Contents
- Hypogonadotropic hypogonadism
- Hypogonadotropic hypogonadism causes
- Hypogonadotropic hypogonadism symptoms
- Hypogonadotropic hypogonadism complications
- Hypogonadotropic hypogonadism diagnosis
- Hypogonadotropic hypogonadism treatment
- Hypogonadotropic hypogonadism prognosis
Hypogonadotropic hypogonadism
Hypogonadotropic hypogonadism also called secondary hypogonadism is a form of hypogonadism that is due to a problem with the pituitary gland or hypothalamus (due to a problem with the ) 1. Hypogonadism is a condition in which the male testes or the female ovaries produce little or no sex hormones 1. In hypogonadotropic hypogonadism the problem could be anywhere with the hypothalamic-pituitary-gonadal axis (HPG axis) can result from failure of the hypothalamic luteinizing-hormone releasing hormone (LHRH) pulse generator or from the inability of the pituitary gland to respond with secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) and is diagnosed in the setting of a low testosterone level and sperm count (oligospermia) in association with low or inappropriately normal serum luteinizing hormone (LH) and follicle-stimulating hormone (FSH) concentrations 2. Increased prevalence of hypogonadotropic hypogonadism has been observed among obese population and also among patients with type 2 diabetes 3, 4, 5, 6, 7. Approximately one-third of men with obesity, type 2 diabetes, or metabolic syndrome have subnormal free testosterone concentrations 8, 9, 10. Testosterone concentrations are inversely related to body mass index (BMI) and insulin resistance 10. One-third of obese young males (14–35 years of age) also have subnormal free testosterone concentrations.
Men with hypogonadotropic hypogonadism have low levels of androgens (testosterone) in the plasma as well as a lack or delay of sexual maturity, which can cause symptoms such as a lack of libido, depression, increase in adipose tissue, and diminished erectile function or impotence (not being able to obtain or keep an erection that is sufficient for sexual intercourse) 11.
Patients with hypogonadotropic hypogonadism usually have an issue with gonadotropin-releasing hormone (GnRH) signaling, which then causes a decrease in follicle-stimulating hormone (FSH) and luteinizing hormone (LH) secretion. This decreased FSH and LH contributes to decreased androgen levels and reduced spermatogenesis. Studies have shown that giving these patients pulsatile GnRH or LH (or hCG) and FSH can help increase spermatogenesis and thus increase the sperm concentration in the ejaculate. Even then, most couples will need assisted reproductive technology (ART) to achieve pregnancy 12.
Testosterone therapy, whether by exogenous testosterone replacement or induction of endogenous testosterone production by human chorionic gonadotropin (hCG) is needed in all hypogonadotropic hypogonadism patients 2. Testosterone plays a number of important physiologic roles in the human and are required not only for virilization and normal sexual function, but also for maintenance of both muscle and bone mass, as well as normal mood and cognition.
While testosterone is the primary treatment modality used to induce and maintain secondary sexual characteristics and sexual function in men with hypogonadism, treatment with testosterone does not restore fertility. Therefore, in patients in whom fertility is the treatment goal, induction of gonadotropin secretion by pulsatile GnRH or exogenous gonadotropins is necessary. While hormone therapy is the mainstay of treatment for congenital hypogonadotropic hypogonadism; undescended testicle (cryptorchidism), if present, should be treated surgically with orchidopexy, ideally before the age of 2 years to improve fertility outcomes and reduce the risk of future testicular malignancy 13.
Hypothalamic-Pituitary-Gonadal Axis
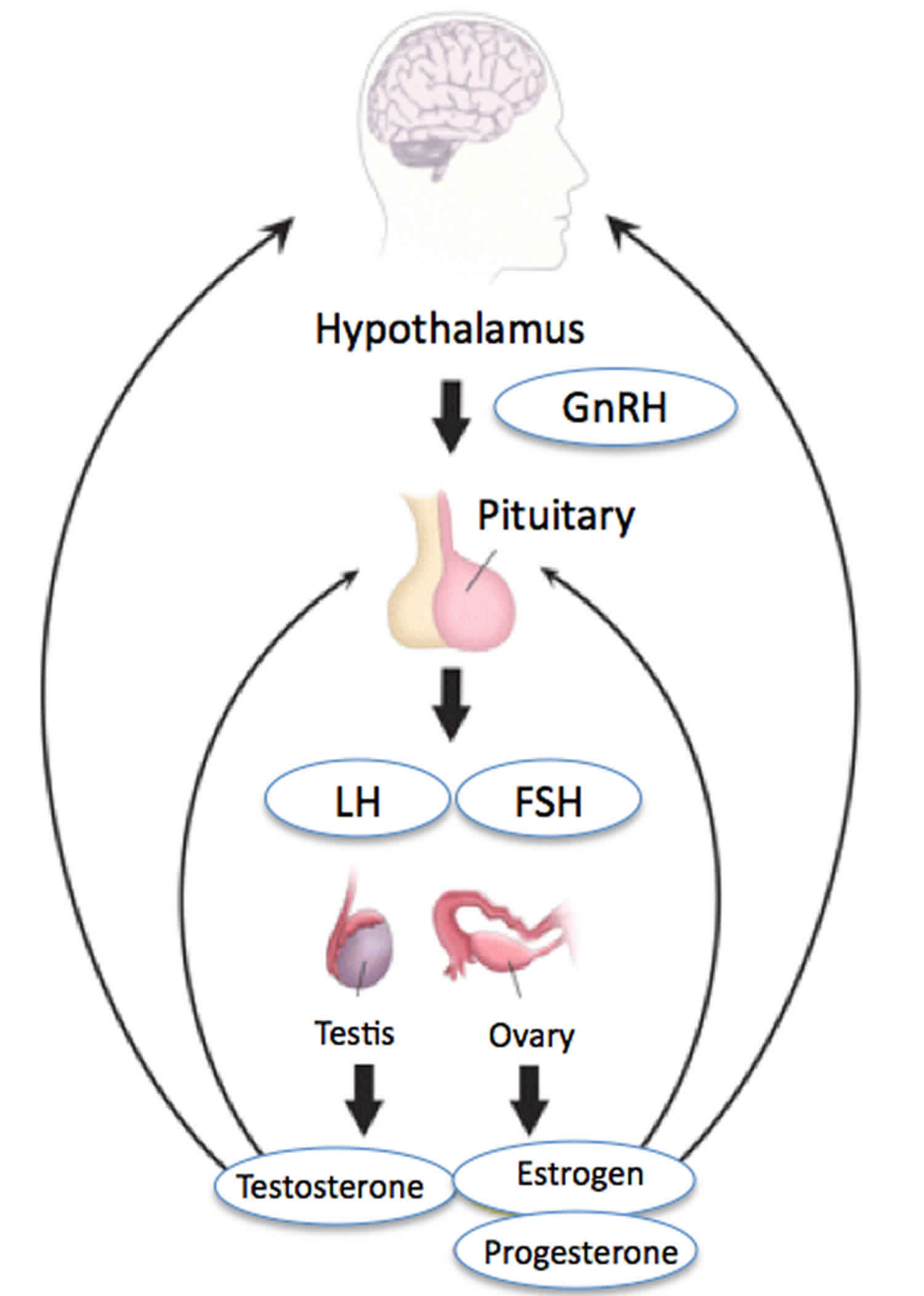
Testicular and ovarian functions are regulated by hormones of the central hypothalamic–pituitary–gonadal axis (HPG axis) also known as the hypothalamic–pituitary–ovarian/testicular axis. Hypothalamic–pituitary–gonadal axis (HPG axis) refers to the hypothalamus, pituitary gland, and gonadal glands (testes in males and ovaries in females) as if these individual endocrine glands were a single entity. The hypothalamus is a special area in the brain that is responsible for control of several hormones in the body. Reproductive function in humans is under the control of a group of ~ 1,200-1,500 specialized neurons called GnRH (Gonadotropin-Releasing Hormone) neurons or “GnRH pulse generator” in the hypothalamus 14. The hypothalamic control of reproduction is coordinated through the release of gonadotropin-releasing hormone (GnRH). At the time of puberty, GnRH (Gonadotropin-Releasing Hormone) neurons coordinately secrete gonadotropin-releasing hormone (GnRH), in a series of discrete series of bursts or pulses. Pulsatile secretion of gonadotropin-releasing hormone (GnRH) from the hypothalamus is required for both the initiation and maintenance of the hypothalamic–pituitary–ovarian/testicular axis in human. GnRH pulses exert a stimulating effect on GnRH receptors that are located on gonadotropic cells of the anterior pituitary gland. In response, the gonadotropins luteinizing hormone (LH) and follicle-stimulating hormone (FSH) are cosecreted into the bloodstream from the anterior pituitary gonadotropes. The intensity of pituitary secretion of both pituitary hormones is differentially modulated by the amplitude and frequency of GnRH pulses. Male and female gonads respond to gonadotropin-stimulation by secreting sex steroid hormones [i.e., testosterone (T), estradiol (primarily 17β-estradiol, E2), and progesterone (P4)].
In females, ovarian follicles are stimulated by follicle-stimulating hormone (FSH) to grow and mature; luteinizing hormone (LH) stimulates ovulation and corpus luteum formation. In females, ovarian theca cells produce androstenedione and testosterone. These hormones diffuse to the neighboring granulosa cells, where they are used as precursors for estrogen synthesis 15. Follicle-stimulating hormone (FSH) stimulates estrogen secretion by granulosa cells, which in turn induces a peak of pituitary LH secretion that induces ovulation. Thereafter, luteinized granulosa and theca cells produce progesterone and estrogens on stimulation by both gonadotropins.
In males, testicular Leydig cells produce and secrete testosterone (T) on luteinizing hormone (LH) stimulation. Testosterone, together with 5-dihydrotestosterone (DHT), are the key bioactive androgens 16. Follicle-stimulating hormone (FSH) stimulates testicular Sertoli cells with a 2-fold effect 17. Specifically, during mini-puberty and early puberty, Sertoli cells respond to FSH stimulation by undergoing mitotic divisions. During later puberty and adulthood, the role of Sertoli cells is to form the niche that nurtures germ cells on FSH stimulation, to enable spermatogenesis. Spermatogenesis includes meiosis of diploid spermatogonia to haploid pachytene spermatocytes and completion of spermatogenesis through development of elongated spermatids from round spermatids (spermiogenesis). This results in the production of mature sperms 18.
Human HPG axis function is modulated by feedback mechanisms of the gonads to the hypothalamus and pituitary, including both positive and negative mechanisms. While testosterone, estradiol, progesterone, and inhibins act as inhibitors on GnRH, LH, and FSH secretion, activins exert a positive feedback 19.
In females, the feedback of estradiol on pituitary LH secretion is 2-fold: While stimulatory in the first phase of the menstrual cycle, to allow for the LH surge at ovulation, it is inhibitory in the second phase 20.
In both men and women, gonadal failure results in increased LH, because of loss of the negative feedback of estrogen at the hypothalamus and pituitary in women and from decreases in both androgen and estrogen feedback in men. In response to decreased levels of sex steroids as well as the loss of inhibin, FSH levels are also elevated following gonadal damage. Serum gonadotropin [luteinizing hormone (LH) and follicle-stimulating hormone (FSH)] and sex steroid values will differentiate between reproductive failure at the gonadal level or at the hypothalamic/pituitary level. Sex steroid levels will be low in both, but serum gonadotropin levels will be high in primary gonadal failure and low in those with hypothalamic or pituitary disease. The hallmark of primary gonadal failure from any cause is elevation of gonadotropin [luteinizing hormone (LH) and follicle-stimulating hormone (FSH)] levels, and this is the usual state in postpubertal patients receiving substantial doses of chemotherapy agents. Cranial radiation, on the other hand, may result in significant hypothalamic-pituitary dysfunction and secondary gonadal failure with low serum levels of gonadotropins.
There are 3 physiologic waves of central HPG axis activity over the lifetime. The first occurs during fetal life, the second in the first months after birth (“mini-puberty”), and the third at puberty. From then on, the HPG axis remains active all through adulthood 21.
Figure 1. Hypothalamic–pituitary–gonadal axis (HPG axis)
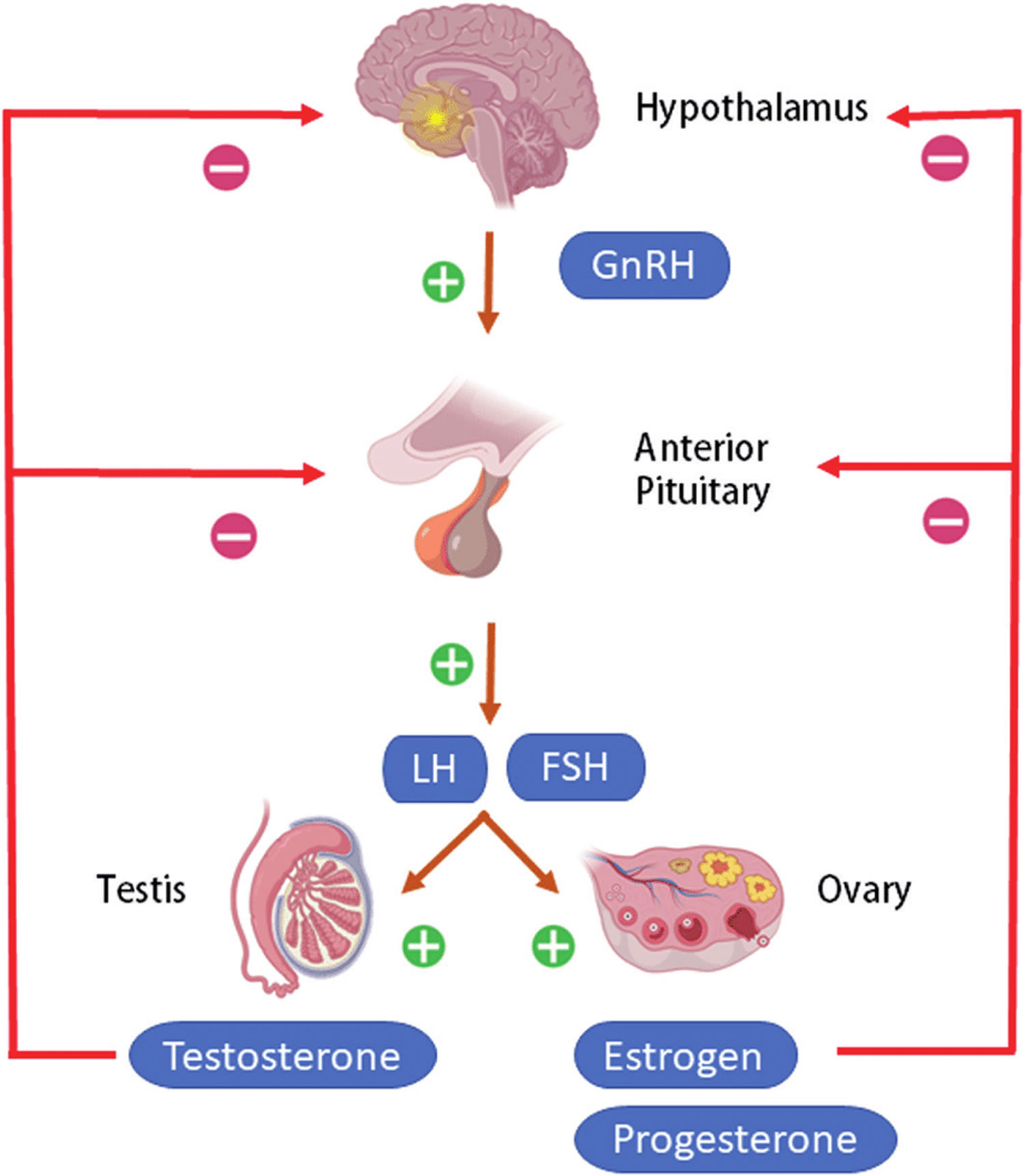
Figure 2. The hypothalamus and pituitary gland (anterior and posterior) endocrine pathways and target organs
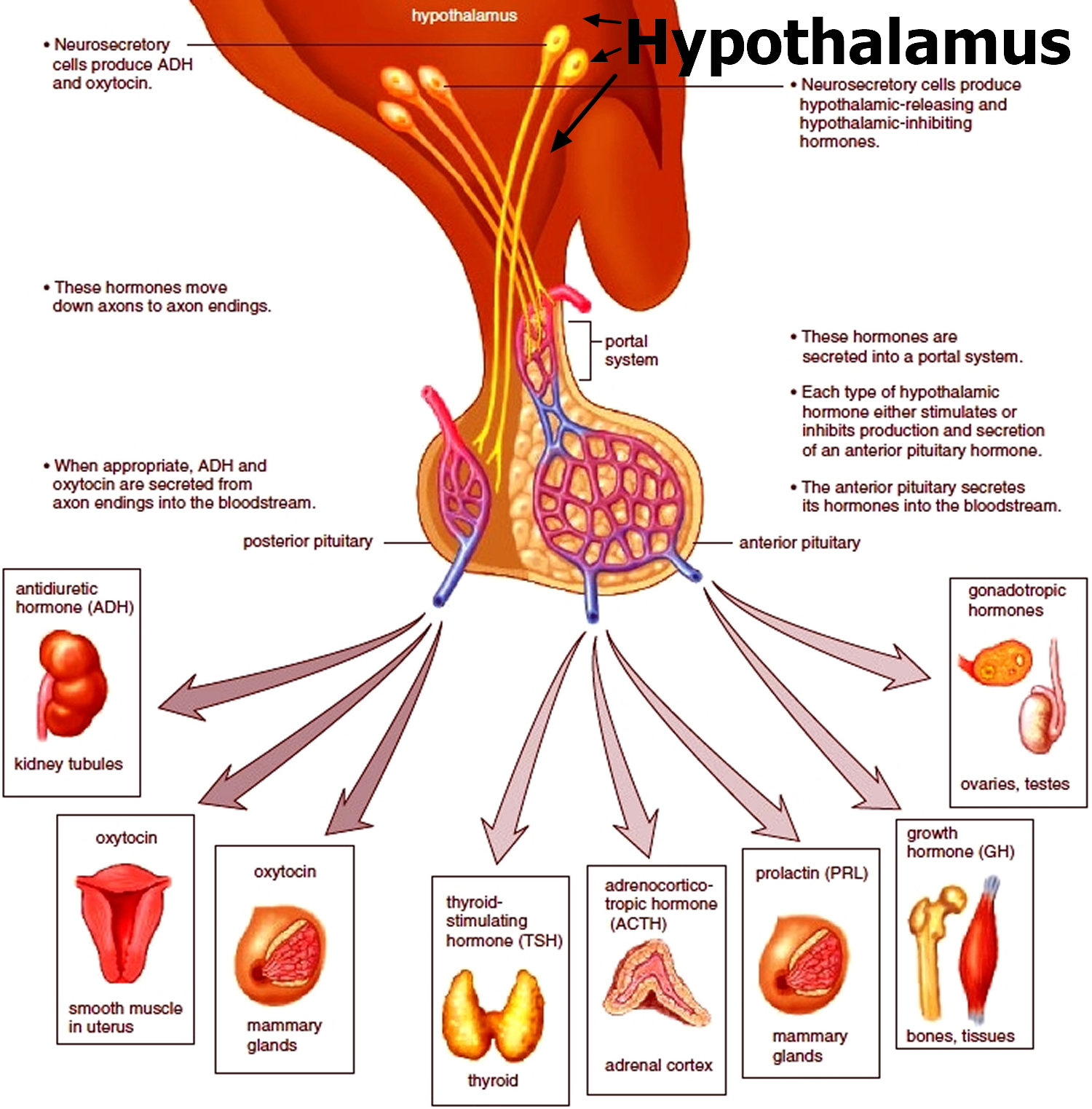
Figure 3. Testosterone production in men
Footnotes: Drawing shows that testosterone (androgen) production is regulated by luteinizing hormone (LH) and luteinizing hormone-releasing hormone (LHRH). The hypothalamus releases luteinizing hormone-releasing hormone (LHRH), which stimulates the release of LH (luteinizing hormone) from the pituitary gland. Luteinizing hormone (LH) acts on Leydig cells in the testes to produce the majority of testosterone in the body. Most of the remaining testosterone are produced by the adrenal glands. Testosterone are taken up by prostate cells, where they either bind to the androgen receptor (AR) directly or are converted to dihydrotestosterone (DHT), which has a greater binding affinity for the androgen receptor than testosterone.
Figure 4. Estrogen and progesterone production in premenopausal women
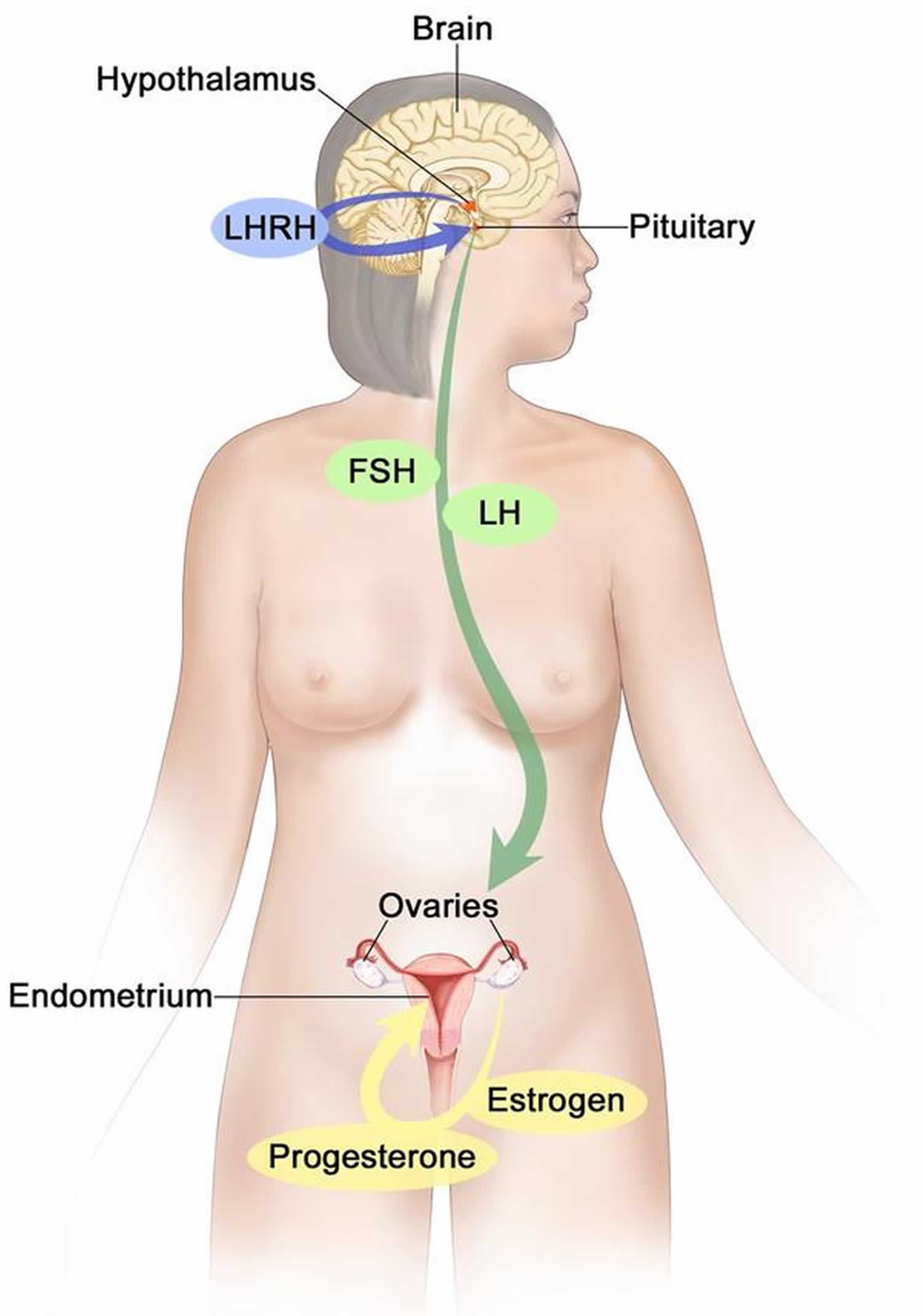
Footnotes: Drawing shows that in premenopausal women, estrogen and progesterone production by the ovaries is regulated by luteinizing hormone (LH) and luteinizing hormone-releasing hormone (LHRH). The hypothalamus releases LHRH, which then causes the pituitary gland to make and secrete LH and follicle-stimulating hormone (FSH). LH and FSH cause the ovaries to make estrogen and progesterone, which act on the endometrium (inner lining of the uterus). When estrogen and progesterone production reaches a certain level during the menstrual cycle, these hormones act on the hypothalamus and pituitary to turn off production of LHRH, LH, and FSH. Estrogen is a steroid hormone that is responsible for the growth and regulation of the female reproductive system and secondary sex characteristics. Estrogen is produced by the granulosa cells of the developing follicle and exerts negative feedback on LH production in the early part of the menstrual cycle. However, once estrogen levels reach a critical level as oocytes mature within the ovary in preparation for ovulation, estrogen begins to exert positive feedback on LH production, leading to the LH surge through its effects on GnRH pulse frequency. Estrogen also has many other effects that are important for bone health and cardiovascular health in premenopausal patients 23.
Hypogonadotropic hypogonadism causes
There are two basic types of hypogonadism:
- Primary hypogonadism also known as primary gonadal failure or HYPERgonadotropic hypogonadism originates from a problem in the testicles or ovaries.
- Secondary hypogonadism also known as HYPOgonadotropic hypogonadism indicates a problem in the hypothalamus or the pituitary gland — parts of the brain that signal the testicles to produce testosterone. The hypothalamus produces gonadotropin-releasing hormone (GnRH), which signals the anterior pituitary gland to make follicle-stimulating hormone (FSH) and luteinizing hormone (LH). Luteinizing hormone (LH) then signals the testes to produce testosterone.
Either type of hypogonadism can be caused by an inherited (congenital) trait or something that happens later in life (acquired), such as an injury or an infection. At times, primary and secondary hypogonadism can occur together.
In hypogonadotropic hypogonadism, the testicles in males or ovaries in females are normal but don’t function properly due to a problem with your pituitary gland or the hypothalamus. Hypogonadotropic hypogonadism (secondary hypogonadism) is caused by a lack of gonadotropin-releasing hormone (GnRH), follicle stimulating hormone (FSH) and luteinizing hormone (LH) that normally stimulate the ovaries or testes.
Hypogonadotropic hypogonadism is caused by a lack of hormones that normally stimulate the ovaries or testes. These hormones include:
- Gonadotropin-releasing hormone (GnRH)
- Follicle stimulating hormone (FSH)
- Luteinizing hormone (LH)
Normally:
- The hypothalamus in the brain releases gonadotropin-releasing hormone (GnRH).
- This gonadotropin-releasing hormone (GnRH) stimulates the anterior pituitary gland to release follicle-stimulating hormone (FSH) and luteinizing hormone (LH).
- The follicle-stimulating hormone (FSH) and luteinizing hormone (LH) tell the female ovaries or the male testes to release hormones that lead to normal sexual development in puberty, normal menstrual cycles, estrogen levels and fertility in adult women, and normal testosterone production and sperm production in adult men.
- Any change in this hormone release chain causes a lack of sex hormones. This prevents normal sexual maturity in children and normal function of the testicles or ovaries in adults.
Hypogonadotropic hypogonadism common causes include 1, 24:
- Congenital hypogonadotropic hypogonadism (CHH) or normosmic congenital hypogonadotropic hypogonadism (nCHH) 25
- Kallmann’s syndrome. Kallmann’s syndrome is a type of congenital hypogonadotropic hypogonadism with loss of smell (anosmia) or decreased sense of smell (hyposmia). This feature distinguishes Kallmann syndrome from most other forms of hypogonadotropic hypogonadism, which do not affect the sense of smell. Kallmann syndrome is a rare genetic disorder that is defined by a delay/absence of signs of puberty along with an absent/impaired sense of smell. A closely related disorder, normosmic idiopathic hypogonadotropic hypogonadism (nIHH), refers to patients with pubertal failure but with a normal sense of smell. Both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism (nIHH) are due to an isolated deficiency of a key reproductive hormone called gonadotropin-releasing hormone (GnRH). Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism (nIHH) occurs in both sexes but males are more commonly diagnosed with this condition. Many people with Kallmann syndrome are not aware that they are unable to detect odors until the impairment is discovered through testing. Kallmann syndrome can cause red-green color blindness. Changes in more than 20 genes have been associated with Kallmann syndrome. Among the most common causes of the condition are mutations in the ANOS1, CHD7, FGF8, FGFR1, PROK2, or PROKR2 gene. In some cases, affected individuals have mutations in more than one of these genes. Additionally, researchers have identified mutations in other genes that may contribute to the development and features of Kallmann syndrome, but are unlikely to cause the disease on their own 26, 27.
- Idiopathic hypogonadotropic hypogonadism
- Damage to the pituitary gland or hypothalamus from surgery, injury, tumor, infection, or radiation. An abnormality in the pituitary gland can impair the release of hormones from the pituitary gland to the testicles, affecting normal testosterone production. A pituitary tumor or other type of brain tumor located near the pituitary gland may cause testosterone or other hormone deficiencies. Also, treatment for a brain tumor, such as surgery or radiation therapy, can affect the pituitary gland and cause hypogonadism.
- Congenital disorders such as Prader-Willi syndrome, Laurence-Moon syndrome, Bardet-Biedl syndrome 28 and Gaucher disease
- High prolactin level (a different hormone released by the pituitary)
- Inflammatory disease. Certain inflammatory diseases, such as sarcoidosis, histiocytosis and tuberculosis, involve the hypothalamus and pituitary gland and can affect testosterone production.
- HIV/AIDS. HIV/AIDS can cause low levels of testosterone by affecting the hypothalamus, the pituitary and the testes.
- Medications. The use of certain drugs, such as opioid pain medications and some steroid (glucocorticoid) medicines, can affect testosterone production.
- Drug use, such as heroin
- Obesity. Being significantly overweight at any age might be linked to hypogonadism.
- Aging. As men age, there’s a slow, progressive decrease in testosterone production. The rate varies greatly.
- Certain medical conditions, such as iron overload or hemochromatosis. Too much iron in the blood can cause testicular failure or pituitary gland dysfunction, affecting testosterone production.
- Severe stress
- Nutritional problems (both rapid weight gain or weight loss).
Congenital hypogonadotropic hypogonadism
Congenital hypogonadotropic hypogonadism is a rare genetic disorder characterized by isolated deficiency or dysfunction of gonadotropin-releasing hormone (GnRH) 21, 29. Congenital hypogonadotropic hypogonadism consists of hypogonadotropic hypogonadism with anosmia or hyposmia called Kallmann syndrome, and the normosmic idiopathic hypogonadotropic hypogonadism 30. Changes in more than 40 genes have been associated with congenital hypogonadotropic hypogonadism 31.
Congenital hypogonadotropic hypogonadism is clinically characterized by absent or incomplete development during puberty, resulting in small testes by the age of 18 years and infertility in adults 32. Congenital hypogonadotropic hypogonadism patients have low circulating testosterone levels with low gonadotropin levels, whereas other pituitary hormones are normal 33.
According to the degree of pubertal development, patients with congenital hypogonadotropic hypogonadism (CHH) can be divided into partial congenital hypogonadotropic hypogonadism (PCHH) and complete congenital hypogonadotropic hypogonadism (CCHH) 34. Patients who present with partial pubertal development can be diagnosed with partial congenital hypogonadotropic hypogonadism (PCHH). There is currently no definite diagnostic standard for partial congenital hypogonadotropic hypogonadism (PCHH). Partial congenital hypogonadotropic hypogonadism (PCHH) typically presents with mild gonadotropin deficiency and partial pubertal development 25, 35. Mutations in gonadotropin-releasing hormone receptor gene (GNRHR), fibroblast growth factor receptor 1 gene (FGFR1), tachykinin 3 (TAC3), and tachykinin receptor 3 gene (TACR3) cause partial congenital hypogonadotropic hypogonadism (PCHH) 36. Because of lack of clear diagnostic criteria, previous studies of PCHH were mainly limited to description of symptoms and characterization of mutant genes.
Idiopathic hypogonadotropic hypogonadism
Idiopathic hypogonadotropic hypogonadism refers to hypogonadotropic hypogonadism with no apparent causes resulting gonadotropin-releasing hormone (GnRH) insufficiency or deficiency causing delayed or absent puberty and infertility 37, 38, 39, 33. Traditionally, idiopathic hypogonadotropic hypogonadism (IHH) is divided into two major categories: Kallmann syndrome (KS) and normosmic idiopathic hypogonadotropic hypogonadism (nIHH).
- Kallmann’s syndrome is a type of congenital hypogonadotropic hypogonadism with loss of smell (anosmia) or decreased sense of smell (hyposmia). This feature distinguishes Kallmann syndrome from most other forms of hypogonadotropic hypogonadism, which do not affect the sense of smell. Kallmann syndrome is often due to the embryonic maldevelopment and/or interrupted migration of GnRH specific neurons. Since the embryonic migration of GnRH neurons from the nasal placode towards their final destination in the hypothalamus occurs in association with olfactory receptor neurons, the resulting phenotype includes anosmia in addition to hypogonadotropic hypogonadism. Kallmann syndrome cases often have additional congenital anomalies such as cleft palate, unilateral renal agenesis, split hands and feet, short metacarpals, deafness, and mirror movements (synkinesia).
- Normosmic idiopathic hypogonadotropic hypogonadism (nIHH) refers to those idiopathic hypogonadotropic hypogonadism cases not associated with loss of smell (anosmia) 40. Normosmic idiopathic hypogonadotropic hypogonadism (nIHH) results from the dysfunction of the normally sited GnRH neurons in the hypothalamus. These cases typically do not have any accompanying congenital lesions.
Idiopathic hypogonadotropic hypogonadism can be sporadic or familial in occurrence. The great majority of hereditary causes of idiopathic hypogonadotropic hypogonadism are congenital (present at birth). To date, mutations in more than 50 genes have been reported to cause idiopathic hypogonadotropic hypogonadism 37. These genetic mutations are estimated to account for up to 50% of all apparently hereditary idiopathic hypogonadotropic hypogonadism cases 41, 42, 43.
Pubertal delay is the most typical presentation of idiopathic hypogonadotropic hypogonadism. Pubertal delay is defined as absence of breast development (Tanner breast stage 1) in a girl at age 13 or failure to achieve a testicular volume of 4 mL in a boy by age 14 44.
Typically, in girls there is no signs or symptoms of idiopathic hypogonadotropic hypogonadism before the early teen years. In boys, since the HPG axis is very active roughly between the 16th and 22nd week of gestation and androgenic end products of this period are required for normal virilization of the 46,XY fetus, male infants with idiopathic hypogonadotropic hypogonadism may have unusually small penis (micropenis) and/or undescended testicle (cryptorchidism) at birth. A slight and temporary reactivation of the HPG axis in early infancy (around four to sixteen weeks) is called “minipuberty” and provides a unique opportunity to diagnose both male and female infants with congenital idiopathic hypogonadotropic hypogonadism 45.
With appropriate HRT, patients with CHH can develop secondary sexual characteristics, maintain normal sex hormone levels and a healthy sexual life, and achieve fertility. Several regimens of treatment with different administrative routes exist. The choice of treatment depends on the therapeutic goal, the timing of treatment, and the personal preference of each patient. It is important to know that randomized controlled trials on hormonal treatment in CHH are scarce, and data on clinical observational studies are also limited. There is no uniform treatment regimen used internationally.
The majority of idiopathic hypogonadotropic hypogonadism men respond well to induction of spermatogenesis with gonadotropins or pulsatile gonadotropin releasing hormone (GnRH).
Functional hypogonadotropic hypogonadism
Functional hypothalamic hypogonadism is a reversible form of gonadotropin releasing hormone (GnRH) deficiency commonly triggered by stressors such as excessive exercise, nutritional deficits, or psychological distress. Stressors such as weight loss, excessive exercise, eating disorders, and psychological distress suppress the hypothalamic–pituitary–gonadal axis by inhibiting hypothalamic pulsatile secretion of GnRH. Functional hypothalamic hypogonadism is associated with a spectrum of abnormal GnRH-secretion patterns, and administration of exogenous pulsatile GnRH can restore functionality of the hypothalamic–pituitary–gonadal axis (HPG axis). It was observed that in a male patient with functional hypogonadotropic hypogonadism, levels of both testosterone (T) and serum estradiol (E2) increased dramatically after prolonged clomiphene medication and in response to human chorionic gonadotropin.
Hypogonadism risk factors
Risk factors for hypogonadism include:
- HIV/AIDS
- Damage to the pituitary gland or hypothalamus from surgery, injury, tumor, infection, or radiation
- Previous chemotherapy or radiation therapy
- Aging
- Obesity
- Malnutrition
Hypogonadism can be inherited. If any of these risk factors are in your family health history, tell your doctor.
Hypogonadotropic hypogonadism symptoms
Hypogonadism can begin during fetal development, before puberty or during adulthood. Signs and symptoms depend on when the condition develops.
The clinical hallmark of hypogonadotropic hypogonadism is the failure of onset of puberty. This lack of pubertal maturation occurs in both sexes and is characterized by reduced blood levels of the sex hormone levels (testosterone and estrogen) as well as gonadotropins (LH and FSH) and infertility. In boys, the onset of normal pubertal development is signaled by testicular enlargement that is then followed by penile growth and the appearance of pubic hair. Affected men complain of absence of secondary sexual characteristics (facial hair growth, body hair growth, decreased pubic hair growth and genital enlargement) and a delayed growth spurt in comparison to their peers. In addition, an absence of sexual interest (libido) and poor sexual function (inability to attain or sustain an erection) may also be present. Unusual growth of breasts may also be rarely seen in these subjects although this more typically occurs during treatment of this condition and is often transient.
Clinical examinations in these subjects usually confirms the incomplete sexual maturation (e.g. prepubertal testicular volume [< 4ml]), a eunuchoid body habitus (disproportionally long arms when compared to height) and decreased muscle mass. The degree of pubertal maturation can vary considerably with some individuals lacking any sign of puberty whereas others may have partial pubertal features that do not progress normally. Although hypogonadotropic hypogonadism in males is typically diagnosed at puberty, this diagnosis can be made in infancy due to a small genital size (micropenis/microphallus) and/or lack of descent of testes (undescended testes or referred to as cryptorchidism). As mentioned earlier, pulsatile GnRH secretion and evidence of a normal reproductive axis occurs during the neonatal period. Therefore, timely biochemical testing during the first 6 months or so of life may also confirm the presence of hypogonadism with low gonadotropin levels, i.e. the biochemical hallmarks of this condition during this critical window of normal development. However, if this brief developmental window of diagnostic testing is missed, a definite diagnostic confirmation may have to wait until the expected time of puberty.
Most often, the diagnosis of hypogonadotropic hypogonadism is delayed until adolescence, when there is a failure to go through puberty 2. Hypogonadotropic hypogonadism signs and symptoms include lack of development of secondary sex characteristics, eunuchoidal body proportions (upper/lower body ratio <1 with an arm span 6 cm > standing height), a high-pitched voice, mild anemia, delayed bone age, and pre-pubertal testes 46. However, hypogonadotropic hypogonadism is clinically heterogeneous in that some patients have evidence of partial spontaneous pubertal development reflected by larger gonadal size despite hypogonadal testosterone levels and inappropriately low gonadotropin levels. Gynecomastia (enlarged breasts in males) is not a typical feature of GnRH deficiency given the hypogonadotropic state and is most commonly seen in patients treated with gonadotropins 47, 48. Hypogonadotropic hypogonadism may also present after completion of puberty resulting in a disruption in reproductive function in adulthood characterized by decreased libido, erectile dysfunction (not being able to obtain or keep an erection that is sufficient for sexual intercourse) and low sperm count (oligospermia) or complete absence of sperm (azoospermia) 49.
Fetal development
If the body doesn’t produce enough testosterone during fetal development, the result may be impaired growth of the external sex organs. Depending on when hypogonadism develops and how much testosterone is present, a child who is genetically male may be born with:
- Female genitals
- Genitals that are neither clearly male nor clearly female (ambiguous genitals)
- Underdeveloped male genitals
Children
- Lack of growth and sexual development at the standard age for puberty (development may be very late or incomplete)
- In girls, a lack of breast development and menstrual periods
- In boys, no development of sex characteristics, such as enlargement of the testes and penis, deepening of the voice, and facial hair
- Inability to smell (in some cases)
- Short stature (in some cases)
Adults
- Loss of interest in sex (libido) in men
- Loss of menstrual periods (amenorrhea) in women
- Decreased energy and interest in activities
- Loss of muscle mass in men
- Weight gain
- Mood changes
- Infertility
Hypogonadotropic hypogonadism symptoms female
Hypogonadism in females can affect the ovaries and their secretion of hormones. Hypogonadism in females can cause:
- Delayed puberty
- Lack of menstruation (amenorrhea)
- Absence of breast development
- Absence of pubic hair
- Vaginal agenesis
- Hot flashes
- Breast discharge
- Short stature
- Infertility
In girls, the first sign of normal puberty is the onset of breast budding (thelarche), followed by a growth spurt, the appearance of pubic hair growth, and then only later, the onset of menstrual flow, i.e. menarche (first menstrual period). Hypogonadotropic hypogonadism females typically report absence of breast development, reduced growth spurt, decreased pubic hair growth, and lack of initiation of menstrual period (primary amenorrhea). However, some females may exhibit some evidence of a partial puberty with onset of breast budding (thelarche) that fails to progress. Very occasionally, some hypogonadotropic hypogonadism females may report onset of menstrual period at the appropriate time period in adolescence that ceases after a few cycles. Clinical exam in hypogonadotropic hypogonadism females usually confirms their immature sexual characteristics and enuchoid habitus.
Hypogonadotropic hypogonadism symptoms male
Male hypogonadism can delay puberty or cause incomplete or lack of normal development. It can hamper:
- Development of muscle mass
- Voice deepening
- Growth of body and facial hair
- Growth of the penis and testicles
And it can cause:
- Excessive growth of the arms and legs in relation to the trunk of the body
- Development of breast tissue (gynecomastia)
In adult males, hypogonadism can alter certain masculine physical characteristics and impair normal reproductive function. Early signs and symptoms might include:
- Decreased sex drive (low libido)
- Decreased energy
- Depression
Over time, men with hypogonadism can develop:
- Erectile dysfunction (not being able to obtain or keep an erection that is sufficient for sexual intercourse)
- Infertility
- Decrease in hair growth on the face and body
- Decrease in muscle mass
- Development of breast tissue (gynecomastia)
- Loss of bone mass (osteoporosis)
Severe hypogonadism can also cause mental and emotional changes. As testosterone decreases, some men have symptoms similar to those of menopause in women. These can include:
- Difficulty concentrating
- Hot flashes.
Hypogonadotropic hypogonadism complications
The complications of untreated hypogonadism differ depending on when it develops — during fetal development, puberty or adulthood.
Hypogonadotropic hypogonadism complications may include:
- Abnormal genitalia
- Delayed puberty
- Enlarged male breasts (gynecomastia)
- Infertility
- Erectile dysfunction (not being able to obtain or keep an erection that is sufficient for sexual intercourse)
- Sexual problems, such as low libido
- Osteoporosis or low bone density and fractures later in life
- Low self-esteem due to late start of puberty (emotional support may be helpful)
- Early menopause (in females)
Early hypogonadism detection in boys can help prevent problems from delayed puberty. Early diagnosis and treatment in men offer better protection against osteoporosis and other related conditions.
Hypogonadotropic hypogonadism diagnosis
Your doctor will conduct a physical exam and note whether your sexual development, such as your pubic hair, muscle mass and size of your testes, is consistent with your age.
Your doctor will test your blood level of testosterone if you have signs or symptoms of hypogonadism. Because testosterone levels vary and are generally highest in the morning, blood testing is usually done early in the day, before 10 a.m., possibly on more than one day.
If tests confirm that you have low testosterone, further testing can determine if a testicular disorder or a pituitary abnormality is the cause. These studies might include:
- Hormone testing
- Semen analysis
- Pituitary imaging
- Genetic studies
- Testicular biopsy
If hypogonadism is suspected, measurement of serum LH and FSH concentrations can be used to distinguish between hypogonadotropic hypogonadism (problem in the hypothalamus or the pituitary gland) and hypergonadotropic hypogonadism (primary gonadal failure) and guide further evaluation and management. Serum LH and FSH levels are elevated in cases of hypergonadotropic hypogonadism (primary gonadal failure).
Further hormonal testing (ie, measurement of serum prolactin or thyroid function tests) can be ordered based on presentation and history. Obtaining a karyotype can be considered in certain cases.
In the absence of specific symptoms or signs to direct the workup, evaluation for chronic illness includes a complete white blood count, a sedimentation rate, a comprehensive metabolic panel, a celiac screening, thyroid function tests, and a urinalysis.
Males physical exam
Examination of the testes is the most important feature of the physical examination. Determine whether both testes are palpable, their position in the scrotum, and their consistency. Testes size can be quantitated by comparison with testicular models (orchidometer), or their length and width may be measured. Before puberty, testes usually are 1-3 cm³ in volume (approximately 2 cm in length). During puberty, testes grow up to 25 cm³ in size.
Examining the genitalia for hypospadias (a condition in which the opening of the penis is on the underside rather than the tip) is the next important step. Check the scrotum to see if it is completely fused. Hypospadias is usually not related to an endocrine abnormality, but it may be seen in disorders associated with a testosterone biosynthesis defect, partial androgen insensitivity syndrome, or a defect in testicular determination. Finally, evaluate the extent of the biological development of adult male characteristics in young males (virilization).
The presence of micropenis (an atypically small penis that’s diagnosed at birth) suggests Kallmann syndrome or panhypopituitarism.
Puberty should be staged using the Tanner criteria for genitalia, pubic hair, and axillary hair.
Look for signs of Klinefelter syndrome, such as tall stature (especially if the legs are disproportionately long), gynecomastia, small or soft testes, and a eunuchoid body habitus.
Males Lab Tests
For males after puberty, the guidelines of the Endocrine Society require that the diagnosis of hypogonadism be based on symptoms and signs of hypogonadism plus the presence of a low testosterone level measured on at least two occasions 50. Because of the circadian variation in testosterone levels, a morning sample is recommended.
For postpubertal males with total testosterone concentrations near the lower limit of the normal range, measurement of free or bioavailable testosterone using a reliable assay is suggested if there is suspicion of sex hormone binding globulin (SHBG) changes 50.
Obesity decreases serum sex hormone binding globulin (SHBG) concentrations to a degree that is proportionate to the severity of the obesity, thereby decreasing total testosterone values without usually affecting free testosterone. However, morbid obesity (BMI >40 kg/m²) may be associated with a low free testosterone concentration because of hypothalamic hypogonadism. Decreased sex hormone binding globulin (SHBG) concentrations are also seen in insulin resistance, type 2 diabetes, hypothyroidism, glucocorticoid use, and nephrotic syndrome. Increased sex hormone binding globulin (SHBG) concentrations can occur with aging, hyperthyroidism, liver disease, and anticonvulsant use.
Examination of seminal fluid analysis, karyotyping (a type of genetic testing that looks at the size, shape, and number of chromosomes in a sample of cells from your body), and testicular biopsy may also be helpful.
Females physical exam
Examination of the genitalia is important. Determine the extent of the development of secondary male sexual characteristics, which may be adrenal or ovarian in origin and is demonstrated in pubic and armpit hair.
Determine the extent of estrogenization, as evidenced by breast development and maturation of the vaginal mucosa.
Look for signs of Turner syndrome, such as short stature, webbing of the neck (eg, pterygium colli), a highly arched palate, short fourth metacarpals, widely spaced nipples, or multiple pigmented nevi.
Females Lab Tests
Karyotyping (a type of genetic testing that looks at the size, shape, and number of chromosomes in a sample of cells from your body) may be helpful. In females with normal karyotype and elevated gonadotropin levels, measure antiovarian antibody levels to rule out autoimmune disease.
In postpubertal females with signs of acne, hirsutism, and/or amenorrhea or irregular menses, measurement of total and free testosterone and 17-hydroxyprogesterone concentrations is indicated.
Imaging Studies
Magnetic resonance imaging (MRI) of the brain should be considered in cases of loss of smell (anosmia) and suspected hypogonadotropic hypogonadism (problem in the hypothalamus or the pituitary gland). Absence of the olfactory bulbs is associated with Kallmann syndrome.
Furthermore, brain MRI should be ordered in cases of hypogonadotropic hypogonadism that are either isolated or occurring in combination with pituitary defects.
MRI of the pelvis is usually performed in disorders of sex development (DSD) cases, such as androgen insensitivity or ovotesticular DSD, to help delineate the anatomy of internal genitalia. Uterine aplasia in a pubertal girl with normal gonadal function and amenorrhea can be seen in Mayer-Rokitansky-Küster-Hauser syndrome.
Pelvic ultrasonography may be helpful in females.
Bone age may be helpful in the evaluation of adolescents with delayed puberty and provides insight into their growth potential.
Other Tests
Adrenocorticotropic hormone (ACTH) stimulation testing: In patients in whom a form of congenital adrenal hyperplasia is suspected, adrenal steroid synthesis is best evaluated by performing a cosyntropin (ACTH 1-24) stimulation test. Baseline serum adrenocortical hormone levels are measured, then 0.25 mg of cosyntropin is intravenously injected, and serum hormone levels are remeasured after 60 minutes. Precursor product ratios are compared with those in age-matched control subjects to determine whether a steroidogenic defect is involved in sex hormone synthesis.
Luteinizing-hormone releasing hormone (LHRH) stimulation testing: To distinguish between true hypogonadotropic hypogonadism and constitutional delay in growth and maturation, performing a stimulation test with LHRH may be helpful.
- LHRH is intravenously injected, and LH and FSH levels are determined at 15-minute intervals following LHRH administration.
- A shortened version of the study has been used, in which LHRH is subcutaneously injected, and the specimen for LH and FSH levels is taken at 30-40 minutes.
- Obtaining LHRH for testing over the past several years has been difficult. Some centers have substituted testing LH response to aqueous leuprolide.
Testicular tissue testing: If testes are not palpable and whether any testicular tissue is present is unclear, administering human chorionic gonadotropin (hCG) and measuring testosterone response may be helpful.
Procedures
In prepubertal males with delayed puberty, priming with testosterone (usually testosterone enanthate 50 mg IM monthly for a total of 3 months) may lead to puberty initiation and help in the differential diagnosis of hypogonadotropic hypogonadism.
In postpubertal females with amenorrhea, withdrawal bleeding after a 5-10 day course of progestin (such as medroxyprogesterone 10mg hs) indicates adequate estrogen secretion and implies intact gonadal function.
Occasionally, testicular biopsy findings are helpful, particularly if azoospermia or oligospermia is present.
Hypogonadotropic hypogonadism treatment
Hypogonadotropic hypogonadism treatment depends on the source of the problem, but may involve:
- Injections of testosterone (in males)
- Slow-release testosterone skin patch (in males)
- Testosterone gels (in males)
- Estrogen and progesterone pills or skin patches (in females)
- Gonadotropin-releasing hormone (GnRH) injections
- Human chorionic gonadotropin (hCG) injections
Patients with hypogonadism are typically treated with sex steroid replacement. The goals of treatment are:
- To promote the development of and maintain secondary sexual characteristics and normal sexual function
- To build and sustain normal bone and muscle mass
- To assist in the proper psychosocial adjustment of adolescents with hypogonadism
Consultation with a reproductive endocrinologist or urologist is required for patients with hypogonadotropic hypogonadism who would like to become fertile. Pulsatile luteinizing-hormone releasing hormone (LHRH) or gonadotropin therapy can induce fertility in individuals with hypogonadotropic hypogonadism. Administration of pulsatile LHRH or gonadotropins in females results in ovulation in 95% of the cases. In males, pulsatile LHRH therapy or hCG alone or in combination with gonadotropins can induce spermatogenesis (the production of sperm) and results in normal adult male testosterone levels.
In Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism, for treatment, initially, hormone replacement therapy (testosterone in males; estrogen and progesterone in females) is used to induce secondary sexual characteristics 51. Once pubertal maturation is achieved, if Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism individuals wish to be fertile, either injections of pituitary hormones (the gonadotropins, LH and FSH) or in some instances, therapy with the synthetic peptide, GnRH, whose deficiency causes these syndromes, are required to induce the sex organs (testes or ovaries) to make sperm (males) or eggs (females). While both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism are usually life-long in their nature, about 10-15% of patients may experience a recovery of their hormonal system, the reasons for which currently remain unclear 51.
Treatment of hypogonadotropic hypogonadism in males
Male with hypogonadotropic hypogonadism usually is treated with testosterone replacement to return testosterone levels to normal. Testosterone can help counter the signs and symptoms of male hypogonadism, such as decreased sexual desire, libido, sexual function, decreased energy, decreased facial and body hair, and loss of muscle mass and bone density and general well-being. While you’re taking testosterone, the Endocrine Society recommends that your doctor monitor you for treatment effectiveness and side effects several times during your first year of treatment and yearly after that.
Testosterone can be given as an injectable formulation (aromatizable androgen such as enanthate, cypionate, or undecanoate) or transdermal application 52, 53, 54. The maintenance dose of testosterone is usually 250 mg of testosterone enanthate IM every 2 to 4 weeks, 1 g of testosterone undecanoate IM every 3 to 4 months, or 50 to 80 mg of testosterone gel daily (Table 1). The surveillance of trough serum testosterone levels is important, as there exists considerable variation regarding the metabolism of exogenous testosterone products among patients with hypogonadotropic hypogonadism 55. For testosterone injections, the frequency of injections should be assessed according to the trough serum testosterone measurement, targeting the lower end of the normal range. IM testosterone injections may cause substantial differences between the peak and trough testosterone levels. Pilot studies have shown that a weekly subcutaneuous injection of low doses of testosterone cypionate or testosterone enanthate can induce a more steady profile of plasma testosterone 56, 57. For patients treated with testosterone gel, the target for random serum testosterone level is the middle of the normal range. The advantage of testosterone gel is its pharmacokinetics with a more stable testosterone concentration within the normal adult range, and the lack of minimally invasive injections. However, patients on testosterone gel should avoid skin contact with others (partners or children), as there are known risks for hyperandrogenism in women 58 or for precocious puberty in children 59. Among the reported disadvantages of transdermal testosterone are the high cost and the lack of reimbursement in some countries. Whichever treatment is used, men with hypogonadotropic hypogonadism are challenged to adhere to long-term treatment, and poor adherence may contribute to adverse effects on bone, sexual, and psychological health 60.
Types of testosterone replacement therapy
Several testosterone formulations are available. The different regimens of testosterone replacement therapy are summarized in Table 1. Direct comparisons among different testosterone products are still lacking. Patients who are considering testosterone therapy should be adequately informed about the possible risks and benefits of all available testosterone preparations. The final choice should be based on the clinical situation, testosterone formulation availability, and patient needs and expectations 61, 62.
One Food and Drug Administration-approved oral testosterone replacement preparation, testosterone undecanoate (Jatenzo), is absorbed by the lymph system. It might avoid the liver problems seen with other oral forms of testosterone.
Other preparations you might choose, depending on convenience, cost and your insurance coverage, include:
- Testosterone Gel. There are several gels and solutions available, with different ways of applying them. Depending on the brand, you rub the testosterone into your skin on your upper arm or shoulder (AndroGel, Testim, Vogelxo) or apply it to the front and inner thigh (Fortesta). Your body absorbs testosterone through your skin. Don’t shower or bathe for several hours after a gel application, to be sure it gets absorbed. Side effects include skin irritation and the possibility of transferring the medication to another person. Avoid skin-to-skin contact until the gel is completely dry, or cover the area after an application.
- Testosterone Injection. Testosterone cypionate (Depo-Testosterone) and testosterone enanthate are given in a muscle or under the skin. Your symptoms might waver between doses depending on the frequency of injections. You or a family member can learn to give testosterone injections at home. If you’re uncomfortable giving yourself injections, member of your care team can give the injections.
- Testosterone undecanoate (Aveed) is given by deep intramuscular injection, typically every 10 weeks. It must be given at your provider’s office and can have serious side effects.
- Testosterone Patch. A patch containing testosterone (Androderm) is applied each night to your thighs or torso. A possible side effect is severe skin reaction.
- Testosterone Pellets (Testopel): Testosterone-containing pellets are surgically implanted under the skin every three to six months for consistent and long-term dosages. Side effects: pellet coming out through skin, site infection/ bleeding (rare), dose decreasing over time and hypogonadism symptoms possibly returning towards the end of dose period.
- Testosterone Gum and Cheek (buccal cavity). A small putty-like substance, gum-and-cheek testosterone replacement delivers testosterone through the natural depression above your top teeth where your gum meets your upper lip (buccal cavity). This product, taken three times a day, sticks to your gumline and allows testosterone to be absorbed into your bloodstream. It can cause gum irritation.
- Testosterone Nasal. This testosterone gel (Natesto) can be pumped into the nostrils. This option reduces the risk that medication will be transferred to another person through skin contact. Nasal-delivered testosterone must be applied twice in each nostril, three times daily, which might be more inconvenient than other delivery methods.
Testosterone therapy carries various risks, including:
- Increased production of red blood cells (polycythemia or erythrocytosis)
- Acne
- Enlarged breasts
- Sleep disturbances
- Prostate enlargement
- Limited sperm production
Table 1. Males with hypogonadotropic hypogonadism treatment
| Treatment | Dosing and Administration | Advantages | Disadvantages |
|---|---|---|---|
| Induction of puberty in boys | |||
| Testosterone enanthate | Initial dose: 50 mg IM monthly | Standard care with long clinical experience | Premature epiphyseal closure (high dose) |
| ↑ 50 mg increments every 6–12 mo | Aromatizable to estradiol (E2): promote bone maturation | Could inhibit testicular volume and spermatogenesis | |
| Up to 250 mg/mo | Impact on future fertility unknown | ||
| Gonadotropin | hCG: initial dose 250 IU subcutaneously twice weekly, | Stimulate testicular volume growth and spermatogenesis | Not standard treatment |
| ↑ 250–500 IU increments every 6 mo | Pre-FSH treatment can be beneficial in patients with testicular volume <4 mL or history of cryptorchidism | Need good compliance in adolescent patients | |
| Up to 1500 IU three times weekly | Need studies in larger cohorts | ||
| rFSH: dose 75–150 IU subcutaneously three times weekly, | |||
| Hypogonadism treatment in adult males | |||
| Testosterone enanthate | 250 mg IM every 2 to 4 wk | Cost-effective | Relatively frequent IM injection |
| Interval adjusted based on trough testosterone | Available around the world | Subcutaneous route under investigation (302) | |
| Self-injection | |||
| Testosterone undecanoate | 1000 mg IM every 10 to 14 wk | Cost-effective | Interval of treatment highly variable; follow-up of trough testosterone is important |
| Interval adjusted based on trough testosterone | Infrequent injection | Injections by nurses | |
| Testosterone gel | 50–80 mg/d transdermally | Noninvasive | Risk of transmission by skin contact |
| Self-administered | |||
| Treatment of infertility in adult males | |||
| Pulsatile GnRH | SC pump: 25 ng/kg per pulse every 120 min | Most physiological treatment | Not available in many countries |
| Dose adapted based on serum testosterone | Require centers with expertise | ||
| Up to 600 ng/kg per pulse | Pituitary resistance (rare) | ||
| Gonadotropin | hCG: dose 500–1500 IU subcutaneously three times weekly, | Available around the world | Relatively expensive for rFSH |
| Dose adjusted based on trough testosterone | For patients with absent puberty (testicular volume <4 mL): | Frequent injections | |
| rFSH: dose 75–150 IU subcutaneously three times weekly, | Pre-rFSH treatment increases fertility prognosis | ||
| Dose adjusted based on serum FSH, sperm count | |||
Treatment of male infertility
Hypogonadotropic hypogonadism is one of the few medically treatable causes of male infertility, and fertility treatments have very good outcomes. Fertility induction can be accomplished either by long-term pulsatile GnRH therapy or with combined gonadotropin therapy. A pituitary tumor may require surgical removal, medication, radiation or the replacement of other hormones.
Both pulsatile GnRH and gonadotropin therapy are effective to induce spermatogenesis and fertility in men with hypogonadotropic hypogonadism 64, 65, 66. However, no clear superiority of GnRH vs gonadotropins was observed. Similarly, none of the available FSH preparations appears to differ in terms of sperm output.
The overall success rate in terms of sperm output is variable across studies (64% to 95% success), with sperm counts ranging from zero to several hundred million per milliliter. The weighted average median time to achieve sperm production was slightly more than a year 63. It is well established that even low sperm concentrations in men with hypogonadotropic hypogonadism are sufficient to impregnate partners 67. Pregnancy was successfully achieved in 175 partners of patients with hypogonadotropic hypogonadism and successful pregnancies were reported in 16% to 57% of patients with hypogonadotropic hypogonadism desiring fertility 63. As reported 63, most pregnancies obtained were by natural conception. In a minority, in vitro fertilization (IVF) was necessary because of the existence of concomitant ovarian or uterotubal abnormalities in the partner 63. Conversely, 192 patients were not able to produce sperm despite long-term gonadotropin treatment (median, 24 months), corresponding to 12% to 40% depending on the study. In patients with azoospermia after treatment or poor sperm quality, more invasive treatments such as testicular sperm extraction were proposed followed by intracytoplasmic spermatozoid injection 68.
Pulsatile GnRH treatment
Pulsatile GnRH treatment is a logical approach in patients with hypogonadotropic hypogonadism seeking fertility. Physiological GnRH secretion is episodic, and therefore GnRH treatment requires intravenous (IV) or subcutaneous (SC) GnRH administration in a pulsatile manner via a mini-infusion pump 69. This therapy will stimulate pituitary gonadotropin secretion and in turn intragonadal testosterone production, resulting in the initiation and maintenance of spermatogenesis as evidenced by increased testicular volume and sperm output by 12 months of treatment on average. The common initial dose is 25 ng/kg per pulse every 2 hours, with a subsequent titration to normalize serum testosterone to the adult normal range 70, 71, 72, 73. Response to treatment varies according to the degree of GnRH deficiency, with normalization of testicular volume and successful induction of spermatogenesis for all patients with partial puberty. On the contrary, testicular volume and sperm counts are lower in patients with absent puberty, and 20% of these patients remained azoospermic despite 12 to 24 months of pulsatile GnRH treatment 70.
Gonadotropin treatment
Gonadotropin treatment (hCG alone or combined with rFSH) is another treatment option for fertility induction in male patients with hypogonadotropic hypogonadism. Whereas intramuscular (IM) injections were prescribed in the past, subcutaneous (SC) gonadotropin injections are currently preferred, and various formulations are used. Typical doses vary from 500 to 2500 IU two to three times a week for hCG, and from 75 to 225 IU two to three times a week for FSH preparations, namely hMG, highly purified urinary FSH, or rFSH. The dosage of hCG is adjusted based on trough serum T, and rFSH dosage is titrated based on serum FSH levels and sperm counts.
Neonatal treatment
To date, hormonal therapy during the neonatal period is only applied in male patients exhibiting micropenis/cryptorchidism and hypogonadotropic hypogonadism 74, 75, 76, 77, 78, 79. An equivalent therapy is not proposed in female patients, as the consequences of severe GnRH deficiency during the late fetal period and minipuberty in females are unclear 63.
In male infants with severe GnRH deficiency, the main goals of hormonal treatment are to increase the penile size and to stimulate testicular growth. Early reports in 1999 and in 2000 described the benefit of early androgen therapy in boys with either hypogonadotropic hypogonadism or combined pituitary hormone deficiency (CPHD) 80, 76. Testosterone treatment can increase penile size and stimulate scrotal development.
Human chorionic gonadotropin (hCG) therapy with or without a combination of nasal spray of GnRH has been shown to be effective to treat cryptorchidism in neonates and prepubertal boys 81, 82. This finding could represent a further benefit of neonatal treatment of children with hypogonadotropic hypogonadism, as cryptorchidism is a factor of poor prognosis for adult fertility and is also a risk factor for testicular cancer. Alternatively, orchidopexy—surgery to move an undescended testicle into the scrotum—is the current treatment of choice of cryptorchidism. Some publications point to a deleterious effect of isolated hCG therapy in boys with cryptorchidism 83. A concern for high-dose hCG treatment is its potentially deleterious effect on germ cells with increased apoptosis, and thus negative consequences for future fertility 83. However, the deleterious effect of hCG has not been demonstrated in males with hypogonadotropic hypogonadism with cryptorchidism.
In 2002, Main et al. 79 reported the effects of subcutaneous (SC) injections of recombinant human LH (rLH) and recombinant human FSH (rFSH) during the first year of life in an infant with hypogonadotropic hypogonadism born with micropenis. This treatment led to an increase in penile length (1.6 to 2.4 cm), as well as a 170% increase in testicular volume accompanied by an increase in inhibin B levels. Similarly, Bougnères et al. 78 reported the use of gonadotropin infusion in two neonates, one diagnosed with hypogonadotropic hypogonadism and the other with combined pituitary hormone deficiency (CPHD). In this study, recombinant human LH (rLH) and recombinant human FSH (rFSH) were administered subcutaneously via a pump for 6 months. This treatment not only corrected the micropenis in both patients (8 to 30 mm and 12 to 48 mm, respectively), but also induced testicular growth (0.57 to 2.1 mL and 0.45 to 2.1 mL, respectively). Serum LH and FSH levels increased to normal or supranormal levels, leading to an endogenous secretion of testosterone, inhibin B, and anti-mullerian hormone (AMH). Similarly, Sarfati et al. 75 reported another case with a perinatal diagnosis of Kallmann syndrome based on the presence of an ANOS1 (KAL1) mutation, the detection of kidney agenesis during fetal life, and the presence of micropenis at birth. The combined gonadotropins infusion from 1 to 7 months of age induced the normalization of testicular size (0.33 to 2.3 mL) and penis length (15 to 38 mm). Recently, Lambert and Bougnères 77 reported the effect of combined recombinant human LH (rLH) and recombinant human FSH (rFSH) injections in a series of eight male infants with either hypogonadotropic hypogonadism or combined pituitary hormone deficiency (CPHD). All patients presented with either cryptorchidism or high scrotal testis at diagnosis and were treated with gonadotropin infusion. Apart from the increase in both penile length and testicular size, the authors observed complete testicular descent in six out of eight cases. However, the effect of combined gonadotropin treatment on cryptorchidism in hypogonadotropic hypogonadism infants will need to be formally assessed by randomized controlled trials. Furthermore, the effect of such treatment on males with cryptorchidism without hypogonadism remains unknown.
Collectively, these studies suggest that combined gonadotropin therapy in male patients with hypogonadotropic hypogonadism during the neonatal period can have a beneficial effect on both testicular endocrine function and genital development. This treatment may be superior to androgen therapy, as it stimulates Sertoli cell proliferation and the growth of seminiferous tubules, as evidenced by the marked increase in testicular volume and in serum inhibin B concentrations 74.
It is possible that the normalization of penis size in the neonate will lead to a normal adult penis size during subsequent pubertal virilization with exogenous testosterone or hCG, thus preventing the feeling of inadequacy often reported by males with hypogonadotropic hypogonadism with micropenis 84. In parallel, the increase in testicular size, which correlates with the increase in Sertoli cell mass, could lead to better outcomes in terms of sperm output during fertility induction in adolescence or adulthood 74. Taken together, these data imply that combined gonadotropin therapy in males during the neonate period may reduce the psychological effects of micropenis later in adolescence, and potentially improve fertility in adulthood.
Randomized controlled trials with a larger number of patients are needed to rigorously assess the effect of gonadotropins on cryptorchidism in male neonates. Furthermore, longitudinal studies are warranted to determine the long-term benefits on reproductive function of hormonal intervention during infancy. However, there are no data to support such a treatment in female patients with hypogonadotropic hypogonadism 63.
Treatment for boys
Therapeutic goals in the adolescent male with hypogonadotropic hypogonadism are: to induce virilization, to reach optimal adult height, to acquire normal bone mass and body composition, to achieve normal psychosocial development, and to gain fertility. However, available treatment regimens may not always cover all of these aspects. The hormonal treatment options for the induction of puberty in males with hypogonadotropic hypogonadism are presented in Table 1.
Treatment of delayed puberty in boys depends on the underlying cause. Three to six months of testosterone ester such as testosterone enanthate given as an injection can stimulate puberty and the development of secondary sex characteristics, such as increased muscle mass, beard and pubic hair growth, and growth of the penis 52, 85, 53.
Pediatric endocrinologists treating younger patients from 12 years of age typically begin treatment with low-dose testosterone (e.g., 50 mg of testosterone enanthate monthly) and gradually increase to full adult dose (250 mg every 2 to 4 weeks) during the course of ∼24 months 63. For patients with hypogonadotropic hypogonadism seeking treatment in later adolescence or early adulthood, a higher dose of testosterone can be used to induce rapid virilization. Initial testosterone doses such as 100 mg of testosterone enanthate monthly can be quickly increased to 250 mg intramuscularly (IM) monthly 63. Such regimens induce secondary sexual characteristics and maximize final height 85, 86. Side effects for testosterone treatment include erythrocytosis (too much red blood cells), premature closure of the epiphysis (when doses are too high during the first year of treatment), and occasional pain and redness at the injection site. Testosterone treatment does not stimulate testicular growth or spermatogenesis 52, 53, because intragonadal testosterone production is needed to stimulate spermatogenesis. In contrast, increased testicular growth during testosterone treatment indicates hypogonadotropic hypogonadism reversal and requires treatment withdrawal followed by hormone profiling 87.
Induction of testicular maturation
Gonadotropins are used for fertility treatments in adult patients with hypogonadotropic hypogonadism, but can also be used to induce pubertal maturation in adolescent males with hypogonadotropic hypogonadism. An additional advantage of gonadotropin treatment compared with testosterone treatment is the stimulation of testicular growth and spermatogenesis. Therefore, gonadotropin treatment may offer important psychological reassurance in adolescents and enhance self-confidence. Varying treatment protocols including human chorionic gonadotropin (hCG) alone or in combination with FSH have been used to induce puberty in boys 88, 89, 90, 91, 92. In a retrospective analysis of boys with hypogonadotropic hypogonadism, Bistritzer et al. 88 showed a comparable virilizing effect of monthly testosterone injections and weekly human chorionic gonadotropin (hCG) injections (5000 IU/wk), but testicular growth was significantly larger in boys treated with hCG.
Rohayem et al. 92 studied a relatively large group of adolescents with delayed puberty, most of them with absent puberty (n = 34). The adolescents received low-dose hCG (250 to 500 IU twice weekly) with increasing increments of 250 to 500 IU every 6 months, and recombinant human FSH (rFSH) was added once serum testosterone achieved targeted pubertal level (5.2 nmol/L). This treatment led to a substantial increase in testicular volume (bitesticular volumes, 5 ± 5 to 34 ± 3 mL) and induction of spermatogenesis in 91% of patients 92.
Pretreatment with FSH in adolescents with severe GnRH deficiency
The idea behind priming with FSH alone in patients with severe GnRH deficiency is that the mass of Sertoli cells is a predictor of future sperm output. FSH induces proliferation of immature Sertoli cells prior to seminiferous tubule maturation in rats 93, juvenile non-human primate (Macaca mulatta) 94 and probably also in humans 95.
In contrast, adult men with mutation of the follicle-stimulating hormone (FSH) receptor gene exhibit small testicular size and variable degrees of spermatogenesis failure 96. Additionally, it has been suggested that patients with hypogonadotropic hypogonadism with absent puberty with/without micropenis and cryptorchidism likely have a suboptimal Sertoli cell complement due to lack of minipuberty, as evidenced by low serum inhibin B levels, and could thus benefit from pretreatment with FSH. A study of 14 boys with gonadotropin deficiency treated with recombinant human FSH (rFSH) priming showed significant increases in inhibin B and testicular volume in the absence of an increase in intragonadal testosterone production consistent with proliferation of Sertoli cells 97. Spermatogenesis was achieved in six out of seven boys who provided semen samples, with a maximal sperm count ranging from 2.9 to 92 million/mL (median, 8.5 million/mL) 97. A subsequent randomized controlled study showed similar results in young adults 98. Therefore, pretreatment with FSH prior to testicular maturation appears to compensate for the suboptimal Sertoli cell proliferation during late fetal life and minipuberty, and might be beneficial in adolescent males for future fertility 63. However, this treatment is intensive, requires frequent injections and close follow-up, and might not be optimal for all adolescent patients with hypogonadotropic hypogonadism. A large multicenter study to evaluate the benefits and cost-effectiveness of pretreatment with FSH in severe cases of adolescents and adults with hypogonadotropic hypogonadism is warranted 63.
Treatment of hypogonadotropic hypogonadism in females
Female hypogonadotropic hypogonadism medical treatment involve hormone replacement therapies and this is usually tailored the clinical need of the patients. Typically, once the diagnosis is made, female hypogonadotropic hypogonadism usually is treated with estrogen and progesterone pills or skin patches to return estrogen and progesterone levels to normal levels. Gonadotropins or pulsatile GnRH therapy can be utilized to stimulate production of mature egg cells (folliculogenesis). Therapy with sex steroid replacement (estrogen and progestin therapy) ensures development of secondary sex characteristics and maintenance of normal sexual function. Introduction of sex steroids in such cases starts with the use of small, escalating doses over a period of a couple of years 99. In females, introduction of puberty can begin with administration of small doses of estrogen given either orally or transdermally. One traditional regimen uses conjugated estrogen starting at doses as low as 0.15 mg daily and titrating upwards in 6-12 month intervals to typically 0.625 mg daily, at which point menses can be induced with the introduction of a progestin 99. Alternatively, transdermal 17β-estradiol (0.08 to 0.12 mcg estradiol/kg) can be used 99.
The main side effect of estrogen therapy is increased risk of thromboembolic events. When taken without progesterone, unopposed estrogen therapy can also increase the risk of endometrial hyperplasia and endometrial cancer. Although hormone replacement therapy is effective in inducing puberty, or as lifelong replacement therapy in those with permanent hypogonadism, it does not induce fertility. Subsequently, treatment may also be need for inducing fertility for achieving pregnancy.
If fertility is desired, fertility options can be explored in consultation with a reproductive endocrinologist or urologist. Pulsatile LHRH or gonadotropin therapy can induce fertility in individuals with hypogonadotropic hypogonadism. If spontaneous pregnancy fails to occur despite normal folliculogenesis, in vitro fertilization (IVF) may be considered with conception rates reported to be approximately 30% per ovulatory cycle.
In addition to treating hypogonadism, potential deterioration in bone health that may have resulted from periods of low circulating sex hormones should be addressed. Depending on the history (the timing of puberty, duration of hypogonadism, and other osteoporotic risk factors [e.g., glucocorticoid excess, smoking) and bone mineral density measurement, measurement, specific treatment for decreased bone mass should be considered.
Table 2. Females with hypogonadotropic hypogonadism treatment
| Treatment | Dosing and Administration | Advantages | Disadvantages |
|---|---|---|---|
| Induction of puberty in girls | |||
| 17β-estradiol (tablets) | Initial dose: 5 µg/kg daily orally | Natural estrogen | Less preferable than transdermal route |
| ↑ 5 µg/kg increments every 6–12 months | |||
| Up to 1–2 mg/d | |||
| 17β-estradiol (patch) | Inital dose: 0.05–0.07 µg/kg, only nocturnal | Natural estrogen | Small dose patch not available; need to cut the patch of 25 μg/24 h |
| ↑ to 0.08–0.12 µg/kg every 6 months | No hepatic passage (decrease thromboembolic risk) | ||
| Up to 50–100 µg/24 h | |||
| Progesterone | Added after full breast development or breakthrough bleeding, during the last 14 d of menstrual cycle | ||
| Treatment of hypogonadism in adult females | |||
| Estroprogestin therapy (tablets) | 17β-Estradiol 1 or 2 mg | Mimic the physiological hormone changes | |
| Progestin: during the last 14 d of the month micronized progestin at 200 mg/d orally, or dydrogesterone at 10 mg/d orally | |||
| Estroprogestin therapy (patch or gel) | 17β-Estradiol patch 50–100 μg/24 h daily, OR | Mimic the physiological hormone changes | |
| 17β-Estradiol gel 7.5–15 mg daily | |||
| Progestin: during the last 14 d of the month, micronized progestin at 200 mg/d orally, or dydrogesterone at 10 mg/d orally | |||
| Treatment of fertility in adult females | |||
| Pulsatile GnRH | IV pump: 75 ng/kg per pulse every 90 min | Most physiological treatment | Not available in many countries |
| Dose adapted based on response, up to 500 ng/kg per pulse | Possibility to adjust pulse frequency in IV pump | Require centers with expertise | |
| Subcutaneous pump: 15 µg per pulse every 90 min | High success rate | Risk of phlebitis for IV treatment (rare) | |
| Dose adapted based on response, up to 30 µg per pulse | Less risk in multiple pregnancy | Pituitary resistance (rare) | |
| Luteal phase: continue GnRH pump, OR | |||
| hCG 1500 U every 3 d for three times | |||
| Gonadotropins | hMG (FSH + LH) 75 to 150 IU SC daily, dose adapted based on follicular growth | Available around the world | More expensive |
| Induction of ovulation by hCG 6500 IU subcutaneous injection | Self-injection | Higher risk of overstimulation | |
| Luteal phase: | Requires close monitoring of E2 and ultrasound | ||
| hCG 1500 U every 3 d for three times | Higher risk of multiple pregnancy | ||
| Progesterone 200 mg intravaginally daily | |||
Treatment of female infertility
Infertility in women with hypogonadotropic hypogonadism is caused by impaired pituitary secretion of both gonadotropins, LH and FSH, leading to an impaired ovarian stimulation 63. Specifically, GnRH deficiency leads to an impairment in follicular terminal growth and maturation, resulting in chronic anovulation. However, there is no evidence of a decreased ovarian reserve (egg reserve) 100. Ovarian reserve refers to the quantity of an individual’s eggs (ova) that are stored in the ovaries. Ovarian reserve (egg reserve) is a critical factor in a woman’s fertility and reproductive potential. Understanding your ovarian reserve is essential when making decisions about family planning, fertility treatments, and assisted reproductive technologies as it can help plan for potential fertility treatments, if needed.
The combination of small ovaries, decreased antral follicular count, and low circulating AMH concentrations observed in women with hypogonadotropic hypogonadism could wrongly suggest an alteration in ovarian reserve and a poor fertility prognosis 100. In contrast, these patients should be informed that ovulation induction will lead to a fairly good outcome in terms of fertility in the absence of a male factor of infertility or advanced age (>35 years) 100, 101, 102, 103, 104.
Before considering ovulation induction, sono-hysterosalpingography or traditional hysterosalpingography could be performed to evaluate both the integrity and the permeability of the uterine cavity and fallopian tubes 105. Alternatively, sono-hysterosalpingography could be performed after a couple of cycles of successful ovulation in the absence of pregnancy. Additionally, an associated male infertility factor should be ruled out by obtaining a semen analysis 104. Couples should be advised on the optimal timing of sexual intercourse during the ovulation induction, as this first-line therapy does not require in vitro fertilization (IVF) 100, 101, 102, 103.
The goal of ovulation induction therapy in female patients with hypogonadotropic hypogonadism is to obtain a mono-ovulation to avoid multiple pregnancies. Ovulation can be achieved either with pulsatile GnRH therapy or stimulation with gonadotropins. The latter includes either extractive or rFSH treatment followed by hCG or rLH to trigger ovulation 106. The therapeutic choice will depend on the expertise of each center and the local availability of the different medical therapeutics.
Pulsatile GnRH treatment
Pulsatile GnRH therapy via a pump was first proposed by Leyendecker et al. 107, 108, 109 to induce ovulation in women with different causes of hypogonadotropic amenorrhea. Given its remarkable efficiency in acquired forms of hypogonadotropic hypogonadism, pulsatile GnRH was successfully applied to women with hypogonadotropic hypogonadism 110 and other causes of acquired hypogonadotropic hypogonadism 111, 112, 113. Both SC and IV routes for GnRH administration are appropriate to restore fertility 111, 114. Pulsatile GnRH restores the physiological secretion of pituitary gonadotropins, which in turn induces ovulation in patients with hypogonadotropic hypogonadism 115, 116, 117, 118, 119. The major advantage of pulsatile GnRH therapy compared with gonadotropin treatment is the decreased risk of multiple pregnancy or ovarian hyperstimulation 119. Consequently, it requires less monitoring and surveillance during treatment. Therefore, when pulsatile GnRH treatment is available within the region the patient is being treated, it should be considered the first line of therapy in females with hypogonadotropic hypogonadism, given that it is the most physiological regimen and results in fewer side effects.
Physiologically, GnRH pulse intervals vary throughout the menstrual cycle, as evidenced by LH pulse studies in a large series of women with regular menses 120. Based on this study, the frequency of GnRH pulses is set for every 90 minutes during the early follicular phase of treatment, and subsequently accelerated to every 60 minutes during the middle and late follicular phase. After ovulation, the frequency is reduced to every 90 minutes. Finally, during the late luteal phase, there is a further decrease to every 4 hours that will favor FSH secretion over LH. However, pulsatile GnRH at a constant frequency of 90 minutes also induces maturation of ovarian follicles, an LH surge, and ovulation 114.
The dosage of GnRH required to restore normal ovulation has been well studied in females with hypogonadotropic hypogonadism or functional hypothalamic amenorrhea. IV doses of 75 ng/kg per pulse are considered a physiological dose to induce adequate pituitary gonadotropin secretion and ovarian stimulation 121. In 30% of females with hypogonadotropic hypogonadism, pituitary resistance is present at the first cycle, requiring increased GnRH doses and longer stimulation 118. Once ovulation is achieved, the corpus luteum must be stimulated to produce progesterone, which is mandatory for embryo implantation. The pulsatile GnRH pump is able to maintain endogenous pulsatile LH secretion sufficient to ensure progesterone release by the corpus luteum until the endogenous secretion of hCG from the placenta begins (355, 358). Another treatment option for luteal support is hCG (SC injections of 1500 IU every 3 days for three times), which is less costly and well tolerated. The success rate of ovulation induction is excellent in females with hypogonadotropic hypogonadism, reaching 90% ovulation per cycle, and 27.6% conception per ovulatory cycle. The number of cycles needed to obtain a pregnancy is quite variable, ranging from one to six cycles 119, 122. The multiple pregnancy rate is slightly higher than the general population at 5% to 8% 121, but much lower than with gonadotropin therapy. The pulsatile GnRH pump can be effective even in the presence of GnRH resistance, such as in women with hypogonadotropic hypogonadism who harbor partial loss-of-function GnRH receptor mutations 118, 115.
When administered SC, higher doses (15 µg per pulse) are needed, and typically the frequency of pulses are kept at one every 90 minutes. The success rate is slightly lower at 70% of ovulation rate per cycle 123. However, the SC administration has no risk of phlebitis and is more convenient.
GnRH pulse treatment is discontinued when pregnancy occurs, and adverse effects in early pregnancy have not been reported 124. After several unsuccessful cycles of GnRH stimulation, gonadotropin therapy should be proposed 102, 103 to bypass a potential pituitary resistance associated or not with loss-of-function GnRH receptor mutations 125, 118.
Gonadotropin treatment
In women with hypogonadotropic hypogonadism, ovulation can also be achieved with FSH treatment followed by hCG or rLH to trigger ovulation. However, women with severe GnRH deficiency have very low gonadotropin levels, thus requiring both FSH and LH during the follicular phase. LH stimulates the ovarian theca cells to produce androgen substrates, allowing sufficient secretion of estradiol (E2) by the maturing follicles 100, 126, 102, 127. Estradiol (E2) is necessary for optimal endometrial thickness and cervical mucus production, which in turn are needed for sperm transit and embryo implantation 100. Typically, SC human menopausal gonadotropins (hMGs; FSH plus hCG) doses of 75 to 150 IU/d are sufficient to induce ovulation. Usually, a dominant follicle (>18 mm) will mature in ∼12 days. The starting dose of hMG is often increased or decreased depending on the ovarian response, as assessed by repeated serum estradiol (E2) measurements or by using ultrasonography to count and measure maturing follicles every other day. This regimen minimizes the risk of multiple pregnancy and ovarian hyperstimulation syndrome. After ovulation, progesterone production can be stimulated by repeated hCG injections, or direct administration of progesterone during the postovulatory phase until the end of the luteal phase.
In vitro fertilization (IVF)
If conception fails after repeated successful ovulation induction in females with hypogonadotropic hypogonadism, in vitro fertilization (IVF) may be an alternative 128, 129.
Long-Term Monitoring
Patients with hypogonadism require lifelong treatment, with the exception of persons with congenital hypogonadotropic hypogonadism (spontaneous recovery having been described in 10-20% of these individuals) 130. Patients with hypogonadism receiving hormone replacement therapy are typically evaluated every 6-12 months. Monitoring may include measurement of testosterone concentrations in males, evaluation of bone mass by dual radiographic absorptiometry, and assessment of cardiovascular risk factors.
An increase in red blood cell count (polycythemia) can be a complication of testosterone replacement. For older adult men with testosterone deficiency, the Endocrine Society clinical guidelines recommend monitoring hematocrit values to avoid polycythemia. Also in these individuals, prostate examination and prostate-specific antigen (PSA) measurements should be performed before testosterone therapy and periodically after treatment is instituted 50. Referral to a urologist can be considered based on individual assessment for prostate cancer.
Hypogonadotropic hypogonadism prognosis
In men, complications of untreated hypogonadism include loss of libido, failure to achieve physical strength, the social implications of failing to go through puberty with peers (if hypogonadism occurs before puberty), and osteoporosis. In addition, if hypogonadism occurs before epiphyseal closure, the result is usually tall stature with a eunuchoid body habitus. Males with hypergonadotropic hypogonadism are typically infertile, although procedures such as testicular sperm extraction have resulted in fertility in Klinefelter syndrome. Men who have hypogonadotropic hypogonadism due to hypothalamic or pituitary dysfunction can potentially become fertile with administration of gonadotropins.
A retrospective study by Baillargeon et al 131 indicated that males with untreated hypogonadism are at increased risk for the development of any rheumatic autoimmune disease, as well as for rheumatoid arthritis and lupus.
In women with hypogonadism, complications include the social implication of failing to go through puberty with peers (if hypogonadism occurs before puberty). An additional concern for untreated women is osteoporosis, which can be avoided with estrogen replacement. Women who have hypogonadotropic hypogonadism because of hypothalamic or pituitary dysfunction can potentially become fertile with administration of gonadotropins. Women with primary hypogonadism (hypergonadotropic hypogonadism) are infertile; however, with in vitro fertilization (IVF) using a donor ovum, these women can carry an infant to term.
Osteoporosis has an earlier onset in individuals with hypogonadism; hence, bone mineral density should be compared with age-matched normative standards, and followed longitudinally. Prescribe treatment using appropriate therapeutic interventions.
The right hormone treatment will cause puberty to start in children and may restore fertility in adults. If the condition begins after puberty or in adulthood, symptoms will often improve with treatment.
- Hypogonadism. https://emedicine.medscape.com/article/922038-overview[↩][↩][↩]
- Hayes F, Dwyer A, Pitteloud N. Hypogonadotropic Hypogonadism (HH) and Gonadotropin Therapy. [Updated 2013 Nov 25]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279078[↩][↩][↩]
- Russo V, Chen R, Armamento-Villareal R. Hypogonadism, Type-2 Diabetes Mellitus, and Bone Health: A Narrative Review. Front Endocrinol (Lausanne). 2021 Jan 18;11:607240. doi: 10.3389/fendo.2020.607240[↩]
- Dhindsa S, Ghanim H, Batra M, Dandona P. Hypogonadotropic Hypogonadism in Men With Diabesity. Diabetes Care. 2018 Jul;41(7):1516-1525. doi: 10.2337/dc17-2510[↩]
- Rabijewski M, Papierska L, Zgliczyński W, Piątkiewicz P. The incidence of hypogonadotropic hypogonadism in type 2 diabetic men in Polish population. Biomed Res Int. 2013;2013:767496. doi: 10.1155/2013/767496[↩]
- Dandona P, Dhindsa S, Chaudhuri A, Bhatia V, Topiwala S. Hypogonadotrophic hypogonadism in type 2 diabetes. Aging Male. 2008 Sep;11(3):107-17. doi: 10.1080/13685530802317934[↩]
- Dandona P, Dhindsa S, Chaudhuri A, Bhatia V, Topiwala S, Mohanty P. Hypogonadotrophic hypogonadism in type 2 diabetes, obesity and the metabolic syndrome. Curr Mol Med. 2008 Dec;8(8):816-28. doi: 10.2174/156652408786733658[↩]
- Dandona P, Dhindsa S. Update: Hypogonadotropic hypogonadism in type 2 diabetes and obesity. J Clin Endocrinol Metab. 2011 Sep;96(9):2643-51. doi: 10.1210/jc.2010-2724[↩]
- Dhindsa S, Prabhakar S, Sethi M, Bandyopadhyay A, Chaudhuri A, Dandona P. Frequent occurrence of hypogonadotropic hypogonadism in type 2 diabetes. J Clin Endocrinol Metab. 2004 Nov;89(11):5462-8. doi: 10.1210/jc.2004-0804[↩]
- Dhindsa S, Miller MG, McWhirter CL, Mager DE, Ghanim H, Chaudhuri A, Dandona P. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care. 2010 Jun;33(6):1186-92. doi: 10.2337/dc09-1649. Epub 2010 Mar 3. Erratum in: Diabetes Care. 2010 Aug;33(8):1911.[↩][↩]
- Fraietta R, Zylberstejn DS, Esteves SC. Hypogonadotropic hypogonadism revisited. Clinics (Sao Paulo). 2013;68 Suppl 1(Suppl 1):81-8. doi: 10.6061/clinics/2013(sup01)09[↩]
- Clavijo RI, Hsiao W. Update on male reproductive endocrinology. Transl Androl Urol. 2018 Jul;7(Suppl 3):S367-S372. doi: 10.21037/tau.2018.03.25[↩]
- Canavese F, Mussa A, Manenti M, Cortese MG, Ferrero L, Tuli G, Macchieraldo R, Lala R. Sperm count of young men surgically treated for cryptorchidism in the first and second year of life: fertility is better in children treated at a younger age. Eur J Pediatr Surg. 2009 Dec;19(6):388-91. doi: 10.1055/s-0029-1241171[↩]
- Herbison AE. The Gonadotropin-Releasing Hormone Pulse Generator. Endocrinology. 2018 Nov 1;159(11):3723-3736. doi: 10.1210/en.2018-00653[↩]
- Wilson EA, Erickson GF, Zarutski P, Finn AE, Tulchinsky D, Ryan KJ. Endocrine studies of normal and polycystic ovarian tissues in vitro. Am J Obstet Gynecol. 1979 May 1;134(1):56-63. doi: 10.1016/0002-9378(79)90796-8[↩]
- Zirkin BR, Papadopoulos V. Leydig cells: formation, function, and regulation. Biol Reprod. 2018 Jul 1;99(1):101-111. doi: 10.1093/biolre/ioy059[↩]
- Griswold MD. The central role of Sertoli cells in spermatogenesis. Semin Cell Dev Biol. 1998 Aug;9(4):411-6. doi: 10.1006/scdb.1998.0203[↩]
- O’Donnell L, Smith LB, Rebourcet D. Sertoli cells as key drivers of testis function. Semin Cell Dev Biol. 2022 Jan;121:2-9. doi: 10.1016/j.semcdb.2021.06.016[↩]
- Jin JM, Yang WX. Molecular regulation of hypothalamus-pituitary-gonads axis in males. Gene. 2014 Nov 1;551(1):15-25. doi: 10.1016/j.gene.2014.08.048[↩]
- Wintermantel TM, Campbell RE, Porteous R, Bock D, Gröne HJ, Todman MG, Korach KS, Greiner E, Pérez CA, Schütz G, Herbison AE. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron. 2006 Oct 19;52(2):271-80. doi: 10.1016/j.neuron.2006.07.023[↩]
- Rohayem J, Alexander EC, Heger S, Nordenström A, Howard SR. Mini-Puberty, Physiological and Disordered: Consequences, and Potential for Therapeutic Replacement. Endocr Rev. 2024 Jul 12;45(4):460-492. doi: 10.1210/endrev/bnae003[↩][↩]
- Gupta, Priya & Mahapatra, Archisman & Suman, Anjali & Singh, Rahul. (2021). Effect of Endocrine Disrupting Chemicals on HPG Axis: A Reproductive Endocrine Homeostasis. https://www.researchgate.net/publication/350037153_Effect_of_Endocrine_Disrupting_Chemicals_on_HPG_Axis_A_Reproductive_Endocrine_Homeostasis[↩]
- Tsutsumi R, Webster NJ. GnRH pulsatility, the pituitary response and reproductive dysfunction. Endocr J. 2009;56(6):729-37. doi: 10.1507/endocrj.k09e-185[↩]
- Hypogonadotropic hypogonadism. https://medlineplus.gov/ency/article/000390.htm[↩]
- de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, Milgrom E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997 Nov 27;337(22):1597-602. doi: 10.1056/NEJM199711273372205[↩][↩]
- Kallmann syndrome. https://medlineplus.gov/genetics/condition/kallmann-syndrome[↩]
- Kallmann Syndrome. https://rarediseases.org/rare-diseases/kallmann-syndrome[↩]
- Abdulla AB, Niloy AA, Shah TA, Biswas SK, Imran AK, Murshed KM, Ahmed M. Laurence Moon Bardet Biedl Syndrome. Mymensingh Med J. 2009 Jan;18(1 Suppl):S124-128.[↩]
- Bonomi M, Vezzoli V, Krausz C, et al. Italian Network on Central Hypogonadism; Italian Network on Central Hypogonadism (NICe group). Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (IHH). Eur J Endocrinol. 2018 Jan;178(1):23-32. doi: 10.1530/EJE-17-0065[↩]
- Lee HS, Shim YS, Hwang JS. Treatment of congenital hypogonadotropic hypogonadism in male patients. Ann Pediatr Endocrinol Metab. 2022 Sep;27(3):176-182. doi: 10.6065/apem.2244208.104[↩]
- Amato LGL, Montenegro LR, Lerario AM, Jorge AAL, Guerra Junior G, Schnoll C, Renck AC, Trarbach EB, Costa EMF, Mendonca BB, Latronico AC, Silveira LFG. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism. Eur J Endocrinol. 2019 Aug 1;181(2):103-119. doi: 10.1530/EJE-18-0764[↩]
- Hao M, Nie M, Yu BQ, Gao YJ, Wang X, Ma WL, Huang QB, Zhang R, Mao JF, Wu XY. Gonadotropin treatment for male partial congenital hypogonadotropic hypogonadism in Chinese patients. Asian J Androl. 2020 Jul-Aug;22(4):390-395. doi: 10.4103/aja.aja_88_19[↩]
- Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R, Young J. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015 Sep;11(9):547-64. doi: 10.1038/nrendo.2015.112[↩][↩]
- Beneduzzi D, Trarbach EB, Min L, Jorge AA, Garmes HM, Renk AC, Fichna M, Fichna P, Arantes KA, Costa EM, Zhang A, Adeola O, Wen J, Carroll RS, Mendonça BB, Kaiser UB, Latronico AC, Silveira LF. Role of gonadotropin-releasing hormone receptor mutations in patients with a wide spectrum of pubertal delay. Fertil Steril. 2014 Sep;102(3):838-846.e2. doi: 10.1016/j.fertnstert.2014.05.044[↩]
- Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol. 2011 Oct 22;346(1-2):21-8. doi: 10.1016/j.mce.2011.04.018[↩]
- Hietamäki J, Hero M, Holopainen E, Känsäkoski J, Vaaralahti K, Iivonen AP, Miettinen PJ, Raivio T. GnRH receptor gene mutations in adolescents and young adults presenting with signs of partial gonadotropin deficiency. PLoS One. 2017 Nov 28;12(11):e0188750. doi: 10.1371/journal.pone.0188750[↩]
- Topaloğlu AK. Update on the Genetics of Idiopathic Hypogonadotropic Hypogonadism. J Clin Res Pediatr Endocrinol. 2017 Dec 30;9(Suppl 2):113-122. doi: 10.4274/jcrpe.2017.S010[↩][↩]
- Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2009 Oct;5(10):569-76. doi: 10.1038/nrendo.2009.177[↩]
- Young J, Xu C, Papadakis GE, Acierno JS, Maione L, Hietamäki J, Raivio T, Pitteloud N. Clinical Management of Congenital Hypogonadotropic Hypogonadism. Endocr Rev. 2019 Apr 1;40(2):669-710. doi: 10.1210/er.2018-00116[↩]
- Semple RK, Topaloglu AK. The recent genetics of hypogonadotrophic hypogonadism – novel insights and new questions. Clin Endocrinol (Oxf). 2010 Apr;72(4):427-35. doi: 10.1111/j.1365-2265.2009.03687.x[↩]
- Crowley WF Jr, Pitteloud N, Seminara S. New genes controlling human reproduction and how you find them. Trans Am Clin Climatol Assoc. 2008;119:29-37; discussion 37-8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2394706[↩]
- Maione L, Dwyer AA, Francou B, Guiochon-Mantel A, Binart N, Bouligand J, Young J. GENETICS IN ENDOCRINOLOGY: Genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol. 2018 Mar;178(3):R55-R80. doi: 10.1530/EJE-17-0749[↩]
- Cangiano B, Swee DS, Quinton R, Bonomi M. Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease. Hum Genet. 2021 Jan;140(1):77-111. doi: 10.1007/s00439-020-02147-1[↩]
- Palmert MR, Dunkel L. Clinical practice. Delayed puberty. N Engl J Med. 2012 Feb 2;366(5):443-53. doi: 10.1056/NEJMcp1109290[↩]
- Grumbach MM. A window of opportunity: the diagnosis of gonadotropin deficiency in the male infant. J Clin Endocrinol Metab. 2005 May;90(5):3122-7. doi: 10.1210/jc.2004-2465[↩]
- Van Dop C, Burstein S, Conte FA, Grumbach MM. Isolated gonadotropin deficiency in boys: clinical characteristics and growth. J Pediatr. 1987 Nov;111(5):684-92. doi: 10.1016/s0022-3476(87)80243-3[↩]
- Boyar RM, Finkelstein JW, Witkin M, Kapen S, Weitzman E, Hellman L. Studies of endocrine function in “isolated” gonadotropin deficiency. J Clin Endocrinol Metab. 1973 Jan;36(1):64-72. doi: 10.1210/jcem-36-1-64[↩]
- Lieblich JM, Rogol AD, White BJ, Rosen SW. Syndrome of anosmia with hypogonadotropic hypogonadism (Kallmann syndrome): clinical and laboratory studies in 23 cases. Am J Med. 1982 Oct;73(4):506-19. doi: 10.1016/0002-9343(82)90329-1[↩]
- Nachtigall LB, Boepple PA, Pralong FP, Crowley WF Jr. Adult-onset idiopathic hypogonadotropic hypogonadism–a treatable form of male infertility. N Engl J Med. 1997 Feb 6;336(6):410-5. doi: 10.1056/NEJM199702063360604[↩]
- Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, Montori VM; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010 Jun;95(6):2536-59. doi: 10.1210/jc.2009-2354. Erratum in: J Clin Endocrinol Metab. 2021 Jun 16;106(7):e2848. doi: 10.1210/clinem/dgab311[↩][↩][↩]
- Kallmann Syndrome. https://rarediseases.org/rare-diseases/kallmann-syndrome/[↩][↩]
- Young J. Approach to the male patient with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2012 Mar;97(3):707-18. doi: 10.1210/jc.2011-1664[↩][↩][↩]
- Han TS, Bouloux PM. What is the optimal therapy for young males with hypogonadotropic hypogonadism? Clin Endocrinol (Oxf). 2010 Jun;72(6):731-7. doi: 10.1111/j.1365-2265.2009.03746.x[↩][↩][↩]
- Santhakumar A, Miller M, Quinton R. Pubertal induction in adult males with isolated hypogonadotropic hypogonadism using long-acting intramuscular testosterone undecanoate 1-g depot (Nebido). Clin Endocrinol (Oxf). 2014 Jan;80(1):155-7. doi: 10.1111/cen.12160[↩]
- Gan EH, Bouloux PM, Quinton R. Unexpectedly prolonged washout period of exogenous testosterone after discontinuation of intramuscular testosterone undecanoate depot injection (Nebido(®) or Reandron(®) ) in men with congenital hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf). 2016 Jun;84(6):947-50. doi: 10.1111/cen.13018[↩]
- Kaminetsky J, Jaffe JS, Swerdloff RS. Pharmacokinetic Profile of Subcutaneous Testosterone Enanthate Delivered via a Novel, Prefilled Single-Use Autoinjector: A Phase II Study. Sex Med. 2015 Sep 17;3(4):269-79. doi: 10.1002/sm2.80[↩]
- Spratt DI, Stewart II, Savage C, Craig W, Spack NP, Chandler DW, Spratt LV, Eimicke T, Olshan JS. Subcutaneous Injection of Testosterone Is an Effective and Preferred Alternative to Intramuscular Injection: Demonstration in Female-to-Male Transgender Patients. J Clin Endocrinol Metab. 2017 Jul 1;102(7):2349-2355. doi: 10.1210/jc.2017-00359[↩]
- de Ronde W. Hyperandrogenism after transfer of topical testosterone gel: case report and review of published and unpublished studies. Hum Reprod. 2009 Feb;24(2):425-8. doi: 10.1093/humrep/den372[↩]
- Martinez-Pajares JD, Diaz-Morales O, Ramos-Diaz JC, Gomez-Fernandez E. Peripheral precocious puberty due to inadvertent exposure to testosterone: case report and review of the literature. J Pediatr Endocrinol Metab. 2012;25(9-10):1007-12. doi: 10.1515/jpem-2012-0124[↩]
- Dwyer AA, Tiemensma J, Quinton R, Pitteloud N, Morin D. Adherence to treatment in men with hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf). 2017 Mar;86(3):377-383. doi: 10.1111/cen.13236[↩]
- Rastrelli G, Vignozzi L, Corona G, Maggi M. Pharmacotherapy of male hypogonadism. Curr Opin Pharmacol. 2023 Feb;68:102323. doi: 10.1016/j.coph.2022.102323[↩]
- Rastrelli G, Maggi M, Corona G. Pharmacological management of late-onset hypogonadism. Expert Rev Clin Pharmacol. 2018 Apr;11(4):439-458. doi: 10.1080/17512433.2018[↩]
- Jacques Young, Cheng Xu, Georgios E Papadakis, James S Acierno, Luigi Maione, Johanna Hietamäki, Taneli Raivio, Nelly Pitteloud, Clinical Management of Congenital Hypogonadotropic Hypogonadism, Endocrine Reviews, Volume 40, Issue 2, April 2019, Pages 669–710, https://doi.org/10.1210/er.2018-00116[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- HELLER CG, NELSON WO. Classification of male hypogonadism and a discussion of the pathologic physiology, diagnosis and treatment. J Clin Endocrinol Metab. 1948 May;8(5):345-66. doi: 10.1210/jcem-8-5-345[↩]
- Lytton B, Kase N. Effects of human menopausal gonadotrophin on a eunuchoidal male. N Engl J Med. 1966 May 12;274(19):1061-4. doi: 10.1056/NEJM196605122741905[↩]
- Johnsen SG. Maintenance of spermatogenesis induced by HMG treatment by means of continuous HCG treatment in hypogonadotrophic men. Acta Endocrinol (Copenh). 1978 Dec;89(4):763-9. doi: 10.1530/acta.0.0890763[↩]
- Burris AS, Clark RV, Vantman DJ, Sherins RJ. A low sperm concentration does not preclude fertility in men with isolated hypogonadotropic hypogonadism after gonadotropin therapy. Fertil Steril. 1988 Aug;50(2):343-7. doi: 10.1016/s0015-0282(16)60084-5[↩]
- Zorn B, Pfeifer M, Virant-Klun I, Meden-Vrtovec H. Intracytoplasmic sperm injection as a complement to gonadotrophin treatment in infertile men with hypogonadotrophic hypogonadism. Int J Androl. 2005 Aug;28(4):202-7. doi: 10.1111/j.1365-2605.2004.00519.x[↩]
- Hoffman AR, Crowley WF Jr. Induction of puberty in men by long-term pulsatile administration of low-dose gonadotropin-releasing hormone. N Engl J Med. 1982 Nov 11;307(20):1237-41. doi: 10.1056/NEJM198211113072003[↩]
- Pitteloud N, Hayes FJ, Dwyer A, Boepple PA, Lee H, Crowley WF Jr. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002 Sep;87(9):4128-36. doi: 10.1210/jc.2002-020518[↩][↩]
- Spratt DI, Finkelstein JS, O’Dea LS, Badger TM, Rao PN, Campbell JD, Crowley WF Jr. Long-term administration of gonadotropin-releasing hormone in men with idiopathic hypogonadotropic hypogonadism. A model for studies of the hormone’s physiologic effects. Ann Intern Med. 1986 Dec;105(6):848-55. doi: 10.7326/0003-4819-105-6-848[↩]
- Aulitzky W, Frick J, Galvan G. Pulsatile luteinizing hormone-releasing hormone treatment of male hypogonadotropic hypogonadism. Fertil Steril. 1988 Sep;50(3):480-6. doi: 10.1016/s0015-0282(16)60137-1[↩]
- Whitcomb RW, Crowley WF Jr. Clinical review 4: Diagnosis and treatment of isolated gonadotropin-releasing hormone deficiency in men. J Clin Endocrinol Metab. 1990 Jan;70(1):3-7. doi: 10.1210/jcem-70-1-3[↩]
- Bouvattier C, Maione L, Bouligand J, Dodé C, Guiochon-Mantel A, Young J. Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2011 Oct 18;8(3):172-82. doi: 10.1038/nrendo.2011.164[↩][↩][↩]
- Sarfati J, Bouvattier C, Bry-Gauillard H, Cartes A, Bouligand J, Young J. Kallmann syndrome with FGFR1 and KAL1 mutations detected during fetal life. Orphanet J Rare Dis. 2015 Jun 9;10:71. doi: 10.1186/s13023-015-0287-9[↩][↩]
- Bin-Abbas B, Conte FA, Grumbach MM, Kaplan SL. Congenital hypogonadotropic hypogonadism and micropenis: effect of testosterone treatment on adult penile size why sex reversal is not indicated. J Pediatr. 1999 May;134(5):579-83. doi: 10.1016/s0022-3476(99)70244-1[↩][↩]
- Lambert AS, Bougneres P. Growth and descent of the testes in infants with hypogonadotropic hypogonadism receiving subcutaneous gonadotropin infusion. Int J Pediatr Endocrinol. 2016;2016:13. doi: 10.1186/s13633-016-0031-9[↩][↩]
- Bougnères P, François M, Pantalone L, Rodrigue D, Bouvattier C, Demesteere E, Roger D, Lahlou N. Effects of an early postnatal treatment of hypogonadotropic hypogonadism with a continuous subcutaneous infusion of recombinant follicle-stimulating hormone and luteinizing hormone. J Clin Endocrinol Metab. 2008 Jun;93(6):2202-5. doi: 10.1210/jc.2008-0121[↩][↩]
- Main KM, Schmidt IM, Toppari J, Skakkebaek NE. Early postnatal treatment of hypogonadotropic hypogonadism with recombinant human FSH and LH. Eur J Endocrinol. 2002 Jan;146(1):75-9. doi: 10.1530/eje.0.1460075[↩][↩]
- Main KM, Schmidt IM, Skakkebaek NE. A possible role for reproductive hormones in newborn boys: progressive hypogonadism without the postnatal testosterone peak. J Clin Endocrinol Metab. 2000 Dec;85(12):4905-7. doi: 10.1210/jcem.85.12.7058[↩]
- Nane I, Ziylan O, Esen T, Kocak T, Ander H, Tellaloglu S. Primary gonadotropin releasing hormone and adjunctive human chorionic gonadotropin treatment in cryptorchidism: a clinical trial. Urology. 1997 Jan;49(1):108-11. doi: 10.1016/S0090-4295(96)00359-7[↩]
- Christiansen P, Müller J, Buhl S, Hansen OR, Hobolth N, Jacobsen BB, Jørgensen PH, Kastrup KW, Nielsen K, Nielsen LB, et al. Treatment of cryptorchidism with human chorionic gonadotropin or gonadotropin releasing hormone. A double-blind controlled study of 243 boys. Horm Res. 1988;30(4-5):187-92. doi: 10.1159/000181058[↩]
- Dunkel L, Taskinen S, Hovatta O, Tilly JL, Wikström S. Germ cell apoptosis after treatment of cryptorchidism with human chorionic gonadotropin is associated with impaired reproductive function in the adult. J Clin Invest. 1997 Nov 1;100(9):2341-6. doi: 10.1172/JCI119773[↩][↩]
- Dwyer AA, Quinton R, Morin D, Pitteloud N. Identifying the unmet health needs of patients with congenital hypogonadotropic hypogonadism using a web-based needs assessment: implications for online interventions and peer-to-peer support. Orphanet J Rare Dis. 2014 Jun 11;9:83. doi: 10.1186/1750-1172-9-83[↩]
- Dunkel L, Quinton R. Transition in endocrinology: induction of puberty. Eur J Endocrinol. 2014 Jun;170(6):R229-39. doi: 10.1530/EJE-13-0894[↩][↩]
- Lawaetz JG, Hagen CP, Mieritz MG, Blomberg Jensen M, Petersen JH, Juul A. Evaluation of 451 Danish boys with delayed puberty: diagnostic use of a new puberty nomogram and effects of oral testosterone therapy. J Clin Endocrinol Metab. 2015 Apr;100(4):1376-85. doi: 10.1210/jc.2014-3631[↩]
- Dwyer AA, Raivio T, Pitteloud N. MANAGEMENT OF ENDOCRINE DISEASE: Reversible hypogonadotropic hypogonadism. Eur J Endocrinol. 2016 Jun;174(6):R267-74. doi: 10.1530/EJE-15-1033[↩]
- Bistritzer T, Lunenfeld B, Passwell JH, Theodor R. Hormonal therapy and pubertal development in boys with selective hypogonadotropic hypogonadism. Fertil Steril. 1989 Aug;52(2):302-6. doi: 10.1016/s0015-0282(16)60859-2[↩][↩]
- Barrio R, de Luis D, Alonso M, Lamas A, Moreno JC. Induction of puberty with human chorionic gonadotropin and follicle-stimulating hormone in adolescent males with hypogonadotropic hypogonadism. Fertil Steril. 1999 Feb;71(2):244-8. doi: 10.1016/s0015-0282(98)00450-6[↩]
- Zacharin M, Sabin MA, Nair VV, Dabadghao P. Addition of recombinant follicle-stimulating hormone to human chorionic gonadotropin treatment in adolescents and young adults with hypogonadotropic hypogonadism promotes normal testicular growth and may promote early spermatogenesis. Fertil Steril. 2012 Oct;98(4):836-42. doi: 10.1016/j.fertnstert.2012.06.022. Epub 2012 Jul 3. Erratum in: Fertil Steril. 2013 May;99(6):1798. Dagabdhao, Preeti [corrected to Dabadghao, Preeti].[↩]
- Sinisi AA, Esposito D, Maione L, Quinto MC, Visconti D, De Bellis A, Bellastella A, Conzo G, Bellastella G. Seminal anti-Müllerian hormone level is a marker of spermatogenic response during long-term gonadotropin therapy in male hypogonadotropic hypogonadism. Hum Reprod. 2008 May;23(5):1029-34. doi: 10.1093/humrep/den046[↩]
- Rohayem J, Hauffa BP, Zacharin M, Kliesch S, Zitzmann M; “German Adolescent Hypogonadotropic Hypogonadism Study Group”. Testicular growth and spermatogenesis: new goals for pubertal hormone replacement in boys with hypogonadotropic hypogonadism? -a multicentre prospective study of hCG/rFSH treatment outcomes during adolescence. Clin Endocrinol (Oxf). 2017 Jan;86(1):75-87. doi: 10.1111/cen.13164[↩][↩][↩]
- Meachem SJ, McLachlan RI, de Kretser DM, Robertson DM, Wreford NG. Neonatal exposure of rats to recombinant follicle stimulating hormone increases adult Sertoli and spermatogenic cell numbers. Biol Reprod. 1996 Jan;54(1):36-44. doi: 10.1095/biolreprod54.1.36[↩]
- Arslan M, Weinbauer GF, Schlatt S, Shahab M, Nieschlag E. FSH and testosterone, alone or in combination, initiate testicular growth and increase the number of spermatogonia and Sertoli cells in a juvenile non-human primate (Macaca mulatta). J Endocrinol. 1993 Feb;136(2):235-43. doi: 10.1677/joe.0.1360235[↩]
- Raivio T, Toppari J, Perheentupa A, McNeilly AS, Dunkel L. Treatment of prepubertal gonadotrophin-deficient boys with recombinant human follicle-stimulating hormone. Lancet. 1997 Jul 26;350(9073):263-4. doi: 10.1016/S0140-6736(05)62227-1[↩]
- Tapanainen JS, Aittomäki K, Min J, Vaskivuo T, Huhtaniemi IT. Men homozygous for an inactivating mutation of the follicle-stimulating hormone (FSH) receptor gene present variable suppression of spermatogenesis and fertility. Nat Genet. 1997 Feb;15(2):205-6. doi: 10.1038/ng0297-205[↩]
- Raivio T, Wikström AM, Dunkel L. Treatment of gonadotropin-deficient boys with recombinant human FSH: long-term observation and outcome. Eur J Endocrinol. 2007 Jan;156(1):105-11. doi: 10.1530/eje.1.02315[↩][↩]
- Dwyer AA, Sykiotis GP, Hayes FJ, Boepple PA, Lee H, Loughlin KR, Dym M, Sluss PM, Crowley WF Jr, Pitteloud N. Trial of recombinant follicle-stimulating hormone pretreatment for GnRH-induced fertility in patients with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2013 Nov;98(11):E1790-5. doi: 10.1210/jc.2013-2518[↩]
- Hypogonadism Treatment & Management. https://emedicine.medscape.com/article/922038-treatment#d7[↩][↩][↩]
- Bry-Gauillard H, Larrat-Ledoux F, Levaillant JM, Massin N, Maione L, Beau I, Binart N, Chanson P, Brailly-Tabard S, Hall JE, Young J. Anti-Müllerian Hormone and Ovarian Morphology in Women With Isolated Hypogonadotropic Hypogonadism/Kallmann Syndrome: Effects of Recombinant Human FSH. J Clin Endocrinol Metab. 2017 Apr 1;102(4):1102-1111. doi: 10.1210/jc.2016-3799[↩][↩][↩][↩][↩][↩]
- Tang RY, Chen R, Ma M, Lin SQ, Zhang YW, Wang YP. Clinical characteristics of 138 Chinese female patients with idiopathic hypogonadotropic hypogonadism. Endocr Connect. 2017 Nov;6(8):800-810. doi: 10.1530/EC-17-0251[↩][↩]
- Shoham Z, Smith H, Yeko T, O’Brien F, Hemsey G, O’Dea L. Recombinant LH (lutropin alfa) for the treatment of hypogonadotrophic women with profound LH deficiency: a randomized, double-blind, placebo-controlled, proof-of-efficacy study. Clin Endocrinol (Oxf). 2008 Sep;69(3):471-8. doi: 10.1111/j.1365-2265.2008.03299.x[↩][↩][↩][↩]
- Kaufmann R, Dunn R, Vaughn T, Hughes G, O’Brien F, Hemsey G, Thomson B, O’Dea LS. Recombinant human luteinizing hormone, lutropin alfa, for the induction of follicular development and pregnancy in profoundly gonadotrophin-deficient women. Clin Endocrinol (Oxf). 2007 Oct;67(4):563-9. doi: 10.1111/j.1365-2265.2007.02925.x[↩][↩][↩]
- Tournaye H, Krausz C, Oates RD. Concepts in diagnosis and therapy for male reproductive impairment. Lancet Diabetes Endocrinol. 2017 Jul;5(7):554-564. doi: 10.1016/S2213-8587(16)30043-2[↩][↩]
- Dreyer K, van Rijswijk J, Mijatovic V, Goddijn M, Verhoeve HR, van Rooij IAJ, Hoek A, Bourdrez P, Nap AW, Rijnsaardt-Lukassen HGM, Timmerman CCM, Kaplan M, Hooker AB, Gijsen AP, van Golde R, van Heteren CF, Sluijmer AV, de Bruin JP, Smeenk JMJ, de Boer JAM, Scheenjes E, Duijn AEJ, Mozes A, Pelinck MJ, Traas MAF, van Hooff MHA, van Unnik GA, de Koning CH, van Geloven N, Twisk JWR, Hompes PGA, Mol BWJ. Oil-Based or Water-Based Contrast for Hysterosalpingography in Infertile Women. N Engl J Med. 2017 May 25;376(21):2043-2052. doi: 10.1056/NEJMoa1612337[↩]
- Youssef MA, Abou-Setta AM, Lam WS. Recombinant versus urinary human chorionic gonadotrophin for final oocyte maturation triggering in IVF and ICSI cycles. Cochrane Database Syst Rev. 2016 Apr 23;4(4):CD003719. doi: 10.1002/14651858.CD003719.pub4[↩]
- Leyendecker G, Struve T, Plotz EJ. Induction of ovulation with chronic intermittent (pulsatile) administration of LH-RH in women with hypothalamic and hyperprolactinemic amenorrhea. Arch Gynecol. 1980;229(3):177-90. doi: 10.1007/BF02108310[↩]
- Leyendecker G, Wildt L. From physiology to clinics–20 years of experience with pulsatile GnRH. Eur J Obstet Gynecol Reprod Biol. 1996 Apr;65 Suppl:S3-12. doi: 10.1016/0301-2115(96)02411-6[↩]
- Leyendecker G, Wildt L, Hansmann M. Pregnancies following chronic intermittent (pulsatile) administration of Gn-RH by means of a portable pump (“Zyklomat”)–a new approach to the treatment of infertility in hypothalamic amenorrhea. J Clin Endocrinol Metab. 1980 Nov;51(5):1214-6. doi: 10.1210/jcem-51-5-1214[↩]
- Crowley WF Jr, McArthur JW. Simulation of the normal menstrual cycle in Kallman’s syndrome by pulsatile administration of luteinizing hormone-releasing hormone (LHRH). J Clin Endocrinol Metab. 1980 Jul;51(1):173-5. doi: 10.1210/jcem-51-1-173[↩]
- Mason P, Adams J, Morris DV, Tucker M, Price J, Voulgaris Z, Van der Spuy ZM, Sutherland I, Chambers GR, White S, et al. Induction of ovulation with pulsatile luteinising hormone releasing hormone. Br Med J (Clin Res Ed). 1984 Jan 21;288(6412):181-5. doi: 10.1136/bmj.288.6412.181[↩][↩]
- Martin KA, Hall JE, Adams JM, Crowley WF Jr. Comparison of exogenous gonadotropins and pulsatile gonadotropin-releasing hormone for induction of ovulation in hypogonadotropic amenorrhea. J Clin Endocrinol Metab. 1993 Jul;77(1):125-9. doi: 10.1210/jcem.77.1.8325934[↩]
- Santoro N, Elzahr D. Pulsatile gonadotropin-releasing hormone therapy for ovulatory disorders. Clin Obstet Gynecol. 1993 Sep;36(3):727-36. doi: 10.1097/00003081-199309000-00029[↩]
- Christin-Maitre S, de Crécy M; Groupe Français des pompes à GnRH. Grossesses obtenues par administration pulsatile de GnRH: résultats d’une large étude rétrospective multicentrique [Pregnancy outcomes following pulsatile GnRH treatment: results of a large multicenter retrospective study]. J Gynecol Obstet Biol Reprod (Paris). 2007 Feb;36(1):8-12. French. doi: 10.1016/j.jgyn.2006.12.001[↩][↩]
- Seminara SB, Beranova M, Oliveira LM, Martin KA, Crowley WF Jr, Hall JE. Successful use of pulsatile gonadotropin-releasing hormone (GnRH) for ovulation induction and pregnancy in a patient with GnRH receptor mutations. J Clin Endocrinol Metab. 2000 Feb;85(2):556-62. doi: 10.1210/jcem.85.2.6357[↩][↩]
- Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, Lombès M, Millar RP, Guiochon-Mantel A, Young J. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009 Jun 25;360(26):2742-8. doi: 10.1056/NEJMoa0900136[↩]
- Francou B, Bouligand J, Voican A, Amazit L, Trabado S, Fagart J, Meduri G, Brailly-Tabard S, Chanson P, Lecomte P, Guiochon-Mantel A, Young J. Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations. PLoS One. 2011;6(10):e25614. doi: 10.1371/journal.pone.0025614[↩]
- Abel BS, Shaw ND, Brown JM, Adams JM, Alati T, Martin KA, Pitteloud N, Seminara SB, Plummer L, Pignatelli D, Crowley WF Jr, Welt CK, Hall JE. Responsiveness to a physiological regimen of GnRH therapy and relation to genotype in women with isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2013 Feb;98(2):E206-16. doi: 10.1210/jc.2012-3294[↩][↩][↩][↩]
- Christou F, Pitteloud N, Gomez F. The induction of ovulation by pulsatile administration of GnRH: an appropriate method in hypothalamic amenorrhea. Gynecol Endocrinol. 2017 Aug;33(8):598-601. doi: 10.1080/09513590.2017.1296948[↩][↩][↩]
- Filicori M, Santoro N, Merriam GR, Crowley WF Jr. Characterization of the physiological pattern of episodic gonadotropin secretion throughout the human menstrual cycle. J Clin Endocrinol Metab. 1986 Jun;62(6):1136-44. doi: 10.1210/jcem-62-6-1136[↩]
- Martin K, Santoro N, Hall J, Filicori M, Wierman M, Crowley WF Jr. Clinical review 15: Management of ovulatory disorders with pulsatile gonadotropin-releasing hormone. J Clin Endocrinol Metab. 1990 Nov;71(5):1081A-1081G. doi: 10.1210/jcem-71-5-1081[↩][↩]
- Letterie GS, Coddington CC, Collins RL, Merriam GR. Ovulation induction using s.c. pulsatile gonadotrophin-releasing hormone: effectiveness of different pulse frequencies. Hum Reprod. 1996 Jan;11(1):19-22. doi: 10.1093/oxfordjournals.humrep.a019015[↩]
- Homburg R, Eshel A, Armar NA, Tucker M, Mason PW, Adams J, Kilborn J, Sutherland IA, Jacobs HS. One hundred pregnancies after treatment with pulsatile luteinising hormone releasing hormone to induce ovulation. BMJ. 1989 Mar 25;298(6676):809-12. doi: 10.1136/bmj.298.6676.809[↩]
- Messinis IE. Ovulation induction: a mini review. Hum Reprod. 2005 Oct;20(10):2688-97. doi: 10.1093/humrep/dei128[↩]
- de Roux N, Young J, Brailly-Tabard S, Misrahi M, Milgrom E, Schaison G. The same molecular defects of the gonadotropin-releasing hormone receptor determine a variable degree of hypogonadism in affected kindred. J Clin Endocrinol Metab. 1999 Feb;84(2):567-72. doi: 10.1210/jcem.84.2.5449[↩]
- Schoot DC, Coelingh Bennink HJ, Mannaerts BM, Lamberts SW, Bouchard P, Fauser BC. Human recombinant follicle-stimulating hormone induces growth of preovulatory follicles without concomitant increase in androgen and estrogen biosynthesis in a woman with isolated gonadotropin deficiency. J Clin Endocrinol Metab. 1992 Jun;74(6):1471-3. doi: 10.1210/jcem.74.6.1592896[↩]
- Couzinet B, Lestrat N, Brailly S, Forest M, Schaison G. Stimulation of ovarian follicular maturation with pure follicle-stimulating hormone in women with gonadotropin deficiency. J Clin Endocrinol Metab. 1988 Mar;66(3):552-6. doi: 10.1210/jcem-66-3-552[↩]
- Shimoda M, Iwayama H, Ishiyama M, Nakatani A, Yamashita M. Successful pregnancy by vitrified-warmed embryo transfer for a woman with Kallmann syndrome. Reprod Med Biol. 2015 Jul 23;15(1):45-49. doi: 10.1007/s12522-015-0214-8[↩]
- Kuroda K, Ezoe K, Kato K, Yabuuchi A, Segawa T, Kobayashi T, Ochiai A, Katoh N, Takeda S. Infertility treatment strategy involving combined freeze-all embryos and single vitrified-warmed embryo transfer during hormonal replacement cycle for in vitro fertilization of women with hypogonadotropic hypogonadism. J Obstet Gynaecol Res. 2018 May;44(5):922-928. doi: 10.1111/jog.13597[↩]
- Hypogonadism Treatment & Management. https://emedicine.medscape.com/article/922038-treatment#d6[↩]
- Baillargeon J, Al Snih S, Raji MA, Urban RJ, Sharma G, Sheffield-Moore M, Lopez DS, Baillargeon G, Kuo YF. Hypogonadism and the risk of rheumatic autoimmune disease. Clin Rheumatol. 2016 Dec;35(12):2983-2987. doi: 10.1007/s10067-016-3330-x[↩]





{kind=link}