Contents
- What is fibromatosis
- Fibromatosis classification
- Juvenile fibromatosis
- Adult fibromatosis
- Plantar fibromatosis
- Desmoid fibromatosis
- What are the features of fibromatosis?
- What causes fibromatosis?
- Fibromatosis treatment
What is fibromatosis
Fibromatosis refers to a group of benign (non-cancerous) conditions characterized by overgrowths of skin and fibrous connective tissue tumors called fibromas 1. Fibromatosis is a histologically benign growth of fibroblastic and myofibroblastic cells with a potential to recur and invade local organs 2. Fibromatosis or fibrous tissue tumors have features in between fibrosarcoma (a rare malignant tumor or cancer that develops in fibrous connective tissue) and benign tumors such as fibromas 3. Fibromas are usually benign (non-cancerous) 4. There appear to be many different ways to classify fibromatosis. One classification system used is based on age (i.e., juvenile vs adult fibromatoses) and localization (i.e., superficial vs deep fibromatoses) 4. Fibromatosis can be classified by a person’s age or by the location of the fibromas (e.g., in your arms, legs, abdomen, head, and neck). Fibromatosis are also called desmoid tumors also known as aggressive fibromatosis or desmoid-type fibromatosis, as well as by the more scientific name musculoaponeurotic fibromatosis 3. Whilst most fibromatoses are benign tumors and do not metastasize (spread to other parts of the body), the desmoid tumors although they do not metastasize like malignant cancers can be locally aggressive. They can grow quickly into large tumors that can obstruct vital structures such as major blood vessels, nerves and organs 4.
Types of fibromatosis:
- Desmoid-type fibromatosis also known as aggressive fibromatosis or desmoid tumor, this type of fibromatosis is usually benign but can recur and spread to nearby tissue.
- Superficial fibromatosis: This type of fibromatosis usually remains small and doesn’t tend to recur.
- Deep fibromatosis: This type of fibromatosis can be locally invasive.
Fibromatosis tend to grow slowly but often steadily. They rarely, if ever, spread to distant parts of the body (metastasize), but they can cause problems by growing into nearby tissues 3. Fibromatosis can sometimes even be fatal. Some doctors consider them a type of low-grade fibrosarcoma, but others believe they are a unique type of fibrous tissue tumor. Certain hormones, like estrogen, make some desmoid tumors grow. Anti-estrogen drugs are sometimes useful in treating desmoid tumors that cannot be removed completely by surgery.
The cause of fibromatosis is often unknown, and treatment depends on the individual disease 5.
Fibromatosis treatment options:
- Surgical excision. The main treatment for fibromatosis is surgical resection with a wide margin.
- Radiotherapy or radiation therapy. Radiotherapy or radiation therapy can be used to reduce the chance of local recurrence after surgery, or to treat inoperable tumors.
- Systemic therapy. Tamoxifen (a selective estrogen receptor modulator [SERM] is a medicine that blocks the effects of estrogen hormone that is used to prevent breast cancer) and non-steroidal anti-inflammatory agents (NSAIDs) are sometimes used to treat lesions that don’t respond to surgery or radiotherapy.
Fibromatosis classification
There appear to be many different ways to classify fibromatosis. One classification system used is based on age (i.e., juvenile vs adult fibromatoses) and localization (i.e., superficial vs deep fibromatoses) 4. Fibromatosis can be classified by a person’s age or by the location of the fibromas (e.g., in your arms, legs, abdomen, head, and neck).
Table 1. Fibromatosis classification
| Juvenile | Adult |
|---|---|
| Congenital generalised fibromatosis (infantile myofibromatosis) | Superficial (fascial) fibromatoses |
| Aponeurotic fibroma * Infantile digital fibromatosis | Palmar (Dupuytren contracture) and plantar (Ledderhose disease) fibromatosis * Pachydermodactyly |
| Aggressive infantile fibromatosis | Penile fibromatosis (Peyronie disease) – Knuckle pads |
| Fibromatosis colli | Dermatofibroma |
| Dermatofibrosis lenticularis (Buschke-Ollendorf syndrome) | Nodular fasciitis |
| Elastofibroma | |
| Fibrous papule of the face | |
| Deep (musculoaponeurotic) fibromatoses | |
| Desmoid tumours (aggressive fibromatoses) | |
| Extra-abdominal fibromatosis | |
| Abdominal fibromatosis | |
| Intra-abdominal fibromatosis (eg, pelvic fibromatosis) |
Juvenile fibromatosis
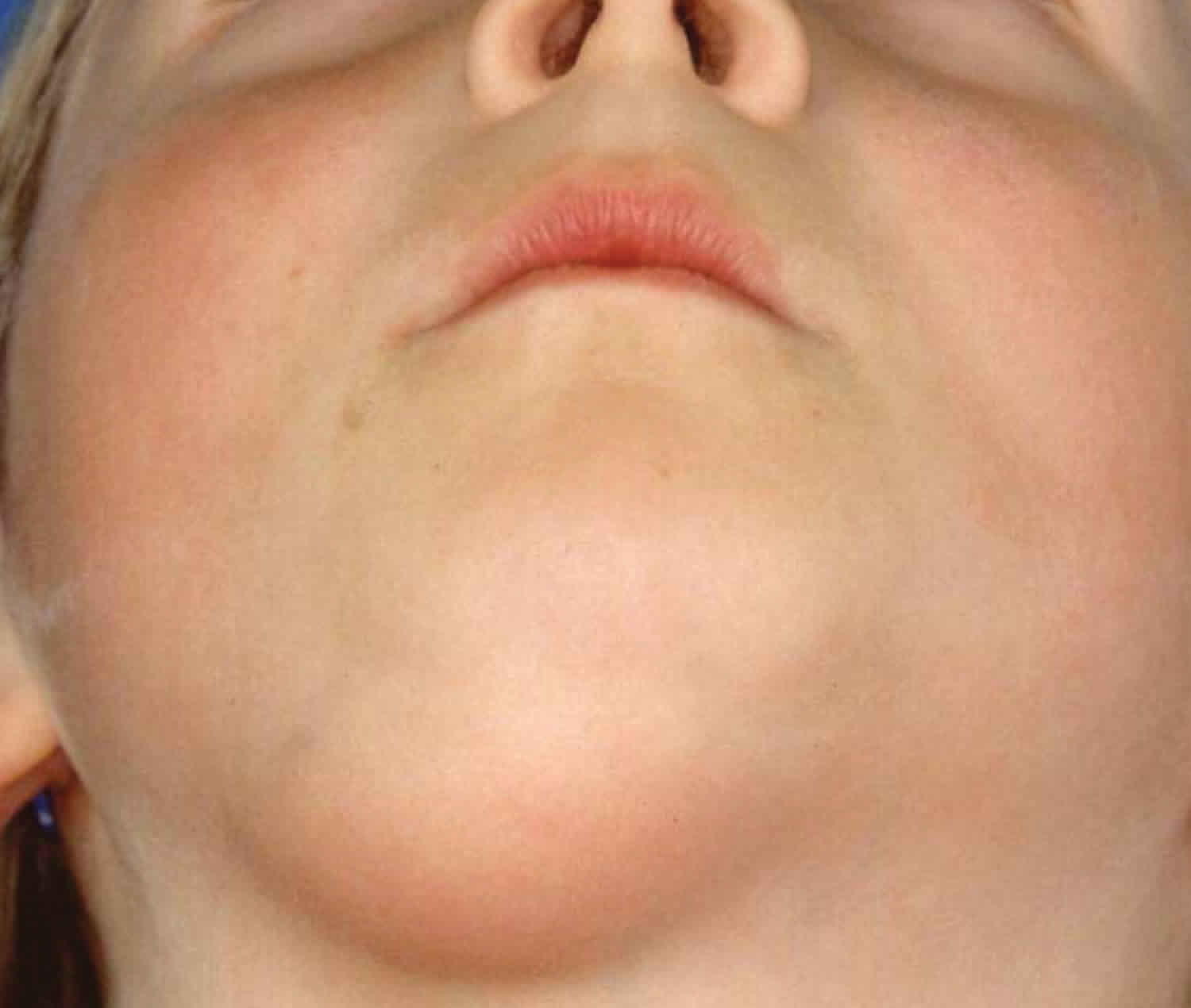
- Congenital generalized fibromatosis (infantile myofibromatosis): Congenital fibromatosis (infantile myofibromatosis) is characterized by single or multiple benign tumours that appear to be derived from connective tissue and smooth muscle cells. These tumours may involve the skin and underlying tissues, bones, and/or certain internal organs. They are present at birth or develop within the first few weeks of life. Lesions usually resolve spontaneously, however severe or widespread involvement of internal organs may cause potentially life-threatening complications.
- Aponeurotic fibroma
- Infantile digital fibromatosis: Infantile digital fibromatosis (or fibroma) presents as single or multiple gelatinous or firm pinkish nodules on the fingers or toes of an infant. Similar lesions are occasionally diagnosed elsewhere on the hands, feet, arms or elsewhere on the body. In one-third of cases, they are present at birth. They are rare and are seen in both males and females. Although infantile digital fibromatosis may grow to a size of 2cm, they are harmless and do not usually cause any symptoms unless they rub on the neighboring toe or footwear. The cause of infantile digital fibromatosis is unknown.
- Aggressive infantile fibromatosis
- Fibromatosis colli
- Dermatofibrosis lenticularis (Buschke-Ollendorf syndrome)
Adult fibromatosis
Superficial (fascial) fibromatoses
- Palmar (Dupuytren contracture) and plantar (Ledderhose disease) fibromatosis
- Penile fibromatosis (Peyronie disease)
- Knuckle pads
- Dermatofibroma
- Nodular fasciitis
- Elastofibroma
- Fibrous papule of the face
Deep (musculoaponeurotic) fibromatoses
- Desmoid tumors (aggressive fibromatoses)
- Extra-abdominal fibromatosis
- Abdominal fibromatosis
- Intra-abdominal fibromatosis (eg, pelvic fibromatosis)
Plantar fibromatosis
The term plantar fibromatosis represents not a single entity, but is used for different group of conditions with the common characteristics of plantar location and histologic features of mature collagen and fibroblasts with no malignant cytologic features 6, 7, 8:
- A relatively common plantar equivalent of Dupuytren palmar contracture most commonly termed Ledderhose disease (named after George Ledderhose, a German physician, initially described the disorder in 1897 after observing 50 patients with painful sole lesions), but also referred to as Morbus Ledderhose 9, 10, 11. Ledderhose’s disease occurs in up to 14.85% of patients with Dupuytren’s contracture of the hand 12. Ledderhose disease or Morbus Ledderhose is an uncommon plantar superficial fibromatosis that the deep fibromatosis (e.g, abdominal, extra-abdominal, and visceral fibromatosis) and generally has a less aggressive and recurrent tendency;
- An extremely rare, benign cerebriform mesodermal hamartomatous proliferation that in a plantar location, appears to be a clinicopathologic marker of Proteus syndrome.
- Juvenile aponeurotic fibroma and aggressive infantile fibromatosis can also be considered to be in the plantar fibromatosis group when lesions are present on the sole of the foot in children.
The plantar fascia is a broad fibrous aponeurosis that originates from the medial and anterior aspects of the calcaneus, divides into five digital slips at the metatarsophalangeal joints, and inserts distally into the periosteum at the base of the proximal phalanges (Figure 1) 13, 14. The plantar fascia is composed of three separate bands of dense connective tissue: central, medial, and lateral 15.
The main function of the plantar fascia is to maintain the longitudinal plantar arch of your foot 16. This is accomplished in part due to the plantar fascia’s great tensile strength, especially with weight bearing. When you dorsiflex your toes, the plantar fascia tightens, the distance between calcaneus and metatarsals is decreased, and the medial longitudinal arch is elevated. This dynamic mechanism has been described as the windlass mechanism, reflecting its similarity to tightening a cable in an efficient, predictable manner. This series of events is critical for maintenance of the gait cycle, where even minor arch collapse can cause great inefficiency in walking 13
A plantar fibromatosis is a benign fibrous knot (nodule) that grows on in the arch of your foot (the superficial plantar aponeurosis) on the bottom of your foot and usually appears in the second through sixth decade of life 17, 11, 18. However, plantar fibromatosis in the heel of children less than 16 years old to as young as 9 months have been published 19.
Plantar fibromatosis is embedded within the plantar fascia, a band of tissue that extends from the heel to the toes on the bottom of the foot. Plantar fibromatosis is most often present on the medial and central bands 20, 21. Approximately 25% of cases affect both feet with males being more affected than females 22, 6.
Plantar fibromatosis is rare, affecting less than 200,000 people in the United States 6. Plantar fibromatosis symptoms consist of a painful mass on the bottom of the foot, roughly in the middle of the arch or instep, between the heel pad and the forefoot pad. The mass will cause a soft convexity in the contour of the bottom of the foot that may be painful with pressure or shoewear. A plantar fibroma can develop in one or both feet, is benign (nonmalignant) and usually will not go away or get smaller without treatment. It is usually slow growing and measures less than an inch in size. More invasive, rapid-growing and multi-plantar fibromas are considered plantar fibromatosis. Both of them are benign tumors made up of cells found in ligaments. Definitive causes for this condition have not been clearly identified.
The most common symptom of a plantar fibroma is pain on the bottom of your foot, usually in the arch. You’ll likely notice this pain for the first time when wearing shoes that put pressure on the plantar fibroma under your skin. Depending on how big it is, a plantar fibroma can cause pressure on your foot. It might feel like there’s a stone in your shoe, but when you try to shake it out, there’s nothing there. You might be able to see the plantar fibroma. It might look like there’s a tiny marble — less than an inch across — embedded in your skin. The skin on your foot’s arch will curve out around it, or slightly bulge in a way that’s unusual for the shape of your foot.
Nonsurgical treatment may help relieve the pain of a plantar fibroma, although it will not make the mass disappear. The foot and ankle surgeon may select one or more of the following nonsurgical options 23:
- Over-the-counter medicines to reduce pain. Over-the-counter non-steroidal anti-inflammatory drugs (NSAIDs) like aspirin or ibuprofen are typically all you’ll need to reduce the pain caused by a plantar fibroma. Non-steroidal anti-inflammatory drugs (NSAIDs) will also reduce inflammation around the fibroma, which could reduce the pressure it puts on your foot. You can take them as needed to reduce pain, but talk to your doctor before starting, stopping or changing any regular use of medications. You shouldn’t take NSAIDs for longer than 10 days in a row without your doctor’s approval.
- Verapamil (a cream you put on the bottom of your foot). Your doctor might prescribe verapamil, a cream you apply to the bottom of your foot. Verapamil is usually used to manage blood pressure, but a topical verapamil cream can reduce inflammation and shrink the fibroma.
- Stretching. Your doctor might recommend you stretch your foot, ankle and calf. This can relieve tension and pressure on your plantar fascia and lessen the severity of your plantar fibroma symptoms.
- Steroid injections. Injecting corticosteroid medication into the mass may help shrink it and thereby relieve the pain that occurs when walking. This reduction may only be temporary and the fibroma could slowly return to its original size.
- Orthotic devices (inserts for your shoes). If the fibroma is stable, meaning it is not changing in size, custom orthotic devices (shoe inserts) may relieve the pain by distributing the patient’s weight away from the fibroma. You can buy generic, over-the-counter orthotics or they can be custom-made for your feet. You may also need to wear different, more supportive shoes.
- Physical therapy. The pain is sometimes treated through physical therapy methods that deliver anti-inflammatory medication into the fibroma without the need for injection.
If the mass increases in size or pain, you should see your foot surgeon for further evaluation.
Surgical treatment to remove the fibroma is considered if the patient continues to experience pain following nonsurgical approaches. Surgical removal of a plantar fibroma may result in a flattening of the arch or development of hammer toes. Orthotic devices may be prescribed to provide support to the foot. Due to the high incidence of recurrence with this condition, continued follow-up with the foot and ankle surgeon is recommended.
Figure 1. Plantar fascia anatomy
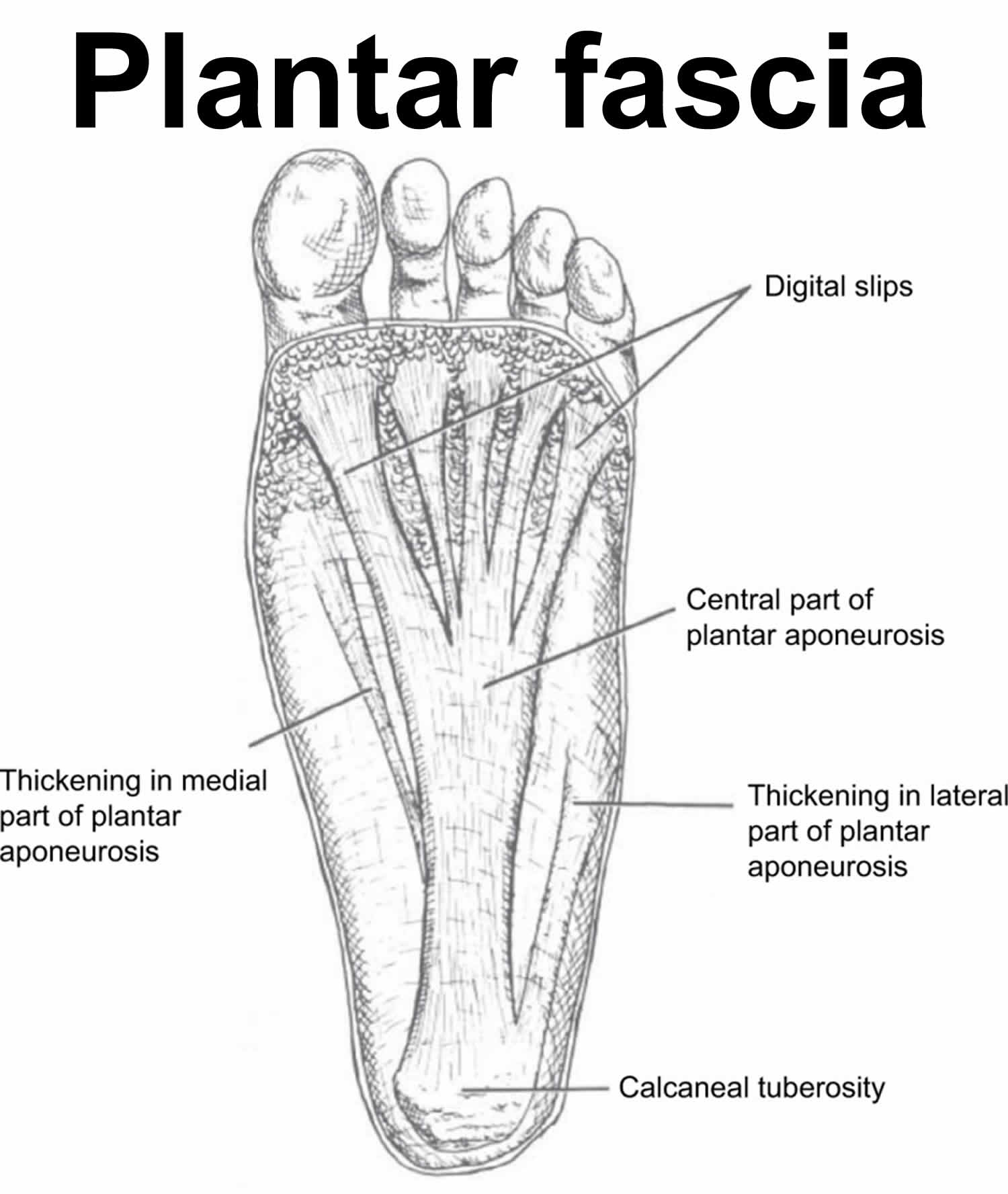
Figure 2. Plantar fibromatosis
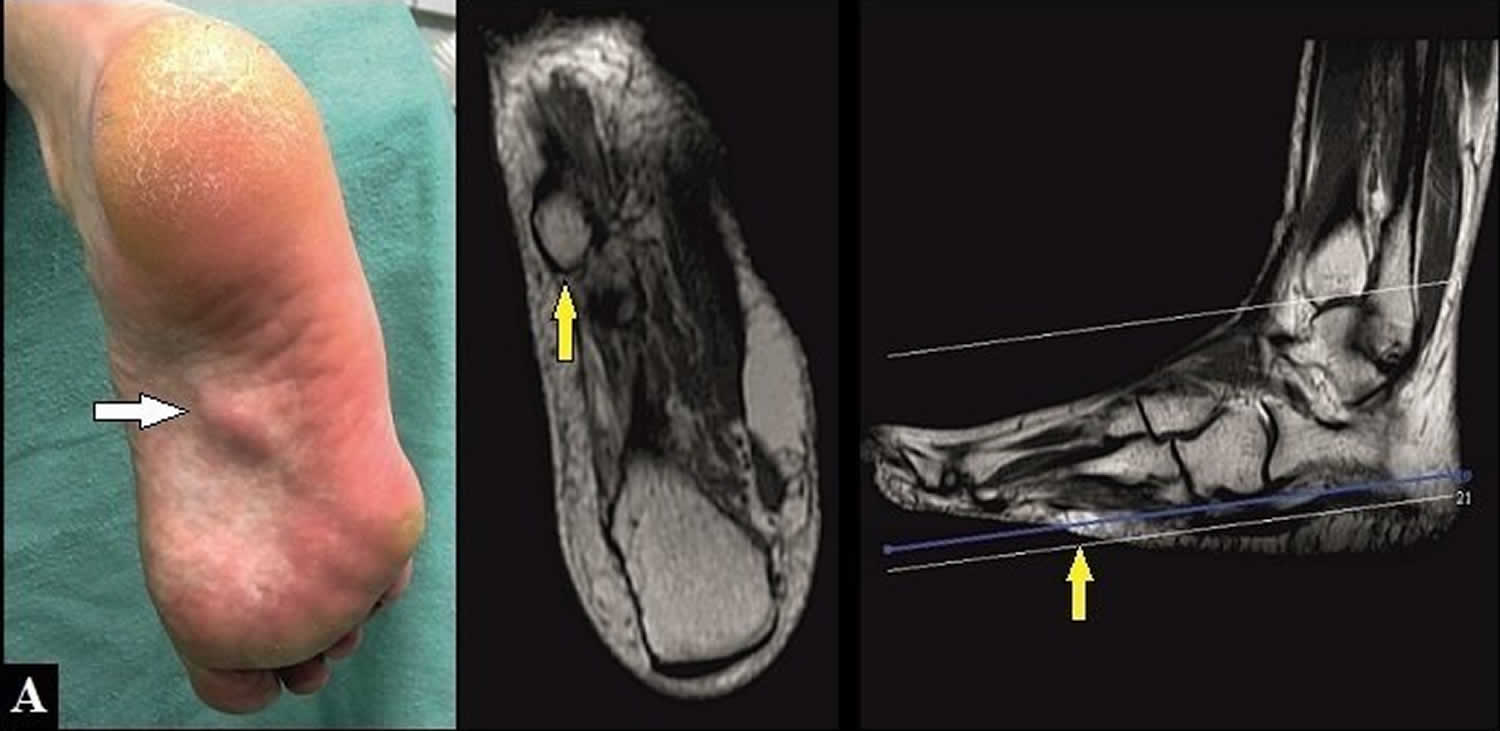
Footnotes: (A) 54-year-old female with plantar fibromatosis in her right foot. Preoperative clinical findings and magnetic resonance imaging (MRI), the nodule is partially stuck to the skin (arrows).
[Source 24 ]Figure 3. Plantar fibroma MRI
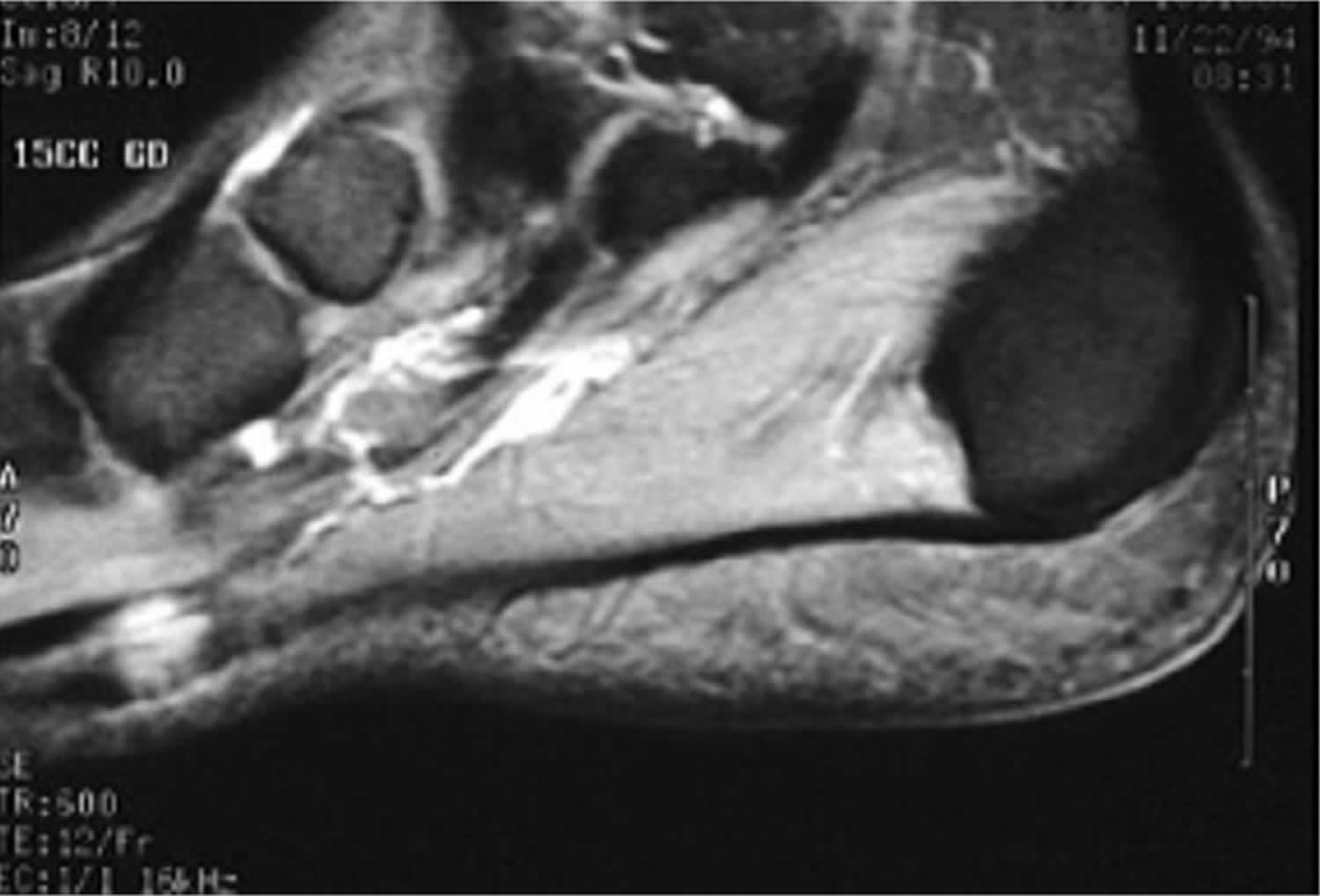
Footnotes: Sagittal T2 MRI demonstrating a plantar fascia fibroma. The fibroma has low-to-intermediate signal relative to muscle.
[Source 22 ]Figure 4. Plantar fibroma ultrasound
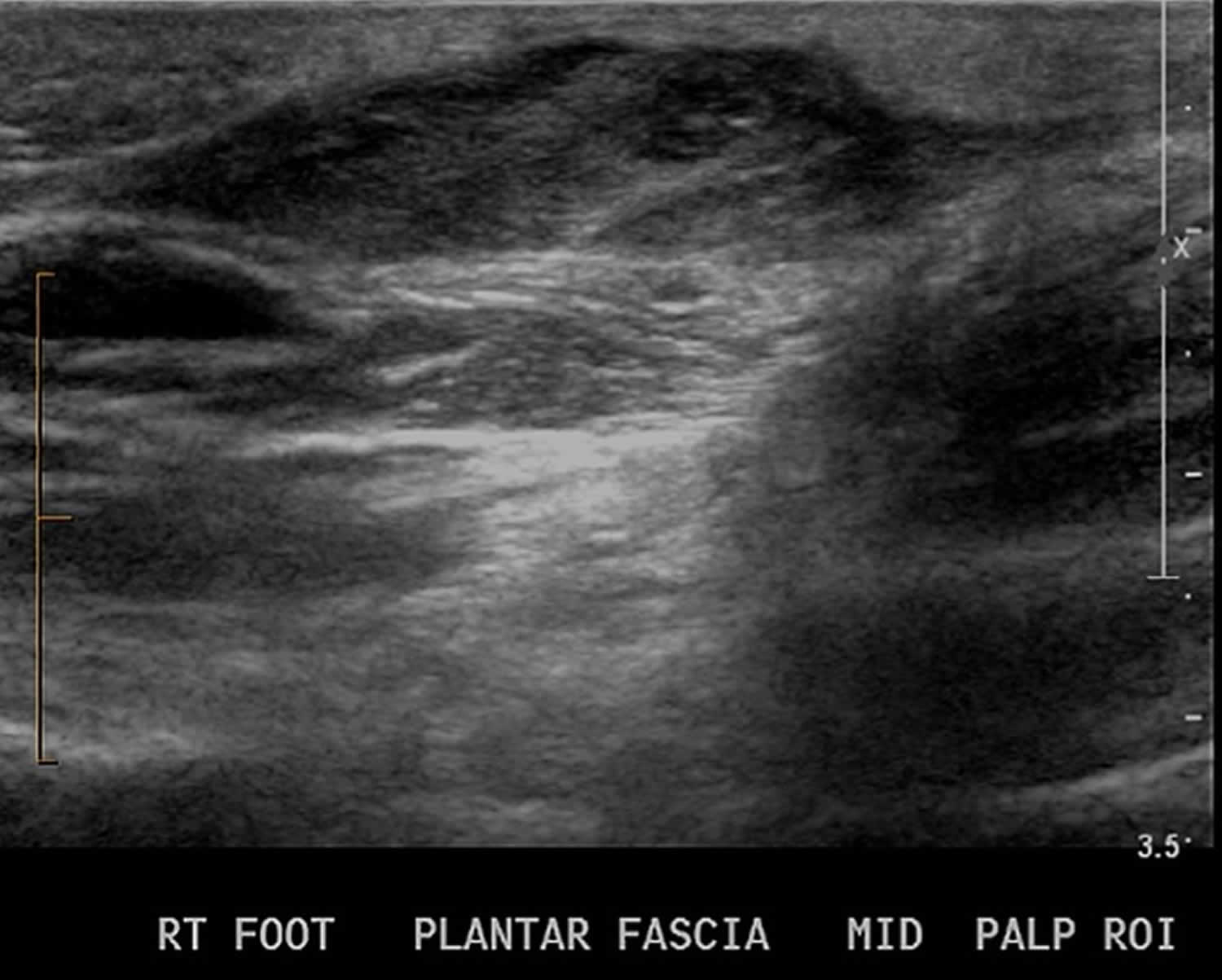
Footnote: RT= right
[Source 22 ]See your doctor as soon as you notice any new growths or changes in the shape of your foot, or if you’re experiencing new pain. Never assume any mass on your foot or anywhere else on your body is harmless, especially if you’ve never noticed it before.
Your doctor will help you understand if the growth on the arch of your foot is a plantar fibroma or something more serious. No matter what’s causing it, anything that affects your ability to walk needs to be examined.
Plantar fibroma vs. plantar fasciitis
Both plantar fibromas and plantar fasciitis affect your plantar fascia. A plantar fibroma is a rare benign (non-cancerous) growth on your plantar fascia, the rubber band-like fibrous connective tissue (ligament) that stretches from your heel to your toes (see Figure 1). Plantar fibromas are small — usually less than an inch and grow on the arch of your foot. You might not even notice one at first, but eventually a plantar fibroma can cause foot pain, especially when you’re wearing shoes.
Plantar fasciitis is a painful inflammation of the plantar fascia, the tissue that runs from the heel to the ball of the foot. Plantar fasciitis is a common cause of heel pain, especially when taking your first steps in the morning or after resting
Even though both conditions cause pain on the arch of your foot, the reason for your discomfort is different. No matter what’s causing it, see your doctor or foot specialist about any new or unusual foot pain.
What causes plantar fibroma?
The cause is unknown but thought to have a genetic component 25, 22, 26, 27, 6, 7. A possible genetic predisposition has been suggested in some cases as 2 genetic variants were found in a genome wide association study where one indel (chr5:118704153:D) and one single nucleotide polymorphism (rs62051384) were detected 28. Individuals with these variants may have an increased risk for plantar fibromatosis 28.
Trauma to the foot does not seem to be a factor. Owing to the high concurrence rate with Dupuytren contracture, there is suspicion that a shared defect in wound repair could be to blame, but no definitive caused has been identified to date 29, 30.
Who gets plantar fibromas?
Plantar fibromas can appear in anyone, and they have no confirmed cause, but you might be more likely to develop one if you are:
- Older than 40: Older adults — especially those between 40 and 60 — seem more likely to develop plantar fibromas than younger people.
- A man: Men are twice as likely to develop plantar fibromatosis than women.
- Of European descent: People of European descent are more prone to plantar fibromas than other ethnicities 31.
- Share a genetic predisposition: Studies suggest the tendency to develop plantar fibromas may be genetically inherited, meaning you might be more likely to if someone in your immediate family gets them.
You may also be at increased risk of developing plantar fibromas if you have other health conditions, including 32, 33:
- Diabetes.
- Epilepsy with long-term phenobarbital use for epilepsy
- Alcohol use disorder.
- Alcoholic liver disease.
- Smoking.
Penile fibromatosis or Peyronie’s disease has also been associated with plantar fibroma 11.
Plantar fibroma prevention
Because plantar fibromas develop at random, there aren’t many identifiable risk factors for plantar fibromas. And there’s nothing you can do to prevent a plantar fibroma. However, wearing supportive shoes, exercising regularly and stretching your plantar fascia can help alleviate symptoms. The better your overall foot health is, the less a plantar fibroma will impact your day-to-day routine.
Plantar fibromatosis signs and symptoms
The characteristic sign of a plantar fibroma is a noticeable lump in the arch of your foot that feels firm to the touch. This mass can remain the same size or get larger over time or additional fibromas may develop.
People who have a plantar fibroma may or may not have pain. When pain occurs, it is often caused by shoes pushing against the lump in the arch, although it can also arise when walking or standing barefoot.
Even though plantar fibromas themselves are not dangerous, you should talk to your doctor as soon as you notice any new growths on your foot or changes to its shape. Your doctor will rule out other, more serious issues with a physical exam and imaging tests.
Plantar fibromatosis stages
Plantar fibromatosis development advances through 3 phases 6:
- Proliferative phase: Increased fibroblast activity and cellular proliferation (cell number increases) is observed.
- Active or involution phase where nodule formation occurs: Fibroblast maturation, myofibroblast differentiation, and increased collagen production occur.
- Residual phase: Residual phase consisting of collagen deposition, scar formation and tissue contracture in the plantar foot.
The factors stimulating the hyperactive proliferating fibroblasts involved in the disease process are unknown. However, an increased release of growth factors, such as insulin-like growth factor-1, fibroblast growth factor, platelet-derived growth factor, and transforming growth factor-β, has been implicated. Increased interleukin-1-α and β have also been reported 6.
Sammarco and Mangone severity classification
Plantar fibromatosis severity may be evaluated using the following Sammarco and Mangone grading scheme 6:
- Grade 1: has a focal lesion but no extension and skin or muscle involvement.
- Grade 2: involves multiple areas and can extend distally or proximally; has no skin or muscle encroachment.
- Grade 3: involves multiple areas and can extend distally or proximally; has skin or muscle involvement.
- Grade 4: encompasses multiple areas and can extend distally or proximally; involves both skin and muscle.
Plantar fibromatosis diagnosis
There are a few conditions that can cause soft-tissue masses in the foot, including cysts, swollen tendons or tendon ruptures, nerve tumors (neurilemomas) or fat tumors. Foreign body reactions from previous penetrating trauma can also cause a mass in the bottom of the foot, as can an infection. A more serious synovial cell sarcoma, a cancer, will usually show calcification on X-ray and a more worrisome appearance on MRI. Clinical exam, X-ray and sometimes an MRI may be needed for diagnosis.
To diagnose a plantar fibroma, the foot and ankle surgeon will examine the foot and press on the affected area. Sometimes this can produce pain that extends down to the toes. An MRI or biopsy may be performed to further evaluate the lump and aid in diagnosis.
Plantar fibroma tests
If you need additional tests, your foot surgeon will explain what they’re checking for. The most common tests you’ll need include:
- Magnetic resonance imaging (MRI): An MRI will confirm the mass is a fibroma and not any other type of benign growth like a cyst. On MRI, a plantar fibroma may appear as a focal, round area or lobulated, disorganized tissue in the plantar fascia with low signal intensity or a signal similar to muscle 34. Nodules appear well-defined, with signal intensity low on T1 images and low to moderate on T2 images. MRI with contrast, such as gadolinium, may enhance the nodules’ intensity. MRI may be obtained for excision planning and identifying the plantar contractures’ extent.
- X-ray or bone scan: These tests will show any issues or changes in the bones in your foot around the growth. Your doctor will check for more serious conditions like a cancerous sarcoma.
- Ultrasound: A musculoskeletal ultrasound is like an X-ray for soft tissue. Ultrasound is a time- and cost-efficient modality, providing your doctor with a quick office diagnostic tool during your appointment. Your doctor can use it to take pictures of the area around the growth. Ultrasound can show the nodule depth, size, and number. Nodules typically appear isoechoic and heterogenous on ultrasound. Hyperechoic septa may also be present. Vascular flow is not usually appreciable when using Doppler ultrasound 35.
- Biopsy. A biopsy may be used to rule out cancer 36.
Plantar fibroma histopathology
The gross and microscopic characteristics of plantar fibromatosis are similar to its palmar fibromatosis 25. Palmar fibromatosis presents macroscopically as small nodules or nodular masses associated with aponeuroses and subcutaneous fat with a gray to yellow to white cut surface, the exact nature of the color depending on the amount of collagen content 25. These are usually hypocellular with clusters of bland spindle cells with oval to elongated nuclei. Nuclear atypia and mitotic activity are not present 25. Evans described the presence of a variable number of multinucleated giant cells found during the proliferative phase 37. Proliferating fibroblasts produce the pathologic plantar fascia nodules of plantar fibromatosis. These fibroblasts are found in areas surrounded by less cellular, modestly collagenous fascia. The cells are the same size, randomly arranged in a matrix, and have little vascular supply. The extracellular matrix and cytoskeleton have abundant fibronectin and myofibroblasts and a predominance of type 3 collagen 38. The formation of nodules is seen during the active phase while the less cellular, more collagenous residual phase often has a prominent chronic inflammatory component and hemosiderin deposition 25.
Immunohistochemical studies are usually not necessary for diagnosis due to the characteristic histology of these lesions 25. Vimentin is uniformly reactive while muscle specific and smooth muscle actins are variable. Infrequently, desmin may show reactivity. Keratins, CD34, epithelial membrane antigen, and S-100 should all be negative 25. Beta-catenin is negative in plantar fibromatosis, but aberrant nuclear staining in palmar fibromatosis is common 39, 21.
Plantar fibromatosis treatment
Nonsurgical treatment may help relieve the pain of a plantar fibroma, although it will not make the mass disappear. The foot and ankle surgeon may select one or more of the following nonsurgical options 23:
- Over-the-counter medicines to reduce pain. Over-the-counter non-steroidal anti-inflammatory drugs (NSAIDs) like aspirin or ibuprofen are typically all you’ll need to reduce the pain caused by a plantar fibroma. Non-steroidal anti-inflammatory drugs (NSAIDs) will also reduce inflammation around the fibroma, which could reduce the pressure it puts on your foot. You can take them as needed to reduce pain, but talk to your doctor before starting, stopping or changing any regular use of medications. You shouldn’t take NSAIDs for longer than 10 days in a row without your doctor’s approval.
- Verapamil (a cream you put on the bottom of your foot). Your doctor might prescribe verapamil, a cream you apply to the bottom of your foot. Verapamil is a calcium channel blocker typically used for blood pressure management, but it also plays a vital role in the metabolism of the extracellular matrix. Verapamil inhibits collagen production and increases the activity of collagenase, which, in turn, decreases the contractile function of fibroblasts and myofibroblasts. Verapamil has also been shown to exhibit anti-inflammatory properties by altering the release of pro-inflammatory cytokines interleukin (IL)-6 and IL-8 27. Verapamil cream can reduce inflammation and shrink the fibroma, however, there is little published data assessing its efficacy 27. One study has shown that 15% transdermal verapamil cream and intralesional verapamil can decrease plaque size in Peyronie’s disease by up to 55%–85%, with the only adverse effect observed being contact dermatitis 27. Treatment recommendations for Peyronie’s disease include using the transdermal cream twice a day for 9 months, or one intralesional injection every other week 40. Based on the similar pathophysiology of Peyronie’s disease and plantar fibroma, it is reasonable to consider verapamil as an initial primary or add-on treatment in conservative management of plantar fibroma.
- Stretching. Your doctor might recommend you stretch your foot, ankle and calf. This can relieve tension and pressure on your plantar fascia and lessen the severity of your plantar fibroma symptoms.
- Steroid injections. Injecting corticosteroid medication into the mass may help shrink it and thereby relieve the pain that occurs when walking. This reduction may only be temporary and the fibroma could slowly return to its original size.
- Orthotic devices (inserts for your shoes). If the fibroma is stable, meaning it is not changing in size, custom orthotic devices (shoe inserts) may relieve the pain by distributing the patient’s weight away from the fibroma. You can buy generic, over-the-counter orthotics or they can be custom-made for your feet. You may also need to wear different, more supportive shoes.
- Physical therapy. The pain is sometimes treated through physical therapy methods that deliver anti-inflammatory medication into the fibroma without the need for injection.
- Extracorporeal shock wave therapy (ESWT) is a relatively new type of treatment for many musculoskeletal disorders 41, 42. Shock waves mimic various mechanical loading conditions and cause a biochemical response in tendon fibroblasts. It is thought that extracorporeal shock wave therapy (ESWT) plays a role in tendon metabolism by stimulating the biosynthesis of the extracellular matrix in tenocytes. After treatment with extracorporeal shock wave therapy (ESWT), biochemical signals like TGF-β and insulin-like growth factor 1 become overexpressed, which suggests that tendon tissue can convert shock wave stimulation into biochemical signals 41. It is this increased production of extracellular matrix components that helps in counteracting the maturation process of myofibroblasts and leads to decreased tissue contraction 41, 43. Like many of the other conservative therapies, ESWT has been shown to be effective in Peyronie’s disease and Dupuytren’s contracture, but there is limited published data supporting its use in plantar fibroma. ESWT has not been shown to change the physical size of the nodules, but has been able to reduce pain and soften the fascia and nodules as early as 2 weeks after initiation of treatment 41, 42, 27, 44, 45. Although there is variability in the protocol for the treatment in terms of devices, energy, and frequency, focused shock waves can be considered a valid therapeutic option and an effective tool for pain relief and improved functionality 41, 42, 44.
- Tamoxifen (a selective estrogen receptor modulator [SERM] is a medicine that blocks the effects of estrogen hormone that is used to prevent breast cancer). Estrogen plays many roles in the body, including that of increasing the contractile properties of certain cell types. For this reason, antiestrogen therapy has been proposed as a treatment for plantar fibroma 27. Although there have been no in vivo studies assessing its efficacy, tamoxifen, a selective estrogen-receptor modulator (SERM), has been successfully studied in vitro 27, 44. Fibroblasts were isolated from patients with Dupuytren’s and exposed to tamoxifen for 5 days. After the treatment period, those cells showed decreased rates of contractures compared with cells not treated with an antiestrogen 27, 44. Studies have also shown that tamoxifen is effective in inhibiting the release of TGF-β, which, in turn, reduces the proliferative activity of fibroblasts 44. The decreases in both contracture rates and proliferative activity of fibroblasts show that antiestrogen therapy has promise as a conservative treatment for plantar fibroma. Another study involving Dupuytren’s contracture patients showed that 15%–20% reported nodule size regression and 25%–30% reported no further increase in nodule growth after treatment with an antiestrogen 46. Therefore, using antiestrogens like tamoxifen for patients with plantar fibroma may help to prevent the progression of the disease.
If the mass increases in size or pain, you should see your foot surgeon for further evaluation.
Surgical treatment to remove the fibroma is considered if the patient continues to experience pain following nonsurgical approaches. Surgical removal of a plantar fibroma may result in a flattening of the arch or development of hammer toes. Orthotic devices may be prescribed to provide support to the foot. Due to the high incidence of recurrence with this condition, continued follow-up with the foot and ankle surgeon is recommended.
How soon after treatment will I feel better?
A combination of NSAIDs and changing your footwear should relieve your symptoms almost right away. In general, increasing your foot’s flexibility and wearing the right kind of shoes will reduce the symptoms of a plantar fibroma and prevent additional issues from developing.
NSAIDs should reduce your pain within a few hours of taking them. Orthotics will relieve pressure, but might take longer to make a noticeable difference. Stretching is a longer-term solution that you should work into your regular routine.
Plantar fibromatosis surgery
Plantar fibroma surgery is done for symptomatic fibromas when conservative treatment fails to give adequate pain relief. It’s very rare for you to need surgery to remove (excise) a plantar fibroma, but it is an option if your symptoms don’t clear up or aren’t manageable with more conservative treatments. Talk to your foot surgeon if you feel like the pain or discomfort from a plantar fibroma is affecting your quality of life.
Your surgeon will explain how much of your foot’s tissues they’ll remove. There are a few techniques they can use for a plantar fibroma surgery:
- Local excision: Only the plantar fibroma itself is removed. Recurrence rate is 60% to 100% 6.
- Wide excision: The plantar fibroma and an area between 2 and 3 millimeters — less than one-tenth of an inch — around it is removed. Recurrence rate is up to 60% 6.
- Plantar fasciectomy: The entire plantar fascia ligament is removed. This is extremely rare, in addition to how rarely people need plantar fibroma surgery in the first place. Recurrence rate is up to 25% 6.
- Percutaneous fasciectomy (endoscopic subtotal plantar fasciectomy technique): Similar to a full plantar fasciectomy. Your doctor will use an ultrasound to guide them and remove only a portion of your plantar fascia near your heel. This modality is indicated for recalcitrant fibromas. However, the treatment is contraindicated for nodules attaching superficially to the skin, invading deep foot musculature, or involving neurovasculature 36 Compared to open excision, endoscopy is technically more challenging but results in better wound healing and minimal scarring due to the smaller incisions required.
Which type of surgery you will need depends on the severity of your symptoms, the size and exact location the plantar fibroma, and whether or not you’ve developed past plantar fibromas.
The recurrence rate following plantar fibroma surgery is low for fibromas and significantly higher for plantar fibromatosis and in revision cases. Risks of surgery include wound complications, injury to local structures such as the digital nerves, and recurrence.
Local recurrence is the most common concern in any surgical treatment. Some consider total or complete fasciectomy to be the primary plantar fibromatosis surgical treatment since it seems to have the lowest recurrence 47.
Overall, studies have demonstrated a 60% nodular recurrence rate when surgery is chosen as the therapeutic option for plantar fibroma 44, 48. This risk is increased with bilateral foot involvement, multiple nodules, and a positive family history of plantar fibroma 49. Apart from recurrence, other surgical risks include impaired wound healing, skin necrosis, painful scarring, nerve entrapment, and loss of arch height 27.
Surgical planning should pay particular attention to incisions in weight-bearing areas. Longitudinal incisions can predispose the skin to hypertrophic scarring. Medial-to-midline transverse or zigzag incisions can put the skin at risk for necrosis due to plantar arterial supply disruption 23.
Recovery from surgery depends on which type of surgery you need. It can be anywhere from a few weeks to a few months, or as long as it takes for your plantar fascia and incision to heal fully. Your doctor or surgeon will provide you with a customized recovery timeline based on your surgery.
Recovery may be hastened by elevation of the your foot and diligent control of swelling to help prevent blood clot formation and delayed wound healing. You may need to take one to two weeks off work after surgery, if you can keep your foot elevated and stay on crutches, or longer if this is not possible. Return to unrestricted activity and shoewear is in the one- to two-month range.
Recurrence is rare for fibromas but more common in multiple lesions or if invasive lesions are encountered. Adjuvant radiotherapy (radiation therapy that is used after surgery to help keep the tumor from coming back) has been proposed as a solution to fibroma recurrence. de Bree et al 50 noted that recurrence after surgical excision was rarely observed following adjuvant radiotherapy. Others have demonstrated that a recurrence rate less than 50% can be expected with wide excision and adjuvant radiotherapy 8, 27, 51. While these results are promising, the rare but significant risks of radiation, including impaired function of the foot, impaired wound healing, lymphedema, marked fibrosis, fracture of irradiated bone, and radiation-induced cancer must be balanced with the benefit of recurrence prevention 50, 52.
Potential complications include wound drainage or infection, a healed but painful wound, the return of a mass, and chronic neuritic pain, especially for an invasive lesion or in revision surgery.
Sammarco and Mangone operative staging system
Sammarco and Mangone published an operative staging system taking into account the extent of plantar fascia involvement, skin adherence, and nodule depth.
- Grade 1: has a focal lesion but no extension and skin or muscle involvement
- Grade 2: involves multiple areas and can extend distally or proximally; has no skin or muscle encroachment
- Grade 3: involves multiple areas and can extend distally or proximally; has skin or muscle involvement
- Grade 4: encompasses multiple areas and can extend distally or proximally; involves both skin and muscle
Plantar fibroma prognosis
The good news is that plantar fibromas are always benign (non-cancerous), which means even if you do develop one, it won’t ever be cancerous. Most people will not be impacted much by a plantar fibroma. Even if you have plantar fibromatosis and develop fibromas more often, the pain and discomfort are manageable at home with readily available medicines and treatments. If you develop a plantar fibroma, you’re likely to get more throughout your life.
Untreated plantar fibromatosis’ main complication is chronic pain due to continued lesion growth 6. Additional complications include wound healing problems, hypertrophic scarring, and recurrence after treatment 6.
Surgery, particularly local excision, has high recurrence rates 6. Recurrence is more frequent in patients with multiple lesions and positive family history. Besides recurrence, potential surgical complications include nerve entrapment and postoperative wound-related issues, such as dehiscence and painful scarring 6. A loss of medial longitudinal foot arch height has also been reported after subtotal fasciectomy. Plantar nerve damage can lead to numbness or neuroma formation 23.
How long does a plantar fibroma last?
There’s no known duration for a plantar fibroma. In many cases, they shrink or disappear on their own, sometimes as suddenly as they appear. If you have one, it’s much more likely to be a minor, temporary inconvenience than a major disruption.
When can I go back to work or school?
You shouldn’t need to miss any work or school if your pain and discomfort are manageable with non-steroidal anti-inflammatory drugs (NSAIDs) like aspirin or ibuprofen and other at-home treatments.
If you need surgery to remove a plantar fibroma, your surgeon will explain the procedure and everything you need to know to recover.
Plantar fibromas can be annoying and uncomfortable, but they’re not life-threatening and won’t spread. Once you’ve confirmed that you have a plantar fibroma and not any other kind of growth in your foot, you should focus on treating your symptoms.
If you’re not experiencing pain or other symptoms, you should be able to do all the activities and exercises you normally do, including running and playing sports. However, if you notice new pain or pressure after running or working out, make sure to see your doctor.
Desmoid fibromatosis
Desmoid fibromatosis also known as desmoid tumor or aggressive fibromatosis is an abnormal growth that arises from connective tissue that contains fibroblast or myofibroblast cells, which is the tissue that provides strength and flexibility to structures such as bones, ligaments, fascia and muscles 53, 54, 55, 56, 57, 58, 59, 60. The term “desmoid” is from the Greek word “desmos,” which means band or tendon, to illustrate the band- or tendon-like consistency of the tumor 57. According to World Health Organization (WHO), desmoid tumor is a “clonal fibroblastic proliferation that arises in the deep soft tissues and is characterized by infiltrative growth and a tendency toward local recurrence but an inability to metastasize”, even though it may be multifocal in the same limb or body part 61. Typically, a single tumor develops, although some people have multiple tumors. Desmoid fibromatosis can occur anywhere in your body. Desmoid fibromatosis that form in your abdominal wall are called abdominal desmoid tumors; those that arise from the tissue that connects the abdominal organs are called intra-abdominal desmoid tumors; and desmoid fibromatosis tumors found in other regions of your body are called extra-abdominal desmoid tumors. Extra-abdominal desmoid tumors occur most often in the shoulders, upper arms, and upper legs.
Desmoid tumors are fibrous, much like scar tissue 62, 63, 64. Desmoid tumors are generally not considered cancerous (malignant) because they do not spread to other parts of the body (metastasize); however, they can aggressively invade the surrounding tissue, extend along fascial planes, attach to and erode bones, and compress and engulf vessels, nerves, and other hollow organs and can be very difficult to remove surgically 65, 55, 53. Desmoid fibromatosis often recur, even after complete removal 66.
The most common symptom of aggressive fibromatosis or desmoid fibromatosis is pain. Other signs and symptoms of desmoid tumors, which are often caused by growth of the tumor into surrounding tissue, vary based on the size and location of the tumor. Intra-abdominal desmoid tumors can block the bowel, causing constipation. Extra-abdominal desmoid tumors can restrict the movement of affected joints and cause limping or difficulty moving the arms or legs.
Desmoid tumors are rare, affecting an estimated 1 to 2 per 500,000 people worldwide. In the United States, 900 to 1,500 new cases are diagnosed per year 67. In Finland, the estimated incidence of aggressive fibromatosis is less than 4 cases per 1,000,000 population per year 68. Sporadic desmoid tumors are more common than those associated with familial adenomatous polyposis.
Desmoid tumors occur frequently in people with an inherited form of colon cancer called familial adenomatous polyposis (FAP). These individuals typically develop intra-abdominal desmoid tumors in addition to abnormal growths called polyps and cancerous tumors in the colon. Desmoid tumors that are not part of an inherited condition are described as sporadic.
Generally, desmoid tumors can be classified into 2 categories based on their genetics:
- Sporadic desmoid tumors are associated with mutations in the catenin beta 1 (CTNNB1) gene which codes for beta-catenin. Sporadic desmoid tumors are more common than those associated with familial adenomatous polyposis (FAP).
- Associated with adenomatous polyposis coli (APC) gene mutations — tumors occurring as part of syndromic conditions such as in patients with a family history of desmoid tumors, known familial adenomatous polyposis (FAP) or Gardner syndrome.
Other proposed risk factors for desmoid tumor development include trauma, recent surgery, and high estrogen states.
Because of its rarity, desmoid tumor may be misdiagnosed in as many as 30%–40% of cases, resulting in inappropriate or delayed care 69, 70. In one study, the time from patient‐reported symptom onset to desmoid tumor diagnosis exceeded one year for 54% of patients 71.
Standard treatment for aggressive fibromatosis or desmoid fibromatosis ranges from active surveillance for asymptomatic lesions to surgery, radiotherapy, and systemic therapies such as nonsteroidal anti-inflammatory drugs (NSAIDs), anti-estrogens, and tyrosine kinase inhibitors (TKI) 54. Since desmoid tumors do not spread (metastasize), many approaches are nonoperative in nature 72. Many cases of desmoid tumors may remain stable or even regress spontaneously, active observation and regular follow-up are often the initial management approach 73, 74. However, the most successful primary treatment modality for desmoid tumor in patients with active symptoms (ie, volumetric progression and compression symptoms) is surgery with negative surgical margins 74. Positive margins after surgery reflect a high risk for recurrence 75. Surgery plus the administration of nonsteroidal anti-inflammatory medication (NSAIDs), hormonal therapy, targeted biologics, and cytotoxic chemotherapy is sometimes used for patients with rapidly growing tumors or where surgery would be inappropriate 76. Radiotherapy (radiation therapy) is a second-line option where surgery is contraindicated or an adjuvant (add-on) therapy in post-surgical patients with positive margins on initial resection.
Figure 5. Desmoid tumor (aggressive fibromatosis) MRI
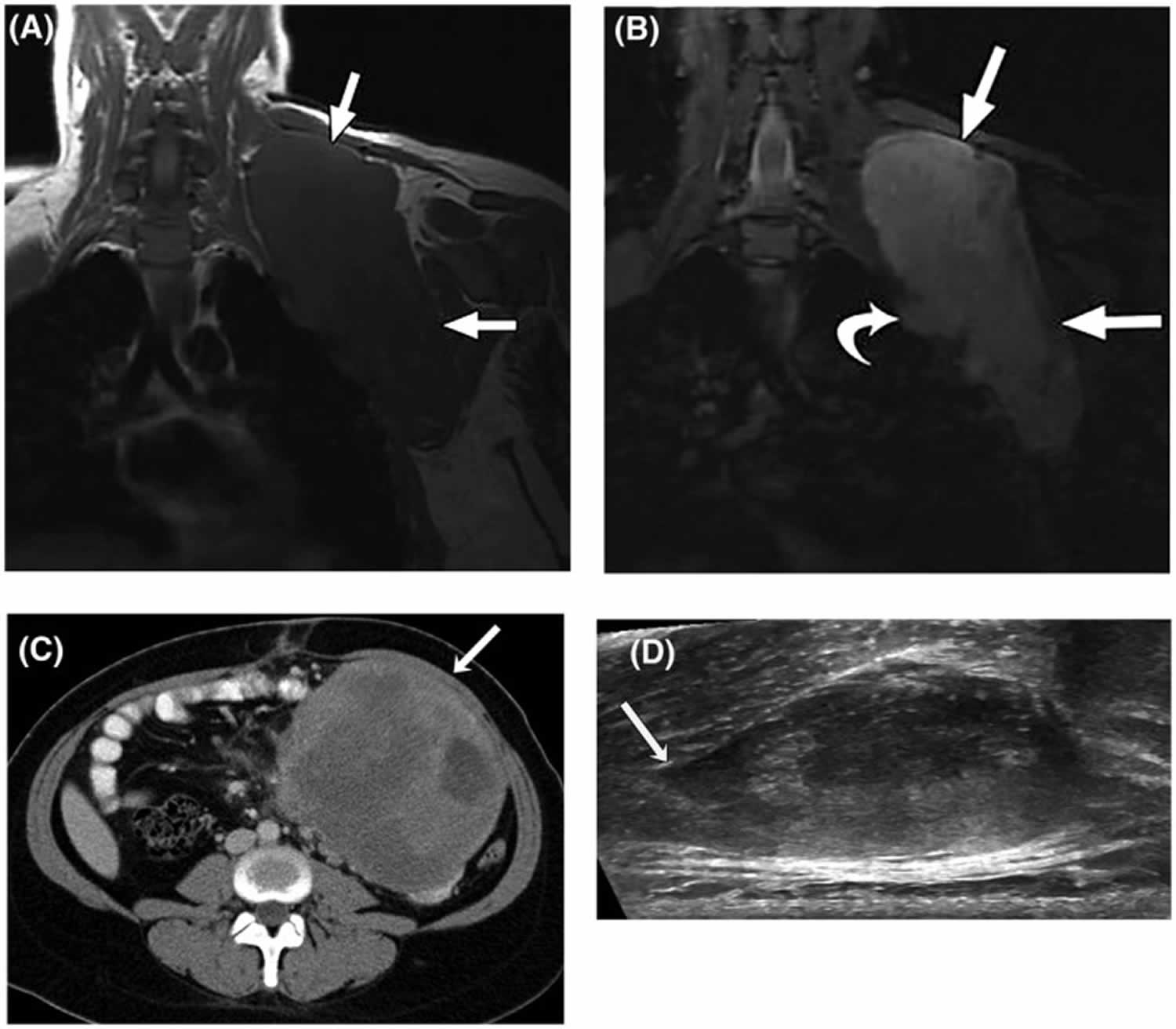
Footnotes: (A) T1‐weighted and (B) T2‐weighted fat‐suppressed magnetic resonance images (MRI) in the coronal plain of a 28‐year‐old woman with a large desmoid tumor (aggressive fibromatosis) in the shoulder region (straight arrows). The curved arrow indicates a nodular protrusion that raises concern for pleural invasion. (C) Axial, contrast‐enhanced computed tomography image from a 27‐year‐old woman with a nonresectable, solitary intra‐abdominal aggressive fibromatosis not associated with familial adenomatous polyposis. An arrow indicates a large, well defined mass adherent to the small bowel and mesenteric vessels. (D) Transverse ultrasound of a sporadic right paraspinal musculature extra‐abdominal desmoid tumor in a 26‐year‐old woman. Linear fascial extension (tail sign) is indicated by the arrow.
[Source 73 ]Figure 6. Aggressive fibromatosis arm MRI
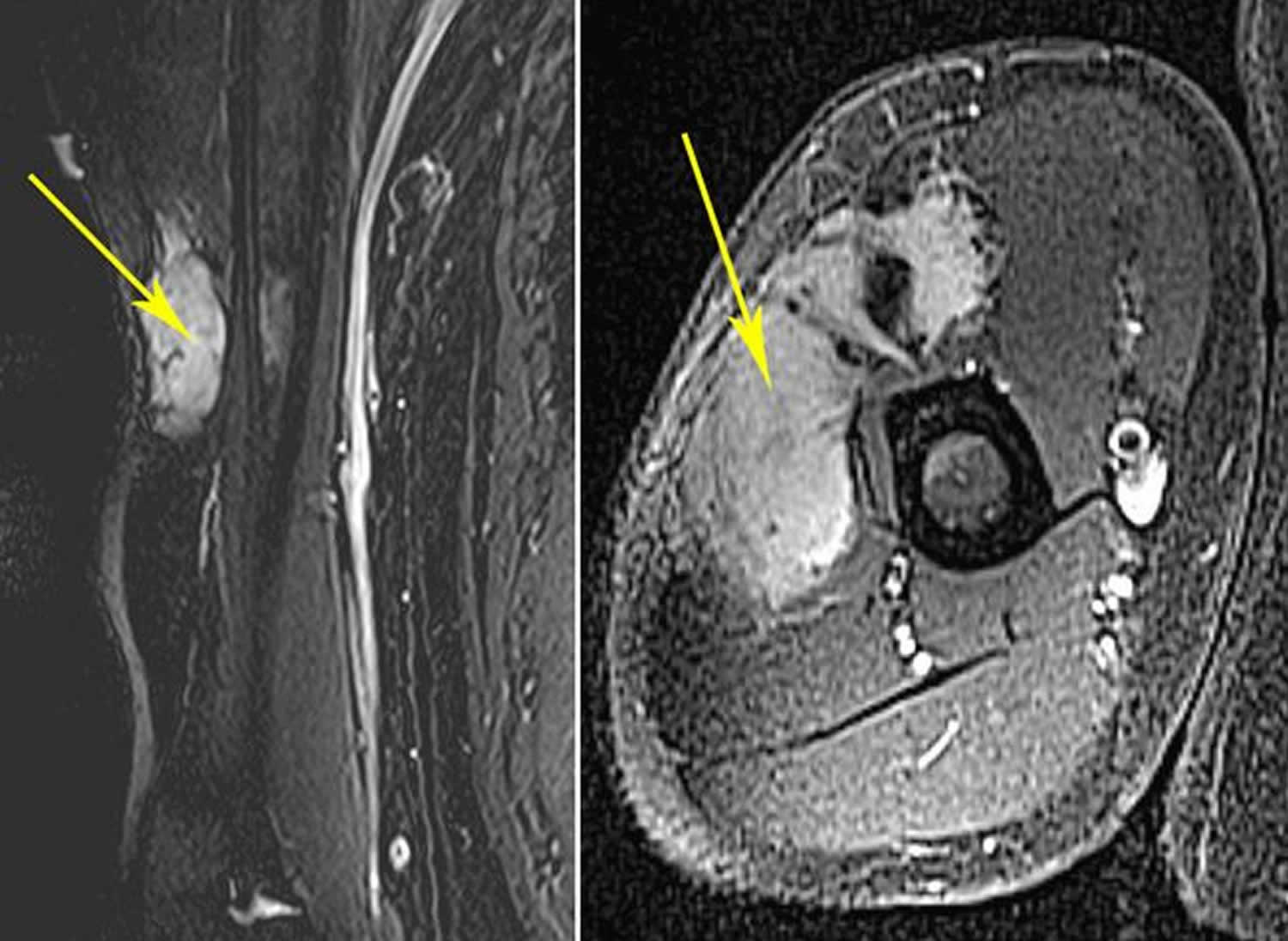
Footnotes: (Left) This MRI shows an extra-abdominal desmoid tumor in the arm. (Right) This MRI shows the same tumor in a cross-section view.
[Source 77 ]Who gets desmoid tumors?
Desmoid tumors or aggressive fibromatosis are rare, affecting an estimated 1 to 2 per 500,000 people worldwide 78. In the United States, 900 to 1,500 new cases are diagnosed per year 78. Typically, desmoid tumors affect young adults between the ages of 35 to 40 years and are more common in women.
Desmoid tumors may develop in 5 to 10% of patients with an inherited form of colon cancer called familial adenomatous polyposis (FAP) and in patients with conditions related to adenomatous polyposis coli (APC) gene mutations.
What causes desmoid fibromatosis?
The cause of desmoid fibromatosis or aggressive fibromatosis is multifactorial, although trauma, hormonal factors and genetics are thought to play a central role, including mutations in the Wnt/APC/beta-catenin pathway 79, 80, 81, 82.
Mutations in the catenin beta 1 (CTNNB1) gene or the adenomatous polyposis coli (APC) gene cause desmoid tumors 83. CTNNB1 (catenin beta 1) gene mutations particularly at codons 41A, 45F, and 45 (with p.Thr41Ala and p.Ser45Phe being the most common mutations) account for around 90 to 95 percent of sporadic desmoid tumors 54, 67, 84. Mutation 45F is associated with a high risk of recurrence 61, 85, 86. The 5-year recurrence-free survival rate has been reported to be 23% for patients with a 45F mutation, 57% for patients with a 41A mutation, and 65% for those with no mutations 53.
Conversely, hereditary cases, often associated with familial adenomatous polyposis (FAP) or Gardner syndrome as well as 10 to 15 percent of sporadic desmoid tumors, are linked to germline APC (adenomatous polyposis coli) gene mutations 84. Both the CTNNB1 and the APC genes are involved in an important cell signaling pathway that controls the growth and division (proliferation) of cells and the process by which cells mature to carry out specific functions (differentiation).
The CTNNB1 gene provides instructions for making a protein called beta-catenin 87. Beta-catenin protein is present in many types of cells and tissues, where it is primarily found at junctions that connect neighboring cells (adherens junctions) 87. Beta-catenin plays an important role in sticking cells together (cell adhesion) and in communication between cells 87.
The beta-catenin protein is also involved in cell signaling as an essential part of the Wnt signaling pathway 87. Certain proteins in Wnt signaling attach (bind) to beta-catenin, which triggers a multistep process that allows the protein to move into the cell nucleus. Once in the nucleus, beta-catenin interacts with other proteins to control the activity (expression) of particular genes. The Wnt signaling pathway promotes the growth and division (proliferation) of cells and helps determine the specialized functions a cell will have (differentiation). Wnt signaling is known to be involved in many aspects of development before birth. In adult tissues, Wnt signaling plays a role in the maintenance and renewal of stem cells, which are cells that help repair tissue damage and can give rise to other types of cells.
The adenomatous polyposis coli (APC) gene provides instructions for making the APC protein, which plays a critical role in several cellular processes 88. The APC protein acts as a tumor suppressor, which means that it keeps cells from growing and dividing too fast or in an uncontrolled way 88. The APC protein helps control how often a cell divides, how it attaches to other cells within a tissue, and whether a cell moves within or away from a tissue. The APC protein also helps ensure that the number of chromosomes in a cell is correct following cell division. The APC protein accomplishes these tasks mainly through association with other proteins, especially those that are involved in cell attachment and signaling 88.
One protein with which APC associates is beta-catenin 88. Beta-catenin helps control the activity (expression) of particular genes and promotes the growth and division (proliferation) of cells and the process by which cells mature to carry out specific functions (differentiation). Beta-catenin also helps cells attach to one another and is important for tissue formation. Association of APC protein with beta-catenin signals for beta-catenin to be broken down when it is no longer needed 88. When beta-catenin is no longer needed, the APC protein attaches (binds) to it, which signals for it to be broken down. Mutations in the APC gene that cause desmoid tumors lead to a short APC protein that is unable to interact with beta-catenin. As a result, beta-catenin is not broken down and, instead, accumulates in cells. Excess beta-catenin, whether caused by CTNNB1 or APC gene mutations, promotes uncontrolled growth and division of cells, allowing the formation of desmoid tumors.
Most desmoid tumors are sporadic and are not inherited 78. Sporadic tumors result from gene mutations that occur during a person’s lifetime, called somatic mutations. A somatic mutation in one copy of the gene is sufficient to cause the disorder. Somatic mutations (changes in DNA that occur sporadically or randomly) in either the CTNNB1 or the APC gene can cause sporadic desmoid tumors 78.
A history of trauma often surgical to the site of the desmoid tumor is elicited in 1 in 4 cases 89. Desmoid tumors most commonly arise from the rectus abdominis muscle in postpartum women and in scars due to abdominal surgery 90. Implant-associated breast desmoid tumors may also occur 91. An endocrine cause is suggested. Desmoid tumors most commonly appear in young women during or after pregnancy. The tumors regress during menopause 92 and after tamoxifen (a selective estrogen-receptor modulator [SERM]) treatment to reduce the risk of developing breast cancer 93. Desmoid tumors may regress after exposure to oral contraceptives 94.
An inherited mutation in one copy of the APC gene causes familial adenomatous polyposis (FAP) and predisposes affected individuals to develop desmoid tumors. The desmoid tumors occur when a somatic mutation occurs in the second copy of the APC gene. In these cases, the condition is sometimes called hereditary desmoid disease 78.
Desmoid fibromatosis signs and symptoms
Desmoid tumors can develop at virtually any site in your body. Desmoid tumors most commonly present in the abdominal wall; however, they can also occur in your head, neck, arms and legs, and pelvis. Retroperitoneal desmoid fibromatosis are more common in familial polyposis coli and Gardner syndrome after abdominal surgery than in other conditions 95. Desmoid tumors can have a wide range of clinical symptoms or no symptoms at all. Many desmoid tumors are accidentally picked up on a scan or a routine physical exam done for other medical reasons. Many people have no symptoms at the time of diagnosis or even after many years. Some people feel a range of symptoms that range from slight to severe pain, decrease in their movement or range of motion, swelling of the area affected by the desmoid tumor, loss of sleep, anxiety and many other symptoms.
Desmoids deep inside the abdomen or pelvis can also be entirely without symptoms or they can cause bloating, severe pain, rupture of intestines, compression of the kidneys or ureters or rectal bleeding. They can compress critical blood vessels such as the mesenteric vessels and the vena cava. It is important to know that the desmoid tumors that present superficially on the abdominal wall behave much differently than the ones that are deep inside the abdomen or pelvis.
If the desmoid fibromatosis tumor is large enough, it can mimic cancer of the affected organ (eg, breast cancer). In these cases, the only way to establish a definitive diagnosis is to perform a biopsy.
Intra-abdominal and extra-abdominal desmoid fibromatosis
Intra-abdominal aggressive fibromatosis may be seen. Extra-abdominal aggressive fibromatosis may also be seen (rarely) in the urological system, including in the bladder and scrotum 96, 97. Intra-abdominal desmoid tumors remain asymptomatic until their growth and infiltration cause visceral compression. Symptoms of intestinal, vascular, ureteric, or neural involvement may be the initial manifestations. An ethmoidal desmoid tumor has been described in a child 98.
Breast desmoid fibromatosis
Aggressive fibromatosis account for 0.2% of primary breast tumors, developing from muscular fasciae and aponeuroses 99. Desmoid fibromatosis may mimic breast cancer, either in the breasts or, less commonly, in the armpits 100, 101. When a desmoid tumor involves the breast, it may mimic breast cancer on physical examination, mammography, and breast ultrasonography 100.
Desmoid fibromatosis complications
The complications of aggressive fibromatosis or desmoid fibromatosis depends on their location, extent, and infiltrative capacity.
- Intra-abdominal desmoid tumors — may cause intestinal or ureteral obstruction or involve the mesenteric vessels.
- Pelvis desmoid tumors — may infiltrate the urinary bladder or cause hydrosalpinx.
- Extra-abdominal desmoid tumors — may invade the pleura if occurring in the chest or may compress nearby vessels and nerves.
Pain management is crucial due to the potential for the tumor to invade the surrounding tissue and can be very difficult to remove surgically.
Desmoid tumors can be more complex in pregnancy – a combination of hormonal signalling, trauma, and mechanical constraints may worsen symptoms during this time. Pregnant patients need to be managed in experienced centers with multi-disciplinary team.
Desmoid fibromatosis diagnosis
Desmoid fibromatosis or aggressive fibromatosis are usually diagnosed through a combination of imaging, including magnetic resonance imaging (MRI), and biopsy. Magnetic resonance imaging (MRI) is the preferred imaging modality, showing mixed hyperintense/isointense T2 signals 102, 103, 104, 105. CT scans can reveal a soft tissue mass, which typically is sharply marginated in abdominal wall tumors or has poorly defined, infiltrative margins in extra‐abdominal or mesenteric tumors and are useful for diagnosis and follow‐up of intra‐abdominal desmoid fibromatosis and associated complications, such as small bowel obstruction 106, 63. Ultrasound can be used with the palpable lesions of extremities, abdominal, chest wall, and breast. The sonographic appearance of desmoid fibromatosis is variable hyperechogenic areas within the tumor and well to poorly defined margins.
The preferred diagnostic test is biopsy of the tumor. A fine-needle aspiration biopsy specimen may be considered 107.
Histologically, desmoid tumor is usually poorly circumscribed and composed of abundant collagen surrounding poorly circumscribed bundles of spindle cells. The dense bundles of eosinophilic spindle cells contain regular nuclei and pale cytoplasm with neither mitoses nor giant cells. Macrophages, giant cells, and lymphocytes are present peripherally. In most patients, staining for nuclear beta-catenin, vimentin, cyclooxygenase‐2 (COX‐2), c-KIT, PDGFRb, androgen receptors and beta estrogen receptors using immunohistochemistry will be positive and help establish a definitive diagnosis. They are negative for desmin, S-100, h-caldesmon, CD34 and CKIT 108, 109.
Given the high rate of misdiagnosis that occurs, guidelines recommend a second opinion by an expert pathologist for confirmation.
Desmoid fibromatosis differential diagnosis
Differential diagnoses for extra-abdominal desmoid tumors:
- Fibroblastic sarcoma
- Gardner fibroma.
Differential diagnoses for intra-abdominal desmoid tumors:
- Gastrointestinal stromal tumor
- Solitary fibrous tumor
- Inflammatory myofibroblastic tumor
- Sclerosing mesenteritis
- Retroperitoneal fibrosis
- Secondary to certain drugs or an underlying cancer.
Desmoid fibromatosis treatment
For aggressive fibromatosis or desmoid fibromatosis that are asymptomatic or nonprogressive, some prefer a wait-and-see approach 110. However, aggressive, wide surgical resection is the treatment of choice for desmoid fibromatosis or aggressive fibromatosis 111. Complete surgical excision of desmoid fibromatosis or aggressive fibromatosis is the most effective method of cure. This sometimes necessitates removal of most of an anterior compartment of a leg. Extensive cases may require excision plus adjuvant (add-on) treatment including chemotherapy and repeat surgery 112. Positive margins (incomplete resections) after surgery reflect a high risk for recurrence 113. Surgery plus the administration of nonsteroidal anti-inflammatory medication (NSAIDs), hormonal therapy, and cytotoxic chemotherapy is sometimes used, the latter being the most effective pharmacological approach 114. In selected patients, radical resection with intraoperative margin evaluation by frozen sections followed by immediate mesh reconstruction may be a safe and effective procedure providing definitive cure yet minimizing functional limitations 115.
Lesions involving the extremities and deep soft tissues of the trunk have a higher risk of recurrence, as do Gardner syndrome–associated lesions in other locations 116.
In those patients who refuse surgery or are not surgical candidates, the options below may be considered.
Radiation therapy may be used as a treatment for recurrent disease or as primary therapy to avoid mutilating surgical resection. It may be used postoperatively, preoperatively, or as the sole treatment 117.
Systemic therapy is appropriate if a primary complete resection is not feasible or if there is relapse or progression after resection 118. There are a number of novel drug therapy candidates for desmoid tumors 119. Pazopanib, a potent tyrosine kinase inhibitor, represents a promising new therapy for desmoid tumors in adolescent and young adult patients 120.
In November 2023, the US Food and Drug Administration (FDA) approved the first drug for desmoid tumor, nirogacestat (Ogsiveo) 121 . Nirogacestat, a gamma secretase inhibitor, cleaves multiple transmembrane proteins, including Notch, that are believed to play a role in activating pathways that contribute to growth of desmoid tumors 121. Nirogacestat (Ogsiveo) is indicated for adults with progressing tumors who require systemic treatment 121. The approval was supported by results from the phase 3 DeFi clinical trial 122. In the DeFi clinical trial 122, 70 patients were assigned to receive nirogacestat and 72 to receive placebo. Nirogacestat showed a significant progression-free survival (PFS) benefit over placebo; the likelihood of being event-free at 2 years was 76% with nirogacestat and 44% with placebo 122. Between-group differences in progression-free survival (PFS) were consistent across prespecified subgroups. The percentage of patients who had an objective response was significantly higher with nirogacestat than with placebo (41% vs 8%), with a median time to response of 5.6 months and 11.1 months, respectively; the percentage of patients with a complete response was 7% and 0%, respectively 122.
There are a number of new drug therapy candidates for aggressive fibromatosis 123. Pazopanib, a potent tyrosine kinase inhibitor, represents a promising new therapy for aggressive fibromatosis in adolescent and young adult patients 124. Another drug option is sorafenib 125.
Drug therapy with antiestrogens and prostaglandin inhibitors may also be used. Drug therapy result in objective response rates of approximately 40-50% of aggressive fibromatosis; the duration of response is variable 126.
In cases of recurrent extra-abdominal desmoid tumors in which surgery is contraindicated or in cases of recurrence, a chemotherapeutic regimen of doxorubicin, dacarbazine, and carboplatin may be effective. Intra-abdominal desmoid tumors as a part of Gardner syndrome may respond to systemic doxorubicin, and ifosfamide can be useful for patients with complications from the tumor 127. Polychemotherapy has been used 128 and can be combined with targeted therapy with imatinib 129.
Expanded knowledge of familial adenomatosis polyposis–desmoid tumor molecular underpinnings may aid in the development of novel therapeutic strategies 130.
Magnetic resonance‒guided high-intensity focused ultrasound may prove a safe and effective option for selected desmoid tumors 131.
Split-course radiotherapy in patients with desmoid tumors was well tolerated with good outcomes 132.
Active surveillance
In recent years, as a result of the unsatisfactory results obtained with surgery, the initial approach for desmoid tumors has turned toward a conservative, non-operative strategy, which is now the strategy of choice. It is recommended that all cases have a period of active surveillance. It allows for better predictions concerning the natural history and biology of desmoid tumors and allows the clinician to plan the next step in the therapeutic sequence, since a high percentage of cases reach a stabilization period and some patients even present regression of the tumor 133. Active surveillance was first proposed for patients that had recurrences not amenable for limb salvage; being a benign tumor, the main goal of observation was to avoid mutilating procedures. Under the surveillance period, it was noted that more than half of the tumors stabilized. Afterwards, the same strategy was offered to patients with resectable tumors and the same results were obtained, setting the foundations for what is now the initial treatment of choice; active surveillance 134.
The rationale for active surveillance is based on the fact that most patients will be able to avoid an unnecessary surgical procedure. Approximately 50% of the cases enter a stabilization period in an average of 14 to 19 months. When progressive disease presents, it is usually in the first months of observation and is rarely seen after three years of follow-up. Following this approach, only 14–16% of cases will require a surgical intervention and a quarter of the patients will show tumor regression. It has been demonstrated that the surveillance period can be safely carried out without detrimental outcomes in those patients who progress 135, 136. Recently, Duhil de Bénazé et al 137, reported encouraging outcomes in young patients with an initial wait and see approach. It was proved to be a safe and feasible option, not associated with impair in long term functionality when compared with other treatment strategies 138, 137. Another case-series, carried out by Grignol et al 138, included 142 patients (74 with primary tumors and 68 with recurrent tumors), in which a total of 83 patients were managed with active surveillance and reached a 5-year progression-free survival of 49.9%.
A study of patients with desmoid tumors managed at a referral center in the United Kingdom demonstrated a shift in the trend of treatment over time. The authors reported an increase from 10% in 1998 to 40% in 2016 of patients managed with active surveillance initially 139. In the whole series, they had a 36% of stable disease with 27% of either partial or complete response and 36% of progressive disease. They recorded older age (> 50 years old) as a risk factor for progression when compared with younger patients, as well as upper extremity and chest wall location 139.
The period of active surveillance should include close monitoring of patients with MRI or computed tomography every month for the first two months, then every three months for the first year followed by every six months until the fifth year, and yearly after. The intensity of the surveillance regimen, especially during the first years serves for early identification of rapidly progressive cases. Patients who have tumors in life-threatening locations as well as those with severe pain may avoid the surveillance period.
Systemic therapy
Current indications for systemic treatment include rapidly progressive desmoid fibromatosis or patient rejection to active surveillance. Systemic treatment options for desmoid fibromatosiss include non-steroid anti-inflammatory drugs (NSAIDs), anti-hormonal therapies, tyrosine kinase inhibitors (TKI), and conventional “low dose” chemotherapeutic regimens, including liposomal doxorubicin.
Anti-inflammatories have shown the ability to block the beta-catenin pathway mediated by cyclooxygenase‐2 (COX‐2) or prostaglandins, and thus induce an objective response and improve pain control. The proposed mechanism of this response is due to cyclooxygenase‐2 (COX‐2) overexpression in the tumor microenvironment, causing increased expression of platelet-derived growth factor (PDGF) that contributes to tumor growth, stimulates angiogenesis and promotes pathways of resistance to apoptosis. Among the most widely used non-steroid anti-inflammatory drugs (NSAIDs) are sulindac, indomethacin, meloxicam and celecoxib 140.
Anti-hormonal agents have been used with favorable results even though experts do not know precisely its mechanism of action. The pathway of transforming growth factor beta may be implicated. Tamoxifen and toremifene are the most widely used agents. In one study, progression-free survival was 90% at 12 and 24 months. According to RECIST, partial response, stable disease, and disease progression were observed in 25%, 65% and 10% of patients, respectively. They can be used alone or in combination with anti-inflammatory (anti-COX2) drugs. They are generally the first line of treatment due to their good tolerance and low toxicity profile. Unfortunately, response is usually poor, mostly achieving stabilization of the disease and improvement in pain, which can be observed promptly after treatment has started 141.
In November 2023, the US Food and Drug Administration (FDA) approved the first drug for desmoid tumor, nirogacestat (Ogsiveo) 121 . Nirogacestat, a gamma secretase inhibitor, cleaves multiple transmembrane proteins, including Notch, that are believed to play a role in activating pathways that contribute to growth of desmoid tumors 121. Nirogacestat (Ogsiveo) is indicated for adults with progressing tumors who require systemic treatment 121. The approval was supported by results from the phase 3 DeFi clinical trial 122. In the DeFi clinical trial 122, 70 patients were assigned to receive nirogacestat and 72 to receive placebo. Nirogacestat showed a significant progression-free survival (PFS) benefit over placebo; the likelihood of being event-free at 2 years was 76% with nirogacestat and 44% with placebo 122. Between-group differences in progression-free survival (PFS) were consistent across prespecified subgroups. The percentage of patients who had an objective response was significantly higher with nirogacestat than with placebo (41% vs 8%), with a median time to response of 5.6 months and 11.1 months, respectively; the percentage of patients with a complete response was 7% and 0%, respectively 122.
Controversy exists for the use of systemic chemotherapy in a disease that does not carry a metastatic potential; however, it should be considered as the first line treatment in patients with rapidly progressive or unresectable symptomatic desmoid fibromatosis. Drugs used include doxorubicin (alone or in combination with dacarbazine), vinorelbine, vinblastine, and methotrexate. The anthracycline-based regimen is similar to the one used in sarcomas and is associated with high response rates. It is administered for 6–8 cycles, and regardless of the combination used, objective response or stabilization of the disease is achieved in 80% of the cases, with a lasting response in 45% of patients 142.
Tyrosine kinase inhibitors (TKI) have shown objective responses, despite the fact that their mechanism of action is not fully understood in this circumstance. Still, by blocking the receptor phosphorylation, activation, and proliferation of the kinase, they inhibit growth and block cell proliferation. Agents used include imatinib, nilotinib, sorafenib, sunitinib, and pazopanib. Pazopanib, a potent tyrosine kinase inhibitor, represents a promising new therapy for aggressive fibromatosis in adolescent and young adult patients 124. Imatinib, a selective tyrosine kinase inhibitor (TKI), inhibits several receptors including ABL, PDGFR, and CKIT and has demonstrated a 3-year progression-free survival of 58% with 6% regression after 19–26 months of treatment with a clinical benefit in 84% of the patients 143, 144, 145.
In the DESMOPAZ randomized open phase 2 trial, where patients with progressive desmoid fibromatosis were included, and randomly assigned to pazopanib or vinblastine and methotrexate. The primary objective was the proportion of patients who did not progress in the first six months with 83.7% for pazopanib and 45% for methotrexate-vinblastine. Adverse events were well tolerated 146.
Gounder et al 147 conducted a phase 3, double-blind study in 87 patients with desmoid fibromatosis and progressive symptomatic or recurrent disease. One arm received sorafenib 400 mg orally daily and was compared with placebo. Crossover to the sorafenib group was allowed for patients in the placebo group with progressive disease. The primary objective was progression-free survival. Objective response rates and adverse events were also evaluated. The 2-year progression-free survival rate was 81% in the sorafenib group and 36% in the placebo group. Before crossover, the objective response rate was 33% in the sorafenib group and 20% in the placebo group. Among the patients receiving sorafenib, the most frequently reported adverse events were grade 1 or 2 events, namely rash (73%), fatigue (67%), hypertension (55%), and diarrhea (51%). The high response rate in the placebo group is to be noted and can suggest a proportion of patients with spontaneous regression 147.
Finally, several new pieces of evidence support the concept that deregulation of the mammalian target of the rapamycin (mTOR) cell proliferation/survival pathway may play an important role in tumor biology when the APC/beta-catenin pathway is disrupted. Sirolimus, a drug that inhibits the mammalian target of rapamycin (mTOR), is currently being evaluated as an anti-cancer agent in desmoid tumor 148.
Desmoid fibromatosis surgery
According to the latest European consensus on desmoid tumors, surgery should be considered in cases of progression to medical or radiation therapies, always considering location and age. When surgery is carried out, it should always be done trying to preserve function and after considering all the alternatives for it. Cases of mesenteric or retroperitoneal tumors not associated with familial polyposis can be treated initially with surgery due to the morbidity and symptoms they cause.
Until a few years ago, aggressive, wide surgical resection is the treatment of choice for desmoid fibromatosis or aggressive fibromatosis 110, 149. Complete surgical excision of aggressive fibromatosis is the most effective method of cure. This sometimes necessitates removal of most of an anterior compartment of a leg. However, the 5-year recurrence rate was high, ranging from 25% to 60%, and extensive or disabling procedures were common 62.
Extensive cases may require excision plus adjuvant (add-on) treatment including chemotherapy and repeat surgery 150. In selected patients, radical resection with intraoperative margin evaluation by frozen sections followed by immediate mesh reconstruction may be a safe and effective procedure providing definitive cure yet minimizing functional limitations 151. However, there has been a recent tendency to more conservative management 79. Active surveillance MRI has become a popular option 152.
Amputations should seldom be used for aggressive fibromatosis and only in cases of unresectable and severe, untreatable symptomatic recurrent disease or when some treatment-related side effects (surgery +/− radiation therapy) cause significant loss of function or disabling chronic symptoms 153, 154, 155.
Evidence suggests that pregnancy does not adversely affect surgical outcomes 156.
Lesions involving the extremities and deep soft tissues of the trunk have a higher risk of recurrence, as do Gardner syndrome–associated lesions in other locations 157.
Radiotherapy
The role of radiotherapy is controversial. Indications for its use in desmoid fibromatosis are debated due to its toxicity, especially in the young population. In the Italian-French consensus it is recommended in progressive disease or in the absence of other therapeutic alternatives 143. In a retrospective review of 22 articles the local control rate was 75% when radiotherapy + surgery was used, 78% for radiotherapy and 61% for surgery, including patients with positive and negative margins; therefore they suggest its use in anatomical sites where surgery can generate considerable morbidity, such as head and neck 158.
The recommended dose is 50–56 Gy in 2 Gy fractions. Other studies have published doses greater than 56 Gy, but they have failed to demonstrate improvement in local control and are associated with greater toxicity including edema, pathological fractures, fibrosis, soft tissue necrosis or vascular complications as well as radio-induced neoplasms 159. In 2017 a meta-analysis was published and concluded that adjuvant radiotherapy should be considered especially in those patients with R1 or R2 resections, since they are at greatest risk of recurrence 160.
Long-Term Monitoring
It is recommended to monitor patients in an outpatient setting, to perform a physical examination and imaging every 3–4 months, for the first 2 years. Subsequently, the intervals may be longer, and the clinician may choose to alternate MR imaging and ultrasound. When active surveillance is chosen, it should be performed with magnetic resonance imaging (MRI) and with CT for intra-abdominal tumors every month the first 2 months, then every 3 months for a year followed by every 6 months for 5 years and annually thereafter.
Desmoid fibromatosis prognosis
The clinical outcome of aggressive fibromatosis or desmoid tumors may be unpredictable 73, 161, 162, 65, 163. Some tumors resolve spontaneously and others frequently recur despite surgical intervention.
The clinical outcome of aggressive fibromatosis or desmoid tumors can be divided into 4 main groups based on tumor progression:
- Remains stable after diagnosis (most common). Frequently, an initial growth phase is followed by stabilization 164, 165, 166.
- Spontaneously resolves. Spontaneous regressions occur in 20%–30% of patients who are followed for 2–3 years 167, 168. The prognostic value of miRNA expression profiling has been suggested as a way to delineate surgical candidates from those who might be monitored without treatment 169.
- Undergoes cycles of progression and resolution.
- Progresses rapidly causing local infiltration with potential obstruction of vital structures and organs.
Intra-abdominal desmoid tumors carry the highest risk of complications, and patients with syndromic conditions such as familial adenomatous polyposis (FAP) are more likely to have more invasive, symptomatic tumors. Treatment focuses on preventing symptomatic disease rather than total cure; in 70% of patients, the tumor recurs following definitive management, such as surgery or radiotherapy.
Local desmoid tumor recurrence rates are reported to be as high as 70% 170. A positive surgical margin is a significant risk factor for recurrence 171, 172.
Factors significantly associated with shorter progression‐free survival (PFS) include age (younger than 37 years), tumor size (>7 cm), and tumor location (extra‐abdominal) 173, 174, 175, 176.
Intra-abdominal desmoid tumors may kill patients with familial adenomatous polyposis (FAP) 177. Five-year survival rates of such patients with stage 1, 2, 3, and 4 intra-abdominal desmoid tumors were found to be 95%, 100%, 89%, and 76%, respectively 170. The 5-year survival rate of stage 4 patients with severe pain or narcotic dependency, tumor size larger than 10 cm, and need for total parenteral nutrition was only 53% 170.
A study of 179 patients with primary, sporadic desmoid tumors who had complete surgical resection found that those with the S45F mutation had a greater tendency for local recurrence than those without it 178.
What are the features of fibromatosis?
The following table lists the distinguishing features between superficial and deep fibromatoses 4.
Superficial fibromatoses
- Slow growing tumor
- Small size
- Arise from fascia or aponeurosis
- Less aggressive
Deep fibromatoses
- Rapidly growing pseudotumor
- Usually, they reach a large size
- Often involve deeper structures (muscles of the trunk and extremities)
What causes fibromatosis?
The cause of fibromatosis remains unclear. In some types of fibromatosis such as desmoid tumors, it is thought that the condition may be related to trauma, hormonal factors, or have a genetic association. Superficial fibromatoses such as palmar, plantar and penile fibromatosis have sometimes been linked to certain diseases such as diabetes, liver disease and high blood pressure 4.
Fibromatosis treatment
Management of fibromatosis depends on individual disease.
- Congenital generalized fibromatosis (infantile myofibromatosis)
- Aponeurotic fibroma
- Infantile digital fibromatosis: Eventually, many fibromatosis resorb and disappear by themselves over 2 to 3 years so that a conservative wait and see approach is best if they are not causing problems. If desired, they can be removed by a simple surgical procedure in which the lump is shaved off. However, many recur after surgery.
- Aggressive infantile fibromatosis
- Fibromatosis colli
- Dermatofibrosis lenticularis (Buschke-Ollendorf syndrome)
- Palmar (Dupuytren disease) and plantar (Ledderhose disease) fibromatosis
- Penile fibromatosis (Peyronie disease)
- Knuckle pads
- Dermatofibroma
- Nodular fasciitis
- Elastofibroma
- Fibrous papule of the nose or face
- Desmoid tumours (aggressive fibromatoses).
- Tolan S, Shanks JH, Loh MY, Taylor B, Wylie JP. Fibromatosis: benign by name but not necessarily by nature. Clin Oncol (R Coll Radiol). 2007 Jun;19(5):319-26. doi: 10.1016/j.clon.2007.03.002[↩]
- Lacka DE, Nasierowska-Guttmejer A. Fibromatosis – immunohistochemical evaluation, differential diagnosis from gastrointestinal tumors, and other mesenchymal tumours. Prz Gastroenterol. 2019;14(1):79-85. doi: 10.5114/pg.2019.83429[↩]
- What Is a Soft Tissue Sarcoma? https://www.cancer.org/cancer/types/soft-tissue-sarcoma/about/soft-tissue-sarcoma.html[↩][↩][↩]
- Fibromatosis. https://dermnetnz.org/topics/fibromatosis[↩][↩][↩][↩][↩][↩][↩]
- Fibromatosis. https://www.dermnetnz.org/topics/fibromatosis/[↩]
- Meyers AL, Marquart MJ. Plantar Fibromatosis. [Updated 2024 Feb 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557674[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Plantar Fibromatosis. https://emedicine.medscape.com/article/1061903-overview[↩][↩]
- Dürr HR, Krödel A, Trouillier H, Lienemann A, Refior HJ. Fibromatosis of the plantar fascia: diagnosis and indications for surgical treatment. Foot Ankle Int. 1999;20(1):13–17. doi: 10.1177/107110079902000103[↩][↩]
- Carroll P, Henshaw RM, Garwood C, Raspovic K, Kumar D. Plantar Fibromatosis: Pathophysiology, Surgical and Nonsurgical Therapies: An Evidence-Based Review. Foot Ankle Spec. 2018 Apr;11(2):168-176. doi: 10.1177/1938640017751184[↩]
- Banerjee S., Muhammad M., Nath C., Kumar Pal D. Plantar fibromatosis: a case report. The Foot and Ankle Online Journal. 2009;2(12, article 3) doi: 10.3827/faoj.2009.0212.0003[↩]
- de Bree E., Zoetmulder F. A. N., Keus R. B., Peterse H. L., Van Coevorden F. Incidence and treatment of recurrent plantar fibromatosis by surgery and postoperative radiotherapy. The American Journal of Surgery. 2004;187(1):33–38. doi: 10.1016/j.amjsurg.2002.11.002[↩][↩][↩]
- Trybus M, Bednarek M, Budzyński P, Gniadek M, Lorkowski J. Współistnienie choroby Ledderhose’a z przykurczem dupuytrena. Doświadczenia własne [Concomitance of Ledderhose’s disease with Dupuytren’s contracture. Own experience]. Przegl Lek. 2012;69(9):663-6. Polish.[↩]
- Rosenbaum AJ, Dipreta JA, Misener D. Plantar heel pain. Med Clin North Am. 2014;98(2):339–352. doi: 10.1016/j.mcna.2013.10.009[↩][↩]
- Lareau CR, Sawyer GA, Wang JH, Digiovanni CW. Plantar and medial heel pain: diagnosis and management. J Am Acad Orthop Surg. 2014;22(6):372–380. doi: 10.5435/JAAOS-22-06-372[↩]
- Neufeld SK, Cerrato R. Plantar fasciitis: evaluation and treatment. J Am Acad Orthop Surg. 2008;16(6):338–346. doi: 10.5435/00124635-200806000-00006[↩]
- Jeswani T, Morlese J, Mcnally EG. Getting to the heel of the problem: plantar fascia lesions. Clin Radiol. 2009;64(9):931–939. doi: 10.1016/j.crad.2009.02.020[↩]
- Lee TH, Wapner KL, Hecht PJ. Plantar fibromatosis. J Bone Joint Surg Am. 1993;75(7):1080–1084. doi: 10.2106/00004623-199307000-00016[↩]
- Oliveira EA, Pato T, Barcelos A. Doença de Ledderhose – um caso clínico [Ledderhose disease – a clinical case report]. Acta Reumatol Port. 2010 Oct-Dec;35(5):529-30. Portuguese.[↩]
- Godette GA, O’Sullivan M, Menelaus MB. Plantar fibromatosis of the heel in children: a report of 14 cases. J Pediatr Orthop. 1997 Jan-Feb;17(1):16-7. https://journals.lww.com/pedorthopaedics/abstract/1997/01000/plantar_fibromatosis_of_the_heel_in_children__a.5.aspx[↩]
- Johnston FE, Collis S, Peckham NH, Rothstein AR. Plantar fibromatosis: literature review and a unique case report. J Foot Surg. 1992;31(4):400–406.[↩]
- Fausto de Souza D, Micaelo L, Cuzzi T, Ramos-E-Silva M. Ledderhose disease: an unusual presentation. J Clin Aesthet Dermatol. 2010 Sep;3(9):45-7. https://pmc.ncbi.nlm.nih.gov/articles/PMC2945849[↩][↩]
- Young JR, Sternbach S, Willinger M, Hutchinson ID, Rosenbaum AJ. The etiology, evaluation, and management of plantar fibromatosis. Orthop Res Rev. 2018 Dec 17;11:1-7. doi: 10.2147/ORR.S154289[↩][↩][↩][↩][↩]
- Souza BG, de Souza Júnior GZ, Rodrigues RM, Dias DS, de Oliveira VM. Surgical Treatment of a Case of Ledderhose’s Disease: A Safe Plantar Approach to Subtotal Fasciectomy. Case Rep Orthop. 2015;2015:509732. doi: 10.1155/2015/509732[↩][↩][↩][↩]
- Schmidt I. Ledderhose’s Disease-What do We Know and What do We not know. SM Musculoskelet Disord. 2019; 4(1): 1033. DOI:10.36876/smmd.1033[↩]
- Stewart BD, Nascimento AF. Palmar and plantar fibromatosis: a review. J Pathol Transl Med. 2021 Jul;55(4):265-270. doi: 10.4132/jptm.2021.06.14[↩][↩][↩][↩][↩][↩][↩]
- Gudmundsson KG, Jónsson T, Arngrímsson R. Association of Morbus Ledderhose with Dupuytren’s contracture. Foot Ankle Int. 2013;34(6):841–845. doi: 10.1177/1071100713475352[↩]
- Carroll P, Henshaw RM, Garwood C, Raspovic K, Kumar D. Plantar fibromatosis: pathophysiology, surgical and nonsurgical therapies: an evidence-based review. Foot Ankle Spec. 2018;11(2):168–176. doi: 10.1177/1938640017751184[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Kim SK, Ioannidis JPA, Ahmed MA, Avins AL, Kleimeyer JP, Fredericson M, Dragoo JL. Two Genetic Variants Associated with Plantar Fascial Disorders. Int J Sports Med. 2018 Apr;39(4):314-321. doi: 10.1055/s-0044-100280[↩][↩]
- Espert M, Anderson MR, Baumhauer JF. Current Concepts Review: Plantar Fibromatosis. Foot Ankle Int. 2018 Jun;39(6):751-757. doi: 10.1177/1071100718768051[↩]
- Hu FZ, Nystrom A, Ahmed A, Palmquist M, Dopico R, Mossberg I, Gladitz J, Rayner M, Post JC, Ehrlich GD, Preston RA. Mapping of an autosomal dominant gene for Dupuytren’s contracture to chromosome 16q in a Swedish family. Clin Genet. 2005 Nov;68(5):424-9. doi: 10.1111/j.1399-0004.2005.00504.x[↩]
- Veith NT, Tschernig T, Histing T, Madry H. Plantar fibromatosis–topical review. Foot Ankle Int. 2013 Dec;34(12):1742-6. doi: 10.1177/1071100713505535[↩]
- Carroll P, Henshaw RM, Garwood C, Raspovic K, Kumar D. Plantar fibromatosis: pathophysiology, surgical and nonsurgical therapies: an evidence-based review. Foot Ankle Spec. 2018;11:168–76. doi: 10.1177/1938640017751184[↩]
- Strzelczyk A, Vogt H, Hamer HM, Kramer G. Continuous phenobarbital treatment leads to recurrent plantar fibromatosis. Epilepsia. 2008;49:1965–8. doi: 10.1111/j.1528-1167.2008.01684.x[↩]
- McNally EG, Shetty S. Plantar fascia: imaging diagnosis and guided treatment. Semin Musculoskelet Radiol. 2010 Sep;14(3):334-43. doi: 10.1055/s-0030-1254522[↩]
- Draghi F, Gitto S, Bortolotto C, Draghi AG, Ori Belometti G. Imaging of plantar fascia disorders: findings on plain radiography, ultrasound and magnetic resonance imaging. Insights Imaging. 2017 Feb;8(1):69-78. doi: 10.1007/s13244-016-0533-2[↩]
- Lui TH. Endoscopic Subtotal Fasciectomy of the Foot. Arthrosc Tech. 2016 Dec 5;5(6):e1387-e1393. doi: 10.1016/j.eats.2016.08.005[↩][↩]
- Evans HL. Multinucleated giant cells in plantar fibromatosis. Am J Surg Pathol. 2002;26:244–8. doi: 10.1097/00000478-200202000-00012[↩]
- Omor Y, Dhaene B, Grijseels S, Alard S. Ledderhose Disease: Clinical, Radiological (Ultrasound and MRI), and Anatomopathological Findings. Case Rep Orthop. 2015;2015:741461. doi: 10.1155/2015/741461[↩]
- Carlson JW, Fletcher CD. Immunohistochemistry for beta-catenin in the differential diagnosis of spindle cell lesions: analysis of a series and review of the literature. Histopathology. 2007;51:509–14. doi: 10.1111/j.1365-2559.2007.02794.x[↩]
- Jordan GH, Carson CC, Lipshultz LI. Minimally invasive treatment of Peyronie’s disease: evidence-based progress. BJU Int. 2014;114(1):16–24. doi: 10.1111/bju.12634[↩]
- Frizziero A, Barazzuol M, Vittadini F, Bellon G, Masiero S, Meneghini A. Plantar fascial fibromatosis: two cases treated with low-energy focused shock waves. J Clin Rheumatol. 2017;23(1):63–65. doi: 10.1097/RHU.0000000000000462[↩][↩][↩][↩][↩]
- Yin MC, Ye J, Yao M, et al. Is extracorporeal shock wave therapy clinical efficacy for relief of chronic, recalcitrant plantar fasciitis? A systematic review and meta-analysis of randomized placebo or active-treatment controlled trials. Arch Phys Med Rehabil. 2014;95(8):1585–1593. doi: 10.1016/j.apmr.2014.01.033[↩][↩][↩]
- Frairia R, Berta L. Biological effects of extracorporeal shock waves on fibroblasts. A review. Muscles Ligaments Tendons J. 2012 Apr 1;1(4):138-47. https://pmc.ncbi.nlm.nih.gov/articles/PMC3666484[↩]
- Veith NT, Tschernig T, Histing T, Madry H. Plantar fibromatosis–topical review. Foot Ankle Int. 2013;34(12):1742–1746. doi: 10.1177/1071100713505535[↩][↩][↩][↩][↩][↩]
- Knobloch K, Vogt PM. High-energy focussed extracorporeal shockwave therapy reduces pain in plantar fibromatosis (Ledderhose’s disease) BMC Res Notes. 2012;5:542. doi: 10.1186/1756-0500-5-542[↩]
- Patel SR, Benjamin RS. Desmoid tumors respond to chemotherapy: defying the dogma in oncology. J Clin Oncol. 2006;24(1):11–12. doi: 10.1200/JCO.2005.03.6566[↩]
- Beckmann J, Kalteis T, Baer W, Grifka J, Lerch K. Plantarfibromatose: Therapie mit totaler Plantarfasziektomie [Plantar fibromatosis: therapy by total plantarfasciectomy]. Zentralbl Chir. 2004 Jan;129(1):53-7. German. doi: 10.1055/s-2004-816231[↩]
- van der Veer WM, Hamburg SM, de Gast A, Niessen FB. Recurrence of plantar fibromatosis after plantar fasciectomy: single-center long-term results. Plast Reconstr Surg. 2008;122(2):486–491. doi: 10.1097/PRS.0b013e31817d61ab[↩]
- Kadir HKA, Chandrasekar CR. Partial fasciectomy is a useful treatment option for symptomatic plantar fibromatosis. Foot. 2017;31:31–34. doi: 10.1016/j.foot.2017.02.002[↩]
- de Bree E, Zoetmulder FA, Keus RB, Peterse HL, van Coevorden F. Incidence and treatment of recurrent plantar fibromatosis by surgery and postoperative radiotherapy. Am J Surg. 2004;187(1):33–38. doi: 10.1016/j.amjsurg.2002.11.002[↩][↩]
- Aluisio FV, Mair SD, Hall RL. Plantar fibromatosis: treatment of primary and recurrent lesions and factors associated with recurrence. Foot Ankle Int. 1996;17(11):672–678. doi: 10.1177/107110079601701105[↩]
- Lui TH. Endoscopic Subtotal Fasciectomy of the Foot. Arthrosc Tech. 2016;5(6):e1387–e1393. doi: 10.1016/j.eats.2016.08.005[↩]
- Master SR, Mangla A, Shah C. Desmoid Tumor. [Updated 2024 Mar 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459231[↩][↩][↩]
- Fritchie KJ, Crago AM. Soft Tissue and Bone Tumours, 5th edition. Lyon, France: International Agency for Research on Cancer; 2020. Desmoid Fibromatosis.[↩][↩][↩]
- Ito M, Yokoyama K, Ariizumi Y, Kano Y, Yamamoto K, Asakage T, Tateishi U. Desmoid Fibromatosis Involving the Retropharyngeal and Danger Spaces: A Case Report. Cureus. 2024 Dec 18;16(12):e75969. doi: 10.7759/cureus.75969[↩][↩]
- Murli D, Smriti V, Yadav S, Trivedi B, Qureshi SS. Desmoid Fibromatosis of the Oesophagus Creating an Oesophageal Diverticulum in a 2-year-old Girl. Afr J Paediatr Surg. 2024 Jul 1;21(3):210-212. doi: 10.4103/ajps.ajps_120_22[↩]
- Zhao J, Cheng F, Yao Z, Zheng B, Niu Z, He W. Surgical Management of a Giant Desmoid Fibromatosis of Abdominal Wall With Vessels Invasion in a Young Man: A Case Report and Review of the Literature. Front Surg. 2022 Apr 11;9:851164. doi: 10.3389/fsurg.2022.851164[↩][↩]
- Jang J, Cavallo K, Lee J. Complex Surgical Management of Extensive Chest-Wall Desmoid Fibromatosis. Cureus. 2024 Oct 16;16(10):e71670. doi: 10.7759/cureus.71670[↩]
- Subhawong TK, Feister K, Sweet K, Alperin N, Kwon D, Rosenberg A, Trent J, Wilky BA. MRI Volumetrics and Image Texture Analysis in Assessing Systemic Treatment Response in Extra-Abdominal Desmoid Fibromatosis. Radiol Imaging Cancer. 2021 Jul;3(4):e210016. doi: 10.1148/rycan.2021210016[↩]
- Kangas-Dick A, Ali M, Poss M, Khoury T, Takabe K. Diagnosis and Management of Desmoid Fibromatosis of the Breast. World J Oncol. 2024 Jun;15(3):394-404. doi: 10.14740/wjon1844[↩]
- Ganeshan D, Amini B, Nikolaidis P, Assing M, Vikram R. Current Update on Desmoid Fibromatosis. J Comput Assist Tomogr. 2019 Jan-Feb;43(1):29-38. doi: 10.1097/RCT.0000000000000790[↩][↩]
- Garcia-Ortega DY, Martín-Tellez KS, Cuellar-Hubbe M, Martínez-Said H, Álvarez-Cano A, Brener-Chaoul M, Alegría-Baños JA, Martínez-Tlahuel JL. Desmoid-Type Fibromatosis. Cancers (Basel). 2020 Jul 9;12(7):1851. doi: 10.3390/cancers12071851[↩][↩]
- Braschi-Amirfarzan M, Keraliya AR, Krajewski KM, Tirumani SH, Shinagare AB, Hornick JL, Baldini EH, George S, Ramaiya NH, Jagannathan JP. Role of Imaging in Management of Desmoid-type Fibromatosis: A Primer for Radiologists. Radiographics. 2016 May-Jun;36(3):767-82. doi: 10.1148/rg.2016150153[↩][↩]
- Leithner A, Gapp M, Radl R, Pascher A, Krippl P, Leithner K, Windhager R, Beham A. Immunohistochemical analysis of desmoid tumours. J Clin Pathol. 2005 Nov;58(11):1152-6. doi: 10.1136/jcp.2005.026278[↩]
- Kasper B, Ströbel P, Hohenberger P. Desmoid tumors: clinical features and treatment options for advanced disease. Oncologist. 2011;16(5):682-93. doi: 10.1634/theoncologist.2010-0281[↩][↩]
- Zhao CX, Dombrowski ND, Perez-Atayde AR, Robson CD, Afshar S, Janeway KA, Rahbar R. Desmoid tumors of the head and neck in the pediatric population: Has anything changed? Int J Pediatr Otorhinolaryngol. 2021 Jan;140:110511. doi: 10.1016/j.ijporl.2020.110511[↩]
- Extra-abdominal desmoid tumor fibromatosis: a multicenter EMSOS study. Cuomo P, Scoccianti G, Schiavo A, et al. BMC Cancer. 2021;21:437. doi: 10.1186/s12885-021-08189-6[↩][↩]
- The desmoid tumor. I. Incidence, sex-, age- and anatomical distribution in the Finnish population. Reitamo JJ, Häyry P, Nykyri E, Saxén E. Am J Clin Pathol. 1982;77:665–673. doi: 10.1093/ajcp/77.6.665[↩]
- Lucas A, Braggio D, Hernandez L, Mercier K. A retrospective collection of diagnostic data from the Desmoid Tumor Research Foundation natural history study [abstract]. J Clin Oncol. 2021;39(15 suppl):e23549.[↩]
- Kasper B, Baumgarten C, Garcia J, Bonvalot S, et al; Desmoid Working Group. An update on the management of sporadic desmoid-type fibromatosis: a European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol. 2017 Oct 1;28(10):2399-2408. doi: 10.1093/annonc/mdx323[↩]
- Mercier KA, Hernandez L, Boulanger V, Seebald A, Rossov S, Milligan K. Quality of life and tumor location in patients with desmoid tumors: data from the Desmoid Tumor Research Foundation natural history study [abstract]. J Clin Oncol. 2019;37(15 suppl):e18291.[↩]
- Fiore M, Crago A, Gladdy R, Kasper B. The Landmark Series: Desmoid. Ann Surg Oncol. 2021 Mar;28(3):1682-1689. doi: 10.1245/s10434-020-09395-5[↩]
- Riedel RF, Agulnik M. Evolving strategies for management of desmoid tumor. Cancer. 2022 Aug 15;128(16):3027-3040. doi: 10.1002/cncr.34332[↩][↩][↩]
- Desmoid Tumor. https://emedicine.medscape.com/article/1060887-overview[↩][↩]
- Buitendijk S, van de Ven CP, Dumans TG, den Hollander JC, Nowak PJ, Tissing WJ, Pieters R, van den Heuvel-Eibrink MM. Pediatric aggressive fibromatosis: a retrospective analysis of 13 patients and review of literature. Cancer. 2005 Sep 1;104(5):1090-9. doi: 10.1002/cncr.21275[↩]
- Inoue Y, Ishida H, Ueno H, Kobayashi H, Yamaguchi T, Konishi T, Tomita N, Matsubara N, Ishida F, Hinoi T, Kanemitsu Y, Watanabe T, Sugihara K. The treatment of desmoid tumors associated with familial adenomatous polyposis: the results of a Japanese multicenter observational study. Surg Today. 2017 Oct;47(10):1259-1267. doi: 10.1007/s00595-017-1500-3[↩]
- Extra-Abdominal Desmoid Tumors. https://orthoinfo.aaos.org/en/diseases–conditions/extra-abdominal-desmoid-tumors[↩]
- Desmoid tumor. https://medlineplus.gov/genetics/condition/desmoid-tumor[↩][↩][↩][↩][↩]
- Desmoid Tumor Working Group. The management of desmoid tumours: A joint global consensus-based guideline approach for adult and paediatric patients. Eur J Cancer. 2020 Mar;127:96-107. doi: 10.1016/j.ejca.2019.11.013[↩][↩]
- Crago AM, Chmielecki J, Rosenberg M, O’Connor R, Byrne C, Wilder FG, Thorn K, Agius P, Kuk D, Socci ND, Qin LX, Meyerson M, Hameed M, Singer S. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer. 2015 Oct;54(10):606-15. doi: 10.1002/gcc.22272[↩]
- Martinez Trufero J., Pajares Bernad I., Torres Ramon I., Hernando C.J., Pazo Cid R. Desmoid Type Fibromatosis: Who, When, and How to Treat. Curr. Treat. Options Oncol. 2017;18:29. doi: 10.1007/s11864-017-0474-0[↩]
- Fiore M., MacNeill A., Gronchi A., Colombo C. Desmoid-Type Fibromatosis: Evolving Treatment Standards. Surg. Oncol. Clin. N. Am. 2016;25:803–826. doi: 10.1016/j.soc.2016.05.010[↩]
- Dômont J, Salas S, Lacroix L, et al. High frequency of beta-catenin heterozygous mutations in extra-abdominal fibromatosis: a potential molecular tool for disease management. Br J Cancer. 2010;102:1032–6. doi: 10.1038/sj.bjc.6605557[↩]
- Immunohistochemistry for beta-catenin in the differential diagnosis of spindle cell lesions: analysis of a series and review of the literature. Carlson JW, Fletcher CD. Histopathology. 2007;51:509–514. doi: 10.1111/j.1365-2559.2007.02794.x[↩][↩]
- Timbergen MJM, van de Poll-Franse LV, Grünhagen DJ, van der Graaf WT, Sleijfer S, Verhoef C, Husson O. Identification and assessment of health-related quality of life issues in patients with sporadic desmoid-type fibromatosis: a literature review and focus group study. Qual Life Res. 2018 Dec;27(12):3097-3111. doi: 10.1007/s11136-018-1931-3[↩]
- Couto Netto SD, Teixeira F, Menegozzo CAM, Leão-Filho HM, Albertini A, Ferreira FO, Akaishi EH, Utiyama EM. Sporadic Abdominal Wall Desmoid type Fibromatosis: treatment paradigm after thirty two years. BMC Surg. 2018 Jun 7;18(1):37. doi: 10.1186/s12893-018-0367-6[↩]
- CTNNB1 gene. https://medlineplus.gov/genetics/gene/ctnnb1[↩][↩][↩][↩]
- APC gene. https://medlineplus.gov/genetics/gene/apc[↩][↩][↩][↩][↩]
- Lopez R, Kemalyan N, Moseley HS, Dennis D, Vetto RM. Problems in diagnosis and management of desmoid tumors. Am J Surg. 1990 May;159(5):450-3. doi: 10.1016/s0002-9610(05)81243-7[↩]
- Desmoid Tumor. https://emedicine.medscape.com/article/1060887-overview#a5[↩]
- Jeong WS, Oh TS, Sim HB, Eom JS. Desmoid tumor following augmentation mammoplasty with silicone implants. Arch Plast Surg. 2013 Jul;40(4):470-2. doi: 10.5999/aps.2013.40.4.470[↩]
- Lotfi AM, Dozois RR, Gordon H, Hruska LS, Weiland LH, Carryer PW, Hurt RD. Mesenteric fibromatosis complicating familial adenomatous polyposis: predisposing factors and results of treatment. Int J Colorectal Dis. 1989;4(1):30-6. doi: 10.1007/BF01648547[↩]
- Wilcken N, Tattersall MH. Endocrine therapy for desmoid tumors. Cancer. 1991 Sep 15;68(6):1384-8. doi: 10.1002/1097-0142(19910915)68:6<1384::aid-cncr2820680634>3.0.co;2-f[↩]
- Waddell WR. Treatment of intra-abdominal and abdominal wall desmoid tumors with drugs that affect the metabolism of cyclic 3′,5′-adenosine monophosphate. Ann Surg. 1975 Mar;181(3):299-302. doi: 10.1097/00000658-197503000-00009[↩]
- Raynham WH, Louw JH. Desmoid tumours in familial polyposis of the colon. S Afr J Surg. 1971 Jul-Sep;9(3):133-40.[↩]
- Shi B, Zhu Y, Xu Z, Liu Y, Zheng B, Qi T. Aggressive fibromatosis in the urological system. Report of two adult patients and review of the literature. Urol Int. 2007;78(1):93-6. doi: 10.1159/000096945[↩]
- Kohno J, Sumiyoshi T, Tsutsumi N, Maeno A, Okubo K, Mitsumori K, Nishimura K, Shintaku M. [Scrotal desmoid tumor in a patient with familial adenomatous polyposis]. Hinyokika Kiyo. 2015 Jan;61(1):27-31. Japanese.[↩]
- Neri HA, Villagra EJ, Alvarez AC, Valencia P, Jiménez E, de la Torre C, Rodríguez MA, Espinosa R. Ethmoidal desmoid tumor in a pediatric patient. Otolaryngol Head Neck Surg. 2007 Jan;136(1):137-8. doi: 10.1016/j.otohns.2005.10.056[↩]
- Muller M, Dessogne P, Baron M, Picquenot JM, Riopel C, Diologent B, Dupre PF, Collet M. Fibromatose mammaire chez une fillette de neuf ans [Desmoid tumor of the breast in a 9 years old little girl]. Ann Pathol. 2011 Feb;31(1):41-5. French. doi: 10.1016/j.annpat.2010.09.011[↩]
- Pajares B, Galera I, Ribelles N, Polo M. Insidious mastalgia hiding a desmoid tumour of the breast. Clin Transl Oncol. 2010 Jan;12(1):63-5. doi: 10.1007/s12094-010-0468-x[↩][↩]
- Mekhail FG, Montgomery JR, Spicer PJ. Imaging findings of a biopsy-proven desmoid tumor of the axilla in a young female. Radiol Case Rep. 2022 Feb 3;17(4):1050-1053. doi: 10.1016/j.radcr.2022.01.054[↩]
- New concepts in understanding evolution of desmoid tumors: MR imaging of 30 lesions. Vandevenne JE, De Schepper AM, De Beuckeleer L, et al. Eur Radiol. 1997;7:1013–1019. doi: 10.1007/s003300050243[↩]
- Casillas J, Sais GJ, Greve JL, Iparraguirre MC, Morillo G. Imaging of intra- and extra-abdominal desmoid tumors. Radiographics. (1991) 11:959–68. 10.1148/radiographics.11.6.1749859[↩]
- Dinauer PA, Brixey CJ, Moncur JT, Fanburg-Smith JC, Murphey MD. Pathologic and MR imaging features of benign fibrous soft-tissue tumors in adults. Radiographics. 2007 Jan-Feb;27(1):173-87. doi: 10.1148/rg.271065065[↩]
- Einstein DM, Tagliabue JR, Desai RK. Abdominal desmoids: CT findings in 25 patients. AJR Am J Roentgenol. 1991 Aug;157(2):275-9. doi: 10.2214/ajr.157.2.1853806[↩]
- Shinagare AB, Ramaiya NH, Jagannathan JP, Krajewski KM, Giardino AA, Butrynski JE, Raut CP. A to Z of desmoid tumors. AJR Am J Roentgenol. 2011 Dec;197(6):W1008-14. doi: 10.2214/AJR.11.6657[↩]
- Jakowski JD, Mayerson J, Wakely PE Jr. Fine-needle aspiration biopsy of the distal extremities: a study of 141 cases. Am J Clin Pathol. 2010 Feb;133(2):224-31. doi: 10.1309/AJCPBWJP3CG6JZKA[↩]
- Kotiligam D., Lazar A.J.F., Pollock R., Lev D. Desmoid tumor: A disease opportune for molecular insights. Histol. Histopathol. 2008;23:117–126. doi: 10.14670/HH-23.117[↩]
- Owens C.L., Sharma R., Ali S.Z. Deep fibromatosis (desmoid tumor): Cytopathologic characteristics, clinicoradiologic features, and immunohistochemical findings on fine-needle aspiration. Cancer. 2007;111:166–172. doi: 10.1002/cncr.22689[↩]
- Oudot C, Defachelles AS, Minard-Colin V, Olschwang S, Fourcade L, Helfre S, Orbach D. Les tumeurs desmoïdes en pédiatrie : état des connaissances actuelles [Desmoid tumors in children: current strategy]. Bull Cancer. 2013 May;100(5):518-28. French. doi: 10.1684/bdc.2013.1747[↩][↩]
- Soto-Miranda MA, Sandoval JA, Rao B, Neel M, Krasin M, Spunt S, et al. Surgical Treatment of Pediatric Desmoid Tumors. A 12-Year, Single-Center Experience. Ann Surg Oncol. 2013 Jul 10.[↩]
- Ramirez RN, Otsuka NY, Apel DM, Bowen RE. Desmoid tumor in the pediatric population: a report of two cases. J Pediatr Orthop B. 2009 May. 18(3):141-4[↩]
- Buitendijk S, van de Ven CP, Dumans TG, et al. Pediatric aggressive fibromatosis: a retrospective analysis of 13 patients and review of literature. Cancer. 2005 Sep 1. 104(5):1090-9.[↩]
- Inoue Y, Ishida H, Ueno H, Kobayashi H, Yamaguchi T, Konishi T, et al. The treatment of desmoid tumors associated with familial adenomatous polyposis: the results of a Japanese multicenter observational study. Surg Today. 2017 Mar 1.[↩]
- Bertani E, Chiappa A, Testori A, et al. Desmoid tumors of the anterior abdominal wall: results from a monocentric surgical experience and review of the literature. Ann Surg Oncol. 2009 Jun. 16(6):1642-9[↩]
- Cates JM, Stricker TP, Sturgeon D, Coffin CM. Desmoid-type fibromatosis-associated Gardner fibromas: prevalence and impact on local recurrence. Cancer Lett. 2014 Oct 28. 353(2):176-81.[↩]
- El-Haddad M, El-Sebaie M, Ahmad R, et al. Treatment of aggressive fibromatosis: the experience of a single institution. Clin Oncol (R Coll Radiol). 2009 Dec. 21(10):775-80.[↩]
- Sparber-Sauer M, Seitz G, von Kalle T, Vokuhl C, Leuschner I, Scheer M, et al. Systemic therapy of aggressive fibromatosis in children and adolescents: Report of the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer. 2018 Jan 5.[↩]
- Mercier KA, Al-Jazrawe M, Poon R, Acuff Z, Alman B. A Metabolomics Pilot Study on Desmoid Tumors and Novel Drug Candidates. Sci Rep. 2018 Jan 12. 8 (1):584.[↩]
- Agresta L, Kim H, Turpin BK, Nagarajan R, Plemmons A, Szabo S, et al. Pazopanib therapy for desmoid tumors in adolescent and young adult patients. Pediatr Blood Cancer. 2018 Jan 31.[↩]
- Desmoid Tumor Treatment & Management. https://emedicine.medscape.com/article/1060887-treatment[↩][↩][↩][↩][↩][↩]
- Gounder M, Ratan R, Alcindor T, et al. Nirogacestat, a γ-Secretase Inhibitor for Desmoid Tumors. N Engl J Med. 2023 Mar 9;388(10):898-912. doi: 10.1056/NEJMoa2210140[↩][↩][↩][↩][↩][↩][↩][↩]
- Mercier KA, Al-Jazrawe M, Poon R, Acuff Z, Alman B. A Metabolomics Pilot Study on Desmoid Tumors and Novel Drug Candidates. Sci Rep. 2018 Jan 12;8(1):584. doi: 10.1038/s41598-017-18921-7[↩]
- Agresta L, Kim H, Turpin BK, Nagarajan R, Plemmons A, Szabo S, Dasgupta R, Sorger JI, Pressey JG. Pazopanib therapy for desmoid tumors in adolescent and young adult patients. Pediatr Blood Cancer. 2018 Jun;65(6):e26968. doi: 10.1002/pbc.26968[↩][↩]
- Johns MS, Merritt WT 3rd, Rhodes L, Ford CN, Thompson M, Lee WM, Sheldon Y, Petrelli NJ, Tiesi GJ. A cost analysis of sorafenib for desmoid tumors. J Oncol Pharm Pract. 2023 Apr;29(3):663-668. doi: 10.1177/10781552221077927[↩]
- Mendenhall WM, Zlotecki RA, Morris CG, Hochwald SN, Scarborough MT. Aggressive fibromatosis. Am J Clin Oncol. 2005 Apr;28(2):211-5. doi: 10.1097/01.coc.0000144817.78549.53[↩]
- Bhama PK, Chugh R, Baker LH, Doherty GM. Gardner’s syndrome in a 40-year-old woman: successful treatment of locally aggressive desmoid tumors with cytotoxic chemotherapy. World J Surg Oncol. 2006 Dec 17. 4:96.[↩]
- Constantinidou A, Jones RL, Scurr M, Al-Muderis O, Judson I. Advanced aggressive fibromatosis: Effective palliation with chemotherapy. Acta Oncol. 2010 Aug 30.[↩]
- Knechtel G, Stoeger H, Szkandera J, Dorr K, Beham A, Samonigg H. Desmoid tumor treated with polychemotherapy followed by imatinib: a case report and review of the literature. Case Rep Oncol. 2010 Aug 6. 3(2):287-93.[↩]
- Colombo C, Foo WC, Whiting D, Young ED, Lusby K, Pollock RE, et al. FAP-related desmoid tumors: a series of 44 patients evaluated in a cancer referral center. Histol Histopathol. 2012 May. 27(5):641-9.[↩]
- Avedian RS, Bitton R, Gold G, Butts-Pauly K, Ghanouni P. Is MR-guided High-intensity Focused Ultrasound a Feasible Treatment Modality for Desmoid Tumors?. Clin Orthop Relat Res. 2016 Mar. 474 (3):697-704.[↩]
- Luo J, Jin K, Qian S, Ma X, Pan Z, Yao W, et al. Single institution experience of split course radiotherapy in patients with desmoid tumors. Onco Targets Ther. 2019. 12:1741-1748.[↩]
- Improta L., Tzanis D., Bouhadiba T., Abdelhafidh K., Bonvalot S. Desmoid tumours in the surveillance era: What are the remaining indications for surgery? Eur. J. Surg. Oncol. 2020;46:1310–1314. doi: 10.1016/j.ejso.2020.04.025[↩]
- Al-Jazrawe M., Au M., Alman B. Optimal therapy for desmoid tumors: Current options and challenges for the future. Expert Rev. Anticancer Ther. 2015;15:1443–1458. doi: 10.1586/14737140.2015.1096203[↩]
- Bonvalot S., Eldweny H., Haddad V., Rimareix F., Missenard G., Oberlin O., Vanel D., Terrier P., Blay J.Y., Le Cesne A., et al. Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur. J. Surg. Oncol. 2008;34:462–468. doi: 10.1016/j.ejso.2007.06.006[↩]
- Salas S., Dufresne A., Bui B., Blay J.Y., Terrier P., Ranchere-Vince D., Bonvalot S., Stoeckle E., Guillou L., Le Cesne A., et al. Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: A wait-and-see policy according to tumor presentation. J. Clin. Oncol. 2011;29:3553–3558. doi: 10.1200/JCO.2010.33.5489[↩]
- de Bénazé G.D., Vigan M., Corradini N., Minard-Colin V., Marie-Cardine A., Verite C., Defachelles A.S., Thebaud E., Castex M.P., Sirvent N., et al. Functional analysis of young patients with desmoid-type fibromatosis: Initial Surveillance Does not Jeopardize Long Term Quality of life. Eur. J. Surg. Oncol. 2020;46:1294–1300. doi: 10.1016/j.ejso.2020.02.028[↩][↩]
- Grignol V.P., Pollock R., Howard J.H.H. Management of Desmoids. Surg. Clin. N. Am. 2016;96:1015–1030. doi: 10.1016/j.suc.2016.05.008[↩][↩]
- van Houdt W.J., Husson O., Patel A., Jones R.L., Smith M.J., Miah A.B., Messiou C., Moskovic E., Al-Muderis O., Benson C., et al. Outcome of primary desoid tumors at all anatomic locations initially managed with active surveillance. Ann. Surg. Oncol. 2019;26:4699–4706. doi: 10.1245/s10434-019-07826-6[↩][↩]
- Crago A.M., Denton B., Salas S., Dufresne A., Mezhir J.J., Hameed M., Gonen M., Singer S., Brennan M.F. A prognostic nomogram for prediction of recurrence in desmoid fibromatosis. Ann. Surg. 2013;258:347–353. doi: 10.1097/SLA.0b013e31828c8a30[↩]
- Yang S., Wang X., Jiang H., Wang Y., Li Z., Lu H. Effective treatment of aggressive fibromatosis with celecoxib guided by genetic testing. Cancer Biol. Ther. 2017;18:757–760. doi: 10.1080/15384047.2017.1373215[↩]
- Janinis J., Patriki M., Vini L., Aravantinos G., Whelan J.S. The pharmacological treatment of aggressive fibromatosis: A systematic review. Ann. Oncol. 2003;14:181–190. doi: 10.1093/annonc/mdg064[↩]
- Garbay D., Le Cesne A., Penel N., Chevreau C., Marec-Berard P., Blay J.Y., Debled M., Isambert N., Thyss A., Bompas E., et al. Chemotherapy in patients with desmoid tumors: A study from the French Sarcoma Group (FSG) Ann. Oncol. 2012;23:182–186. doi: 10.1093/annonc/mdr051[↩][↩]
- Skubitz K.M., Manivel J.C., Clohisy D.R., Frolich J.W. Response of imatinib-resistant extra-abdominal aggressive fibromatosis to sunitinib: Case report and review of the literature on response to tyrosine kinase inhibitors. Cancer Chemother Pharmacol. 2009;64:635–640. doi: 10.1007/s00280-009-1010-0[↩]
- Chugh R., Wathen J.K., Patel S.R., Maki R.G., Meyers P.A., Schuetze S.M., Priebat D.A., Thomas D.G., Jacobson J.A., Samuels B.L., et al. Efficacy of imatinib in aggressive fibromatosis: Results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin. Cancer Res. 2010;16:4884–4891. doi: 10.1158/1078-0432.CCR-10-1177[↩]
- Toulmonde M., Pulido M., Ray-Coquard I., Andre T., Isambert N., Chevreau C., Penel N., Bompas E., Saada E., Bertucci F., et al. Pazopanib or methotrexate–vinblastine combination chemotherapy in adult patients with progressive desmoid tumours (DESMOPAZ): A non-comparative, randomised, open-label, multicentre, phase 2 study. Lancet. 2019;20:1263–1272. doi: 10.1016/S1470-2045(19)30276-1[↩]
- Gounder M.M., Mahoney M.R., Van Tine B.A., Ravi V., Attia S., Deshpande H.A., Gupta A.A., Milhem M.M., Conry R.M., Movva S., et al. Sorafenib for Advanced and Refractory Desmoid Tumors. J. Med. 2018;379:2417–2428. doi: 10.1056/NEJMoa1805052[↩][↩]
- Skubitz K.M. Biology and Treatment of Aggressive Fibromatosis or Desmoid Tumor. Mayo Clin. Proc. 2017;92:947–964. doi: 10.1016/j.mayocp.2017.02.012[↩]
- Soto-Miranda MA, Sandoval JA, Rao B, Neel M, Krasin M, Spunt S, Jenkins JJ, Davidoff AM, Ver Halen JP. Surgical treatment of pediatric desmoid tumors. A 12-year, single-center experience. Ann Surg Oncol. 2013 Oct;20(11):3384-90. doi: 10.1245/s10434-013-3090-7[↩]
- Ramirez RN, Otsuka NY, Apel DM, Bowen RE. Desmoid tumor in the pediatric population: a report of two cases. J Pediatr Orthop B. 2009 May;18(3):141-4. doi: 10.1097/BPB.0b013e3283298923[↩]
- Bertani E, Chiappa A, Testori A, Mazzarol G, Biffi R, Martella S, Pace U, Soteldo J, Vigna PD, Lembo R, Andreoni B. Desmoid tumors of the anterior abdominal wall: results from a monocentric surgical experience and review of the literature. Ann Surg Oncol. 2009 Jun;16(6):1642-9. doi: 10.1245/s10434-009-0439-z[↩]
- Ben Haj Amor M, Ploton L, Ceugnart L, Taïeb S. Imagerie par résonance magnétique des tumeurs desmoïdes : critères d’évaluations actuels [Magnetic resonance imaging of desmoid-type fibromatosis: Current evaluation criteria]. Bull Cancer. 2020 Mar;107(3):359-363. French. doi: 10.1016/j.bulcan.2019.11.009[↩]
- Stojadinovic A., Leung D.H., Hoos A., Jaques D.P., Lewis J.J., Brennan M.F. Analysis of the prognostic significance of microscopic margins in 2084 localized primary adult soft tissue sarcomas. Ann. Surg. 2002;235:424–434. doi: 10.1097/00000658-200203000-00015[↩]
- Gronchi A., Casali P.G., Mariani L., Lo Vullo S., Colecchia M., Lozza L., Bertulli R., Fiore M., Olmi P., Santinami M., et al. Quality of surgery and outcome in extra-abdominal aggressive fibromatosis: A series of patients surgically treated at a single institution. J. Clin. Oncol. 2003;21:1390–1397. doi: 10.1200/JCO.2003.05.150[↩]
- Lewis J.J., Boland P.J., Leung D.H., Woodruff J.M., Brennan M.F. The enigma of desmoid tumors. Ann. Surg. 1999;229:866–872. doi: 10.1097/00000658-199906000-00014[↩]
- Cates JM. Pregnancy does not increase the local recurrence rate after surgical resection of desmoid-type fibromatosis. Int J Clin Oncol. 2015 Jun;20(3):617-22. doi: 10.1007/s10147-014-0743-x[↩]
- Cates JM, Stricker TP, Sturgeon D, Coffin CM. Desmoid-type fibromatosis-associated Gardner fibromas: prevalence and impact on local recurrence. Cancer Lett. 2014 Oct 28;353(2):176-81. doi: 10.1016/j.canlet.2014.07.020[↩]
- A Pilot Study Evaluating the Use of mTor Inhibitor Sirolimus in Children and Young Adults With Desmoid-Type Fibromatosis. https://clinicaltrials.gov/study/NCT01265030[↩]
- Gronchi A., Colombo C., Le Péchoux C., Dei Tos A.P., Le Cesne A., Marrari A., Penel N., Grignani G., Blay J.Y., Casali P.G., et al. Sporadic desmoid-type fibromatosis: A stepwise approach to a non-metastasising neoplasm–A position paper from the Italian and the French Sarcoma Group. Ann. Oncol. 2014;25:578–583. doi: 10.1093/annonc/mdt485[↩]
- Janssen M.L., van Broekhoven D.L., Cates J.M., Salas S., Bonvalot S., Grünhagen D.J., Verhoef C. Meta-analysis of the influence of surgical margin and adjuvant radiotherapy on local recurrence after resection of sporadic desmoid-type fibromatosis. Br. J. Surg. 2017;104:347–357. doi: 10.1002/bjs.10477[↩]
- A prognostic nomogram for prediction of recurrence in desmoid fibromatosis. Crago AM, Denton B, Salas S, et al. Ann Surg. 2013;258:347–353. doi: 10.1097/SLA.0b013e31828c8a30[↩]
- Penel N, Coindre JM, Bonvalot S, Italiano A, Neuville A, Le Cesne A, Terrier P, Ray-Coquard I, Ranchere-Vince D, Robin YM, Isambert N, Ferron G, Duffaud F, Bertucci F, Rios M, Stoeckle E, Le Pechoux C, Guillemet C, Courreges JB, Blay JY. Management of desmoid tumours: A nationwide survey of labelled reference centre networks in France. Eur J Cancer. 2016 May;58:90-6. doi: 10.1016/j.ejca.2016.02.008[↩]
- Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: a wait-and-see policy according to tumor presentation. Salas S, Dufresne A, Bui B, et al. J Clin Oncol. 2011;29:3553–3558. doi: 10.1200/JCO.2010.33.5489[↩]
- Gronchi A, Raut CP. Optimal approach to sporadic desmoid tumors: from radical surgery to observation. Time for a consensus? Ann Surg Oncol. 2012 Dec;19(13):3995-7. doi: 10.1245/s10434-012-2636-4[↩]
- Kim Y, Rosario MS, Cho HS, Han I. Factors Associated with Disease Stabilization of Desmoid-Type Fibromatosis. Clin Orthop Surg. 2020 Mar;12(1):113-119. doi: 10.4055/cios.2020.12.1.113[↩]
- Stoeckle E, Coindre JM, Longy M, Binh MB, Kantor G, Kind M, de Lara CT, Avril A, Bonichon F, Bui BN. A critical analysis of treatment strategies in desmoid tumours: a review of a series of 106 cases. Eur J Surg Oncol. 2009 Feb;35(2):129-34. doi: 10.1016/j.ejso.2008.06.1495[↩]
- Gounder MM, Mahoney MR, Van Tine BA, et al. Sorafenib for advanced and refractory desmoid tumors. N Engl J Med. 2018;379(25):2417‐2428[↩]
- Bonvalot S, Ternès N, Fiore M, Bitsakou G, Colombo C, Honoré C, Marrari A, Le Cesne A, Perrone F, Dunant A, Gronchi A. Spontaneous regression of primary abdominal wall desmoid tumors: more common than previously thought. Ann Surg Oncol. 2013 Dec;20(13):4096-102. doi: 10.1245/s10434-013-3197-x[↩]
- Dufresne A, Paturel M, Alberti L, Philippon H, Duc A, Decouvelaere AV, Cassier P, Blay JY. Prediction of desmoid tumor progression using miRNA expression profiling. Cancer Sci. 2015 May;106(5):650-5. doi: 10.1111/cas.12640[↩]
- Desmoid Tumor. https://emedicine.medscape.com/article/1060887-overview#a2[↩][↩][↩]
- Huang PW, Tzen CY. Prognostic factors in desmoid-type fibromatosis: a clinicopathological and immunohistochemical analysis of 46 cases. Pathology. 2010 Feb;42(2):147-50. doi: 10.3109/00313020903494078[↩]
- Meazza C, Bisogno G, Gronchi A, Fiore M, Cecchetto G, Alaggio R, Milano GM, Casanova M, Carli M, Ferrari A. Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer. 2010 Jan 1;116(1):233-40. doi: 10.1002/cncr.24679[↩]
- Constantinidou A, Scurr M, Judson I, Litchman C. Clinical presentation of desmoid tumor. In: Litchman C, ed. Desmoid Tumor. Springer Science; 2012:5‐16.[↩]
- Salas S, Dufresne A, Bui B, Blay JY, Terrier P, Ranchere-Vince D, Bonvalot S, Stoeckle E, Guillou L, Le Cesne A, Oberlin O, Brouste V, Coindre JM. Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: a wait-and-see policy according to tumor presentation. J Clin Oncol. 2011 Sep 10;29(26):3553-8. doi: 10.1200/JCO.2010.33.5489[↩]
- Crago AM, Denton B, Salas S, Dufresne A, Mezhir JJ, Hameed M, Gonen M, Singer S, Brennan MF. A prognostic nomogram for prediction of recurrence in desmoid fibromatosis. Ann Surg. 2013 Aug;258(2):347-53. doi: 10.1097/SLA.0b013e31828c8a30[↩]
- Bishop AJ, Zarzour MA, Ratan R, Torres KE, Feig BW, Wang WL, Lazar AJ, Moon BS, Roland CL, Guadagnolo BA. Long-Term Outcomes for Patients With Desmoid Fibromatosis Treated With Radiation Therapy: A 10-Year Update and Re-evaluation of the Role of Radiation Therapy for Younger Patients. Int J Radiat Oncol Biol Phys. 2019 Apr 1;103(5):1167-1174. doi: 10.1016/j.ijrobp.2018.12.012[↩]
- Quintini C, Ward G, Shatnawei A, Xhaja X, Hashimoto K, Steiger E, Hammel J, Diago Uso T, Burke CA, Church JM. Mortality of intra-abdominal desmoid tumors in patients with familial adenomatous polyposis: a single center review of 154 patients. Ann Surg. 2012 Mar;255(3):511-6. doi: 10.1097/SLA.0b013e31824682d4[↩]
- Colombo C, Miceli R, Lazar AJ, Perrone F, Pollock RE, Le Cesne A, Hartgrink HH, Cleton-Jansen AM, Domont J, Bovée JV, Bonvalot S, Lev D, Gronchi A. CTNNB1 45F mutation is a molecular prognosticator of increased postoperative primary desmoid tumor recurrence: an independent, multicenter validation study. Cancer. 2013 Oct 15;119(20):3696-702. doi: 10.1002/cncr.28271[↩]





{kind=link}