Contents
What is homocystinuria
Homocystinuria is an inherited disorder in which the body is unable to process the amino acid methionine properly. Methionine from ingested protein is normally converted to homocysteine. In classical homocystinuria due to cystathionine beta-synthase (CBS) deficiency, homocysteine cannot be converted to cystathionine. As a result, the concentration of homocysteine and its precursor, methionine, will become elevated in the blood and urine.
There are multiple forms of homocystinuria, which are distinguished by their signs and symptoms and genetic cause. The most common form of homocystinuria, called cystathionine beta-synthase (CBS) deficiency, is characterized by nearsightedness (myopia), dislocation of the lens at the front of the eye, an increased risk of abnormal blood clotting, and brittle bones that are prone to fracture (osteoporosis) or other skeletal abnormalities. Some affected individuals also have developmental delay and learning problems. Most states in the United States test for homocystinuria due to cystathionine beta-synthase (CBS) deficiency at birth by newborn screening.
Less common forms of homocystinuria are caused by different missing or non-working enzymes, can cause intellectual disability, failure to grow and gain weight at the expected rate (failure to thrive), seizures, problems with movement, and a blood disorder called megaloblastic anemia. Megaloblastic anemia occurs when a person has a low number of red blood cells (anemia), and the remaining red blood cells are larger than normal (megaloblastic).
The signs and symptoms of homocystinuria typically develop within the first year of life, although some mildly affected people may not develop features until later in childhood or adulthood.
Homocystinuria symptoms may occur as mildly delayed development or failure to thrive. Increasing visual problems may lead to diagnosis of homocystinuria.
Mutations in the MTHFR, MTR, MTRR and MMADHC genes can cause homocystinuria. All these forms of homocystinuria are inherited in an autosomal recessive manner. Treatment and long-term outlook varies depending upon the cause of homocystinuria.
Approximately 1 in 200,000 to 1 in 300,000 people in the US has the most common type of homocystinuria (homocystinuria due to CBS deficiency). The most common form of homocystinuria affects at least 1 in 200,000 to 335,000 people worldwide. Homocystinuria appears to be more common in some countries, such as Ireland (1 in 65,000), Germany (1 in 17,800), Norway (1 in 6,400), and Qatar (1 in 1,800). The rarer forms of homocystinuria each have a small number of cases reported in the scientific literature. It is unclear how many people have homocystinuria due to other gene mutations. Worldwide, it is thought that about 1 in 150,000 people has homocystinuria due to either a CBS or an MTHFR gene mutation 1.
Lowering the level of homocysteine in the blood, either with diet or supplements or both, can prevent symptoms. With treatment, people with the most severe form of homocystinuria can have normal growth and development. Some may still have eye problems or blood clots and should be monitored 2. Blood clots can be serious and cause organ damage.
Treatment for milder forms of homocystinuria may depend on clinical symptoms and the level of homocysteine in the blood 3.
People who have the most severe form of homocystinuria are put on a special protein-restricted diet to reduce the blood levels of homocysteine and methionine. In addition, they may be given supplements including vitamin B6, vitamin B12, folate and betaine. The recommendation is that these people stay on the protein-restricted diet for life. People with milder forms may be treated with supplements depending on the level of homocysteine in their blood.
The medication listed below have been approved by the Food and Drug Administration (FDA) as orphan products for treatment of this condition.
- Betaine (Brand name: Cystadane) – Manufactured by Orphan Europe SARL
- FDA-approved indication: Treatment of homocystinuria to decrease elevated homocysteine blood levels.
My daughter has homocystinuria and wants to get pregnant. Is there any information about homocystinuria and pregnancy?
Pregnancy increases the risk for blood clots, stroke, and heart disease in women with homocystinuria, especially in the post-partum period 4. Most pregnancies, however, are uncomplicated 4. Prophylactic anticoagulation (preventing blood clots) during the third trimester of pregnancy and post partum in women with homocystinuria is recommended to reduce risk of thromboembolism 4. Women are often given blood thinning medication (such as herapin) during the last few months of pregnancy until about 6 weeks after delivery. Aspirin in low doses has also been given throughout pregnancy 4. The usual treatments for homocystinuria are typically continued during pregnancy. In addition to blood clots, untreated women are at higher risk for miscarriage and stillbirth 2.
Maternal homocystinuria does not appear to have major teratogenic effects (effects that can harm the development of the embryo or fetus) requiring additional counseling or, with respect to the fetus, more stringent management. Nevertheless, treatment with pyridoxine or methionine-restricted diet or both should be continued during pregnancy. Betaine may also be continued and appears not to be teratogenic 4.
A 2002 study in the Journal of Inherited Metabolic Disease 5 obtained information on 11 women with maternal homocystinuria, their pregnancies (15 total), and their offspring. 5 women were pyridoxine-nonresponsive and 6 were pyridoxine-responsive. The authors reported there was no relationship between the severity of the homocystinuria or the therapies during pregnancy to either the pregnancy complications or the offspring outcomes. They stated that the infrequent occurrences of pregnancy complications, offspring abnormalities and maternal thromboembolic events in the series suggest that pregnancy and outcome in maternal homocystinuria are usually normal. Nevertheless, a cautious approach would include careful monitoring of these pregnancies with attention to metabolic therapy and possibly anticoagulation 5.
Fertility
There is no compelling evidence that CBS deficiency affects fertility. There is also no convincing evidence that CBS deficient patients have an increased risk of fetal loss or fetal malformations.
The only published studies looking at fertility in CBS deficiency have been in mice. These showed changes in the estrus cycle, possible oocyte differences, increased fetal loss and lactation disturbances 6. Mouse studies also indicate that CBS is important (via its role in H2S production) in the maintenance of labour 7.
Increased malformations have not been reported in the children of women with CBS deficiency 8. It is more difficult to analyse fetal loss because it is not always reported and early losses are not always recognised. Although data from several studies link miscarriage to increases in homocystine 9, studies in treated patients with CBS deficiency have not shown an increased risk of miscarriage compared to the general population 10.
Males are fertile and in one study 21 males sired 34 fetuses of which 33 were healthy and one spontaneously aborted 10.
Homocystinuria causes
Homocystinuria can be caused by mutations in several different genes. All of these genes are responsible for making enzymes that are involved in the way our body uses and processes amino acids. The most common gene associated with homocystinuria is the CBS (cystathionine beta-synthase) gene that causes a lack of the enzyme, cystathionine beta-synthase. Rarer causes of homocystinuria include mutations in the MTHFR, MTR, MTRR and MMADHC genes 11. It is not clear why high levels of homocysteine cause the symptoms seen in homocystinuria.
Mutations in the CBS gene cause the most common form of homocystinuria. The CBS gene provides instructions for producing an enzyme called cystathionine beta-synthase. This enzyme acts in a chemical pathway and is responsible for converting the amino acid homocysteine to a molecule called cystathionine. As a result of this pathway, other amino acids, including methionine, are produced. Mutations in the CBS gene disrupt the function of cystathionine beta-synthase, preventing homocysteine from being used properly. As a result, this amino acid and toxic byproducts substances build up in the blood. Some of the excess homocysteine is excreted in urine.
Rarely, homocystinuria can be caused by mutations in several other genes. The enzymes made by the MTHFR, MTR, MTRR, and MMADHC genes play roles in converting homocysteine to methionine. Mutations in any of these genes prevent the enzymes from functioning properly, which leads to a buildup of homocysteine in the body. Researchers have not determined how excess homocysteine and related compounds lead to the signs and symptoms of homocystinuria.
There are other, non-genetic causes of high levels of homocysteine 12. Non-genetic homocystinuria is not a rare condition. Some of the non-genetic causes are listed here:
- Vitamin B6 or vitamin B12 deficiency
- Folate deficiency
- Low thyroid hormones (hypothyroidism)
- Obesity
- Diabetes
- High cholesterol
- Physical inactivity
- High blood pressure
- Certain medications (such as carbamazepine, atorvastatin, fenofibrate, methotrexate, phenytoin, and nicotinic acid)
- Smoking
- Advanced age
Homocystinuria inheritance pattern
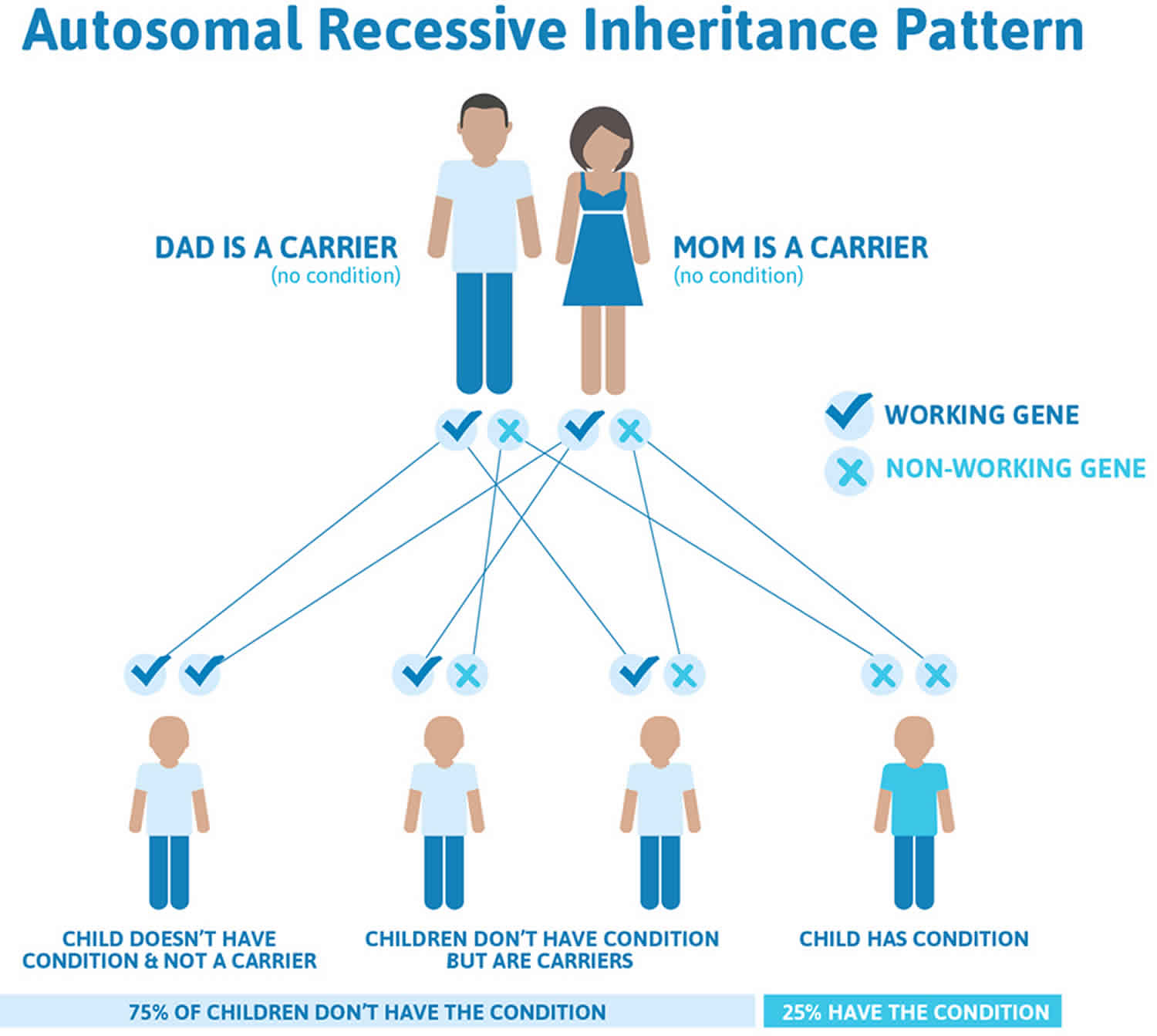
Homocystinuria is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. This means that the child must inherit a non-working copy of the gene from each parent to be seriously affected. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition.
Although people who carry one mutated copy and one normal copy of the cystathionine beta-synthase (CBS) gene do not have homocystinuria, they are more likely than people without a cystathionine beta-synthase (CBS) mutation to have shortages (deficiencies) of vitamin B12 and folic acid.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition
Key points to remember
- A person must inherit two copies of a abnormal gene, one from each parent, in order to be affected by the condition (25% chance). If a person inherits only one abnormal gene then they will be a carrier (50% chance). These outcomes occur randomly. They remain the same in every pregnancy and are the same for boys and girls.
- A abnormal gene cannot be corrected – it is present for life.
- A abnormal gene is not something that can be caught from other people. They can still be a blood donor, for example.
- People often feel guilty about a genetic condition which runs in the family. It is important to remember that it is no-one’s fault and no-one has done anything to cause it to happen.
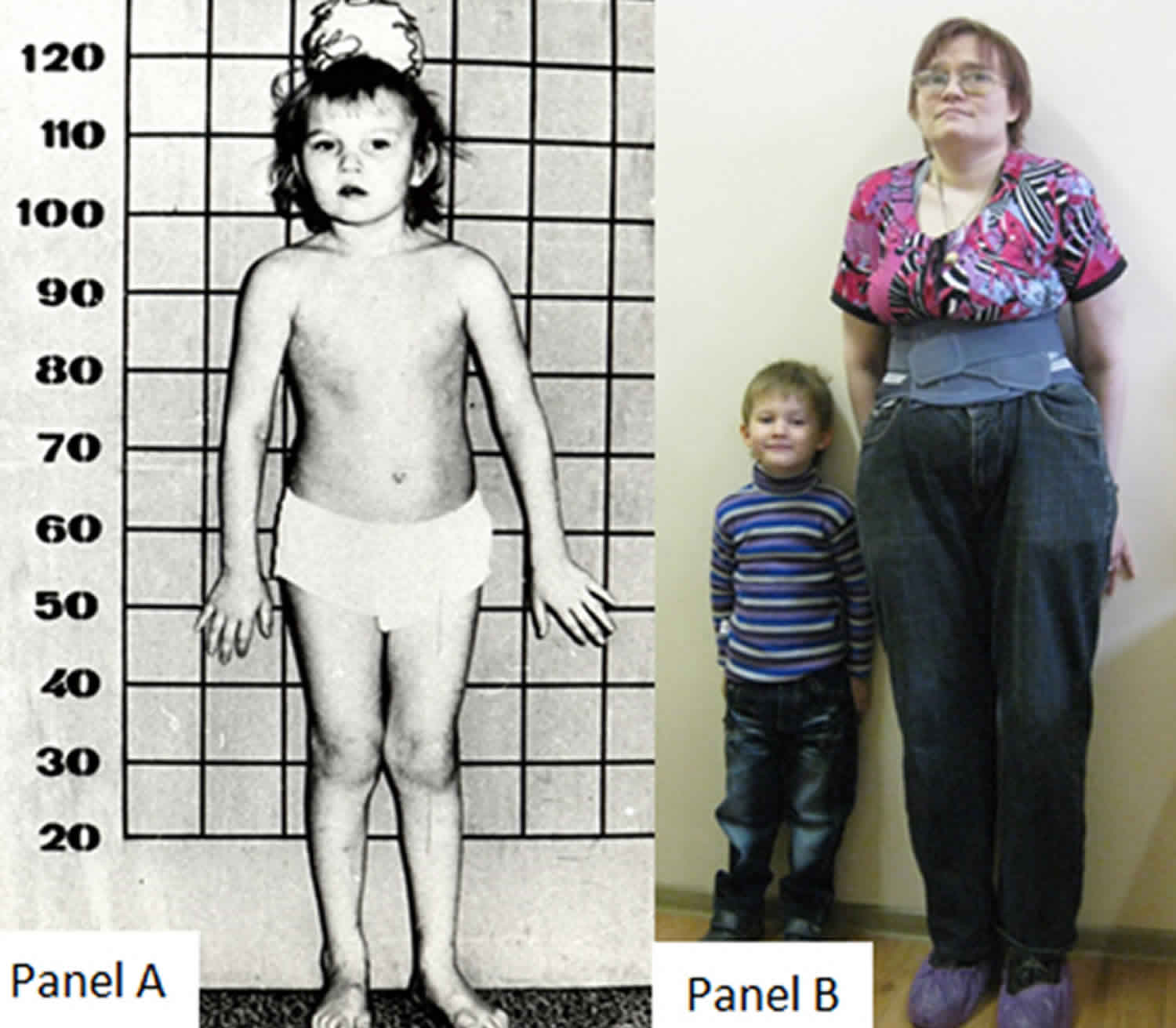
Figure 1. Homocystinuria autosomal recessive inheritance pattern
Homocystinuria symptoms
Homocystinuria is usually asymptomatic in the neonate and newborn infants appear healthy. Early symptoms, if present, are not obvious. Homocystinuria has several features in common with Marfan syndrome, including skeletal and eye changes.
If untreated, these children eventually develop mental retardation, ectopia lentis, a marfanoid appearance including arachnodactyly, osteoporosis, other skeletal deformities and thromboembolism.
Symptoms of the most severe form of homocystinuria will start in infancy or early childhood. The first of these symptoms may be poor growth and failure to gain weight 2. Other people with homocystinuria may not have any symptoms until adulthood. The most common symptom seen in adults with homocystinuria is an abnormal blood clot 13.
Other homocystinuria symptoms include:
- Chest deformities (pectus carinatum, pectus excavatum)
- Flush across the cheeks
- High arches of the feet
- Intellectual disability
- Knock knees
- Long limbs
- Mental disorders
- Nearsightedness
- Spidery fingers (arachnodactyly)
- Tall, thin build
Other symptoms of untreated homocystinuria can include:
Eye:
- Dislocation of the lens of the eye
- Nearsightedness
Skeletal:
- Caved-in chest (pectus excavatum)
- Curvature of the spine (scoliosis)
- Long, thin bones
- Osteoporosis (weak, brittle bones)
Central nervous system:
- Learning and intellectual disabilities
- Psychiatric problems
Blood and heart:
- Cardiovascular disease
- Abnormal blood clots anywhere including the brain
Many of the symptoms can be prevented by early and aggressive therapy.
- For those who are B6 responsive, beginning B6 supplementation will improve their homocysteine levels.
- Most will be on a special diet low in homocysteine and methionine and will take the medication Betaine (Cystadane ®).
- Most will need additional vitamin supplements to prevent deficiencies.
Homocystinuria diagnosis
Most states in the US test for homocystinuria due to CBS deficiency at birth by newborn screening. A baby that has a positive newborn screening test needs to have additional blood testing to look for high levels of homocysteine and methionine in the blood. Genetic testing can also be helpful for diagnosis 2.
A child or an adult with dislocation of the lens of the eye may also get tested for homocystinuria using blood and urine testing. In addition, a child or adult who has a blood clot, especially at an early age, may also get tested for homocystinuria.
Quantitative plasma amino acids will show increased homocystine and methionine in CBS deficiency homocystinuria but only increased methionine in the other disorders. Plasma homocysteine analysis will show markedly increased homocysteine in CBS deficiency homocystinuria and normal or only slightly increased homocysteine in the other disorders. Urine homocysteine is markedly increased in CBS deficiency homocystinuria.
Homocystinuria life expectancy
Without treatment, life expectancy is markedly reduced in pyridoxine-unresponsive patients. The life expectancy of pyridoxine-responsive patients is uncertain due to under-ascertainment in studies of this group. With early life-long adequate treatment, outcomes are generally good, although the very long-term outcomes are not yet known. Late teenagers and young adults are at high risk of non-compliance and complications, including death. Outcomes are determined primarily by pyridoxine responsiveness, adequacy of treatment and age of detection.
If untreated, the prognosis of pyridoxine-unresponsive CBS deficiency is bleak 10. The consequences of untreated, or partially treated, CBS deficiency include thromboembolic events, mental retardation, ocular and skeletal manifestations. An international study that documented the natural history of 629 untreated CBS patients showed that the risk of complications increases with age, and that pyridoxine-unresponsive patients are more severely affected 10. Pyridoxine responsive patients had significantly better mental capacities (n = 107, mean 79) than pyridoxine-unresponsive patients (n = 115, mean 57). The results of this study, however, were subject to ascertainment bias as milder cases, associated with pyridoxine responsiveness, were probably under-represented 14. Pyridoxine-responsive patients who present with thromboembolism as adults usually have a normal IQ and no other complications.
Long-term treatment, with good biochemical control, prevents complications from developing but it does not reverse complications already present. Moreover, loss of biochemical control at any age is associated with serious complications, which may be life-threatening. Thus, for pyridoxine unresponsive patients, optimal outcomes require newborn screening and treatment shortly after birth. No complications were observed in 15 such patients from Ireland, aged up to 25 years, whose lifetime median free homocystine was maintained below 11 μmol/L 15. They all had excellent vision, with full scale IQ ranging from 84-120 (mean 105.8) in 13 of the early treated and compliant patients 16. These patients are now aged up to 43 years and outcomes remain good in those whose plasma fHcy has remained less than 11 μmol/L with only brief rises above this. Similar findings were reported in 11 pyridoxine unresponsive, early treated patients from Manchester, aged up to 25 years 17, with full scale IQ ranging from 84-117 (median 100). The outcome for pyridoxine non-responsive patients diagnosed clinically (and therefore later) was poor with a median IQ of 58 (n = 2).
Homocystinuria treatment
The treatment of homocystinuria due to CBS deficiency is directed toward preventing or reducing the symptoms commonly associated with the disorder by controlling the levels of homocystine in the fluid portion of the blood (plasma). Treatment may include therapy with pyridoxine (vitamin B6), a diet that restricts the intake of protein and methionine, betaine therapy, and supplementation with folate (vitamin B9) or cobalamin (vitamin B12).
The goal is to keep one’s total homocysteine below 100 mcmol/L at all times and to not have any further complications. Some clinics like homocysteine levels to be even lower, and the guidelines recommend keeping below 50 mcmol/L for patients who are responsive to pyridoxine, as those patients can more easily reach that lower level without compromising their nutritional status. You need to talk to your metabolic geneticist and metabolic dietitian for your specific risks and goals.
Affected individuals may first undergo a pyridoxine response assessment. In approximately 50 percent of individuals, pyridoxine therapy is effective in reducing the levels of homocystine and methionine in the body. In order to determine whether an individual is responsive to pyridoxine therapy, folate levels must be normal and some individuals may require folate supplementation.
There is debate within the medical community as whether individuals who respond to pyridoxine therapy require additional treatment. Some physicians believe such individuals should also be placed on a diet low in protein and methionine or receive supplementary treatment with betaine (see below).
Individuals who are not diagnosed with homocystinuria due to CBS deficiency until childhood or adolescence or individuals who do not respond to therapy with pyridoxine require a restricted diet that is low in protein and methionine. Individuals on this diet require a methionine-free supplemental formula to provide them with other essential amino acids. A low protein, low methionine diet when started during infancy before any complications have developed has been effective in preventing or delaying the onset of symptoms.
A low protein, low methionine diet may be combined with cysteine supplementation. Cysteine is an amino acid that is often low in individuals with homocystinuria due to CBS deficiency. When methionine is broken down (metabolized) it produces cystine. Since individuals with homocystinuria cannot properly breakdown methionine, this may cause low levels of cysteine in some individuals.
When individuals who are not responsive to pyridoxine therapy are diagnosed later during childhood or adolescence, maintaining the dietary restrictions often proves difficult. The diet is usually not well-tolerated when it is begun in individuals diagnosed in childhood or adolescence.
Individuals with homocystinuria due to CBS deficiency, especially those who do not respond to pyridoxine therapy may be treated with betaine, which can be used to lower the levels of homocystine in the body. Betaine is often used an adjunct to individuals on a low protein, low methionine treatment. Betaine for oral solution (Cystadane®) has received marketing approval from the Food and Drug Administration (FDA) as a treatment for homocystinuria due to CBS deficiency and is manufactured by Rare Disease Therapeutics.
Specific symptoms of homocystinuria due to CBS deficiency are treated as appropriate. For example, dislocation of the lenses of the eyes (ectopia lentis) or certain skeletal malformations may be treated surgically. However, affected individuals who undergo any surgery should receive particular care because homocystinuria due to CBS deficiency may increase the risk of post-surgical thromboembolic complications.
Genetic counseling is recommended for affected individuals and their families.
Homocystinuria diet
Dietary treatment should be considered for all patients with CBS deficiency unless target homocysteine levels are achieved entirely by pyridoxine supplementation 18. Diet may be used either as a sole treatment or adjunctive therapy along with pyridoxine and/or betaine 18. Most pyridoxine-unresponsive patients require a diet that is very low in natural protein, with supplements of a methionine-free L-amino acid mixture. Lifelong treatment is required 18.
Dietary management of CBS deficiency can be highly successful. It should be considered for all pyridoxine unresponsive patients and as additional treatment in individuals who are partially pyridoxine responsive 19.
Restricting intake of the essential amino acid, methionine, reduces the precursor load on the transsulfuration pathway, thereby reducing homocysteine production. In most pyridoxine-unresponsive patients, the biochemical targets can only be achieved by a diet that is very low in natural protein, with supplements of a methionine-free L-amino acid mixture. The approach is analogous to the management of phenylketonuria (PKU) for which there is a greater body of published evidence.
1. Low-methionine diet:
The special diet is made up of foods that are very low in methionine. This means your child must not have cow’s milk, regular formula, meat, fish, cheese, or eggs. Regular flour, dried beans, nuts, and peanut butter also contain methionine and must be avoided or strictly limited.
Many vegetables and fruits have only small amounts of methionine and can be eaten in carefully measured amounts. There are other medical foods such as special low-protein
or low-methionine flours, breads, and pastas that are made especially for people with homocystinuria.
Your metabolic doctor and dietician will decide on the best food plan for your child. Your child’s diet will depend on many things such as his or her age, weight, and blood test results. Your dietician will fine-tune your child’s diet over time. The diet is usually needed throughout life.
2. Medical Foods and Formula
In addition to a low-methionine diet, some children are given a special medical formula as a substitute for milk. This formula will give your child the correct amount of nutrients and protein while helping to keep his or her methionine and homocystine levels within a safe range. Your metabolic doctor and dietician will tell you what type of formula is best for your child and how much to use.
Some states offer help with payment, or require private insurance coverage for formula and other special medical foods.
There are very few reported complications with well managed dietary treatment 20, however the diet is complex and difficult so poor adherence is common 21. Problems can be reduced by starting dietary treatment in individuals as young as possible and utilizing the skills of an experienced metabolic dietitian 21. Treatment for CBS deficiency must be continued throughout life, as loss of biochemical control in later life is associated with serious complications 17. Compliance with treatment often deteriorates, particularly in adolescence, as in other disorders 17. Initiating dietary restrictions in late diagnosed individuals is more challenging than in neonates but it can reduce the risk of further complications and lead to improvement, for example in seizures and behaviour 22.
Additional treatment with betaine can help patients who find it difficult to adhere to dietary restrictions and to attain good metabolic control (see Betaine treatment). Betaine lowers homocysteine levels, potentially allowing an increase in methionine intake 17. Methionine restriction in individuals treated with betaine can also prevent excessively raised methionine levels and the possible risks associated with these—see side effects of betaine 23. A recent European survey of pyridoxine unresponsive patients found that a combination of dietary restriction and betaine was the commonest treatment 19.
Supplements
Vitamin B6
Some children are helped by vitamin B6 supplements. In children who benefit from this treatment, the supplements help prevent intellectual disabilities and behavior problems. Vitamin B6 may also reduce the risk for blood clotting and eye and bone problems.
Ask your metabolic doctor whether your child would benefit from vitamin B6 supplements. Your doctor can do special tests to figure out whether your child will respond to vitamin B6.
Betaine
Betaine (N,N,N-trimethylglycine) is a vitamin-like substance found in grains and other foods. It can also be bought in pill form as a supplement. Betaine can help lower the amount of homocystine in the blood, and may be especially helpful for children who do not respond to vitamin B6. It may also lessen the risk of blood clots.
Your metabolic doctor will decide whether your child needs betaine. Unless you are advised otherwise, use only betaine prescribed by your doctor.
Vitamin B12
Some people with homocystinuria have low levels of vitamin B12 in their blood. They may need to have vitamin B12 injections. Ask your doctor whether your child needs extra vitamin B12.
Folic Acid
Some people have low levels of folic acid, a type of B vitamin, in their blood. They may need to take folic acid supplements by mouth. This vitamin can help lower the level of homocystine in the blood. Ask your doctor whether your child needs folic acid supplements.
L-cystine
People with homocystinuria may have low levels of another amino acid called L-cystine. L-cystine may already be part of the special medical formula. If not, it can be taken by mouth as a supplement. Unless you are advised otherwise, use only L-cystine prescribed by your doctor.
Do not use any supplements or medications without checking with your metabolic doctor.
Betaine treatment
Betaine (N,N,N-trimethylglycine) should be considered as adjunctive treatment in patients who cannot achieve target levels of homocysteine by other means.
Betaine is formed in the body from choline and small amounts are present in the normal diet 24. It lowers homocysteine concentrations in CBS deficiency by donating a methyl group and converting homocysteine to methionine 25. Betaine may also act as a chemical chaperone and correct partial mis-folding of CBS mutants 26. Betaine can increase cysteine levels 27 but this is probably secondary to decreased protein bound homocysteine.
Betaine treatment alone seldom achieves target homocysteine levels in patients with pyridoxine-unresponsive CBS deficiency. Studies of CBS-deficient mice gave similar results 28. This may be because betaine treatment raises the Met concentration. Individuals with plasma Met concentrations greater than 80 μmol/L respond less well to betaine 29, though in practice some response is usually seen. For these reasons, betaine is best used as adjunctive treatment in patients who are partially responsive to pyridoxine or who are on dietary treatment but cannot achieve good control.
Betaine recommended doses
Patients’ responses to betaine are variable and optimal doses have to be individualized. For children, the initial betaine dose is 50 mg/kg twice daily. For adults, the starting dose is 3 grams twice a day. The dose and frequency are adjusted according to response. There is unlikely to be any benefit in exceeding a dose of 150-200 mg/kg/day.
The published doses of betaine vary and very few studies are consistent. Betaine has a half-life of 14 hours so twice daily dosing is adequate 30.
In children, the initial dose is 100 mg/kg/day, divided into twice daily doses, and then adjusted according to response (typically increased weekly by 50 mg/kg increments). Studies based on pharmacokinetic and pharmacodynamic modelling after single doses of 50-100 mg/kg betaine suggest there is unlikely to be any additional benefit from using doses higher than 150-200 mg/kg/day 30.
The maximum licensed dose is 3 grams twice daily and this is the usual dose in adults but higher doses have sometimes been used with anecdotal evidence of biochemical benefit.
Betaine side effects
Generally betaine is well tolerated and safe. Higher doses have been associated with a fishy odor. Cerebral edema is a very rare side effect.
Betaine is generally safe but some people dislike the taste and compliance may be poor 17. It can result in a fishy odor 31. This is probably due to inadequate activity of flavin containing monooxygenase 3 and may respond to riboflavin 31.
Acute cerebral edema has been reported in two CBS deficient patients treated with betaine. The plasma Met concentration was above 2000 μmol/L in one patient 32 and 1190 μmol/L in the other patient 33. In both patients, problems resolved after withdrawing betaine and lowering the plasma Met concentration. Two other patients treated with betaine have developed similar white matter abnormalities without evidence of raised intracranial pressure; their plasma Met concentrations were 904 and 1282 μmol/L. One patient made a full recovery after the plasma Met decreased 34; neurological deficits persisted in the other patient, who was encephalopathic for more than 2 months before the plasma Met was lowered 35. A number of other CBS deficient patients on betaine treatment have had Met levels above 1000 μmol/L and have not experienced cerebral edema. Cerebral edema has also been seen in a few non-CBS-deficient patients with high levels of Met. Further research is required but the current recommendation is to avoid Met levels above 1000 μmol/L in patients treated with betaine.
Monitoring
Monitoring of plasma total homocystine, amino acid, folate and vitamin B 12 is recommended in all patients. The frequency depends on the severity of CBS deficiency, treatment, age and clinical condition of the patient. These factors also determine the need for additional monitoring; for example, patients on dietary treatment require regular nutritional assessment.
Total homocysteine, plasma amino acids (including methionine), vitamin B12 and folate should be monitored regularly in all patients with CBS deficiency. There is, however, little evidence concerning the optimal frequency of monitoring. This will vary depending on the severity of the disorder (e.g. pyridoxine-responsiveness), the patient’s treatment, compliance, age and previous complications (e.g. thrombosis). In adult patients who are fully pyridoxine-responsive it may be adequate to monitor total homocystine levels every six months. In contrast, in children on dietary treatment for pyridoxine-unresponsive CBS deficiency, total homocysteine and methionine will need to be monitored much more frequently.
The method of analysis may also influence the frequency of analysis. If total homocystine is monitored in dry blood spots sent in from home, it is reasonable to request samples every week during infancy (as in PKU) but this technique is not yet widely available. In most centres, patients will need to attend a hospital for total homocystine monitoring samples to be taken from liquid blood and samples will be taken less often.
The serum vitamin B12 and folate levels should be measured annually; if the vitamin B12 is low, an intramuscular supplement is generally given and levels repeated every 3-6 months thereafter.
Patients on dietary treatment require regular nutritional assessment and additional tests, depending on the patient’s age and clinical condition. Some suggestions are listed in Table 1. One should consider annual monitoring of the blood count, renal profile, liver profile, copper, zinc and selenium, vitamin D and essential fatty acids as well as plasma amino acids. There is no specific evidence relating to CBS deficiency but there are reports of micronutrient deficiency in patients on similar dietary treatment for PKU. Tests should be done more frequently if there is poor adherence to diet, inadequate medical food consumption, poor growth or clinical evidence of malnutrition. More extensive monitoring can be done if clinically indicated. Supplements should be given if nutritional deficiencies are identified.
Bone density scans (DEXA) should be done every 3-5 years from adolescence with additional scans in individuals who have frequent fractures and/or low vitamin D levels. Neuroimaging (MRI) is only indicated in individuals who have abnormal neurological signs.
Table 1. Homocystinuria monitoring recommendations
Area | Tests | Frequency |
|---|---|---|
Anthropometry | Height & weight | Every clinic visit |
Dietary | Dietary intake analysis | Every clinic visit if on dietary treatment |
Biochemical–metabolic control | total homocystine, methionine | See text |
Nutritional | Vitamin B12, folate | At least annually |
Blood count, albumin, plasma AA, ferritin, zinc, 25-hydroxyvitamin D | At least annually if on dietary treatment | |
Selenium, essential fatty acids | If concerns about intake | |
Neurodevelopmental/neurological | Clinical examination | Annually |
MRI/EEG | Only if new CNS symptoms | |
Ophthalmological | Eye examination | At least annually |
Neuropsychological ffunction | IQ | At least every 5 years during childhood |
Psychological | Clinical psychology or psychiatric assessment | As required |
Bone density | DEXA | Every 3-5 years from adolescence—unless clinically indicated earlier |
Cardiovascular | Lipid profile, cardiovascular risk factor review | Once in childhood, annually in adulthood |
- Wasim M, Awan FR, Khan HN, Tawab A, Iqbal M, Ayesha H. Aminoacidopathies: Prevalence, Etiology, Screening, and Treatment Options. Biochem Genet. Apr 2018; 56(1-2):7-21. https://www.ncbi.nlm.nih.gov/pubmed/29094226[↩]
- Homocystinuria. Screening, Technology and Research in Genetics (STAR-G). http://www.newbornscreening.info/Parents/aminoaciddisorders/CBS.html[↩][↩][↩][↩]
- Morris AA, Kozich V, Santra S et al. Guidelines for the diagnosis and treatment of cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2017; 40(1):49-74. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5203861/pdf[↩]
- Sacharow SJ, Picker JD, Levy HL. Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency. 2004 Jan 15 [Updated 2017 May 18]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1524[↩][↩][↩][↩][↩]
- Levy HL et al. Reproductive fitness in maternal homocystinuria due to cystathionine beta-synthase deficiency. Journal of Inherited Metabolic Disease. August 2002; 25(4):299-314. http://www.ncbi.nlm.nih.gov/pubmed/12227460[↩][↩]
- Nuno-Ayala M, Guillen N, Navarro MA et al (2010) Cysteinemia, rather than homocysteinemia, is associated with plasma apolipoprotein A-I levels in hyperhomocysteinemia: lipid metabolism in cystathionine beta-synthase deficiency. Atherosclerosis 212:268–273[↩]
- You XJ, Xu C, Lu JQ et al (2011) Expression of cystathionine beta-synthase and cystathionine gamma-lyase in human pregnant myometrium and their roles in the control of uterine contractility. PLoS One 6:e23788[↩]
- Levy HL, Vargas JE, Waisbren SE et al (2002) Reproductive fitness in maternal homocystinuria due to cystathionine beta-synthase deficiency. J Inherit Metab Dis 25:299–314[↩]
- Owen EP, Human L, Carolissen AA, Harley EH, Odendaal HJ (1997) Hyperhomocysteinemia—a risk factor for abruptio placentae. J Inherit Metab Dis 20:359–362[↩]
- Mudd SH, Skovby F, Levy HL et al (1985) The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet 37:1–31[↩][↩][↩][↩]
- Homocystinuria. https://ghr.nlm.nih.gov/condition/homocystinuria[↩]
- Varga E and Moll S. Homocysteine and MTHFR Mutations. Circulation. 2015; 132:e6-e9. https://www.ahajournals.org/doi/pdf/10.1161/CIRCULATIONAHA.114.013311[↩]
- Morris AA, Kozich V, Santra S et al. Guidelines for the diagnosis and treatment of cystathionine beta-synthase deficiency. J Inherit Metab Dis. 2017; 40(1):49-74. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5203861/pdf/10545_2016_Article_9979.pdf[↩]
- Skovby F, Gaustadnes M, Mudd SH (2010) A revisit to the natural history of homocystinuria due to cystathionine beta-synthase deficiency. Mol Genet Metab 99:1–3[↩]
- Yap S, Naughten E (1998) Homocystinuria due to cystathionine beta-synthase deficiency in Ireland: 25 years’ experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J Inherit Metab Dis 21:738–747[↩]
- Yap S, Rushe H, Howard PM, Naughten ER (2001c) The intellectual abilities of early-treated individuals with pyridoxine-nonresponsive homocystinuria due to cystathionine beta-synthase deficiency. J Inherit Metab Dis 24:437–447[↩]
- Walter JH, Wraith JE, White FJ, Bridge C, Till J (1998) Strategies for the treatment of cystathionine beta-synthase deficiency: the experience of the Willink Biochemical Genetics Unit over the past 30 years. Eur J Pediatr 157(Suppl 2):S71–S76[↩][↩][↩][↩][↩]
- Morris, A.A.M., Kožich, V., Santra, S. et al. J Inherit Metab Dis (2017) 40: 49. https://doi.org/10.1007/s10545-016-9979-0[↩][↩][↩]
- Adam S, Almeida MF, Carbasius Weber E et al (2013) Dietary practices in pyridoxine non-responsive homocystinuria: a European survey. Mol Genet Metab 110:454–459[↩][↩]
- Perry TL, Hansen S, Love DL, Crawford LE, Tischler B (1968) Treatment of homocystinuria with a low-methionine diet, supplemental cystine, and a methyl donor. Lancet 2:474–478[↩]
- Schiff M, Blom HJ (2012) Treatment of inherited homocystinurias. Neuropediatrics 43:295–304[↩][↩]
- Kabra M (2002) Dietary management of inborn errors of metabolism. Indian J Pediatr 69:421–426[↩]
- Lawson-Yuen A, Levy HL (2006) The use of betaine in the treatment of elevated homocysteine. Mol Genet Metab 88:201–207[↩]
- Zeisel SH, Mar MH, Howe JC, Holden JM (2003) Concentrations of choline-containing compounds and betaine in common foods. J Nutr 133:1302–1307[↩]
- Singh RH, Kruger WD, Wang L, Pasquali M, Elsas LJ (2004) Cystathionine beta-synthase deficiency: effects of betaine supplementation after methionine restriction in B6-nonresponsive homocystinuria. Genet Med 6:90–95[↩]
- Kopecka J, Krijt J, Rakova K, Kozich V (2011) Restoring assembly and activity of cystathionine beta-synthase mutants by ligands and chemical chaperones. J Inherit Metab Dis 34:39–48[↩]
- Wilcken DE, Dudman NP, Tyrrell PA (1985) Homocystinuria due to cystathionine beta-synthase deficiency—the effects of betaine treatment in pyridoxine-responsive patients. Metabolism 34:1115–1121[↩]
- Gupta S, Wang L, Kruger WD (2016) Betaine supplementation is less effective than methionine restriction in correcting phenotypes of CBS deficient mice. J Inherit Metab Dis 39:39–46[↩]
- Sakamoto A, Sakura N (2003) Limited effectiveness of betaine therapy for cystathionine beta synthase deficiency. Pediatr Int 45:333–338[↩]
- Schwahn BC, Hafner D, Hohlfeld T, Balkenhol N, Laryea MD, Wendel U (2003) Pharmacokinetics of oral betaine in healthy subjects and patients with homocystinuria. Br J Clin Pharmacol 55:6–13[↩][↩]
- Manning NJ, Allen EK, Kirk RJ, Sharrard MJ, Smith EJ (2012) Riboflavin-responsive trimethylaminuria in a patient with homocystinuria on betaine therapy. JIMD Rep 5:71–75[↩][↩]
- Yaghmai R, Kashani AH, Geraghty MT et al (2002) Progressive cerebral edema associated with high methionine levels and betaine therapy in a patient with cystathionine beta-synthase (CBS) deficiency. Am J Med Genet 108:57–63[↩]
- Devlin AM, Hajipour L, Gholkar A, Fernandes H, Ramesh V, Morris AA (2004) Cerebral edema associated with betaine treatment in classical homocystinuria. J Pediatr 144:545–548[↩]
- Sasai H, Shimozawa N, Asano T et al (2015) Successive MRI findings of reversible cerebral white matter lesions in a patient with cystathionine beta-synthase deficiency. Tohoku J Exp Med 237:323–327[↩]
- Vatanavicharn N, Pressman BD, Wilcox WR (2008) Reversible leukoencephalopathy with acute neurological deterioration and permanent residua in classical homocystinuria: a case report. J Inherit Metab Dis 31(Suppl 3):477–81[↩]



{kind=link}