Contents
- Familial combined hyperlipidemia
- What is cholesterol?
- What are Cholesterol levels?
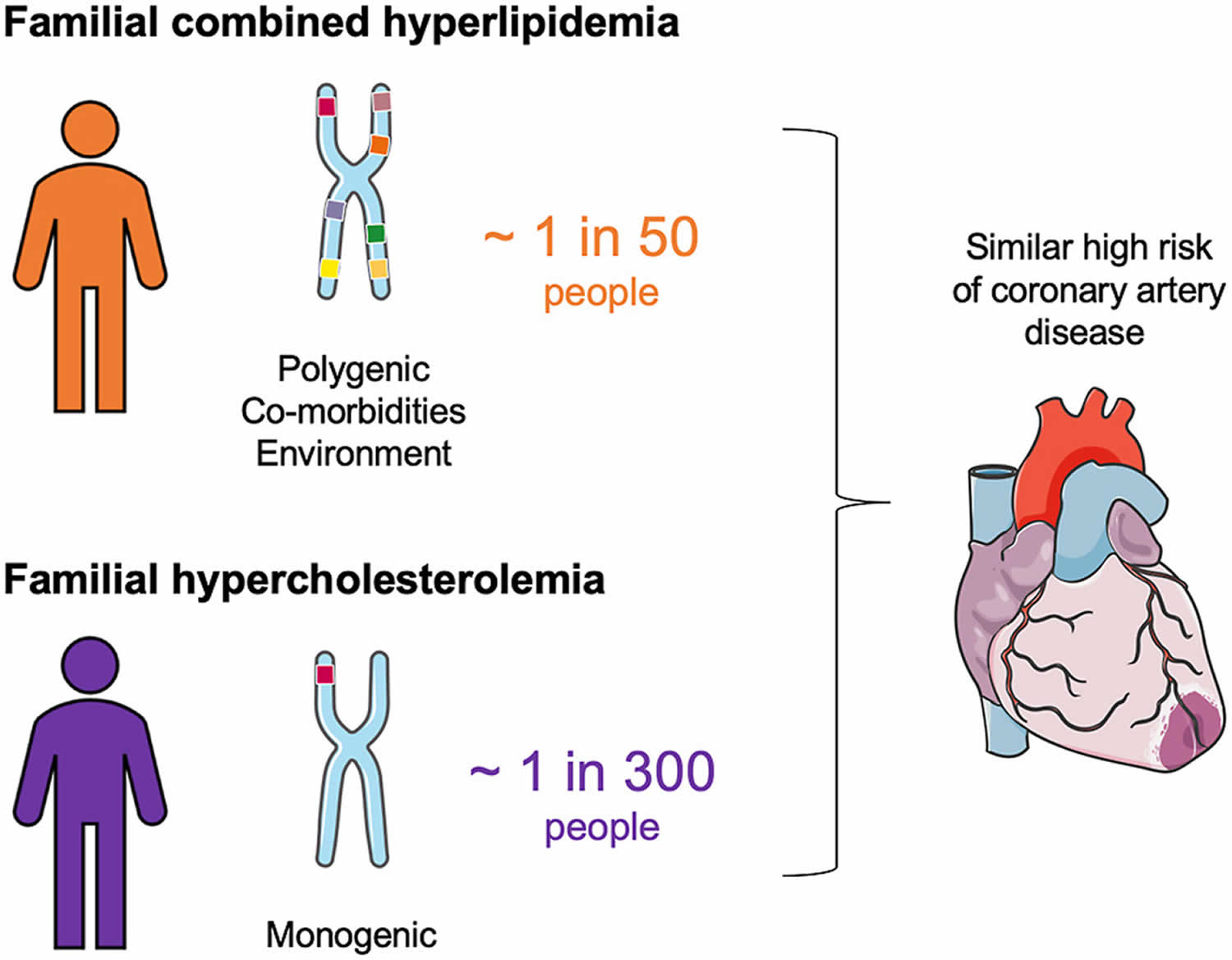
- What is the difference between familial combined hyperlipidemia and familial hypercholesterolemia?
- Familial combined hyperlipidemia cause
- Familial combined hyperlipidemia pathophysiology
- Familial combined hyperlipidemia signs and symptoms
- Familial combined hyperlipidemia diagnosis
- Familial combined hyperlipidemia treatment
- Familial combined hyperlipidemia prognosis
Familial combined hyperlipidemia
Familial combined hyperlidemia (FCHL) is a common inherited lipid metabolic disorder characterized by: (a) increase in blood cholesterol level (hypercholesterolemia) and/or increased levels of triglycerides (triglyceridemia) in at least two members of the same family, (b) intra-individual and intrafamilial variability of the lipid phenotype, and (c) increased risk of premature coronary artery disease 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,. The laboratory abnormalities most frequently found in familial combined hyperlidemia (FCHL) are an increase of plasma triglycerides (triglyceridemia) and/or cholesterol levels (hypercholesterolemia), and a high prevalence of small very-low-density lipoproteins (VLDLs) and/or low-density lipoproteins (LDLs), mainly related to an increased plasma level of apolipoprotein B100 (apo B) 12. Some familial combined hyperlidemia patients can present with a decrease in high-density lipoprotein (HDL) cholesterol level, often inversely correlated to the triglycerides (TG) plasma level 13. There is a predominance of small and dense LDL so-called atherogenic LDL “B” pattern, poor in cholesterol, and with a high apolipoprotein B (apo B)/cholesterol ratio. The main determinants of LDLs size appear to be the triglyceride (TG) and HDL plasma levels 14. The synthesis of LDL-apo B increases due to uncontrolled overproduction of apo B 15. No major alterations in the LDL liver catabolic rate have been described.
In familial combined hyperlipidemia patients the activity of the LDL receptor (with a high affinity for apo B100) is normal 16. The reduction in lipid levels after diet and lipid-lowering drugs does not normalize the kinetic and structural characteristics of the LDLs, at least in a large percentage of patients 17. Some studies suggest that a relative deficit of liver lipoprotein lipase (LPL) can reduce the liver uptake of apo B to simulate the increased synthesis of these apolipoproteins 18. Moreover, LDL from familial combined hyperlipidemia patients, irrespective of lipid phenotypes, are more susceptible to oxidation in vitro than LDL from healthy controls 8. This increased susceptibility of LDL to oxidation in vitro seems to be a consequence of the abundance of small dense LDL particles and not to defects of antioxidant capacity in familial combined hyperlipidemia 19. In familial combined hyperlipidemia patients with very high LDL cholesterol plasma levels of lipoprotein-a [Lp(a)] may be high as well 20.
Reduced levels of HDL cholesterol are a frequent finding in familial combined hyperlipidemia patients 8. HDL cholesterol and HDL2 cholesterol reduction could be due to triglyceride-enrichment of HDL particles and enhanced hepatic lipase, while the role of lipoprotein lipase (LPL) and activities of cholesterol ester transfer protein (CETP) and phospholipid transfer protein (PLTP) appears to be less evident 21. Recent data suggest that HDL cholesterol values are lower in subjects with predominantly small and dense LDL and are associated with a very high concentration of VLDL-1 (with low apo AI and apo E content) 8. LDL pattern is suggested to be the main determinant of the phenotype expressed by familial combined hyperlipidemia patients 22.
An increase in the synthesis of VLDL-apo B is usually present 23, but the reason why is yet to be fully understood. Some authors suggest that this increase is related to alterations in the incorporation of fatty acids in the triglyceride (TG) 17 and/or alterations of the after meal metabolism of the VLDLs, with greater conversion to small and dense LDLs and/or reduced turnover of the VLDLs themselves 24. However, other authors showed that VLDL increase in familial combined hyperlipidemia patients is mainly related to defects in activity of lipoprotein lipase 25, lecithin:cholesterol acyltransferase 26, and/or hepatic lipase 27. On the other hand, Evans et al 28 recently used stable isotope techniques combined with tissue-specific measurements in adipose tissue and forearm muscle to investigate fatty acid handling by these tissues in the fasting and postprandial states of familial combined hyperlipidemia patients. They found that the major defect appeared to be overproduction of triacylglycerol (TAG) by the liver due to decreased fatty acid oxidation, with fatty acids directed to triglyceride (TG) synthesis, while evidence of decreased lipoprotein lipase action or impaired fatty acid re-esterification in adipose tissue was observed 28.
An impaired after meal plasma component C3 response has been observed in familial combined hyperlipidemia patients, most likely as a result of a delayed response by C3, as the precursor for the biologically active acylation-stimulating protein, acting on free fatty acid (FFA) metabolism 29. Therefore, an impaired postprandial C3 response may be associated with impaired peripheral postprandial free fatty acid (FFA) uptake and, consequently, lead to increased liver free fatty acid (FFA) flux and VLDL overproduction 17.
In familial combined hyperlipidemia patients, the VLDL triglyceride (TG) content is inversely related to the LDL cholesterol plasma level: the redistribution of apoB and plasma cholesterol could be a key process in development of various phenotypes. The plasma apoB and cholesterol in VLDL particles, when in abundance, are associated with significantly lower cholesterol levels in the bigger and more buoyant LDL particles. This effect is reversible by reducing plasma triglyceride (TG) levels (by diet, by drugs, and/or by physical activity), which in turn may result in redistribution of apoB and total cholesterol from the VLDL particles to LDL particles 30.
In a recent study, de Graaf et al 31 point to high remnant-like particles cholesterol (RLP-C) as a potential biomarker of familial combined hyperlipidemia. They observed that patients with familial combined hyperlipidemia have 2-fold elevated plasma remnant-like particles cholesterol (RLP-C) levels, which add to the atherogenic lipid profile and contribute to the increased risk for cardiovascular disease 31. Plasma remnant-like particles cholesterol (RLP-C) levels above the 90th percentile predicted prevalent cardiovascular disease, independently of non-lipid cardiovascular risk factors and triglyceride (TG) levels. However, in both familial combined hyperlipidemia patients and controls, plasma remnant-like particles cholesterol (RLP-C) did not provide additional information about prevalent cardiovascular disease over and above non-HDL cholesterol levels 8.
The prevalence of familial combined hyperlipidemia is estimated at 1 in 100 in the general population. However, familial combined hyperlipidemia can be higher in patients with premature coronary artery disease, being the most frequent in patients affected by coronary artery disease (10%) and among survivors of heart attacks aged less than 60 (11.3%) 32, 8. According to a conservative estimate (whole population: 0–99 years), over 3.5 million people are affected by familial combined hyperlipidemia in EU and 2.7 million in the US; familial combined hyperlipidemia is the cause of approximately 30,000–70,000 heart attacks/year in the EU and more or less the same number in US, often prematurely (onset at a younger age <50 years of age) 33.
Figure 1. Familial combined hyperlipidemia diagnostic algorithm
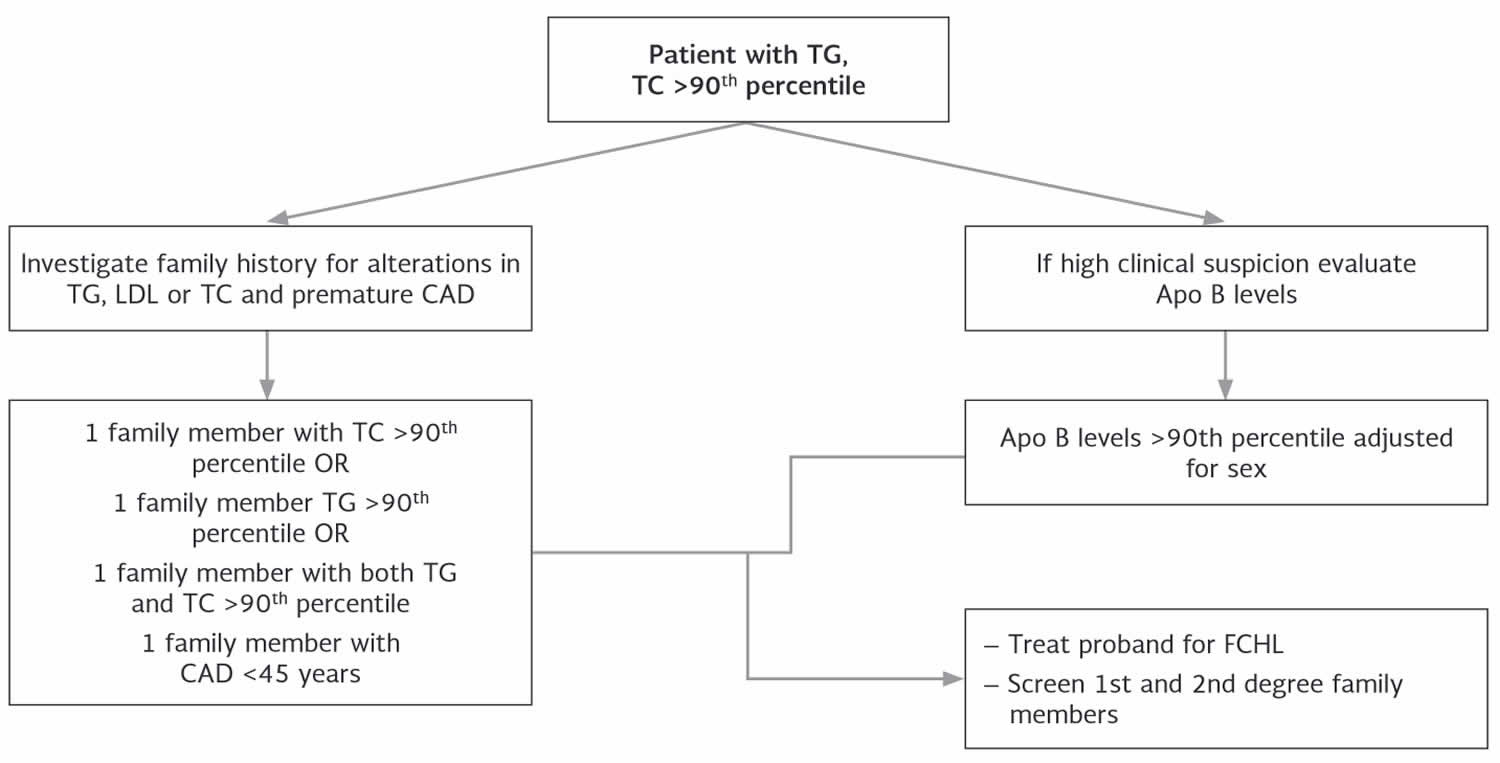
Abbreviations: TC = total cholesterol; TG = triglycerides; apoB = apolipoprotein B-100; CAD = coronary artery disease, FCHL = familial combined hyperlipidemia
[Source 6 ]Figure 2. Familial combined hyperlipidemia genetics
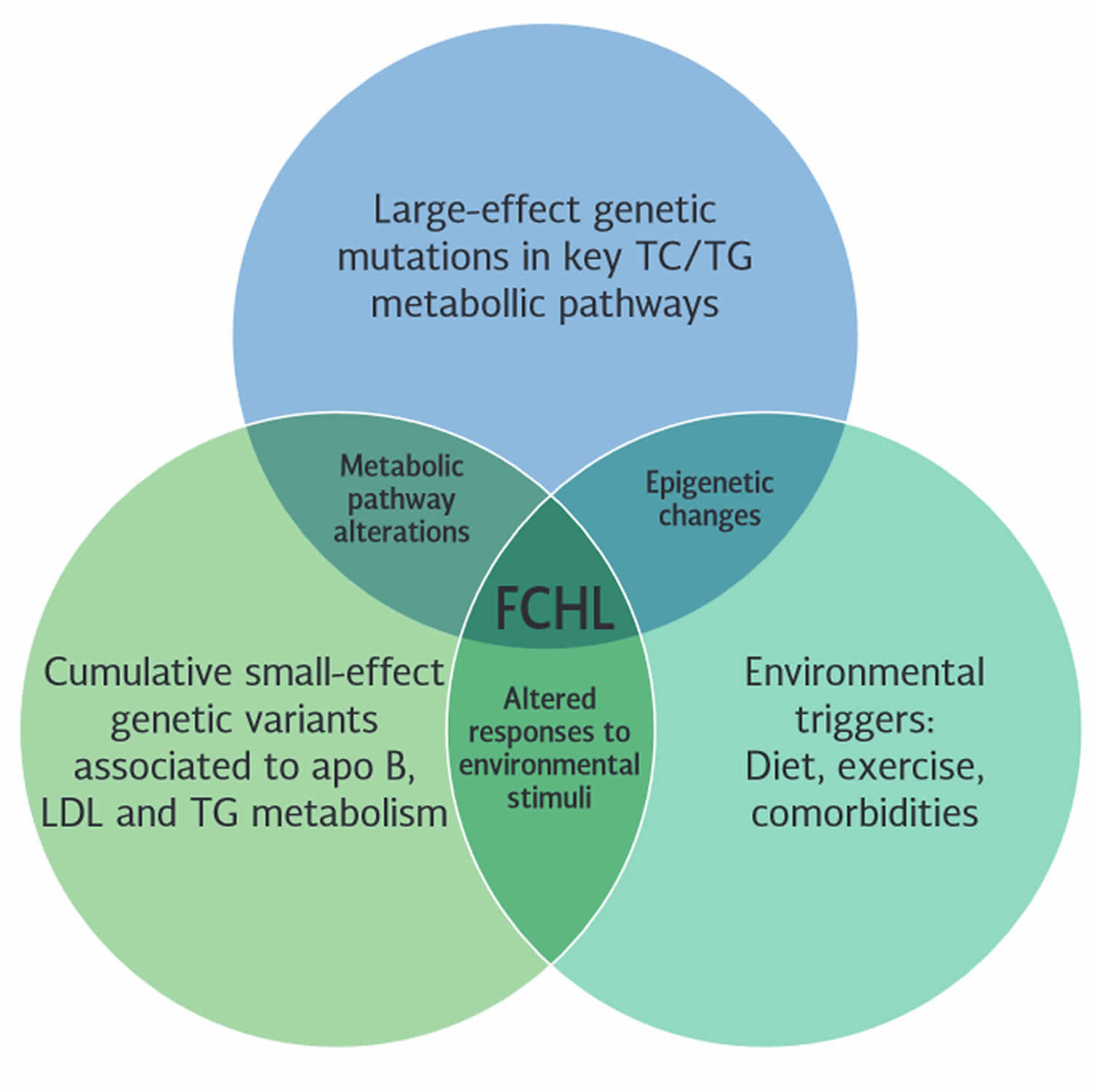
Footnotes: Genetics of familial combined hyperlipidemia (FCHL). The interplay of large-effect genetic mutations, cumulative small-effect genetic variants, and environmental triggers contribute to developing the familial combined hyperlipidemia phenotype.
[Source 6 ]What is cholesterol?
Cholesterol is a waxy, fat-like substance that’s found in all the cells in your body. Cholesterol is produced by your liver, adrenal glands, intestines, and in gonads and 20 to 25% of cholesterol comes from your diet (foods you eat) 34. Cholesterol is an essential component of all cell membranes – it helps to maintain structural integrity and fluidity of cell membranes, allowing your cells to change shapes easily without cell walls 35. More than 90% of cellular cholesterol is located at the plasma membrane 36. Your body needs some cholesterol to make hormones such as testosterone and estrogen, vitamin D and the biosynthesis of bile acids in your liver that help you digest foods 37, 38, 39.
Cholesterol comes from two sources. Your liver makes all of the cholesterol your body needs to form cell membranes and to make certain hormones. Cholesterol is also found in foods from animal sources, such as egg yolks, meat, and cheese, which is called dietary cholesterol. Although we often blame the cholesterol found in foods that we eat for raising blood cholesterol, the main culprit is actually saturated fat. Foods rich in saturated fat include butter fat in milk products, fat from red meat, and tropical oils such as coconut oil.
Cholesterol travels through your blood on proteins called lipoproteins. One type, LDL (low-density lipoprotein), is sometimes called the “bad” cholesterol. A high LDL level leads to a buildup of cholesterol in your arteries. Another type, HDL (high-density lipoprotein), is sometimes called the “good” cholesterol. HDL carries cholesterol from other parts of your body back to your liver. Then your liver removes the cholesterol from your body.
Types of cholesterol:
- HDL stands for high-density lipoprotein or HDL-C (high-density lipoprotein cholesterol). HDL is sometimes called “good cholesterol” because it carries harmful cholesterol from other parts of your body including your arteries back to your liver. Your liver then removes the cholesterol from your body and helps protect you from heart attack and stroke. A healthy HDL-cholesterol level may protect against heart attack and stroke. If you have low HDL levels, you have a greater heart disease risk, even if your total cholesterol is below 200 mg/dL. Your doctor will evaluate your HDL and other cholesterol levels and other factors to assess your risk for heart attack or stroke. People with high blood triglycerides usually also have lower levels of HDL. Genetic factors, Type 2 diabetes, smoking, being overweight and being sedentary can all lower HDL cholesterol. Women tend to have higher levels of HDL cholesterol than men do, because the female hormone estrogen raises HDL, but this can change after menopause.
- LDL stands for low-density lipoprotein or LDL-C (low-density lipoprotein cholesterol). LDL is sometimes called “bad cholesterol” because a high LDL level leads to the buildup of plaque in your arteries. LDL is the most important lipid for predicting your heart disease risk. Low-density lipoprotein (LDL or ‘bad’) cholesterol can join with fats and other substances to build up (also known as plaque) in the inner walls of your arteries, which starts a disease process called atherosclerosis. The arteries can become clogged and narrow, and blood flow is reduced. When plaque builds up in your coronary arteries that supply blood to your heart, you are at greater risk of having a heart attack. Since LDL is the bad kind of cholesterol, a low LDL level is considered good for your heart health. A diet high in saturated and trans fat is unhealthy because it tends to raise LDL cholesterol levels. Your LDL levels may be high if you eat a diet with a lot of saturated fat, cholesterol, or both. Sometimes, an under-active thyroid called hypothyroidism may also increase LDL levels.
- VLDL stands for very low-density lipoprotein or VLDL-C (very low-density lipoprotein cholesterol). Some people also call VLDL a “bad cholesterol” because it too contributes to the buildup of plaque in your arteries. But VLDL and LDL are different; VLDL mainly carries triglycerides and LDL mainly carries cholesterol. VLDL particles are released into the blood by the liver and circulate in the bloodstream, ultimately being converted into LDL as they lose triglyceride, having carried it to other parts of the body. According to the National Heart, Lung and Blood Institute’s National Cholesterol Education Program Guidelines ATP III, there is growing evidence that VLDL plays an important role in atherogenesis, in which plaques form on the interior walls of arteries, narrowing these passageways and restricting blood flow, which can lead to heart disease and increase the risk of stroke. Currently, direct measurement of VLDL cholesterol requires specialized testing. However, since VLDL-C contains most of the circulating triglyceride (if a person is fasting) and since the composition of the different particles is relatively constant, it is possible to estimate the amount of VLDL-C based on the triglyceride value. To estimate VLDL-C, divide the triglyceride value by 5 if the value is in mg/dL or divide by 2.2 if the value is in mmol/L. In most cases, this formula provides a good estimate of VLDL-C. However, this formula becomes less accurate with increased triglyceride levels when, for example, a person has not fasted before having blood drawn. The calculation is not valid when the triglyceride level is greater than 400 mg/dl (4.5 mmol/L) because other lipoproteins are usually present. In this situation, VLDL-C may be measured directly using specialized testing.
- Triglycerides. Triglycerides are the most common type of fat in your blood. Triglycerides come from food, and your body also makes them. When you eat, your body converts calories it doesn’t need into triglycerides, which are stored in fat cells. Triglycerides are fats that provide energy for your muscles. If you eat foods with a lot of saturated fat or carbohydrates, you will raise your triglyceride levels. High triglyceride levels are associated with several factors, including being overweight, eating too many sweets or drinking too much alcohol, smoking, being sedentary, or having diabetes with elevated blood sugar levels. Elevated triglycerides levels are thought to lead to a greater risk of heart disease, but scientists do not agree that high triglycerides alone are a risk factor for heart disease. Normal triglyceride levels vary by age and sex. People with high triglycerides often have a high total cholesterol level, including a high LDL (bad) cholesterol level and a low HDL (good) cholesterol level. Many people with metabolic syndrome or diabetes also have high triglyceride levels. Extremely high triglyceride levels (more than 1000 mg/dL) can lead to abdominal pain and a life-threatening disorder of the pancreas called pancreatitis. Factors that can contribute to elevated triglyceride levels:
- Overweight or obesity
- Insulin resistance or metabolic syndrome
- Diabetes mellitus, especially with poor glucose control
- Alcohol consumption, especially in excess
- Excess sugar intake, especially from processed foods
- High saturated fat intake
- Hypothyroidism
- Chronic kidney disease
- Physical inactivity
- Pregnancy (especially in the third trimester)
- Inflammatory diseases (such as rheumatoid arthritis, systemic lupus erythematosus
- Some medications may also increase triglycerides.
Your body naturally produces all the LDL (bad) cholesterol it needs. However, the genes you inherit and your lifestyle habits play a major role in your cholesterol levels. The most common cause of high cholesterol is an unhealthy lifestyle. An unhealthy lifestyle makes your body produce more LDL cholesterol than it needs. This can include:
- Unhealthy eating habits or unhealthy diet, such as eating lots of bad fats. One type, saturated fat, is found in some meats, dairy products, chocolate, baked goods, and deep-fried and processed foods. Eating a lot of foods high in saturated fats raises “bad” LDL cholesterol levels. Another type, trans fat, is in some fried and processed foods. Eating these fats can raise your LDL (bad) cholesterol. No more than 10% of your daily calories should come from saturated fats.
- Lack of physical activity, with lots of sitting and little exercise. This lowers your HDL (good) cholesterol.
- Smoking or exposure to tobacco smoke, which lowers HDL cholesterol, especially in women. It also raises your LDL cholesterol.
- Being overweight or obese.
- Stress may raise levels of certain hormones, such as corticosteroids. These can cause your body to make more cholesterol.
- Drinking too much alcohol (more than two drinks a day for men or one drink a day for women) can raise your total cholesterol level.
- Getting little or low-quality sleep has been linked to lower cardiovascular health.
Genetics may also cause people to have high cholesterol. For example, some people inherit genes from their mother, father or even grandparents that cause them to have too much cholesterol. This is called familial hypercholesterolemia (FH). The severity of familial hypercholesterolemia is related to the duration and degree of LDL cholesterol in the blood. Familial hypercholesterolemia is dangerous because it can cause premature atherosclerotic heart disease. If you have a family history of familial hypercholesterolemia or problems related to high cholesterol, get your cholesterol levels checked.
High cholesterol usually causes no signs or symptoms. Cholesterol travels through your blood silently. And it turns into atherosclerotic plaque (hardened deposits) silently. Plaque buildup is like someone tip-toeing on carpet. You might not see or notice its presence for a long time. You may have no symptoms until you have a heart attack or stroke. At that point, the plaque is like high heels on a hardwood floor. And it’s already caused serious damage to your body.
You can live for many years with high cholesterol and not even know it. That’s why it’s essential to get your cholesterol levels checked on a regular basis. If your cholesterol levels are too high (hyperlipidemia), that’s a red flag for you and your doctor. High cholesterol is a major risk factor for heart disease. But catching it early gives you a chance to make changes and get your cholesterol to a healthy level.
You can find out your cholesterol level with a cholesterol or lipid profile blood test. If you are concerned about your cholesterol level, talk to your doctor. You will need to stop eating for 10 to 12 hours before a cholesterol or lipid profile blood test, and the only liquid you may drink is water.
Lifestyle changes such as regular physical activity, losing excess weight, quitting if you smoke and healthy eating are the first line of defense against high cholesterol. Eating lots of fruit and vegetables, whole grains (especially oats), and beans and lentils can help lower your cholesterol. You can also help by losing weight, avoiding foods that are high in saturated fat, quitting smoking and being active. But, if you’ve made these important lifestyle changes and your cholesterol levels remain high, your doctor might recommend medication.
To reduce your risk with high cholesterol, it’s important to:
- Quit cigarette smoking.
- Do regular aerobic exercise.
- Identify and treat high blood pressure.
- Maintain a healthy weight.
- Diagnose and treat diabetes.
- Have a healthy diet.
High cholesterol is one of the major controllable risk factors for coronary heart disease, heart attack and stroke. If you have other risk factors such as smoking, high blood pressure or diabetes, your risk increases even more. The more risk factors you have and the more severe they are, the higher your overall risk.
Remember, making even modest changes now can help to prevent significant medical issues later. Do all you can to reduce your risk for the serious effects of heart attack and stroke.
Figure 3. Cholesterol molecular structure
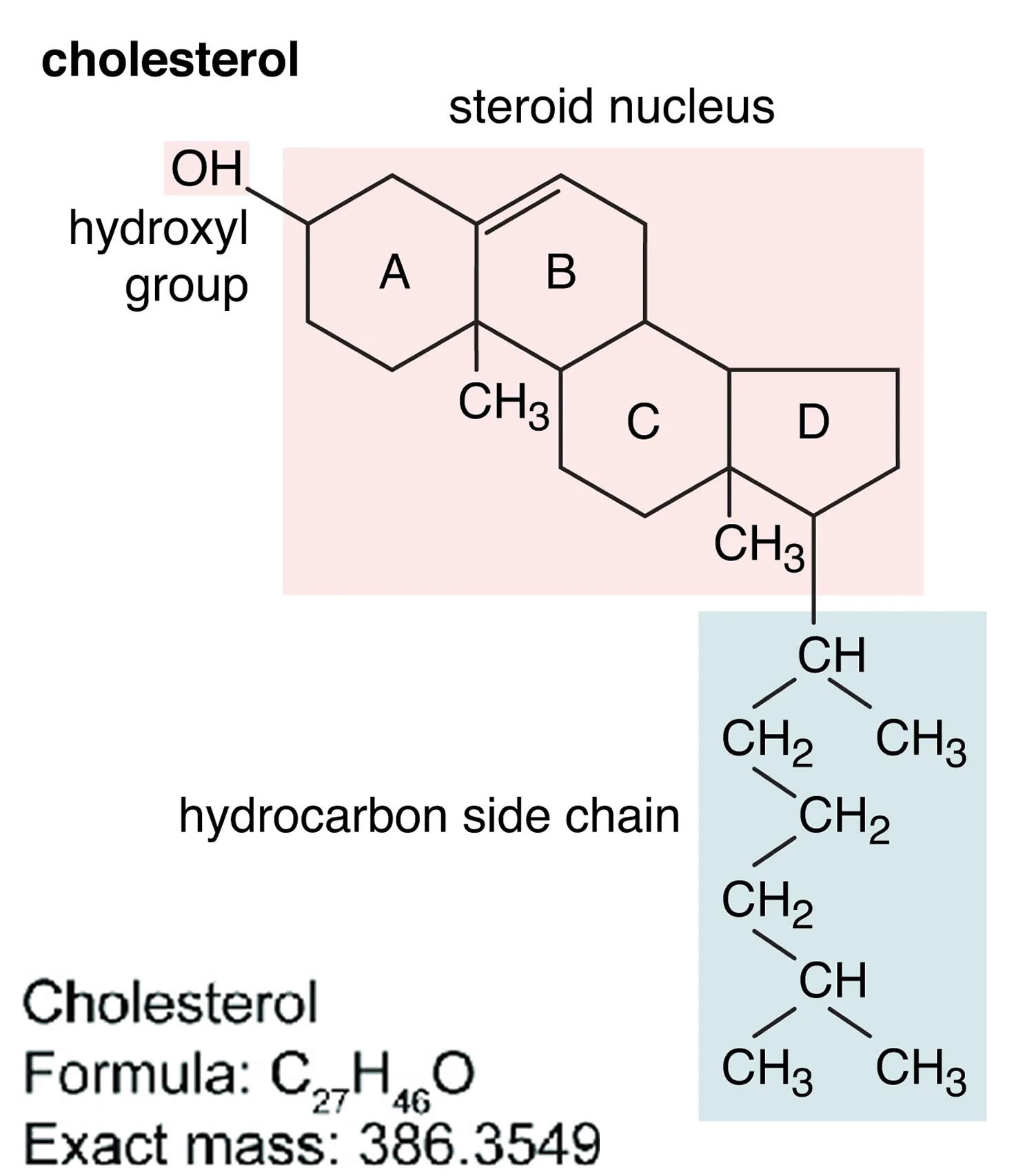
Footnotes: A cholesterol molecule contains three major parts: 1) tetracyclic carbon ring (A, B, C and D) as the core of steroids, 2) polar hydroxyl (OH) group attached to ring A, and 3) short non-polar carbon chain attached to ring D 40, 41. All four rings (A, B, C and D) of the sterol group are in a trans conformation, making cholesterol a planner molecule. The double bond between C5 and C6 helps to keep the rigidity of cholesterol.
[Source 42 ]What are Cholesterol levels?
Your cholesterol levels show how much cholesterol is circulating in your blood. Your blood cholesterol levels tell you how much lipid or fat is in your blood and your cholesterol levels are expressed in milligrams per deciliter (mg/dL). High cholesterol usually has no symptoms. You can find out your blood cholesterol levels with a cholesterol or lipid profile blood test. You will need to stop eating for 10 to 12 hours before a cholesterol or lipid profile blood test, and the only liquid you may drink is water.
Total blood or serum cholesterol is a composite of different measurements. Your “total blood cholesterol” is calculated by adding your HDL (“good” cholesterol) and LDL (“bad” cholesterol) cholesterol levels, plus 20% of your triglyceride level. Here’s the formula for calculating your “total blood cholesterol”:
Total cholesterol = HDL + LDL + 20% triglycerides.
- “Total cholesterol” is the total amount of cholesterol that’s circulating in your blood. Your “Total cholesterol” should be below 200 milligrams per deciliter of blood (less than 200 mg/dL) or 5.18 mmol/L.
- Your HDL “good” cholesterol is the one number you want to be high, ideally above 60 mg/dL (1.55 mmol/L) or higher.
- Your LDL “bad” cholesterol should be below 100 mg/dL (less than 2.59 mmol/L).
- Your triglycerides should be below 150 mg/dL (less than 1.70 mmol/L). Triglycerides are the most common type of fat in your blood. Triglycerides come from food, and your body also makes them. When you eat, your body converts calories it doesn’t need into triglycerides, which are stored in fat cells. High triglyceride levels are associated with several factors, including being overweight, eating too many sweets or drinking too much alcohol, smoking, being sedentary, or having diabetes with elevated blood sugar levels.
- According to the American Heart Association, more than 43% of American adults have cholesterol levels of 200 milligrams (mg) per deciliter (dL) or higher. Talk with your doctor about what your results mean for you and how to manage your cholesterol.
“Normal cholesterol levels” are less important than your overall cardiovascular risk. Like HDL and LDL cholesterol levels, your total blood cholesterol level should be considered in context with your other known risk factors. To determine your cardiovascular risk, your doctor will consider your cholesterol test results in context with your age, sex and family history. Other risk factors, such as smoking, diabetes and high blood pressure, will be considered as well. If your risk remains uncertain, and treatment options are unclear, your doctor may consider other factors and/or request a coronary artery calcium measurement to provide greater insight into your risk and help in decision-making.
In general, you want to have a total cholesterol level below 200 mg/dL or 5.18 mmol/L. Between 200 mg/dL and 239 mg/dL, your cholesterol level is elevated or borderline-high and should be lowered if you can. With a total cholesterol level of 240 mg/dL or above, your cholesterol level is high, and there is a need for action. For example, changing your diet, beginning an exercise program, and taking statins or other cholesterol-lowering medicines are all ways to lower your cholesterol level.
Factors that can increase your risk of bad cholesterol include:
- Poor diet. Eating saturated fat, found in animal products, and trans fats, found in some commercially baked cookies and crackers and microwave popcorn, can raise your cholesterol level. Foods that are high in cholesterol, such as red meat and full-fat dairy products, will also increase your cholesterol.
- Age. Your cholesterol levels tend to rise as you get older. For instance, as you age, your liver becomes less able to remove LDL cholesterol. Even though it is less common, younger people, including children and teens, can also have high cholesterol.
- Sex. Between ages 20 and 39, men have a greater risk for high total cholesterol than women. A woman’s risk goes up after menopause. Menopause lowers levels of female hormones that may protect against high blood cholesterol. After menopause, women’s levels of total and “bad” LDL cholesterol usually go up, while their levels of “good” HDL cholesterol go down.
- Heredity. High blood cholesterol can run in families.
- Weight. Being overweight or having obesity raises your cholesterol level. Having a body mass index (BMI) of 30 or greater puts you at risk of high cholesterol.
- Race. Certain races may have an increased risk of high cholesterol.
- Overall, non-Hispanic White people are more likely than other groups to have high levels of total cholesterol.
- Asian Americans, including those of Indian, Filipino, Japanese, and Vietnamese descent, are more likely to have high levels of “bad” LDL cholesterol than other groups.
- Hispanic Americans are more likely to have lower levels of “good” HDL cholesterol than other groups.
- African Americans are more likely than other groups to have high levels of “good” HDL cholesterol.
- Lack of exercise. Being physically inactive contributes to overweight and can raise LDL and lower HDL. Exercise helps boost your body’s HDL, or “good,” cholesterol while increasing the size of the particles that make up your LDL, or “bad,” cholesterol, which makes it less harmful.
- Smoking. Cigarette smoking damages the walls of your blood vessels, making them more prone to accumulate fatty deposits. Smoking might also lower your level of HDL, or “good,” cholesterol.
- Diabetes. High blood sugar contributes to higher levels of a dangerous cholesterol called very-low-density lipoprotein (VLDL) and lower HDL cholesterol. High blood sugar also damages the lining of your arteries.
The following foods can lower your bad cholesterol.
- Vegetables such as leafy greens (spinach, collard greens, kale, cabbage), broccoli, and carrots
- Fruits such as apples, bananas, oranges, pears, grapes, and prunes
- Whole grains such as plain oatmeal, brown rice, and whole-grain bread or tortillas
- Fat-free or low-fat dairy foods such as milk, cheese, or yogurt
- Protein-rich foods:
- Fish high in omega-3 fatty acids (salmon, tuna, and trout)
- Lean meats such as 95% lean ground beef or pork tenderloin or skinless chicken or turkey
- Eggs
- Nuts, seeds, and soy products (tofu)
- Legumes such as kidney beans, lentils, chickpeas, black-eyed peas, and lima beans
- Oils and foods high in monounsaturated and polyunsaturated fats:
- Canola, corn, olive, safflower, sesame, sunflower, and soybean oils (not coconut or palm oil)
- Nuts such as walnuts, almonds, and pine nuts
- Nut and seed butters
- Salmon and trout
- Seeds (sesame, sunflower, pumpkin, or flax)
- Avocados
- Tofu
There are usually no signs or symptoms that you have high cholesterol. A blood test is the only way to detect if you have it. The American Heart Association recommends all adults age 20 or older with no other risk factors for heart disease should have their cholesterol (and other traditional risk factors) checked every four to six years. If certain factors put you at high risk, or if you already have heart disease, your doctor may ask you to check it more often. Work with your doctor to determine your risk for cardiovascular disease and stroke and create a plan to reduce your risk.
If you have risk factors or if previous testing showed that you had a high cholesterol level, more frequent testing with a full lipid panel is recommended.
Examples of risk factors other than high LDL include:
- Cigarette smoking
- Being overweight or obese
- Unhealthy diet
- Being physically inactive—not getting enough exercise
- Age (if you are a male 45 years or older or a female 50-55 years or older)
- Hypertension (blood pressure of 140/90 or higher or taking high blood pressure medications)
- Family history of premature heart disease (heart disease in a first-degree male relative under age 55 or a first-degree female relative under age 65)
- Pre-existing heart disease or already having had a heart attack
- Diabetes or prediabetes
For people who are age 20 or older:
- Younger adults should have the test every 5 years
- Men ages 45 to 65 and women ages 55 to 65 should have it every 1 to 2 years.
Children, teens, and young adults (ages 2 to 24 years old) with no risk factors should have a lipid panel once between the ages of 9 and 11 and again between 17 and 21, according to the American Academy of Pediatrics.
For people who are age 19 or younger:
- The first test should be between ages 9 to 11
- Children should have the test again every 5 years
- Some children may have this test starting at age 2 if there is a family history of high blood cholesterol, heart attack, or stroke.
Children, teens, and young adults with an increased risk of developing heart disease as adults should have earlier and more frequent screening with lipid panels. Some of the risk factors are similar to those in adults and include a family history of heart disease or health problems such as diabetes, high blood pressure, or being overweight. High-risk children should be tested between 2 and 8 years old with a fasting lipid panel, according to the American Academy of Pediatrics.
Children younger than 2 years old are too young to be tested.
Figure 4. Cholesterol levels
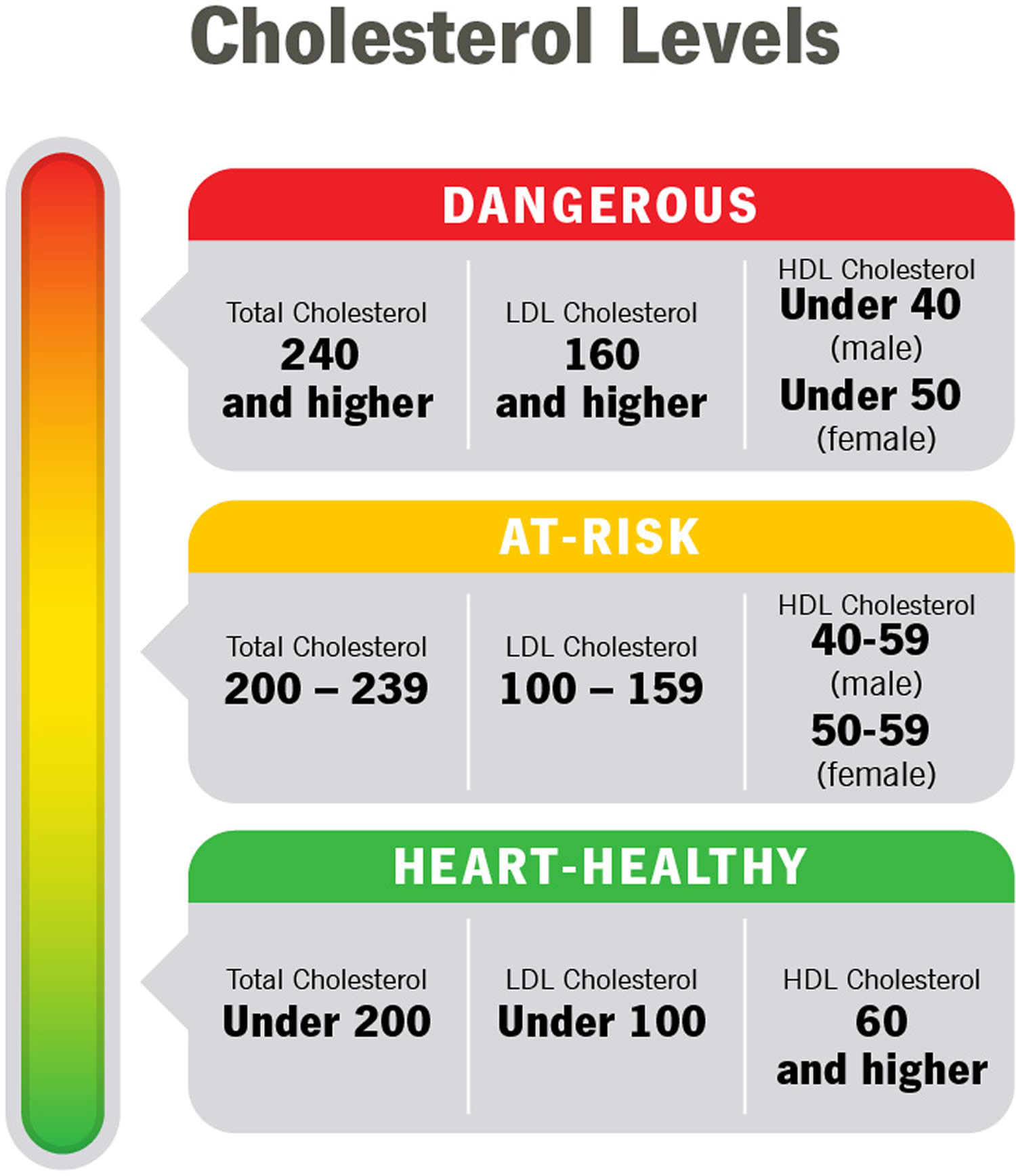
Table 1. Normal Cholesterol Levels
| Age | Total cholesterol | Non-HDL cholesterol | LDL (“bad” cholesterol) | HDL (“good” cholesterol) |
|---|---|---|---|---|
| 19 and younger | Below 170 milligrams per deciliter of blood (mg/dL) | Below 120 mg/dL | Below 110 mg/dL | Above 45 mg/dL |
| 20 and older Male | 125 to 200 milligrams per deciliter of blood (mg/dL) | Below 130 mg/dL | Below 100 mg/dL | 40 mg/dL or higher |
| 20 and older Female | 125 to 200 milligrams per deciliter of blood (mg/dL) | Below 130 mg/dL | Below 100 mg/dL | 50 mg/dL or higher |
Footnotes: As you review your results, remember that you want your LDL to be low and your HDL to be high. Ideally, your HDL should be above 60 mg/dL (1.55 mmol/L). It’s the helpful cholesterol. An HDL above 60 mg/dL (greater than 1.55 mmol/L) offers you protection against heart disease.
Table 2. High Cholesterol Levels
| Age | Total cholesterol | Non-HDL cholesterol | LDL (“bad” cholesterol) |
|---|---|---|---|
| 19 and younger | Borderline high: 170-199 mg/dL High: 200 mg/dL or higher | Borderline high: 120-144 mg/dL High: 145 mg/dL or higher | Borderline high: 110-129 mg/dL High: 130 mg/dL or higher |
| 20 and older | Borderline high: 200-239 mg/dL High: 240 mg/dL or higher | High: 130 mg/dL or higher | Near-optimal: 100-129 mg/dL Borderline high: 130-159 mg/dL High: 160-189 mg/dL Very high: 190 mg/dL or higher |
Footnotes: High cholesterol generally means your total cholesterol is 200 mg/dL (greater than 5.18 mmol/L) or higher. But doctors use additional categories like “borderline high” and “near optimal” to break down your results. If your numbers are close to normal levels, they may be easier to manage through lifestyle and dietary changes.
Table 3. Desirable Cholesterol Levels
| Desirable Cholesterol Levels | |
|---|---|
| Total cholesterol | Less than 200 milligrams per deciliter of blood (mg/dL) or 5.18 mmol/L |
| LDL (“bad” cholesterol) | Less than 100 mg/dL (2.59 mmol/L) |
| HDL (“good” cholesterol) | 60 mg/dL (1.55 mmol/L) or higher |
| Triglycerides | Less than 150 mg/dL (1.70 mmol/L) |
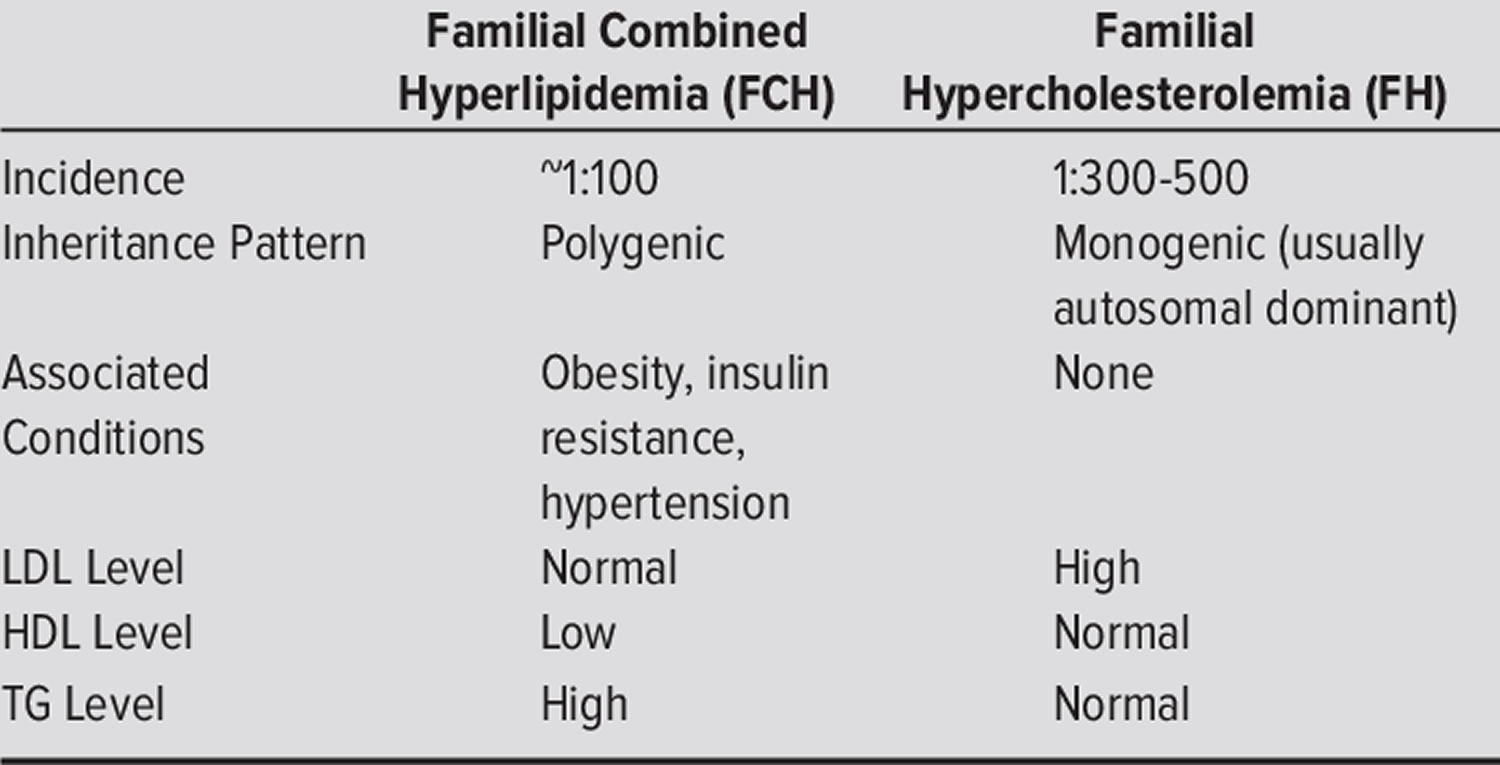
What is the difference between familial combined hyperlipidemia and familial hypercholesterolemia?
Familial combined hyperlidemia (FCHL) is a common inherited metabolic disorder characterized by: (a) increase in blood cholesterol level (hypercholesterolemia) and/or increased levels of triglycerides (triglyceridemia) in at least two members of the same family, (b) intra-individual and intrafamilial variability of the lipid phenotype, and (c) increased risk of premature coronary artery disease 2, 7, 8, 9. The laboratory abnormalities most frequently found in familial combined hyperlidemia (FCHL) are an increase of plasma triglycerides (triglyceridemia) and/or cholesterol levels (hypercholesterolemia), and a high prevalence of small very-low-density lipoproteins (VLDLs) and/or low-density lipoproteins (LDLs), mainly related to an increased plasma level of apolipoprotein B100 (apo B) 12. Some familial combined hyperlidemia patients can present with a decrease in high-density lipoprotein (HDL) cholesterol level, often inversely correlated to the triglycerides (TG) plasma level 13. There is a predominance of small and dense LDL so-called atherogenic LDL “B” pattern, poor in cholesterol, and with a high apolipoprotein B (apo B)/cholesterol ratio. The main determinants of LDLs size appear to be the triglyceride (TG) and HDL plasma levels 14. The synthesis of LDL-apo B increases due to uncontrolled overproduction of apo B 15. No major alterations in the LDL liver catabolic rate have been described.
Familial hypercholesterolemia also known as familial hyperlipoproteinemia type 2A or Fredrickson type 2A hyperlipidemia is an autosomal dominant genetic lipid disorder where your liver can’t process cholesterol properly and this leads to a severely elevated low-density lipoprotein (LDL) “bad” cholesterol in your blood that lead to atherosclerotic plaque deposition in the coronary arteries and proximal aorta at an early age, leading to an increased risk for premature atherosclerotic cardiovascular disease , the leading cause of preventable death in the United States 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54. The gene that causes familial hypercholesterolemia is inherited due to a defect (mutation) in a gene changes how your body processes cholesterol. Familial hypercholesterolemia is present from birth but symptoms may not appear until adulthood and a significant number of people remain undiagnosed. In familial hypercholesterolemia patients, genetic mutations make the liver incapable of metabolizing or removing excess low-density lipoprotein (LDL) “bad” cholesterol, from your blood. The result is very high low-density lipoprotein (LDL) “bad” cholesterol levels which can lead to atherosclerotic plaques deposition in the coronary arteries and proximal aorta occurring at an early age, leading over time to an increased risk for cardiovascular disease and even narrowing of your heart valves. As a result, people with familial hypercholesterolemia have a higher risk of premature coronary artery disease (coronary heart disease) manifesting as angina, early heart attack (myocardial infarction) and stroke 55. Familial hypercholesterolemia likely accounts for 2% to 3% of heart attacks (myocardial infarctions) in individuals younger than age 60 years. Untreated familial hypercholesterolemia, men are at a 50% risk for a fatal or non-fatal coronary event by age 50 years; untreated women are at a 30% risk by age 60 years (see Figure 5 below) 56.
Familial hypercholesterolemia is the most common inherited cardiovascular disease:
- About 1 in 200-250 people worldwide have familial hypercholesterolemia 57
- In the United States alone, an estimated 1.3 million people live with familial hypercholesterolemia. Yet only 10% of them are diagnosed. Nearly 2 million people in the US might have familial hypercholesterolemia and not even know it. Perhaps they won’t know it until they have a heart attack.
- Over 90% of people with familial hypercholesterolemia have not been properly diagnosed 57
- Familial hypercholesterolemia runs in families. If one parent has familial hypercholesterolemia, each child has a 50% chance of having familial hypercholesterolemia 58
- If left untreated, men have a 50% rise of having a heart attack by age 50. Untreated women have a 30% risk by age 60 59
- 1 in 160,000 to 1 in 1 million people have homozygous familial hypercholesterolemia 57. Homozygous familial hypercholesterolemia is more likely to occur in countries where the prevalence of heterozygous familial hypercholesterolemia is very high, especially those where consanguinity (marriage between relatives) is common.
- Familial hypercholesterolemia is even more common in certain populations such as French Canadians, Ashkenazi Jews, Lebanese, and South African Afrikaners. Lebanese Christians (1 in every 85 people), Afrikaners in South Africa (1/72 – 1/100), French Canadians (1 in every 270 people), and Ashkenazi Jews originating from Lithuania (1 in every 67 people) known as a founder affect.
There are two forms of familial hypercholesterolemia:
Heterozygous familial hypercholesterolemia
- If you have inherited this genetic mutation (one of three genes: low density lipoprotein receptor (LDLR) gene, apolipoprotein B-100 (APOB) gene and proprotein convertase subtilisin/kexin type 9 (PCSK9) gene) from one parent, then you will have Heterozygous familial hypercholesterolemia (HeFH). Heterozygous familial hypercholesterolemia occurs in 1 in 250 people worldwide. Most people with familial hypercholesterolemia have one affected gene and one normal gene (heterozygous familial hypercholesterolemia). An estimated 70%-95% of familial hypercholesterolemia results from a heterozygous pathogenic variant in one of three genes (apolipoprotein B [ApoB], low density lipoprotein receptor (LDLR), or PCSK9 gene).
- Until recently, it was believed that 1/500 people (630,000 people in the U.S.) had heterozygous familial hypercholesterolemia while about 1/1,000,000 people (300 people in the U.S.) had homozygous familial hypercholesterolemia. New studies suggest that heterozygous familial hypercholesterolemia and homozygous familial hypercholesterolemia are more common. It is now believed that 1/250 people (or 1.3 million people in the U.S. alone) have heterozygous familial hypercholesterolemia and 1/160,000 (or 2,000 people in the U.S.) have homozygous familial hypercholesterolemia. Even though homozygous familial hypercholesterolemia is still rare, it is a lot more common than we once thought. If you think that you or someone you know may have familial hypercholesterolemia, talk to your doctor.
- Heterozygous familial hypercholesterolemia: LDL-C > 160 mg/dL (4 mmol/L) for children and >190 mg/dL (5 mmol/L) for adults and with one first degree relative similarly affected or with positive genetic testing for an LDL-C raising gene defect (LDL receptor [LDLR], apolipoprotein-B [apo B] or proprotein convertase subtilisin/kexin type 9 [PCSK9])
Homozygous familial hypercholesterolemia
- If you inherit familial hypercholesterolemia from both parents, it is much more severe in its consequences. This form of familial hypercholesterolemia is called Homozygous familial hypercholesterolemia (HoFH). Homozygous familial hypercholesterolemia is very rare, occurring in about 1 in 160,000 to one million people worldwide. Most individuals with homozygous familial hypercholesterolemia experience severe coronary artery disease by their mid-20s and the rate of either death or coronary bypass surgery by the teenage years is high. Severe aortic stenosis is also common.
- Homozygous familial hypercholesterolemia: LDL-C > 400 mg/dL (10 mmol/L) and one or both parents:
- having clinically diagnosed familial hypercholesterolemia,
- positive genetic testing for an LDL-C raising (LDL receptor, apo B or PCSK9) gene defect, or
- autosomal recessive familial hypercholesterolemia If LDL-C > 560 mg/dL (14 mmol/L) or LDL-C > 400 mg/dL (10 mmol/L) with aortic valve disease or xanthomata at less than 20 years of age, homozygous familial hypercholesterolemia is highly likely.
Both types of familial hypercholesterolemia can be diagnosed with a physical examination and lab results, as well as personal and family history. Familial hypercholesterolemia also can be discovered through molecular diagnosis, genetic diagnosis or genetic testing. It’s helpful when genetic testing reveals familial hypercholesterolemia because it can alert relatives to their risk.
Some people with familial hypercholesterolemia have physical symptoms, but many don’t. One symptom of Familial hypercholesterolemia is cholesterol deposits in the Achilles tendons or the tendons of the hands or elbows. People with homozygous familial hypercholesterolemia also can develop cholesterol deposits in other areas, such as the skin surrounding the eyes or on the outer edge of the cornea.
If one person in a family has familial hypercholesterolemia, then all immediate relatives — parents, brothers, sisters and children — should be checked for it. Similarly, if someone in a family has an early heart attack, it’s a good idea for other family members to get tested.
Children with increased risk for familial hypercholesterolemia should be screened beginning at age 2. All children should have their cholesterol checked between ages 9 and 11 and again between ages 17 and 21.
Family history of early heart disease + High LDL “bad” cholesterol = Familial Hypercholesterolemia
Therefore, if you have familial hypercholesterolemia, your LDL-cholesterol levels will be very high, leading to narrowing or blockage of blood vessels (atherosclerosis). This process starts prior to birth and can ultimately result in heart disease, heart attack, or stroke. Because people with familial hypercholesterolemia have excessive cholesterol levels since before their birth, their risk of heart disease is 20 times greater than that of the general population. If a child has Homozygous familial hypercholesterolemia (inherited the familial hypercholesterolemia gene from both parents), she is exposed to an even higher risk, because her LDL-cholesterol levels are extraordinarily elevated and lead to progressive heart disease very early in life (often in the teens). Other cardiovascular disease risk factors include smoking, over-weight, high blood pressure, and a sedentary life. If you have familial hypercholesterolemia, eliminate smoking, control your weight and blood pressure, and lead a physically active lifestyle. All these are important factors, together with medical therapy and a healthful diet, in lowering your risk of an early heart attack.
All individuals with familial hypercholesterolemia should be considered “high risk” (i.e., increased ~20-fold) for coronary artery disease (coronary heart disease). Recent data suggest that individuals with an LDL “bad” cholesterol >190 mg/dL (>4.9 mmol/L) and a pathogenic variant in one of the genes (APOB, LDLR, PCSK9 & unknown genes) have a 22-fold increased risk for coronary heart disease, while those without a pathogenic variant have a sixfold increased risk for coronary artery disease over the general population 60.
Familial hypercholesterolemia is a lifelong condition. It’s not a temporary condition, like the common cold; it’s in your genes. When you have familial hypercholesterolemia, the most important step to take is therapies prescribed by your physician. But while this is the most essential measure, it’s not the only one. Familial hypercholesterolemia also means controlling weight, not smoking, eating a balanced diet low in saturated fat, and exercising. What is more, Familial hypercholesterolemia means bringing your family together to understand the impact of this disease. Familial hypercholesterolemia means living healthy, as individuals and as a family.
The good news is familial hypercholesterolemia is treatable! If familial hypercholesterolemia is found early, serious problems of the heart and blood vessels may be prevented or dramatically delayed by taking steps to protect yourself. These include:
- Not smoking.
- Exercising regularly.
- Eating a healthy diet low in saturated and trans fats.
- Taking medications.
- Going on LDL-apheresis.
Familial hypercholesterolemia in almost all cases requires aggressive treatment through a combined approach – medication, low-fat diet, exercise, weight control and not smoking. Familial hypercholesterolemia involves heart-healthy meals, regular exercise to get the blood flowing, controlling your weight and eliminating smoking. Because obesity and smoking are risk factors for heart disease and those with familial hypercholesterolemia are already at a 20 times higher risk, it is important to adapt to a healthy lifestyle.
Nearly 100% of people with familial hypercholesterolemia will require cholesterol-lowering medications. For some people with familial hypercholesterolemia, more aggressive measures are needed, including LDL-apheresis (a very simple procedure in which LDL-C cholesterol is removed from the blood on a weekly or biweekly basis.)
The American Academy of Pediatrics recommends that if a family has a pattern of early heart attacks or heart disease defined as before age 55 for men and 65 for women, children in that family should have cholesterol testing after the age of 2 years and before age 10.
It is important to find familial hypercholesterolemia and take action at any age, because when treated, the risk of heart disease can be reduced to levels similar to those of the general population.
Figure 5. Features of Familial Combined Hyperlipidemia and Familial Hypercholesterolemia
Footnotes: Features of Familial Combined Hyperlipidemia and Familial Hypercholesterolemia in Children.
Abbreviations: LDL = low-density lipoprotein; HDL = high-density lipoprotein; TG = triglycerides.
[Sources 4, 61 ]Figure 6. Familial hypercholesterolemia inheritance pattern
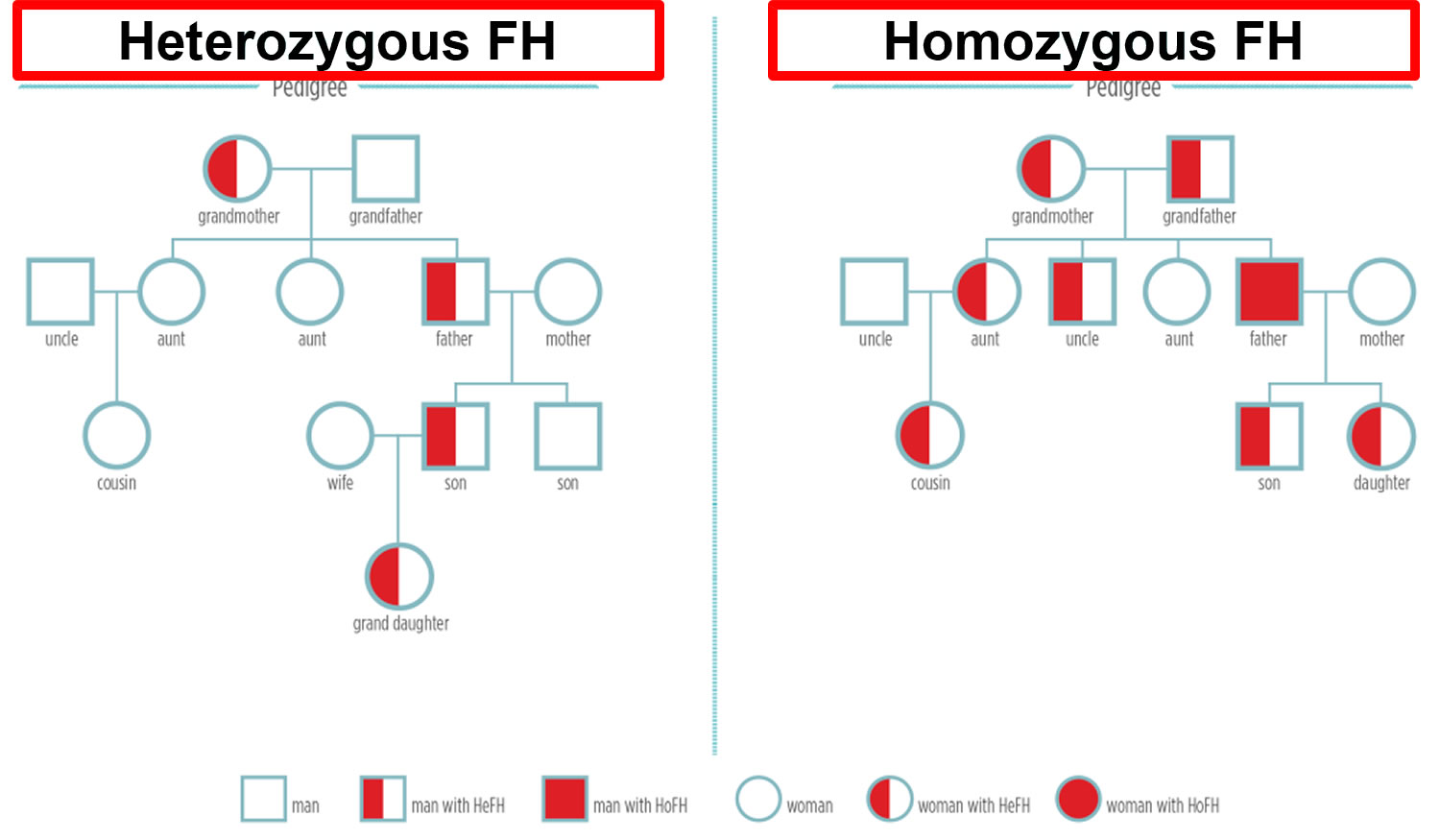
Note: HeFH = Heterozygous familial hypercholesterolemia; HoFH = Homozygous familial hypercholesterolemia
How is Familial Hypercholesterolemia different from Hypercholesterolemia or Hyperlipidemia?
Your clue here is “Familial”. By definition, hypercholesterolemia (or hyperlipidemia) simply means high cholesterol. However, Familial Hypercholesterolemia is a lifelong condition that is inherited. Otherwise put, your genes cause it. Therefore, it is a lot more serious than simply having high cholesterol caused from diet and it requires more aggressive treatment. familial hypercholesterolemia is a life-threatening disorder.
One of the most common scenarios in patients with familial hypercholesterolemia is being told that they have high cholesterol and that they need to change their diet. However, with familial hypercholesterolemia, eliminating fried foods and cheesecake from your diet is not enough. While saturated foods should be completely avoided, your high LDL-cholesterol mainly has to do with your genes/ your family’s medical history. With familial hypercholesterolemia, the cause of your high cholesterol is NOT your diet. Even if you ate nothing but oats and fruits, you would still have high LDL-cholesterol because of your liver’s inability to keep up with recycling and removing it.
Familial combined hyperlipidemia cause
The genetic inheritance of familial combined hyperlipidemia remains poorly understood. Familial combined hyperlipidemia (FCHL) was initially suggested to have a autosomal dominant monogenic mode of inheritance 62, 3. Later, some authors hypothesized that familial combined hyperlipidemia has a complex pattern related to multiple genetic variants with variable penetrance, environmental factors, and lifestyles 63, 64, 1. Certain ethnic groups are particularly susceptible to familial combined hyperlipidemia, as demonstrated by Paramsothy et al. 65 in a multiethnic cohort of 5923 participants in the United States, reporting a prevalence of 4.8% within Hispanics 66, 67.
Familial combined hyperlipidemia coexists with other metabolic diseases such as obesity, insulin resistance, type 2 diabetes, high blood pressure (hypertension), non-alcoholic fatty liver disease (NAFLD), and metabolic syndrome 68. Familial combined hyperlipidemia cases with metabolic comorbidities have remarkably higher apoB plasma levels compared to cases with a similar severity of insulin resistance. In addition, subjects with FCHL have a greater susceptibility to developing type 2 diabetes and are at a higher cardiovascular risk in comparison to matched controls 68, 11.
Pajukanta et al 69, 70 identified genetic loci linked to familial combined hyperlipidemia from different ethnic backgrounds on chromosome 1 long arm (1q21-1q23) in Finnish families with the disease. This region has also been linked to familial combined hyperlipidemia in families from other populations 71, 5, 72, 73, 74 and to type 2 diabetes 75, 76. The region in chromosome 1q21-1q23 includes several genes which might contribute to familial combined hyperlipidemia phenotype, including the upstream transcription factor 1 gene (USF1) 5. The gene encoding upstream transcription factor 1 (USF1) has appeared to be specifically linked to familial combined hyperlipidemia in 60 extended families with familial combined hyperlipidemia, including 721 genotyped individuals, especially males with high triglyceride (hypertriglyceridemia). The upstream transcription factor 1 (USF1) gene encodes a transcription factor that regulates nearly 40 genes implicated in lipid, lipoprotein and glucose metabolism, as well as immune response, and is located 1.5Mb away from TXNIP, a gene linked to mixed hyperlipidemia in mice 5, 11. The upstream transcription factor 1 (USF1) gene encodes for a basic helix-loop-helix leucine zipper transcription factor located in chromosome 1q23.3, which binds to a palindromic E-box sequence.
USF1 was first described by Sawadogo et al. 77 as a key component in adenovirus replication and its role as a regulator of lipid and glucose metabolism was later reported. USF1 has been shown to regulate expression of L-pyruvate kinase, fatty acid synthase, and glucokinase, as well as apoA-V, apoC-III, apoA-II, apoE, hormone-sensitive lipase, and other enzymes involved in lipid and carbohydrate metabolism 78, 79, 80.
The concept that USF1 affects the complex lipid phenotype of familial combined hyperlipidemia, and not only one lipid trait, is supported by the findings of the same authors on allelic associations of the usf1s1-usf1s2 risk haplotype with triglyceride (TG), apo B, total cholesterol, and LDL peak particle size 81. Pajukanta et al. 69 characterized USF1 as the major genetic trait of familial combined hyperlipidemia which was further demonstrated by Huertas-Vázquez et al. in Mexican population 82, 83.
A gene-hunting technique that traces patterns of disease in high-risk families (linkage studies) and association analysis suggested that the association of the newly discovered apo AV gene with APOAI/CIII/AIV cluster contributes to familial combined hyperlipidemia transmission in a case report of 128 European families 84.
Other authors proposed that LDL size in familial combined hyperlipidemia patients is a trait influenced by multiple loci located to chromosome 9 short arm [p], chromosome 16 long arm [q], and chromosome 11 long arm [q] 85.
Familial combined hyperlipidemia pathophysiology
Familial combined hyperlipidemia comprises both hyperapobetalipoproteinemia and normal or elevated apoB synthesis 86, 87. Alterations in both secretion and degradation of apoB particles have been encountered in familial combined hyperlipidemia patients and have been linked to insulin resistance, decreased apoB clearance rate, and increased expression of molecules that downregulate the LDL receptor 88, 64, 3. Imbalance between de novo lipogenesis and β-oxidation is a hallmark of familial combined hyperlipidemia, resulting in liver fat accumulation and very low-density lipoprotein (VLDL) overproduction 89. Adipose tissue dysfunction has been linked with an increase in free-fatty acid (FFA) levels and efflux of free-fatty acid (FFA) toward the liver, leading to an increased rate of lipoprotein synthesis 3, 11. It is known that increased levels and production of apoC-II and C-III are determinants of kinetics and plasma concentrations of triglyceride-rich lipoproteins (TRLs), including VLDL1 and 2 90. The APOC III gene has also been linked to states of insulin resistance and type 2 diabetes, both of which are frequent in familial combined hyperlipidemia patients.
Familial combined hyperlipidemia has also been characterized by lower intestinal cholesterol absorption and higher cholesterol synthesis independent of body mass index (BMI) in comparison to primary hypercholesterolemia of genetic origin 91. Unfavorable lipid profiles and increased postprandial lipemia have been linked to higher cardiovascular risk in familial combined hyperlipidemia 92. A study by Almeda-Valdes et al. 93 determined that the incremental area under the curve of postprandial lipemia in familial combined hyperlipidemia patients is determined by fasting apoB-48 levels and potentiated by the presence of abdominal obesity 94. This study also proposed that apoA-V was associated with VLDL and chylomicron production in familial combined hyperlipidemia subjects 93.
The role of USF1 in the pathogenesis of familial combined hyperlipidemia has not been completely explained. Inactivation of USF1 in mice leads to protection for diet-induced dyslipidemia, obesity, NAFLD, and atherosclerosis; the proposed mechanism has been linked to increased triglyceride uptake by brown adipose tissue through an LPL-dependent mechanism, which increases adrenergic response and thermogenesis 95. USF1 knockout mice (USF1−/−) preserved a normal lipid profile when exposed to a high-fat and -carbohydrate diet; in addition, this group showed an enhanced insulin sensitivity and reduced liver steatosis compared with USF1+/+ type mice 95. The findings of USF-1 downregulation in animal models are similar to those observed in humans in whom improved insulin sensitivity, atheroprotective lipid profiles, and decreased atherosclerosis were associated with reduced USF1 mRNA expression 95. Plaisier et al. 5 compared USF1 expression patterns in subcutaneous adipose tissue from familial combined hyperlipidemia patients compared to healthy controls, demonstrating higher USF1 expression in affected subjects. Wu et al. 96 developed two overexpression USF1 models in mice; both liver and systemic USF1 overexpression models showed adverse metabolic phenotypes including obesity, worsened lipid profile, and higher glucose/insulin ratio. These observations suggest a role for USF1 in the pathophysiology of familial combined hyperlipidemia; however, identification of precise mechanisms requires functional studies on human subjects with familial combined hyperlipidemia with and without USF1 variants that can be later confirmed in controlled studies in animal models.
In familial combined hyperlipidemia, there is an upregulation of thioredoxins, which are disulfide reductases responsible for regulating redox reactions, and confers an increased oxidative stress with reduced glutathione levels associated to insulin resistance. Both the increase in oxidative stress damage and insulin resistance contribute to atherosclerosis and potentially to increased cardiovascular risk in familial combined hyperlipidemia patients 97. Most familial combined hyperlipidemia patients have increased small dense low-density lipoprotein (sdLDL) and apoB levels for all levels of insulin resistance in comparison to controls, adjusted by HOMA-IR and BMI 98; this supports the concept that the cause of the lipid phenotype in familial combined hyperlipidemia is a result of additive effects of genetic determinants with modulation by BMI and insulin resistance. Cardiovascular risk has also been associated to insulin resistance in familial combined hyperlipidemia. Subjects with familial combined hyperlipidemia have been reported to have increased vascular inflammation and metabolic activity in spleen, bone marrow, and liver as measured by 18F-fluorodeoxyglucose positron-emission tomography/computed tomography imaging 99. In addition, Carratala et al. 100 showed that subjects with familial combined hyperlipidemia had increased plasminogen activator inhibitor type 1 (PAI-1) levels, which correlated with insulin resistance, metabolic syndrome components and increased carotid intima thickness, all of which are markers of increased cardiovascular risk 99, 101, 102, 103, 79.
Along with triglyceride-rich lipoproteins (TRLs), diminished lipoprotein clearance associated to insulin resistance-mediated decreased LPL activity, leads to small dense low-density lipoprotein (sdLDL) and intermediate-density lipoprotein particle accumulation, both of which are highly atherogenic and easily oxidized, contributing to its entry into subendothelial pathways 104. Decreased adiponectin levels in the setting of insulin resistance have been linked to higher levels of apoB and VLDL particles, further contributing to atherogenesis 105, which might be feasible in the setting of familial combined hyperlipidemia. Fibroblast growth factor 21 (FGF-21) is also implicated in the metabolism and kinetics of triglyceride-rich lipoproteins (TRLs), causing an increase in insulin-induced CD36 and LPL-mediated catabolism of triglyceride-rich lipoproteins (TRLs) in white and brown adipose tissue and a reduction of serum triglyceride concentrations. It could be hypothesized that the FGF-21 physiologic activity may be decreased in familial combined hyperlipidemia, but this has not been shown in animal or human subjects 106. There is also evidence that PCSK9 concentrations are elevated in familial combined hyperlipidemia and contribute to the impaired catabolism of apoB12. PSCK9 induces degradation and downregulation of the LDL receptor through resistin and other pro-inflammatory cytokines 3, 64 and one of the factors contributing to the hyperapolipoproteinemia in familial combined hyperlipidemia.
Familial combined hyperlipidemia signs and symptoms
Common features familial combined hyperlipidemia are:
- Frequent hypertriglyceridemia (high blood triglyceride level) and/or low plasma HDL cholesterol level. HDL is sometimes called the “good” cholesterol because it helps remove cholesterol from your arteries.
- Frequent association with non-lipid cardiovascular risk factors as high blood pressure (hypertension), abdominal obesity, reduced glucose tolerance or diabetes
- Frequent association with metabolic syndrome. Metabolic syndrome is the name for a group of risk factors for heart disease, stroke, type 2 diabetes, and other health problems. You can have just one risk factor, but people often have several of them together. When you have at least three of them, it is called metabolic syndrome. These risk factors include:
- A large waistline, also called abdominal obesity or “having an apple shape.” Too much fat around the stomach is a greater risk factor for heart disease than too much fat in other parts of the body.
- Having a high triglyceride level.
- Having a low HDL cholesterol level.
- Having high blood pressure. If your blood pressure stays high over time, it can damage your heart and lead to other health problems.
- Having a high fasting blood sugar. Mildly high blood sugar may be an early sign of diabetes.
- The more factors you have, the higher your risk for heart disease, diabetes, and stroke is.
- Strongly increased cardiovascular disease risk. Familial combined hyperlipidemia is strongly associated with premature coronary artery disease, with up to 10-14% of patients with premature coronary artery disease having comorbid familial combined hyperlipidemia 107. A patient diagnosed with familial combined hyperlipidemia has 1.7-10-fold higher risk of coronary artery disease compared to the average population 20 years after the initial diagnosis 107, 108. Wierzbicki et al. 109 demonstrated that 38% of premature heart attack survivors had familial combined hyperlipidemia, and a similar study including 706 participants with familial combined hyperlipidemia reported a coronary artery disease prevalence of 15.3%, describing that disease presentation was independent of age, sex, or presence of type 2 diabetes 68. Cardiovascular risk in patients with hypertriglyceridemia is also increased, especially in the setting of older age, tobacco use, and hypertension and decreased HDL cholesterol levels 63. Among familial combined hyperlipidemia patients, males are more susceptible to inherit and develop the lipid disorder independent of lipid profile, which might also account for the increased risk 110. Elevated expression of CD11b, a marker of fasting and postprandial leucocyte activation, has been previously reported for familial combined hyperlipidemia subjects and has been associated to increased cardiovascular risk in subjects with familial combined hyperlipidemia and comorbid type 2 diabetes 111, 112, 113, 114, 115.
The National Institutes of Health guidelines define metabolic syndrome as having three or more of the following traits, including traits for which you may be taking medication to control:
- Large waist — A waistline that measures at least 35 inches (89 centimeters) for women and 40 inches (102 centimeters) for men
- High triglyceride level — 150 milligrams per deciliter (mg/dL), or 1.7 millimoles per liter (mmol/L), or higher of this type of fat found in blood
- Reduced high-density lipoprotein (HDL) cholesterol or “good” cholesterol — Less than 40 milligrams per deciliter (mg/dL) (1.04 millimoles per liter (mmol/L)) in men or less than 50 mg/dL (1.3 mmol/L) in women of high-density lipoprotein (HDL) cholesterol
- Increased blood pressure — 130/85 millimeters of mercury (mm Hg) or higher
- Elevated fasting blood sugar — 100 mg/dL (5.6 mmol/L) or higher
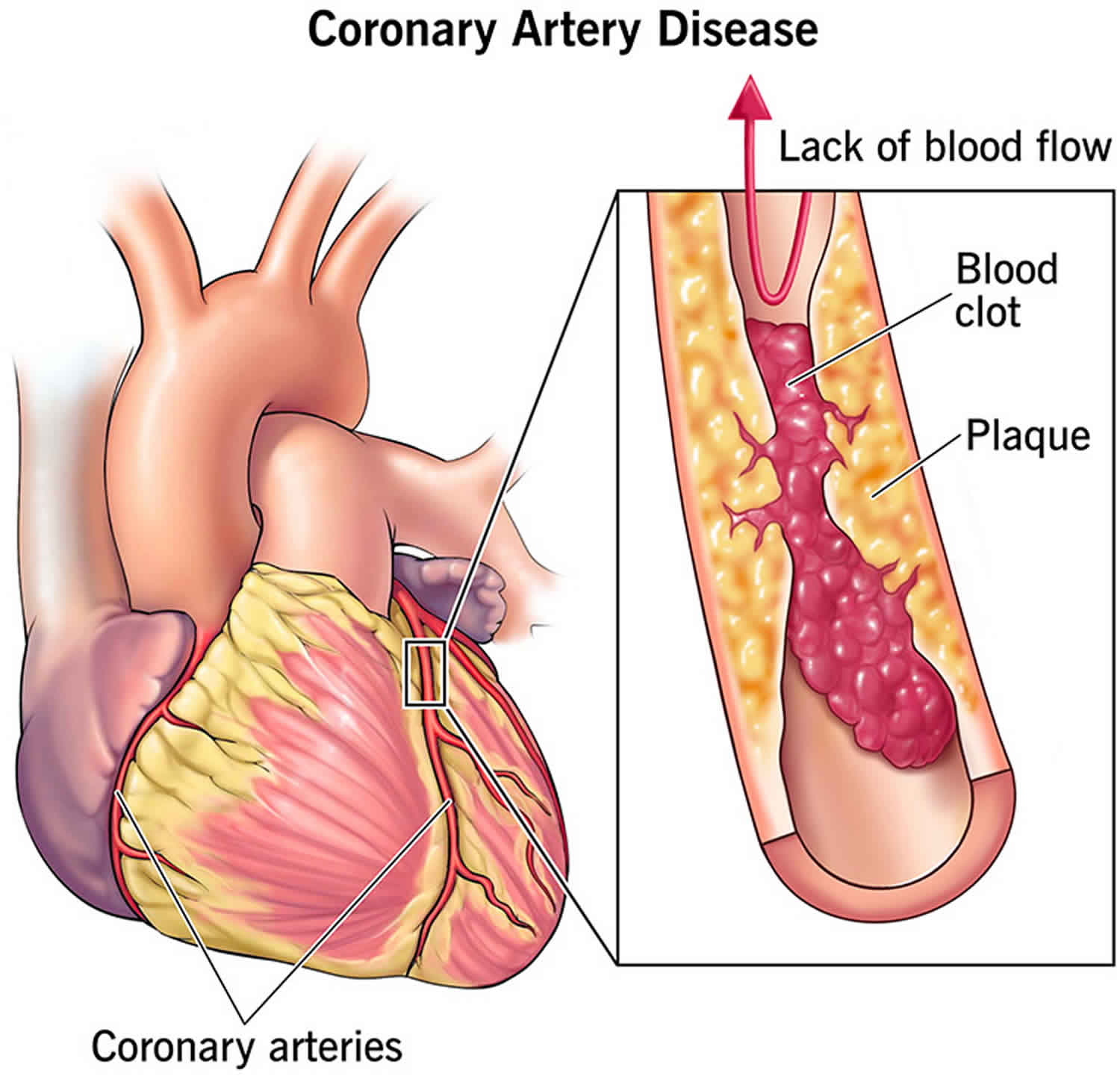
Cholesterol can enter your artery wall, damage its integrity and lead to the formation of atherosclerotic plaque (hardened deposits). This process of plaque buildup is called atherosclerosis. It can lead to serious problems like:
- Coronary artery disease: Blocked blood flow to your heart.
- Peripheral artery disease: Blocked blood flow to your legs and arms.
- Carotid artery disease: Blocked blood flow to your brain.
Familial combined hyperlipidemia diagnosis
Due to the lack of agreement among researchers because of a lack in specific laboratory or clinical marker of familial combined hyperlipidemia, a high degree of diagnostic uncertainty exists in the categorization familial combined hyperlipidemia 109, 6. Classically, to establish the diagnosis of familial combined hyperlipidemia comprised either isolated hypercholesterolemia or hypertriglyceridemia or a mixed lipid profile along with the first-degree family history of premature coronary artery disease, excluding other causes of hyperlipidemia 11. More recent criteria have also included elevated apoB levels as highly suggestive of familial combined hyperlipidemia (see Table 4) 116.
Due to the polygenic nature of familial combined hyperlipidemia, genetic testing is not yet a possibility 11. But familial combined hyperlipidemia diagnosis can be made based on a fluctuating lipid profile, increased apoB levels, and first-degree family history of mixed lipid disorders and premature cardiovascular disease 98, 117, 63. Some limitations on these criteria include the low practicality of apoB measurements in everyday clinical settings in addition to interethnic differences in establishing the 90th percentile in both lipid and apoB measurements, which require population-specific percentiles that might not always be available.
The following diagnostic criteria are suggested for familial combined hyperlipidemia:
First level diagnosis
- In the patient: primary hyperlipoproteinemia (LDL > 160 mg/dL and/or triglyceride > 200 mg/dL), PLUS
- In the patient and in at least one member of the family: primary variability of the lipid phenotype (hypercholesterolemia, hypertriglyceridemia, both, or even a “normal” phenotype) evaluated on the basis of at least 3 consecutive (bimonthly) controls (the repetition of lipid analysis before to define a diagnosis of dyslipidemia is in agreement with the international guidelines) 118.
Second level diagnosis (specialized labs only)
- Evaluation of apo B100 plasma level: preferably by standardized immunoturbidimetric assay 119
- Detection of small and dense LDL particles (LDL pattern B): there is not yet a standardized method to dose small dense LDLs; different methods have been tested (from preparative and non-equilibrium density gradient ultracentifugation to nuclear magnetic resonance) but the most frequently used is the gradient gel electrophoresis 120
- Genetic tests to exclude similar more rare forms of familial dyslipidaemias, when indicated (unclear situations, suggestive clinical and laboratory condition)
Specific cases
If family data are not available, the presence of unexplained (primary) IIb phenotype (eg, not related to significant change in dietary habits or body-weight gain or by an evident double heterozygosis for familial hypercholesterolemia and familial hypertriglyceridemia) may suggest the diagnosis 33. The presence of early onset atherosclerosis (increased carotid artery intima-media thickness (IMT) included) and/or clinical complications (coronary artery disease, cardiovascular disease or peripheral artery disease) in the patient and/or in relatives (probably carriers of the disease on the basis of genealogical tree) is not strictly diagnostic of familial combined hyperlipidemia, but it could suggest an aggressive dyslipidemia or, in any case, a condition at high risk of cardiovascular disease 8. Lipid abnormalities including presence of small and dense LDL in non-controlled diabetes will be regarded with caution and have to be re-evaluated after improvement of diabetes control 8.
Table 4. Changes in diagnostic criteria for familial combined hyperlipidemia throughout the years
| Year | Study/Author | Triglycerides (mmol/l) | Total cholesterol (mmol/l) | ApoB (g/l) | Family history | |
|---|---|---|---|---|---|---|
| 1973 | Goldstein | > 95th percentile | And | > 95th percentile | – | Coronary artery disease < 60 years |
| 1983 | Brunzell | 6.42 ± 1.19 | And | 2.53 ± 1.17 | 1.44 ± 0.36 | Coronary artery disease < 60 years |
| 1999 | EuroFam/Pajukanta | > 90th percentile | Or | > 90th percentile | – | Mixed hyperlipidemia |
| 1999 | Dutch/Aouizerat | > 6.5 | And | > 2.3 | > 1.2 | Differing hyperlipidemia in relative, coronary artery disease age < 60 years |
| 2001 | Consensus/Sniderman | – | – | > 1.5 | > 75th percentile | Hyperlipidemia in 1st degree relative |
| 2003 | British mapping/Naoumova | > 95th percentile | And | > 90th percentile | – | Hyperlipidemia in 1st and 2nd degree relatives |
| 2004 | Dutch clinical/Veerkamp | > 6.0 | And | > 1.5 | > 1.2 | Hyperlipidemia in 1st degree relative |
| 2004 | Huertas-Vazquez | > 90th percentile | Or | > 90th percentile | > 90th percentile | Coronary artery disease (i.e, heart attack) < 60 years in proband or 1st degree relative and One 1st degree relative triglycerides or total cholesterol > 90th percentile |
| 2004 | Aguilar-Salinas | > 150 mg/dL | Or | > 200 mg/dL | > 90th percentile | Coronary artery disease (i.e, heart attack) < 60 years At least three different family members: one with hypercholesterolemia, one with hypertriglyceridemia, and one with mixed hyperlipidemia |
| 2005 | GEM study/Wyszynski | – | – | > 75th percentile | – | Index case and one relative with relevant profile |
| 2014 | Mata | > 200 mg/dL | And/or | < 240 mg/dL (LDL > 160 mg/dL) | – | Two or more family members with hypercholesterolemia, hypertriglyceridemia, or mixed hyperlipidemia |
Familial combined hyperlipidemia treatment
The most important treatment for familial combined hyperlipidemia is a heart-healthy lifestyle, which includes:
- A heart-healthy diet, which limits the amount of saturated and trans fats that you eat. It encourages you to choose a variety of nutritious foods, including fruits, vegetables, whole grains, and lean meats. Healthy-eating plans include the Dietary Approaches to Stop Hypertension (DASH) diet and the Mediterranean diet, emphasize eating vegetables, fruits, high-fiber whole grains and lean protein. Healthy-eating plans tend to recommend limiting sugar-sweetened beverages, alcohol, salt, sugar and fat, especially saturated fat and trans fat.
- Healthy weight. Losing 7% of your body weight can reduce insulin resistance and blood pressure and decrease your risk of diabetes. Mateo-Gallego et al. 121 showed that a weight loss of 5% of total weight in overweight adults with familial combined hyperlipidemia significantly reduces triglyceride and non-HDL cholesterol levels at 3 and 6 months. This justifies the role of weight loss in overweight patients with familial combined hyperlipidemia to complement lipid-lowering therapy in familial combined hyperlipidemia 122. In fact, any amount of weight loss is beneficial. It’s also important to maintain your weight loss. If you’re struggling with losing weight and keeping it off, talk to your doctor about what options might be available to help you, such as medications or weight-loss surgery.
- Managing stress. Physical activity, meditation, yoga and other programs can help you handle stress and improve your emotional and physical health.
- Getting regular physical activity. Health experts recommend getting at least 30 minutes of exercise, such as brisk walking, daily. But you don’t have to do that activity all at once. Look for ways to increase activity any chance you get, such as walking instead of driving and using the stairs instead of an elevator.
- Quitting smoking or not starting if you don’t already smoke. Giving up cigarettes greatly improves your overall health. Talk to your doctor if you need help quitting.
If making lifestyle changes is not enough, you may need to take medicines. Your doctor might suggest medications to help control your blood pressure, cholesterol and blood sugar levels.
With regard to medicines, some small clinical trials have been conducted on patients defined as affected by “combined” or “mixed” hyperlipoproteinemia 123, 124, 125. Other small trials conducted on subjects selected as being affected by familial combined hyperlipidemia suggest some efficacy of statins 126, 127, fibrates 128, omega 3 polyunsaturated fatty acids 129, and pioglitazone 130 on secondary outcomes (eg, endothelial function, LDL composition, oxidation markers, inflammation markers). Atorvastatin and fenofibrate displayed comparable efficiency in decreasing oxysterols, but they decreased lipid-corrected alpha-tocopherol levels in plasma, which are already low in familial combined hyperlipidemia patients 131. However a full-dosage of a powerful statin such as rosuvastatin was not able to improve endothelial function of familial combined hyperlipidemia patients 132. Moreover, pioglitazone 30 mg/day in patients on conventional lipid-lowering therapy acts favorably on several metabolic parameters, such as TG/HDL (atherogenic index of plasma, plasma glucose, alanine-aminotransferase [ALT], and adiponectin) 133. Because of the lack of specific long-term data on drug efficacy on strong outcomes of familial combined hyperlipidemia patients, the main proposed recommendations for familial combined hyperlipidemia therapy are based on the results obtained from long-term clinical trials with hard outcomes on cardiovascular morbidity and mortality. However, the majority of available trial analyses are on the same group of patients with familial combined hyperlipidemia, with mixed/multigenic hyperlipoproteinemia (from random association of different genetic factors in the same subjects), metabolic syndrome, secondary hyperlipoproteinemia, etc, and data obtained might be not strictly representative of the effect of tested drugs/lifestyle changes on familial combined hyperlipidemia patients.
In any case, the effectiveness of statins to reduce cardiovascular risk suggests that statins should be the first-line treatment for familial combined hyperlipidemia also 134, perhaps with a preference for those with a stronger triglyceride-lowering activity 135. The triglyceride-lowering effect, which is mainly through an increase in the liver reuptake of VLDL, ILDL, and LDL is, however, less than that of fibrates, which increase lipoprotein lipase activity by a mechanism involving peroxisome proliferators activator receptors alpha and gamma 136.
The fibrates’ cholesterol-lowering effect is, however, smaller than that of statins. Omega-3 polyunsaturated fatty acids also lower VLDL triglycerides, slightly increasing LDL cholesterol and HDL cholesterol 137. The association of statins with drugs more active on triglyceride plasma levels (omega-3 polyunsaturated fatty acids, fibrates, nicotinic acid) could be an efficacious way to treat this kind of patients 125, 138. Ezetimibe, a selective inhibitor of the bowel cholesterol adsorption, might be an optimal drug to be associated to statins or fibrates instead of the prescribed resins 139.
Slow-release nicotinic acid is another very interesting and plausible therapeutic weapon to be associated to the standard statin and/or to fibrate therapy 140; probably, the dose-dependent effects of nicotinic acid derivatives and the good safety and interaction profiles, will open a new therapeutic approach even for more severe (and drug-resistant) familial combined hyperlipidemia patients.
Familial combined hyperlipidemia prognosis
Familial combined hyperlipidemia patients appear to be associated with an increased risk of coronary artery disease and cardiovascular disease 2, 7, 141, 142. Until now, no adequately designed trials on familial combined hyperlipidemia patients have been carried out to estimate their peculiar cardiovascular disease risk 8. Some authors suggest that it is at least as elevated as that of heterozygous familial hypercholesterolemia patients 143. Familial combined hyperlipidemia is clearly also a risk factor for increased carotid artery intima-media thickness (IMT): the increased carotid artery intima-media thickness (IMT) observed in familial combined hyperlipidemia patients corresponds, on average, to a 7-year increase in carotid artery intima-media thickness (IMT) 144. The parameter best correlated with IMT is the plasma apo B level and consequently the LDL particle size (but not LDL susceptibility to oxidation) 19. A worse prognostic factor appears to be the constant association of hypercholesterolemia to hypertriglyceridemia; young people with this kind of lipid phenotype have a reduced coronary flow reserve compared with age-matched hypercholesterolemic non-hypertriglyceridemic subjects 145. Hypertriglyceridemia per se appears in fact to be a significant predictor of cardiovascular disease in proportion to the baseline triglyceride levels 146.
- Taghizadeh, E., Farahani, N., Mardani, R. et al. Genetics of Familial Combined Hyperlipidemia (FCHL) Disorder: An Update. Biochem Genet 60, 453–481 (2022). https://doi.org/10.1007/s10528-021-10130-2[↩][↩]
- Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973 Jul;52(7):1544-68. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC302426/pdf/jcinvest00183-0022.pdf[↩][↩][↩]
- van Greevenbroek MM, Stalenhoef AF, de Graaf J, Brouwers MC. Familial combined hyperlipidemia: from molecular insights to tailored therapy. Curr Opin Lipidol. 2014 Jun;25(3):176-82. doi: 10.1097/MOL.0000000000000068[↩][↩][↩][↩][↩]
- Trinder M, Vikulova D, Pimstone S, Mancini GBJ, Brunham LR. Polygenic architecture and cardiovascular risk of familial combined hyperlipidemia. Atherosclerosis. 2022 Jan;340:35-43. https://doi.org/10.1016/j.atherosclerosis.2021.11.032[↩][↩]
- Brouwers MC, van Greevenbroek MM, Stehouwer CD, de Graaf J, Stalenhoef AF. The genetics of familial combined hyperlipidaemia. Nat Rev Endocrinol. 2012 Feb 14;8(6):352-62. doi: 10.1038/nrendo.2012.15[↩][↩][↩][↩][↩]
- Bello-Chavolla OY, Kuri-García A, Ríos-Ríos M, Vargas-Vázquez A, Cortés-Arroyo JE, Tapia-González G, Cruz-Bautista I, Aguilar-Salinas CA. FAMILIAL COMBINED HYPERLIPIDEMIA: CURRENT KNOWLEDGE, PERSPECTIVES, AND CONTROVERSIES. Rev Invest Clin. 2018;70(5):224-236. doi: 10.24875/RIC.18002575 https://clinicalandtranslationalinvestigation.com/frame_esp.php?id=187[↩][↩][↩][↩][↩]
- Sniderman AD, Castro Cabezas M, Ribalta J, Carmena R, de Bruin TW, de Graaf J, Erkelens DW, Humphries SE, Masana L, Real JT, Talmud PJ, Taskinen MR. A proposal to redefine familial combined hyperlipidaemia — third workshop on FCHL held in Barcelona from 3 to 5 May 2001, during the scientific sessions of the European Society for Clinical Investigation. Eur J Clin Invest. 2002 Feb;32(2):71-3. doi: 10.1046/j.1365-2362.2002.00941.x[↩][↩][↩]
- Gaddi A, Cicero AF, Odoo FO, Poli AA, Paoletti R; Atherosclerosis and Metabolic Diseases Study Group. Practical guidelines for familial combined hyperlipidemia diagnosis: an up-date. Vasc Health Risk Manag. 2007;3(6):877-86. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2350131[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Peterson AL, McBride PE. A review of guidelines for dyslipidemia in children and adolescents. WMJ. 2012 Dec;111(6):274-81; quiz 282.[↩][↩]
- Brouwers, M., van Greevenbroek, M., Stehouwer, C. et al. The genetics of familial combined hyperlipidaemia. Nat Rev Endocrinol 8, 352–362 (2012). https://doi.org/10.1038/nrendo.2012.15[↩]
- Mata P, Alonso R, Ruíz-Garcia A, Díaz-Díaz JL, González N, Gijón-Conde T, Martínez-Faedo C, Morón I, Arranz E, Aguado R, Argueso R, Perez de Isla L. Hiperlipidemia familiar combinada: documento de consenso [Familial combined hyperlipidemia: consensus document]. Semergen. 2014 Oct;40(7):374-80. Spanish. doi: 10.1016/j.semerg.2014.07.007[↩][↩][↩][↩][↩][↩]
- Sniderman AD, Ribalta J, Castro Cabezas M. How should FCHL be defined and how should we think about its metabolic bases? Nutr Metab Cardiovasc Dis. 2001 Aug;11(4):259-73. Erratum in: Nutr Metab Cardiovasc Dis 2001 Dec;11(6):412.[↩][↩]
- Hokanson JE, Austin MA, Zambon A, Brunzell JD. Plasma triglyceride and LDL heterogeneity in familial combined hyperlipidemia. Arterioscler Thromb. 1993 Mar;13(3):427-34. doi: 10.1161/01.atv.13.3.427[↩][↩]
- Vakkilainen J, Pajukanta P, Cantor RM, Nuotio IO, Lahdenperä S, Ylitalo K, Pihlajamäki J, Kovanen PT, Laakso M, Viikari JS, Peltonen L, Taskinen MR. Genetic influences contributing to LDL particle size in familial combined hyperlipidaemia. Eur J Hum Genet. 2002 Sep;10(9):547-52. doi: 10.1038/sj.ejhg.5200844[↩][↩]
- Kissebah AH, Alfarsi S, Evans DJ. Low density lipoprotein metabolism in familial combined hyperlipidemia. Mechanism of the multiple lipoprotein phenotypic expression. Arteriosclerosis. 1984 Nov-Dec;4(6):614-24. doi: 10.1161/01.atv.4.6.614[↩][↩]
- Kane JP, Havel RJ. Disorders of biogenesis and secretion of lipoproteins containing the B apolipoproteins. In: Scriver CR, Beaudet AL, Sly W, Valle D, editors. The metabolic basis of inherited disease. New York: McGraw-Hill Inc.; 1989. pp. 1129–64.[↩]
- Meijssen S, Derksen RJ, Bilecen S, Erkelens DW, Cabezas MC. In vivo modulation of plasma free fatty acids in patients with familial combined hyperlipidemia using lipid-lowering medication. J Clin Endocrinol Metab. 2002 Apr;87(4):1576-80. doi: 10.1210/jcem.87.4.8408[↩][↩][↩]
- Williams KJ, Petrie KA, Brocia RW, Swenson TL. Lipoprotein lipase modulates net secretory output of apolipoprotein B in vitro. A possible pathophysiologic explanation for familial combined hyperlipidemia. J Clin Invest. 1991 Oct;88(4):1300-6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC295599/pdf/jcinvest00063-0250.pdf[↩]
- Liu ML, Ylitalo K, Nuotio I, Salonen R, Salonen JT, Taskinen MR. Association between carotid intima-media thickness and low-density lipoprotein size and susceptibility of low-density lipoprotein to oxidation in asymptomatic members of familial combined hyperlipidemia families. Stroke. 2002 May;33(5):1255-60. doi: 10.1161/01.str.0000014924.29238.e1[↩][↩]
- Cicero AF, Panourgia MP, Linarello S, D’Addato S, Sangiorgi Z, Gaddi A. Serum lipopotein (a) levels in a large sample of subjects affected by familial combined hyperlipoproteinaemia and in general population. J Cardiovasc Risk. 2003 Apr;10(2):149-51. doi: 10.1097/01.hjr.0000060835.46105.75[↩]
- Soro A, Jauhiainen M, Ehnholm C, Taskinen MR. Determinants of low HDL levels in familial combined hyperlipidemia. J Lipid Res. 2003 Aug;44(8):1536-44. doi: 10.1194/jlr.M300069-JLR200[↩]
- Georgieva AM, van Greevenbroek MM, Krauss RM, Brouwers MC, Vermeulen VM, Robertus-Teunissen MG, van der Kallen CJ, de Bruin TW. Subclasses of low-density lipoprotein and very low-density lipoprotein in familial combined hyperlipidemia: relationship to multiple lipoprotein phenotype. Arterioscler Thromb Vasc Biol. 2004 Apr;24(4):744-9. doi: 10.1161/01.ATV.0000119681.47218.a4[↩]
- Venkatesan S, Cullen P, Pacy P, Halliday D, Scott J. Stable isotopes show a direct relation between VLDL apoB overproduction and serum triglyceride levels and indicate a metabolically and biochemically coherent basis for familial combined hyperlipidemia. Arterioscler Thromb. 1993 Jul;13(7):1110-8. doi: 10.1161/01.atv.13.7.1110[↩]
- Verseyden C, Meijssen S, Castro Cabezas M. Postprandial changes of apoB-100 and apoB-48 in TG rich lipoproteins in familial combined hyperlipidemia. J Lipid Res. 2002 Feb;43(2):274-80. https://doi.org/10.1016/S0022-2275(20)30169-3[↩]
- Campagna F, Montali A, Baroni MG, Maria AT, Ricci G, Antonini R, Verna R, Arca M. Common variants in the lipoprotein lipase gene, but not those in the insulin receptor substrate-1, the beta3-adrenergic receptor, and the intestinal fatty acid binding protein-2 genes, influence the lipid phenotypic expression in familial combined hyperlipidemia. Metabolism. 2002 Oct;51(10):1298-305. doi: 10.1053/meta.2002.35197[↩]
- Aouizerat BE, Allayee H, Cantor RM, Dallinga-Thie GM, Lanning CD, de Bruin TW, Lusis AJ, Rotter JI. Linkage of a candidate gene locus to familial combined hyperlipidemia: lecithin:cholesterol acyltransferase on 16q. Arterioscler Thromb Vasc Biol. 1999 Nov;19(11):2730-6. doi: 10.1161/01.atv.19.11.2730[↩]
- Pihlajamäki J, Karjalainen L, Karhapää P, Vauhkonen I, Taskinen MR, Deeb SS, Laakso M. G-250A substitution in promoter of hepatic lipase gene is associated with dyslipidemia and insulin resistance in healthy control subjects and in members of families with familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol. 2000 Jul;20(7):1789-95. doi: 10.1161/01.atv.20.7.1789[↩]
- Evans K, Burdge GC, Wootton SA, Collins JM, Clark ML, Tan GD, Karpe F, Frayn KN. Tissue-specific stable isotope measurements of postprandial lipid metabolism in familial combined hyperlipidaemia. Atherosclerosis. 2008 Mar;197(1):164-70. doi: 10.1016/j.atherosclerosis.2007.03.009[↩][↩]
- Meijssen S, van Dijk H, Verseyden C, Erkelens DW, Cabezas MC. Delayed and exaggerated postprandial complement component 3 response in familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol. 2002 May 1;22(5):811-6. doi: 10.1161/01.atv.0000014079.98335.72[↩]
- Ayyobi AF, McGladdery SH, McNeely MJ, Austin MA, Motulsky AG, Brunzell JD. Small, dense LDL and elevated apolipoprotein B are the common characteristics for the three major lipid phenotypes of familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol. 2003 Jul 1;23(7):1289-94. doi: 10.1161/01.ATV.0000077220.44620.9B[↩]
- de Graaf J, van der Vleuten GM, ter Avest E, Dallinga-Thie GM, Stalenhoef AF. High plasma level of remnant-like particles cholesterol in familial combined hyperlipidemia. J Clin Endocrinol Metab. 2007 Apr;92(4):1269-75. doi: 10.1210/jc.2006-1973[↩][↩]
- Pappan N, Awosika AO, Rehman A. Dyslipidemia. [Updated 2024 Mar 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560891[↩]
- Gaddi A, Galetti C, Pauciullo P, Arca M. Familial combined hyperlipoproteinemia: experts panel position on diagnostic criteria for clinical practice. Committee of experts of the Atherosclerosis and Dysmetabolic Disorders Study Group. Nutr Metab Cardiovasc Dis. 1999 Dec;9(6):304-11.[↩][↩]
- Norum KR, Berg T, Helgerud P, Drevon CA. Transport of cholesterol. Physiol Rev. 1983 Oct;63(4):1343-419. https://doi.org/10.1152/physrev.1983.63.4.1343[↩]
- Maekawa M, Fairn GD. Complementary probes reveal that phosphatidylserine is required for the proper transbilayer distribution of cholesterol. J Cell Sci. 2015;128:1422–33. doi: 10.1242/jcs.164715[↩]
- Goluszko P, Nowicki B. Membrane cholesterol: a crucial molecule affecting interactions of microbial pathogens with mammalian cells. Infect Immun. 2005;73:7791–6. doi: 10.1128/IAI.73.12.7791-7796.2005[↩]
- Mauvais-Jarvis F, editor. Sex and gender factors affecting metabolic homeostasis, diabetes and obesity. Vol. 1043. Cham: Springer International Publishing; 2017.[↩]
- Faridi KF, Lupton JR, Martin SS, Banach M, Quispe R, Kulkarni K, et al. Vitamin D deficiency and non-lipid biomarkers of cardiovascular risk. Arch Med Sci. 2017;13:732–7. doi: 10.5114/aoms.2017.68237[↩]
- Boyer JL. Bile formation and secretion. Compr Physiol. 2013 Jul;3(3):1035-78. doi: 10.1002/cphy.c120027[↩]
- Zhou Y, Briand V, Sharma N, Ahn S, Kasi R. Polymers comprising cholesterol: synthesis, self-assembly, and applications. Materials (Basel) 2009;2:636–60. doi: 10.3390/ma2020636[↩]
- Song Y, Kenworthy AK, Sanders CR. Cholesterol as a co-solvent and a ligand for membrane proteins. Protein Sci. 2014;23:1–22. doi: 10.1002/pro.2385[↩]
- Li LH, Dutkiewicz EP, Huang YC, Zhou HB, Hsu CC. Analytical methods for cholesterol quantification. J Food Drug Anal. 2019 Apr;27(2):375-386. doi: 10.1016/j.jfda.2018.09.001[↩]
- National Cholesterol Education Program. Third Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. https://www.nhlbi.nih.gov/files/docs/guidelines/atp3xsum.pdf[↩]
- van den Bosch SE, Hutten BA, Corpeleijn WE, Kusters DM. Familial hypercholesterolemia in children and the importance of early treatment. Curr Opin Lipidol. 2024 Jun 1;35(3):126-132. doi: 10.1097/MOL.0000000000000926[↩]
- Ontario Health (Quality). Genetic Testing for Familial Hypercholesterolemia: Health Technology Assessment. Ont Health Technol Assess Ser. 2022 Aug 23;22(3):1-155. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9470216[↩]
- Ison HE, Clarke SL, Knowles JW. Familial Hypercholesterolemia. 2014 Jan 2 [Updated 2022 Jul 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK174884[↩]
- Vaezi Z, Amini A. Familial Hypercholesterolemia. [Updated 2022 Sep 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK556009[↩]
- Campbell-Salome G, Jones LK, Masnick MF, et al. Developing and Optimizing Innovative Tools to Address Familial Hypercholesterolemia Underdiagnosis: Identification Methods, Patient Activation, and Cascade Testing for Familial Hypercholesterolemia. Circ Genom Precis Med. 2021 Feb;14(1):e003120. doi: 10.1161/CIRCGEN.120.003120[↩]
- Abul-Husn NS, Manickam K, Jones LK, et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science. 2016 Dec 23;354(6319):aaf7000. doi: 10.1126/science.aaf7000[↩]
- Varghese MJ. Familial hypercholesterolemia: A review. Ann Pediatr Cardiol. 2014 May;7(2):107-17. doi: 10.4103/0974-2069.132478[↩]
- Newman WP 3rd, Freedman DS, Voors AW, Gard PD, Srinivasan SR, Cresanta JL, Williamson GD, Webber LS, Berenson GS. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N Engl J Med. 1986 Jan 16;314(3):138-44. doi: 10.1056/NEJM198601163140302[↩]
- Warden BA, Fazio S, Shapiro MD. Familial Hypercholesterolemia: Genes and Beyond. [Updated 2021 Oct 23]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK343488[↩]
- Guirguis-Blake JM, Evans CV, Coppola EL, et al. Screening for Lipid Disorders in Children and Adolescents: An Evidence Update for the U.S. Preventive Services Task Force [Internet]. Rockville (MD): Agency for Healthcare Research and Quality (US); 2023 Jul. (Evidence Synthesis, No. 229.) Chapter 1, Introduction. Available from: https://www.ncbi.nlm.nih.gov/books/NBK593597[↩]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986 Apr 4;232(4746):34-47. doi: 10.1126/science.3513311[↩]
- Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262–8. https://www.ncbi.nlm.nih.gov/pubmed/22398274[↩]
- Reiner Ž, Catapano AL, De Backer G, Graham I, Taskinen MR, Wiklund O, Agewall S, Alegria E, Chapman MJ, Durrington P, Erdine S, Halcox J, Hobbs R, Kjekshus J, Filardi PP, Riccardi G, Storey RF, Wood D., Clinical Practice Guidelines Committee of the Spanish Society of Cardiology. ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J. 2011;32:1769–818 https://www.ncbi.nlm.nih.gov/pubmed/21712404[↩]
- Nordestgaard B G et al. Eur Heart J: 2013, 34:3478-3490[↩][↩][↩]
- Goldstein JL et al. Familial hypercholesterolemia. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001:2863-2913[↩]
- Marks D, Thorogood M, Neil HA et al. A review on the diagnosis, natural history, and treatment of familial hypercholesterolemia. Athersclerosis 2003;168:1-14.[↩]
- Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, Morrison AC, Brody JA, Gupta N, Nomura A, Kessler T, Duga S, Bis JC, van Duijn CM, Cupples LA, Psaty B, Rader DJ, Danesh J, Schunkert H, McPherson R, Farrall M, Watkins H, Lander E, Wilson JG, Correa A, Boerwinkle E, Merlini PA, Ardissino D, Saleheen D, Gabriel S, Kathiresan S. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–89 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5405769/[↩]
- Peterson AL, McBride PE. A review of guidelines for dyslipidemia in children and adolescents. WMJ. 2012 Dec;111(6):274-81; quiz 282. https://wmjonline.org/wp-content/uploads/2012/111/6/274.pdf[↩]
- Austin MA, Brunzell JD, Fitch WL, Krauss RM. Inheritance of low density lipoprotein subclass patterns in familial combined hyperlipidemia. Arteriosclerosis. 1990 Jul-Aug;10(4):520-30. doi: 10.1161/01.atv.10.4.520[↩]
- Brahm AJ, Hegele RA. Combined hyperlipidemia: familial but not (usually) monogenic. Curr Opin Lipidol. 2016 Apr;27(2):131-40. doi: 10.1097/MOL.0000000000000270[↩][↩][↩]
- Ellis KL, Hooper AJ, Burnett JR, Watts GF. Progress in the care of common inherited atherogenic disorders of apolipoprotein B metabolism. Nat Rev Endocrinol. 2016 Aug;12(8):467-84. doi: 10.1038/nrendo.2016.69[↩][↩][↩]
- Paramsothy P, Knopp R, Bertoni AG, Tsai MY, Rue T, Heckbert SR. Combined hyperlipidemia in relation to race/ethnicity, obesity, and insulin resistance in the Multi-Ethnic Study of Atherosclerosis. Metabolism. 2009 Feb;58(2):212-9. doi: 10.1016/j.metabol.2008.09.016[↩]
- Escobedo-de la Peña J, de Jesús-Pérez R, Schargrodsky H, Champagne B. Prevalencia de dislipidemias en la ciudad de México y su asociación con otros factores de riesgo cardiovascular. Resultados del estudio CARMELA [Prevalence of dyslipidemias in Mexico city and Its relation to other cardiovascular risk factors. Results from the CARMELA study]. Gac Med Mex. 2014 Mar-Apr;150(2):128-36. Spanish.[↩]
- Jacobson TA, Maki KC, Orringer CE, Jones PH, et al. NLA Expert Panel. National Lipid Association Recommendations for Patient-Centered Management of Dyslipidemia: Part 2. J Clin Lipidol. 2015 Nov-Dec;9(6 Suppl):S1-122.e1. doi: 10.1016/j.jacl.2015.09.002. Epub 2015 Sep 18. Erratum in: J Clin Lipidol. 2016 Jan-Feb;10(1):211. Underberg, James A [added].[↩]
- Skoumas I, Masoura C, Aznaouridis K, Metaxa V, Tsokanis A, Papadimitriou L, Tousoulis D, Pitsavos C, Stefanadis C. Impact of cardiometabolic risk factors on major cardiovascular events in patients with familial combined hyperlipidemia. Circ J. 2013;77(1):163-8. doi: 10.1253/circj.cj-12-0320[↩][↩][↩]
- Pajukanta P, Nuotio I, Terwilliger JD, Porkka KV, Ylitalo K, Pihlajamäki J, Suomalainen AJ, Syvänen AC, Lehtimäki T, Viikari JS, Laakso M, Taskinen MR, Ehnholm C, Peltonen L. Linkage of familial combined hyperlipidaemia to chromosome 1q21-q23. Nat Genet. 1998 Apr;18(4):369-73. doi: 10.1038/ng0498-369[↩][↩]
- Aguilar-Salinas CA, Tusie-Luna T, Pajukanta P. Genetic and environmental determinants of the susceptibility of Amerindian derived populations for having hypertriglyceridemia. Metabolism. 2014 Jul;63(7):887-94. doi: 10.1016/j.metabol.2014.03.012[↩]
- Sentinelli F, Minicocci I, Montali A, Nanni L, Romeo S, Incani M, Cavallo MG, Lenzi A, Arca M, Baroni MG. Association of RXR-Gamma Gene Variants with Familial Combined Hyperlipidemia: Genotype and Haplotype Analysis. J Lipids. 2013;2013:517943. doi: 10.1155/2013/517943[↩]
- Coon H, Myers RH, Borecki IB, Arnett DK, Hunt SC, Province MA, Djousse L, Leppert MF. Replication of linkage of familial combined hyperlipidemia to chromosome 1q with additional heterogeneous effect of apolipoprotein A-I/C-III/A-IV locus. The NHLBI Family Heart Study. Arterioscler Thromb Vasc Biol. 2000 Oct;20(10):2275-80. doi: 10.1161/01.atv.20.10.2275[↩]
- Pei W, Baron H, Müller-Myhsok B, Knoblauch H, Al-Yahyaee SA, Hui R, Wu X, Liu L, Busjahn A, Luft FC, Schuster H. Support for linkage of familial combined hyperlipidemia to chromosome 1q21-q23 in Chinese and German families. Clin Genet. 2000 Jan;57(1):29-34. doi: 10.1034/j.1399-0004.2000.570105.x[↩]
- Allayee H, Krass KL, Pajukanta P, Cantor RM, van der Kallen CJ, Mar R, Rotter JI, de Bruin TW, Peltonen L, Lusis AJ. Locus for elevated apolipoprotein B levels on chromosome 1p31 in families with familial combined hyperlipidemia. Circ Res. 2002 May 3;90(8):926-31. doi: 10.1161/01.res.0000015885.27134.f0[↩]
- Elbein SC, Hoffman MD, Teng K, Leppert MF, Hasstedt SJ. A genome-wide search for type 2 diabetes susceptibility genes in Utah Caucasians. Diabetes. 1999 May;48(5):1175-82. doi: 10.2337/diabetes.48.5.1175[↩]
- Wiltshire S, Hattersley AT, Hitman GA, Walker M, Levy JC, Sampson M, O’Rahilly S, Frayling TM, Bell JI, Lathrop GM, Bennett A, Dhillon R, Fletcher C, Groves CJ, Jones E, Prestwich P, Simecek N, Rao PV, Wishart M, Bottazzo GF, Foxon R, Howell S, Smedley D, Cardon LR, Menzel S, McCarthy MI. A genomewide scan for loci predisposing to type 2 diabetes in a U.K. population (the Diabetes UK Warren 2 Repository): analysis of 573 pedigrees provides independent replication of a susceptibility locus on chromosome 1q. Am J Hum Genet. 2001 Sep;69(3):553-69. doi: 10.1086/323249. Epub 2001 Aug 1. Erratum in: Am J Hum Genet 2002 Apr;70(4):1075.[↩]
- Kurokawa R. Initiation of Transcription Generates Divergence of Long Noncoding RNA, Long Noncoding RNAs. Switzerland:Springer;2015. 69-91[↩]
- Laurila PP, Soronen J, Kooijman S, et al. USF1 deficiency activates brown adipose tissue and improves cardiometabolic health. Sci Transl Med. 2016 Jan 27;8(323):323ra13. doi: 10.1126/scitranslmed.aad0015[↩]
- Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol. 2015 Nov;16(11):678-89. doi: 10.1038/nrm4074. Erratum in: Nat Rev Mol Cell Biol. 2016 Jan;17(1):64.[↩][↩]
- Di Taranto MD, Staiano A, D’Agostino MN, D’Angelo A, Bloise E, Morgante A, Marotta G, Gentile M, Rubba P, Fortunato G. Association of USF1 and APOA5 polymorphisms with familial combined hyperlipidemia in an Italian population. Mol Cell Probes. 2015 Feb;29(1):19-24. doi: 10.1016/j.mcp.2014.10.002[↩]
- Pajukanta P, Lilja HE, Sinsheimer JS, Cantor RM, Lusis AJ, Gentile M, Duan XJ, Soro-Paavonen A, Naukkarinen J, Saarela J, Laakso M, Ehnholm C, Taskinen MR, Peltonen L. Familial combined hyperlipidemia is associated with upstream transcription factor 1 (USF1). Nat Genet. 2004 Apr;36(4):371-6. doi: 10.1038/ng1320[↩]
- Auer S, Hahne P, Soyal SM, Felder T, Miller K, Paulmichl M, Krempler F, Oberkofler H, Patsch W. Potential role of upstream stimulatory factor 1 gene variant in familial combined hyperlipidemia and related disorders. Arterioscler Thromb Vasc Biol. 2012 Jun;32(6):1535-44. doi: 10.1161/ATVBAHA.112.245639). This finding might explain both the “monogenic”-like transmission of the trait and the intra-individual and intra-family variability of the phenotype. However, the gene–environment interaction could strongly influence the laboratory and clinical features of familial combined hyperlipidemia, complicating the disease detection by specialized lipidologists ((Stalenhoef AF. Interaction between genes and environment in inherited lipid disorders determines clinical presentation. Cardiovasc Drugs Ther. 2002 Jul;16(4):271-2. doi: 10.1023/a:1021717506730[↩]
- Corella D, Ordovas JM. SINGLE NUCLEOTIDE POLYMORPHISMS THAT INFLUENCE LIPID METABOLISM: Interaction with Dietary Factors. Annu Rev Nutr. 2005;25:341-90. doi: 10.1146/annurev.nutr.25.050304.092656[↩]
- Eichenbaum-Voline S, Olivier M, Jones EL, Naoumova RP, Jones B, Gau B, Patel HN, Seed M, Betteridge DJ, Galton DJ, Rubin EM, Scott J, Shoulders CC, Pennacchio LA. Linkage and association between distinct variants of the APOA1/C3/A4/A5 gene cluster and familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol. 2004 Jan;24(1):167-74. doi: 10.1161/01.ATV.0000099881.83261.D4[↩]
- Badzioch MD, Igo RP Jr, Gagnon F, Brunzell JD, Krauss RM, Motulsky AG, Wijsman EM, Jarvik GP. Low-density lipoprotein particle size loci in familial combined hyperlipidemia: evidence for multiple loci from a genome scan. Arterioscler Thromb Vasc Biol. 2004 Oct;24(10):1942-50. doi: 10.1161/01.ATV.0000143499.09575.93[↩]
- Lapierre L, McLeod R. Regulation of hepatic production of lipoproteins containing apolipoprotein B by ER-associated degradation. Future Lipidol. 2007;2:173-84.[↩]
- Morita SY. Metabolism and Modification of Apolipoprotein B-Containing Lipoproteins Involved in Dyslipidemia and Atherosclerosis. Biol Pharm Bull. 2016;39(1):1-24. doi: 10.1248/bpb.b15-00716[↩]
- Johansen RF, Søndergaard E, Sørensen LP, Jurik AG, Christiansen JS, Nielsen S. Basal and insulin-regulated VLDL1 and VLDL2 kinetics in men with type 2 diabetes. Diabetologia. 2016 Apr;59(4):833-43. doi: 10.1007/s00125-015-3856-5[↩]
- Lewis GF, Xiao C, Hegele RA. Hypertriglyceridemia in the genomic era: a new paradigm. Endocr Rev. 2015 Feb;36(1):131-47. doi: 10.1210/er.2014-1062[↩]
- Ooi EM, Chan DC, Hodson L, Adiels M, Boren J, Karpe F, Fielding BA, Watts GF, Barrett PH. Triglyceride-rich lipoprotein metabolism in women: roles of apoC-II and apoC-III. Eur J Clin Invest. 2016 Aug;46(8):730-6. doi: 10.1111/eci.12657[↩]
- Baila-Rueda L, Cenarro A, Lamiquiz-Moneo I, Perez-Calahorra S, Bea AM, Marco-Benedí V, Jarauta E, Mateo-Gallego R, Civeira F. Cholesterol oversynthesis markers define familial combined hyperlipidemia versus other genetic hypercholesterolemias independently of body weight. J Nutr Biochem. 2018 Mar;53:48-57. doi: 10.1016/j.jnutbio.2017.10.005[↩]
- Cruz-Bautista I, Mehta R, Cabiedes J, García-Ulloa C, Guillen-Pineda LE, Almeda-Valdés P, Cuevas-Ramos D, Aguilar-Salinas CA. Determinants of VLDL composition and apo B-containing particles in familial combined hyperlipidemia. Clin Chim Acta. 2015 Jan 1;438:160-5. doi: 10.1016/j.cca.2014.08.018[↩]
- Almeda-Valdes P, Cuevas-Ramos D, Mehta R, Muñoz-Hernandez L, Cruz-Bautista I, Perez-Mendez O, Tusie-Luna MT, Gomez-Perez FJ, Pajukanta P, Matikainen N, Taskinen MR, Aguilar-Salinas CA. Factors associated with postprandial lipemia and apolipoprotein A-V levels in individuals with familial combined hyperlipidemia. BMC Endocr Disord. 2014 Nov 25;14:90. doi: 10.1186/1472-6823-14-90[↩][↩]
- Klop B, Verseyden C, Ribalta J, Salazar J, Masana L, Cabezas MC. MTP gene polymorphisms and postprandial lipemia in familial combined hyperlipidemia: effects of treatment with atorvastatin. Clin Investig Arterioscler. 2014 Mar-Apr;26(2):49-57. doi: 10.1016/j.arteri.2013.11.006[↩]
- Laurila PP, Soronen J, Kooijman S, Forsström S, et al. USF1 deficiency activates brown adipose tissue and improves cardiometabolic health. Sci Transl Med. 2016 Jan 27;8(323):323ra13. doi: 10.1126/scitranslmed.aad0015[↩][↩][↩]
- Roman TS, Marvelle AF, Fogarty MP, Vadlamudi S, Gonzalez AJ, Buchkovich ML, Huyghe JR, Fuchsberger C, Jackson AU, Wu Y, Civelek M, Lusis AJ, Gaulton KJ, Sethupathy P, Kangas AJ, Soininen P, Ala-Korpela M, Kuusisto J, Collins FS, Laakso M, Boehnke M, Mohlke KL. Multiple Hepatic Regulatory Variants at the GALNT2 GWAS Locus Associated with High-Density Lipoprotein Cholesterol. Am J Hum Genet. 2015 Dec 3;97(6):801-15. doi: 10.1016/j.ajhg.2015.10.016[↩]
- Martinez-Hervas S, Artero A, Martinez-Ibañez J, Tormos MC, Gonzalez-Navarro H, Priego A, Martinez-Valls JF, Saez GT, Real JT, Carmena R, Ascaso JF. Increased thioredoxin levels are related to insulin resistance in familial combined hyperlipidaemia. Eur J Clin Invest. 2016 Jul;46(7):636-42. doi: 10.1111/eci.12642[↩]
- Sahebkar A, Watts GF. New therapies targeting apoB metabolism for high-risk patients with inherited dyslipidaemias: what can the clinician expect? Cardiovasc Drugs Ther. 2013 Dec;27(6):559-67. doi: 10.1007/s10557-013-6479-4[↩][↩]
- Toutouzas K, Skoumas J, Koutagiar I, Benetos G, Pianou N, Georgakopoulos A, Galanakos S, Antonopoulos A, Drakopoulou M, Oikonomou EK, Kafouris P, Athanasiadis E, Metaxas M, Spyrou G, Pallantza Z, Galiatsatos N, Aggeli C, Antoniades C, Keramida G, Peters AM, Anagnostopoulos CD, Tousoulis D. Vascular inflammation and metabolic activity in hematopoietic organs and liver in familial combined hyperlipidemia and heterozygous familial hypercholesterolemia. J Clin Lipidol. 2018 Jan-Feb;12(1):33-43. doi: 10.1016/j.jacl.2017.10.019[↩][↩]
- Carratala A, Martinez-Hervas S, Rodriguez-Borja E, Benito E, Real JT, Saez GT, Carmena R, Ascaso JF. PAI-1 levels are related to insulin resistance and carotid atherosclerosis in subjects with familial combined hyperlipidemia. J Investig Med. 2018 Jan;66(1):17-21. doi: 10.1136/jim-2017-000468[↩]
- Brouwers MC, de Graaf J, van Greevenbroek MM, Schaper N, Stehouwer CD, Stalenhoef AF. Novel drugs in familial combined hyperlipidemia: lessons from type 2 diabetes mellitus. Curr Opin Lipidol. 2010 Dec;21(6):530-8. doi: 10.1097/MOL.0b013e32833ea9ec[↩]
- Jiang ZG, de Boer IH, Mackey RH, Jensen MK, Lai M, Robson SC, Tracy R, Kuller LH, Mukamal KJ. Associations of insulin resistance, inflammation and liver synthetic function with very low-density lipoprotein: The Cardiovascular Health Study. Metabolism. 2016 Mar;65(3):92-9. doi: 10.1016/j.metabol.2015.10.017[↩]
- Conlon DM, Thomas T, Fedotova T, Hernandez-Ono A, Di Paolo G, Chan RB, Ruggles K, Gibeley S, Liu J, Ginsberg HN. Inhibition of apolipoprotein B synthesis stimulates endoplasmic reticulum autophagy that prevents steatosis. J Clin Invest. 2016 Oct 3;126(10):3852-3867. doi: 10.1172/JCI86028[↩]
- Castro Cabezas M. Postprandial lipaemia in familial combined hyperlipidaemia. Biochem Soc Trans. 2003;31:1090-3.[↩]
- Dallinga-Thie GM, Kroon J, Borén J, Chapman MJ. Triglyceride-Rich Lipoproteins and Remnants: Targets for Therapy? Curr Cardiol Rep. 2016 Jul;18(7):67. doi: 10.1007/s11886-016-0745-6[↩]
- Schlein C, Talukdar S, Heine M, Fischer AW, Krott LM, Nilsson SK, Brenner MB, Heeren J, Scheja L. FGF21 Lowers Plasma Triglycerides by Accelerating Lipoprotein Catabolism in White and Brown Adipose Tissues. Cell Metab. 2016 Mar 8;23(3):441-53. doi: 10.1016/j.cmet.2016.01.006[↩]
- Minicocci I, Prisco C, Montali A, Di Costanzo A, Ceci F, Pigna G, Arca M. Contribution of mutations in low density lipoprotein receptor (LDLR) and lipoprotein lipase (LPL) genes to familial combined hyperlipidemia (FCHL): a reappraisal by using a resequencing approach. Atherosclerosis. 2015 Oct;242(2):618-24. doi: 10.1016/j.atherosclerosis.2015.06.036[↩][↩]
- Mehta R, Reyes-Rodríguez E, Yaxmehen Bello-Chavolla O, Guerrero-Díaz AC, Vargas-Vázquez A, Cruz-Bautista I, A Aguilar-Salinas C. Performance of LDL-C calculated with Martin’s formula compared to the Friedewald equation in familial combined hyperlipidemia. Atherosclerosis. 2018 Oct;277:204-210. doi: 10.1016/j.atherosclerosis.2018.06.868[↩]
- Wierzbicki AS, Graham CA, Young IS, Nicholls DP. Familial combined hyperlipidaemia: under – defined and under – diagnosed? Curr Vasc Pharmacol. 2008 Jan;6(1):13-22. doi: 10.2174/157016108783331268[↩][↩]
- De Castro-Orós I, Cenarro A, Tejedor MT, Baila-Rueda L, Mateo-Gallego R, Lamiquiz-Moneo I, Pocoví M, Civeira F. Common genetic variants contribute to primary hypertriglyceridemia without differences between familial combined hyperlipidemia and isolated hypertriglyceridemia. Circ Cardiovasc Genet. 2014 Dec;7(6):814-21. doi: 10.1161/CIRCGENETICS.114.000522[↩]
- de Vries MA, Alipour A, Klop B, van de Geijn GJ, Janssen HW, Njo TL, van der Meulen N, Rietveld AP, Liem AH, Westerman EM, de Herder WW, Cabezas MC. Glucose-dependent leukocyte activation in patients with type 2 diabetes mellitus, familial combined hyperlipidemia and healthy controls. Metabolism. 2015 Feb;64(2):213-7. doi: 10.1016/j.metabol.2014.10.011[↩]
- Alipour A, Valdivielso P, Elte JW, Janssen HW, Rioja J, van der Meulen N, van Mechelen R, Njo TL, González-Santos P, Rietveld AP, Cabezas MC. Exploring the value of apoB48 as a marker for atherosclerosis in clinical practice. Eur J Clin Invest. 2012 Jul;42(7):702-8. doi: 10.1111/j.1365-2362.2011.02635.x[↩]
- Fan YM, Hernesniemi J, Oksala N, Levula M, Raitoharju E, Collings A, Hutri-Kähönen N, Juonala M, Marniemi J, Lyytikäinen LP, Seppälä I, Mennander A, Tarkka M, Kangas AJ, Soininen P, Salenius JP, Klopp N, Illig T, Laitinen T, Ala-Korpela M, Laaksonen R, Viikari J, Kähönen M, Raitakari OT, Lehtimäki T. Upstream Transcription Factor 1 (USF1) allelic variants regulate lipoprotein metabolism in women and USF1 expression in atherosclerotic plaque. Sci Rep. 2014 Apr 11;4:4650. doi: 10.1038/srep04650[↩]
- Amor AJ, Ortega E, Perea V, Cofán M, Sala-Vila A, Nuñez I, Gilabert R, Ros E. Relationship Between Total Serum Bilirubin Levels and Carotid and Femoral Atherosclerosis in Familial Dyslipidemia. Arterioscler Thromb Vasc Biol. 2017 Dec;37(12):2356-2363. doi: 10.1161/ATVBAHA.117.310071[↩]
- Averna M, Stroes E; lipid alterations beyond LDL expert working group. How to assess and manage cardiovascular risk associated with lipid alterations beyond LDL. Atheroscler Suppl. 2017 Apr;26:16-24. doi: 10.1016/S1567-5688(17)30021-1[↩]
- Relimpio F, Losada F, Pumar A, Mangas MA, Morales F, Astorga R. Relationships of apolipoprotein B(100) with the metabolic syndrome in Type 2 diabetes mellitus. Diabetes Res Clin Pract. 2002 Sep;57(3):199-207. doi: 10.1016/s0168-8227(02)00096-7[↩]
- Ripatti P, Rämö JT, Söderlund S, Surakka I, Matikainen N, Pirinen M, Pajukanta P, Sarin AP, Service SK, Laurila PP, Ehnholm C, Salomaa V, Wilson RK, Palotie A, Freimer NB, Taskinen MR, Ripatti S. The Contribution of GWAS Loci in Familial Dyslipidemias. PLoS Genet. 2016 May 26;12(5):e1006078. doi: 10.1371/journal.pgen.1006078[↩]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. 2001 May 16;285(19):2486-97. doi: 10.1001/jama.285.19.2486[↩]
- Plan and operation of the Third National Health and Nutrition Examination Survey, 1988-94. Series 1: programs and collection procedures. Vital Health Stat 1. 1994 Jul;(32):1-407.[↩]
- Rizzo M, Berneis K. Low-density lipoprotein size and cardiovascular risk assessment. QJM. 2006 Jan;99(1):1-14. doi: 10.1093/qjmed/hci154. Erratum in: QJM. 2007 Feb;100(2):147.[↩]
- Mateo-Gallego R, Perez-Calahorra S, Cofán M, Baila-Rueda L, Cenarro A, Ros E, Puzo J, Civeira F. Serum lipid responses to weight loss differ between overweight adults with familial hypercholesterolemia and those with familial combined hyperlipidemia. J Nutr. 2014 Aug;144(8):1219-26. doi: 10.3945/jn.114.191775[↩]
- Martin SS, Abd TT, Jones SR, Michos ED, Blumenthal RS, Blaha MJ. 2013 ACC/AHA cholesterol treatment guideline: what was done well and what could be done better. J Am Coll Cardiol. 2014 Jun 24;63(24):2674-8. doi: 10.1016/j.jacc.2014.02.578[↩]
- Forster LF, Stewart G, Bedford D, Stewart JP, Rogers E, Shepherd J, Packard CJ, Caslake MJ. Influence of atorvastatin and simvastatin on apolipoprotein B metabolism in moderate combined hyperlipidemic subjects with low VLDL and LDL fractional clearance rates. Atherosclerosis. 2002 Sep;164(1):129-45. doi: 10.1016/s0021-9150(02)00052-7[↩]
- Wang TD, Chen WJ, Lin JW, Cheng CC, Chen MF, Lee YT. Efficacy of fenofibrate and simvastatin on endothelial function and inflammatory markers in patients with combined hyperlipidemia: relations with baseline lipid profiles. Atherosclerosis. 2003 Oct;170(2):315-23. doi: 10.1016/s0021-9150(03)00296-x[↩]
- Grundy SM, Vega GL, Yuan Z, Battisti WP, Brady WE, Palmisano J. Effectiveness and tolerability of simvastatin plus fenofibrate for combined hyperlipidemia (the SAFARI trial). Am J Cardiol. 2005 Feb 15;95(4):462-8. doi: 10.1016/j.amjcard.2004.10.012. Erratum in: Am J Cardiol. 2006 Aug 1;98(3):427-8.[↩][↩]
- Blanco-Colio LM, Martín-Ventura JL, Sol JM, Díaz C, Hernández G, Egido J. Decreased circulating Fas ligand in patients with familial combined hyperlipidemia or carotid atherosclerosis: normalization by atorvastatin. J Am Coll Cardiol. 2004 Apr 7;43(7):1188-94. doi: 10.1016/j.jacc.2003.10.046[↩]
- Sirtori CR, Calabresi L, Pisciotta L, Cattin L, Pauciullo P, Montagnani M, Manzato E, Bittolo Bon G, Fellin R. Effect of statins on LDL particle size in patients with familial combined hyperlipidemia: a comparison between atorvastatin and pravastatin. Nutr Metab Cardiovasc Dis. 2005 Feb;15(1):47-55. doi: 10.1016/j.numecd.2004.08.001[↩]
- Bredie SJ, Westerveld HT, Knipscheer HC, de Bruin TW, Kastelein JJ, Stalenhoef AF. Effects of gemfibrozil or simvastatin on apolipoprotein-B-containing lipoproteins, apolipoprotein-CIII and lipoprotein(a) in familial combined hyperlipidaemia. Neth J Med. 1996 Aug;49(2):59-67. doi: 10.1016/0300-2977(96)00015-0[↩]
- Tatò F, Keller C, Wolfram G. Effects of fish oil concentrate on lipoproteins and apolipoproteins in familial combined hyperlipidemia. Clin Investig. 1993 Apr;71(4):314-8. doi: 10.1007/BF00184734[↩]
- Abbink EJ, De Graaf J, De Haan JH, Heerschap A, Stalenhoef AF, Tack CJ. Effects of pioglitazone in familial combined hyperlipidaemia. J Intern Med. 2006 Jan;259(1):107-16. doi: 10.1111/j.1365-2796.2005.01579.x[↩]
- Arca M, Natoli S, Micheletta F, Riggi S, Di Angelantonio E, Montali A, Antonini TM, Antonini R, Diczfalusy U, Iuliano L. Increased plasma levels of oxysterols, in vivo markers of oxidative stress, in patients with familial combined hyperlipidemia: reduction during atorvastatin and fenofibrate therapy. Free Radic Biol Med. 2007 Mar 1;42(5):698-705. doi: 10.1016/j.freeradbiomed.2006.12.013[↩]
- ter Avest E, Abbink EJ, Holewijn S, de Graaf J, Tack CJ, Stalenhoef AF. Effects of rosuvastatin on endothelial function in patients with familial combined hyperlipidaemia (FCH). Curr Med Res Opin. 2005 Sep;21(9):1469-76. doi: 10.1185/030079905X61910[↩]
- Thomas EL, Potter E, Tosi I, Fitzpatrick J, Hamilton G, Amber V, Hughes R, North C, Holvoet P, Seed M, Betteridge DJ, Bell JD, Naoumova RP. Pioglitazone added to conventional lipid-lowering treatment in familial combined hyperlipidaemia improves parameters of metabolic control: relation to liver, muscle and regional body fat content. Atherosclerosis. 2007 Nov;195(1):e181-90. doi: 10.1016/j.atherosclerosis.2007.03.043[↩]
- Bays H, Stein EA. Pharmacotherapy for dyslipidaemia–current therapies and future agents. Expert Opin Pharmacother. 2003 Nov;4(11):1901-38. doi: 10.1517/14656566.4.11.1901[↩]
- Verseyden C, Meijssen S, Cabezas MC. Effects of atorvastatin on fasting plasma and marginated apolipoproteins B48 and B100 in large, triglyceride-rich lipoproteins in familial combined hyperlipidemia. J Clin Endocrinol Metab. 2004 Oct;89(10):5021-9. doi: 10.1210/jc.2003-032171[↩]
- Insua A, Massari F, Rodríguez Moncalvo JJ, Rubén Zanchetta J, Insua AM. Fenofibrate of gemfibrozil for treatment of types IIa and IIb primary hyperlipoproteinemia: a randomized, double-blind, crossover study. Endocr Pract. 2002 Mar-Apr;8(2):96-101. doi: 10.4158/EP.8.2.96[↩]
- Calabresi L, Villa B, Canavesi M, Sirtori CR, James RW, Bernini F, Franceschini G. An omega-3 polyunsaturated fatty acid concentrate increases plasma high-density lipoprotein 2 cholesterol and paraoxonase levels in patients with familial combined hyperlipidemia. Metabolism. 2004 Feb;53(2):153-8. doi: 10.1016/j.metabol.2003.09.007[↩]
- Koh KK, Quon MJ, Han SH, Chung WJ, Ahn JY, Seo YH, Choi IS, Shin EK. Additive beneficial effects of fenofibrate combined with atorvastatin in the treatment of combined hyperlipidemia. J Am Coll Cardiol. 2005 May 17;45(10):1649-53. doi: 10.1016/j.jacc.2005.02.052[↩]
- Jeu L, Cheng JW. Pharmacology and therapeutics of ezetimibe (SCH 58235), a cholesterol-absorption inhibitor. Clin Ther. 2003 Sep;25(9):2352-87. doi: 10.1016/s0149-2918(03)80281-3[↩]
- Elam MB, Hunninghake DB, Davis KB, Garg R, Johnson C, Egan D, Kostis JB, Sheps DS, Brinton EA. Effect of niacin on lipid and lipoprotein levels and glycemic control in patients with diabetes and peripheral arterial disease: the ADMIT study: A randomized trial. Arterial Disease Multiple Intervention Trial. JAMA. 2000 Sep 13;284(10):1263-70. doi: 10.1001/jama.284.10.1263[↩]
- Ayyobi AF, Brunzell JD. Lipoprotein distribution in the metabolic syndrome, type 2 diabetes mellitus, and familial combined hyperlipidemia. Am J Cardiol. 2003 Aug 18;92(4A):27J-33J. doi: 10.1016/s0002-9149(03)00613-1[↩]
- Cicero AFG, Martini C, Nativio V, et al. Association between lipidic phenotype variability and CHD/CVD in a large rural population: the Brisighella Study. Atherosclerosis. 2000;151:105.[↩]
- Skoumas I, Masoura C, Pitsavos C, Tousoulis D, Papadimitriou L, Aznaouridis K, Chrysohoou C, Giotsas N, Toutouza M, Tentolouris C, Antoniades C, Stefanadis C. Evidence that non-lipid cardiovascular risk factors are associated with high prevalence of coronary artery disease in patients with heterozygous familial hypercholesterolemia or familial combined hyperlipidemia. Int J Cardiol. 2007 Oct 1;121(2):178-83. doi: 10.1016/j.ijcard.2006.11.005[↩]
- Keulen ET, Kruijshoop M, Schaper NC, Hoeks AP, de Bruin TW. Increased intima-media thickness in familial combined hyperlipidemia associated with apolipoprotein B. Arterioscler Thromb Vasc Biol. 2002 Feb 1;22(2):283-8. doi: 10.1161/hq0202.104100[↩]
- Pitkänen OP, Nuutila P, Raitakari OT, Porkka K, Iida H, Nuotio I, Rönnemaa T, Viikari J, Taskinen MR, Ehnholm C, Knuuti J. Coronary flow reserve in young men with familial combined hyperlipidemia. Circulation. 1999 Apr 6;99(13):1678-84. doi: 10.1161/01.cir.99.13.1678[↩]
- Austin MA, McKnight B, Edwards KL, Bradley CM, McNeely MJ, Psaty BM, Brunzell JD, Motulsky AG. Cardiovascular disease mortality in familial forms of hypertriglyceridemia: A 20-year prospective study. Circulation. 2000 Jun 20;101(24):2777-82. doi: 10.1161/01.cir.101.24.2777[↩]





{kind=link}