin for the treatment of Dubin-Johnson syndrome is 10 mg/kg/day, or 600 mg/day, administered orally and in divided doses 9. However, rifampicin should be used with caution in Dubin-Johnson syndrome because it can cause liver toxicity and interact with other medications 34, 35.
In many Dubin Johnson syndrome cases, patients may require no treatment even though they have recurrent mild jaundice. However, metabolism of certain drugs may be affected in patients with Dubin-Johnson syndrome since many pharmaceutical products are metabolized in the liver. Therefore, medications should be carefully supervised by a physician. Genetic counseling may be of benefit for patients and their families affected by Dubin Johnson syndrome.
Dubin Johnson syndrome prognosis
Dubin-Johnson syndrome is a benign condition, and life expectancy among patients is normal 19. An interesting case report describes an infant who received a living related liver transplant donor graft from his mother, who had Dubin-Johnson syndrome. One year after transplantation there were no unexpected issues with the donor or the child who had “inherited” Dubin-Johnson syndrome from his mother 36.
- Talaga ZJ, Vaidya PN. Dubin-Johnson Syndrome. [Updated 2023 Jul 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK536994[↩][↩]
- Dubin-Johnson syndrome. https://medlineplus.gov/genetics/condition/dubin-johnson-syndrome/[↩][↩][↩]
- ABCC2 gene. https://medlineplus.gov/genetics/gene/abcc2/[↩][↩]
- Dubin Johnson Syndrome. https://rarediseases.org/rare-diseases/dubin-johnson-syndrome/[↩][↩]
- Barnett S, Nyein AC, Galler M, Jamieson D, Davies M, Connor P, Veal GJ. Excessive vincristine exposure in a child being treated for acute lymphoblastic leukaemia with underlying Dubin-Johnson syndrome: a case report. Cancer Chemother Pharmacol. 2023 Oct;92(4):325-328. doi: 10.1007/s00280-023-04565-0[↩][↩]
- Kim JH, Kang MW, Kim S, Han JW, Jang JW, Choi JY, Yoon SK, Sung PS. Genotype-Phenotype Association in ABCC2 Exon 18 Missense Mutation Leading to Dubin-Johnson Syndrome: A Case Report. Int J Mol Sci. 2022 Dec 18;23(24):16168. doi: 10.3390/ijms232416168[↩][↩]
- Zhao C, Shi X, Zhang Y, Huang H. Case Report: Three novel pathogenic ABCC2 mutations identified in two patients with Dubin-Johnson syndrome. Front Genet. 2022 Aug 25;13:895247. doi: 10.3389/fgene.2022.895247[↩][↩]
- Kamal NM, Saadah O, Alghamdi H, Algarni A, El-Shabrawi MHF, Sherief LM, Abosabie SAS. Case Report: Dubin-Johnson Syndrome Presenting With Infantile Cholestasis: An Overlooked Diagnosis in an Extended Family. Front Pediatr. 2022 May 25;10:855210. doi: 10.3389/fped.2022.855210[↩][↩]
- Siddiqui AH, Alsabe MR, Tehseen Z, Hatamleh MI, Taslim S, Abdelrahman A, Saleem F. Dubin-Johnson Syndrome: A Case Report. Cureus. 2023 Mar 14;15(3):e36115. doi: 10.7759/cureus.36115[↩][↩][↩][↩][↩]
- Chronic idiopathic jaundice with unidentified pigment in liver cells; a new clinicopathologic entity with a report of 12 cases. Dubin IN, Johnson FB. Medicine (Baltimore) 1954;33:155–197. doi: 10.1097/00005792-195409000-00001[↩][↩][↩]
- Bosia JD, D’Ascenzo MV, Borzi S, Cozzi S, Defelitto JR, Curciarello JO. Síndrome de Dubin Johnson: presentación de un caso y revisión de la literatura [The Dubin-Johnson syndrome: case report and review of literature]. Acta Gastroenterol Latinoam. 2008 Sep;38(3):194-8. Spanish.[↩][↩]
- Sinusoidal obstruction syndrome and nodular regenerative hyperplasia are frequent oxaliplatin-associated liver lesions and partially prevented by bevacizumab in patients with hepatic colorectal metastasis. Rubbia-Brandt L, Lauwers GY, Wang H, et al. Histopathology. 2010;56:430–439. doi: 10.1111/j.1365-2559.2010.03511.x[↩]
- A recurrent ABCC2 p.G693R mutation resulting in loss of function of MRP2 and hyperbilirubinemia in Dubin-Johnson syndrome in China. Wu L, Li Y, Song Y, et al. Orphanet J Rare Dis. 2020;15:74–76. doi: 10.1186/s13023-020-1346-4[↩][↩]
- Fardel O, Jigorel E, Le Vee M, Payen L. Physiological, pharmacological and clinical features of the multidrug resistance protein 2. Biomed Pharmacother. 2005 Apr;59(3):104-14. doi: 10.1016/j.biopha.2005.01.005[↩][↩]
- Shani M, Seligsohn U, Gilon E, Sheba C, Adam A. Dubin-Johnson syndrome in Israel. I. Clinical, laboratory, and genetic aspects of 101 cases. Q J Med. 1970 Oct;39(156):549-67.[↩]
- DUBIN IN. Chronic idiopathic jaundice; a review of fifty cases. Am J Med. 1958 Feb;24(2):268-92. doi: 10.1016/0002-9343(58)90315-2[↩]
- Cohen L, Lewis C, Arias IM. Pregnancy, oral contraceptives, and chronic familial jaundice with predominantly conjugated hyperbilirubinemia (Dubin-Johnson syndrome). Gastroenterology. 1972 Jun;62(6):1182-90.[↩]
- Mutation analysis of the ABCC2 gene in Chinese patients with Dubin-Johnson syndrome. Wu L, Zhang W, Jia S, et al. Exp Ther Med. 2018;16:4201–4206. doi: 10.3892/etm.2018.6682[↩][↩]
- Memon N, Weinberger BI, Hegyi T, Aleksunes LM. Inherited disorders of bilirubin clearance. Pediatr Res. 2016 Mar;79(3):378-86. doi: 10.1038/pr.2015.247[↩][↩][↩][↩][↩]
- Jemnitz K, Heredi-Szabo K, Janossy J, Ioja E, Vereczkey L, Krajcsi P. ABCC2/Abcc2: a multispecific transporter with dominant excretory functions. Drug Metab Rev. 2010 Aug;42(3):402-36. doi: 10.3109/03602530903491741[↩][↩]
- Erlinger S, Arias IM, Dhumeaux D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology. 2014 Jun;146(7):1625-38. doi: 10.1053/j.gastro.2014.03.047[↩]
- Arrese M. Identificación de defectos moleculares en las enfermedades hepáticas. Ejemplos recientes [Identification of molecular defects in liver diseases. Recent advances]. Rev Med Chil. 1999 Sep;127(9):1112-20. Spanish.[↩]
- Zhou L, Liu C, Bai J, Dong S, Wei J. Dubin-Johnson syndrome with cholecystolithiasis and choledocholithiasis. Int J Surg Case Rep. 2013. 4(7):587-8.[↩]
- Genomic structure of the canalicular multispecific organic anion-transporter gene (MRP2/cMOAT) and mutations in the ATP-binding-cassette region in Dubin-Johnson syndrome. Toh S, Wada M, Uchiumi T, et al. Am J Hum Genet. 1999;64:739–746. doi: 10.1086/302292[↩]
- The Dubin-Johnson syndrome: electron microscopic observation of hepatic pigment–a case study. Baba N, Ruppert RD. Am J Clin Pathol. 1972;57:306–310. doi: 10.1093/ajcp/57.3.306[↩]
- Mor-Cohen R, Zivelin A, Fromovich-Amit Y, Kovalski V, Rosenberg N, Seligsohn U. Age estimates of ancestral mutations causing factor VII deficiency and Dubin-Johnson syndrome in Iranian and Moroccan Jews are consistent with ancient Jewish migrations. Blood Coagul Fibrinolysis. 2007 Mar. 18(2):139-44.[↩]
- Mayatepek E, Lehmann WD. Defective hepatobiliary leukotriene elimination in patients with the Dubin-Johnson syndrome. Clin Chim Acta. 1996 May 30. 249(1-2):37-46.[↩]
- Frank M, Doss M, de Carvalho DG. Diagnostic and pathogenetic implications of urinary coproporphyrin excretion in the Dubin-Johnson syndrome. Hepatogastroenterology. 1990 Feb. 37(1):147-51.[↩]
- Respaud R, Benz-de Bretagne I, Blasco H, Hulot JS, Lechat P, Le Guellec C. Quantification of coproporphyrin isomers I and III in urine by HPLC and determination of their ratio for investigations of multidrug resistance protein 2 (MRP2) function in humans. J Chromatogr B Analyt Technol Biomed Life Sci. 2009 Nov 15. 877(30):3893-8.[↩]
- Rocchi E, Balli F, Gibertini P, et al. Coproporphyrin excretion in healthy newborn babies. J Pediatr Gastroenterol Nutr. 1984 Jun. 3(3):402-7.[↩]
- Shimizu T, Tawa T, Maruyama T, Oguchi S, Yamashiro Y, Yabuta K. A case of infantile Dubin-Johnson syndrome with high CT attenuation in the liver. Pediatr Radiol. 1997 Apr. 27(4):345-7.[↩]
- LeVee A, Cooper C, Russell MB, Sterling M. Dubin-Johnson Syndrome Presenting During Cardiac Transplantation Evaluation. Cureus. 2020 Jan 8;12(1):e6594. doi: 10.7759/cureus.6594[↩]
- Viñuela M, Sotomayor C, Morales E, Pérez M, Torres J, Barrera F, Martínez JA. An Incidental Finding of an Indigo-Blue Liver in a Patient With Dubin-Johnson Syndrome Confirmed via Genetic Testing. Cureus. 2025 Feb 23;17(2):e79505. doi: 10.7759/cureus.79505[↩]
- Loscalzo J. New York: Access Medicine; 2018. Harrison’s Principles of Internal Medicine, 20e.[↩]
- Title: Rifampin treatment in patients with Dubin-Johnson syndrome Authors. Schiodt FV, Balko J, Schilsky M, Harrison ME, Thornton J, Lee WM, Squires RH Jr. J Liver Transplant Year. 2002;3:365–369.[↩]
- Liu C, Niu DM, Hsia CY, et al. Living donor liver transplantation using a graft from a donor with Dubin-Johnson syndrome. Pediatr Transplant. 2012 Feb. 16(1):E25-9.[↩]





{kind=link}