Contents
What is Gaucher disease
Gaucher disease is an inherited disorder where your body does not produce enough of an enzyme called glucocerebrosidase that leads to the build up of fatty deposits in multiple organs within the body, including the spleen, liver, bone marrow and, rarely, the brain. Gaucher disease is a type of lipid metabolism disorder. The signs and symptoms of Gaucher disease vary widely among affected individuals. Researchers have described several types of Gaucher disease based on their characteristic features.
Gaucher disease is a genetically inherited autosomal recessive disorder. To pass it on to a child, both parents need to be either affected by Gaucher disease or by asymptomatic carriers of the disease.
Gaucher disease is known to have a higher prevalence amongst certain cultural and religious groups, including Ashkenazi Jews, and a number of non-Jewish European populations, including Northern Sweden.
Gaucher disease occurs in 1 in 40,000 to 100,000 people in the general population. According to the USA National Gaucher Foundation, 2,500 Americans suffer from Gaucher disease. Type 1 is the most common form of the disorder; it occurs more frequently in people of Ashkenazi (eastern and central European) Jewish heritage than in those with other backgrounds. This form of the condition affects 1 in 500 to 1,000 people of Ashkenazi Jewish heritage. The other forms of Gaucher disease, types 2 and 3, are rare and affect only 1 in 100,000 to 1 in 150,000 births and do not occur more frequently in people of Ashkenazi Jewish descent.
There are three subtypes of Gaucher disease:
- Type 1 Gaucher disease is the most common form, and is the least serious.
- Type 2 Gaucher disease is rare, and fatal in the early stages of life.
- Type 3 Gaucher disease is an intermediate form between types 1 and 2.
Type 1 Gaucher disease is also known as non-neuronopathic Gaucher disease, whilst Gaucher disease type 2 and type 3 Gaucher disease are commonly classified as neuronopathic Gaucher disease. Non-neuronopathic Gaucher disease (type 1 Gaucher disease) can be acute or subacute. Neuronopathic Gaucher disease can be subclassified on the basis of central nervous system involvement.
Gaucher disease has no cure. Treatment options for types 1 and 3 include medicine and enzyme replacement therapy, which is usually very effective. There is no good treatment for the brain damage of types 2 and 3.
Other names for Gaucher disease
- cerebroside lipidosis syndrome
- Gaucher splenomegaly
- Gaucher syndrome
- Gaucher’s disease
- Gauchers disease
- GD
- glucocerebrosidase deficiency
- glucocerebrosidosis
- glucosyl cerebroside lipidosis
- glucosylceramidase deficiency
- glucosylceramide beta-glucosidase deficiency
- glucosylceramide lipidosis
- kerasin histiocytosis
- kerasin lipoidosis
- kerasin thesaurismosis
- lipoid histiocytosis (kerasin type)
Gaucher disease types
The progression of Gaucher disease is heavily dependent upon the subtype of the disease that an individual inherits, as well as the age of onset of the disease.
The most severe type of Gaucher disease is called the perinatal lethal form. This condition causes severe or life-threatening complications starting before birth or in infancy. Features of the perinatal lethal form can include extensive swelling caused by fluid accumulation before birth (hydrops fetalis); dry, scaly skin (ichthyosis) or other skin abnormalities; hepatosplenomegaly; distinctive facial features; and serious neurological problems. As its name indicates, most infants with the perinatal lethal form of Gaucher disease survive for only a few days after birth.
Another form of Gaucher disease is known as the cardiovascular type because it primarily affects the heart, causing the heart valves to harden (calcify). People with the cardiovascular form of Gaucher disease may also have eye abnormalities, bone disease, and mild enlargement of the spleen (splenomegaly).
Gaucher disease type 1 (Nonneuropathic)
Gaucher disease type 1 is the most common form of this condition. Type 1 is also called non-neuronopathic Gaucher disease because the brain and spinal cord (the central nervous system) are usually not affected. The features of this condition range from mild to severe and may appear anytime from childhood to adulthood. People with Type 1 Gaucher disease can display a wide variation of clinical signs – even brothers and sisters with Gaucher disease can be affected very differently – and symptoms may occur at any age.
Common symptoms include:
- General fatigue – lack of energy and stamina, tiredness even after a full night’s sleep due to anemia
- Abdominal irregularities – enlarged spleen and liver, pain, compression of the lungs
- Skeletal irregularities – growth retardation in children, pain and degeneration of joints and bone-covering tissue, loss of bone density leading to widening of bones along the knee joint, curvature of the bones and spontaneous fractures, acute bone infarctions (‘bone crisis’), and bone necrosis (death of bone tissue)
- Blood problems – Increased bleeding tendency such as nosebleeds and bruising, subnormal levels of blood platelets and red and white blood cells, elevated levels of acid phosphatase and plasma proteins.
Rarer signs and symptoms include:
- Decreased ability of the lungs to provide oxygen to the blood
- Loss of appetite and intestinal complaints
- Disruption of normal kidney function
- Yellow-brown pigmentation of the skin and/or non-raised, round, purplish-red spots, especially around the eyes.
Gaucher disease type 1 has a slow and indolent progression. Many individuals are able to live a full life using only symptomatic relief for their symptoms. However, if type 1 Gaucher disease manifests early in life, progression of the disease tends to be more abrupt, and more extensive treatment is usually required. Major signs and symptoms include enlargement of the liver and spleen (hepatosplenomegaly), a low number of red blood cells (anemia), easy bruising caused by a decrease in blood platelets (thrombocytopenia), lung disease, and bone abnormalities such as bone pain, fractures, and arthritis.
Gaucher disease type 2 (acute infantile neuropathic Gaucher disease)
Type 2 Gaucher Disease, called the acute neuropathic form, is characterized by brainstem abnormalities and is usually fatal during the first few years of life.
Types 2 and 3 Gaucher disease are known as neuronopathic forms of the disorder because they are characterized by problems that affect the central nervous system. Type 2 Gaucher disease is a very rare form of the disease that affects the brain as well as the organs affected by Type 1. In addition to the signs and symptoms described in Gaucher disease type 1, type 2 Gaucher disease can cause abnormal eye movements, seizures, spasticity, poor ability to suck and swallow, and enlarged liver and spleen and extensive brain damage. Type 2 Gaucher disease usually causes life-threatening medical problems beginning in infancy (3 months of age). Gaucher disease type 2 usually manifests before 2 years of age and is universally fatal within 2 years.
Gaucher disease type 3 (chronic neuropathic Gaucher disease)
Type 3 Gaucher disease (also very rare) is characterized by slowly progressive neurological disease, though this may vary markedly in severity. Other than nervous system involvement, Type 3 Gaucher symptoms (which usually appear in early childhood) resemble those of Type 1, but it tends to worsen more slowly than type 2.
Gaucher disease type 3 has the most varied course and outcomes, with a life expectancy of 20–40 years. In terms of symptoms and progression, it lies in between type 1 and type 2 Gaucher disease. It has an earlier onset and a more pronounced progression than Gaucher disease type 1.
Three subtypes of type 3 Gaucher disease have been classified:
- Type 3a: Found in Northern Sweden, typically showing symptoms of early dementia.
- Type 3b: Predominantly bone, liver and spleen involvement with some neurological symptoms.
- Type 3c: Rare, with cardiovascular and neurological symptoms.
Gaucher disease prognosis
Individuals with type 1 Gaucher disease are usually able to live a normal lifespan. Recent evidence suggests that individuals with type 1 Gaucher disease may, on average, have a life expectancy nine years less than the normal population.
Type 2 Gaucher disease is universally fatal within two years.
Type 3 Gaucher disease has a life expectancy of 20 to 40 years. However, recent advances in medical therapy are likely to extend the length and quality of life of individuals with type 3 Gaucher disease.
Gaucher disease complications
Gaucher disease can result in:
- Delays in growth and puberty in children
- Gynecological and obstetric problems
- Parkinson’s disease
- Cancers such as myeloma, leukemia and lymphoma
Gaucher disease cause
Mutations in the GBA gene cause Gaucher disease. The GBA gene provides instructions for making an enzyme called beta-glucocerebrosidase. This enzyme breaks down a fatty substance called glucocerebroside into a sugar (glucose) and a simpler fat molecule (ceramide). Mutations in the GBA gene greatly reduce or eliminate the activity of beta-glucocerebrosidase. Without enough of this enzyme, glucocerebroside and related substances can build up to toxic levels within cells. Tissues and organs are damaged by the abnormal accumulation and storage of these substances, causing the characteristic features of Gaucher disease.
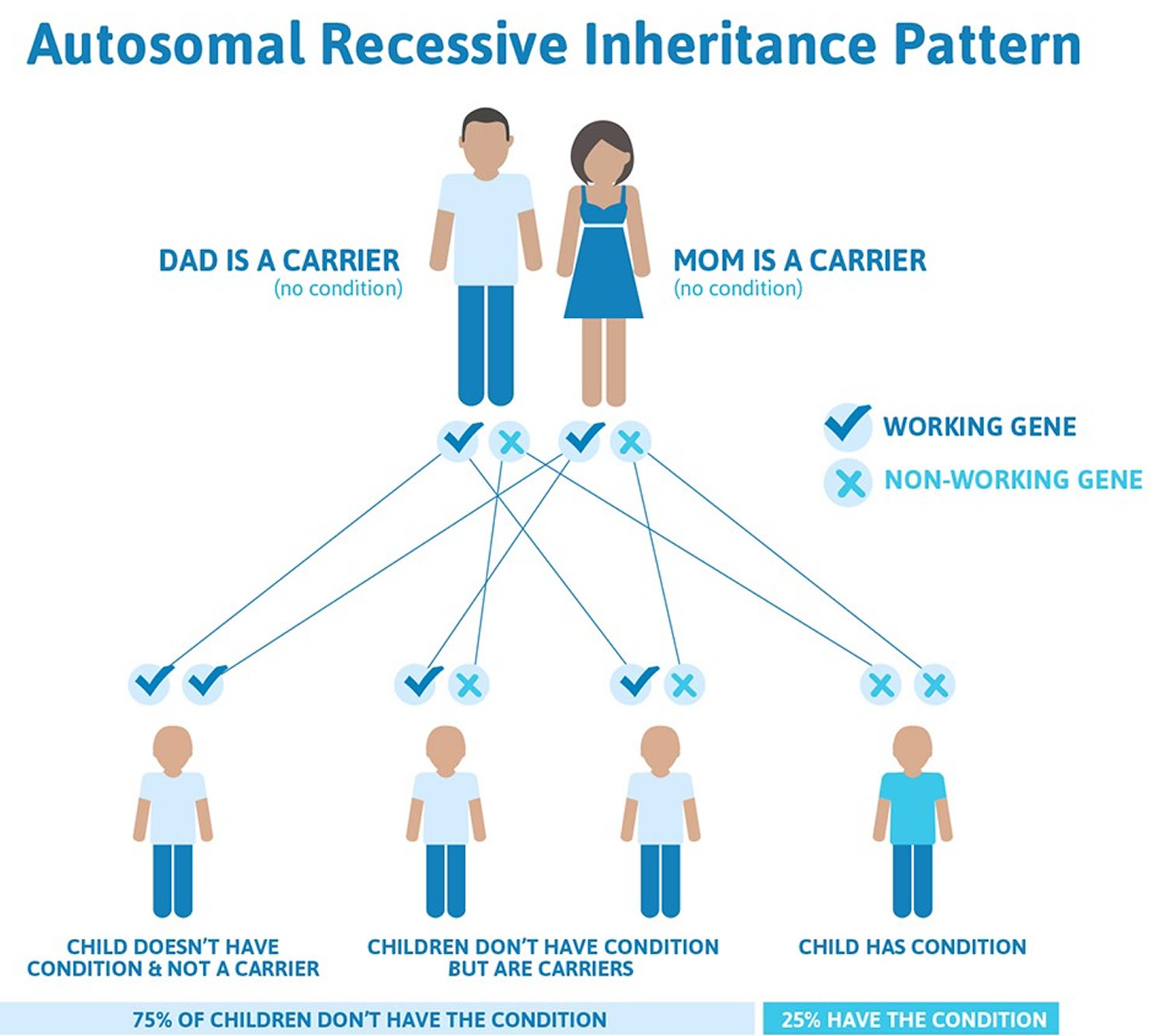
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. As in other recessive disorders, a couple where both people are carriers of the Gaucher disease gene face a 25% chance in each pregnancy that their child will inherit two copies of the altered gene and, in all probability, have the disease.
If you are a Gaucher disease carrier, it means you have just a single gene mutation associated with the disorder. To have the actual disease, you need to have two mutations in the GBA gene; one from your mother and one from your father.
Figure 1. Gaucher disease autosomal recessive inheritance pattern
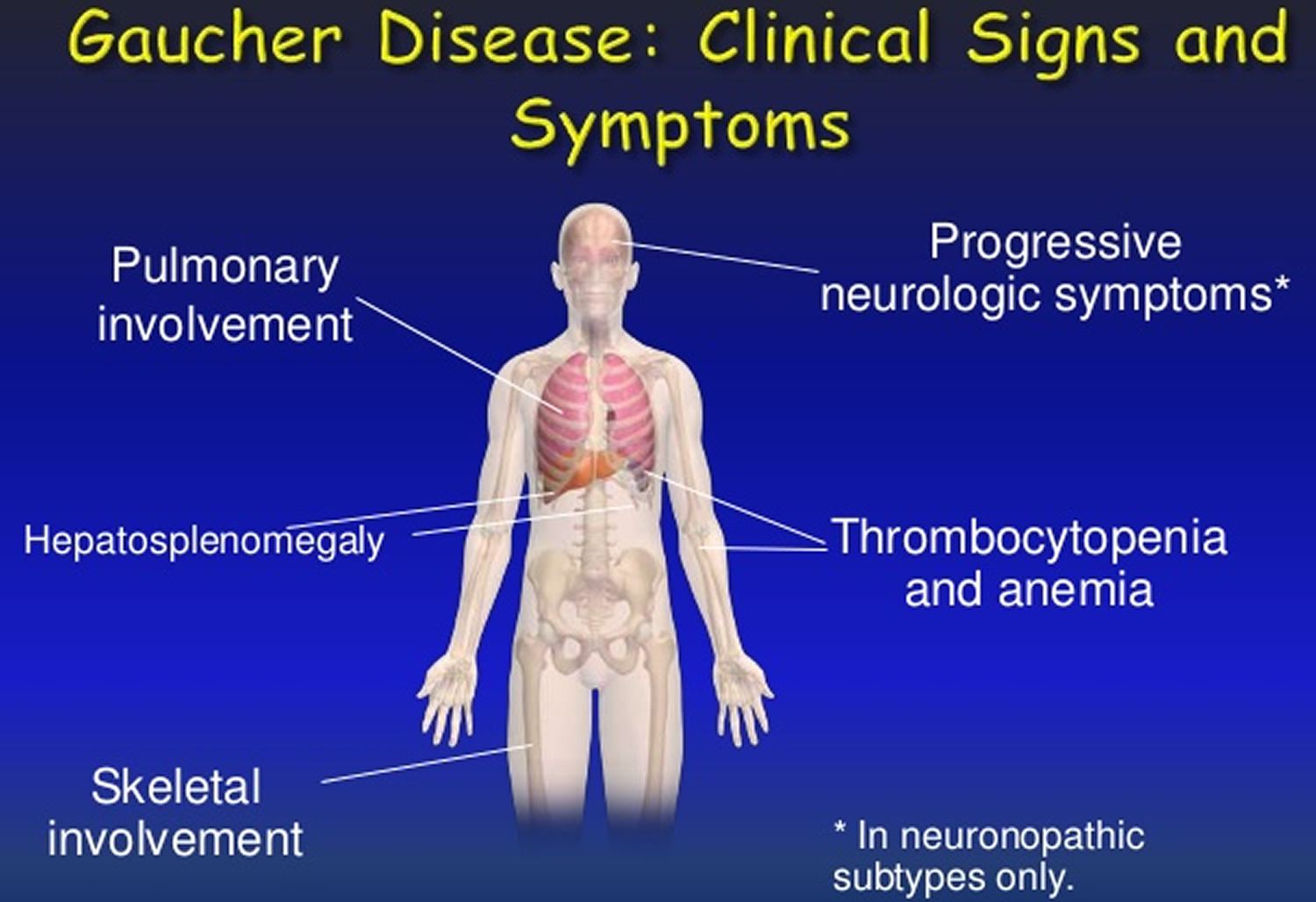
Gaucher disease symptoms
Gaucher disease has a widespread effect on the body, including the enlargement of the liver and spleen which may or may not be symptomatic. This can appear as early satiety, abdominal bloating or discomfort, weight gain or increase in abdominal girth.
Gaucher disease can cause bone pain, fatigue due to anemia, recurrent bleeding disorders (e.g. nose bleeds, heavy periods), painful and enlarged lymph nodes, and recurrent fractures. Affected individuals may also feel tired and generally unwell.
In type 3 Gaucher disease, there may be neurological symptoms affecting vision and cognition.
Blood and organ symptoms and signs
Gaucher disease symptoms and signs involving the blood and organs include:
- Spleen and liver enlargement: When Gaucher cells build up in the spleen and/or liver, these organs become enlarged and can cause your belly to become swollen and painful.
- Low platelet count: A normally functioning spleen disposes of old blood cells. A spleen enlarged by Gaucher disease destroys blood cells too rapidly, including platelets that are responsible for clotting even after minor injuries.
- Bleeding and clotting problems: With fewer platelets, patients with Gaucher disease can have bleeding issues. Low platelet counts can cause problems like frequent nosebleeds, gum bleeding and easy bruising. Low platelets can also result in more serious bleeding problems, particularly after dental work, surgery, trauma and delivering a baby.
- Anemia: Gaucher cells in bone marrow reduce production of blood cells, and the spleen quickly destroys blood cells the body does make. These processes can cause anemia, or low levels of red blood cells that carry energy-producing oxygen to all parts of the body. Patients can also become anemic for other reasons such as iron deficiency or vitamin B12 deficiency. A Gaucher specialist can help you understand and address anemia problems.
- Fatigue: Anemia causes fatigue, and it’s common for patients with Gaucher disease to experience excessive tiredness. However, not all fatigue in Gaucher disease is due to anemia.
- Lungs: In some cases, glucocerebroside may accumulate in the lungs, causing respiratory problems.
Bone symptoms and signs
Bone problems are common in people with Gaucher disease. With early treatment, you can minimize any permanent harm to your bones and joints.
Gaucher disease symptoms and signs affecting the bones include:
- Bone pain and bone crisis: Patients often experience bone pain, including severe episodes called “bone crisis” resulting from reduced blood flow to the bones.
- Bone infarction or avascular necrosis (AVN): This condition occurs when parts of the bone don’t get enough oxygen, causing bone tissue to deteriorate and die. Bone infarction often leads to hip or shoulder problems, severe arthritis and increased fracture risk.
- Osteopenia and osteoporosis: Gaucher disease causes loss of calcium and mineral content in the bones (osteopenia and osteoporosis) in male and female patients of all ages. Smoking, excessive alcohol use, lack of physical activity and certain medications can add to the risk of osteoporosis in patients with Gaucher disease.
- Spontaneous fractures: Osteopenia (bone loss) and osteoporosis weaken the bones, making them more likely to break. Bone fractures in patients with Gaucher disease can occur even without trauma.
- Joint pain, arthritis and joint damage: It is common for patients with Gaucher disease to experience joint pain. Gaucher disease can cause severe arthritis and joint damage, which can be permanent if the disease is untreated.
Neurological signs and symptoms
Neurological (brain stem) symptoms and signs are present only in patients with type 2 or type 3 Gaucher disease. These symptoms and signs can be severe and may cause early death.
Gaucher disease diagnosis
A thorough clinical examination is required if a medical practitioner is exploring the possibility of Gaucher disease. This includes an abdominal examination to look for an enlarged liver or spleen, a cardiorespiratory examination to look for signs in the lungs, and an examination of lymph nodes. Blood tests should also be performed to investigate fatigue and easy bleeding and bruising.
If neurological involvement is suspected, a specialist eye and neurological assessment will also be required.
The definitive method of diagnosing Gaucher disease is by performing a blood test to look at the levels of the defective enzyme.
A number of methods can provide useful information, including:
- Positive clinical examination findings;
- Laboratory investigations (e.g. blood tests, biochemical markers, genotyping and DNA analysis);
- Specialized MRI scans; and
- Bone marrow biopsy if required.
Preconception screening and prenatal testing
You might want to consider genetic screening before starting a family if you or your partner is of Ashkenazi Jewish heritage or if either of you have a family history of Gaucher disease. In some cases, doctors recommend prenatal testing to see if the fetus is at risk of Gaucher disease.
Gaucher disease treatment
Treatments for Gaucher disease include symptomatic relief, enzyme replacement therapy (ERT), substrate reduction therapy (SRT) and bone marrow transplantation. Treatment for Type I Gaucher Disease has traditionally included periodic blood transfusions, partial or total spleen removal, and the use of pain relievers. Other individuals may require joint replacement surgery to improve mobility and quality of life.
More recently enzyme replacement therapy has been successful in slowing and reversing the progression of many symptoms of Gaucher disease. The treatment consists of a modified form of the glucocerebrosidase enzyme given intravvenously. Performed on an outpatient basis, the treatment takes about 1-2 hours and is given every two weeks. Enzyme replacement therapy can stop and often reverse the symptoms of Gaucher disease, allowing patients to enjoy a better quality of life, but is ongoing – probably for life.
Osteoporosis drugs. These types of medication can help rebuild bone weakened by Gaucher disease.
There is no effective treatment for severe brain damage that may occur in persons with types 2 and 3 Gaucher disease.
Symptomatic relief
In the event that adult patients with type 1 Gaucher disease are only mildly affected, symptomatic relief (e.g. pain control, blood transfusions, and treatment of infective exacerbations) may be all that is required. Careful and regular monitoring should be conducted to ensure that irreversible complications do not result from the disease.
Enzyme replacement therapy (ERT)
Enzyme replacement therapy is the current standard of care treatment for moderately to severely affected individuals with Gaucher disease. It aims to supplement the low levels of enzymes that are needed to break down the fat deposits caused by Gaucher disease.
Enzyme replacement therapy is available for most people with types 1 and 3 Gaucher disease. Given intravenously every two weeks, this therapy decreases liver and spleen size, reduces skeletal abnormalities, and reverses other symptoms of the disorder.
Enzyme Replacement Therapy Drugs for Gaucher Disease
The FDA has approved enzyme replacement therapy treatments for Gaucher Disease including the following enzyme replacement therapy drugs:
- Cerezyme® (imiglucerase)
- VPRIV® (velaglucerase alfa)
Talk to a Gaucher specialist to find out which enzyme replacement therapy drug and treatment option is right for you.
The U.S. Food and Drug Administration (FDA) has approved eligustat tartrate for Gaucher treatment, which works by administering small molecules that reduce the action of the enzyme that catalyzes glucose to ceramide.
Enzyme replacement therapy balances low levels of glucocerebrosidase (GCase) enzyme with a modified version of the enzyme. This enzyme breaks down glucocerebroside, the fatty chemical that accumulates in the body of patients with Gaucher disease. (This compensates for the missing enzyme, which is why the therapy is called enzyme replacement.) Patients receive enzyme replacement therapy via IV infusion, which usually takes 1 to 2 hours. The U.S. Food and Drug Administration (FDA) approved the first enzyme replacement therapy in 1991.
Patients typically need an enzyme replacement therapy infusion every 2 weeks, depending on the individual. You can receive enzyme replacement therapy at an infusion center, a Gaucher disease treatment center or at home, and each setting has benefits and drawbacks.
Infusion centers
Many patients receive enzyme replacement therapy at infusion centers, which are medical facilities specializing in IV infusions. Patients travel to the infusion center where they wait for medical staff to prepare the medication. The process can take an hour or more, although some centers will start the preparation when patients are on their way.
Many people find the travel and waiting associated with infusion centers time-consuming and inconvenient. It can also be difficult for children, who often must miss activities and events to receive infusions. Some infusion centers also cannot care for children. Even so, many patients prefer infusion centers because they are familiar, insurance covers their enzyme replacement therapy treatment and they enjoy good relationships with the staff.
Gaucher disease treatment centers
Patients may also receive enzyme replacement therapy infusions at some hospitals and physicians’ offices. This allows Gaucher specialists to more easily monitor key health markers and adjust your medication dosage.
You will still need to travel to your appointment and wait for medication to be prepared if you go to a Gaucher disease treatment center. However, patients may find it more convenient if they can combine appointments and may prefer having experts on hand.
Home infusions
Some patients prefer getting enzyme replacement therapy infusions at home with the help of a home health nurse. The nurse prepares the medication and assists with the IV, so you can receive enzyme replacement therapy treatment in the comfort of your own home.
The main benefit of home infusions is that you don’t have to travel or spend time waiting at infusion centers. Home infusions can improve your quality of life and offer children a more normal childhood.
Drawbacks include not having extra staff on hand if complications occur, like if the nurse has trouble inserting the IV. While getting insurance to cover home infusions can be a hurdle, pharmaceutical case managers can help you navigate your coverage.
Oral Treatment for Gaucher Disease
In addition to enzyme replacement therapy infusion drugs, oral substrate reduction therapy is also available for treating Gaucher disease. This medication works differently from enzyme replacement therapy, and only certain patients can receive it.
Working with a Gaucher specialist is key to determining which treatment is right for you.
Advantages and Disadvantages of Enzyme Replacement Therapy
When deciding between enzyme replacement therapy and substrate reduction therapy, it’s important to weigh the pros and cons of taking enzyme replacement therapy.
Pros of using enzyme replacement therapy to treat Gaucher disease include:
- Fewer side effects: Compared to substrate reduction therapy, enzyme replacement therapy has fewer negative side effects.
- Longer drug history: substrate reduction therapy is newer than enzyme replacement therapy, and some patients prefer taking medications that have been on the market for a longer period of time.
- Wider availability: Unlike substrate reduction therapy, enzyme replacement therapy is available for both children and adults, including pregnant and breastfeeding women.
Cons of taking enzyme replacement therapy as a Gaucher disease treatment include:
- Inconvenience: Many people find taking an substrate reduction therapy pill is more convenient than having to schedule IV infusions.
- More invasive: enzyme replacement therapy requires an IV or a port, a device installed under the skin for repeat use in delivering IV medications. IV access can be a problem for some patients, and the long-term safety of ports over many years of therapy is a potential concern.
- Ups and downs: Some patients feel best right after an infusion and often feel fatigued when they are due for an infusion. Compared to enzyme replacement therapy, substrate reduction therapy provides a more consistent dose.
Paying for Enzyme Replacement Therapy
Enzyme replacement therapy can be very expensive, totaling up to $200,000 or more each year. Insurance may cover most of this cost, and resources are available if you need help paying for enzyme replacement therapy treatments.
These resources include:
- Pharmaceutical companies: Dedicated case managers at pharmaceutical companies can help you navigate your insurance coverage if you have received a Gaucher disease diagnosis.
- CARE and CARE+PLUS Programs: The National Gaucher Foundation offers financial grants for people with Gaucher disease and their families. The CARE Program [care-program] provides assistance with health insurance premiums, while the CARE+PLUS Program can help with genetic testing and other medical expenses not covered by insurance.
- Nonprofits: Organizations like the Patient Access Network, Patient Services and NeedyMeds may also be able to help you with the cost of medication.
- Learn more about financial support for help paying for Gaucher disease treatment 1.
Substrate reduction therapy (SRT)
For individuals who cannot receive enzyme replacement therapy because of treatment failure or unsuitability, an alternative medication is available that aims to reduce the production of fats that are deposited in organs around the body.
The U.S. Food and Drug Administration (FDA) approved the first oral substrate reduction therapy medication for Gaucher disease in 2003. There are currently 2 FDA-approved oral substrate reduction therapy drugs for patients with Gaucher disease:
- Cerdelga® (eliglustat): Approved by the Food and Drug Administration in 2014 for treating the most common form of Gaucher disease, this drug also seems to inhibit the production of fatty substances that build up in people with this this condition. Possible side effects include fatigue, headache, nausea and diarrhea.
- Zavesca® (miglustat): This oral medication appears to interfere with the production of fatty substances that build up in people with Gaucher disease. Diarrhea and weight loss are common side effects.
These drugs act differently in the body and are only approved for use in certain patients. If you are eligible to receive substrate reduction therapy, a Gaucher specialist can help you identify which medication is right for you.
How Substrate Reduction Therapy Works
Substrate reduction therapy partially blocks the body from producing glucocerebroside, the fatty chemical that builds up in the bodies of patients with Gaucher disease. The drug works differently from enzyme replacement therapy for Gaucher disease, which breaks down excess glucocerebroside.
If you think of this process as recycling, enzyme replacement therapy helps the body recycle more waste (glucocerebroside), while substrate reduction therapy helps the body produce less of it to begin with. It does not completely shut down production, but it allows the enzymes you have to keep your body in balance.
Who Can Receive Substrate Reduction Therapy for Gaucher Disease?
Substrate reduction therapy is approved only for certain patients.
The drug is NOT approved for use in:
- Children or teens
- Pregnant or breastfeeding women
- Very elderly patients
- People with severe kidney or liver disease
To receive eliglustat, your metabolic profile (rate at which your body breaks down the drug) must fall within a certain range. Free testing is available from the pharmaceutical manufacturer to determine whether you are eligible. A Gaucher specialist can help you determine the treatment that is right for you, and you should always discuss any side effects with your physician.
Advantages and Disadvantages of Substrate Reduction Therapy for Gaucher Disease
Before you start taking oral substrate reduction therapy, it is important to consider the pros and cons compared with enzyme replacement therapy.
Pros of using substrate reduction therapy to treat Gaucher disease include:
- Convenience: Many people find it more convenient to take a pill than to travel to an infusion center.
- Less invasive: substrate reduction therapy doesn’t require an IV or port, a device installed under your skin for long-term use in delivering IV medication.
- Consistent dosage: substrate reduction therapy provides a consistent dose over time. In comparison, patients receive enzyme replacement therapy only every 2 weeks or so. As a result, some report feeling fatigued right before an infusion and feeling best right after one.
Cons of taking substrate reduction therapy as a Gaucher disease treatment include:
- Side effects: Certain substrate reduction therapy drugs may have unpleasant side effects such as diarrhea, stomach problems and even neuropathy. Neuropathy causes numbness, pain and weakness in areas like your hands and feet.
- Drug interactions: Some substrate reduction therapy drugs can interact with medications that include antibiotics, cardiac drugs and antidepressants. However, you can stop taking substrate reduction therapy to take antibiotics. Make sure your primary care physician and Gaucher specialist know about all the medications you take and whenever you change your medications.
- Sticking to your drug regimen: Some people stop taking their oral medication or change the recommended dose due to the side effects. Some may also forget to take their medication simply because they feel better. It is very important for people with Gaucher disease to continue receiving the treatment to prevent permanent damage to their bodies.
Bone marrow transplantation
Bone marrow transplantation is rarely used now, because complications were high and alternative treatments with enzyme replacement therapy and substrate reduction therapy have proven to be just as effective.
Gene therapy
Gene therapy is a novel approach to treating Gaucher disease. It uses a virus to deliver the missing genes to blood stem cells. This method of treatment has only been trialed in mice and is yet to be proven in humans.
Genetic testing is an important consideration if Gaucher disease is a concern, or if individuals are aware of the disorder occurring in their family. Testing can be arranged to assess for the presence of genetic mutations which may lead to Gaucher disease in the tested individual. Testing can determine if an individual is a carrier of the disease and therefore estimate the risk of passing the disorder on to children.
- National Gaucher Care Foundation Financial Support. https://www.gaucherdisease.org/financial-support/[↩]





{kind=link}