Contents
- What is familial hypercholesterolemia
- Is there a cure for familial hypercholesterolemia?
- What is the difference between Heterozygous and Homozygous Familial Hypercholesterolemia?
- How is Familial Hypercholesterolemia different from Hypercholesterolemia/Hyperlipidemia?
- How is familial hypercholesterolemia diagnosed? Is there a specific blood test for it?
- I have been diagnosed with Familial hypercholesterolemia but I don’t want to take medication. Can I lower my cholesterol through a low-fat diet?
- What is Cholesterol
- What is the cause of familial hypercholesterolemia
- Familial hypercholesterolemia prevention
- Homozygous familial hypercholesterolemia
- Women with familial hypercholesterolemia and pregnancy
- What will happen to my cholesterol level during pregnancy?
- What if I become pregnant while on a statin?
- Can I continue to take my cholesterol medications during pregnancy?
- How can I keep my cholesterol under control during pregnancy?
- Is my baby likely to inherit familial hypercholesterolemia?
- Should I become pregnant?
- Should I Nurse My Baby?
- If I have trouble getting pregnant should I consider infertility medications?
- Men with familial hypercholesterolemia
- Why do men tend to experience heart disease a decade earlier than the women in their family?
- Are men less likely to be treated for their elevated LDL than women?
- I have been diagnosed with familial hypercholesterolemia, are there any other lab studies I should have?
- Can anything be done to lower Lipoprotein-A?
- How much can I lower my risk if I take cholesterol lowering medications?
- I know statins can lower my risk for a heart attack, but every time I take a statin my muscles and joints ache what can I do?
- My doctor has asked me to begin LDL-apheresis. Is this likely to lower my risk of a heart attack?
- I have familial hypercholesterolemia. How likely is it that my child will also have familial hypercholesterolemia?
- Familial hypercholesterolemia life expectancy
- Familial hypercholesterolemia symptoms
- Familial hypercholesterolemia complications
- Familial hypercholesterolemia diagnosis
- Familial hypercholesterolemia treatment
- Lifestyle and dietary changes
- Medications
- Treatment Guidelines for Heterozygous familial hypercholesterolemia
- Treatment Guidelines for Homozygous familial hypercholesterolemia
- Familial Hypercholesterolemia and Pregnancy
- Children or Adults with Homozygous familial hypercholesterolemia
- Children and cholesterol treatment
- Surgical care
- Familial hypercholesterolemia prognosis
What is familial hypercholesterolemia
Familial hypercholesterolemia also known as familial hyperlipoproteinemia type 2A or Fredrickson type 2A hyperlipidemia is an autosomal dominant genetic lipid disorder where your liver can’t process cholesterol properly and this leads to a severely elevated low-density lipoprotein (LDL) “bad” cholesterol in your blood that lead to atherosclerotic plaque deposition in the coronary arteries and proximal aorta at an early age, leading to an increased risk for premature atherosclerotic cardiovascular disease , the leading cause of preventable death in the United States 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11. The gene that causes familial hypercholesterolemia is inherited due to a defect (mutation) in a gene changes how your body processes cholesterol. Familial hypercholesterolemia is present from birth but symptoms may not appear until adulthood and a significant number of people remain undiagnosed. In familial hypercholesterolemia patients, genetic mutations make the liver incapable of metabolizing or removing excess low-density lipoprotein (LDL) “bad” cholesterol, from your blood. The result is very high low-density lipoprotein (LDL) “bad” cholesterol levels which can lead to atherosclerotic plaques deposition in the coronary arteries and proximal aorta occurring at an early age, leading over time to an increased risk for cardiovascular disease and even narrowing of your heart valves. As a result, people with familial hypercholesterolemia have a higher risk of premature coronary artery disease (coronary heart disease) manifesting as angina, early heart attack (myocardial infarction) and stroke 12. Familial hypercholesterolemia likely accounts for 2% to 3% of heart attacks (myocardial infarctions) in individuals younger than age 60 years. Untreated familial hypercholesterolemia, men are at a 50% risk for a fatal or non-fatal coronary event by age 50 years; untreated women are at a 30% risk by age 60 years (see Figure 2 below) 13.
Familial hypercholesterolemia is the most common inherited cardiovascular disease:
- About 1 in 200-250 people worldwide have familial hypercholesterolemia 14
- In the United States alone, an estimated 1.3 million people live with familial hypercholesterolemia. Yet only 10% of them are diagnosed. Nearly 2 million people in the US might have familial hypercholesterolemia and not even know it. Perhaps they won’t know it until they have a heart attack.
- Over 90% of people with familial hypercholesterolemia have not been properly diagnosed 14
- Familial hypercholesterolemia runs in families. If one parent has familial hypercholesterolemia, each child has a 50% chance of having familial hypercholesterolemia 15
- If left untreated, men have a 50% rise of having a heart attack by age 50. Untreated women have a 30% risk by age 60 16
- 1 in 160,000 to 1 in 1 million people have homozygous familial hypercholesterolemia 14. Homozygous familial hypercholesterolemia is more likely to occur in countries where the prevalence of heterozygous familial hypercholesterolemia is very high, especially those where consanguinity (marriage between relatives) is common.
- Familial hypercholesterolemia is even more common in certain populations such as French Canadians, Ashkenazi Jews, Lebanese, and South African Afrikaners. Lebanese Christians (1 in every 85 people), Afrikaners in South Africa (1/72 – 1/100), French Canadians (1 in every 270 people), and Ashkenazi Jews originating from Lithuania (1 in every 67 people) known as a founder affect.
The vast majority of the cholesterol circulating in a person’s body is produced by the liver. Cholesterol is a necessary component in the structure and function of human cells. Individuals with familial hypercholesterolemia are unable to recycle this natural supply of cholesterol that their bodies are constantly producing. Therefore, the cholesterol levels of an individual with familial hypercholesterolemia are exceedingly high. Over time the elevated blood cholesterol can lead to blockages in the arteries of the heart and/or brain. The longer a person experiences high low-density lipoprotein (LDL) “bad” cholesterol, the more likely he or she will be to experience angina and myocardial infarctions (heart attacks) and strokes. As familial hypercholesterolemia is a genetic disorder, even when affected children are still in their mother’s womb they are “bathing” in their own high cholesterol. The process of blood vessels disease therefore can have its origins even prior to your children’s birth. Familial hypercholesterolemia gene mutations are passed from parent to child. To have the condition, children need to inherit an altered copy of the gene from one parent.
There are two forms of familial hypercholesterolemia:
Heterozygous familial hypercholesterolemia
- If you have inherited this genetic mutation (one of three genes: low density lipoprotein receptor (LDLR) gene, apolipoprotein B-100 (APOB) gene and proprotein convertase subtilisin/kexin type 9 (PCSK9) gene) from one parent, then you will have Heterozygous familial hypercholesterolemia (HeFH). Heterozygous familial hypercholesterolemia occurs in 1 in 250 people worldwide. Most people with familial hypercholesterolemia have one affected gene and one normal gene (heterozygous familial hypercholesterolemia). An estimated 70%-95% of familial hypercholesterolemia results from a heterozygous pathogenic variant in one of three genes (apolipoprotein B [ApoB], low density lipoprotein receptor (LDLR), or PCSK9 gene).
- Until recently, it was believed that 1/500 people (630,000 people in the U.S.) had heterozygous familial hypercholesterolemia while about 1/1,000,000 people (300 people in the U.S.) had homozygous familial hypercholesterolemia. New studies suggest that heterozygous familial hypercholesterolemia and homozygous familial hypercholesterolemia are more common. It is now believed that 1/250 people (or 1.3 million people in the U.S. alone) have heterozygous familial hypercholesterolemia and 1/160,000 (or 2,000 people in the U.S.) have homozygous familial hypercholesterolemia. Even though homozygous familial hypercholesterolemia is still rare, it is a lot more common than we once thought. If you think that you or someone you know may have familial hypercholesterolemia, talk to your doctor.
- Heterozygous familial hypercholesterolemia: LDL-C > 160 mg/dL (4 mmol/L) for children and >190 mg/dL (5 mmol/L) for adults and with one first degree relative similarly affected or with positive genetic testing for an LDL-C raising gene defect (LDL receptor [LDLR], apolipoprotein-B [apo B] or proprotein convertase subtilisin/kexin type 9 [PCSK9])
Homozygous familial hypercholesterolemia
- If you inherit familial hypercholesterolemia from both parents, it is much more severe in its consequences. This form of familial hypercholesterolemia is called Homozygous familial hypercholesterolemia (HoFH). Homozygous familial hypercholesterolemia is very rare, occurring in about 1 in 160,000 to one million people worldwide. Most individuals with homozygous familial hypercholesterolemia experience severe coronary artery disease by their mid-20s and the rate of either death or coronary bypass surgery by the teenage years is high. Severe aortic stenosis is also common.
- Homozygous familial hypercholesterolemia: LDL-C > 400 mg/dL (10 mmol/L) and one or both parents:
- having clinically diagnosed familial hypercholesterolemia,
- positive genetic testing for an LDL-C raising (LDL receptor, apo B or PCSK9) gene defect, or
- autosomal recessive familial hypercholesterolemia If LDL-C > 560 mg/dL (14 mmol/L) or LDL-C > 400 mg/dL (10 mmol/L) with aortic valve disease or xanthomata at less than 20 years of age, homozygous familial hypercholesterolemia is highly likely.
Both types of familial hypercholesterolemia can be diagnosed with a physical examination and lab results, as well as personal and family history. Familial hypercholesterolemia also can be discovered through molecular diagnosis, genetic diagnosis or genetic testing. It’s helpful when genetic testing reveals familial hypercholesterolemia because it can alert relatives to their risk.
Some people with familial hypercholesterolemia have physical symptoms, but many don’t. One symptom of Familial hypercholesterolemia is cholesterol deposits in the Achilles tendons or the tendons of the hands or elbows. People with homozygous familial hypercholesterolemia also can develop cholesterol deposits in other areas, such as the skin surrounding the eyes or on the outer edge of the cornea.
If one person in a family has familial hypercholesterolemia, then all immediate relatives — parents, brothers, sisters and children — should be checked for it. Similarly, if someone in a family has an early heart attack, it’s a good idea for other family members to get tested.
Children with increased risk for familial hypercholesterolemia should be screened beginning at age 2. All children should have their cholesterol checked between ages 9 and 11 and again between ages 17 and 21.
Doctors measure blood cholesterol in milligrams per deciliter (mg/dL).
Adults with familial hypercholesterolemia may have untreated low-density lipoprotein (LDL-C) “bad” cholesterol levels that range from 190mg/dL to 400mg/dL or even higher. Children with familial hypercholesterolemia generally have low-density lipoprotein (LDL-C) “bad” cholesterol levels above 160mg/dL, but in pre-teens, levels can be even lower.
- High-density lipoprotein (HDL-C) “good” cholesterol levels are desirable and somewhat protect against coronary heart disease.
- High LDL-C “bad” cholesterol levels are undesirable and contribute to coronary heart disease risk.
Triglycerides, another blood fat, come from the diet and are also produced in the liver. When extremely elevated, triglycerides can cause pancreatitis and can also increase the risk of developing coronary heart disease. High triglycerides are generally not present in people with familial hypercholesterolemia.
All individuals with familial hypercholesterolemia should be considered “high risk” (i.e., increased ~20-fold) for coronary artery disease (coronary heart disease). Recent data suggest that individuals with an LDL “bad” cholesterol >190 mg/dL (>4.9 mmol/L) and a pathogenic variant in one of the genes (APOB, LDLR, PCSK9 & unknown genes) have a 22-fold increased risk for coronary heart disease, while those without a pathogenic variant have a sixfold increased risk for coronary artery disease over the general population 17.
Figure 1. Familial hypercholesterolemia inheritance pattern
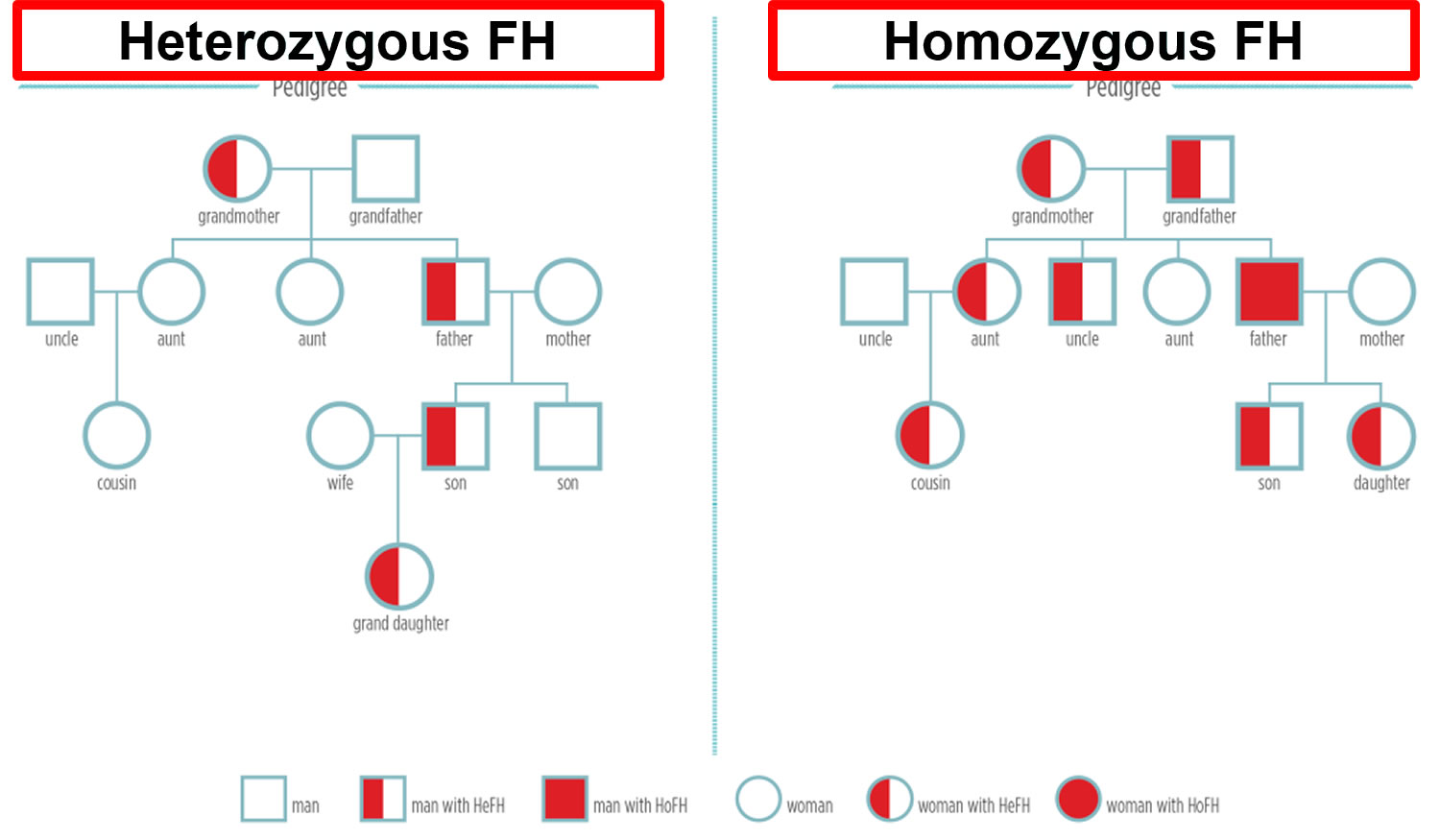
Note: HeFH = Heterozygous familial hypercholesterolemia; HoFH = Homozygous familial hypercholesterolemia
Figure 2. LDL cholesterol burden in individuals with or without familial hypercholesterolemia as a function of the age of initiation of Statin therapy
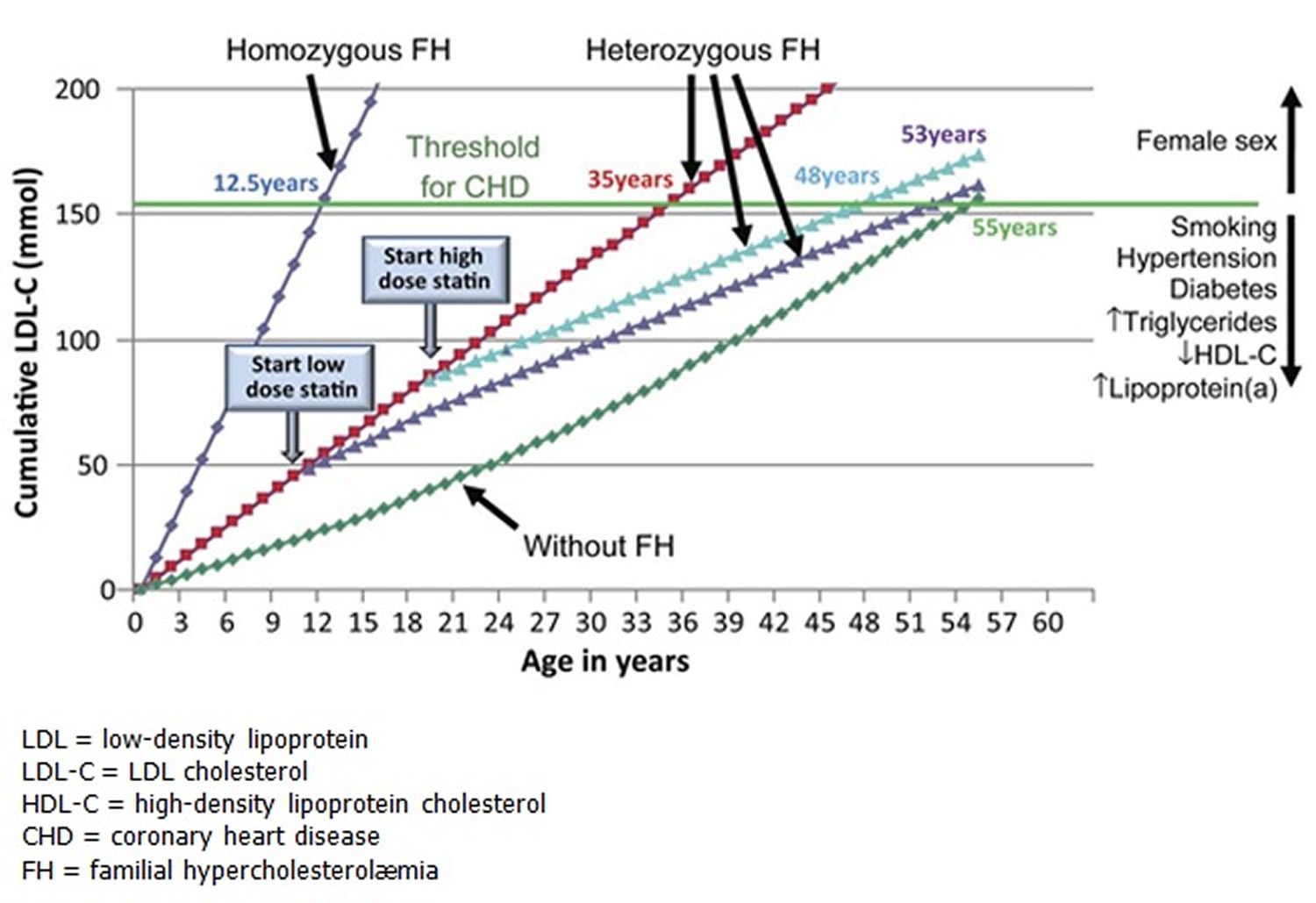
Figure 3. Estimated number of individuals diagnosed with familial hypercholesterolemia in different countries
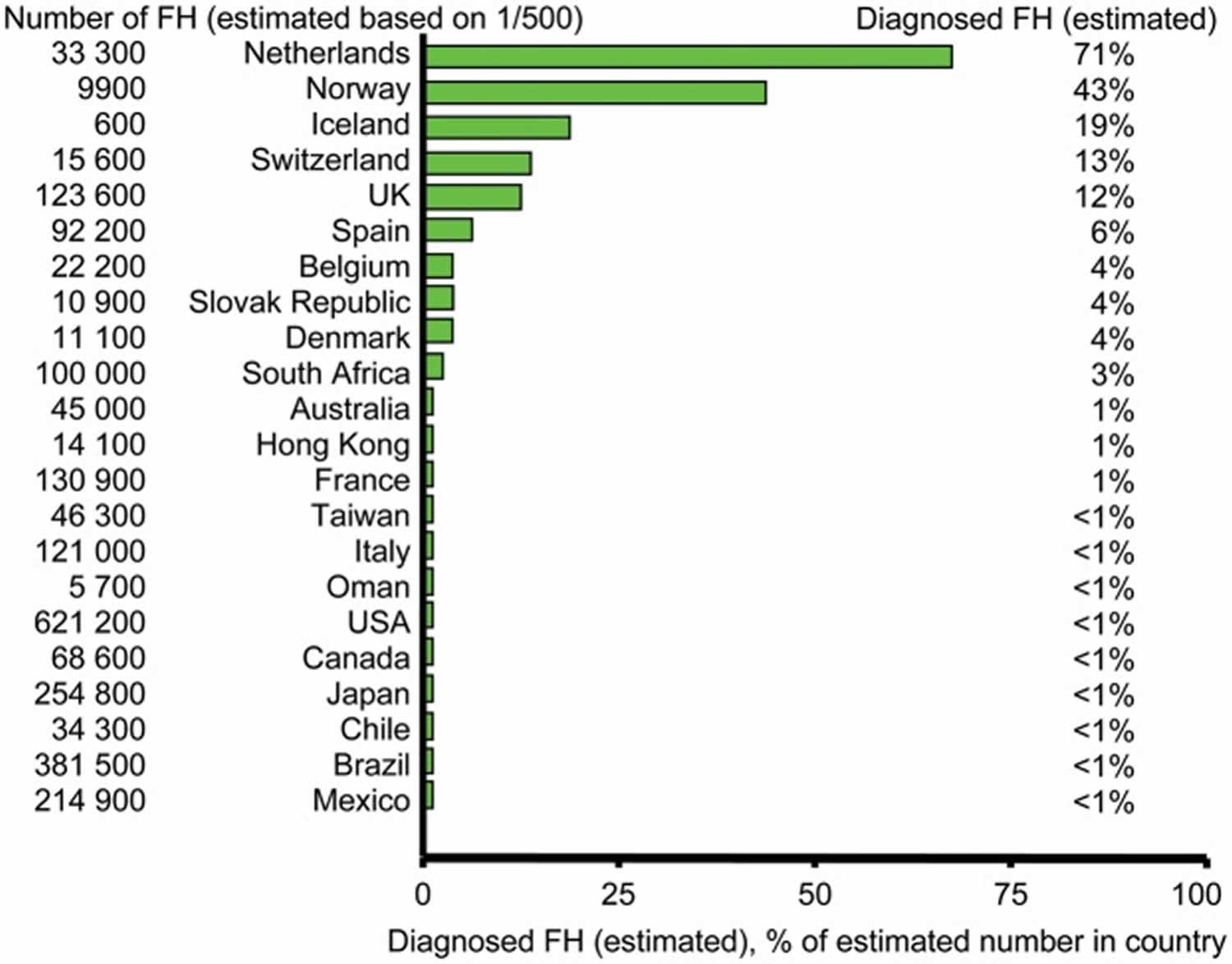
Footnotes: As most countries do not have valid nationwide registries for familial hypercholesterolemia, several values in this figure represent informed estimates from clinicians/scientists with recognized expertise in and knowledge of familial hypercholesterolemia in their respective countries.
[Source 18 ]Family history of early heart disease + High LDL “bad” cholesterol = Familial Hypercholesterolemia
Therefore, if you have familial hypercholesterolemia, your LDL-cholesterol levels will be very high, leading to narrowing or blockage of blood vessels (atherosclerosis). This process starts prior to birth and can ultimately result in heart disease, heart attack, or stroke. Because people with familial hypercholesterolemia have excessive cholesterol levels since before their birth, their risk of heart disease is 20 times greater than that of the general population. If a child has Homozygous familial hypercholesterolemia (inherited the familial hypercholesterolemia gene from both parents), she is exposed to an even higher risk, because her LDL-cholesterol levels are extraordinarily elevated and lead to progressive heart disease very early in life (often in the teens). Other cardiovascular disease risk factors include smoking, over-weight, high blood pressure, and a sedentary life. If you have familial hypercholesterolemia, eliminate smoking, control your weight and blood pressure, and lead a physically active lifestyle. All these are important factors, together with medical therapy and a healthful diet, in lowering your risk of an early heart attack.
Familial hypercholesterolemia is a lifelong condition. It’s not a temporary condition, like the common cold; it’s in your genes. When you have familial hypercholesterolemia, the most important step to take is therapies prescribed by your physician. But while this is the most essential measure, it’s not the only one. Familial hypercholesterolemia also means controlling weight, not smoking, eating a balanced diet low in saturated fat, and exercising. What is more, Familial hypercholesterolemia means bringing your family together to understand the impact of this disease. Familial hypercholesterolemia means living healthy, as individuals and as a family.
The good news is familial hypercholesterolemia is treatable! If familial hypercholesterolemia is found early, serious problems of the heart and blood vessels may be prevented or dramatically delayed by taking steps to protect yourself. These include:
- Not smoking.
- Exercising regularly.
- Eating a healthy diet low in saturated and trans fats.
- Taking medications.
- Going on LDL-apheresis.
Familial hypercholesterolemia in almost all cases requires aggressive treatment through a combined approach – medication, low-fat diet, exercise, weight control and not smoking. Familial hypercholesterolemia involves heart-healthy meals, regular exercise to get the blood flowing, controlling your weight and eliminating smoking. Because obesity and smoking are risk factors for heart disease and those with familial hypercholesterolemia are already at a 20 times higher risk, it is important to adapt to a healthy lifestyle.
Nearly 100% of people with familial hypercholesterolemia will require cholesterol-lowering medications. For some people with familial hypercholesterolemia, more aggressive measures are needed, including LDL-apheresis (a very simple procedure in which LDL-C cholesterol is removed from the blood on a weekly or biweekly basis.)
The American Academy of Pediatrics recommends that if a family has a pattern of early heart attacks or heart disease defined as before age 55 for men and 65 for women, children in that family should have cholesterol testing after the age of 2 years and before age 10.
It is important to find familial hypercholesterolemia and take action at any age, because when treated, the risk of heart disease can be reduced to levels similar to those of the general population.
Is there a cure for familial hypercholesterolemia?
Familial hypercholesterolemia is not curable. However, it is treatable. Now, more than ever, there are various treatment options for people with Familial hypercholesterolemia. This realistically means that individuals with Familial hypercholesterolemia could lead normal lives, if diagnosed early and treated adequately.
What is the difference between Heterozygous and Homozygous Familial Hypercholesterolemia?
Heterozygous Familial Hypercholesterolemia (HeFH) is the most common form of familial hypercholesterolemia (1 in 250 worldwide have it) caused when a child inherits one copy of a “bad gene” from one parent. Homozygous Familial Hypercholesterolemia (HoFH) is a very rare, but extremely severe form of familial hypercholesterolemia (roughly 1 in 160,000 have it) caused when a child gets a “bad gene” from both parents. Typically, Heterozygous familial hypercholesterolemia remains invisible for longer and Homozygous familial hypercholesterolemia manifests itself earlier, with more visible signs such as xanthomas (bumps or lumps in the skin which are deposits of excess fat) and corneal arcus (a white arc near the colored part of the eye, which is often found by an ophthalmologist).
How is Familial Hypercholesterolemia different from Hypercholesterolemia/Hyperlipidemia?
Your clue here is “Familial”. By definition, hypercholesterolemia (or hyperlipidemia) simply means high cholesterol. However, Familial Hypercholesterolemia is a lifelong condition that is inherited. Otherwise put, your genes cause it. Therefore, it is a lot more serious than simply having high cholesterol caused from diet and it requires more aggressive treatment. familial hypercholesterolemia is a life-threatening disorder.
One of the most common scenarios in patients with familial hypercholesterolemia is being told that they have high cholesterol and that they need to change their diet. However, with familial hypercholesterolemia, eliminating fried foods and cheesecake from your diet is not enough. While saturated foods should be completely avoided, your high LDL-cholesterol mainly has to do with your genes/ your family’s medical history. With familial hypercholesterolemia, the cause of your high cholesterol is NOT your diet. Even if you ate nothing but oats and fruits, you would still have high LDL-cholesterol because of your liver’s inability to keep up with recycling and removing it.
How is familial hypercholesterolemia diagnosed? Is there a specific blood test for it?
Diagnosis can be strongly suspected based on cholesterol levels (also called a lipid test or lipid panel). The diagnosis can be confirmed through additional information about your personal and family medical history, and certain physical exam findings (like xanthomas, bumps or lumps of cholesterol deposits on the skin). Learn more about familial hypercholesterolemia diagnosis in a section below.
I have been diagnosed with Familial hypercholesterolemia but I don’t want to take medication. Can I lower my cholesterol through a low-fat diet?
Familial hypercholesterolemia causes excessively high LDL-cholesterol levels. This is dangerous as it leads to cholesterol getting built up in your blood vessels, leading to atherosclerosis, heart attacks, and even death. While it is important to be mindful of your diet, this is almost never enough to manage your condition.
In all cases, familial hypercholesterolemia requires aggressive treatment. Consult an familial hypercholesterolemia specialist to find the best therapy regime for you.
What is Cholesterol
Cholesterol is a waxy, fat-like substance that’s found in all the cells in your body. Cholesterol comes from two sources. Cholesterol is produced by your body and also found in some foods. Your body makes most of its own cholesterol in the liver. You also get a small amount of cholesterol from some foods such as egg yolks, meat, liver, kidneys, cheese and seafood such as prawns, which is called dietary cholesterol. Although we often blame the cholesterol found in foods that we eat for raising blood cholesterol, the main culprit is actually saturated fat. Foods rich in saturated fat include butter fat in milk products, fat from red meat, and tropical oils such as coconut oil.
Cholesterol plays a vital role in how every cell in your body works. It’s also the material that your body uses to make other vital chemicals such as vitamin D, bile to aid digestion, and some hormones such as cortisol, estrogen and testosterone. Cholesterol is especially important for growth when many new cells are being formed very quickly, for example, in children or during pregnancy. However, too much cholesterol in your blood can increase your risk of getting diseases of the heart and circulation.
Cholesterol travels to cells through your bloodstream in special carriers called lipoproteins. Two of the most important lipoproteins are low-density lipoprotein (LDL) and high-density lipoprotein (HDL).
There are different types of cholesterol:
- HDL stands for high-density lipoprotein or HDL-C (high-density lipoprotein cholesterol). HDL is sometimes called “good cholesterol” because it carries harmful cholesterol from other parts of your body including your arteries back to your liver. Your liver then removes the cholesterol from your body and helps protect you from heart attack and stroke. A healthy HDL-cholesterol level may protect against heart attack and stroke. If you have low HDL levels, you have a greater heart disease risk, even if your total cholesterol is below 200 mg/dL. Your doctor will evaluate your HDL and other cholesterol levels and other factors to assess your risk for heart attack or stroke. People with high blood triglycerides usually also have lower levels of HDL. Genetic factors, Type 2 diabetes, smoking, being overweight and being sedentary can all lower HDL cholesterol. Women tend to have higher levels of HDL cholesterol than men do, because the female hormone estrogen raises HDL, but this can change after menopause.
- LDL stands for low-density lipoprotein or LDL-C (low-density lipoprotein cholesterol). LDL is sometimes called “bad cholesterol” because a high LDL level leads to the buildup of plaque in your arteries. LDL is the most important lipid for predicting your heart disease risk. Low-density lipoprotein (LDL or ‘bad’) cholesterol can join with fats and other substances to build up (also known as plaque) in the inner walls of your arteries, which starts a disease process called atherosclerosis. The arteries can become clogged and narrow, and blood flow is reduced. When plaque builds up in your coronary arteries that supply blood to your heart, you are at greater risk of having a heart attack. Since LDL is the bad kind of cholesterol, a low LDL level is considered good for your heart health. A diet high in saturated and trans fat is unhealthy because it tends to raise LDL cholesterol levels. Your LDL levels may be high if you eat a diet with a lot of saturated fat, cholesterol, or both. Sometimes, an under-active thyroid called hypothyroidism may also increase LDL levels.
- VLDL stands for very low-density lipoprotein or VLDL-C (very low-density lipoprotein cholesterol). Some people also call VLDL a “bad cholesterol” because it too contributes to the buildup of plaque in your arteries. But VLDL and LDL are different; VLDL mainly carries triglycerides and LDL mainly carries cholesterol. VLDL particles are released into the blood by the liver and circulate in the bloodstream, ultimately being converted into LDL as they lose triglyceride, having carried it to other parts of the body. According to the National Heart, Lung and Blood Institute’s National Cholesterol Education Program Guidelines ATP III, there is growing evidence that VLDL plays an important role in atherogenesis, in which plaques form on the interior walls of arteries, narrowing these passageways and restricting blood flow, which can lead to heart disease and increase the risk of stroke. Currently, direct measurement of VLDL cholesterol requires specialized testing. However, since VLDL-C contains most of the circulating triglyceride (if a person is fasting) and since the composition of the different particles is relatively constant, it is possible to estimate the amount of VLDL-C based on the triglyceride value. To estimate VLDL-C, divide the triglyceride value by 5 if the value is in mg/dL or divide by 2.2 if the value is in mmol/L. In most cases, this formula provides a good estimate of VLDL-C. However, this formula becomes less accurate with increased triglyceride levels when, for example, a person has not fasted before having blood drawn. The calculation is not valid when the triglyceride level is greater than 400 mg/dl (4.5 mmol/L) because other lipoproteins are usually present. In this situation, VLDL-C may be measured directly using specialized testing.
- Triglycerides. Triglycerides are the most common type of fat in your blood. Triglycerides come from food, and your body also makes them. When you eat, your body converts calories it doesn’t need into triglycerides, which are stored in fat cells. Triglycerides are fats that provide energy for your muscles. If you eat foods with a lot of saturated fat or carbohydrates, you will raise your triglyceride levels. High triglyceride levels are associated with several factors, including being overweight, eating too many sweets or drinking too much alcohol, smoking, being sedentary, or having diabetes with elevated blood sugar levels. Elevated triglycerides levels are thought to lead to a greater risk of heart disease, but scientists do not agree that high triglycerides alone are a risk factor for heart disease. Normal triglyceride levels vary by age and sex. People with high triglycerides often have a high total cholesterol level, including a high LDL (bad) cholesterol level and a low HDL (good) cholesterol level. Many people with metabolic syndrome or diabetes also have high triglyceride levels. Extremely high triglyceride levels (more than 1000 mg/dL) can lead to abdominal pain and a life-threatening disorder of the pancreas called pancreatitis. Factors that can contribute to elevated triglyceride levels:
- Overweight or obesity
- Insulin resistance or metabolic syndrome
- Diabetes mellitus, especially with poor glucose control
- Alcohol consumption, especially in excess
- Excess sugar intake, especially from processed foods
- High saturated fat intake
- Hypothyroidism
- Chronic kidney disease
- Physical inactivity
- Pregnancy (especially in the third trimester)
- Inflammatory diseases (such as rheumatoid arthritis, systemic lupus erythematosus
- Some medications may also increase triglycerides.
“Total cholesterol” is the total amount of cholesterol that’s circulating in your blood. Your “total blood cholesterol” is calculated by adding your HDL (“good” cholesterol) and LDL (“bad” cholesterol) cholesterol levels, plus 20% of your triglyceride level.
Here’s the formula for calculating your “total blood cholesterol”:
- Total cholesterol = HDL + LDL + 20% triglycerides.
In the United States, cholesterol levels are measured in milligrams (mg) of cholesterol per deciliter (dL) of blood (mg/dL). In Canada and many European countries, cholesterol levels are measured in millimoles per liter (mmol/L).
Your “Total cholesterol” should be below 200 milligrams per deciliter of blood (mg/dL) or 5.18 mmol/L.
- Your HDL “good” cholesterol is the one number you want to be high, ideally above 60 mg/dL (1.55 mmol/L) or higher.
- Your LDL “bad” cholesterol should be below 100 mg/dL (less than 2.59 mmol/L).
- Your triglycerides should be below 150 mg/dL (less than 1.70 mmol/L). Triglycerides are the most common type of fat in your blood. Triglycerides come from food, and your body also makes them. When you eat, your body converts calories it doesn’t need into triglycerides, which are stored in fat cells. High triglyceride levels are associated with several factors, including being overweight, eating too many sweets or drinking too much alcohol, smoking, being sedentary, or having diabetes with elevated blood sugar levels.
- Talk with your doctor about what your results mean for you and how to manage your cholesterol.
High cholesterol usually has no symptoms. You can find out the levels of these in your blood and also your total cholesterol level with a cholesterol or lipid profile blood test. If you are concerned about your cholesterol level, talk to your doctor. You will need to stop eating for 10 to 12 hours before a cholesterol or lipid profile blood test, and the only liquid you may drink is water.
According to the National Heart, Lung and Blood Institute, a person’s first cholesterol screening should occur between the ages of 9 and 11 and then be repeated every five years after that 19. The National Heart, Lung and Blood Institute recommends that cholesterol screenings occur every 1 to 2 years for men ages 45 to 65 and for women ages 55 to 65. People over 65 should receive cholesterol tests annually 19. More-frequent cholesterol testing might be needed if your initial cholesterol test results were abnormal or if you already have coronary artery disease, you’re taking cholesterol-lowering medications or you’re at higher risk of coronary artery disease because you:
- Have a family history of high cholesterol or heart attacks
- Are overweight
- Are physically inactive
- Have diabetes
- Eat an unhealthy diet
- Smoke cigarettes
People undergoing treatment for high cholesterol require regular cholesterol testing to monitor the effectiveness of their treatments.
LDL cholesterol and HDL cholesterol
Cholesterol and other fats have a special way of reaching all the cells in your body that need it. They use your blood circulation as their ‘road system’ and are carried on ‘vehicles’ made up of proteins. These combinations of fats and proteins are called lipoproteins.
There are two main types of lipoproteins – LDL (low-density lipoprotein) and HDL (high-density lipoprotein).
- Low-density lipoproteins sometimes called LDL cholesterol (LDL-C) or ‘bad cholesterol’ – carry most of the cholesterol from your liver, through the bloodstream, to where it is needed. About 70 per cent of the cholesterol in your body is carried by LDL. The lower the density of the lipoprotein, the more fats it contains, so having high LDL is harmful to you.
- High-density lipoproteins sometimes called HDL cholesterol (HDL-C) or ‘good cholesterol’ – return the extra cholesterol, that isn’t needed, from your cells and your bloodstream to your liver for recycling. HDL cholesterol is a ‘good’ type of cholesterol because it removes cholesterol from your bloodstream. This helps prevent the cholesterol from being deposited in the arteries and causing atheroma (atherosclerosis).
Triglycerides
Triglycerides are another type of fatty substance found in your blood. You get some triglycerides from foods such as dairy products, butter, meat, cooking oils and other fats. Some triglycerides are important for good health. However, high triglyceride levels in your blood or hypertriglyceridemia can raise your risk of heart disease and stroke. How severe hypertriglyceridemia is can vary based on sex, age, hormone use, and dietary factors. If you are very overweight, eat a lot of fatty and sugary foods, or drink too much alcohol, you are more likely to have a high triglyceride level (hypertriglyceridemia). Triglyceride levels increase after a meal and are normally cleared from the bloodstream over the following hours. They are usually at their lowest first thing in the morning before breakfast.
Triglycerides can also be produced in your body, either by your body’s fat stores or in the liver. Triglycerides mainly come from extra calories your body does not need right away. Unused calories (energy you don’t need right away) are stored as triglycerides in fat cells, therefore triglycerides is a major source of energy for your body. When your body needs energy, it releases the triglycerides. In between meals, triglycerides are released from fat tissue to be used as an energy source for your body. Most triglycerides are carried in the blood by lipoproteins called very low-density lipoproteins (VLDL).
Your doctor will usually check for high triglycerides as part of a cholesterol test, which is sometimes called a lipid panel or lipid profile. You’ll have to fast before blood can be drawn for an accurate triglyceride measurement.
For adults, triglyceride levels results are categorized as follows:
- Normal: Less than 150 mg/dL (less than 1.7 mmol/L)
- Mild high: 150 to 199 mg/dL (1.7-2.2 mmol/L)
- Moderate high: 200 to 999 mg/dL (2.3-11.3 mmol/L)
- Severe high: 1,000 to 1,999 mg/dL (11.3-22.6 mmol/L)
- Very severe: Greater than 2,000 mg/dL (more than 22.6 mmol/L)
Note: For improved metabolic health and protection to the heart and blood vessels, the American Heart Association now recommends an optimum fasting triglyceride level of 100 mg/dL (1.12 mmol/L). This puts an even stronger emphasis on lifestyle change which has been the recommended therapy for mildly elevated triglycerides. However, the American Heart Association does not recommend people use drug therapy to achieve this optimal level because there has not been adequate study to show that drug therapy to lower triglycerides to this level is helpful. Many people will be able to reduce their triglycerides as well as other metabolic risk factors such as elevated blood sugar and elevated blood pressure with diet, weight loss and increased physical activity.
What is the cause of familial hypercholesterolemia
Familial hypercholesterolemia is an inherited genetic condition affecting cholesterol metabolism. Genes that are related to monogenic familial hypercholesterolemia include low density lipoprotein receptor (LDLR), apolipoprotein B-100 (APOB), proprotein convertase subtilisin/kexin type 9 (PCSK9), STAP1, APOE, LDLRAP1, LIPA, ABCG5, and ABCG8 28.
Familial hypercholesterolemia is commonly caused by a mutation in the gene for the low density lipoprotein receptor (LDLR), which is involved in passing LDL from the blood into cells for use by, or removal from, the body. Mutations in other genes can also cause inherited high cholesterol. Those genes include the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene and the gene for Apolipoprotein B-100 (APOB). If you inherit a specific type of mutation in any of these three genes, you can develop familial hypercholesterolemia.
One in about 200 adults have the familial hypercholesterolemia genetic mutation. Including children, familial hypercholesterolemia affects about 1.3 million people in the U.S. But only about 10% are aware they have it.
Familial hypercholesterolemia is an autosomal dominant disorder with essentially complete penetrance. What this means is that if you have inherited the familial hypercholesterolemia gene, you will have high LDL-cholesterol (and will require therapy to lower your LDL-cholesterol). In addition, each of your children will have a 50% chance of having familial hypercholesterolemia. If someone else in your family has familial hypercholesterolemia (like a brother or a sister) and has high LDL cholesterol but you have totally normal LDL-cholesterol, then you will not pass on the familial hypercholesterolemia gene to your children. However, if someone in your family has familial hypercholesterolemia and your cholesterol is “borderline” or mildly elevated (ask your physician what LDL-cholesterol levels would be considered borderline or elevated in your case), there is a chance that you have familial hypercholesterolemia and could pass it along to your children. In this case a consultation with a lipid specialist should be considered. Generally, if an adult’s LDL-cholesterol is less than 160 mg/dL without taking cholesterol-lowering medication, there is a very low chance that they have familial hypercholesterolemia. If an adult has an LDL cholesterol level > 190mg/dL there is a stronger suspicion for Familial hypercholesterolemia.
In familial hypercholesterolemia there is a “dose effect” so that homozygous familial hypercholesterolemia (two defective genes – one from each parent) is more severe than heterozygous familial hypercholesterolemia (one defective and one normal gene).
- Heterozygous familial hypercholesterolemia (HeFH) occurs when a child inherits a nonfunctional copy of one of their familial hypercholesterolemia genes from an affected parent and a functional copy from their unaffected parent. Each egg or sperm they produce has a 50% chance of getting the nonfunctional copy (passing down familial hypercholesterolemia) and a 50% chance of getting the functional copy, Therefore, the risk of passing the familial hypercholesterolemia from the affected parent to a child is 50% in each pregnancy. New, spontaneous variants appear to be very rare.
- Most individuals with homozygous familial hypercholesterolemia (HoFH) have inherited one mutated gene from each parent, such that each parent has heterozygous familial hypercholesterolemia. These parents have a 25% risk in each pregnancy to have a child with homozygous familial hypercholesterolemia, a 50% chance of having a child with heterozygous familial hypercholesterolemia and a 25% chance that the child will inherit a normal gene from each parent. The risk is the same for males and females.
If you have familial hypercholesterolemia, every child you have has a 50% chance of having familial hypercholesterolemia.
- When one parent has heterozygous familial hypercholesterolemia and the other has homozygous familial hypercholesterolemia, there is a 50% chance with each pregnancy to have a child with heterozygous familial hypercholesterolemia and a 50% chance to have a child with homozygous familial hypercholesterolemia.
- When one parent has homozygous familial hypercholesterolemia and the other has two normal genes, all children will have heterozygous familial hypercholesterolemia.
- When both parents have homozygous familial hypercholesterolemia, all children will have homozygous familial hypercholesterolemia.
Figure 4. Familial hypercholesterolemia autosomal dominant inheritance pattern
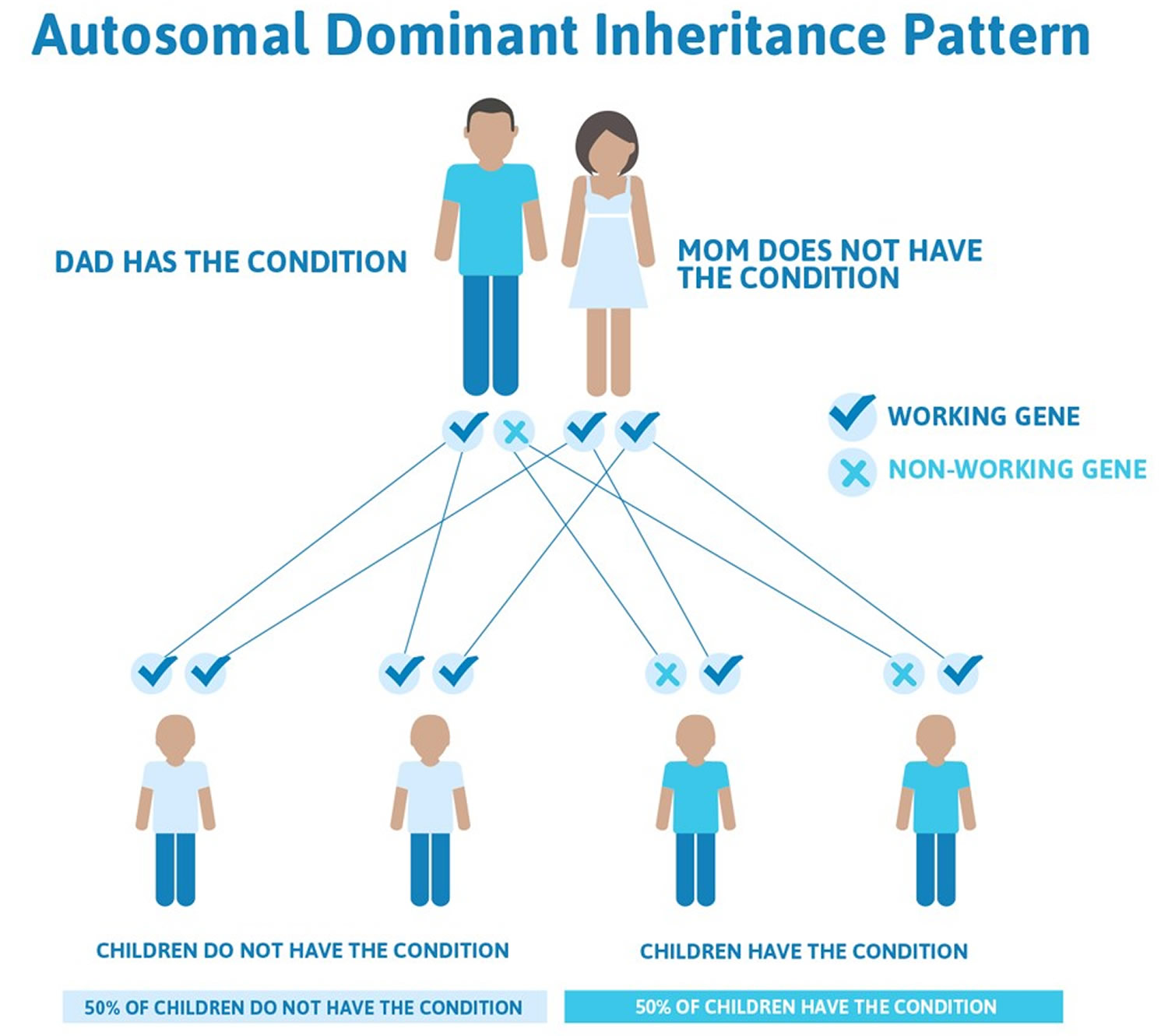
There are two forms of Familial hypercholesterolemia.
- Heterozygous (HeFH) – Heterozygous Familial hypercholesterolemia inherited from only 1 parent
- Homozygous (HoFH) – Homozygous Familial hypercholesterolemia inherited from both parents (patients with Homozygous Familial hypercholesterolemia are at an even higher risk of early heart disease)
If Familial hypercholesterolemia is left untreated, it can lead to heart disease, heart attack, or stroke.
Familial hypercholesterolemia genetics
Familial hypercholesterolemia is inherited in families in an autosomal dominant manner. In autosomal dominant inherited conditions, a parent who carries an altered gene that causes the condition has a 1 in 2 (50 percent) chance to pass on that altered gene to each of his or her children. If you have familial hypercholesterolemia, every child you have has a 50% chance of inheriting familial hypercholesterolemia.
Parents can give a disorder to their children through their DNA, which is made up of genes. Genes have all the characteristics that a person gets from their parents (such as eye color, hair color, height, etc.), and some may be mutated and not work properly. When a person gets a mutated gene, they can have mild or severe problems depending on the type of mutation.
LDL is generated in the circulation by the delipidation and modification of very low density lipoproteins (VLDL) secreted by the liver. There are three different genes that may be mutated in familial hypercholesterolemia. The most common gene codes for a protein called the LDL receptor (LDLR). LDL receptors in the liver remove LDL-C “bad cholesterol” from the blood. A LDL receptor (LDLR) gene variant is the most common cause of heterozygous familial hypercholesterolemia, accounting for ~90% of pathogenic variants. If a person has a mutated gene for the LDL receptor, the level of LDL-C “bad cholesterol” in his or her blood will increase. Genes that make other proteins, such as proprotein convertase subtilisin/kexin type 9 (PCSK9) and apolipoprotein B (ApoB), may also be mutated and decrease the removal of LDL-C “bad cholesterol” from the blood. If a person has a mutated gene for any of these proteins, the level of cholesterol in her or her blood will increase.
Apolipoprotein B100 (apoB100) is the major structural apoprotein of VLDL and LDL. LDL is cleared from the circulation by hepatic LDL-receptor with apoB100 acting as the ligand for the receptor. A pathogenic variant in the APOB gene is responsible for ~10% of familial hypercholesterolemia cases; this variation is seen most commonly in those of European Caucasian ancestry. Apolipoprotein B-100 is a protein that binds to LDL receptors, which enables uptake of lipoproteins by the liver and reduces the cholesterol level in the blood. Pathogenic variants in the APOB gene lead to faulty uptake and increased cholesterol level. The major pathophysiological abnormality in homozygous familial hypercholesterolemia is decreased LDL clearance although hepatic overproduction of apoB100-containing lipoproteins may further exacerbate the hyperlipidaemia.
PCSK9 gene variants are responsible for only a small percentage of familial hypercholesterolemia cases. The normal PCSK9 gene codes for an enzyme that breaks down the cholesterol receptors after they have done their job. A pathogenic variant in this gene is unlike most variants, which cause dysfunction of the affected gene. The PCSK9 variant increases the gene’s function, leading to too few remaining LDL receptors and thus an increase in the LDL cholesterol level.
People can have one or two of these mutated genes. When a person has one of these mutated genes (either for the LDL receptor, PCSK9, or ApoB), he or she has heterozygous familial hypercholesterolemia. So, the difference between heterozygous familial hypercholesterolemia and homozygous familial hypercholesterolemia is that a person with homozygous familial hypercholesterolemia has two mutated genes. Two mutated genes greatly increase a person’s blood cholesterol level and the risk of a heart attack.
I have been diagnosed with Familial hypercholesterolemia. I want to get my family tested, but they refuse. How do I convince them?
First and foremost it is important to remember that familial hypercholesterolemia runs in families. If you have familial hypercholesterolemia, the question isn’t “Does anyone else in my family have it?”. The question is “Who in my family has it?”. Of course, only you know how to best approach your relatives. But make sure they fully understand that this is a lifelong condition that requires consistent treatment and the sooner they rule it out, the better.
I have heard the term “cascade screening” in relation to familial hypercholesterolemia. What is cascade screening?
Cascade screening is not a certain type of blood test or certain kind of examination. It is simply a method of finding individuals with Familial Hypercholesterolemia. This method consists of screening entire families for familial hypercholesterolemia. Cascade screening involves screening close relatives of a person diagnosed with a specific genetic condition to determine if they also have the same genetic mutation. Because familial hypercholesterolemia is genetic condition, this means that more than one person in a family has it (because they inherited it from someone else). Cascade screening means that when a health professional diagnoses someone with familial hypercholesterolemia, they need to test the rest of their immediate family members for familial hypercholesterolemia too.
Risk factors for familial hypercholesterolemia
The risk of familial hypercholesterolemia is higher if one or both of your parents have the gene alteration that causes it. Most people who have familial hypercholesterolemia receive one affected gene. But in rare cases, a child can get the affected gene from both parents. This can cause a more severe form of the condition.
Familial hypercholesterolemia may be more common in certain populations, including:
- Ashkenazi Jews
- Some Lebanese groups
- French Canadians
Familial hypercholesterolemia prevention
Familial hypercholesterolemia is not preventable per se. You either inherit the gene (or genes) or you don’t. What you could potentially prevent, however, is the heart disease associated with it. Scientists like to say that with familial hypercholesterolemia you inherit the condition, but not the heart attack. If left untreated, familial hypercholesterolemia leads to early and aggressive heart disease, atherosclerosis (narrowing and blocking of the blood vessels), and heart attacks. This is why early diagnosis and treatment are crucial.
Homozygous familial hypercholesterolemia
Homozygous familial hypercholesterolemia is a rare but exceedingly aggressive form of familial hypercholesterolemia. If both parents have familial hypercholesterolemia, there is a 25% chance that their child or children will have homozygous familial hypercholesterolemia (along with a 50% chance that their children will have heterozygous familial hypercholesterolemia). However, if a child is diagnosed with homozygous familial hypercholesterolemia, that means both parents have some form of familial hypercholesterolemia.
Homozygous familial hypercholesterolemia leads to aggressive atherosclerosis (narrowing and blocking of blood vessels). This process starts even before birth and progresses rapidly. It can affect the coronary arteries, carotid arteries, aorta, and aortic valve . If left untreated, heart attack or sudden death are likely to occur as early as the teenage years and sometimes even in early childhood.
Homozygous familial hypercholesterolemia is much less common than heterozygous familial hypercholesterolemia, but lives can be saved if it is identified and treated early in childhood. When a person with homozygous familial hypercholesterolemia is not treated with medications, the low-density lipoprotein cholesterol (LDL-C, “bad cholesterol”) level is between 500-1000 mg/dL – over 4 times the normal level.
The prevalence of homozygous familial hypercholesterolemia is markedly increased in certain regions of the world and may be as high as one in thirty thousand in some populations. Populations with a very high prevalence of homozygous familial hypercholesterolemia include Afrikaners in South Africa, French Canadians, Christian Lebanese and Japanese from the Hokuriku district. These regions are characterized by a high prevalence of heterozygous familial hypercholesterolemia (estimated to be one in seventy to a hundred for Afrikaners in South Africa) which results from founder effects that occur when small, isolated populations increase in size rapidly with little outside genetic admixture 29.
It is important to remember that homozygous familial hypercholesterolemia is a serious medical condition and is life-threatening if not treated at a young age, preferably beginning in early childhood. A child or adult with homozygous familial hypercholesterolemia needs life-long medications and other specialized treatments to lower the LDL-C and prevent heart attacks. This requires the expertise of a lipid specialist.
My Child has homozygous familial hypercholesterolemia (HoFH) – what do I need to know?
Many couples learn that they both have heterozygous familial hypercholesterolemia (HeFH) when they have a baby who has homozygous familial hypercholesterolemia (HoFH). When two people with heterozygous familial hypercholesterolemia have a child, they have a 1 in 4 (25%) chance that their child will have a completely normal cholesterol profile, a 2 in 4 (50%) chance that their child will have heterozygous familial hypercholesterolemia, in which case the child will have an LDL level similar to his/her parents, and finally a 1 in 4 (25%) chance that the child will have inherited a gene for familial hypercholesterolemia from each parent, in which case the child will have a cholesterol level far exceeding either parent’s level (see Figure 4 – Familial hypercholesterolemia autosomal dominant inheritance pattern – above). These children are often diagnosed at around the age of 2 when they develop xanthomas (orange cholesterol deposits on the elbows, buttocks, legs, ankles, on the tendons of the fingers and between the fingers). These physical findings prompt a cholesterol profile. In children with homozygous familial hypercholesterolemia, the LDL cholesterol is often over 500 and can be as high as 1000 mg/dL.
- Children with homozygous familial hypercholesterolemia must see a lipodologist for immediate cholesterol lowering therapies
If your child has homozygous familial hypercholesterolemia, he or she will require immediate cholesterol lowering medications. Your child’s response to the medications depends on their particular genetic mutation. Some children will respond quite well to high dose statins combined with exetimibe (Zetia®) and a bile acid sequestrant. Other children will require more heroic measures such as LDL-apheresis. This is a procedure in which LDL cholesterol is physically removed from the blood every week. In the past, liver transplant was often considered in homozygous familial hypercholesterolemia patients, but it is rarely necessary today. Children with homozygous familial hypercholesterolemia require close follow-up with a cholesterol specialist. Children with untreated homozygous familial hypercholesterolemia develop very early atherosclerosis, often experiencing heart attacks in their teens. It is therefore crucial to treat children with homozygous familial hypercholesterolemia early and aggressively. Because the aortic valve (all the blood in the heart passes through this valve on its way to the body) can develop cholesterol plaque in homozygous familial hypercholesterolemia, children with this diagnosis often undergo much more involved heart evaluations than do children with heterozygous familial hypercholesterolemia. This can include heart catheterizations and echocardiograms. The future is bright for children with homozygous familial hypercholesterolemia – there are many new treatments being studied which have the potential to lower their LDL level profoundly.
Women with familial hypercholesterolemia and pregnancy
What will happen to my cholesterol level during pregnancy?
Cholesterol increases significantly during pregnancy by about 25-50%. Women with familial hypercholesterolemia experience the same increase, but since they are starting out at a much higher baseline, they can get extremely high cholesterol during pregnancy.
What if I become pregnant while on a statin?
If you become pregnant, stop your statin medications immediately. Other cholesterol lowering medications, including Niacin and ezetimibe (Zetia©), should also be stopped. Fibrates such as feno-brate and gem-brozil are typically not used in patients with familial hypercholesterolemia. These are primarily triglyceride-lowering medications, but if you are on one of these medications, it too should be stopped. In terms of the risk associated with taking cholesterol lowering medications during pregnancy, there are conflicting reports in the literature. In animal studies, some statins have been found to cause skeletal defects. Ezetimibe, niacin, and fibrates have all been associated with birth defects, too. Since we don’t recommend these agents during pregnancy, the human data comes from women who have unplanned pregnancies. In most cases the cholesterol-lowering medications were stopped in the first trimester, and although most babies were perfectly normal, there have been some rare reports of structural birth defects. Should you inadvertently take a statin or other cholesterol-lowering medication during pregnancy we recommend seeing an obstetrician for an immediate assessment.
Can I continue to take my cholesterol medications during pregnancy?
With the exception of the bile acid sequestrants such as Welchol©, cholesterol-lowering medications should be stopped prior to pregnancy to avoid extremely high cholesterol. The bile acid sequestrants are not absorbed into your blood stream so they pose no risk to you or your baby. The guidelines on when to stop cholesterol-lowering medications vary. The National Lipid Association recommends discontinuation of cholesterol-lowering medications 4 weeks prior to attempting to become pregnant. The National Institute of Clinical Excellence (NICE) guidelines, on the other hand, recommend stopping cholesterol-lowering medications 3 months before attempting to conceive. Both the National Lipid Association and National Institute of Clinical Excellence guidelines caution women not to take cholesterol medications during pregnancy or while nursing. Some women with familial hypercholesterolemia might be off their cholesterol-lowering medications for a prolonged period of time in order to become pregnant. The longer you are off your cholesterol-lowering medications, the greater the possibility that you will have plaque build-up in your heart arteries and potentially accumulate extremely high cholesterol as well.
How can I keep my cholesterol under control during pregnancy?
Women with familial hypercholesterolemia must come off their cholesterol lowering medications (except bile acid sequestrants) during pregnancy – this increases your cholesterol immediately. Then, just like everyone else, between the 18-36th week of pregnancy cholesterol can increase by as much as 50%. Eating a low fat, low cholesterol diet during pregnancy is extremely important, and if you have not been on a bile acid sequestrants such as Welchol© in the past it is important to begin. Welchol© is not absorbed into your blood stream so is safe during pregnancy. Some people notice some gastrointestinal disturbances (gas) with Welchol© but this agent does lower the LDL-Cholesterol by about 15%, so it seems worth the eff ort. Again, it is important to emphasize the benefits of diet. We encourage you to work with a registered dietitian who has experience in either a lipid, cardiology, or endocrinology clinic.
Each time you have a baby, you have a 50/50 chance of passing on the abnormal gene.
Is my baby likely to inherit familial hypercholesterolemia?
If you have heterozygous familial hypercholesterolemia, then your baby has a 50% chance of having familial hypercholesterolemia (see Figure 4 above). In the same way that you inherited a gene for LDL cholesterol removal from each of your parents, your baby will inherit one gene for LDL cholesterol removal from you and one from your partner. Because you have one normal gene and one abnormal gene, each time you have a baby you have a 50/50 chance of passing on the abnormal gene. If you have homozygous familial hypercholesterolemia or your partner also has heterozygous familial hypercholesterolemia, then the stakes are much higher and it is recommend you see a lipid specialist or genetic counselor. Familial hypercholesterolemia is a genetic disorder characterized by very high LDL-cholesterol. Familial hypercholesterolemia is an autosomal dominant disorder (see Figure 4 above), which means if you inherit a single gene for familial hypercholesterolemia from one of your parents, you will have familial hypercholesterolemia. Other disorders are autosomal recessive disorders in which you must inherit a gene from each parent in order to display the disease. Although scientists still have much more to learn about the genetics of familial hypercholesterolemia, they know that most people with familial hypercholesterolemia have a defect in the gene for the LDL receptor (LDLR). This is a receptor that sits on your liver cells and allows you to remove LDL cholesterol from your blood stream. Having one defective LDL receptor leads to about a doubling of the normal LDL cholesterol level in the blood. There are other less common genetic defects that can lead to similar LDL levels. These include a defect in apolipoprotein B (Apo B), which is the protein on LDL cholesterol that allows the LDL receptor to recognize and remove LDL-Cholesterol from the blood. Still an even rarer defect is a “Gain of Function” mutation in PCSK9 (proprotein convertase subtilisin/kexin type 9). PCSK9 is a protein that is responsible for destroying LDL receptors. If your PCSK9 works too well it will destroy more LDL receptors and you will be left with an elevated LDL cholesterol. There are probably other genetic defects leading to familial hypercholesterolemia that we have not yet discovered for predicting and regulating extremely high cholesterol.
Should I become pregnant?
This is a personal choice and something you need to discuss with your partner and your health care team. It is important to know that if your cholesterol rises dramatically during pregnancy, there are some options. One such option is LDL-apheresis. This is a dialysis-like procedure that physically removes LDL-cholesterol from the blood stream on a weekly or bi-weekly basis. This 3-hour procedure involves removing blood from one arm and passing it through a special column designed to extract the LDL cholesterol, then returning the blood to the other arm. LDL-apheresis has been successfully performed during pregnancy on many young women with very high cholesterol levels. Other women are successfully managed with diet and bile acid sequestrants. Finally, there are many ways to create a family. Some people choose to adopt and others create a family with the help of a gestational surrogate.
Should I Nurse My Baby?
Although everyone must decide what is best for their family, most lipid specialists would advise limiting breast feeding to 6 months, at which point you can resume your cholesterol lowering medications.
If I have trouble getting pregnant should I consider infertility medications?
Many infertility medications increase cholesterol dramatically. If you are contemplating infertility treatments, it is crucial for you to have a cholesterol specialist, also known as a lipidologist. Your lipidologist should work closely with your infertility physician. You should monitor your cholesterol very closely during these treatments and have a plan for limiting the number of cycles you go through.
Men with familial hypercholesterolemia
Why do men tend to experience heart disease a decade earlier than the women in their family?
Although the answer to this is not 100% certain, there are likely several reasons. The first is puberty. As young men go through puberty, their HDL “good cholesterol” (protective cholesterol) tends to fall. This does not happen to young women. A low HDL is an additional risk factor for early heart disease. Thus not only is a young man with familial hypercholesterolemia at risk due to his elevated LDL, compared to the women in his family, he very likely has a lower HDL. Additionally, women (even women with familial hypercholesterolemia) tend to be relatively protected from symptomatic heart disease until menopause – probably due to estrogen. An aside to women reading this: please notice the word symptomatic. Although it is unusual for a woman with familial hypercholesterolemia to have a heart attack before menopause, your elevated LDL cholesterol means you are still putting cholesterol into your artery wall. Women have smaller heart arteries than men, and it doesn’t take long after going through menopause to block those arteries – so women need to be treated early, too. In the past, but not currently, more men than women smoked. Smoking is poison for everyone, but for people with familial hypercholesterolemia, smoking is doubly poisonous. Smoking can lower the HDL and leads to chemical modifications (oxidation) of LDL. Oxidized LDL is more dangerous than non-oxidized LDL. It is therefore possible that in the past, smoking set men with familial hypercholesterolemia up for earlier heart disease than their non-smoking female relatives. The bottom line is that if you smoke you should quit!
Are men less likely to be treated for their elevated LDL than women?
Once a man develops heart disease, there is no evidence that men are less likely to be aggressively treated for their cholesterol. In fact, in the past there was even evidence that women were less aggressively treated for their heart disease than were men. Familial hypercholesterolemia is treatable if accurately diagnosed. The fact is, the only way you will know if you have high cholesterol is if you get tested. The current national recommendation is that all Americans undergo cholesterol testing between ages 9-11. If you have not yet been tested, especially if you have a strong family history of heart disease, it’s imprtant you get tested. Knowledge is power. If you are found to have familial hypercholesterolemia, get treated, and work to reduce all other risk factors. If you are overweight, try hard to get to a healthier weight. If you smoke, please quit. Both losing weight and quitting smoking can be difficult—but the sooner you do it, the healthier you will be. If you have high blood pressure, then exercise, salt restriction, weight loss, and medications when necessary are all important. Finally, if you have diabetes, work on weight loss through diet and exercise, and, if necessary, take prescribed medications.
I have been diagnosed with familial hypercholesterolemia, are there any other lab studies I should have?
Anyone who has familial hypercholesterolemia should also have a measurement for Lipoprotein-A also known as Lp(a) or lipoprotein(a), which is an inherited lipid found in the blood. Lipoprotein-A or Lp(a) is really the combination of an LDL particle and a clotting particle. People living with familial hypercholesterolemia who also have a high Lp(a) level are at even higher risk for early heart disease than are people who simply have a high LDL level. You can imagine that if Lp(a) contains an LDL particle it can block up the arteries; adding to that a clotting particle makes the risk even higher. Most heart attacks are caused when a cholesterol plaque inside a heart artery ruptures and subsequently a blood clot forms on top of the ruptured plaque. The combination of a ruptured plaque and a blood clot might completely prevent blood from flowing in the heart artery. If the blockage is not opened quickly, a heart attack will ensue. Lp(a) can be measured in two different ways. If your test is measured in mg/dL then a normal level is less than 30 mg/dL. Some labs measure in nmols/L, in which case the goal is a level < 74 nmol/L.
Can anything be done to lower Lipoprotein-A?
Unfortunately, Lipoprotein-A or Lp(a) is quite difficult to treat. If your level is very elevated, it is unlikely to normalize with medication. Although there have not been any formal studies performed to assess whether lowering Lp(a) reduces cardiac risk, it makes sense to try to lower this lipoprotein. Of the currently available cholesterol lowering medications, only niacin significantly lowers Lp(a). Estrogen does have some Lp(a) lowering properties, but estrogen is not used in men. Lp(a) can also be lowered by LDL-apheresis. Although LDL-apheresis, a dialysis like procedure, is typically used to lower LDL cholesterol, sometimes the procedure is approved by insurance companies specifically to lower Lp(a). Some of the cholesterol lowering medications currently in development appear to significantly lower Lp(a).
How much can I lower my risk if I take cholesterol lowering medications?
Again, it is difficult to give you a precise percentage. In any given person, the overall degree of risk reduction will likely depend on the age at which cholesterol-lowering medications are started, and the number of other risk factors a person has for the development of heart disease, such as diabetes, cigarette smoking, high blood pressure, and high Lp(a). Some general statements can be made. In studies (many 5 or more years in length) performed in people with very high cholesterol, including many with familial hypercholesterolemia, statins have been shown to reduce the risk of a future heart attack by over 30%. In observational studies of familial hypercholesterolemia patients performed in Europe, it appears that long-term statin therapy is capable of lowering lifetime risk to that of the general population.
Note that studies show statins reduce chances of a future heart attack by 30%.
I know statins can lower my risk for a heart attack, but every time I take a statin my muscles and joints ache what can I do?
Thankfully, this is not a common problem, but if it is your situation it can be miserable.
First, it is important to make sure that a person does not have an underlying condition known to increase the risk for the development of statin myopathy, such as muscle aches when taking a statin. People with an under active thyroid are significantly more likely to experience statin myopathy than people with a normally functioning thyroid gland, so make sure your health care provider checks this. If you are found to have a hypothyroid problem, once this has been corrected, you may well be able to tolerate statins. Certain medications can lead to an increase blood level of statins. These include, among others, drugs used to treat HIV, certain antibiotics, and another type of cholesterol lowering medication known as gemfibrozil (Lopid®). If this is your situation, your health care provider should make a change. These medications really should not be used with statins. People with underlying liver or kidney disease and people who are frail may be more likely to develop statin myopathy. These people are typically treated with lower doses of statins. Some studies have found Asians with heart attacks in the family are more likely to develop statin myopathy and as such, are also often treated with lower doses of statins.
Finally, as with so many medical conditions, recently genetic factors have been found to play a role in determining who develops statin myopathy. When it comes to statins, they are not all created equal. One large study—The PRIMO Study—found that of the statins simvastatin (Zocor®) was most likely to cause muscle aches and fluvastatin (Lescol®) was least likely. Unfortunately, Lescol® is the least potent statin. That said, other more potent statins including atrovastatin (Lipitor®) and rosuvastatin (Crestor®) are less likely than simvastatin to cause aches. In general, an approach to statin intolerance is to try different statins, one at a time. If a statin causes pain, speak to your doctor about stopping that particular statin medication. Once you feel better you can try a different statin, or a lower dose of the same statin. In some cases, every other day, or even every 3rd or 4th day dosing is effective. If a person with familial hypercholesterolemia can only tolerate low dose, or intermittent statin dosing, he/she will likely need to tighten his/her diet, and almost certainly will require additional medications. Other choices which may be additive to statins include bile acid sequestrants (Welchol®, Questran®, Colestid®), cholesterol absorption inhibitors (Zetia®), or niacin (Niaspan®). Other options for further LDL reduction include LDL apheresis and participation in clinical trials.
My doctor has asked me to begin LDL-apheresis. Is this likely to lower my risk of a heart attack?
LDL-apheresis is a dialysis-like procedure, performed every week in people with homozygous familial hypercholesterolemia and every other week in persons with heterozygous familial hypercholesterolemia. A person has homozygous familial hypercholesterolemia if he/she has inherited 2 genes for familial hypercholesterolemia, one from each parent, and a person with heterozygous familial hypercholesterolemia has inherited one gene for familial hypercholesterolemia. Apheresis literally means “to take out.” In the case of LDL-apheresis, LDL cholesterol is physically removed from the blood. Blood is removed from one arm, specially treated to remove the LDL cholesterol, then returned into the other arm. The procedure, which typically takes between 2-4 hours, can lower LDL by as much as 80% and also dramatically lowers Lp(a). Unfortunately, over a short period of time a person’s level rebounds – this is why it needs to repeated frequently. Although for obvious reasons (scientists will always know who is getting the procedure and who is not), it is impossible to conduct placebo-controlled trials to assess the effectiveness of LDL-apheresis. Several studies have compared the outcomes of patients receiving medications along with LDL-apheresis to those receiving medications without LDL-apheresis. It appears that long term use of LDL-apheresis may reduce cardiac risk anywhere from 44 – 72% above and beyond the use of medications alone. Finally, if your doctor has recommended LDL-apheresis it is likely that your LDL [or Lp(a)] remains very high despite maximally tolerated cholesterol lowering medications.
I have familial hypercholesterolemia. How likely is it that my child will also have familial hypercholesterolemia?
If you have heterozygous familial hypercholesterolemia, as long as your partner doesn’t also have heterozygous familial hypercholesterolemia, your child has a 50 % chance of having familial hypercholesterolemia. In exactly the same way that you inherited a gene for LDL cholesterol removal from each of your parents, your child has inherited one gene for LDL cholesterol removal from you and one from your partner, which reduces the risk of heart attacks in family.
Familial hypercholesterolemia life expectancy
The risk of complications of hypercholesterolaemia can be significantly reduced by therapies that lower serum cholesterol levels. Studies show statins reduce chances of a future heart attack by 30%.
Lowering cholesterol by 1% reduces the risk of coronary artery disease by 2%.
The treatment of other modifiable risk factors such as smoking, high blood pressure and diabetes will further decreases the risk of complications of hypercholesterolaemia.
Maintaining an appropriate weight, eating a low fat diet and exercising can also have a significant impact on cholesterol levels and improve long-term outcomes.
Familial hypercholesterolemia symptoms
Depending on the severity or form of familial hypercholesterolemia you have (Heterozygous vs. Homozygous), symptoms can start appearing in childhood or they might not appear until much later in life. Because familial hypercholesterolemia is caused by a defective gene, it is present in the body from birth. However, this is not necessarily obvious. In fact, this is doctors like to call it the “invisible” disease. Many people with familial hypercholesterolemia just think they have high cholesterol that can be lowered with the right food until, one day, they are only 38 years old and they have a heart attack. Symptoms are not necessarily present.
Signs of Familial hypercholesterolemia are different in every patient and they may or may not include the following:
- Family history of early heart disease or heart attacks (before age 55 in men and before age 65 in women)
- High LDL-cholesterol (above 190 mg/dL in adults and above 160 mg/dL in children)
- Chest pain or angina
- Bumps or lumps on the skin over parts of the hands, elbows, knees, ankles and around the cornea of the eye these fatty skin deposits are called xanthomas
- Cholesterol deposits in the eyelids called xanthelasmas
- Cramping of one or both calves when walking
- Sores on the toes that do not heal
- Sudden stroke-like symptoms such as trouble speaking, drooping on one side of the face, weakness of an arm or leg, and loss of balance.
But remember, you don’t have to have visible symptoms to have Familial hypercholesterolemia. Here’s a formula to help you remember the two main signs of Familial hypercholesterolemia:
- Xanthomas are collections of cholesterol under the skin or tendons. They are yellow bumps that can be small and hard to see. These are often found in the folds of skin and buttocks in children. Also, there can be xanthomas on the tendons at the ankles and hands. Children often have xanthomas without any signs of heart problems in early childhood.
- Tendon xanthoma occurs in three fourth of middle-aged patients and is largely discovered in the Achilles tendon. It may also occur in the extensor area of the dorsum of the hand, heel or the knee. Since xanthomas (patches of yellowish cholesterol buildup) may be manifested only to the extent of thickening of a tendon, familial hypercholesterolemia patients may be overlooked by clinicians without due concern. Xanthelasma or early arcus cornealis may also be observed.
- Xanthomas may worsen with age as a result of extremely high cholesterol levels. Xanthomas can occur around the eyelids and within the tendons of the elbows, hands, knees, and feet.
From figure 5 below:
A) Lateral borders of thickened Achilles’ tendons are shown with arrows.
B) Tendon xanthomas can also occur in the extensor tendons of the hands (shown), feet, elbows and knees.
C) Xanthelasmas are cholesterol deposits in the eyelids.
D) Arcus cornealis (corneal arcus) is a greyish-white ring of cholesterol infiltration around the corneal rim (arrow). A corneal arcus at a young age can mean that the child has homozygous familial hypercholesterolemia.
Figure 5. Familial hypercholesterolemia – Physical signs of heterozygous familial hypercholesterolemia, as a result of cholesterol deposition within macrophages in specific sites.
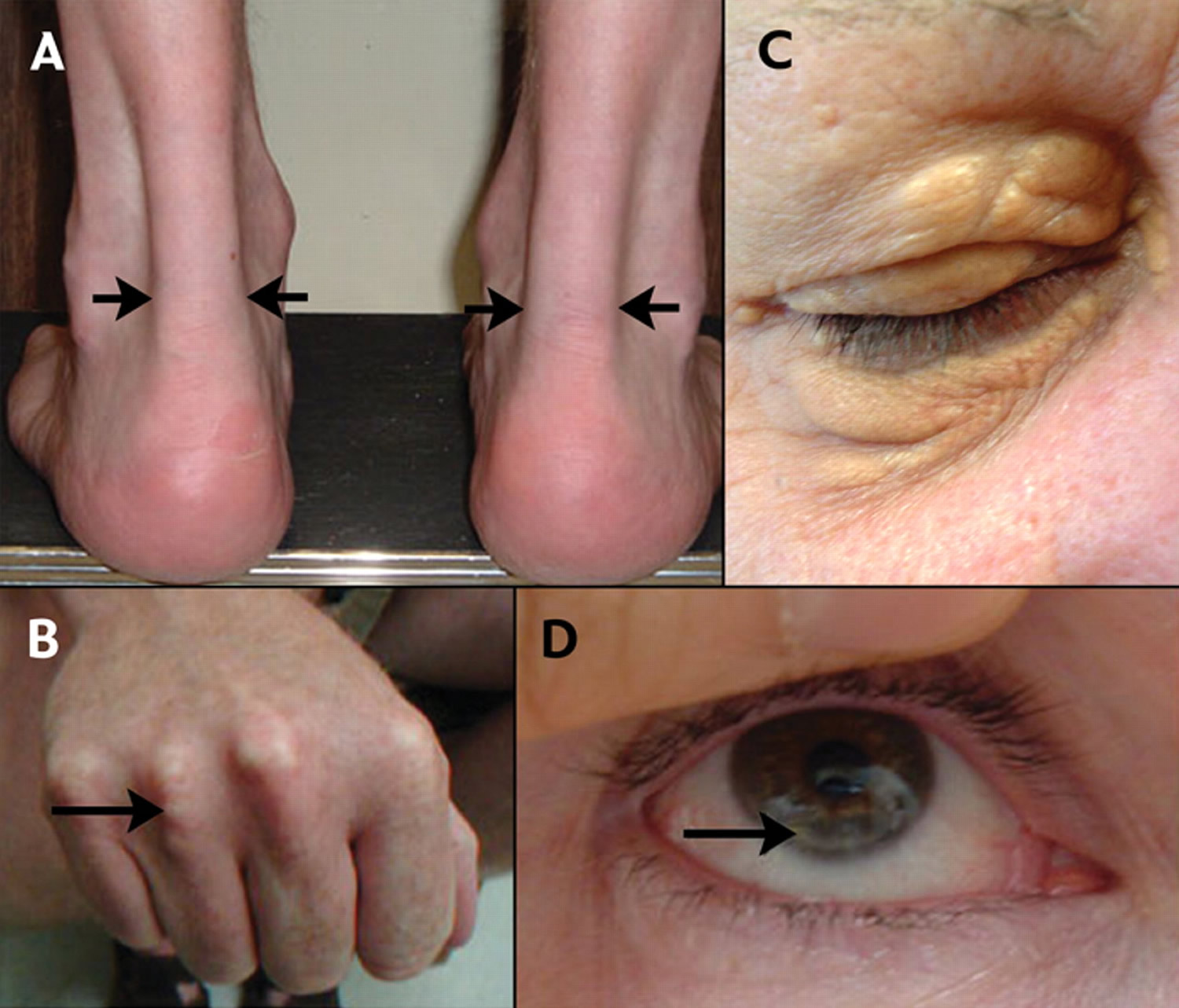
Figure 5. Homozygous familial hypercholesterolemia
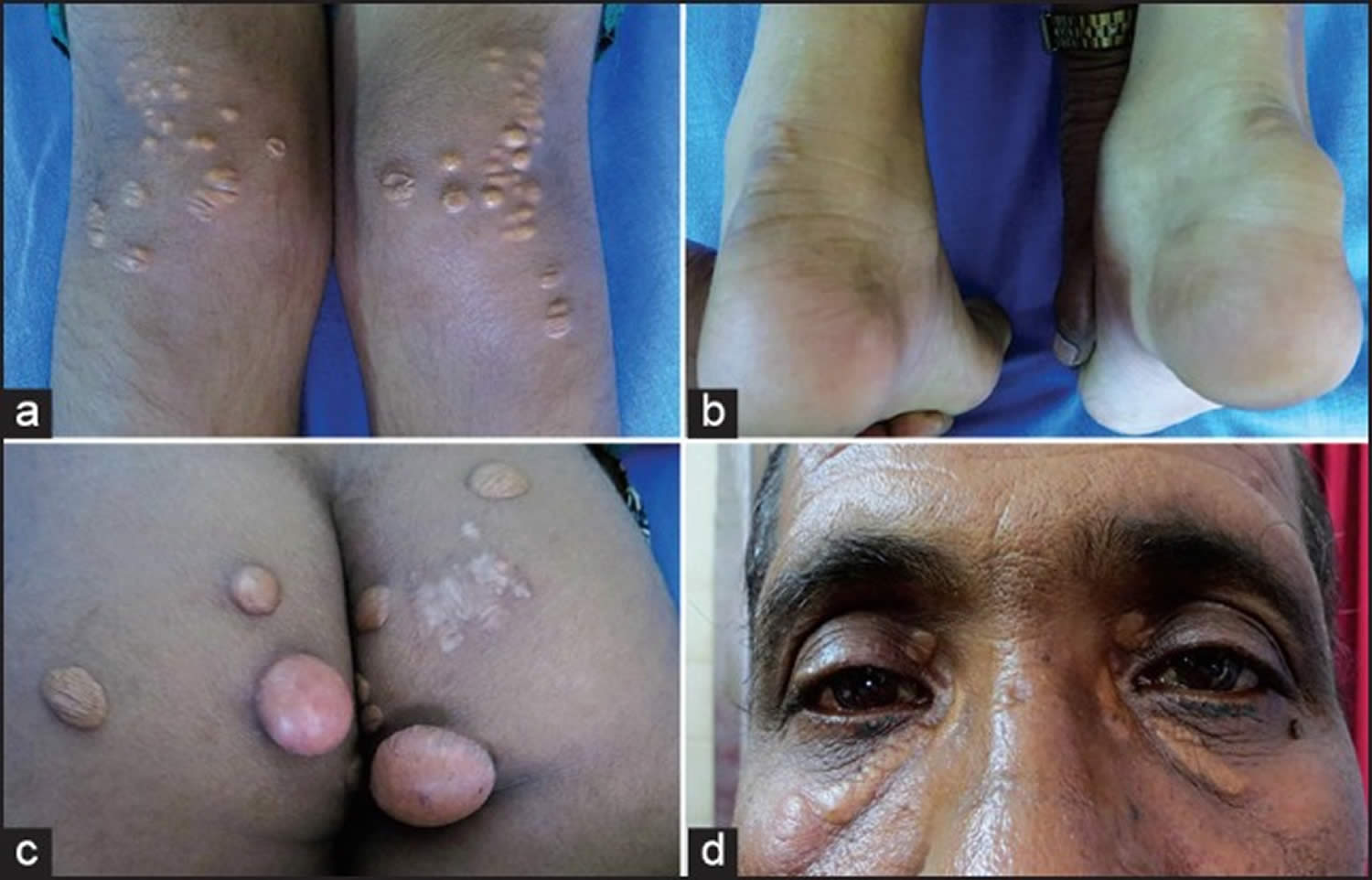
Footnotes: (a) Eruptive xanthoma, (b) tendon xanthoma, (c) tuberous xanthoma and (d) xanthelasma
[Source 7 ]Heart and blood vessel disease
A heart murmur may be the result of narrowing of the opening of the aortic valve by cholesterol buildup. A child with aortic valve disease and high LDL-C may have homozygous familial hypercholesterolemia. Cholesterol plaque builds up in the coronary arteries supplying blood to the heart, the carotids taking blood to the brain, the arteries to the kidneys, and other arteries. Blockage of the flow of blood to the heart may cause chest pain, shortness of breath, dizziness, or irregular heartbeats. Special tests of the heart such as a EKG (electrocardiogram), echocardiogram, CT angiogram or cardiac catheterization are recommended to check the aortic valve and coronary arteries at diagnosis and at least every 5 years.
Familial hypercholesterolemia complications
Possible complications of familial hypercholesterolemia:
- Heart attack at an early age
- Heart disease
- Stroke
- Peripheral vascular disease
People who have familial hypercholesterolemia have a higher risk of heart disease and death at a younger age. Heart attacks may occur before age 50 in men and age 60 in women. The rarer and more severe variety of familial hypercholesterolemia (homozygous familial hypercholesterolemia), if undiagnosed or untreated, can cause death before age 20.
Familial hypercholesterolemia diagnosis
A detailed family history is an important key to diagnosing familial hypercholesterolemia. Doctors will be interested to know if your siblings, parents, aunts, uncles or grandparents ever had high cholesterol levels or heart disease — especially during childhood.
During the physical exam, doctors usually check for cholesterol deposits that may occur in the skin around the hands, knees, elbows and eyes. Tendons in the heel and hand may be thickened, and a gray or white ring may develop around the iris of the eye.
Homozygous familial hypercholesterolemia can be diagnosed through genetic testing (DNA testing) or a clinical diagnosis utilizing one of three well-accepted sets of criteria — Simon Broome (UK), Dutch Lipid Clinic Network (Netherlands), or MEDPED (US).
DNA testing confirms the diagnosis and is considered the “gold standard”, but is not always necessary or feasible. DNA testing should definitely be considered when it’s not clear whether an individual is affected or not, and is very helpful for testing family members. Recent studies also suggest that individual risks for coronary artery disease vary among the affected gene and type of DNA variation (substitution vs. partial deletion of a gene, etc.).
Unfortunately, genetic testing is not commonly used in some countries including the U.S. Also, not everyone with homozygous familial hypercholesterolemia has the same signs and symptoms. If you have any of the above signs or symptoms, please talk to your doctor.
Once an individual is diagnosed with familial hypercholesterolemia (either with or without the use of DNA testing) a process called “cascade screening”, “cascade testing” or “family screening” (testing of close relatives, in a step-wise fashion) is recommended to identify those with familial hypercholesterolemia before symptoms appear, so that early and intensive treatment can be initiated and disease and death prevented 30. Cascade screening involves screening close relatives of a person diagnosed with a specific genetic condition to determine if they also have the same genetic mutation. Because familial hypercholesterolemia is genetic condition, this means that more than one person in a family has it (because they inherited it from someone else). Cascade screening means that when a health professional diagnoses someone with familial hypercholesterolemia, they need to test the rest of their immediate family members for familial hypercholesterolemia too. If a pathogenic variant is identified, risk in the patient’s first degree relatives (parent, sibling, child) and when appropriate, more distant relatives, can be assessed via DNA testing by tracing the altered gene through the family. If DNA testing is not performed, another version of cascade screening can be implemented using cholesterol testing. Cascade screening by either means has been shown to be effective in finding patients with familial hypercholesterolemia who were not being appropriately treated. A genetic counselor can help a family through this process.
Cascade screening has been shown in numerous studies to be cost-effective and has been recommended by the National Institute for Health and Clinical Excellence (NICE) in the UK. The Office of Public Health Genomics at the Centers for Disease Control and Prevention considers cascade screening of relatives of those with FH a “Tier 1 application” which means that there is good evidence that implementation will prevent disease and save lives.
Homozygous familial hypercholesterolemia is easily identified in infants and young children by the presence of planar xanthomas, corneal arcus, and exceedingly high total and LDL-C; LDL-C is usually greater than 400 mg/dL. The parents are “obligate heterozygotes” who are considered to have heterozygous familial hypercholesterolemia until proven otherwise.
Clinical Testing and Workup
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with familial hypercholesterolemia, the following evaluations are recommended in adults and children:
- Pre-treatment lipid values
- Lipoprotein(a) levels when possible as lipoprotein(a) is an additional risk factor for coronary heart disease
- Exclusion of concurrent illnesses (kidney disease, acute myocardial infarction, infection) that can affect lipid values
- Lipid panel including total cholesterol (TC), low density lipoprotein cholesterol (LDL-C), high density lipoprotein cholesterol (HDL-C), and triglycerides
- Consultation with a lipid specialist or clinician with expertise in familial hypercholesterolemia
- Medical genetics or a genetic counseling consultation
In children, noninvasive imaging modalities (e.g., measurement of carotid intima-media thickness) are recommended in some guidelines to help inform treatment decisions.
Level of LDL cholesterol
This is extremely important. Adults who have familial hypercholesterolemia usually have low-density lipoprotein (LDL) cholesterol levels over 190 mg/dL (4.9 mmol/L). Children who have the disorder often have LDL cholesterol levels over 160 mg/dL (4.1 mmol/L). The higher the LDL cholesterol levels, the more likely it is that a person has homozygous familial hypercholesterolemia (LDL > 500 mg/dL [>13 mmol/L]).
LDL cholesterol is also known as “bad” cholesterol because it can build up in the walls of the arteries, making them hard and narrow. This can increase the risk of heart attacks.
Family History
This is key. If both parents have very high LDL cholesterol (>190 mg/dL) and/or history of heart disease before age 55-65, this may suggest that they both have familial hypercholesterolemia and can each pass a mutated gene to their children. When each parent has heterozygous familial hypercholesterolaemia, by chance, 1 of 4 children will have a normal cholesterol level, 2 of 4 children will have heterozygous familial hypercholesterolaemia and 1 of 4 children will have homozygous familial hypercholesterolemia. Prenatal DNA testing may be available if the mutations are identified in the parents. Download our family tree to track your family history.
Response to cholesterol-lowering medications
If a person is living a healthy lifestyle and taking cholesterol-lowering medications like statins, and/or cholesterol absorption inhibitors, and/or bile acid sequestrates and still has high cholesterol level (> 300 mg/dL), they may have homozygous familial hypercholesterolemia.
Genetic testing
A genetic test can confirm familial hypercholesterolemia, but it’s not always necessary. However, a genetic test can help determine whether other family members also may be at risk.
If one parent has familial hypercholesterolemia, each child has a 50% chance of inheriting it. Inheriting the altered gene from both parents can result in a rarer and more severe form of the disease.
If you are diagnosed with familial hypercholesterolemia, doctors usually recommend that your first-degree relatives — such as siblings, parents and children — be checked for the disorder. This will allow treatment to begin early, if needed.
Diagnostic Criteria for Familial Hypercholesterolemia
There are currently 3 accepted resources for familial hypercholesterolemia diagnosis:
- The Simon Broome criteria,
- The MEDPED Criteria, and
- The Familial Hypercholesterolemia Dutch Lipid Clinic Criteria.
Table 1. Simon Broome Diagnostic Criteria For Familial Hypercholesterolemia
| Point | Criteria |
| 1 | Total cholesterol levels > 290mg/dL (7.5 mmol/L) or LDL-C > 190 mg/dL (4.9 mmol/L) in adults. Total cholesterol levels > 260 mg/dL (6.7 mmol/L) or LDL-C > 155 mg/dL (4.0 mmol/L) |
| 2 | Tendon xanthomas in the patient or tendon xanthomas in a first or second degree relative. |
| 3 | DNA-based evidence of an LDL-receptor mutation, familial defective apo B-100, or a PCSK9 mutation. |
| 4 | Family history of myocardial infarction before age 50 years in a second degree relative or before age 60 years in a first degree relative. |
| 5 | Family history of elevated total cholesterol > 290 mg/dL (7.5 mmol/L) in an adult first or second-degree relative. Family history of elevated totacl cholesterol > 260 mg/dL (6.7 mmol/L) in a child, brother, or sister 16 years or younger. |
| DIAGNOSIS |
| Definite familial hypercholesterolemia = 1+2 or 3 Possible familial hypercholesterolemia = 1+4 or 5 |
Table 2. Dutch Lipid Clinic Network Diagnostic Criteria For Familial Hypercholesterolemia
| Group 1: Family history | Points |
| (i) First-degree relative with known premature (<55 years, men; <60 years, women) coronary heart disease (CHD) OR | 1 |
| (ii) First-degree relative with known LDL cholesterol >95th percentile by age and gender for country | 1 |
| (iii) First-degree relative with tendon xanthoma and/or corneal arcus OR | 2 |
| (iv) Child(ren) <18 years with LDL cholesterol >95th percentile by age and gender for country | 2 |
| Group 2: Clinical history | |
| (i) Subject has premature (<55 years, men; <60 years, women) CHD | 2 |
| (ii) Subject has premature (<55 years, men; <60 years, women) cerebral or peripheral vascular disease | 1 |
| Group 3: Physical examination | |
| (i) Tendon xanthoma | 6 |
| (ii) Corneal arcus in a person <45 years | 4 |
| Group 4: Biochemical results (LDL cholesterol) | |
| LDL-cholesterol (mmol/L) >8.5 mmol/L (>325 mg/dL) | 8 |
| LDL-cholesterol (mmol/L) 6.5–8.4 mmol/L (251–325 mg/dL) | 5 |
| LDL-cholesterol (mmol/L) 5.0–6.4 mmol/L (191–250 mg/dL) | 3 |
| LDL-cholesterol (mmol/L) 4.0–4.9 mmol/L (155–190 mg/dL) | 1 |
| Group 5: Molecular genetic testing (DNA analysis) | |
| (i) Causative mutation shown in the LDLR, APOB, or PCSK9 genes | 8 |
Familial hypercholesterolemia diagnosis:
- Definite familial hypercholesterolemia diagnosis can be made if the subject scores >8 points.
- Probable familial hypercholesterolemia diagnosis can be made if the subject scores 6 to 8 points.
- Possible familial hypercholesterolemia diagnosis can be made if the subject scores 3 to 5 points.
- Unlikely familial hypercholesterolemia diagnosis can be made if the subject scores 0 to 2 points.
Footnotes: *Premature = < 55 years in men; < 60 years in women. Use of the per group only one score with the highest applicable. For example, when coronary heart disease and tendon xanthoma as well as dyslipidaemia are present in a family, the highest score for family history is 2. However, if persons with elevated LDL cholesterol levels as well as premature coronary heart disease are present in a family, but no xanthoma or children with elevated LDL cholesterol levels or a causative mutation are found, then the highest score for family history remains 1.
Abbreviations: LDL-C = low density lipoprotein cholesterol; FH, familial hypercholesterolemia; LDLR = low density lipoprotein receptor; Apo B = apolipoprotein B; PCSK9 = Proprotein convertase subtilisin/kexin type 9
[Source 18 ]Table 3. MEDPED Diagnostic Criteria For Familial Hypercholesterolemia
| Familial Hypercholesterolemia is diagnosed if total cholesterol exceeds these cutpoints in mg/dL (mmol/L) | ||||
|---|---|---|---|---|
| Age (years) | First degree relative with FH | Second degree relative with FH | Third degree relative with FH | General population |
| <20 | 220 (5.7) | 230 (5.9) | 240 (6.2) | 270 (7.0) |
| 20-29 | 240 (6.2) | 250 (6.5) | 260 (6.7) | 290 (7.5) |
| 30-39 | 270 (7.0) | 280 (7.2) | 290 (7.5) | 340 (8.8) |
| ≥40 | 290 (7.5) | 300 (7.8) | 310 (8.0) | 360 (9.3) |
Footnote: *The total cholesterol cutpoints for familial hypercholesterolemia is dependent upon the confirmed cases of familial hypercholesterolemia in the family. If familial hypercholesterolemia is not diagnosed in the family, then the cutpoint for diagnosis is as per “general population.”
FH = familial hypercholesterolemia
[Source 32 ]Familial hypercholesterolemia treatment
Familial hypercholesterolemia treatment focuses on reducing the extremely high levels of LDL (bad) cholesterol. This helps lower the risk of heart attack and death.
The treatments for homozygous familial hypercholesterolemia (HoFH) can be quite complex and anyone with this disease is advised to be under the care of a lipid specialist.
There are several important reasons for referring patients with probable familial hypercholesterolemia to a lipid specialist:
- Confirmation of the diagnosis. The Dutch Lipid Clinic Network Criteria (DLNC) score will be confirmed by more extensive clinical and laboratory assessment, including DNA testing for causative mutations in the LDL receptor and other genes. Some patients with classic phenotypic features of familial hypercholesterolemia may still not have a mutation identified (eg. 10% children with definite phenotypic familial hypercholesterolemia), reflecting limitations of the current DNA test and multiple mutations causing the disease 33. However, identification of a specific mutation is important in confirming the diagnosis and in cascade screening
- Cascade screening of family members, either through an identified genetic mutation or phenotypic assessment using the DLNC criteria. The process of cascade screening can be complex, and while general practice may have a role, a centralised service coordinated via a lipid specialist is probably best placed to undertake extended cascade screening of second and third degree relatives.
- Cascade screening may be a feasible option for parents and children under the care of the same family doctor
- Cardiovascular disease assessment and disease prevention. A lipid specialist can determine appropriate investigations to detect existing cardiovascular disease, determine the best drug regimen to manage the abnormal lipid profile, and reinforce the importance of a healthy lifestyle
- If there is no lipid specialist available, referral to a cardiologist or endocrinologist with special interest in lipid disorders would be alternative referral pathways.
Familial hypercholesterolemia management can include a combination approach – statins, cholesterol absorption inhibitors, and bile acid sequestrants. Often, Homozygous familial hypercholesterolemia patients will require even more intensive therapy to bring their LDL cholesterol (“bad cholesterol”) levels down.
Lifestyle and dietary changes
Lifestyle changes, such as exercising and eating a healthy low-fat diet, are the first line of defense against high cholesterol. Specific recommendations include:
- Reducing the amount of saturated fat in your diet to less than 6-10 percent of your daily calories. The most harmful types of fat are saturated fat and trans fat, which are found in foods from animal sources, such as meat and dairy products, as well as packed snacks, fast food, and baked goods (biscuits, cakes, etc). As a rule of thumb, avoid too much processed foods. Try to eat fresh by cooking homemade healthy meals from scratch. LESS! whole milk, butter, egg yolks, snacks (chips), deep-fried fast food (fries, deep-fried chicken), processed meat (sausage, pâté), liver, organ meats, frozen foods (waffles, pies, pizzas, breaded fish). BEWARE OF HIDDEN TRANS FATS (“partially hydrogenated…”)
- Consuming 25 to 35 grams of soluble fiber a day. As a rule of thumb, think plants (not animals): fruits, vegetables, grains, and beans. MORE! barley, oatmeal, sunflower seeds, almonds, soy, tofu, edamame, chickpeas, black beans, kidney beans, lentils, Brussels sprouts, carrots, apples, bananas, pears, oranges, grapefruit, prunes, blackberries.
- Add plant sterols and plant stanols to your diet. They actively reduce LDL-cholesterol and can be found in certain food supplements and substitutes (look for margarine or cheese that states “With added plant sterols/stanols”). An enhanced daily consumption of 2000 to 2500 mg of plant sterols/stanols per day may lower LDL-cholesterol by up to 15%!
- Increasing physical activity.
- Maintaining a healthy body weight.
- Not smoking. And quit smoking if you smoke.
With familial hypercholesterolemia, your doctor likely will also recommend that you take medication to help lower your LDL “bad” cholesterol levels. The specific medication or medications depend on various factors, including your risk factors, your age, your current health and possible side effects.
Table 4. Good and bad fats
| CHOLESTEROL-RAISING FATS | HEALTHIER FATS | ||
| Saturated Fats | Trans Fats | Monounsaturated Fats | Polyunsaturated Fats |
| Animal Sources: butter, cheese, cream, fatty cuts of meat and processed meats (hot dog, bacon, bologna, salami, sausage), ice cream, lard, poultry skin, sour cream, whole milk Plant Sources: coconut, palm, palm kerne | Foods with a high probability of containing partially hydrogenated oils: Baked Goods biscuits, cakes, cookies, doughnuts, muffins, pancake mix, pastries, pie crust, pizza dough Fried Foods French fries, breaded chicken or breaded fish Snack Foods – crackers, microwave popcorn, stick margarine, shortening and non- dairy creamer | Nuts, seeds and natural nut butters: almonds, hazelnuts, pecans, peanuts, pine nuts, pistachios, pumpkin and sesame seeds Avocados, olives Oils: canola, extra virgin olive, peanut, sesame | High omega-3 seafood: Arctic char, Atlantic mackerel, black cod (sablefish), herring, mussels, wild salmon, sardines, trout Nuts and seeds: chia, ground flaxseeds, hemp seeds, soy nuts, sunflower seeds, walnuts Oils: soybean, safflower, corn |
Medications
Most people with familial hypercholesteremia will need to take more than one medication to control their LDL cholesterol levels. Common medication choices include:
- Statins. Statins — among the most commonly prescribed medications for lowering cholesterol — block a substance your liver needs to make cholesterol. This causes your liver to remove cholesterol from your blood. Statins may also help your body reabsorb cholesterol from built-up deposits on your artery walls, potentially reversing coronary artery disease. Choices include atorvastatin (Lipitor), fluvastatin (Lescol), lovastatin (Altoprev,), pitavastatin (Livalo), pravastatin (Pravachol), rosuvastatin (Crestor) and simvastatin (Zocor). Not everyone can take statins especially women who are trying to conceive, pregnant, or nursing. Talk to you doctor about other therapy options.
- Bile-acid-binding resins. Your liver uses cholesterol to make bile acids, a substance needed for digestion. The medications cholestyramine (Prevalite), colesevelam (Welchol) and colestipol (Colestid) lower cholesterol indirectly by binding to bile acids. This prompts your liver to use excess cholesterol to make more bile acids, which reduces the level of cholesterol in your blood.
- Cholesterol absorption inhibitors. Your small intestine absorbs the cholesterol from your diet and releases it into your bloodstream. The drug ezetimibe (Zetia) helps reduce blood cholesterol by limiting the absorption of dietary cholesterol. Zetia can be used in combination with any of the statin drugs. Ezetimibe has been showing to add an extra 10 to 30 percent reduction in LDL levels for patients on maximally tolerated statins 34, 35. Currently, ezetimibe is the drug of choice as the second line in most guidelines 36.
- Combination cholesterol absorption inhibitor and statin. The combination drug ezetimibe-simvastatin (Vytorin) decreases both absorption of dietary cholesterol in your small intestine and production of cholesterol in your liver. It’s unknown whether Vytorin is more effective in reducing heart disease risk than taking simvastatin by itself.
- Injectable medications. A new class of drugs can help the liver absorb more LDL cholesterol — which lowers the amount of cholesterol circulating in your blood. The Food and Drug Administration recently approved alirocumab (Praluent) and evolocumab (Repatha) for people who have a genetic condition that causes very high levels of LDL. These drugs may also be used for people who have had heart attacks or strokes and need additional lowering of their LDL levels. These injectable drugs are administered at home one or two times a month.
- PCSK9 Inhibitor – antibodies that bind the PCSK9 enzyme. Normally, LDL is recycled in the liver by binding to receptors that move it through the cell. The PCSK9 enzyme weakens this process by connecting to and disabling the receptors, which frees up more receptors on liver cells to remove LDL cholesterol from the blood. When the PCSK9 inhibitor is introduced, it binds to the PCSK9 enzyme stopping it from attaching to the receptors and getting in the way of the recycling process. PCSK9 inhibitors drugs such as alirocumab (Praluent) and evolocumab (Repatha) help the liver absorb more LDL cholesterol, which lowers the amount of cholesterol circulating in the blood. They’re injected under the skin every few weeks and are very expensive. PCSK9 inhibitors have shown a great reduction in LDL levels and cardiovascular outcomes in different randomized clinical trials 37, 38. In those trials, PCKS9 inhibitors lowered LDL levels by 50 to 60 percent. While exhibiting higher efficacy than ezetimibe, the use of PCSK9 inhibitors is limited by their high cost and the reluctance of insurance companies to approve their use 39. Hence, PCSK9 inhibitors can be used as second or third-line drugs for patients with familial hypercholesterolemia.
- Lomitapide (marketed as Juxtapid in the U.S.) – Lomitapide is an inhibitor of the microsomal triglyceride transport protein (MTP), a key protein in the assembly and secretion of apolipoprotein (apo) B-containing lipoproteins in the liver and intestine 40. By blocking this protein, lomitapide helps reduce how much fat is absorbed and lowers the level of LDL cholesterol in your blood 41. If you take lomitapide you should consume less than 20 per cent of your calories from fat as this has been shown to help reduce unwanted gastrointestinal (stomach related) side effects. This is lower than normally recommended for a heart healthy diet, but advice and support is available to help you make this change. Lomitapide is approved as an adjunct to a low-fat diet and other lipid lowering treatments. It is an oral capsule.
- Mipomersen (marketed as Kynamro in the U.S.) -Mipomersen is an antisense drug (mRNA inhibition of apolipoprotein B) that leads to the decline of apoB RNA in the liver (apoB is necessary for the formation of LDL particles) 42. Mipomersen is administered as a weekly injection. Mipomersen is fourth line therapy usually reserved if the LDL does not reach the target level using statins, ezetimibe, and PCSK9 inhibitors
Medications for high triglycerides
If you also have high triglycerides, your doctor may prescribe:
- Fibrates. The medications fenofibrate (Tricor) and gemfibrozil (Lopid) decrease triglycerides by reducing your liver’s production of very-low-density lipoprotein (VLDL) cholesterol and by speeding up the removal of triglycerides from your blood. VLDL cholesterol contains mostly triglycerides.
- Niacin. Niacin (Niaspan) decreases triglycerides by limiting your liver’s ability to produce LDL and VLDL cholesterol. But niacin doesn’t usually provide any additional benefit than using statins alone. Niacin has also been linked to liver damage and stroke, so most doctors now recommend it only for people who can’t take statins.
- Omega-3 fatty acid supplements. Omega-3 fatty acid supplements can help lower your triglycerides. They are available by prescription or over-the-counter. If you choose to take over-the-counter supplements, get your doctor’s OK first. Omega-3 fatty acid supplements could affect other medications you’re taking. Fish is a great source of healthy omega-3 fats. The American Heart Association recommends eating 3.5 ounces of fish (especially oily, omega-3 rich fish) at least twice a week. Note: Tilefish, shark, swordfish, and king mackerel have high mercury content and should be eaten only occasionally.
Tolerance varies
Tolerance of medications varies from person to person. The reported side effects are muscle pains, stomach pain, constipation, nausea and diarrhea. If you decide to take cholesterol-lowering medication, your doctor may recommend liver function tests to monitor the medication’s effect on your liver.
LDL Apheresis
LDL apheresis (also called lipoprotein apheresis) can be performed on homozygous familial hypercholesterolemia patients as well as more serious heterozygous familial hypercholesterolemia patients. This procedure involves blood cleansing every other week (or sometimes weekly) with a special machine that looks like a dialysis machine. The procedure takes between one and a half to three hours, and typically drops LDL levels by 70%. Unfortunately, LDL rises over time and, for this reason, repeat treatments are necessary. About 60 lipoprotein apheresis centers exist in the US.
Treatment Guidelines for Heterozygous familial hypercholesterolemia
In patients with heterozygous familial hypercholesterolemia, lifestyle modification is unlikely to result in acceptable LDL levels; therefore, cholesterol-lowering medication is necessary. European Atherosclerosis Society consensus statement for screening and treatment of heterozygous familial hypercholesterolemia includes the following recommendations 18:
- An LDL target of less than 135 mg/dL for children with familial hypercholesterolemia
- An LDL target of less than 100 mg/dL for adults with familial hypercholesterolemia
- An LDL target of less than 70 mg/dL for adults with known coronary artery disease or diabetes
- Lifestyle modifications include a diet that severely limits saturated fats, trans fats, and cholesterol
- Desirable weight should be attained
- Significant weight loss should improve all lipid parameters (LDL, HDL, triglycerides)
- Aerobic and toning exercises improve blood lipid levels if performed for longer than 30 minutes, 4 or more days per week
Treatment Guidelines for Homozygous familial hypercholesterolemia
European Atherosclerosis Society guidelines for the screening and treatment of homozygous familial hypercholesterolemia are summarized as follows 43, 44:
- Treatment of homozygous familial hypercholesterolemia involves a combination of lifestyle changes, statin therapy (first approach), and lipoprotein apheresis for severe cases
- LDL apheresis should begin as early as age 5 years
- For homozygous familial hypercholesterolemia patients, the LDL cholesterol targets are less than 100 mg/dL for adults, less than 70 mg/dL for adults with clinical cardiovascular disease, and less than 135 mg/dL for children
- Other novel agents for LDL cholesterol lowering (eg, lomitapide with or without apheresis) can be considered as adjunctive treatments for patients who do not achieve the recommended LDL cholesterol targets and remain at high cardiovascular risk.
Familial Hypercholesterolemia and Pregnancy
Women with familial hypercholesterolemia planning to get pregnant should discontinue all lipid-lowering agents, including statins, ezetimibe, and PCSK9 inhibitors 4. Cardiovascular risk assessment is recommended before conception. Lipoprotein apheresis may be used if necessary 45.
Children or Adults with Homozygous familial hypercholesterolemia
Early initiation of therapy and monitoring using CT coronary angiography and other imaging are recommended; these patients often require additional treatment strategies, as pharmacological treatment and lifestyle changes may not be sufficient. Statins are usually started as soon as the diagnosis is made (though may not be effective as explained above). Two new drugs (lomitapide and mipomersen) have now been FDA-approved for the treatment of adults with homozygous familial hypercholesterolemia and should be considered for these patients, especially if LDL-C level cannot be controlled using other drugs. A PCSK9 inhibitor (evolocumab) was also approved for homozygous familial hypercholesterolemia. Additional options include LDL apheresis or liver transplantation.
LDL Apheresis
Using a process similar to kidney dialysis, blood is withdrawn from a vein via a catheter and processed to remove LDL particles. Normal blood products are returned via another catheter. LDL-C levels will decrease approximately 50% but will rise between apheresis sessions, so they are necessary approximately weekly or every other week. The procedure is effective and well tolerated though time-consuming and only available in 50-60 sites in the US.
Liver transplantation
Liver transplant is extraordinarily rare and may become even less common with the new medications available. As the donor liver will have normal LDL receptors, the LDL cholesterol quickly normalizes after the procedure, but the risks of any organ transplant are significant and include complications from major surgery and the effects of lifelong suppression of the immune system. Donor organs are often not available. Patients with familial pathogenic APOB or PCSK9 gene variants have normal LDL receptors, so liver transplantation is not an option for them.
Various imaging modalities such as echocardiograms, CT angiograms and cardiac catheterization may be recommended to monitor individuals with homozygous familial hypercholesterolemia.
Children and cholesterol treatment
Diet and exercise are the best initial treatment for children age 2 and older who have high cholesterol, or who are obese. Children age 10 and older might be prescribed cholesterol-lowering drugs, such as statins, if they have extremely high cholesterol levels.
Current recommendations from both the National Lipid Association and American Academy of Pediatrics recommend beginning cholesterol lowering medications at around the age of 8 for children with familial hypercholesterolemia. Statins are the drugs of first choice. By the age of 8, a child should be able to swallow pills. At least one drug company has experimented with a chewable flavored statin, but this is not yet on the market.
Six statins are FDA-approved for children 10 years of age and older (age 8 years and older for pravastatin):
- rosuvastatin (Crestor®),
- atorvastatin (Lipitor®),
- simvastatin (Zocor®),
- pravastatin (Pravachol®),
- lovastatin (Mevacor®),
- fluvastatin (Lescol®)
Depending on which statin is used and at what dose, reductions in LDL of 50% or more can be achieved with the two most potent statins rosuvastatin (Crestor®), atorvastatin (Lipitor®)]. And thankfully, Lipitor® has recently become available as a generic.
National Lipid Association and American Academy of Pediatrics guidelines: Children with familial hypercholesterolemia begin medications around 8 years of age
If your child fails to achieve an LDL of < 130 mg/dL or an LDL reduction from baseline of at least a 50%, consideration should be given to adding an additional medication. In this situation your child’s health care provider might suggest a bile acid sequestrant such as [colesevelam (Welchol®), cholestyramine (Questran®), or colestipol (Colestid®). These medications work in the intestines and are not absorbed into the blood stream. Although these medications can lower cholesterol by 10-20% and appear to be very safe, only one, colesevelam (Welchol®), has been approved for use in the pediatric population. Unfortunately, many children complain of gas while on a bile acid sequestrant.
Finally, ezetimibe (Zetia®) is a medication that inhibits the absorption of cholesterol from the intestines. It can lower the LDL by about 20% and has been studied in children and found to be well tolerated. It is now approved by the FDA for use in children over the age of 10. If your child is not able to achieve his/her goal cholesterol with a single cholesterol lowering medication, it is often appropriate to ask for a referral to a cholesterol (lipid) specialist. Many parents see a cholesterol specialist for the initiation of cholesterol-lowering medications but follow-up with their pediatrician, family doctor or nurse practitioner once their child’s levels are under control.
Once my child begins medication, how often should he/she be seen?
Some doctors typically see a child back 6 weeks after they start a medication. Subsequent visits might occur every 3 to 6 months. To assess liver function, blood should be drawn prior to starting therapy. The need for follow-up liver function tests is generally determined by your child’s health care provider. Repeat cholesterol profiles are usually drawn at least twice a year.
What are some of the side effects of cholesterol lowering medications?
I have already noted that the bile acid sequestrants can cause gas. The statins are typically very well tolerated but in some cases they can cause muscle and joint aches and can rarely cause liver function and muscle function abnormalities. Thankfully, in almost all situations, these side effects can be reversed with either stopping the drug or reducing the dose. And although Zetia® tends also to be very well tolerated, it too can occasionally cause abdominal pain and diarrhea. When Zetia is taken in conjunction with a statin, rare cases of muscle and liver function abnormalities have occurred.
After my child’s cholesterol level normalizes, can he/she come off the medication?
Unfortunately, because familial hypercholesterolemia is caused by a defective gene, there is currently no cure. Medications can help normalize the cholesterol in a person with familial hypercholesterolemia but if they are discontinued, the genetic disorder is again revealed. Cholesterol-lowering medications only work while you are taking them.
Cholesterol-lowering medications only work while you are taking them.
My teen has an awful diet. What should I do?
This is a very common situation that are encountered countless times in clinical practice. Teens with familial hypercholesterolemia are like any other teen, but obviously the stakes are higher for children with familial hypercholesterolemia. It is very frustrating to see your son or daughter’s eating habits deteriorate. Best advice is to set a good example; make the foods you serve at home as healthy as possible.
My child hates to take his/her medications. What can I do?
Stick with it. The medications are crucial – there is strong evidence that statins reduce the risk of heart attack, stroke and total mortality. And heart disease is what kills young adults with familial hypercholesterolemia. Your child is not at risk of a heart attack right now (unless your child has homozygous familial hypercholesterolemia), but he/she is at risk in the future. Just when people with familial hypercholesterolemia should be enjoying their own children (and you, your grandchildren), a heart attack can strike – sometimes these heart attacks can be fatal. You have to stress the importance of getting your child into the habit of taking his/her medicines. Make it a family affair – take your medications together.
Surgical care
Surgery are usually reserved if the LDL does not reach the target level using medications and lifestyle changes (e.g., eating healthy, getting regular exercise, not smoking, total avoidance of tobacco products, limiting of alcohol intake, managing stress, having adequate sleep).
- Liver transplantation for homozygous familial hypercholesterolemia because a new liver provides functional LDL receptors and causes dramatic decreases in LDL levels
- Partial ileal bypass surgery 46
- Portacaval anastomosis for homozygous familial hypercholesterolemia
Familial hypercholesterolemia prognosis
Familial hypercholesterolemia prognosis (outcome) depends on how closely you follow your doctor’s treatment advice and how well you respond to the recommended treatment. Making dietary changes, exercising, and taking your medicines correctly can lower cholesterol level. These changes can help delay a heart attack, especially for people with a milder form of familial hypercholesterolemia.
Men and women with familial hypercholesterolemia typically are at increased risk of early heart attacks. Risk of death varies among people with familial hypercholesterolemia. If you inherit two copies of the defective gene (homozygous familial hypercholesterolemia), you have a poorer prognosis. Homozygous familial hypercholesterolemia does not respond well to treatment and may cause an early heart attack. They usually die before the third decade of life from cardiovascular events 37.
The risk of coronary heart disease before the use of statin in patients with heterozygous familial hypercholesterolemia was very high 47. However, the risk of death in patients with heterozygous familial hypercholesterolemia after acute coronary syndrome within the first year is almost more than two times higher than matched individuals without familial hypercholesterolemia despite high-intensity statins therapy 48. The risk of death or coronary artery disease in relatives of patients with familial hypercholesterolemia was 52% and 32% in males and females, respectively 49.
- van den Bosch SE, Hutten BA, Corpeleijn WE, Kusters DM. Familial hypercholesterolemia in children and the importance of early treatment. Curr Opin Lipidol. 2024 Jun 1;35(3):126-132. doi: 10.1097/MOL.0000000000000926[↩]
- Ontario Health (Quality). Genetic Testing for Familial Hypercholesterolemia: Health Technology Assessment. Ont Health Technol Assess Ser. 2022 Aug 23;22(3):1-155. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9470216[↩]
- Ison HE, Clarke SL, Knowles JW. Familial Hypercholesterolemia. 2014 Jan 2 [Updated 2022 Jul 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK174884[↩]
- Vaezi Z, Amini A. Familial Hypercholesterolemia. [Updated 2022 Sep 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK556009[↩][↩]
- Campbell-Salome G, Jones LK, Masnick MF, et al. Developing and Optimizing Innovative Tools to Address Familial Hypercholesterolemia Underdiagnosis: Identification Methods, Patient Activation, and Cascade Testing for Familial Hypercholesterolemia. Circ Genom Precis Med. 2021 Feb;14(1):e003120. doi: 10.1161/CIRCGEN.120.003120[↩]
- Abul-Husn NS, Manickam K, Jones LK, et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science. 2016 Dec 23;354(6319):aaf7000. doi: 10.1126/science.aaf7000[↩]
- Varghese MJ. Familial hypercholesterolemia: A review. Ann Pediatr Cardiol. 2014 May;7(2):107-17. doi: 10.4103/0974-2069.132478[↩][↩]
- Newman WP 3rd, Freedman DS, Voors AW, Gard PD, Srinivasan SR, Cresanta JL, Williamson GD, Webber LS, Berenson GS. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N Engl J Med. 1986 Jan 16;314(3):138-44. doi: 10.1056/NEJM198601163140302[↩]
- Warden BA, Fazio S, Shapiro MD. Familial Hypercholesterolemia: Genes and Beyond. [Updated 2021 Oct 23]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK343488[↩]
- Guirguis-Blake JM, Evans CV, Coppola EL, et al. Screening for Lipid Disorders in Children and Adolescents: An Evidence Update for the U.S. Preventive Services Task Force [Internet]. Rockville (MD): Agency for Healthcare Research and Quality (US); 2023 Jul. (Evidence Synthesis, No. 229.) Chapter 1, Introduction. Available from: https://www.ncbi.nlm.nih.gov/books/NBK593597[↩]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986 Apr 4;232(4746):34-47. doi: 10.1126/science.3513311[↩]
- Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262–8. https://www.ncbi.nlm.nih.gov/pubmed/22398274[↩]
- Reiner Ž, Catapano AL, De Backer G, Graham I, Taskinen MR, Wiklund O, Agewall S, Alegria E, Chapman MJ, Durrington P, Erdine S, Halcox J, Hobbs R, Kjekshus J, Filardi PP, Riccardi G, Storey RF, Wood D., Clinical Practice Guidelines Committee of the Spanish Society of Cardiology. ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J. 2011;32:1769–818 https://www.ncbi.nlm.nih.gov/pubmed/21712404[↩]
- Nordestgaard B G et al. Eur Heart J: 2013, 34:3478-3490[↩][↩][↩]
- Goldstein JL et al. Familial hypercholesterolemia. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001:2863-2913[↩]
- Marks D, Thorogood M, Neil HA et al. A review on the diagnosis, natural history, and treatment of familial hypercholesterolemia. Athersclerosis 2003;168:1-14.[↩]
- Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, Morrison AC, Brody JA, Gupta N, Nomura A, Kessler T, Duga S, Bis JC, van Duijn CM, Cupples LA, Psaty B, Rader DJ, Danesh J, Schunkert H, McPherson R, Farrall M, Watkins H, Lander E, Wilson JG, Correa A, Boerwinkle E, Merlini PA, Ardissino D, Saleheen D, Gabriel S, Kathiresan S. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–89 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5405769/[↩]
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013 Dec;34(45):3478-90a. doi: 10.1093/eurheartj/eht273. Epub 2013 Aug 15. Erratum in: Eur Heart J. 2020 Dec 14;41(47):4517. doi: 10.1093/eurheartj/ehaa166[↩][↩][↩]
- Blood Cholesterol Diagnosis. https://www.nhlbi.nih.gov/health/blood-cholesterol/diagnosis[↩][↩]
- Chait A, Subramanian S. Hypertriglyceridemia: Pathophysiology, Role of Genetics, Consequences, and Treatment. 2019 Apr 23. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK326743[↩]
- Grønbaek M. The positive and negative health effects of alcohol- and the public health implications. J Intern Med. 2009 Apr;265(4):407-20. doi: 10.1111/j.1365-2796.2009.02082.x[↩]
- Luo WS, Chen F, Ji JM, Guo ZR. Interaction of tobacco smoking and alcohol consumption with obesity on cardiovascular disease in a Chinese cohort. Coron Artery Dis. 2020 Jun;31(4):372-377. doi: 10.1097/MCA.0000000000000837[↩]
- Heidari R, Sadeghi M, Talaei M, Rabiei K, Mohammadifard N, Sarrafzadegan N. Metabolic syndrome in menopausal transition: Isfahan Healthy Heart Program, a population based study. Diabetol Metab Syndr. 2010 Oct 5;2:59. doi: 10.1186/1758-5996-2-59[↩]
- Subramanian S, Chait A. Hypertriglyceridemia secondary to obesity and diabetes. Biochim Biophys Acta. 2012 May;1821(5):819-25. doi: 10.1016/j.bbalip.2011.10.003[↩]
- De Man FH, Cabezas MC, Van Barlingen HH, Erkelens DW, de Bruin TW. Triglyceride-rich lipoproteins in non-insulin-dependent diabetes mellitus: post-prandial metabolism and relation to premature atherosclerosis. Eur J Clin Invest. 1996 Feb;26(2):89-108. doi: 10.1046/j.1365-2362.1996.114256.x[↩]
- Blüher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. 2019 May;15(5):288-298. doi: 10.1038/s41574-019-0176-8[↩]
- Karanchi H, Muppidi V, Wyne K. Hypertriglyceridemia. [Updated 2023 Aug 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459368[↩]
- Hegele RA, Ban MR, Cao H, McIntyre AD, Robinson JF, Wang J. Targeted next-generation sequencing in monogenic dyslipidemias. Curr Opin Lipidol. 2015 Apr;26(2):103-13. doi: 10.1097/MOL.0000000000000163[↩]
- Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: Current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223(2):262-268.[↩]
- Vaseghi G, Arabi S, Haghjooy-Javanmard S, Sabri M, Sadeghi M, Khosravi A, Zarfeshani S, Sarrafzadegan N. CASCADE screening and registry of familial hypercholesterolemia in Iran: Rationale and design. ARYA Atheroscler. 2019 Mar;15(2):53-58. doi: 10.22122/arya.v15i2.1899[↩]
- Melissa A. Austin, Carolyn M. Hutter, Ron L. Zimmern, Steve E. Humphries, Genetic Causes of Monogenic Heterozygous Familial Hypercholesterolemia: A HuGE Prevalence Review, American Journal of Epidemiology, Volume 160, Issue 5, 1 September 2004, Pages 407–420, https://doi.org/10.1093/aje/kwh236[↩]
- Haase A, Goldberg AC. Identification of people with heterozygous familial hypercholesterolemia. Curr Opin Lipidol. 2012 Aug;23(4):282-9. doi: 10.1097/MOL.0b013e3283556c33[↩]
- Familial hypercholesterolaemia. Report of a second WHO Consultation. Geneva, 4 September 1998. Geneva: WHO, 1999.[↩]
- Murphy SA, Cannon CP, Blazing MA, Giugliano RP, White JA, Lokhnygina Y, Reist C, Im K, Bohula EA, Isaza D, Lopez-Sendon J, Dellborg M, Kher U, Tershakovec AM, Braunwald E. Reduction in Total Cardiovascular Events With Ezetimibe/Simvastatin Post-Acute Coronary Syndrome: The IMPROVE-IT Trial. J Am Coll Cardiol. 2016 Feb 2;67(4):353-361. doi: 10.1016/j.jacc.2015.10.077[↩]
- Kastelein JJ, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, Visseren FL, Sijbrands EJ, Trip MD, Stein EA, Gaudet D, Duivenvoorden R, Veltri EP, Marais AD, de Groot E; ENHANCE Investigators. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008 Apr 3;358(14):1431-43. doi: 10.1056/NEJMoa0800742. Epub 2008 Mar 30. Erratum in: N Engl J Med. 2008 May 1;358(18):1977.[↩]
- Grundy SM, Stone NJ, Bailey AL, Beam C, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019 Jun 25;73(24):e285-e350. doi: 10.1016/j.jacc.2018.11.003. Epub 2018 Nov 10. Erratum in: J Am Coll Cardiol. 2019 Jun 25;73(24):3237-3241. doi: 10.1016/j.jacc.2019.05.013[↩]
- Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, et al. European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014 Aug 21;35(32):2146-57. doi: 10.1093/eurheartj/ehu274[↩][↩]
- Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR; FOURIER Steering Committee and Investigators. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017 May 4;376(18):1713-1722. doi: 10.1056/NEJMoa1615664[↩]
- Arrieta A, Hong JC, Khera R, Virani SS, Krumholz HM, Nasir K. Updated Cost-effectiveness Assessments of PCSK9 Inhibitors From the Perspectives of the Health System and Private Payers: Insights Derived From the FOURIER Trial. JAMA Cardiol. 2017 Dec 1;2(12):1369-1374. doi: 10.1001/jamacardio.2017.3655[↩]
- Wetterau JR, Lin MC, Jamil H. Microsomal triglyceride transfer protein. Biochim Biophys Acta. 1997 Apr 1;1345(2):136-50. doi: 10.1016/s0005-2760(96)00168-3[↩]
- Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK, Gaudet D, Stefanutti C, Vigna GB, Du Plessis AM, Propert KJ, Sasiela WJ, Bloedon LT, Rader DJ; Phase 3 HoFH Lomitapide Study investigators. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013 Jan 5;381(9860):40-6. doi: 10.1016/S0140-6736(12)61731-0[↩]
- Duell PB, Santos RD, Kirwan BA, Witztum JL, Tsimikas S, Kastelein JJP. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol. 2016 Jul-Aug;10(4):1011-1021. doi: 10.1016/j.jacl.2016.04.013[↩]
- Mach F, Baigent C, Catapano AL, Koskinas KC, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS), European Heart Journal, Volume 41, Issue 1, 1 January 2020, Pages 111–188, https://doi.org/10.1093/eurheartj/ehz455[↩]
- Raal FJ, Hovingh GK, Catapano AL. Familial hypercholesterolemia treatments: Guidelines and new therapies. Atherosclerosis. 2018 Oct;277:483-492. doi: 10.1016/j.atherosclerosis.2018.06.859[↩]
- Gidding SS, Champagne MA, de Ferranti SD, Defesche J, Ito MK, Knowles JW, McCrindle B, Raal F, Rader D, Santos RD, Lopes-Virella M, Watts GF, Wierzbicki AS; American Heart Association Atherosclerosis, Hypertension, and Obesity in Young Committee of Council on Cardiovascular Disease in Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and Council on Lifestyle and Cardiometabolic Health. The Agenda for Familial Hypercholesterolemia: A Scientific Statement From the American Heart Association. Circulation. 2015 Dec 1;132(22):2167-92. doi: 10.1161/CIR.0000000000000297. Epub 2015 Oct 28. Erratum in: Circulation. 2015 Dec 22;132(25):e397. doi: 10.1161/CIR.0000000000000349[↩]
- Buchwald H, Campos CT. Partial ileal bypass in the therapy of familial hypercholesterolemia. The POSCH Group. Beitr Infusionsther. 1988;23:47-60.[↩]
- Austin MA, Hutter CM, Zimmern RL, Humphries SE. Familial hypercholesterolemia and coronary heart disease: a HuGE association review. Am J Epidemiol. 2004 Sep 1;160(5):421-9. doi: 10.1093/aje/kwh237[↩]
- Nanchen D, Gencer B, Muller O, Auer R, Aghlmandi S, Heg D, Klingenberg R, Räber L, Carballo D, Carballo S, Matter CM, Lüscher TF, Windecker S, Mach F, Rodondi N. Prognosis of Patients With Familial Hypercholesterolemia After Acute Coronary Syndromes. Circulation. 2016 Sep 6;134(10):698-709. doi: 10.1161/CIRCULATIONAHA.116.023007[↩]
- Stone NJ, Levy RI, Fredrickson DS, Verter J. Coronary artery disease in 116 kindred with familial type II hyperlipoproteinemia. Circulation. 1974 Mar;49(3):476-88. doi: 10.1161/01.cir.49.3.476[↩]





{kind=link}