Contents
What is imperforate anus
Imperforate anus is also called anal atresia, is a birth defect (congenital) where the opening to the anus is missing or blocked. The anus, also known as the rectum, is the opening at the end of the intestines through which stool (bowel movement) leaves the body. Imperforate anus is a malformation of the anorectal region that may occur in several forms. Imperforate anus (the rectum) may end in a blind pouch that does not connect with the colon, be too narrow (stenotic or atresic) or open into part of the urinary system, female or male reproductive system (the urethra, bladder, or vagina) or other system of body 1. The estimated incidence of imperforate anus is 1 in 1250 to 1 in 5000 live births 2.
Imperforate anus symptoms may include absence of the first stool within 24 to 48 hours after birth, no anal opening, anal opening in an abnormal place, stool coming out from the vagina, base of penis, scrotum, or urethra, and/or swollen belly.
Most children with an anorectal malformation are identified upon routine newborn physical examination. Delayed presentation is often the result of incomplete initial examination. Newborn anorectal and urogenital examination can be technically challenging and makes many practitioners uncomfortable.
Subtle malformations, such as those in some children with perineal fistula that may look normal to the casual glance, may present months or years after birth when the child presents to a primary care provider for constipation or urinary tract infection and appears to have a small perineal body upon physical examination.
Anorectal malformations in females with a normal-appearing anus who have absent vagina or persistent urogenital sinus may go undiagnosed for years because of examiner reluctance to separate the labia during physical examination. These malformations can be discovered upon evaluation for urinary tract infection or primary amenorrhea (absence of menstruation).
Although the exact cause of imperforate anus is not fully understood, it is believed to be due to the abnormal development of the rectum when the embryo is forming inside the womb. Many forms of imperforate anus occur with other birth defects. Imperforate anus may also be part of a syndrome with multiple birth defects. Imperforate anus treatment may include colostomy and surgery to correct the defect. Prognosis depends on the severity and type of imperforate anus and the severity and type of any other birth defects 3.
See your doctor if your baby treated for imperforate anus has abdominal pain or fails to develop any bowel control by the age of 3.
Imperforate anus types
An imperforate anus can be classified into 3 types depending on the relationship between the distal rectal pouch and the puborectalis muscle 4. These are the high type, in which the distal pouch ends above the puborectalis muscle; the intermediate type, in which the pouch ends at the puborectalis muscle; and the low type, in which the pouch ends through the puborectalis muscle. High‐ and intermediate‐type imperforate anuses are often treated surgically in 2 stages, whereas a low‐type imperforate anus requires perineal anoplasty only. Hence, differentiation of the imperforate anus type is helpful for determining the treatment approach and providing accurate information on postsurgical outcomes to parents. Although there are several methods to determine the type of imperforate anus by transperineal sonography in the postnatal period 5, prenatal determination of the imperforate anus type has not been attempted. Most prenatal series reported only high and intermediate types, with only 1 case of a low type 6.
Figure 1. Imperforate anus types
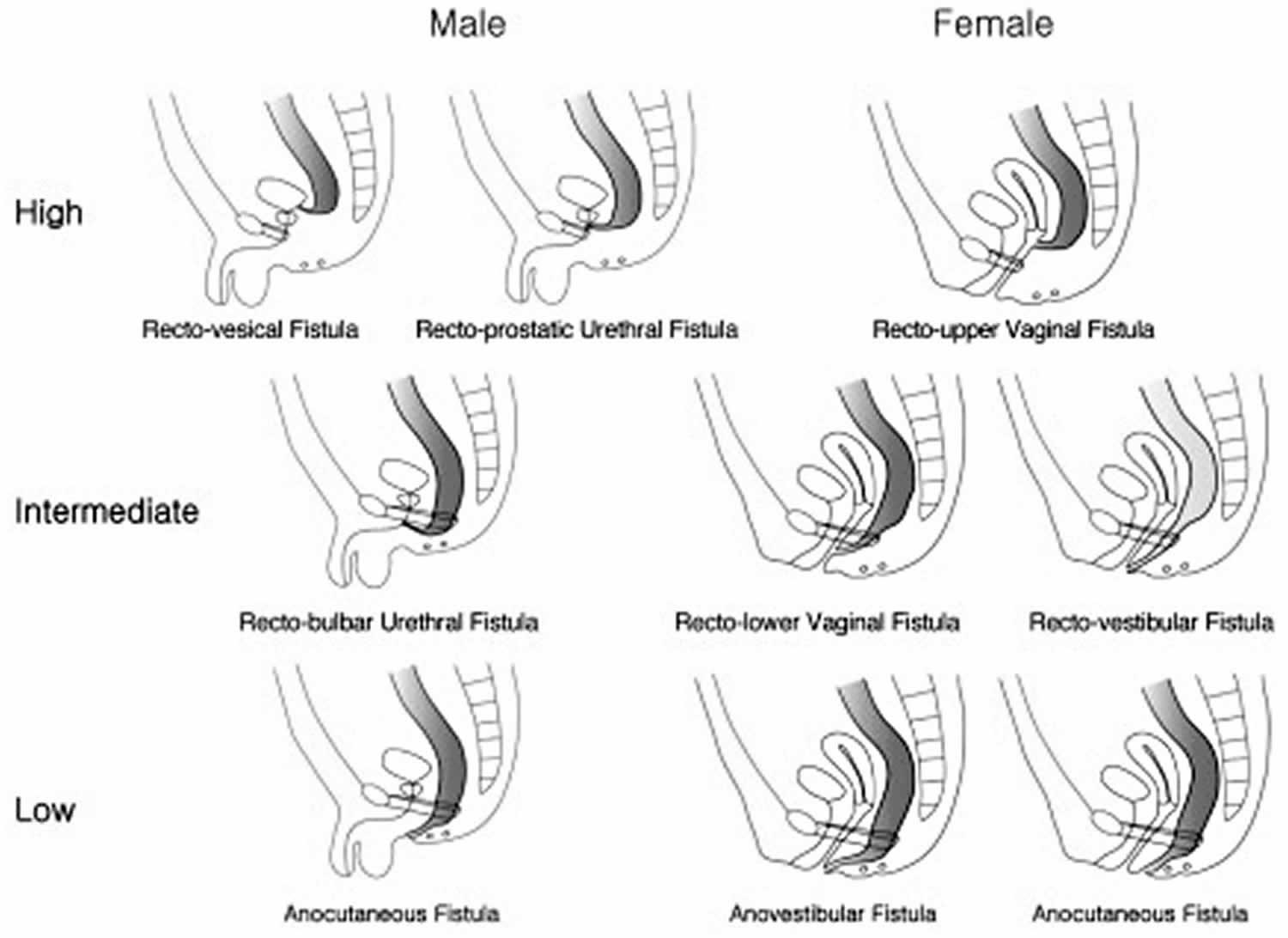
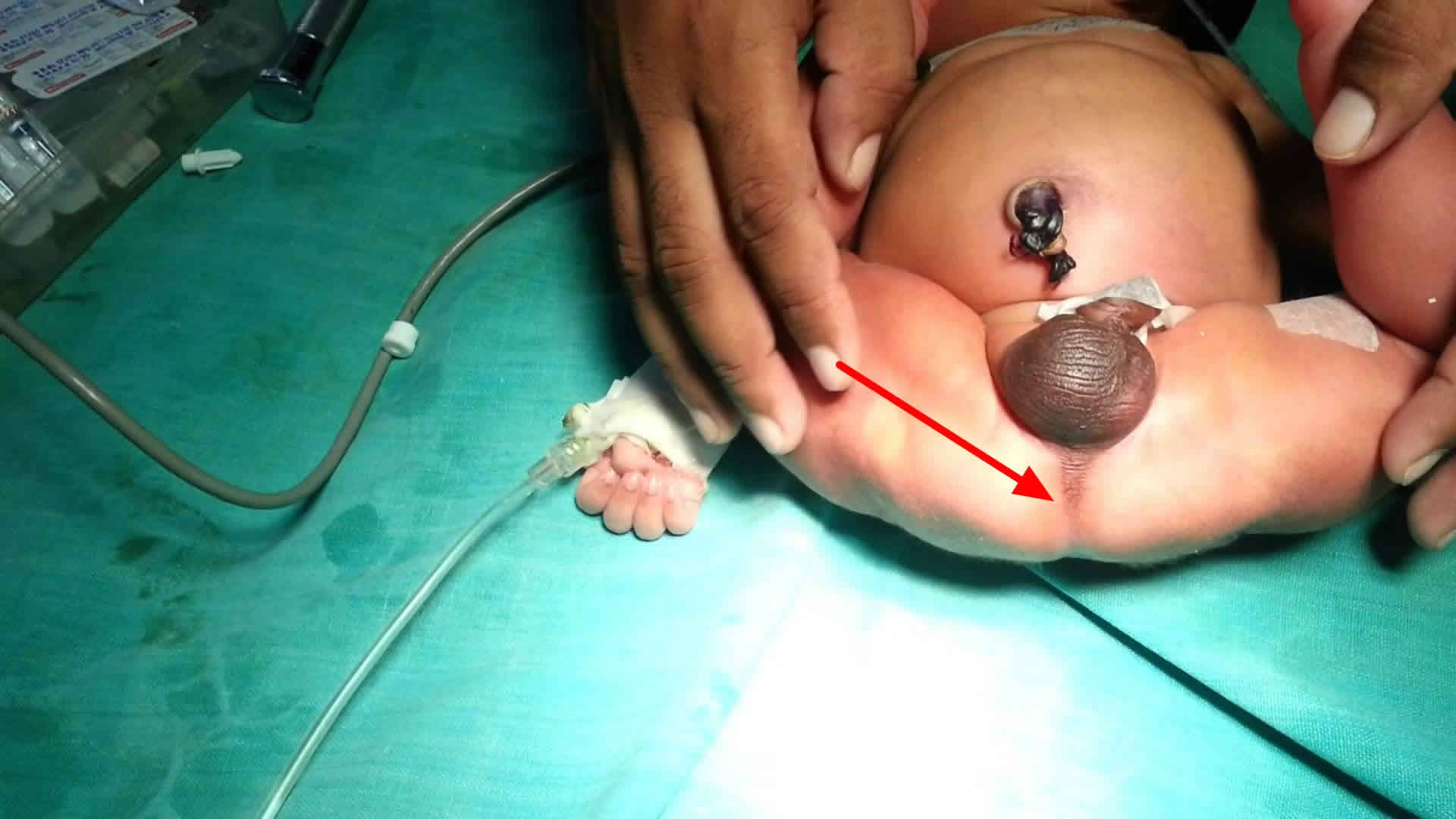
Figure 2. Imperforate anus
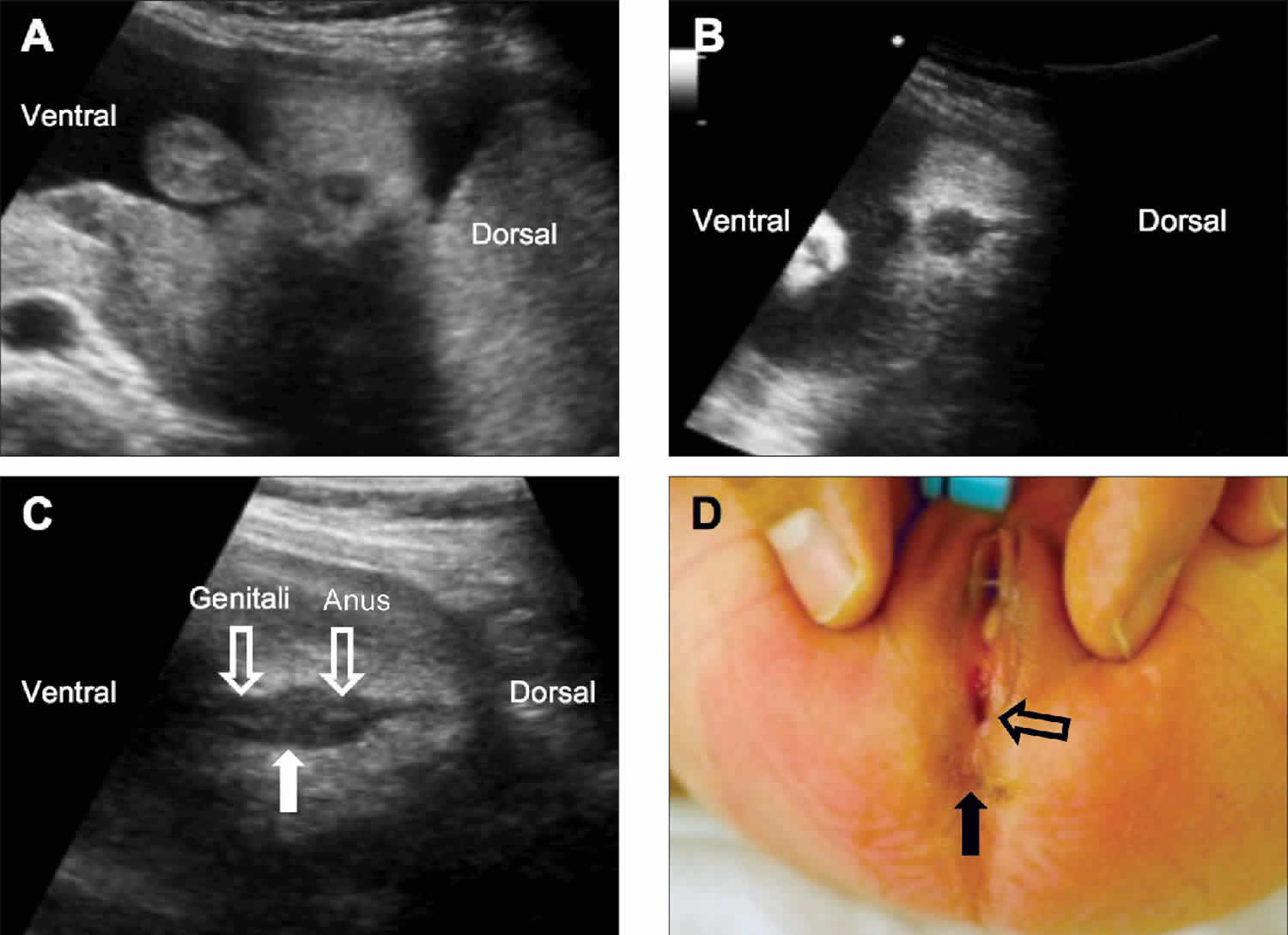
Footnotes: Images from fetuses with a low-type imperforate anus. (A) A 6.2-mm anal sphincter at 33.2 weeks is evident, which is less than 2 SDs from the reference values used in our center. (B) The echogenic center is not visible, which indicates that the anal mucosa is not visualized, although the anal size was normal for the gestational age. (C and D) The genitalia and the anus are shown to be in close proximity without a visible perineal body (C, solid arrow), and the postnatal findings showed no anal opening in the original location (D, solid arrow) and a fistula on the perineal body (D, open arrow).
[Source 7 ]Imperforate anus staging
Imperforate anus was historically classified based on the position of the distal-most aspect of the colon in relation to the levator ani muscles. Malformations at or above the levator muscle complex were defined as high anomalies. Infralevator lesions were termed low and were considered simpler and were associated with better prognosis. This system was based on the now obsolete Wingspread classification.
Information obtained from the posterior sagittal approach has led to an anatomic classification that lists malformations based on their specific anatomy. The following is a list of the most common malformations:
Perineal fistula
This malformation is associated with good prognosis, occurs in either sex, and involves a closed anus with a small connection opening on the perineal body.
Some babies with this malformation have a small loop of skin at the anal opening that resembles a bucket-handle. This is pathognomonic for perineal fistula.
Some boys may have no visible perineal opening but may accumulate mucous or meconium in the fistula, which can extend up the median raphe of the scrotum and resembles a black cord (meconium) or a string of pearls (mucous).
This malformation is amenable to primary neonatal pull-through.
Healthy girls who have normal-sized anal openings and small-appearing perineal bodies do not have perineal fistula. This is easily determined by measuring the size of the anus using Hegar dilators. The anus of an average-term newborn should be approximately 12 mm but varies with the size of the child. The “correct” size of the perineum is largely a matter of experience.
Vestibular fistula
This malformation is associated with good prognosis and is easily diagnosed upon physical examination based on the appearance of a small opening at the posterior aspect of the vestibule. The opening is external to the hymen and is, therefore, not vaginal.
The term vaginal fistula was commonly and incorrectly used to describe vestibular fistula. True solitary congenital rectovaginal fistula is exceedingly rare.
Vestibular fistula is safely treated with diverting colostomy, although some pediatric colorectal surgeons repair this malformation primarily in the newborn period without using colostomy.
Cloaca
Persistent cloaca is a malformation in females that encompasses a spectrum of defects, including the presence of a common channel that incorporates the urethra, vagina, and rectum.
The length of the common channel correlates with complexity and prognosis. Shorter channels (< 3 cm) have fewer associated malformations and carry a better prognosis. Longer channels have more complex malformations and poorer prognosis.
One half of all girls with this malformation have 2 hemivaginas and many have hydrocolpos.
This malformation is easily diagnosed upon physical examination based on the presence of a solitary perineal orifice. Females with this malformation often have very small-appearing labia.
All children with cloacae should undergo colostomy shortly after birth.
Bulbar urethral fistula
This malformation observed in boys is relatively common.
No fistula is observed upon physical examination, and urinalysis often shows meconium.
Colostomy is essential to relieve obstruction, prevent urinary soiling, and to allow for distal colostography, which clarifies the malformation for definitive surgical repair.
Prostatic urethral fistula
This malformation observed in boys is rarer than bulbar fistula and carries a poorer prognosis.
The diagnosis and treatment algorithm are identical to those of bulbar fistula, although the surgical procedures used differ.
Bladder-neck fistula
This rare malformation observed in males (10% of all malformations in males) carries a very poor prognosis.
Most patients with bladder-neck fistula require bowel-management regimens.
The diagnosis and treatment algorithm are identical to those of bulbar and prostatic fistulas, although the surgical procedures used differ.
This fistula is best approached abdominally.
Absent fistula
This malformation can occur in either sex, is somewhat rare, and is associated with a good prognosis. It is commonly associated with trisomy 21.
Diagnosis is primarily by exclusion.
Lateral pelvic radiography is performed in babies who have no external evidence of fistula, who pass no meconium after 24 hours, and who have no meconium in the urine.
If the pelvic rectal pouch is within 1 cm of the anal dimple, a primary pull-through may be performed. In these instances, a fistula is unlikely but should be definitely excluded using the proper surgical technique.
If the surgeon opts for colostomy, the absence of a fistula is confirmed using distal colostography.
Cloacal exstrophy
This extremely rare malformation can occur in either sex but is most common in boys. It encompasses a spectrum that includes variant forms of covered exstrophy. The classic form is devastatingly complex.
Affected children have an omphalocele and a large extrophied cloacal plate on their lower abdominal wall. They have 2 hemibladders separated by an intestinal plate, often with prolapsed terminal ileum that proceeds distally to include an extrophied urethral plate flanked by 2 hemiphallic or hemiclitoral structures.
All children with cloacal exstrophy have some degree of pubic symphysis diastasis and may have a spinal malformation, most commonly myelocystocele.
Thankfully, this complex malformation is rare but it has devastating implications on quality of life.
Imperforate anus causes
The cause of imperforate anus remains unclear. The rectum and anus are believed to develop from the dorsal potion of the hindgut or cloacal cavity when lateral ingrowth of the mesenchyme forms the urorectal septum in the midline. This septum separates the rectum and anal canal dorsally from the bladder and urethra. The cloacal duct is a small communication between the 2 portions of the hindgut. Downgrowth of the urorectal septum is believed to close this duct by 7 weeks’ gestation. During this time, the ventral urogenital portion acquires an external opening; the dorsal anal membrane opens later. The anus develops by a fusion of the anal tubercles and an external invagination, known as the proctodeum, which deepens toward the rectum but is separated from it by the anal membrane. This separating membrane should disintegrate at 8 weeks’ gestation.
Interference with anorectal structure development at varying stages leads to various anomalies, ranging from anal stenosis, incomplete rupture of the anal membrane, or anal agenesis to complete failure of the upper portion of the cloaca to descend and failure of the proctodeum to invaginate. Continued communication between the urogenital tract and rectal portions of the cloacal plate causes rectourethral fistulas or rectovestibular fistulas.
The external anal sphincter, derived from exterior mesoderm, is usually present but has varying degrees of formation, ranging from robust muscle (perineal or vestibular fistula) to virtually no muscle (complex long–common-channel cloaca, prostatic or bladder-neck fistula).
Most cases of imperforate anus are sporadic, with occasional familial forms.
Imperforate anus may occur in several forms:
- The rectum may end in a pouch that does not connect with the colon.
- The rectum may have openings to other structures. These may include the urethra, bladder, base of the penis or scrotum in boys, or vagina in girls.
- There may be narrowing (stenosis) of the anus or no anus.
Imperforate anus is caused by abnormal development of the fetus. Many forms of imperforate anus occur with other birth defects.
Imperforate anus associations
There are frequent associations of imperforate anus with other congenital abnormalities.
- Other atresias: Esophageal atresia 8
- VACTERL association. The acronym VACTERL derives from 9:
- V: vertebral anomalies
- hemivertebrae
- congenital scoliosis
- caudal regression
- spina bifida
- A: anorectal anomalies
- anal atresia
- C: cardiac anomalies; cleft lip
- TE: tracheo-esophageal fistula +/- esophageal atresia
- R: renal anomalies; radial ray anomalies
- L: limb anomalies
- polydactyly
- oligodactyly
- V: vertebral anomalies
- Caudal regression syndrome represents a spectrum of structural defects of the caudal region (sacral agenesis and lower limb hypoplasia). Malformations vary from isolated partial agenesis of the coccyx to lumbosacral agenesis.
- Fistulous tracts to the urethra or vagina may be present or may have a single cloacal opening
Imperforate anus symptoms
Imperforate anus symptoms may include:
- Anal opening very near the vagina opening in girls
- First stool is not passed within 24 to 48 hours after birth
- Missing or moved opening to the anus
- Stool passes out of the vagina, base of penis, scrotum, or urethra
- Swollen belly area
Cardiovascular malformations occur in 12-22% of patients. The most common lesions are tetralogy of Fallot and ventricular septal defects. Transposition of the great arteries and hypoplastic left heart syndrome have been reported but are rare.
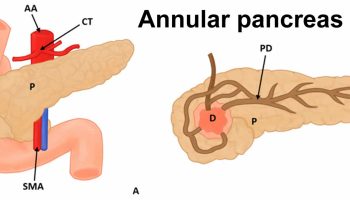
Many gastrointestinal malformations have been described in association with imperforate anus. As many as 10% of patients have tracheoesophageal abnormalities 10. Duodenal obstruction due to annular pancreas or duodenal atresia occurs in a small percentage of patients. Malrotation with Ladd bands that causes obstruction has also been described. Hirschsprung disease has been well described in association with imperforate anus, although the incidence of this combined condition is unknown. Constipation is common.
The association of imperforate anus and vertebral anomalies has been recognized for many years. Patients with high lesions have an increased risk of this association. Lumbosacral anomalies predominate and occur in approximately one third of patients with imperforate anus 11.
The frequency of spinal dysraphism (evaluated with ultrasonography or MRI) had been thought to increase with the severity of the lesion, with higher malformations having greater frequency than lower malformations. Several studies have disputed this and have even shown higher incidence of spinal malformations in children with low malformations. The most common type of dysraphism is tethered spinal cord, which is present in as many as 35% of patients. The normal spinal cord terminates between the first and second lumbar vertebral bodies. In patients with a tethered spinal cord, the cord ends lower in the lumbar spine. Cord lipomas and syringohydromyelia are also common. All lumbosacral spinal malformations negatively affect the child’s prognosis with respect to urinary and fecal incontinence.
Currarino 12 described a triad of sacral defect, presacral mass, and imperforate anus. All patients with an anorectal malformation must be screened for these vertebral abnormalities in the newborn period using sacral radiography and lumbosacral spinal ultrasonography. As many as one half of patients with anorectal malformations have urologic abnormalities. Urinary anomalies are more common in patients with more complex lesions. Improved imaging studies have provided the ability to document an increased range of abnormalities. Mild hydronephrosis is the most common abnormal ultrasonography finding. Vesicoureteric reflux is also a frequent finding, followed by renal agenesis and dysplasia. Cryptorchidism reportedly occurs in 3-19% of males.
Vaginal and uterine abnormalities are common. Bicornate uterus and uterus didelphys occur in 35% of female patients with imperforate anus. A vaginal septum is the most common vaginal abnormality and is seen in as many as one half of girls born with a cloacal malformation. Vaginal duplication and agenesis have also been reported. Vaginal agenesis may be associated with ipsilateral absent ovary and kidney.
Imperforate anus diagnosis
A health care provider can diagnose imperforate anus during a physical exam. The infant should be checked for other problems, such as abnormalities of the genitals, urinary tract, and spine 13.
Imperforate anus in newborns that are missed upon initial examination are often discovered within 24 hours when the newborn is observed to have distention and has failed to pass meconium and a more thorough examination is performed.
The umbilicus should be examined for the absence of an umbilical artery (2-vessel cord), which may suggest an absent kidney. The abdomen should be palpated for masses, which may include a dilated kidney, bladder, hydrocolpos, ectopic kidney, duplication, or other cystic structure.
In males, the testicles must be palpated in the scrotum. The perineum is then examined. Perineal fistulas are diagnosed upon discovery of openings on the perineum, meconium or mucus in a small strip running up into the scrotal median raphe, a perineal groove, or a bucket-handle malformation in the anal dimple skin. If no opening is present, urine is obtained for study, and the child is observed for 24 hours.
In females, a perineal fistula can be directly identified as a small opening on the perineum. If none is present, the labia are separated to search for a vestibular fistula. A fourchette fistula is a type of vestibular fistula that straddles the spectrum of malformation between perineal and vestibular; it is characterized by wet mucosa of the vestibule anteriorly and a dry anoderm posteriorly at the junction of the vestibule and perineum.
If no fistula is visible and only one opening between shortened labia is observed, the child has a cloaca.
If the child has a normal urethra and no vestibular fistula, she may have imperforate anus without fistula. If she appears to have trisomy 21, the likelihood increases that she does not have a fistula. Girls with normal urethra and no visible fistula are observed for 24 hours to allow a perineal fistula to present before operation is required. This waiting period is beneficial in differentiating between children with perineal fistula who may be effectively treated using only a minimal anoplasty from those who require colostomy with further evaluation using distal colostography.
Imaging studies used in imperforate anus include the following:
Sacral radiography: Two views of the sacrum, posteroanterior and lateral, should be obtained to measure sacral ratios and to look for sacral defects, hemivertebrae, and presacral masses. This should be performed before surgery.
Abdominal ultrasonography: This study is specifically used to examine the genitourinary tract and to look for any other masses. Hydronephrosis, hydrocolpos, presacral mass, abdominal mass, or any similar finding can profoundly affect management. This study should be performed before surgery and must be repeated after 72 hours because early ultrasonography findings may be insufficient to rule out hydronephrosis due to vesicoureteral reflux.
Spinal ultrasonography or MRI: All children with any form of anorectal malformation, even those considered minor, should undergo screening for spinal malformations. These lesions can be diagnosed using ultrasonography prior to the ossification of the spine. Ultrasonography should be performed as early as possible but is not essential prior to a newborn surgical procedure. Due to ossification, the use of ultrasound diminishes after 3-4 months of age, and by 6 months MRI is usually necessary. MRI may be required any time an ultrasound is suggestive or nondiagnostic.
Lateral pelvic radiography at 24 hours: Children who could not be diagnosed based solely on physical examination findings, traditionally underwent invertography, which consisted of holding the baby upside down and using lateral radiography to observe the level of gas in the distal rectum. A similar, but more humane, approach is to wait 24 hours after birth to observe for possible maximal pelvic pouch distension and then to use cross-table lateral pelvic radiography with a radio-opaque marker on the anal dimple with the child in the prone position and the hips slightly raised. If the pouch is observed within 1 cm of the marker, some surgeons offer primary repair without colostomy. For pouches farther than 1 cm, colostomy is performed. This 1-cm guideline has been validated only using radiographic measurements and is not directly translatable for measurements made using ultrasonography. Currently, perineal ultrasonography has no role in diagnosis.
Augmented-pressure distal colostography: This is the single most important diagnostic test used to clarify the anatomy in all children with malformations who require colostomy. It is personally performed by the colorectal surgeon in many centers. In a fluoroscopy suite, a balloon catheter is placed into the distal stoma, and the balloon is inflated. The catheter is pulled back, and water-soluble contrast is injected by hand. This pressure is required to overcome the pressure of the levator muscles and to allow the contrast to flow into the lowest part of the colon and reveal any fistula. In patients with a fistula to the urinary tract, the bladder often fills, and the study is continued to obtain as much information as is provided with voiding cystourethrography. If no fistula is present, the distal pouch has a rounded appearance, and no urinary extravasation is visible.
Voiding cystourethrography or micturating cystourethrography: These studies are not necessary if comprehensive distal colostography is performed. They are not recommended for primary evaluation of children with anorectal malformations because of poor sensitivity compared with that of distal colostography.
MRI: All children who have sacral defects on plain radiographs should undergo spine ultrasonography to rule out associated malformations, such as meningocele or meningomyelocele, teratoma, or mixed lesions. If ultrasonography findings are suggestive or nondiagnostic, MRI should be performed. All children who have suggestion of tethered cord on ultrasound, a nondiagnostic ultrasound, or have not had an ultrasound and are older than 6 months should undergo MRI.
CT scanning: CT scanning presently plays no role in the routine evaluation of children with anorectal malformations.
Imperforate anus treatment
Newborns with imperforate anus should not be fed and should receive intravenous hydration. Life-threatening comorbidities take precedence and must be treated first.
If a urinary fistula is suspected, broad-spectrum antibiotics can be administered, although anaerobic coverage is unnecessary within the first 48 hours of life. Any cardiac murmurs identified upon physical examination should be evaluated using echocardiography prior to surgical intervention. The remainder of treatment includes diagnostics and surgical evaluation and management.
Surgery to correct imperforate anus is needed. If the rectum connects with other organs, these organs will also need to be repaired. A temporary colostomy (connecting the end of the large intestine to the abdomen wall so that stool can be collected in a bag) is often needed.
The following consultations may be required:
- Pediatric surgeon: Early consultation with a pediatric surgeon experienced with these anomalies is essential. Ill-conceived procedures during the newborn period may have lifelong consequences for the patient.
- Neurosurgeon: Consultation with a neurosurgeon is warranted if a tethered spinal cord is present (25% of all cases).
- Urologist: The need for consultation with a urologist depends on the malformation and the individual pediatric surgeon.
Imperforate anus surgery
The decision-making process aims to determine which children should undergo primary repair in the neonatal period and which children require colostomy and definitive repair in a staged fashion. Children with anorectal malformations may undergo one or several of the following surgical procedures based on the child’s presentation, physical examination findings, and imaging study findings.
Neonatal colostomy
A colostomy is performed in children who are not amenable to primary pull-through either because of malformation complexity (any urinary fistula in boys, vestibular fistula and cloaca in girls, no fistula in either sex >1 cm from perineal skin) or associated comorbidity.
The colostomy is usually fashioned through a left lower quadrant incision. The colon is divided at the point where the descending colon meets the sigmoid colon, and both ends are brought to the abdominal wall. By fashioning the colostomy at this location, the entire sigmoid colon is kept in place; thus, when the pull-through is eventually performed, a large portion of the colon is available for the surgeon to bring down to the perineal skin.
The mucous fistula (the downstream segment) should be very small, flush with the skin, and far enough from the proximal end to be outside the colostomy appliance (or under the flange) to avoid continued urinary soiling with feces.
During this operation, the distal segment of the colon must be exhaustively irrigated to clean out the impacted meconium, which is always significant. This prevents postdiversion urinary sepsis and allows for effective distal colostography.
Primary neonatal pull-through without colostomy
Many pediatric surgeons opt for primary pull-through in children with perineal fistulas (or no fistulas) and close (< 1 cm) rectal pouches on 24-hour lateral pelvic radiography. Some pediatric surgeons who specialize in colorectal problems often offer the same procedure for girls with vestibular fistulas.
Cystoscopy is usually performed to rule out associated malformation. This is performed immediately prior to the pull-through operation. A Foley catheter is inserted following the cystoscopy.
The preferred surgical approach is the posterior sagittal approach developed by Peña et al. 14:
- The child is placed in the prone position with generous padding under the face and chest and a large bolster under the hips to elevate the area of interest.
- A muscle stimulator is used to show the precise position of the rectal muscle complex to enable exact division at the midline. The midline for this operation is defined by the line that precisely divides the muscle complex in half.
- Dissection proceeds until the rectal pouch is identified. The pouch is then mobilized until adequate length is obtained and the rectum is fully separated from its attachment to the genitourinary tract. Even if the structures do not communicate, they remain intimately associated until fully mobilized. This step ensures that the surgeon does not miss a fistula that was missed on urinalysis findings.
- Once the rectum has been mobilized, the muscle stimulator is used to mark the anterior and posterior limits of the muscle complex. The perineal body is then reconstructed, and the rectum is tacked down in the middle of the muscle complex. The posterior wound is closed and anoplasty is performed.
Posterior sagittal pull-through with colostomy
This approach is used in boys with rectourinary fistula (bulbar, prostatic, or bladder-neck fistula), in girls with cloaca or vestibular fistula, and in patients of either sex who do not have a fistula when the rectal pouch is further than 1 cm on 24-hour lateral prone abdominal radiography.
The approach is also used in children who may have malformations that were amenable to primary neonatal pull-through but were unable to undergo such a procedure because of extreme prematurity or other comorbidity.
Colostomy is performed after 24 hours (or immediately if one of the above diagnoses is made based on either physical examination findings or meconium in the urine).
Several weeks following colostomy, distal colostography is performed, and the specifics of the malformation are clarified.
Cystoscopy is usually performed to clarify anatomy and to rule out associated malformation. This is performed immediately prior to the pull-through operation. A Foley catheter is inserted following the cystoscopy, except in girls with cloaca.
The reconstructive procedure varies based on the malformation, but the essential concepts include identifying and separating the rectum from other structures, dividing and ligating any fistulas, and fully reconstructing the pelvic anatomy with placement of the rectum within the confines of the muscle complex. Procedures for specific malformations are as follows:
- Vestibular fistulas are directly visible but have the longest common wall between the rectum and vagina and require significant delicate mobilization to avoid holes in either structure.
- The posterior sagittal approach is used in boys with bulbar or prostatic urethral fistulas. The rectum is isolated and opened, and the fistula is identified through progressive distal opening. Once the fistula is identified, the rectum proximal to it may be mobilized, and the fistula is then ligated. Reconstruction then proceeds with primary pull-through, as described above.
- Abdominal (open or laparoscopic) and posterior sagittal approaches are best in boys with bladder-neck fistulas because the fistula is best identified in the abdomen.
- Cloaca procedures are complex. A short – common-channel cloaca can be repaired using total urogenital mobilization. The posterior sagittal wound is opened into the cloaca, which is then further proximally opened until the urethral orifice is identified and catheterized. The rectum is then sought. In girls with 2 hemivaginas (50%), the rectum opens in the vaginal septum, although significant asymmetry may be present. After identification, the rectum is separated from the urogenital tract and completely mobilized. The urogenital tract is then mobilized as a solitary structure until the urethral orifice reaches the perineum. This is then reconstructed, and the muscle is marked to enable creation of an adequate vaginal opening and perineal body without impinging on rectal space.
- A long – common-channel cloaca repair often necessitates formal separation of the bladder and vagina, which requires laparotomy and ureteral catheterization. Vaginal replacement is sometimes necessary if the vaginal length is insufficient for reconstruction.
Colostomy closure: Once the wound has completely healed and postoperative dilations have achieved their goal (i.e., the neoanus is at the desired size), the colostomy may be closed in traditional surgical fashion.
Imperforate anus prognosis
Most imperforate anus defects can successfully be corrected with surgery. Most children with mild imperforate anus defects do very well. However, constipation can be a problem.
Children who have more complex surgeries still have control over their bowel movements most of the time. However, they often need to follow a bowel program. This includes eating high-fiber foods, taking stool softeners, and sometimes using enemas.
Some children may need more surgery.
Mortality/Morbidity
Anorectal and urogenital malformations are rarely fatal, although some associated anomalies (cardiac, renal) can be life threatening. Intestinal perforation or postoperative septic complications in a newborn with imperforate anus can result in mortality or severe morbidity. [2]
Morbidity generally arises from the following 2 sources:
Malformation-related morbidity
- Malformation-related morbidity relates to associated malformations of rectal motility, anorectal innervation, and sphincteric musculature. The most common morbidity in this category is constipation. Most children have mild malformations that commonly result in constipation for reasons that remain unclear. If left untreated, chronic constipation results in rectal dilation, which worsens the constipation. This becomes a vicious cycle, which, if untreated, results in fecal impaction and overflow pseudoincontinence, also known as encopresis.
- The most severe forms of malformation-associated morbidity are fecal and urinary incontinence. Higher malformations, such as long–common-channel cloacae and prostatic or bladder-neck fistulas, are associated with poorer nerve and muscle formation, all of which increase the likelihood of fecal or urinary incontinence. Malformations that directly involve urinary sphincteric mechanisms, and, specifically, any malformation in which the rectum or vagina joins the urinary tract at the bladder neck, often results in either urinary incontinence or inability to completely void.
Surgery-related morbidity
- This can include standard complications such as line infections and pneumonia.
- Wound infections or anastomotic breakdowns can occur in any intestinal surgery.
- Children with imperforate anus are at greater risk for injury to surrounding pelvic organs because these organs (such as vagina or urethra and seminal vesicles) are located immediately adjacent to the rectum, and may also be involved in the malformation in some unsuspected way.
- During blind exploration in the pelvis, a dilated ureter can be mistaken for the rectum. Urethras can be opened or transected, and prostates or seminal vesicals can be easily injured. Dissection of these delicate structures can result in ischemia and possible stricture or complete stenosis.
Imperforate anus long term effects
Diet
After the imperforate anus is relieved using colostomy, primary pull-through, or dilation, children do not require special diet. The most common complication of imperforate anus repair is constipation or anal incontinence; therefore, diet can be a crucial part of management. Many patients may require laxatives, enemas, or other medications or irrigations in addition to dietary manipulations 15. Children should avoid constipating foods, such as those included in the bananas, rice, applesauce, and toast (BRAT) diet. High-fiber and laxative foods (whole-grain foods and breads, dairy, fruits, vegetables, greasy foods, spicy foods) should be encouraged. Unfortunately, dietary manipulation is often of limited effectiveness because of the fussy nature of most children regarding diets. Fiber supplements and laxatives can be critically important in avoiding constipation, which can significantly affect prognosis.
Activity
Children with anorectal malformations are often otherwise healthy. Activity limitations are usually related only to the period around their surgical procedures.
Medications
Many children with anorectal malformations require medications for various reasons. Beyond perioperative medications, maintenance medications often include urinary antibiotic prophylaxis or treatment and/or laxatives.
Common laxatives include senna products, milk of magnesia, and propylene glycol solutions (eg, MiraLax, GlycoLax).
Urinary prophylaxis is used to mitigate the risk of urinary infection and urosepsis in children with risk factors for urinary infection such as urinary fistula, vesicoureteral reflux, or continent diversion. Common agents include oral amoxicillin, oral trimethoprim/sulfamethoxazole, and gentamicin bladder irrigations. Comprehensive information on all these medications and others is available in the eMedicine pediatric topic Urinary Tract Infection.
- Imperforate anus. https://medlineplus.gov/ency/article/001147.htm[↩]
- Cuschieri A, EUROCAT Working Group. Descriptive epidemiology of isolated anal anomalies: a survey of 4.6 million births in Europe. Am J Med Genet 2001; 103:207–215.[↩]
- Pediatric Imperforate Anus (Anorectal Malformation) Treatment & Management. https://emedicine.medscape.com/article/929904-treatment#d6[↩]
- Lee, M. , Won, H. , Shim, J. , Lee, P. , Kim, A. , Lee, B. S., Kim, E. A. and Cho, H. J. (2016), Sonographic Determination of Type in a Fetal Imperforate Anus. Journal of Ultrasound in Medicine, 35: 1285-1291. doi:10.7863/ultra.15.08056 https://onlinelibrary.wiley.com/doi/full/10.7863/ultra.15.08056[↩]
- Han TI, Kim IO, Kim WS. Imperforate anus: US determination of the type with infracoccygeal approach. Radiology 2003; 228:226–229.[↩]
- Elchalal U, Yanai N, Valsky DV. Application of 3-dimensional ultrasonography to imaging the fetal anal canal. J Ultrasound Med 2010; 29:1195–1201.[↩]
- Lee, M., Won, H., Kim, A., Lee, P., Lee, B.S., Kim, E.A., & Cho, H.J. (2016). Sonographic Determination of Type in a Fetal Imperforate Anus. Journal of ultrasound in medicine : official journal of the American Institute of Ultrasound in Medicine, 35 6, 1285-91[↩]
- Entezami M, Albig M, Knoll U et-al. Ultrasound Diagnosis of Fetal Anomalies. Thieme. (2003) ISBN:1588902129[↩]
- Stoll C, Alembik Y, Dott B, Roth MP. Associated malformations in patients with anorectal anomalies. Eur J Med Genet 2007; 50:281–290.[↩]
- Rosenbaum DG, Kasdorf E, Renjen P, Brill P, Kovanlikaya A. Sling left pulmonary artery with patent type IIA tracheobronchial anomaly and imperforate anus. Clin Imaging. 2014 Sep-Oct. 38(5):743-6[↩]
- Stathopoulos E, Muehlethaler V, Rais M, Alamo L, Dushi G, Frey P, et al. Preoperative assessment of neurovesical function in children with anorectal malformation: association with vertebral and spinal malformations. J Urol. 2012 Sep. 188(3):943-7.[↩]
- Lee SC, Chun YS, Jung SE, et al. Currarino triad: anorectal malformation, sacral bony abnormality, and presacral mass–a review of 11 cases. J Pediatr Surg. 1997 Jan. 32(1):58-61.[↩]
- Mirza B, Ijaz L, Saleem M, Sharif M, Sheikh A. Anorectal malformations in neonates. Afr J Paediatr Surg. 2011 May-Aug. 8(2):151-4.[↩]
- Pena A, Devries PA. Posterior sagittal anorectoplasty: important technical considerations and new applications. J Pediatr Surg. 1982 Dec. 17(6):796-811.[↩]
- [Guideline] Bischoff A, Levitt MA, Bauer C, Jackson L, Holder M, Pena A. Bowel management for fecal incontinence in patients with anorectal malformations. J Pediatr Surg. 2009 Jun. 44(6):1278-83.[↩]


{kind=link}