
Contents
- What is hemophilia
- How is hemophilia inherited?
- Severity of hemophilia
- Types of hemophilia
- Hemophilia causes
- Hemophilia symptoms
- Hemophilia complications
- Hemophilia diagnosis
- Hemophilia treatment
What is hemophilia
Hemophilia is a bleeding disorder that slows the blood clotting process. People with hemophilia, their blood does not have enough clotting factor. Clotting factor is a protein in blood that controls bleeding.
People with this condition experience prolonged bleeding or oozing following an injury, surgery, or having a tooth pulled. In severe cases of hemophilia, continuous bleeding occurs after minor trauma or even in the absence of injury (spontaneous bleeding). Serious complications can result from bleeding into the joints, muscles, brain, or other internal organs. Milder forms of hemophilia do not necessarily involve spontaneous bleeding, and the condition may not become apparent until abnormal bleeding occurs following surgery or a serious injury.
The major types of this condition are hemophilia A (also known as classic hemophilia or factor VIII deficiency) and hemophilia B (also known as Christmas disease or factor IX deficiency). The result is the same for people with hemophilia A and B; that is, they bleed for a longer time than normal. Although the two types have very similar signs and symptoms, they are caused by mutations in different genes. People with an unusual form of hemophilia B, known as hemophilia B Leyden, experience episodes of excessive bleeding in childhood but have few bleeding problems after puberty.
Hemophilia C (factor XI deficiency) is a very rare inherited autosomal recessive bleeding disorder. Because the body produces less factor XI than it should, or because the factor XI is not working properly, the clotting reaction is blocked prematurely and the blood clot does not form. Patients with Hemophilia C (factor XI deficiency) do not typically show any spontaneous bleeding or specific symptoms unlike those in hemophilia A or hemophilia B. Sometimes those who have this disorder are identified during special situations such as trauma or surgery. Orthognathic surgery is particularly associated with a high bleeding risk. Therefore, great care must be taken when treating patients with bleeding disorders such as Hemophilia C (factor XI deficiency).
- People are born with hemophilia. They cannot catch it from someone like a cold.
- Hemophilia is quite rare. About 1 in 10,000 people are born with it.
Hemophilia is usually inherited, meaning that it is passed on through a parent’s genes. Genes carry messages about the way the body’s cells will develop as a baby grows into an adult. They determine a person’s hair and eye color, for example.
Sometimes hemophilia can occur when there is no family history of it. This is called sporadic hemophilia. About 30% of people with hemophilia did not get it through their parent’s genes. It was caused by a change in the person’s own genes.
Acquired hemophilia
In rare cases, a person can develop hemophilia later in life. The majority of cases involve middle-aged or elderly people, or young women who have recently given birth or are in the later stages of pregnancy. This condition often resolves with appropriate treatment.
How is hemophilia inherited?
The hemophilia gene is passed down from parent to a child. The genes for hemophilia A and B are on the X chromosome. For this reason, hemophilia is called an X-linked (or sex-linked) disorder.
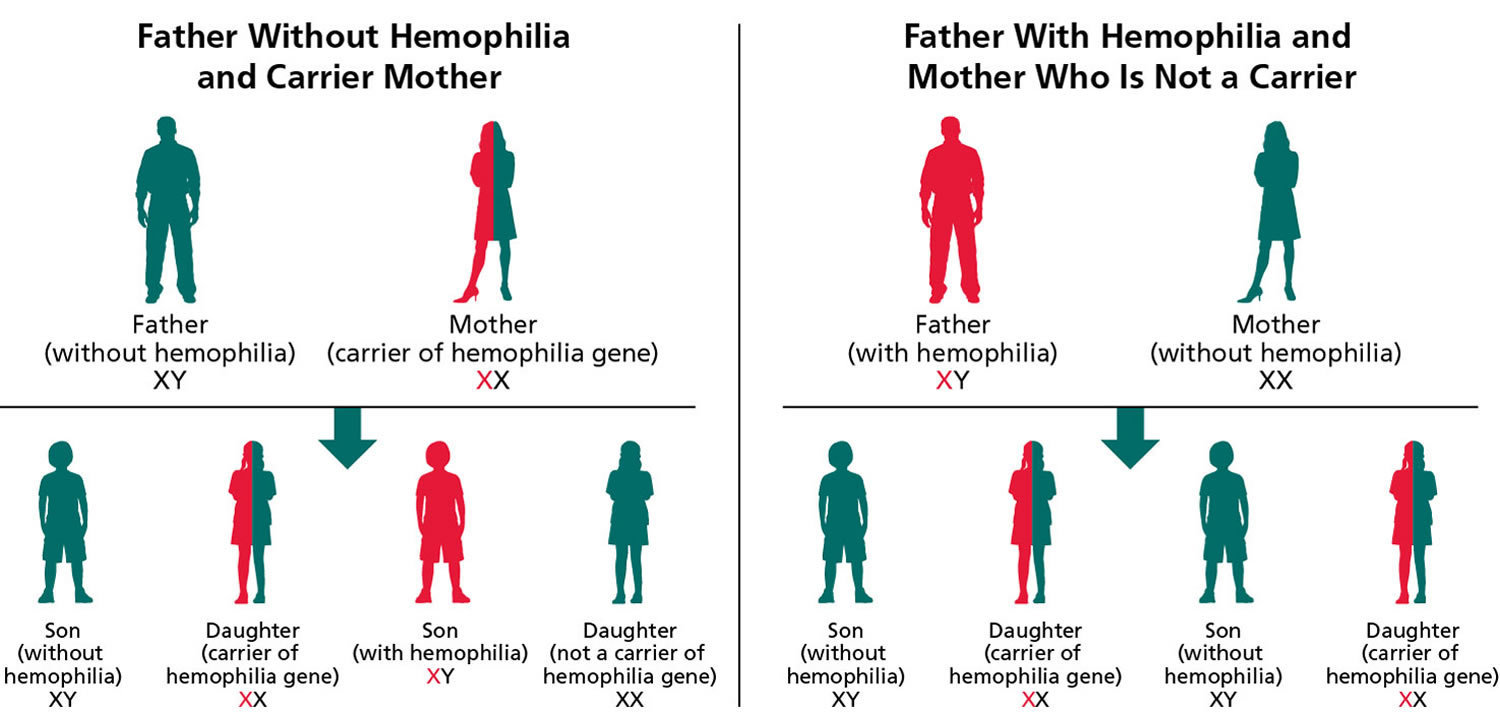
The figure 1 below explains how the hemophilia gene is inherited. When the father has hemophilia but the mother does not, none of the sons will have hemophilia. All the daughters will carry the hemophilia gene.
If a woman inherits a copy of the altered gene from either of her parents, she is said to “carry” the hemophilia gene and is therefore called a “carrier.” In other words, she has one normal and one altered copy of the gene. She can pass either gene onto her children. For each child, there is a 50% chance that a son will have hemophilia and a 50% chance that a daughter will carry the gene. On average, carriers of hemophilia will have about 50 per cent of the normal amount of clotting factor, but some carriers have far lower levels of clotting factor.
Figure 1. How hemophilia inherited – hemophilia A and hemophilia B genetics
Severity of hemophilia
The severity describes how serious a problem is. The level of severity depends on the amount of clotting factor that is missing from a person’s blood.
| Level | Percentage of normal factor activity in blood | Number of international units (IU) per millilitre (ml) of whole blood |
|---|---|---|
| normal range | 50%-150% | 0.50–1.5 IU |
| mild hemophilia | 5%-40% | 0.05–0.40 IU |
| moderate hemophilia | 1%-5% | 0.01–0.05 IU |
| severe hemophilia | less than 1% | less than 0.01 IU |
People with severe hemophilia usually bleed frequently into their muscles or joints. They may bleed one to two times per week. Bleeding is often spontaneous, which means it happens for no obvious reason.
People with moderate hemophilia bleed less frequently, about once a month. They may bleed for a long time after surgery, a bad injury, or dental work. A person with moderate hemophilia will rarely experience spontaneous bleeding.
People with mild hemophilia usually bleed as a result of surgery or major injury. They do not bleed often and, in fact, some may never have a bleeding problem.
Types of hemophilia
Hemophilia A
Hemophilia A is characterized by deficiency in factor VIII clotting activity that results in prolonged oozing after injuries, tooth extractions, or surgery, and delayed or recurrent bleeding prior to complete wound healing. Hemophilia A has also been referred to as “classic hemophilia.”
The birth prevalence of hemophilia A in the United States is approximately 1:6,500 live male births. Worldwide the birth prevalence for hemophilia A and B has been estimated at 1:10,000, although reports vary widely between countries 1.
The birth prevalence is thought to be approximately the same in all countries and all races, presumably because of the high spontaneous mutation rate of clotting factor VIII gene (F8) and its presence on the X chromosome.
The age of diagnosis and frequency of bleeding episodes are related to the level of factor VIII clotting activity.
- Individuals with severe hemophilia A are usually diagnosed during the first two years of life following bleeding from minor mouth injuries and large “goose eggs” from minor head bumps. Without prophylactic treatment, they may average up to two to five spontaneous bleeding episodes each month including spontaneous joint bleeds or deep-muscle hematomas, and prolonged bleeding or excessive pain and swelling from minor injuries, surgery, and tooth extractions or renewed bleeding after initial bleeding has stopped 2.
- Individuals with moderate hemophilia A seldom have spontaneous bleeding; however, they do have prolonged or delayed oozing after relatively minor trauma and are usually diagnosed before age five to six years; the frequency of bleeding episodes varies, usually from once a month to once a year.
- Individuals with mild hemophilia A do not have spontaneous bleeding episodes; however, without pre- and postoperative treatment, abnormal bleeding occurs with surgery or tooth extractions; the frequency of bleeding episodes varies widely, typically from once a year to once every ten years. Individuals with mild hemophilia A are often not diagnosed until later in life. Approximately 30% of heterozygous females have clotting activity below 40% and are at risk for bleeding (even if the affected family member is mildly affected). After major trauma or invasive procedures, prolonged or excessive bleeding usually occurs, regardless of severity.
Hemophilia A signs and symptoms
Hemophilia A should be suspected in an individual with any of the following clinical and/or laboratory features.
Hemophilia A clinical features:
- Hemarthrosis, especially with mild or no antecedent trauma
- Deep-muscle hematomas
- Intracranial bleeding in the absence of major trauma
- Neonatal cephalohematoma or intracranial bleeding
- Prolonged oozing or renewed bleeding after initial bleeding stops following tooth extractions, mouth injury, or circumcision *
- Prolonged or delayed bleeding or poor wound healing following surgery or trauma *
- Unexplained GI bleeding or hematuria *
- Menorrhagia, especially with onset at menarche
- Prolonged nosebleeds, especially recurrent and bilateral *
- Excessive bruising, especially with firm, subcutaneous hematomas
* Of any severity, or especially in more severely affected persons
Muscle hematomas or intracranial bleeding can occur four or five days after the original injury. Intermittent oozing may last for days or weeks after tooth extraction. Prolonged or delayed bleeding or wound hematoma formation after surgery is common. After circumcision, males with hemophilia A of any severity may have prolonged oozing, or they may heal normally without treatment. In severe hemophilia A, spontaneous joint bleeding is the most frequent symptom.
The age of diagnosis and frequency of bleeding episodes in the untreated individual are related to the factor VIII clotting activity (see Table 2). In any affected individual, bleeding episodes may be more frequent in childhood and adolescence than in adulthood. To some extent, this greater frequency is a function of both physical activity levels and vulnerability during more rapid growth.
Individuals with severe hemophilia A are usually diagnosed in the neonatal period due to birth- or neonatal-related procedures or during the first year of life 3. In untreated toddlers, bleeding from minor mouth injuries and large “goose eggs” from minor head bumps are common and are the most frequent presenting symptoms of severe hemophilia A. Intracranial bleeding may also result from head injuries. The untreated child almost always has subcutaneous hematomas; some have been referred for evaluation of possible non-accidental trauma.
As the child grows and becomes more active, spontaneous joint bleeds occur with increasing frequency unless the child is on a prophylactic treatment program. Spontaneous joint bleeds or deep-muscle hematomas initially cause pain or limping before swelling appears. Children and adults with severe hemophilia A who are not treated prophylactically have an average of two to five spontaneous bleeding episodes each month. While joints are the most common sites of spontaneous bleeding, other sites include the kidneys, gastrointestinal tract, and brain. Without prophylactic treatment, individuals with severe hemophilia A have prolonged bleeding or excessive pain and swelling from minor injuries, surgery, and tooth extractions.
Individuals with moderate hemophilia A seldom have spontaneous bleeding but bleeding episodes may be precipitated by relatively minor trauma. Without pretreatment (as for elective invasive procedures) they do have prolonged or delayed oozing after relatively minor trauma and are usually diagnosed before age five to six years. The frequency of bleeding episodes requiring treatment with factor VIII concentrates varies from once a month to once a year. Signs and symptoms of bleeding are otherwise similar to those found in severe hemophilia A.
Individuals with mild hemophilia A do not have spontaneous bleeding. However, without treatment abnormal bleeding occurs with surgery, tooth extractions, and major injuries. The frequency of bleeding may vary from once a year to once every ten years. Individuals with mild hemophilia A are often not diagnosed until later in life when they undergo surgery or tooth extraction or experience major trauma.
Heterozygous females with a factor VIII clotting activity level lower than 40% are at risk for bleeding that is usually comparable to that seen in males with mild hemophilia. However, subtle abnormal bleeding may occur with a baseline factor VIII clotting activity between 35% and 60% or higher 4.
Table 1. Symptoms Related to Severity of Untreated Hemophilia A
- 1. Clinical severity does not always correlate with the in vitro assay result.
Hemophilia A complications
Complications of untreated bleeding. The leading cause of death related to bleeding is intracranial hemorrhage. The major cause of disability from bleeding is chronic joint disease 6. Currently available treatment with clotting factor concentrates is normalizing life expectancy and reducing chronic joint disease for children and adults with hemophilia A. Prior to the availability of such treatment, the median life expectancy for individuals with severe hemophilia A was 11 years (the current life expectancy for affected individuals in several developing countries). Excluding death from HIV, life expectancy for severely affected individuals in the UK receiving adequate treatment was reported in 2007 as 63 years 7.
Since the mid-1960s, the mainstay of treatment of bleeding episodes has been factor VIII concentrates that initially were derived solely from donor plasma. Viral inactivation methods and donor screening of plasmas were introduced by the mid-1980s and recombinant factor VIII concentrates were introduced in the early 1990s, ending the risk of HIV transmission. Many individuals who received plasma-derived factor VIII concentrates from 1979 to 1985 contracted HIV. Approximately half of these individuals died of AIDS prior to the advent of effective HIV therapy.
Hepatitis B transmission from earlier plasma-derived concentrates was eliminated with donor screening and then vaccination in the 1970s. Most individuals exposed to plasma-derived concentrates prior to the late 1980s became chronic carriers of the hepatitis C virus. Viral inactivation methods implemented in concentrate preparation and donor screening assays developed by 1990 have eliminated this complication.
Approximately 30% of individuals with severe hemophilia A develop alloimmune inhibitors to factor VIII, usually within the first 20 exposures to infused factor VIII 8 and, less frequently, in those who have received more than 50 exposures 9. Among individuals with hemophilia A, inhibitors are more prevalent in blacks and Hispanics than whites. Reasons for these disparities are being actively investigated 10.
Hemophilia A diagnosis
The diagnosis of hemophilia A is established in an individual with low factor VIII clotting activity in the presence of a normal, functional von Willebrand factor level. Identification of a hemizygous clotting factor VIII gene (F8) pathogenic variant on molecular genetic testing in a male proband confirms the diagnosis (see Figure 1 below). Identification of a heterozygous clotting factor VIII gene (F8) pathogenic variant on molecular genetic testing in a symptomatic female confirms the diagnosis (see Figure 1 below).
Note:
- Hemizygous refers to a gene normally present in only a single copy; usually an X-linked gene in a male.
- Heterozygous refers to a variant (distinct from the reference sequence) that comprises one of two alleles of a given gene. An individual with two different alleles at a particular locus (one on each chromosome of a pair), one of which is usually pathogenic.
- X-linked inheritance refers to a gene on the X chromosome or to the mode of inheritance in which the causative pathogenic variant is on the X chromosome; hemizygous males will be affected; heterozygous females may or may not be affected depending on the disorder and factors influencing X-chromosome inactivation (see Figure 1 below).
Laboratory features
- Normal platelet count
- Prolonged activated partial thromboplastin time (aPTT)
- Normal prothrombin time (PT)
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with hemophilia A, the following evaluations are recommended if they have not already been completed:
- A personal and family history of bleeding to help predict disease severity
- A joint and muscle evaluation, particularly if the individual describes a history of hemarthrosis or deep-muscle hematomas
- Screening for hepatitis A, B, and C as well as HIV if blood products or plasma-derived clotting factor concentrates were administered prior to 1990
- Baseline complete blood count (CBC) with a platelet count, especially if there is a history of nose bleeds, GI bleeding, mouth bleeding, or (in females) menorrhagia or postpartum hemorrhage
- Referral to a hemophilia treatment center. For locations:
- Worldwide, see World Federation of Haemophilia 11 at https://www.wfh.org/en/page.aspx?pid=1264
- US only, see National Hemophilia Foundation 12 at https://www.hemophilia.org/
- Identification of the specific clotting factor VIII gene (F8) pathogenic variant in an individual to aid in determining disease severity, the likelihood of inhibitor development, and the chance that immune tolerance will be successful if an inhibitor does develop
- Consultation with a clinical geneticist and/or genetic counselor, particularly for a new diagnosis in the family and for females of childbearing years
Hemophilia A genetics
Hemophilia A is inherited in an X-linked manner. The risk to siblings of a proband (index case) depends on the carrier status of the mother. Carrier females have a 50% chance of transmitting the clotting factor VIII gene (F8) pathogenic variant in each pregnancy: sons who inherit the pathogenic variant will be affected; daughters who inherit the pathogenic variant are carriers (see Figure 1 below). Affected males transmit the pathogenic variant to all of their daughters and none of their sons. Carrier testing for at-risk family members and prenatal testing for pregnancies at increased risk are possible if the clotting factor VIII gene (F8) pathogenic variant has been identified or if informative intragenic linked markers have been identified.
Male proband (index case). The diagnosis of hemophilia A is established in a male proband by identification of decreased factor VIII clotting activity and a normal, functional von Willebrand factor level.
- Severe hemophilia A. <1% factor VIII
- Moderate hemophilia A. 1%-5% factor VIII
- Mild hemophilia A. 6%-40% factor VIII
Note: Rarely, in individuals with mild hemophilia A, a standard “one-stage” factor VIII clotting activity assay shows near-normal or low-normal factor VIII clotting activity (40%-80%), whereas in a “two-stage” or chromogenic assay, factor VIII activity is low. Thus, low-normal in vitro clotting activity does not always exclude the presence of mild hemophilia A.
Identification of a hemizygous pathogenic variant in clotting factor VIII gene (F8) by molecular genetic testing can help predict the clinical phenotype, assess the risk of developing a factor VIII inhibitor, and allow family studies (see Table 2 – Molecular Genetic Testing Used in Hemophilia A below).
Female proband (index case). The diagnosis of hemophilia A is established by determination of low factor VIII clotting activity. Carrier status is determined by identification of a heterozygous pathogenic variant in clotting factor VIII gene (F8) by molecular genetic testing (see Table 2). Factor VIII clotting activity is unreliable in the detection of heterozygous females; only approximately 30% of hemophilia A heterozygous females have factor VIII clotting activity lower than 40% 13.
Penetrance
All males with a clotting factor VIII gene (F8) pathogenic variant will be affected and will have approximately the same severity of disease as other affected males in the family. However, other genetic and environmental effects may modify the clinical severity to some extent.
Approximately 30% of females with one clotting factor VIII gene (F8) pathogenic variant and one normal allele have a factor VIII clotting activity lower than 40% and a bleeding disorder; mild bleeding can occur in heterozygous females with low-normal factor VIII activity 13. Overall, carriers have more bleeding than unaffected females 4.
Figure 2. Hemophilia A – X-linked inheritance pattern
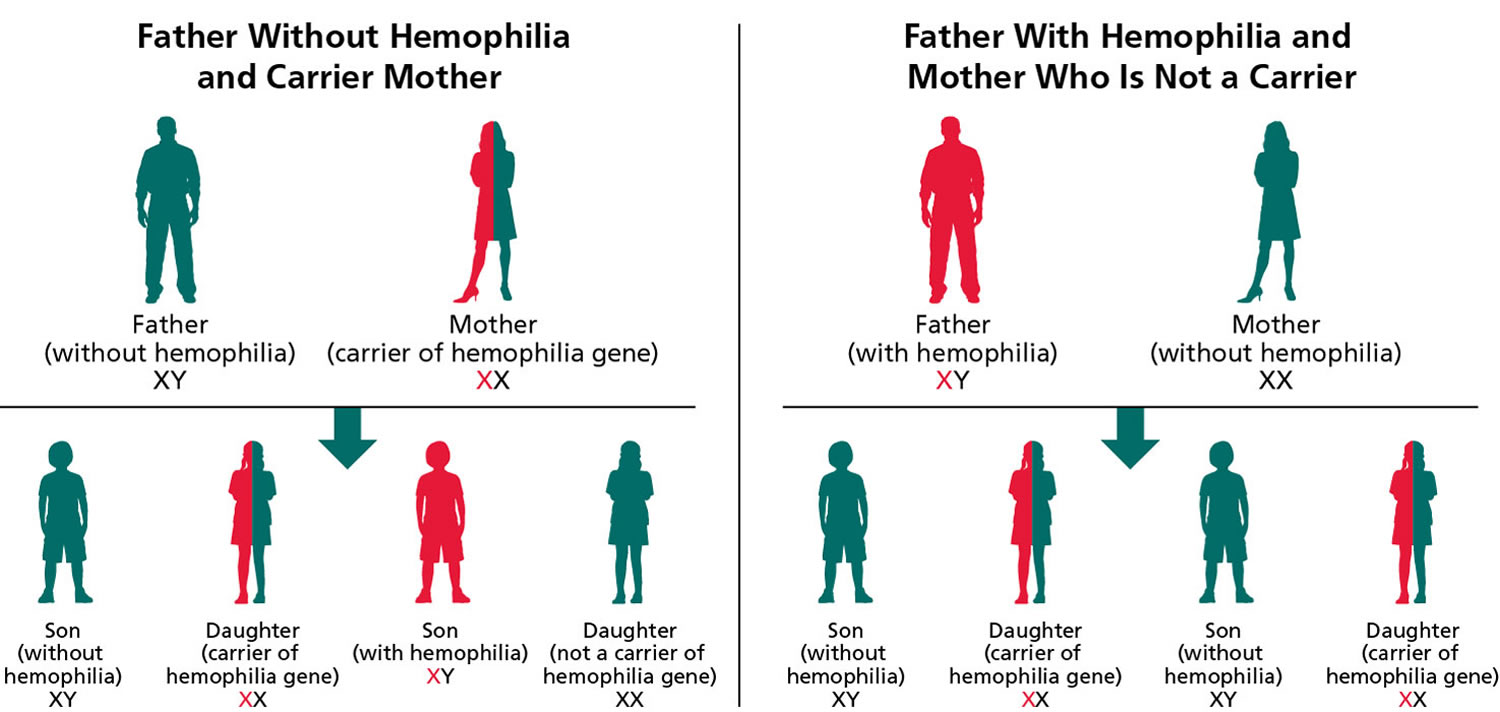
Note: Hemophilia A is inherited in an X-linked manner. Referring to a gene on the X chromosome or to the mode of inheritance in which the causative pathogenic variant is on the X chromosome; hemizygous males will be affected; heterozygous females may or may not be affected depending on the disorder and factors influencing X-chromosome inactivation.
Hemophilia A Molecular Genetic Testing
Molecular genetic testing approaches can include single-gene testing, use of a multi-gene panel, and more comprehensive genomic testing:
- Single-gene testing. Targeted analysis for the intron 22 or intron 1 inversion is frequently performed first in (a) individuals with severe hemophilia A, (b) females with a family history of severe hemophilia A, or (c) females with a family history of hemophilia A of unknown severity in whom the family-specific pathogenic variant is not known. Sequence analysis of clotting factor VIII gene (F8) followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found is performed if the common intron 22 or intron 1 inversion is not detected.
- A multi-gene panel that includes clotting factor VIII gene (F8) and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and over time. (2) Some multi-gene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multi-gene panel provides the best opportunity to identify the genetic cause of the condition at the most reasonable cost while limiting secondary findings. (3) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing based tests. (4) The ability of panels to detect structural variants in clotting factor VIII gene (F8), a common cause of hemophilia A, should be confirmed.
- More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered if serial single-gene testing [and/or use of a multi-gene panel that includes clotting factor VIII gene (F8)] fails to confirm a diagnosis in an individual with features of hemophilia A. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
Table 2. Molecular Genetic Testing Used in Hemophilia A
| Gene | Test Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by This Method | |
|---|---|---|---|
| Severe Hemophilia A | Moderate or Mild Hemophilia A | ||
| F8 | Targeted mutation analysis | ~48% | 0% |
| Sequence analysis | ~43%-51% | 76%-99% | |
| Gene-targeted deletion/duplication analysis | 1.5% | 0.2% | |
Genotype-Phenotype Correlations
Evidence for an association between variant type and disease severity:
- F8 intron (noncoding sequence of DNA removed from mature messenger RNA prior to translation) 22 inversions are associated with severe hemophilia A and account for 45% of individuals with severe hemophilia A 14. Of these, 20% to 30% develop alloimmune inhibitors. Occasionally, individuals considered to have moderate hemophilia A have been found to have F8 inversions. Often their assays have contained either some residual factor VIII clotting activity from a prior transfusion or the assay methods used were inaccurate at low levels.
- An inversion between a 1-kb sequence in intron 1 and an inverted repeat 5′ to F8 15 is also associated with a severe phenotype, and some individuals have developed inhibitors.
- Single-nucleotide variants leading to new stop codons are essentially all associated with a severe phenotype, as are most frameshift variants. (An exception is the insertion or deletion of adenosine bases resulting in a sequence of eight to ten adenosines, which may result in moderate hemophilia A 16.
- Splice site variants often result in severe disease, but can result in mild or moderate disease, depending on the specific change and location.
- Missense variants occur in fewer than 20% of individuals with severe hemophilia A but are found in nearly all of those with a diagnosis of mild or moderate disease.
- A single-base change in the 5’ promoter region of F8 has been associated with mild hemophilia A 17.
Hemophilia A treatment
The World Federation of Hemophilia has published treatment guidelines 18 for the management of individuals with hemophilia. Treatment should be coordinated through a hemophilia treatment center. Individuals in the USA see National Hemophilia Foundation 12; individuals worldwide see World Federation of Hemophilia for locations 19
- Treatment of Hemophilia A manifestations: Referral to a hemophilia treatment center to facilitate treatment; intravenous infusion of factor VIII concentrate is most effective when infused within one hour of the onset of bleeding; training to facilitate home infusions administered by parents; immune tolerance therapy. For those with mild disease, including most symptomatic females, immediate treatment of bleeding with intravenous or nasal desmopressin acetate or factor VIII concentrate.
- Intravenous infusion of plasma-derived or recombinant factor VIII for bleeding episodes within an hour of noticing symptoms:
- Dosing is weight based and target levels and duration of treatment vary by the severity of bleeding and/or the risk associated with the surgery or procedure.
- Staff members who are expert in performing venipunctures in infants and toddlers should be identified, as frequent venipunctures may be necessary.
- Parents of children age two to five years with severe hemophilia A should be trained to administer the infusions. Home treatment allows for prompt treatment and facilitates prophylactic therapy.
- Pediatric issues. Special considerations for care of infants and children with hemophilia A include the following 20:
- Infant males with a family history of hemophilia A should not be circumcised unless hemophilia A is either excluded or, if present, is treated with factor VIII concentrate directly before and after the procedure.
- Immunizations should be administered preferably subcutaneously; intramuscular injections should be avoided unless under factor coverage.
- Effective dosing of factor VIII requires an understanding of different pharmacokinetics in young children.
- DDAVP® (desmopressin acetate). For individuals with mild hemophilia A, including symptomatic females, immediate treatment of bleeding can be achieved with DDAVP®. A single intravenous dose often doubles or triples factor VIII clotting activity. Alternatively, a multi-use nasal formulation of DDAVP® Nasal may be more convenient.Note: Hemophilia genotype influences DDAVP® response 21.
- Immune tolerance therapy. Alloimmune inhibitors to factor VIII greatly compromise the ability to manage bleeding episodes 2. High titer inhibitors can often be eliminated by immune tolerance therapy 22. Individuals with large gene deletions are less likely to respond to immune tolerance than individuals with other types of variants 23.
- Prevention of primary manifestations: For those with severe disease, prophylactic infusions of factor VIII concentrate three times a week or every other day to maintain factor VIII clotting activity higher than 1% nearly eliminates spontaneous bleeding and prevents chronic joint disease. Newer modified recombinant products with longer half-lives allow less frequent infusions.
- Prophylactic treatment is recommended by the National Hemophilia Foundation and the World Federation of Hemophilia for children with severe hemophilia and is usually administered as infusions of factor VIII concentrate three times a week or every other day to maintain factor VIII clotting activity above 1%, although a less intense regimen may provide protection for some affected boys 24. Newer modified recombinant products with longer half-lives allow less frequent infusions.
- Factor VIII concentrate infusions given prophylactically in young boys before or just after their first few joint bleeds can nearly eliminate spontaneous bleeding and prevent chronic joint disease 25.
- The greatest benefit is seen in affected individuals who start therapy before age 2.5 to three years. Routine prophylaxis begun later in childhood or in adults significantly decreases bleeding episodes 26.
- “Secondary” prophylaxis, started after some joint damage has occurred, can be given on a long-term basis or around periods of increased activity or surgical procedures. An increasing number of adults with hemophilia are on long-term prophylaxis and clinical benefit is being documented in short- and long-term studies.
- Prevention of secondary complications: Reduction of bleeding and chronic joint disease is achieved by prophylactic treatment and prompt effective treatment of bleeding, including by use of home therapy. Many recombinant products are now created without human- or animal-derived proteins in the process or final product. Virucidal treatment of plasma-derived concentrates has eliminated the risk of HIV transmission since 1985, and of hepatitis B and C viruses since 1990.
- Surveillance: Persons with hemophilia who are followed at hemophilia treatment centers have lower mortality than those who are not 27. Young children with severe or moderate hemophilia A should be evaluated at a hemophilia treatment center (accompanied by their parents) every six to 12 months to review their history of bleeding episodes and adjust treatment plans as needed. Early signs and symptoms of possible bleeding episodes are reviewed. The assessment should also include a joint and muscle evaluation, an inhibitor screen, and a discussion of any other problems related to the individual’s hemophilia and family and community support.
- For individuals with severe or moderate hemophilia A, assessments including inhibitor screen every six to 12 months at a hemophilia treatment center are recommended; for individuals with mild hemophilia A, assessment at a hemophilia treatment center every one to two years. Comorbidities may require more frequent visits.
- Screening for alloimmune inhibitors is indicated at least once during the first ten to 20 treatment days in children with severe hemophilia, and then every three to six months after treatment with factor VIII concentrates has been initiated either for bleeding or prophylaxis. After 50 to 100 exposure days, annual screening and screening prior to elective surgical procedures is sufficient. Testing for inhibitors should be performed in any individual with hemophilia whenever a suboptimal clinical response to treatment is suspected, regardless of disease severity.
- Older children and adults with severe or moderate hemophilia A benefit from at least yearly contact with a hemophilia treatment center and periodic assessments to review bleeding episodes and treatment plans, evaluate joints and muscles, screen for an inhibitor, perform viral testing if indicated, provide education, and discuss other issues relevant to the individual’s hemophilia.
- Individuals with mild hemophilia A can benefit from an assessment at a hemophilia treatment center every one to two years. Affected individuals with comorbidities and other complications or treatment challenges may require more frequent visits.
- Agents/circumstances to avoid: Circumcision of at-risk males until hemophilia A is either excluded or treated with factor VIII concentrate regardless of severity; intramuscular injections; activities with a high risk of trauma, particularly head injury; cautious, if any, use of medications and herbal remedies that affect platelet function, including aspirin. Medications and herbal remedies that affect platelet function, including aspirin, should be avoided unless there is strong medical indication, such as in individuals with atherosclerotic cardiovascular disease. Individuals with severe hemophilia usually require clotting factor prophylaxis to allow aspirin and other platelet inhibitory drugs to be used safely 28.
- Avoid the following:
- Intramuscular injections
- Activities that involve a high risk of trauma, particularly of head injury.
- Evaluation of relatives at risk: To clarify genetic status of females at risk before pregnancy or early in pregnancy and to facilitate management.
- Identification of at-risk relatives. A thorough family history may identify other relatives who are at risk but have not been tested (particularly in families with mild hemophilia A).
- Early determination of the genetic status of males at risk. Either assay of factor VIII clotting activity from a cord blood sample obtained by venipuncture of the umbilical vein (to avoid contamination by amniotic fluid or placenta tissue) or molecular genetic testing for the family-specific F8 pathogenic variant can establish or exclude the diagnosis of hemophilia A in newborn males at risk. Infants with a family history of hemophilia A should not be circumcised unless hemophilia A is either excluded or, if present, factor VIII concentrate is administered immediately before and after the procedure to prevent delayed oozing and poor wound healing.Note: Ideally, the cord blood for factor VIII clotting activity assay should be drawn into a syringe containing one-tenth volume of sodium citrate to avoid clotting and to provide an optimal mixing of the sample with the anticoagulant. If not available a standard blue top tube can be used.
- Determination of genetic status of females at risk. Approximately 30% of heterozygous females have factor VIII activity lower than 40% and may have abnormal bleeding. In a survey of Dutch heterozygous females, bleeding symptoms correlated with baseline factor clotting activity; there was suggestion of a very mild increase in bleeding even in those with 40% to 60% factor VIII activity [Plug et al 2006]. Therefore, all daughters and mothers of an affected male and other at-risk females should have a baseline factor VIII clotting activity assay to determine if they are at increased risk for bleeding (unless they are known to be non-carriers based on molecular genetic testing). Very occasionally, a female will have particularly low factor VIII clotting activity that may result from heterozygosity for an F8 pathogenic variant associated with skewed X-chromosome inactivation or, on rare occasion, compound heterozygosity for two F8 pathogenic variants 29.It is recommended that the carrier status of a female at risk be established prior to pregnancy or as early in a pregnancy as possible.
- Pregnancy management: Monitor heterozygous females during pregnancy and for delayed bleeding post partum unless it is known that their baseline factor VIII clotting activity is normal.
- Obstetric issues. It is recommended that the carrier status of a female at risk be established prior to pregnancy or as early in a pregnancy as possible. If the female is symptomatic (i.e., has baseline factor VIII clotting activity <40%), she will be somewhat protected by the natural rise of factor VIII clotting activity during pregnancy, which may even double by the end of the third trimester. The factor VIII level should be measured in the third trimester to confirm that the level is in the normal range, and if it is not, a plan for factor replacement therapy should be developed. Postpartum factor VIII clotting activity can return to baseline within 48 hours, and postpartum hemorrhage may ensue 30.
- Newborn males. Controversy remains as to indications for cesarean section versus vaginal delivery 31. In retrospective data analysis of 580 males age 0-2 years with hemophilia, 17 suffered intracranial hemorrhages with delivery, and all but one were delivered vaginally 3. This finding supports the recommendation of cesarean section for hemophilic infants, however, 12 of the 17 were born to women not known to be carriers, suggesting that a planned delivery may mitigate risks. In anticipation of delivery, the relative risks of cesarean section versus vaginal delivery should be considered and discussed with the family and obstetrician in anticipation of delivery so that a coordinated plan can be developed.
- Therapies under investigation: Ongoing clinical trials of longer-acting factor VIII concentrates, bypassing agents, and gene therapy. Two products are FDA approved, one modified by pegylation and one by Fc fusion; others are in clinical trials. These result in half-life extension by approximately 1.5 fold. Products with greater half-life extension are under development.
- Other: Vitamin K does not prevent or control bleeding in hemophilia A; cryoprecipitate contains factor VIII but does not undergo viral inactivation so is no longer used to treat hemophilia A.
Hemophilia B
Hemophilia B is characterized by deficiency in factor IX clotting activity that results in prolonged oozing after injuries, tooth extractions, or surgery, and delayed or recurrent bleeding prior to complete wound healing. The age of diagnosis and frequency of bleeding episodes are related to the level of factor IX clotting activity.
The birth prevalence of hemophilia B is approximately one in 30,000 live male births worldwide. Hemophilia B is about one fifth as prevalent as hemophilia A.
The birth prevalence is the same in all countries and all races, presumably because of the high spontaneous mutation rate of clotting factor IX gene (F9) and its presence on the X chromosome.
In individuals with severe hemophilia B, spontaneous joint or deep-muscle bleeding is the most frequent sign. Individuals with severe hemophilia B are usually diagnosed during the first two years of life; without prophylactic treatment, they may average up to two to five spontaneous bleeding episodes each month.
Individuals with moderate hemophilia B seldom have spontaneous bleeding; however, they do have prolonged or delayed oozing after relatively minor trauma and are usually diagnosed before age five to six years; the frequency of bleeding episodes varies from once a month to once a year.
Individuals with mild hemophilia B do not have spontaneous bleeding episodes; however, without pre- and postoperative treatment, abnormal bleeding occurs with surgery or tooth extractions; the frequency of bleeding may vary from once a year to once every ten years. Individuals with mild hemophilia B are often not diagnosed until later in life.
In any individual with hemophilia B, bleeding episodes may be more frequent in childhood and adolescence than in adulthood. Approximately 30% of heterozygous females have factor IX clotting activity lower than 40% and are at risk for bleeding (even if the affected family member has mild hemophilia B), although symptoms are usually mild. After major trauma or invasive procedures, prolonged or excessive bleeding usually occurs, regardless of severity.
Hemophilia B signs and symptoms
Hemophilia B in the untreated individual is characterized by immediate or delayed bleeding or prolonged oozing after injuries, tooth extractions, or surgery or renewed bleeding after initial bleeding has stopped 32. Muscle hematomas or intracranial bleeding can occur immediately or up to four to five days after the original injury. Intermittent oozing may last for days or weeks after tooth extraction. Prolonged or delayed bleeding or wound hematoma formation after surgery is common. After circumcision, males with hemophilia B of any severity may have prolonged oozing, or they may heal normally. In severe hemophilia B, spontaneous joint bleeding is the most frequent sign.
The age of diagnosis and frequency of bleeding episodes are generally related to the factor IX clotting activity (see Table 3). In any affected individual, bleeding episodes may be more frequent in childhood and adolescence than in adulthood. To some extent, this greater frequency is a function of both physical activity levels and vulnerability during more rapid growth.
Hemophilia B should be suspected in an individual with any of the following clinical and/or laboratory features.
Individuals with severe hemophilia B are usually diagnosed as newborns due to birth- or neonatal-related procedures or during the first year of life 33. In untreated toddlers, bleeding from minor mouth injuries and large “goose eggs” from minor head bumps are common; these are the most frequent presenting symptoms of severe hemophilia B. Intracranial bleeding may also result from head injuries. The untreated child almost always has subcutaneous hematomas; some have been referred for evaluation of possible non-accidental trauma.
As the child grows and becomes more active, spontaneous joint bleeds occur with increasing frequency unless the child is on a prophylactic treatment program. Spontaneous joint bleeds or deep-muscle hematomas initially cause pain or limping before swelling appears. Children and young adults with severe hemophilia B who are not treated have an average of two to five spontaneous bleeding episodes each month. Joints are the most common sites of spontaneous bleeding; other sites include the muscles, kidneys, gastrointestinal tract, brain, and nose. Without prophylactic treatment, individuals with hemophilia B have prolonged bleeding or excessive pain and swelling from minor injuries, surgery, and tooth extractions.
Individuals with moderate hemophilia B seldom have spontaneous bleeding but bleeding episodes may be precipitated by relatively minor trauma. Without pretreatment (as for elective invasive procedures) they do have prolonged or delayed oozing after relatively minor trauma and are usually diagnosed before age five to six years. The frequency of bleeding episodes requiring treatment with factor IX concentrates varies from once a month to once a year. Signs and symptoms of bleeding are otherwise similar to those found in severe hemophilia B.
Individuals with mild hemophilia B do not have spontaneous bleeding. However, without treatment, abnormal bleeding occurs with surgery, tooth extractions, and major injuries. The frequency of bleeding may vary from once a year to once every ten years. Individuals with mild hemophilia B are often not diagnosed until later in life when they undergo surgery or tooth extraction or experience major trauma.
Heterozygous females with a factor IX clotting activity level lower than 40% are at risk for bleeding that is usually comparable to that seen in males with mild hemophilia. However, more subtle abnormal bleeding may occur with baseline factor IX clotting activity levels between 30% and 60% 13.
Table 3. Symptoms Related to Severity of Untreated Hemophilia B
- 1. Clinical severity does not always correlate with the in vitro assay result.
Hemophilia B complications
Complications of untreated bleeding. The leading cause of death related to bleeding is intracranial hemorrhage. The major cause of disability from bleeding is chronic joint disease 6. Currently available treatment with clotting factor concentrates is normalizing life expectancy and reducing chronic joint disease for children and adults with hemophilia B. Prior to the availability of such treatment, the median life expectancy for individuals with severe hemophilia B was 11 years (the current life expectancy for affected individuals in several developing countries). Excluding death from HIV, life expectancy for those severely affected individuals receiving adequate treatment was 63 years in 2000 7, having been greatly improved with factor replacement therapy 35.
Since the late1960s, the mainstay of treatment of bleeding episodes has been factor IX concentrates that initially were derived solely from donor plasma. By the late 1970s, more purified preparations became available, reducing the risk for thrombogenicity. Viral inactivation methods and donor screening of plasmas were introduced by 1990 and a recombinant factor IX concentrate became available shortly thereafter 36. A second recombinant factor IX concentrate was FDA licensed in 2013. Two long-acting modified recombinant factor IX concentrates are now FDA approved, extending the factor IX half-life three- to fivefold compared to unmodified products 37 . HIV transmission from concentrates occurred between 1979 and 1985. Approximately half of these individuals died of AIDS prior to the advent of effective HIV therapy.
Hepatitis B transmission from earlier plasma-derived concentrates was eliminated with donor screening and then vaccination introduced in the 1970s. Most individuals exposed to plasma-derived concentrates prior to the late 1980s became chronic carriers of the hepatitis C virus. Viral inactivation methods implemented in concentrate preparation and donor screening assays developed by 1990 have essentially eliminated hepatitis C transmission from plasma-derived concentrates.
Alloimmune inhibitors occur much less frequently than in hemophilia A. Approximately 2% of individuals with severe hemophilia B develop alloimmune inhibitors to factor IX 38. These individuals usually have partial- or whole-gene deletions or certain nonsense variants. At times, the onset of an alloimmune response has been associated with anaphylaxis to transfused factor IX or development of nephrotic syndrome 39.
Hemophilia B diagnosis
The diagnosis of hemophilia B is established in individuals with low factor IX clotting activity. Identification of a hemizygous clotting factor IX gene (F9) pathogenic variant on molecular genetic testing in a male proband confirms the diagnosis. Identification of a heterozygous F9 pathogenic variant on molecular genetic testing in a symptomatic female confirms the diagnosis (see Figure 3 below).
Note:
- Hemizygous refers to a gene normally present in only a single copy; usually an X-linked gene in a male.
- Heterozygous refers to a variant (distinct from the reference sequence) that comprises one of two alleles of a given gene. An individual with two different alleles at a particular locus (one on each chromosome of a pair), one of which is usually pathogenic.
- X-linked inheritance refers to a gene on the X chromosome or to the mode of inheritance in which the causative pathogenic variant is on the X chromosome; hemizygous males will be affected; heterozygous females may or may not be affected depending on the disorder and factors influencing X-chromosome inactivation (see Figure 3 below).
Laboratory features
- Normal platelet count
- Prolonged activated partial thromboplastin time (aPTT) in severe and moderate hemophilia B. Normal or mildly prolonged aPTT in mild hemophilia B.
- Normal prothrombin time (PT)
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with hemophilia B, the following evaluations are recommended if they have not already been completed:
- A personal and family history of bleeding to help predict disease severity
- A joint and muscle evaluation, particularly if the individual describes a history of hemarthrosis or deep-muscle hematomas
- Screening for hepatitis A, B, and C as well as HIV if blood products or plasma-derived clotting factor concentrates were administered prior to 1990
- Baseline CBC including platelet count and ferritin, especially if there is a history of nose bleeds, GI bleeding, mouth bleeding, or in females, heavy menstrual bleeding or postpartum hemorrhage
- Referral to a hemophilia treatment center. For locations:
- Worldwide, see World Federation of Haemophilia 11 at https://www.wfh.org/en/page.aspx?pid=1264
- US only, see National Hemophilia Foundation 12 at https://www.hemophilia.org/
- Identification of the specific clotting factor IX gene (F9) pathogenic variant in an individual to aid in determining disease severity, the likelihood of inhibitor development, and the risk of anaphylaxis if an inhibitor does develop
- Consultation with a clinical geneticist and/or genetic counselor, particularly if a new diagnosis in the family and for females of childbearing years.
Hemophilia B genetics
Hemophilia B is inherited in an X-linked manner. The risk to siblings of a proband (index case) depends on the carrier status of the mother. Carrier females have a 50% chance of transmitting the clotting factor IX gene (F9) pathogenic variant in each pregnancy. Sons who inherit the pathogenic variant will be affected; daughters who inherit the pathogenic variant are carriers. Affected males transmit the pathogenic variant to all of their daughters and none of their sons. Carrier testing for family members at risk and prenatal testing for pregnancies at increased risk are possible if the clotting factor IX gene (F9) pathogenic variant has been identified in a family member or if informative intragenic linked markers have been identified.
Male proband (index case). The diagnosis of hemophilia B is established in a male proband by identification of decreased factor IX clotting activity.
- Severe hemophilia B. <1% factor IX
- Moderate hemophilia B. 1%-5% factor IX
- Mild hemophilia B. >5%-40% factor IX
Note:
- The normal range for factor IX clotting activity is approximately 50%-150% 40. Individuals with factor IX clotting activity higher than 40% usually have normal coagulation in vivo. However, some increased bleeding can occur with low to low-normal factor IX clotting activity in hemophilia B carrier females 13.
- Somatic mosaicism in males with hemophilia B has been described 41.
Identification of a hemizygous pathogenic variant in clotting factor IX gene (F9) by molecular genetic testing can help predict the clinical phenotype and allow family studies (see Table 4).
Heterozygous females. The diagnosis of hemophilia B is established by determination of low factor IX clotting activity. Approximately 30% of heterozygous females have a factor IX clotting activity below 40%, regardless of the severity of hemophilia B in their family. Bleeding symptoms may be present in those with factor IX activity in the low-normal range 13.
Carrier status is determined by identification of a heterozygous pathogenic variant in clotting factor IX gene (F9) by molecular genetic testing (see Table 4). Factor IX clotting activity is unreliable in the detection of heterozygous females; the majority of obligate carriers, even of severe hemophilia B, have normal factor IX clotting activities.
Penetrance
All males with a clotting factor IX gene (F9) pathogenic variant are affected and will have hemophilia B of approximately the same severity as all other affected males in the family; however, other genetic and environmental effects may modify the clinical severity to some extent.
Approximately 30% of females with one clotting factor IX gene (F9) pathogenic variant and one normal allele have a factor IX clotting activity lower than 40% and a bleeding disorder; mild bleeding can occur in carriers with low-normal factor IX activities 13.
Figure 3. Hemophilia B – X-linked inheritance pattern
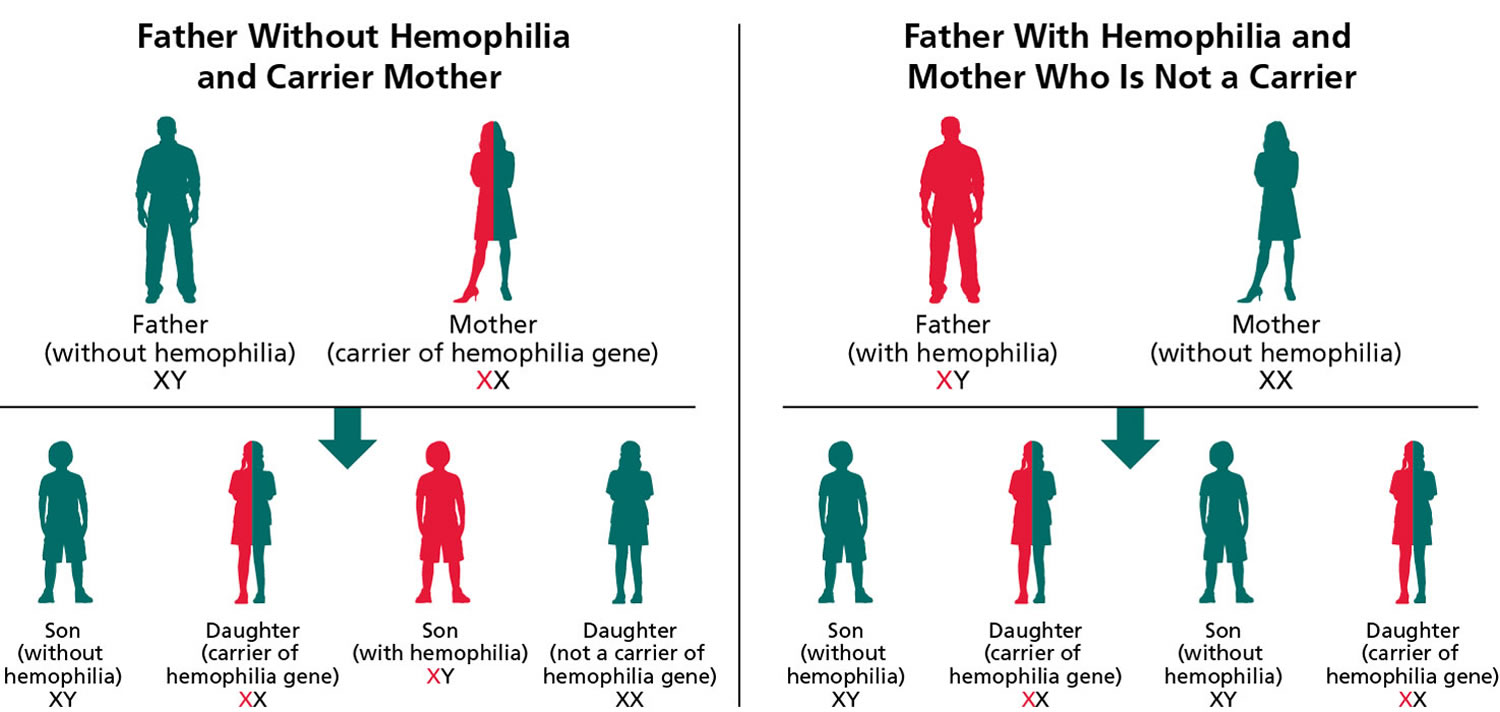
Note: Hemophilia A is inherited in an X-linked manner. Referring to a gene on the X chromosome or to the mode of inheritance in which the causative pathogenic variant is on the X chromosome; hemizygous males will be affected; heterozygous females may or may not be affected depending on the disorder and factors influencing X-chromosome inactivation.
Hemophilia B Molecular Genetic Testing
Molecular genetic testing approaches can include single-gene testing, use of a multi-gene panel, and more comprehensive genomic testing:
- Single-gene testing. Sequence analysis of clotting factor IX gene (F9) is performed first and followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found.
- A multi-gene panel that includes clotting factor IX gene (F9) and other genes of interest may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and over time. (2) Some multi-gene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multi-gene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of pathogenic variants in genes that do not account for the underlying phenotype. (3) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing based tests.
- More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
Table 4. Molecular Genetic Testing Used in Hemophilia B
| Gene | Test Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by This Method |
|---|---|---|
| F9 | Sequence analysis | 97%-100% |
| Gene-targeted deletion/duplication analysis 7 | 2%-3% |
Genotype-Phenotype Correlations
Disease severity
- Large deletions, nonsense variants, and most frameshift variants cause severe disease.
- Missense variants can cause severe, moderate, or mild disease depending on their location and the specific substitutions involved.
Alloimmune inhibitors
- Alloimmune inhibitors occur with the greatest frequency (40%-60%) in individuals with large partial (>50-bp) deletions, whole-gene deletions or early termination (<100 predicted amino acids) variants 42.
- Missense variants are rarely associated with inhibitors.
Unlike hemophilia A, severe hemophilia B is often caused by a missense variant and several of these are associated with normal cross-reacting material (factor IX antigen) levels.
Certain missense variants within the propeptide portion of factor IX enhance sensitivity to warfarin by altering the binding of a gamma-carboxylase responsible for post-translational Gla residue formation 43. Uncommon variants within the carboxylase-binding domain of the propeptide cause increased sensitivity to warfarin anticoagulation in individuals without any baseline bleeding tendency 43.
The variant p.Arg384Leu, a missense gain-of-function change associated with markedly elevated circulating levels of factor IX and venous thrombosis at a young age, has been described in one family 44. This amino acid change has been incorporated into factor IX constructs currently being used in gene therapy clinical trials 45.
In hemophilia B Leyden, more than 20 different causative variants in the proximal clotting factor IX gene (F9) promoter region have been described 46; the severity of disease decreases after puberty; mild disease disappears and severe disease becomes mild, depending on the specific pathogenic variant.
Hemophilia B treatment
The World Federation of Hemophilia has published treatment guidelines 18 for the management of individuals with hemophilia. Treatment should be coordinated through a hemophilia treatment center. Individuals in the USA see National Hemophilia Foundation 12; individuals worldwide see World Federation of Hemophilia for locations 19
Treatment of hemophilia B manifestations: Referral to a hemophilia treatment center for assessment, education, genetic counseling, and treatment. Intravenous infusion of plasma-derived or recombinant factor IX for bleeding episodes within an hour of noticing symptoms. Training and home infusions for those with severe hemophilia B.
Intravenous infusion of plasma-derived or recombinant factor IX for bleeding episodes should be initiated within an hour of noticing symptoms:
- Dosing is weight based and target levels and duration of treatment vary by the severity of bleeding and/or the risk associated with the surgery or procedure.
- Identify staff members who are expert in performing venipunctures in infants and toddlers because frequent venipunctures may be necessary.
- Parents of children age two to five years with severe hemophilia B should be trained to administer the infusions as soon as is feasible. Home treatment allows for prompt treatment and facilitates prophylactic therapy.
Pediatric issues. Special considerations for care of infants and children with hemophilia B include the following 20:
- Infant males with a family history of hemophilia B should not be circumcised unless hemophilia B is either excluded or, if present, treated with factor IX concentrate directly before and after the procedure.
- Immunizations should be administered subcutaneously; intramuscular injections should be avoided unless under factor coverage.
- Effective dosing of factor IX requires an understanding of different pharmacokinetics in young children.
Inhibitors. Alloimmune inhibitors to factor IX, seen in 1%-3% of persons with severe hemophilia B, greatly compromise the ability to manage bleeding episodes 47. Their onset can be associated with anaphylactic reactions to factor IX infusion and nephrotic syndrome 39. Immune tolerance can be challenging and long-term bypassing therapy may be needed for treatment.
Prevention of primary manifestations: For those with severe disease, prophylactic infusions of factor IX concentrate twice weekly to maintain factor IX clotting activity higher than 1% nearly eliminates spontaneous bleeding and prevents chronic joint disease. Some individuals require higher trough levels for this effect. Longer-acting products that allow weekly or biweekly dosing are now available. Choice of product should be individualized based on clinical factors and activity levels. Initiation of prophylactic infusions of factor IX concentrate in young boys before or just after their first few joint bleeds has been shown to nearly eliminate spontaneous bleeding and prevent chronic joint disease 25. Prophylaxis in adults is standard of care in many countries and has been shown to decrease bleeding and improve joint function and quality of life 48.
Prevention of secondary complications: Recombinant factor IX produced without human- or animal-derived proteins and virucidal treatment of plasma-derived concentrates has eliminated the risk of HIV and hepatitis B and C viruses.
Surveillance: For individuals with severe or moderate hemophilia B, assessments every six to 12 months at an hemophilia treatment center; for individuals with mild hemophilia B, assessments at least every two to three years.
Screening for alloimmune inhibitors is usually done in those with severe hemophilia B after treatment with factor IX concentrates has been initiated for either bleeding or prophylaxis. Affected individuals at increased risk for inhibitor formation should be closely monitored during initial infusions and additional screening is usually performed up to a few years of age when the genotype is a large partial deletion, complete F9 deletion, or early termination variant (<100 predicted amino acids). Testing for inhibitors should also be performed in any individual with hemophilia B whenever a suboptimal clinical response to treatment is suspected, regardless of disease severity; with hemophilia B, the onset may be heralded by an allergic reaction to infused factor IX concentrate.
Older children and adults with severe or moderate hemophilia B benefit from at least yearly assessments at an hemophilia treatment center and periodic assessments to review bleeding episodes and treatment plans, evaluate joints and muscles, screen for an inhibitor, perform viral testing if indicated, provide education, and discuss other issues relevant to the individual’s hemophilia.
Agents/circumstances to avoid: Circumcision of at-risk males until hemophilia B is either excluded or treated with factor IX concentrate regardless of severity; intramuscular injections; activities with a high risk of trauma, particularly head injury; aspirin and all aspirin-containing products. Cautious use of other medications and herbal remedies that affect platelet function.
The following should be avoided:
- Circumcision of infant males with a family history of hemophilia B unless hemophilia B is excluded; OR if circumcision is performed on an infant with hemophilia B, the infant should be treated with factor IX concentrate directly before and after the procedure.
- Intramuscular injections
- Activities that involve a high risk of trauma, particularly of head injury
- Medications and herbal remedies that affect platelet function, including aspirin unless there is strong medical indication (e.g., in individuals with atherosclerotic cardiovascular disease). Individuals with severe hemophilia usually require clotting factor prophylaxis to allow aspirin and other platelet inhibitory drugs to be used safely 28.
Older, intermediate purity plasma-derived “prothrombin complex” concentrates should be used cautiously (if at all) in hemophilia B because of their thrombogenic potential.
Evaluation of relatives at risk: To clarify genetic status of females at risk before pregnancy or early in pregnancy in order to facilitate management.
- Identification of at-risk relatives. A thorough family history may identify other male relatives who are at risk but have not been tested (particularly in families with mild hemophilia B).
- Early determination of the genetic status of males at risk. Either assay of factor IX clotting activity from a cord blood sample obtained by venipuncture of the umbilical vein (to avoid contamination by amniotic fluid or placenta tissue) or molecular genetic testing for the family-specific F9 pathogenic variant can establish or exclude the diagnosis of hemophilia B in newborn males at risk. Infants with a family history of hemophilia B should not be circumcised unless hemophilia B is either excluded or, if present, factor IX concentrate is administered immediately before and after the procedure to prevent delayed oozing and poor wound healing. Note: (1) The cord blood for factor IX clotting activity assay should be drawn into a syringe containing one-tenth volume of sodium citrate to avoid clotting and to provide an optimal mixing of the sample with the anticoagulant. (2) Factor IX clotting activity in cord blood in a normal-term newborn is lower than in adults (mean: ~30%; range: 15%-50%); thus, the diagnosis of hemophilia B can be established in an infant with activity lower than 1%, but is equivocal in an infant with moderately low (15%-20%) activity.
- Determination of genetic status of females at risk. Approximately 30% of heterozygous females have factor IX clotting activity lower than 40% and may have abnormal bleeding. In a recent Dutch survey of heterozygous females, bleeding symptoms correlated with baseline factor clotting activity; there was suggestion of a very mild increase in bleeding even in those with 40% to 60% factor IX clotting activity [Plug et al 2006]. Joint range of motion in female carriers with factor VIII or factor IX activity lower than 40% was found to be significantly different from that measured in normal controls and inversely related to factor level [Sidonio et al 2014]. All daughters and mothers of an affected male and other at-risk females should have a baseline factor IX clotting activity assay to determine if they are at increased risk for bleeding (unless they are known to be non-carriers based on molecular genetic testing). Very occasionally, a female will have particularly low factor IX clotting activity that may result from heterozygosity for an F9 pathogenic variant associated with skewed X-chromosome inactivation or, on rare occasion, compound heterozygosity for two F9 pathogenic variants.
It is recommended that the carrier status of a female at risk be established prior to pregnancy or as early in a pregnancy as possible.
Pregnancy management: Maternal factor IX levels do not increase during pregnancy and heterozygous females are more likely to need factor infusion support for delivery or to treat or prevent postpartum hemorrhage; monitor heterozygous mothers for delayed bleeding post partum.
- Obstetric issues. It is recommended that the carrier status of a female at risk be established prior to pregnancy or as early in a pregnancy as possible. Unlike for factor VIII (FVIII), maternal factor IX levels do not increase during pregnancy and carriers are more likely to need factor infusion support for delivery or to treat or prevent postpartum hemorrhage. In carriers, postpartum hemorrhage has been a prominent feature, despite the absence of heavy menstrual bleeding 49. If the female has a baseline factor IX clotting activity below approximately 40%, she by definition has hemophilia and is at risk for excessive bleeding, particularly post partum, and may require therapy with factor IX concentrate 49.
- Newborn males. Controversy remains as to indications for cesarean section versus vaginal delivery 50. For elective deliveries, the relative risks of cesarean section versus vaginal delivery should be considered, especially if a male has been diagnosed with severe hemophilia B prenatally. At birth or in the early neonatal period, intracranial hemorrhage is uncommon (<1%-2%), even in males with severe hemophilia B who are delivered vaginally.
Therapies under investigation: Clinical trials of additional longer-acting recombinant factor IX concentrates and gene therapy using intravenous infusion of an adeno-associated viral vector expressing factor IX are underway. Two products with modifications to prolong half-life (Fc or albumin fusion proteins) are now FDA approved and others, including with site-specific PEGylation, have completed Phase III clinical trials 37. Clinical trial data show a three- to five fold prolongation of FIX half-life with the fusion proteins.
Gene therapy for hemophilia B. Several clinical trials of gene therapy using intravenous infusion of an adeno-associated viral vector expressing factor IX are underway; some have shown sustained factor levels 45. These vectors use liver-restricted promoters to target synthesis to the natural site of FIX synthesis.
Other: Vitamin K does not prevent or control bleeding in hemophilia B. Fresh frozen plasma is no longer recommended to treat hemophilia B because it is not treated with a virucidal agent.
Hemophilia C
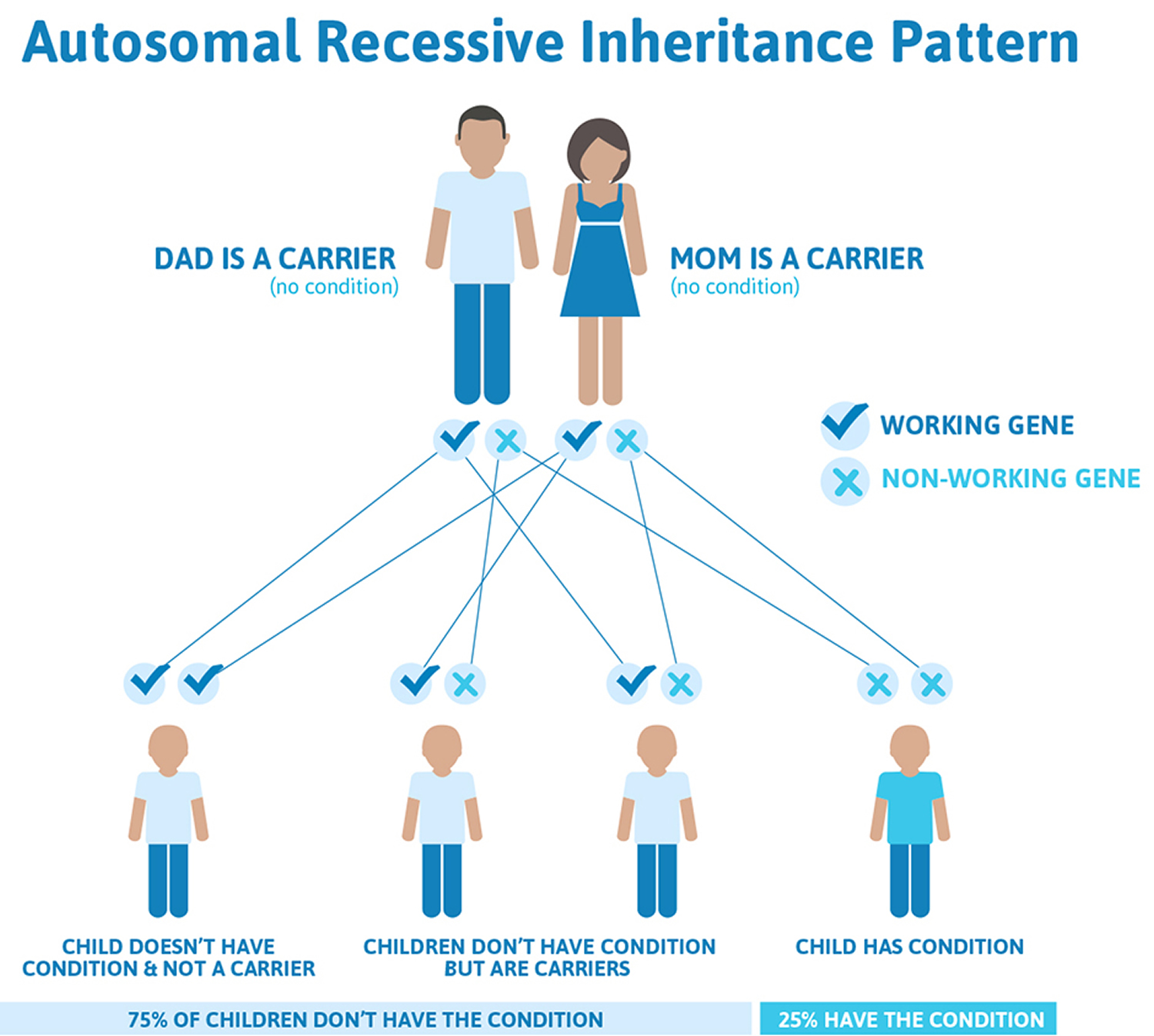
Hemophilia C (factor XI deficiency) is a very rare autosomal recessive bleeding disorder. Hemophilia C (factor XI deficiency) is the most common of the rare bleeding disorders and the second most common bleeding disorder affecting women (after von Willebrand disease). Patients with factor XI deficiency do not typically show any spontaneous bleeding or specific symptoms. Sometimes those who have hemophilia C (factor XI deficiency) are identified during special situations such as trauma or surgery. Orthognathic surgery is particularly associated with a high bleeding risk. Therefore, great care must be taken when treating patients with bleeding disorders such as factor XI deficiency. There are a few reports that address the management of patients with bleeding disorders during orthognathic surgery.
Some people have inherited hemophilia C (factor XI deficiency) when only one parent carries the gene. The disorder is most common among Ashkenazi Jews, that is, Jews of Eastern European ancestry.
Hemophilia C genetics
Figure 4. Hemophilia C autosomal recessive inheritance pattern
Hemophilia C signs and symptoms
Most people with factor XI deficiency will have little or no symptoms at all. The relationship between the amount of factor XI in a person’s blood and the severity of his/her symptoms is unclear; people with only a mild deficiency in factor XI can have serious bleeding episodes. Symptoms of factor XI deficiency vary widely, even among family members, which can make it difficult to diagnose.
Common symptoms
- nosebleeds (epistaxis)
- easy bruising
- heavy or prolonged menstrual bleeding (menorrhagia)
- abnormal bleeding during or after surgery, injury, or childbirth
Other reported symptoms
- bleeding in the gut (gastrointestinal hemorrhage)
- bleeding in the mouth, particularly after dental surgery or tooth extraction
- blood in the urine (hematuria)
emophilia C diagnosis
Factor XI deficiency is diagnosed by a variety of blood tests that should be performed by a specialist at a hemophilia/bleeding disorders treatment center.
Hemophilia C treatment
There are several treatments available to help control bleeding in people with factor XI deficiency.
- Factor XI concentrate. Factor concentrates are the ideal and safest treatment for rare bleeding disorders.Factor concentrates for rare bleeding disorders are usually made from human plasma and are treated to eliminate viruses like HIV and hepatitis B and C. They are made in the laboratory and not from human plasma, so they carry no risk of infectious disease. Factor concentrates are administered intravenously.
- Antifibrinolytic drugs: The antifibrinolytic drugs tranexamic acid and aminocaproic acid are used to hold a clot in place in certain parts of the body, such as the mouth, bladder, and uterus. They are also very useful in many situations, such as during dental work, but are not effective for major internal bleeding or surgery. Antifibrinolytic drugs are particularly useful for patients with factor XI deficiency. They are also used to help control excessive menstrual bleeding. Antifibrinolytic drugs can be administered orally or by injection.
- Fibrin glue: Fibrin glue can be used to treat external wounds and during dental work, such as a tooth extraction. It is not used for major bleeding or surgery. It is applied to the bleeding site.
- Fresh frozen plasma (FFP): Plasma is the portion of blood that contains all the clotting factors, as well as other blood proteins. FFP is used to treat rare bleeding disorders when concentrates of the specific factor that is missing are not available. FFP is the usual treatment for factor V deficiency. However, it usually does not undergo viral inactivation, so the risk of transmission of infectious diseases is higher. Viral-inactivated FFP is available in some countries and is preferable. Circulatory overload is a potential problem with this treatment: since the concentration of each coagulation factor in FFP is low, a large volume of it must be given over several hours in order to achieve an adequate rise in factor level. This large amount of FFP needed can overload the circulatory system and stress the heart. Other complications of treatment with FFP can occur, particularly allergic reactions or lung problems (transfusion-related lung injury [TRALI]). These problems are much less common if viral-inactivated pooled FFP is used. FFP is administered intravenously.
Excessive menstrual bleeding in women with factor XI deficiency may be controlled with hormonal contraceptives (birth control pills), intra-uterine device (IUDs), or antifibrinolytic drugs.
Hemophilia causes
When you bleed, your body normally pools blood cells together to form a clot to stop the bleeding. The clotting process is encouraged by certain blood particles. Hemophilia occurs when you have a deficiency in one of these clotting factors.
There are several types of hemophilia, and most forms are inherited. However, about 30 percent of people with hemophilia have no family history of the disorder. In these people, an unexpected change (spontaneous mutation) occurs in one of the genes associated with hemophilia.
Acquired hemophilia is a rare variety of the condition that occurs when a person’s immune system attacks clotting factors in the blood. It can be associated with:
- Pregnancy
- Autoimmune conditions
- Cancer
- Multiple sclerosis
Hemophilia inheritance
In the most common types of hemophilia, the faulty gene is located on the X chromosome. Everyone has two sex chromosomes, one from each parent. A female inherits an X chromosome from her mother and an X chromosome from her father. A male inherits an X chromosome from his mother and a Y chromosome from his father.
This means that hemophilia almost always occurs in boys and is passed from mother to son through one of the mother’s genes. Most women with the defective gene are simply carriers and experience no signs or symptoms of hemophilia. But some carriers can experience bleeding symptoms if their clotting factors are moderately decreased.
A defect in one of the genes that determines how the body makes blood clotting factor VIII or IX causes hemophilia. These genes are located on the X chromosomes.
Chromosomes come in pairs. Females have two X chromosomes, while males have one X and one Y chromosome. Only the X chromosome carries the genes related to clotting factors.
A male who has a hemophilia gene on his X chromosome will have hemophilia. When a female has a hemophilia gene on only one of her X chromosomes, she is a “hemophilia carrier” and can pass the gene to her children. Sometimes carriers have low levels of clotting factor and have symptoms of hemophilia, including bleeding. Clotting factors are proteins in the blood that work together with platelets to stop or control bleeding.
A female who is a carrier has a 1 in 2 (50 percent) chance to pass on her X chromosome with the gene mutation for hemophilia A or B to a boy who will be affected. She has a 1 in 2 (50 percent) chance to pass on her X chromosome with the normally functioning gene to a boy who will not have hemophilia.
Very rarely, a girl may be born with a very low clotting factor level and have a greater risk for bleeding, similar to boys who have hemophilia and very low levels of clotting factor. There are several hereditary and genetic causes of this much rarer form of hemophilia in females.
Some males who have the disorder are born to mothers who aren’t carriers. In these cases, a mutation (random change) occurs in the gene as it is passed to the child.
Hemophilia symptoms
The major signs and symptoms of hemophilia are excessive bleeding and easy bruising.
Excessive Bleeding
The extent of bleeding depends on how severe the hemophilia is.
Children who have mild hemophilia may not have signs unless they have excessive bleeding from a dental procedure, an accident, or surgery. Males who have severe hemophilia may bleed heavily after circumcision.
Bleeding can occur on the body’s surface (external bleeding) or inside the body (internal bleeding).
Signs of external bleeding may include:
- Bleeding in the mouth from a cut or bite or from cutting or losing a tooth
- Nosebleeds for no obvious reason
- Heavy bleeding from a minor cut
- Bleeding from a cut that resumes after stopping for a short time
Signs of internal bleeding may include:
- Blood in the urine (from bleeding in the kidneys or bladder)
- Blood in the stool (from bleeding in the intestines or stomach)
- Large bruises (from bleeding into the large muscles of the body)
Bleeding in the Joints
Bleeding in the knees, elbows, or other joints is another common form of internal bleeding in people who have hemophilia. This bleeding can occur without obvious injury.
At first, the bleeding causes tightness in the joint with no real pain or any visible signs of bleeding. The joint then becomes swollen, hot to touch, and painful to bend.
Swelling continues as bleeding continues. Eventually, movement in the joint is temporarily lost. Pain can be severe. Joint bleeding that isn’t treated quickly can damage the joint.
Bleeding in the Brain
Internal bleeding in the brain is a very serious complication of hemophilia. It can happen after a simple bump on the head or a more serious injury. The signs and symptoms of bleeding in the brain include:
- Long-lasting, painful headaches or neck pain or stiffness
- Repeated vomiting
- Sleepiness or changes in behavior
- Sudden weakness or clumsiness of the arms or legs or problems walking
- Double vision
- Convulsions or seizures.
Hemophilia complications
Complications of hemophilia may include:
- Deep internal bleeding. Bleeding that occurs in deep muscle can cause your limbs to swell. The swelling may press on nerves and lead to numbness or pain.
- Damage to joints. Internal bleeding may also put pressure on your joints, causing severe pain. Left untreated, frequent internal bleeding may cause arthritis or destruction of the joint.
- Infection. People with hemophilia are likelier to have blood transfusions, increasing their risk of receiving contaminated blood products. Blood products became safer after the mid-1980s due to screening of donated blood for hepatitis and HIV.
- Adverse reaction to clotting factor treatment. In some people with hemophilia, the immune system has a negative reaction to the clotting factors used to treat bleeding. When this happens, the immune system develops proteins (known as inhibitors) that inactivate the clotting factors, making treatment less effective.
Hemophilia diagnosis
For people with a family history of hemophilia, it’s possible to determine during pregnancy if the fetus is affected by hemophilia. However, the testing poses some risks to the fetus. Discuss the benefits and risks of testing with your doctor.
In children and adults, a blood test can reveal a clotting-factor deficiency. Depending on the severity of the deficiency, hemophilia symptoms can first arise at various ages.
Severe cases of hemophilia usually are diagnosed within the first year of life. Mild forms may not be apparent until adulthood. Some people first learn they have hemophilia after they bleed excessively during a surgical procedure.
Hemophilia A and B are diagnosed by measuring factor clotting activity. Individuals who have hemophilia A have low factor VIII clotting activity. Individuals who have hemophilia B have low factor IX clotting activity.
Genetic testing is also available for the factor VIII gene and the factor IX gene. Genetic testing of the FVIII gene finds a disease-causing mutation in up to 98 percent of individuals who have hemophilia A. Genetic testing of the FIX gene finds disease-causing mutations in more than 99 percent of individuals who have hemophilia B.
Genetic testing is usually used to identify women who are carriers of a FVIII or FIX gene mutation, and to diagnose hemophilia in a fetus during a pregnancy (prenatal diagnosis). It is sometimes used to diagnose individuals who have mild symptoms of hemophilia A or B.
Hemophilia treatment
Treatment With Replacement Therapy
The main treatment for hemophilia is called replacement therapy. Concentrates of clotting factor VIII (for hemophilia A) or clotting factor IX (for hemophilia B) are slowly dripped or injected into a vein. These infusions help replace the clotting factor that’s missing or low.
Clotting factor concentrates can be made from human blood. The blood is treated to prevent the spread of diseases, such as hepatitis. With the current methods of screening and treating donated blood, the risk of getting an infectious disease from human clotting factors is very small.
To further reduce the risk, you or your child can take clotting factor concentrates that aren’t made from human blood. These are called recombinant clotting factors. Clotting factors are easy to store, mix, and use at home—it only takes about 15 minutes to receive the factor.
You may have replacement therapy on a regular basis to prevent bleeding. This is called preventive or prophylactic therapy. Or, you may only need replacement therapy to stop bleeding when it occurs. This use of the treatment, on an as-needed basis, is called demand therapy.
Demand therapy is less intensive and expensive than preventive therapy. However, there’s a risk that bleeding will cause damage before you receive the demand therapy.
Complications of Replacement Therapy
Complications of replacement therapy include:
- Developing antibodies (proteins) that attack the clotting factor
- Developing viral infections from human clotting factors
- Damage to joints, muscles, or other parts of the body resulting from delays in treatment
Antibodies to the clotting factor. Antibodies can destroy the clotting factor before it has a chance to work. This is a very serious problem. It prevents the main treatment for hemophilia (replacement therapy) from working.
These antibodies, also called inhibitors, develop in about 20–30 percent of people who have severe hemophilia A. Inhibitors develop in 2–5 percent of people who have hemophilia B.
When antibodies develop, doctors may use larger doses of clotting factor or try different clotting factor sources. Sometimes the antibodies go away.
Researchers are studying new ways to deal with antibodies to clotting factors.
Viruses from human clotting factors. Clotting factors made from human blood can carry the viruses that cause HIV/AIDS and hepatitis. However, the risk of getting an infectious disease from human clotting factors is very small due to:
- Careful screening of blood donors
- Testing of donated blood products
- Treating donated blood products with a detergent and heat to destroy viruses
- Vaccinating people who have hemophilia for hepatitis A and B
Damage to joints, muscles, and other parts of the body. Delays in treatment can cause damage such as:
- Bleeding into a joint. If this happens many times, it can lead to changes in the shape of the joint and impair the joint’s function.
- Swelling of the membrane around a joint.
- Pain, swelling, and redness of a joint.
- Pressure on a joint from swelling, which can destroy the joint.
Home Treatment With Replacement Therapy
You can do both preventive (ongoing) and demand (as-needed) replacement therapy at home. Many people learn to do the infusions at home for their child or for themselves. Home treatment has several advantages:
- You or your child can get quicker treatment when bleeding happens. Early treatment lowers the risk of complications.
- Fewer visits to the doctor or emergency room are needed.
- Home treatment costs less than treatment in a medical care setting.
- Home treatment helps children accept treatment and take responsibility for their own health.
Discuss options for home treatment with your doctor or your child’s doctor. A doctor or other health care provider can teach you the steps and safety procedures for home treatment. Hemophilia treatment centers are another good resource for learning about home treatment.
Doctors can surgically implant vein access devices to make it easier for you to access a vein for treatment with replacement therapy. These devices can be helpful if treatment occurs often. However, infections can be a problem with these devices. Your doctor can help you decide whether this type of device is right for you or your child.
Other Types of Treatment
Desmopressin
Desmopressin (DDAVP) is a man-made hormone used to treat people who have mild hemophilia A. Desmopressin (DDAVP) isn’t used to treat hemophilia B or severe hemophilia A.
Desmopressin (DDAVP) stimulates the release of stored factor VIII and von Willebrand factor; it also increases the level of these proteins in your blood. Von Willebrand factor carries and binds factor VIII, which can then stay in the bloodstream longer.
DDAVP usually is given by injection or as nasal spray. Because the effect of this medicine wears off if it’s used often, the medicine is given only in certain situations. For example, you may take this medicine prior to dental work or before playing certain sports to prevent or reduce bleeding.
Antifibrinolytic Medicines
Antifibrinolytic medicines (including tranexamic acid and epsilon aminocaproic acid) may be used with replacement therapy. They’re usually given as a pill, and they help keep blood clots from breaking down.
These medicines most often are used before dental work or to treat bleeding from the mouth or nose or mild intestinal bleeding.
Hemophilia Gene Therapy
Researchers are trying to find ways to correct the faulty genes that cause hemophilia. Gene therapy hasn’t yet developed to the point that it’s an accepted treatment for hemophilia. However, researchers continue to test gene therapy in clinical trials.
Treatment of a Specific Bleeding Site
Pain medicines, steroids, and physical therapy may be used to reduce pain and swelling in an affected joint. Talk with your doctor or pharmacist about which medicines are safe for you to take.
Which Treatment Is Best for You?
The type of treatment you or your child receives depends on several things, including how severe the hemophilia is, the activities you’ll be doing, and the dental or medical procedures you’ll be having.
- Mild hemophilia—Replacement therapy usually isn’t needed for mild hemophilia. Sometimes, though, Desmopressin (DDAVP) is given to raise the body’s level of factor VIII.
- Moderate hemophilia—You may need replacement therapy only when bleeding occurs or to prevent bleeding that could occur when doing certain activities. Your doctor also may recommend Desmopressin (DDAVP) prior to having a procedure or doing an activity that increases the risk of bleeding.
- Severe hemophilia—You usually need replacement therapy to prevent bleeding that could damage your joints, muscles, or other parts of your body. Typically, replacement therapy is given at home two or three times a week. This preventive therapy usually is started in patients at a young age and may need to continue for life.
For both types of hemophilia, getting quick treatment for bleeding is important. Quick treatment can limit damage to your body. If you or your child has hemophilia, learn to recognize signs of bleeding.
Other family members also should learn to watch for signs of bleeding in a child who has hemophilia. Children sometimes ignore signs of bleeding because they want to avoid the discomfort of treatment.
Lifestyle and home remedies
To avoid excessive bleeding and protect your joints:
- Exercise regularly. Activities such as swimming, bicycle riding and walking can build up muscles while protecting joints. Contact sports — such as football, hockey or wrestling — are not safe for people with hemophilia.
- Avoid certain pain medications. Drugs that can aggravate bleeding include aspirin and ibuprofen (Advil, Motrin IB, others). Instead, use acetaminophen (Tylenol, others), which is a safe alternative for mild pain relief.
- Avoid blood-thinning medications. Medications that prevent blood from clotting include heparin, warfarin (Coumadin, Jantoven), clopidogrel (Plavix) and prasugrel (Effient).
- Practice good dental hygiene. The goal is to prevent tooth extraction, which can lead to excessive bleeding.
- Protect your child from injuries that could cause bleeding. Kneepads, elbow pads, helmets and safety belts all may help prevent injuries from falls and other accidents. Keep your home free of furniture with sharp corners.
Coping and support
To help you and your child cope with hemophilia:
- Get a medical alert bracelet. This bracelet lets medical personnel know that you or your child has hemophilia, and the type of clotting factor that’s best in case of an emergency.
- Talk with a counselor. You may be concerned about striking the right balance between keeping your child safe and encouraging as much normal activity as possible. A social worker or therapist with knowledge of hemophilia can help you cope with your concerns and identify the minimum limitations necessary for your child.
- Let people know. Be sure to tell anyone who will be taking care of your child — baby sitters, workers at your child care center, relatives, friends and teachers — about your child’s condition. If your child plays noncontact sports, be sure to tell coaches, too.
- Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, Ludlam CA, Mahlangu JN, Mulder K, Poon MC, Street A. Guidelines for the management of hemophilia. World Federation of Hemophilia. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2516.2012.02909.x/pdf[↩]
- Fogarty PF, Kessler CM. Hemophilia A and B. In: Kitchens CS, Kessler CM, Konkle BA, eds. Consultative Hemostasis and Thrombosis. 3 ed. Philadelphia, PA: Elsevier Saunders; 2013:45-59.[↩][↩]
- Kulkarni R, Soucie JM, Lusher J, Presley R, Shapiro A, Gill J, Manco-Johnson M, Koerper M, Mathew P, Abshire T, Dimichele D, Hoots K, Janco R, Nugent D, Geraghty S, Evatt B., Haemophilia Treatment Center Network Investigators. Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from The Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Haemophilia. 2009;15:1281–90. https://www.ncbi.nlm.nih.gov/pubmed/19637999[↩][↩]
- Paroskie A, Gailani D, DeBaun MR, Sidonio RF Jr. A cross-sectional study of bleeding phenotype in haemophilia A carriers. Br J Haematol. 2015;170:223–8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4490107/[↩][↩]
- Konkle BA, Huston H, Nakaya Fletcher S. Hemophilia A. 2000 Sep 21 [Updated 2017 Jun 22]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1404/[↩][↩]
- Luck JV Jr, Silva M, Rodriguez-Merchan EC, Ghalambor N, Zahiri CA, Finn RS. Hemophilic arthropathy. J Am Acad Orthop Surg. 2004;12:234–45. https://www.ncbi.nlm.nih.gov/pubmed/15473675[↩][↩]
- Darby SC, Kan SW, Spooner RJ, Giangrande PL, Hill FG, Hay CR, Lee CA, Ludlam CA, Williams M. Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007;110:815–25. http://www.bloodjournal.org/content/110/3/815.long[↩][↩]
- Hay CR, Palmer B, Chalmers E, Liesner R, Maclean R, Rangarajan S, Williams M, Collins PW. Incidence of factor VIII inhibitors throughout life in severe hemophilia A in the United Kingdom. Blood. 2011;117:6367–70. http://www.bloodjournal.org/content/117/23/6367.long[↩]
- Eckhardt CL, van Velzen AS, Peters M, Astermark J, Brons PP, Castaman G, Cnossen MH, Dors N, Escuriola-Ettingshausen C, Hamulyak K, Hart DP, Hay CR, Haya S, van Heerde WL, Hermans C, Holmström M, Jimenez-Yuste V, Keenan RD, Klamroth R, Laros-van Gorkom BA, Leebeek FW, Liesner R, Mäkipernaa A, Male C, Mauser-Bunschoten E, Mazzucconi MG, McRae S, Meijer K, Mitchell M, Morfini M, Nijziel M, Oldenburg J, Peerlinck K, Petrini P, Platokouki H, Reitter-Pfoertner SE, Santagostino E, Schinco P, Smiers FJ, Siegmund B, Tagliaferri A, Yee TT, Kamphuisen PW, van der Bom JG, Fijnvandraat K, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122:1954–62. http://www.bloodjournal.org/content/122/11/1954.long[↩]
- Schwarz J, Astermark J, Menius ED, Carrington M, Donfield SM, Gomperts ED, Nelson GW, Oldenburg J, Pavlova A, Shapiro AD, Winkler CA, Berntorp E, et al. F8 haplotype and inhibitor risk: results from the Hemophilia Inhibitor Genetics Study (HIGS) Combined Cohort. Haemophilia. 2013;19:113–8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3521089/[↩]
- Search the Global Treatment Centre Directory. World Federation of Haemophilia https://www.wfh.org/en/page.aspx?pid=1264[↩][↩]
- National Hemophilia Foundation https://www.hemophilia.org/[↩][↩][↩][↩]
- Plug I, Mauser-Bunschoten EP, Brocker-Vriends AH, van Amstel HK, van der Bom JG, van Diemen-Homan JE, Willemse J, Rosendaal FR. Bleeding in carriers of hemophilia. Blood. 2006;108:52–6. http://www.bloodjournal.org/content/108/1/52.long[↩][↩][↩][↩][↩][↩]
- Kaufman RJ, Fay PJ, Popolo L, Ortel TL. Factor V and factor VIII. In: Marder VJ, Aird WC, Bennett JS, Schulman S, White GC, eds. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 6 ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2013:179-96.[↩]
- Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. 2002;99:168–74. http://www.bloodjournal.org/content/99/1/168.long[↩]
- Nakaya S, Liu ML, Thompson AR. Some factor VIII exon 14 frameshift mutations cause moderately severe haemophilia A. Br J Haematol. 2001;115:977–82. http://onlinelibrary.wiley.com/doi/10.1046/j.1365-2141.2001.03173.x/full[↩]
- Riccardi F, Rivolta GF, Franchini M, Pattacini C, Neri TM, Tagliaferri A. Characterization of a novel mutation in the F8 promoter region associated with mild hemophilia A and resistance to DDAVP therapy. J Thromb Haemost. 2009;7:1234–5. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2009.03468.x/full[↩]
- World Federation of Hemophilia: GUIDELINES FOR THE MANAGEMENT OF HEMOPHILIA. http://www1.wfh.org/publications/files/pdf-1472.pdf[↩][↩]
- Search the Global Treatment Centre Directory. World Federation of Haemophilia https://www.wfh.org/en/page.aspx?pid=1264[↩][↩]
- Chalmers E, Williams M, Brennand J, Liesner R, Collins P, Richards M., Paediatric Working Party of the United Kingdom Haemophilia Doctors’ Organization. Guideline on the management of haemophilia in the fetus and neonate. Br J Haematol. 2011;154:208–15. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2010.08545.x/full[↩][↩]
- Nance D, Fletcher SN, Bolgiano DC, Thompson AR, Josephson NC, Konkle BA. Factor VIII mutation and desmopressin-responsiveness in 62 patients with mild hemophilia A. Haemophilia. 2013;19:720–6. https://www.ncbi.nlm.nih.gov/pubmed/23711294[↩]
- Hay CR, DiMichele DM. The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. 2012;119:1335–44. http://www.bloodjournal.org/content/119/6/1335.long[↩]
- Coppola A, Margaglione M, Santagostino E, Rocino A, Grandone E, Mannucci PM, Di Minno G., AICE PROFIT Study Group. Factor VIII gene (F8) mutations as predictors of outcome in immune tolerance induction of hemophilia A patients with high-responding inhibitors. J Thromb Haemost. 2009;7:1809–15. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2009.03615.x/full[↩]
- Feldman BM, Pai M, Rivard GE, Israels S, Poon MC, Demers C, Robinson S, Luke KH, Wu JK, Gill K, Lillicrap D, Babyn P, McLimont M, Blanchette VS. Tailored prophylaxis in severe hemophilia A: interim results from the first 5 years of the Canadian Hemophilia Primary Prophylaxis Study. J Thromb Haemost. 2006;4:1228–36. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2006.01953.x/full[↩]
- Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ML, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, Matsunaga A, Medeiros D, Nugent D, Thomas GA, Thompson AA, McRedmond K, Soucie JM, Austin H, Evatt BL. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–44. http://www.nejm.org/doi/full/10.1056/NEJMoa067659[↩][↩]
- Manco-Johnson MJ, Kempton CL, Reding MT, Lissitchkov T, Goranov S, Gercheva L, Rusen L, Ghinea M, Uscatescu V, Rescia V, Hong W. Randomized, controlled, parallel-group trial of routine prophylaxis vs. on-demand treatment with sucrose-formulated recombinant factor VIII in adults with severe hemophilia A (SPINART). J Thromb Haemost. 2013;11:1119–27. http://onlinelibrary.wiley.com/doi/10.1111/jth.12202/full[↩]
- Pai M, Key NS, Skinner M, Curtis R, Feinstein M, Kessler C, Lane SJ, Makris M, Riker E, Santesso N, Soucie JM, Yeung CHT, Iorio A, Schünemann HJ. NHF-McMaster guideline on care models for haemophilia management.[↩]
- Angelini D, Konkle BA, Sood SL. Aging among persons with hemophilia: contemporary concerns. Semin Hematol. 2016;53:35–9. https://www.ncbi.nlm.nih.gov/pubmed/26805905[↩][↩]
- Pavlova A, Brondke H, Müsebeck J, Pollmann H, Srivastava A, Oldenburg J. Molecular mechanisms underlying hemophilia A phenotype in seven females. J Thromb Haemost. 2009;7:976–82. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2009.03346.x/full[↩]
- Lee CA, Chi C, Pavord SR, Bolton-Maggs PH, Pollard D, Hinchcliffe-Wood A, Kadir RA. The obstetric and gynaecological management of women with inherited bleeding disorders–review with guidelines produced by a taskforce of UK Haemophilia Centre Doctors’ Organization. Haemophilia. 2006;12:301–36. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2516.2006.01314.x/full[↩]
- James AH, Hoots WK. The optimal mode of delivery for the haemophilia carrier expecting an affected infant is caesarean delivery. Haemophilia. 2010;16:420–4. https://www.ncbi.nlm.nih.gov/pubmed/20028425[↩]
- Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388:187–97. http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(15)01123-X/fulltext[↩]
- Kulkarni R, Soucie JM, Lusher J, Presley R, Shapiro A, Gill J, Manco-Johnson M, Koerper M, Mathew P, Abshire T, Dimichele D, Hoots K, Janco R, Nugent D, Geraghty S, Evatt B., Haemophilia Treatment Center Network Investigators. Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Haemophilia. 2009;15:1281–90. https://www.ncbi.nlm.nih.gov/pubmed/19637999[↩]
- Konkle BA, Huston H, Nakaya Fletcher S. Hemophilia B. 2000 Oct 2 [Updated 2017 Jun 15]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1495/[↩]
- Tagliaferri A, Rivolta GF, Iorio A, Oliovecchio E, Mancuso ME, Morfini M, Rocino A, Mazzucconi MG, Franchini M, Ciavarella N, Scaraggi A, Valdrè L, Tagariello G, Radossi P, Muleo G, Iannaccaro PG, Biasoli C, Vincenzi D, Serino ML, Linari S, Molinari C, Boeri E, La Pecorella M, Carloni MT, Santagostino E, Di Minno G, Coppola A, Rocino A, Zanon E, Spiezia L, Di Perna C, Marchesini M, Marcucci M, Dragani A, Macchi S, Albertini P, D’Incà M, Santoro C, Biondo F, Piseddu G, Rossetti G, Barillari G, Gandini G, Giuffrida AC, Castaman G, et al. Mortality and causes of death in Italian persons with haemophilia, 1990-2007. Haemophilia. 2010;16:437–46. https://www.ncbi.nlm.nih.gov/pubmed/20148978[↩]
- Monahan PE, Di Paola J. Recombinant factor IX for clinical and research use. Semin Thromb Hemost. 2010;36:498–509. https://www.ncbi.nlm.nih.gov/pubmed/20632248[↩]
- Santagostino E, Martinowitz U, Lissitchkov T, Pan-Petesch B, Hanabusa H, Oldenburg J, Boggio L, Negrier C, Pabinger I, von Depka Prondzinski M, Altisent C, Castaman G, Yamamoto K, Alvarez-Roman MT, Voigt C, Blackman N, Jacobs I., PROLONG-9FP Investigators Study Group. Long-acting recombinant coagulation factor IX albumin fusion protein (rIX-FP) in hemophilia B: results of a phase 3 trial. Blood. 2016;127:1761–9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4825413/[↩][↩]
- Puetz J, Soucie JM, Kempton CL, Monahan PE., Hemophilia Treatment Center Network (HTCN) Investigators. Prevalent inhibitors in haemophilia B subjects enrolled in the Universal Data Collection database. Haemophilia. 2014;20:25–31. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4520536/[↩]
- Chitlur M, Warrier I, Rajpurkar M, Lusher JM. Inhibitors in factor IX deficiency a report of O the ISTH-SSC international FIX inhibitor registry (1997-2006). Haemophilia. 2009;15:1027–31. https://www.ncbi.nlm.nih.gov/pubmed/19515028[↩][↩]
- Khachidze M, Buil A, Viel KR, Porter S, Warren D, Machiah DK, Soria JM, Souto JC, Ameri A, Lathrop M, Blangero J, Fontcuberta J, Warren ST, Almasy L, Howard TE. Genetic determinants of normal variation in coagulation factor (F) IX levels: genome-wide scan and examination of the FIX structural gene. J Thromb Haemost. 2006;4:1537–45. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2006.02024.x/full[↩]
- Ketterling RP, Vielhaber E, Li X, Drost J, Schaid DJ, Kasper CK, Phillips JA 3rd, Koerper MA, Kim H, Sexauer C, Gruppo R, Ambriz R, Paredes R, Sommer SS. Germline origins in the human F9 gene: frequent G:C–>A:T mosaicism and increased mutations with advanced maternal age. Hum Genet. 1999;105:629–40. https://www.ncbi.nlm.nih.gov/pubmed/10647899[↩]
- Goodeve AC. Hemophilia B: molecular pathogenesis and mutation analysis. J Thromb Haemost. 2015;13:1184–95. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4496316/[↩]
- Oldenburg J, Kriz K, Wuillemin WA, Maly FE, von Welten A, Siegemun A, Keeling DM, Baker P, Chu K, Konkle BA, Lammie B, Albert T, et al. Genetic predisposition to bleeding during oral anticoagulant therapy: evidence for common founder mutations (FIXVal-10 and FIXThr-10) and an independent CpG hotspot mutation. Thromb Haemost. 2001;85:454–7. https://www.ncbi.nlm.nih.gov/pubmed/11307814[↩][↩]
- Simioni P, Tormene D, Tognin G, Gavasso S, Bulato C, Iacobelli NP, Finn JD, Spiezia L, Radu C, Arruda VR. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med. 2009;361:1671. http://www.nejm.org/doi/full/10.1056/NEJMoa0904377[↩]
- Lheriteau E, Davidoff AM, Nathwani AC. Haemophilia gene therapy: progress and challenges. Blood Rev. 2015;29:321–8. https://www.ncbi.nlm.nih.gov/pubmed/26049173[↩][↩]
- Funnell APW, Crossley M. Hemophilia B Leyden and once mysterious cis-regulatory mutations. Trends Genet. 2014;30:18–23. https://www.ncbi.nlm.nih.gov/pubmed/24138812[↩]
- Hay CR, Brown S, Collins PW, Keeling DM, Liesner R. The diagnosis and management of factor VIII and IX inhibitors: a guideline from the United Kingdom Haemophilia Centre Doctors Organisation. Br J Haematol. 2006;133:591–605. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2006.06087.x/full[↩]
- Josephson N.The hemophilias and their clinical management. Hematology Am Soc Hematol Educ Program. 2013;2013:261-7 http://asheducationbook.hematologylibrary.org/content/2013/1/261.long[↩]
- Yang MY, Ragni MV. Clinical manifestations and management of labor and delivery in women with factor IX deficiency. Haemophilia. 2004;10:483–90. https://www.ncbi.nlm.nih.gov/pubmed/15357775[↩][↩]
- James AH, Hoots K. The optimal mode of delivery for the haemophilia carrier expecting an affected infant is caesarean delivery. Haemophilia. 2010;16:420–4. https://www.ncbi.nlm.nih.gov/pubmed/20028425[↩]





{kind=link}