Contents
- Lymphangiosarcoma
Lymphangiosarcoma
Lymphangiosarcoma is a rare and aggressive cancer of the lymphatic vessels. When lymphangiosarcoma develops after breast cancer surgery and radiation therapy it is called Stewart-Treves syndrome 1, 2, 3, 4, 5, 6. Stewart-Treves syndrome was first described in 1948 by Dr. Fred W. Stewart, Professor of Pathology, and Dr. Norman Treves, Associate Professor of Surgery, both from the Cornell University Medical College, New York, N.Y., USA 1. The authors described 6 cases of angiosarcomas growing in the field of postmastectomic lymphedema in the upper limb, then called “elephantiasis chirurgica” 1. Lymphangiosarcoma begins in the endothelial cells that line the lymphatic vessels 7. Lymphangiosarcoma can also develop in arms or legs with chronic lymphedema (lymphoedema), a chronic (long-term) condition characterized by swelling (edema) from a build-up of protein-rich lymph fluid in the fatty tissues just under your skin due to a compromised lymphatic system, often affecting an arm or leg 8. Certain cancers and cancer treatments can increase your risk for developing lymphedema. Lymphedema (lymphoedema) symptoms include swelling, a feeling of heaviness or tightness, reduced range of motion, and skin changes. On the other hand, lymphangiosarcoma symptoms include skin changes in the form of purple colored hard nodule or mass on the skin which can become sore and ulcerate in a lymphedematous arm or leg within a median of ten years following breast cancer surgery (mastectomy) 9, 10, 11, 12. Lymphangiosarcoma is an aggressive cancer with a poor prognosis due to the cancer’s high likelihood of spreading to other parts of the body 13, 14. Lymphangiosarcoma has a a 5 year survival of less than 5% even with treatment 4. Amputation of the affected limb is a common treatment option to manage lymphangiosarcoma 13, 15. And in advanced cases where the cancer has spread to other parts of the body chemotherapy is used.
Lymphangiosarcoma incidence is reported to be between 0.07 and 0.45% in breast cancer patients surviving at least 5 years after radical mastectomy 16 and it most commonly affects women aged 65–70 years 17. Lymphangiosarcoma incidence in white American women is estimated to be 1.6 per 100,000 with only 25% of these lymphangiosarcomas originating in the upper limb 18. Extremely rare is lymphangiosarcoma in the lower limbs. Here it occurs most often as a result of idiopathic chronic lymphedema 19, 20, sometimes to the appearance of elephantiasis 21. Currently there are 2 million breast cancer survivors in the United States alone and 20% of them suffer from Breast Cancer Related Lymphedema 22, 23, 24, 25. Although a rare cancer, lymphangiosarcoma incidence rates are steadily increasing with over 1000 new cases per year now diagnosed in the United States 26. Estimates of the prevalence of lymphedema vary widely due to differences in the type of cancer, measurement methods, diagnostic criteria, and timing of evaluations relative to cancer diagnosis and treatment. In a survey conducted in 2006 and 2010, 6,593 cancer survivors were asked to identify ongoing concerns 27. Approximately 20% of respondents reported concerns related to lymphedema. Of these individuals, 50% to 60% reported receiving care for lymphedema 27. These results align reasonably well with a survey study of women survivors of ovarian, endometrial, and colorectal cancers, who met criteria for lymphedema based on a validated survey that demonstrated a point prevalence of 37%, 33%, and 31%, respectively 28. Similarly, a randomized intervention study in women with breast cancer demonstrated, by limb volume measurements or physician diagnosis, that 42% of subjects had lymphedema at 18 months after surgery 29.
The exact cause of lymphangiosarcoma is unknown with the majority of lymphangiosarcoma cases reported so far is lymphangiosarcoma following breast cancer surgery (mastectomy) with prolonged chronic lymphedema. Lymphangiosarcoma also develops in other forms of acquired lymphedema and in congenital lymphedema. Causes for secondary lymphedema may include trauma, surgical invasion of the groin for the treatment of penile or cervical cancer, filariasis, idiopathic acquired lymphedema, vascular stasis, and morbid obesity. There are several theories about the cause of lymphangiosarcoma. Lymphatic blockage, resulting in stimuli for growth factors and cytokines with a consequent growth of vessels and lymphatics is one hypothesis that explains the proliferation of lymphatic vessels in the affected edematous tissue 30. Complete axillary lymph node dissection (a surgical procedure to remove lymph nodes and surrounding tissue from the armpit or axilla to check for cancer spread, most commonly in breast cancer) and radiation therapy seem to be the risk factors for developing Breast Cancer Related Lymphedema progressing to lymphangiosarcoma 13, 31. However, some authors argued against the role of radiation therapy in the development of lymphangiosarcoma because most post radiation chest wall sarcomas are not lymphangiosarcomas 10. Review of literature suggests that surgery, particularly radical mastectomy for breast cancer resulting in Breast Cancer Related Lymphedema is the main predisposing factor 10. Other factors such as idiopathic lymphedema, exposure to toxic chemicals such as vinyl chloride, cardiovascular diseases (heart and blood vessel diseases) and high blood pressure (hypertension) have also been described in relation to lymphangiosarcoma 32, 4, 13, 31.
Although originally described in patients after a radical mastectomy (an outdated and rarely performed surgical procedure that removes the entire breast, the overlying skin, the pectoral (chest) muscles underneath, and the lymph nodes from under the arm), lymphangiosarcoma can occur in the following:
- The setting of congenital or hereditary lymphatic malformations, for example, Turner syndrome, Noonan syndrome, Milroy disease, lymphedema praecox, and lymphedema tarda
- Chronic infections
- Chronic venous stasis
- Morbid obesity
- Malignant obstruction
- Surgical procedures that disrupt the lymphatic flow
Patients most commonly present complaining of pain or discomfort. The range of time for the progression of chronic lymphedema to develop lymphangiosarcoma is between 4 to more than 50 years.
Patients with lymphangiosarcoma require referral to medical and surgical oncologist.
CT scan or MRI of the head and neck will help to establish the extent of bone and soft tissue involvement as well as aid in lymph node evaluation.
- Magnetic resonance imaging (MRI) is recommended to evaluate the local extent of angiosarcomas 33. However, its true value is in question because of poor results in delineating the margin of the tumor. It may be low in signal intensity on T2-weighting and short-tau inversion recovery (STIR) imaging, reflecting the densely cellular, fibrous stroma and sparsely vascularized tumor histology 34, 35. Additional administration of intravenous contrast medium may reveal significant enhancement of the tumorous lesions. However, in patients with chronic lymphedema, nodules detected by MRI within the lymphedema should be evaluated for Stewart-Treves syndrome 36, 37.
- Chest CT scanning should be performed to rule out metastatic disease to the lungs before the patient undergoes extensive surgery.
- Chest X-ray can help in identifying pulmonary metastases and pleural effusion.
- A positron-emission tomographic (PET) scan may document the extent of subcutaneous spread and aid in planning surgical management 38. Fluorodeoxyglucose (FDG) PET/CT scanning may delineate tumor spread, including metastases, and detect the possible malignant transformation in patients with chronic lymphedema 39.
Biopsy
Analysis of a biopsy specimen is essential to the diagnosis of lymphangiosarcoma. Fine-needle aspiration (FNA) is inadequate for diagnosis of Stewart-Treves syndrome.
Histologic Findings
Histologically, lymphangiosarcomas in Stewart-Treves syndrome are indistinguishable from lymphangiosarcomas in nonlymphedematous sites. Post-lymphedema lymphangiosarcomas are characterized by proliferating vascular channels, which dissect the dermal collagen and, often, the obliterate appendages. Tumor endothelial cells lining these channels show marked hyperchromatism and pleomorphism. Mitoses are commonly seen in these tumor cells. The vascular endothelial cells appear round or oval, and they are protuberant and often project into the lumen. Erythrocytes can be seen inside these vascular channels. The overlying epidermis may be hyperkeratotic and acanthotic, or it may be atrophic. Prominent proliferation of reticular fibers can be seen in association with lymphangiosarcoma.
At electron microscopic examination, lymphangiosarcoma cells are surrounded by a complete basal lamina. In some tumor cells, pinocytosis, intercellular junctions, and cytoplasmic intermediate filaments are observed. In addition, Weibel-Palade bodies and erythrophagocytosis are often present. These ultrastructural findings suggest a vascular endothelial origin rather than a lymphatic endothelial origin.
The following immunohistologic and ultrastructural findings can be used to confirm that lymphangiosarcoma originates from blood vessels:
- Antibodies against factor VIII–related antigen are markers for endothelial cells. Although malignant endothelial cells may not always show positive staining with this marker, a more sensitive endothelial marker, lectin Ulex europaeus-I, is more likely to react with hemangiosarcoma tumor cells. However, the specificity of this marker is reduced in people with blood group O because normal epithelial cells and carcinomas also bind this lectin in these individuals.
- CD34 antigen is a marker of vascular endothelial cells and does not react with the lymphatic endothelium.
- Antikeratin antibodies show no evidence of keratin in this malignancy; this finding confirms that the tumor cells are nonepithelial in origin.
- Positive staining for laminin, CD31, collagen IV, and vimentin can aid in diagnosing the tumors as lymphangiosarcomas.
There are not many treatment options due to lack of randomized trials. Initial surgical resection with wide margins has been shown to offer patients the best chance of survival 40, 41. All efforts should be made to achieve negative margins even if it calls for repeat resections. The main problem with obtaining wide margins is that most lesions are relatively extensive at the time of diagnosis. Even when margins are found to be negative by histologic studies, the recurrence rate and risk of metastatic disease remain increased. For this reason, a multispecialty approached is often warranted.
Multimodal therapy including hyperthermic isolated limb perfusion with tumor necrosis factor-alpha (TNF-alpha) and melphalan, combined with radical resection of the affected skin and subcutaneous tissue including the fascia, with large safety margins, may provide enhanced survival 42.
In 2000, Grobmyer and associates 43 found no statistical significant difference in the survival rates of patients treated with chemotherapy compared with those treated with radiation therapy. Although long-term survivors after either radiation therapy or systemic chemotherapy have been reported 44, 45, the overall results have been discouraging. A questionable response to weekly paclitaxel has been described 46. As a result of these findings, these treatment options are reserved for patients with inoperable, advanced disease or those who refuse surgery.
Intra-arterial mitoxantrone or paclitaxel was used for angiosarcoma of the lower limb associated with chronic lymphedema (Stewart-Treves syndrome) in a patient with cervical cancer 47.
Complications from metastatic disease, such as pleural effusions, may require hospitalization of the patient. A CT scan may identify bilateral pulmonary involvement. Stewart-Treves syndrome patients may need further inpatient care for pain control. In 1994, Furue et al 48 demonstrated that immunotherapy may be beneficial as palliative treatment for pleural effusions caused by metastatic angiosarcoma.
Expression of VEGF-C makes angiosarcoma a good potential candidate for targeted antilymphangiogenic therapy 49.
Stewart-Treves syndrome occurring in the abdominal wall was successfully treated with eribulin mesylate, a structurally modified analog of halichondrin B, originally isolated from the marine sponge Halichondria okadai 50.
Figure 1. Lymphangiosarcoma arm
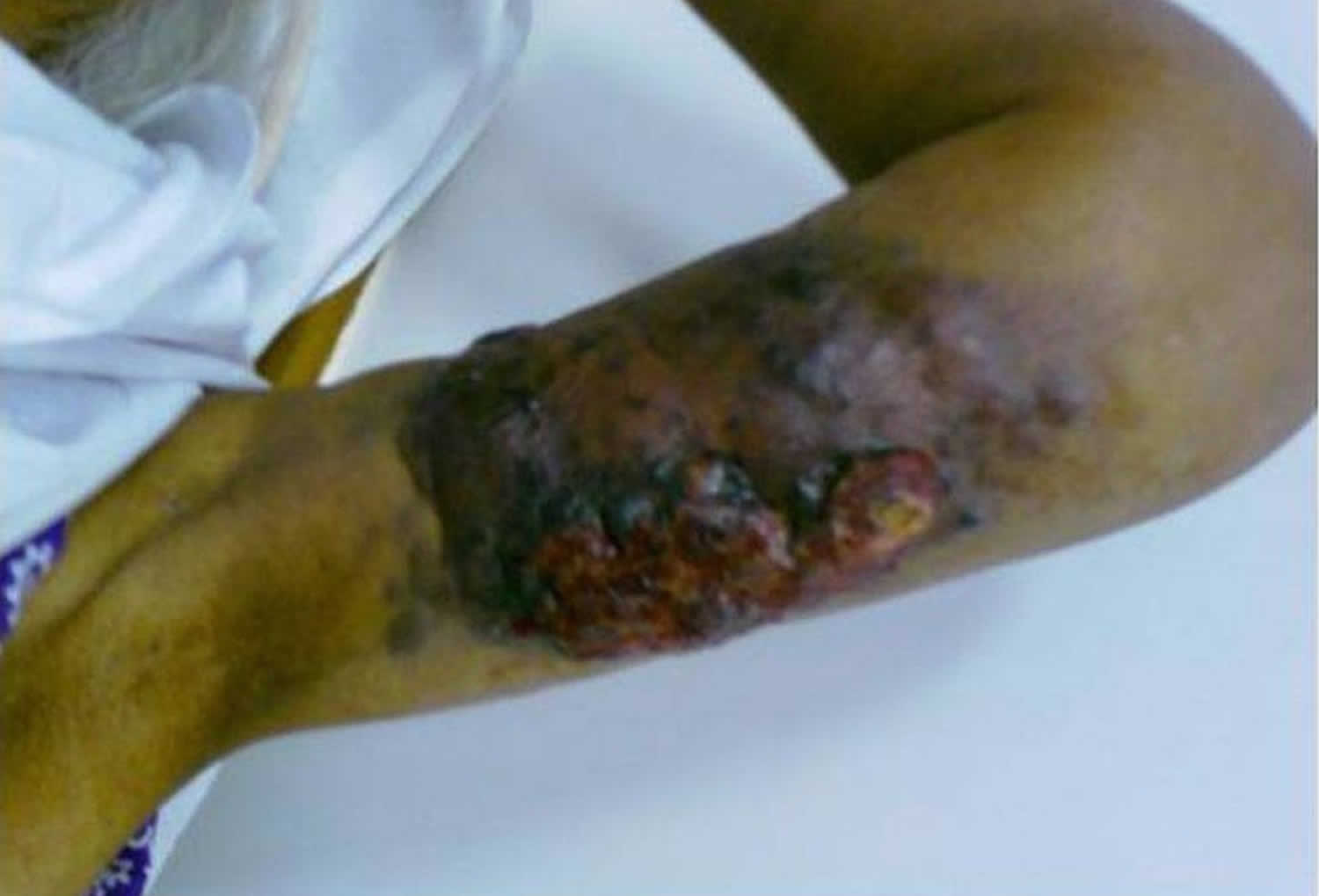
Footnote: Lymphangiosarcoma of the arm presenting with lymphedema in a woman 16 years after mastectomy.
[Source 4 ]Figure 2. Lymphangiosarcoma leg
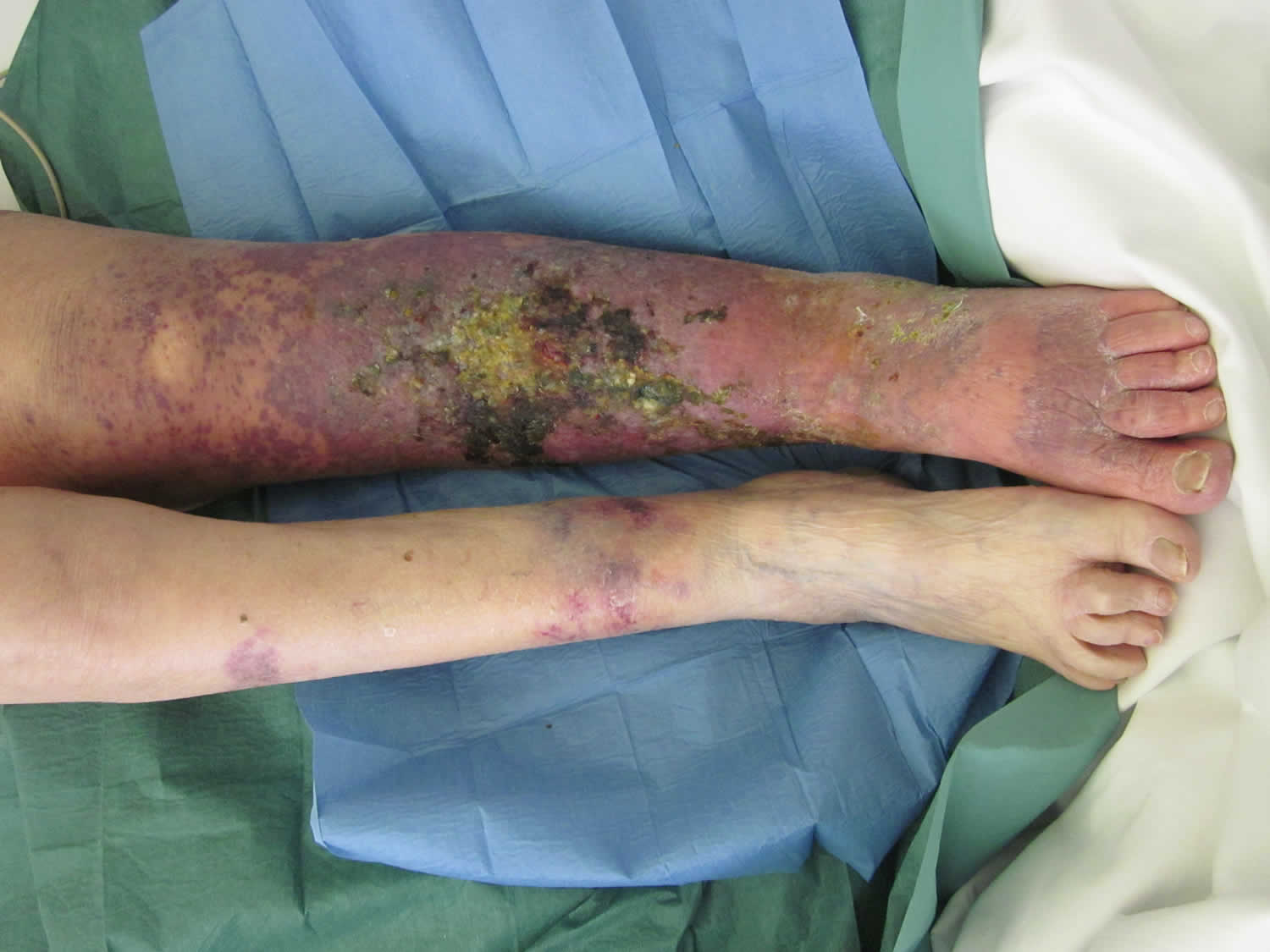
Footnote: An 81-year-old cardiopathic woman with a history of melanoma of the left calf, treated with a radical excision and unilateral inguinal lymph node dissection, presented with a 5-month history of an enlarging ulcerated plaque of coalescing purple papules and bruise-like nodules within a region of chronic lymphedema of the left lower limb. Lymphangiosarcoma developed from well-established lymphedema of the left lower limb.
[Source 41 ]Figure 3. Lymphatic System
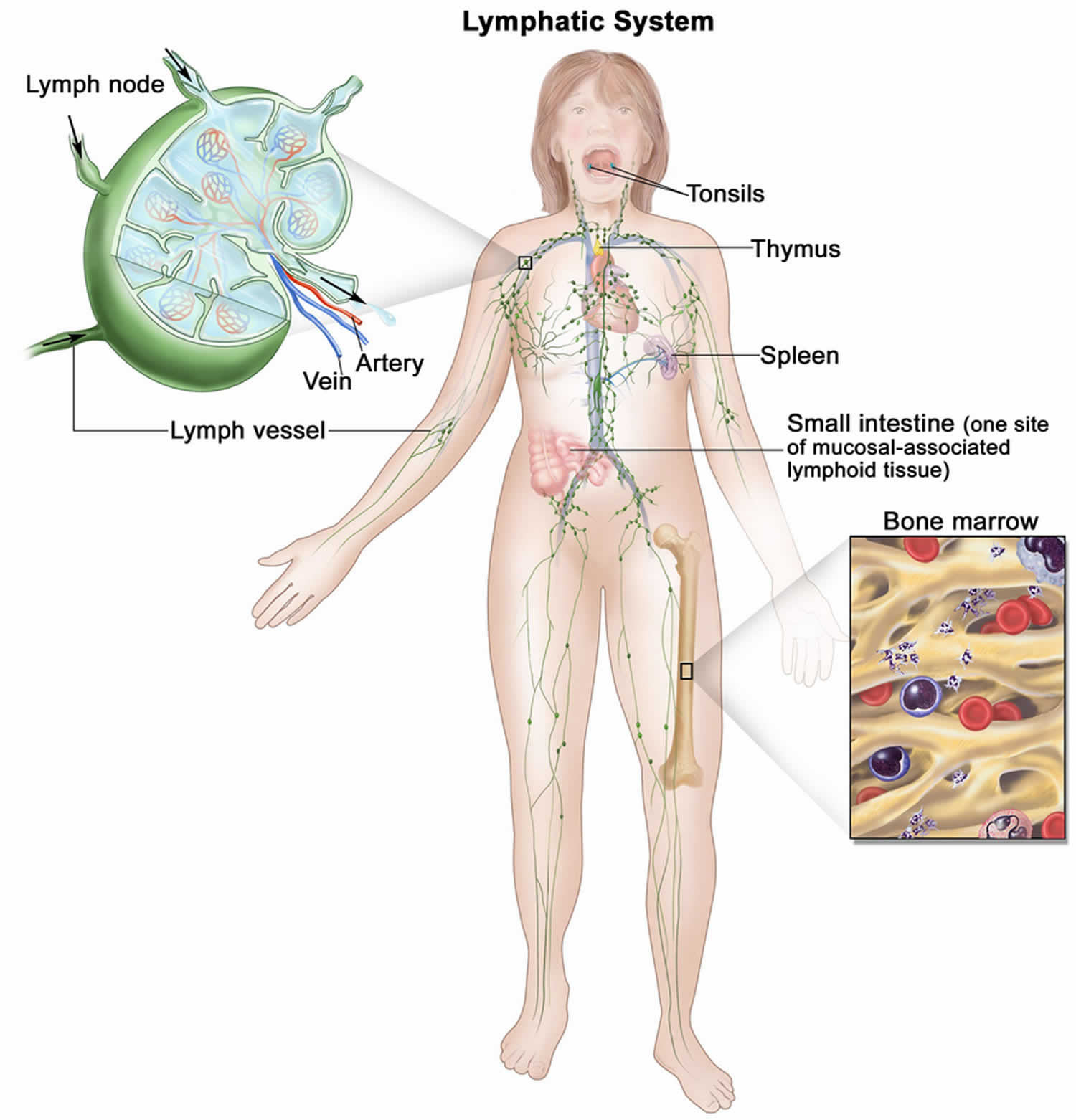
Footnotes: The human lymphatic system generally includes superficial or primary lymphatic vessels that form a complex dermal network of capillary-like channels. Primary lymphatic vessels lack muscular walls and do not have valves. They drain into larger, secondary lymphatic vessels located in the subdermal space. Secondary lymphatic vessels run parallel to the superficial veins and drain into deeper lymphatic vessels located in the subcutaneous fat adjacent to the fascia. Unlike the primary vessels, the secondary and deeper lymphatic vessels have muscular walls and numerous valves to accomplish active and unidirectional lymphatic flow. An intramuscular system of lymphatic vessels that parallels the deep arteries and drains the muscular compartment, joints, and synovium also exists. The superficial and deep lymphatic systems probably function independently, except in abnormal states, although there is evidence that they communicate near lymph nodes 51. Lymph drains from the lower limbs into the lumbar lymphatic trunk. The lumbar lymphatic trunk joins the intestinal lymphatic trunk and cisterna chyli to form the thoracic duct, which empties into the left subclavian vein. The lymphatic vessels of the left arm drain into the left subclavian lymphatic trunk and then into the left subclavian vein. The lymphatic vessels of the right arm drain into the right subclavian lymphatic trunk and then into the right subclavian vein.
[Source 52 ]Figure 4. Lymphedema
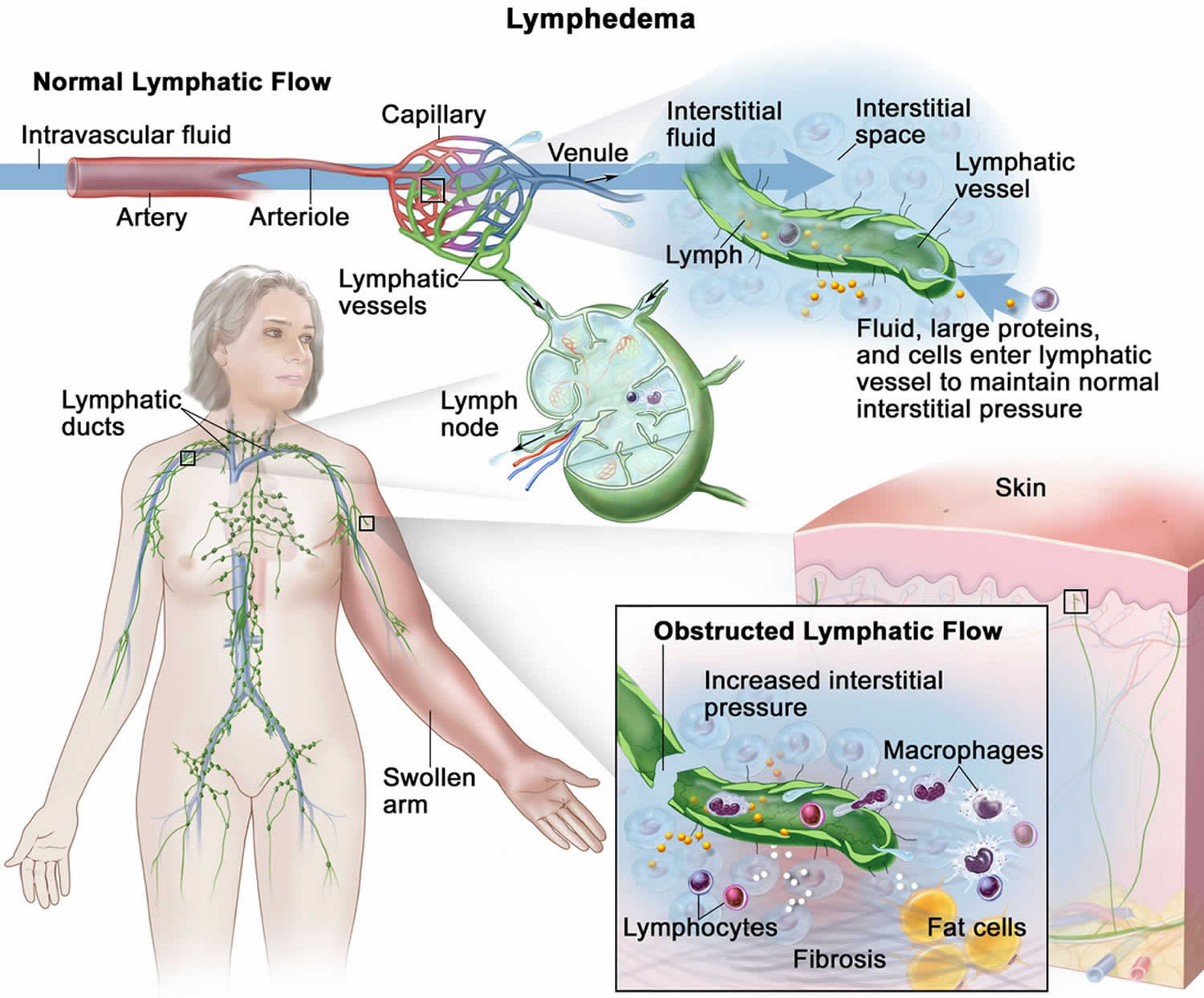
Footnotes: The lymphatic vessels normally maintain normal interstitial pressures by removing the excess interstitial fluid that results from the imbalance between the intravascular fluid that enters from the arterioles and exits into the venules. Large proteins and cells that cannot exit the interstitial space through the venules leave the interstitial fluid through the lymphatic vessels. As the lymph moves through the lymphatic vessels, it passes through lymph nodes and eventually into one of two lymphatic ducts that empty into a large vein near the heart. In lymphedema, the flow of lymph through the lymphatic vessels is disrupted or blocked. This leads to increased interstitial pressure and an accumulation of interstitial fluid, large proteins, and cellular debris in the interstitial space, which induces inflammation. The inflammation may cause further damage to the lymphatic vessels. The macrophages and lymphocytes release inflammatory markers, which causes fibrosis, fat cell hypertrophy, and the classical sign of swelling. Lymphedema may be caused by cancer or cancer treatment.
[Source 52 ]Lymphangiosarcoma causes
The exact cause of lymphangiosarcoma is unknown with the majority of lymphangiosarcoma cases reported so far is lymphangiosarcoma following breast cancer surgery (mastectomy) with prolonged chronic lymphedema. Lymphangiosarcoma also develops in other forms of acquired lymphedema and in congenital lymphedema. Causes for secondary lymphedema may include trauma, surgical invasion of the groin for the treatment of penile or cervical cancer, filariasis, idiopathic acquired lymphedema, vascular stasis, and morbid obesity. Why chronic lymphedema can lead to lymphangiosarcoma is still unclear and controversial, but there are several theories. There are several hypotheses about the cause of lymphangiosarcoma. Lymphatic blockage, resulting in stimuli for growth factors and cytokines with a consequent growth of vessels and lymphatics is one hypothesis that explains the proliferation of lymphatic vessels in the affected edematous tissue 30. Another theory claims that chronic lymphedema causes local immunodeficiency, which indirectly leads to oncogenesis 49. The local immune response in the affected limb is altered by protein-rich interstitial fluid and the lymphatic channels enriched with growth factors, such as vascular endothelial growth factor (VEGF) 5, stimulating lymphangiogenesis and the development of collateral vessels 16. When local immune surveillance mechanisms fail, the lymphedema area becomes an immunologically vulnerable area, predisposed to cancers such as vascular tumors 53 The DNA mutation of the BRCA1 and BRCA2 repair genes also predisposes patients to develop angiosarcoma after breast cancer treatment 54. The interval for developing lymphangiosarcoma in the patient was 8 years 55. Currently, axillary lymph nodes are no longer routinely irradiated after axillary dissection, which is associated with a reduction in the incidence of the chronic lymphedema from 40% to 4% 56. The use of sentinel lymph node biopsy technique is also widespread and the associated risk of lymphedema is further reduced. Therefore, the incidence of lymphangiosarcoma decreased significantly with improved surgical and radiation techniques 19.
Complete axillary lymph node dissection (a surgical procedure to remove lymph nodes and surrounding tissue from the armpit or axilla to check for cancer spread, most commonly in breast cancer) and radiation therapy seem to be the risk factors for developing Breast Cancer Related Lymphedema progressing to lymphangiosarcoma 13, 31. However, some authors argued against the role of radiation therapy in the development of lymphangiosarcoma because most post radiation chest wall sarcomas are not lymphangiosarcomas 10. Review of literature suggests that surgery, particularly radical mastectomy for breast cancer resulting in Breast Cancer Related Lymphedema is the main predisposing factor 10. Other factors such as idiopathic lymphedema, exposure to toxic chemicals such as vinyl chloride, cardiovascular diseases (heart and blood vessel diseases) and high blood pressure (hypertension) have also been described in relation to lymphangiosarcoma 32, 4, 13, 31.
Although originally described in patients after a radical mastectomy (an outdated and rarely performed surgical procedure that removes the entire breast, the overlying skin, the pectoral (chest) muscles underneath, and the lymph nodes from under the arm), lymphangiosarcoma can occur in the following:
- The setting of congenital or hereditary lymphatic malformations, for example, Turner syndrome, Noonan syndrome, Milroy disease, lymphedema praecox, and lymphedema tarda
- Chronic infections
- Chronic venous stasis
- Morbid obesity
- Malignant obstruction
- Surgical procedures that disrupt the lymphatic flow
Patients most commonly present complaining of pain or discomfort. The range of time for the progression of chronic lymphedema to develop lymphangiosarcoma is between 4 to more than 50 years.
What causes lymphedema?
Lymphedema (lymphoedema) can occur when the lymph system is damaged, which can prevent the lymph fluid from moving through your body 52.
The lymph or lymphatic system is part of your body’s immune system. The lymphatic system helps maintain the right balance of fluids in your body, transports immune cells and nutrients, and filters out germs and waste.
- Lymph fluid is the clear fluid inside lymph vessels that travels throughout the body. It contains proteins, salts, water, and white blood cells (infection-fighting cells).
- Lymph vessels carry the lymph fluid throughout the body. One-way valves inside lymph vessels help move the fluid and control the flow.
- Lymph nodes are small, bean-sized glands along the lymph vessels that help filter germs, dead cells, and other waste. Lymph nodes are in many parts of the body, including the neck, armpit, chest, abdomen (belly), and groin.
In some ways, the lymphatic system works like the cardiovascular system (heart and blood vessel system). Both systems transport fluid (blood or lymph) through vessels throughout your body. But your cardiovascular system (heart and blood vessel system) has a powerful pump (the heart) to move blood through the body. The lymph system doesn’t have a pump. Instead, it relies on lymph nodes and the movement of muscles to keep fluid moving. This is why the lymph system is more likely to have poor drainage in some places, especially if lymph nodes are damaged or removed.
Lymphedema occurs when disruption of normal lymphatic drainage leads to accumulation of protein-rich lymph fluid in the interstitial space (the space between cells in tissues). Cancer survivors who experience lymphedema report poor physical functioning, impaired ability to engage in normal activities of daily living, and increased psychological distress 57, 58, 28, 59, 60.
Lymphedema is most common in your arms and legs. But since you have lymph nodes throughout your body, it can develop anywhere.
It’s important to identify and treat lymphedema as early as possible when it’s most likely to be reversible. Lymphedema that isn’t treated can get worse and even become permanent. The goals of lymphedema treatment are to reduce swelling, prevent infection (cellulitis), improve ability to move and function, and relieve discomfort.
Common signs and symptoms of lymphedema can include:
- Swelling, fullness, or heaviness in the breast, chest, shoulder, arm, hand, leg, or foot
- Skin changes such as dryness, discoloration, thickening, or dimpling
- New aching, tingling, numbness, pain, or discomfort
- Less movement or flexibility in your joints
- Trouble putting clothes on, feeling clothes are tighter, or leaving indents on your skin
- Your shirt collar, ring, watch, or bracelet feels tight, even though you haven’t gained weight.
Common treatments for early-stage or mild lymphedema include:
- Elevation of affected area to allow gravity to help drain extra fluid.
- Exercises and movements that promote fluid drainage and improve range of motion.
- Skin and nail care to prevent cuts or injuries to the skin around the affected area, which can make lymphedema worse or lead to cellulitis.
- Manual lymphatic drainage (MLD) is a type of gentle massage that helps move lymph fluid out of the swollen area. A trained therapist does this and might teach you how to do self manual lymphatic drainage (MLD) at home.
- Compression garments are special sleeves or stockings to prevent fluid build-up. A prescription from your doctor is needed. They are fitted to you by a professional and apply different amounts of pressure in different areas. Never wear a compression garment that hasn’t been fitted for you. This can trigger lymphedema or make it worse.
- Promote fluid circulation. Body areas that have damaged or missing lymph nodes can’t move and drain lymph fluid through the area as well. But there are things you can do to promote drainage of lymph fluid:
- Don’t wear tight clothing, jewelry, or other items that constrict or squeeze the affected area.
- If you wear a compression garment, wear it as directed. These garments apply measured and specific amounts of pressure to different areas and are not the same as tight clothing.
- Don’t wear a compression garment that doesn’t fit or that you’ve grown out of. Get re-fitted for a garment every few months.
- Avoid having blood pressure measurements taken on the affected arm, when possible.
- Ask your doctor or therapist what exercises are good for your type and area of lymphedema.
- Raising the affected arm or leg (above the level of your heart if possible) can improve swelling.
- If you have lymphedema in your lower body, avoid crossing your legs.
- Ask how to do manual lymph drainage (MLD) on yourself.
- Note any changes in size, shape, or color of the affected area. Compare to the non-affected side if possible. Tell your doctor right away if you notice any changes.
- Drinking plenty of fluids each day is important for everyone at every age. Drinking enough water helps the body filter and get rid of excess fluid and waste products. Researchers recently studied how people cope with lymphedema. One part of the study looked at their eating and drinking habits. The researchers found:
- About 1 in 3 study participants reported that increasing their daily water intake was helpful for their symptoms.
- About 1 in 4 study participants rated decreased alcohol intake as very helpful in controlling symptoms.
- Water is usually the best choice for daily hydration. In addition to water, choosing unsweetened, low-fat beverages can support fluid intake. Water is also found in many foods, such as vegetables, fruits, broths and soups.
Treating moderate lymphedema
Complete decongestive therapy (CDT) is the combination of manual lymphatic drainage, compression therapy, skin care, exercises, and elevation. CDT is often used to manage mild to moderate lymphedema.
Treating severe lymphedema
For severe lymphedema, intermittent pneumatic compression (IPC) might be added to the treatment plan. Intermittent pneumatic compression (IPC) is a type of compression therapy where a sleeve or stocking is applied to the affected areas and inflated like a blood pressure cuff. It applies specific amounts of pressure to move fluid out of the area. Intermittent pneumatic compression (IPC) might also be used for less severe cases of lymphedema if a person can’t wear compression garments or can’t do manual self-lymphatic drainage.
Surgery might be an option if lymphedema is severe and hasn’t improved with other treatments.
- Liposuction removes extra fat that can develop in areas of lymphedema.
- Lympho-venous or lymphatic bypass takes lymph vessels and attaches them to small veins to improve drainage.
- Vascularized lymph node transfer (VLNT) takes healthy lymph nodes from another part of the body and places them in the area with lymphedema.
Common causes of lymphedema
Any cancer that affects the lymph system can cause lymphedema. But it’s most common in certain cancers including:
- Breast cancer
- Prostate cancer
- Pelvic area cancers (such as bladder, penile, testicular, endometrial, vulvar, or cervical cancer)
- Lymphoma
- Melanoma
- Head and neck cancers
The risk for lymphedema is higher in these cancers because they often require surgery or radiation that involves lymph nodes.
- Surgery or radiation near the abdomen (belly) or pelvis can cause swelling or lymphedema in the abdomen, genitals, or legs.
- Surgery or radiation in the head and neck area can cause swelling or lymphedema in the face and neck.
Common risk factors for developing lymphedema include:
- Extent of local surgery.
- Anatomical location of lymph node dissection.
- Radiation to lymph nodes.
- Localized infection or delayed wound healing.
- Tumor causing lymphatic obstruction of the anterior cervical, thoracic, axillary, pelvic, or abdominal nodes.
- Intrapelvic or intra-abdominal tumors that involve or directly compress lymphatic vessels and/or the cisterna chyli and thoracic duct.
- Having a higher disease stage.
- Overweight (body mass index [BMI] ≥25 kg/m²) or obesity (BMI ≥30 kg/m²) 61
- Black race and Hispanic ethnicity 62
- Rurality 62.
Breast Cancer
A systematic review found the prevalence of lymphedema to be 21.4% (14.9%–29.8%) in patients with breast cancer 63. The incidence increased for up to 2 years after breast cancer diagnosis or surgery, and it was higher in women who underwent axillary lymph node dissection versus sentinel lymph node biopsy (19.9% vs. 5.6%) 63. As a result, not performing axillary lymph node dissection in women with an involved sentinel lymph node is now an accepted practice 52. This practice is a result of a phase 3 randomized study (ACOSOG-Z0011) that showed no difference in overall survival in women who did not undergo a complete axillary dissection, compared with those who did 64. Additional risk factors for lymphedema development included greater number of lymph nodes dissected, having a mastectomy, and overweight or obesity 61, 63. In a prospective study of neoadjuvant chemotherapy (chemotherapy given before surgery) followed by axillary lymph node dissection (ACOSOG-Z1071), the incidence of lymphedema after a median follow-up of 3 years was 37.8% 65. Increasing body mass index (BMI), duration of neoadjuvant chemotherapy (chemotherapy given before surgery), number of lymph nodes removed, and number of involved lymph nodes were associated with lymphedema symptoms 65.
Several risk factors for breast cancer–related lymphedema (BCRL) were demonstrated in a study using data from a 2-year, prospective observational study of 304 patients with breast cancer who had axillary lymph node dissection and radiation therapy 62. Black race and Hispanic ethnicity, receipt of neoadjuvant chemotherapy, older age, and a longer follow-up interval were independently associated with an increased risk for breast cancer–related lymphedema (BCRL) 62.
Another study examined risk factors for breast cancer–related lymphedema (BCRL) related to treatment, comorbidities, and lifestyle in 918 women enrolled in a Prospective Surveillance and Early Intervention (PSEI) trial 66. Women were randomly assigned to either bioimpedance spectroscopy or tape measurement 66. In a secondary analysis, risk factors were used to test for factor associations with outcomes (no lymphedema, subclinical lymphedema, progression to chronic lymphedema after intervention, progression to chronic lymphedema without intervention). Factors associated with BCRL risk included axillary lymph node dissection, taxane-based chemotherapy, regional nodal irradiation, BMI greater than 30 kg/m² and rurality 66.
Gynecological Cancers
A cohort study supports the evidence that a significant proportion of women experience lower-limb lymphedema after treatment of gynecological cancer or colorectal cancer. The highest prevalence (36.5%) among ovarian cancer survivors, followed by endometrial cancer survivors (32.5%) and colorectal cancer survivors (31.4%) 28.
In one study, 802 of 1,774 women diagnosed with a gynecological cancer between 1999 and 2004 responded to a survey about lower limb lymphedema 67. Twenty-five percent of the respondents reported lower-extremity edema; 10% had been diagnosed with lymphedema. Most respondents (75%) had been diagnosed with these conditions within the first year of a cancer diagnosis. Women with vulvar cancer were most likely to have symptoms (36%). Lymph node dissection increased the risk of symptoms in women with cervical cancer but not uterine cancer. The range of symptoms included heavy legs, pain, and skin tightness. Standing all day, long-distance travel, and hot weather were precipitating conditions for lower limb lymphedema. The most common treatments for lower limb lymphedema were compression garments, massage, and lymphatic exercises. The findings related to high prevalence in patients with vulvar cancer and occurrence of symptoms within the first year have been verified 68.
Serial limb volume measurements were obtained from a cohort of women who underwent a lymph node dissection for vulvar (n = 42), endometrial (n = 734), or cervical (n = 138) cancer 4 to 6 weeks after surgery and then every 3 months 69. The incidence of an increase in limb volume of more than 10% was 43% for vulvar cancer, 34% for endometrial cancer, and 33% for cervical cancer. The incidence of severe lymphedema (>40% increase in volume) was less than 2% in all cohorts 69. The peak incidence was at the 4- to 6-week time point, but new cases were identified at all time points. The risk-factor analysis identified a reduced risk in women older than 65 years and a higher risk in women who had more than eight lymph nodes removed in the endometrial cohort 69.
Head and Neck Cancer
Patients with head and neck cancer are susceptible to external and internal lymphedema. External lymphedema typically presents with submental edema or lower neck swelling. Internal lymphedema is more widely distributed in the anatomical regions of the oropharynx. In a small cross-sectional study with video-assisted examinations, 59 of 61 patients had some degree of lymphedema 70. Sixty-one percent of the patients had only internal lymphedema, 35% had internal and external lymphedema, and 4% had only external lymphedema 70. Postoperative radiation therapy was a risk factor for combined lymphedema 70. Chemotherapy was a risk factor for patients with internal lymphedema only 70.
Melanoma
One single-center, cross-sectional study reported on lymphedema after either sentinel lymph node biopsy or lymph node dissection in 435 patients who were treated for melanoma between 1997 and 2015 60. The authors reported a lymphedema prevalence of 25%. Forty-eight patients (44%) had International Society of Lymphology (ISL) stage 1 lymphedema (pitting edema), and 61 patients (56%) had ISL stage 2 or 3 lymphedema 60. Multivariate analyses identified as potential predictive factors the primary site of disease on the affected limb, inguinal surgery, and persistent pain at the site of lymph node surgery. Limb cellulitis was a risk factor for International Society of Lymphology (ISL) stage 2 and 3 lymphedema. The same investigators also reported on health-related quality of life in an earlier publication. In another smaller, single-institution, retrospective study of 66 patients who underwent therapeutic nodal dissection, the rate of permanent lymphedema for inguinal node dissection was 38%, compared with 12% for axillary node dissection 71. Another potentially relevant variable is the type of dissection. A 2017 systematic review did not find an appreciable difference between the rate of lymphedema after therapeutic lymphadenectomy and completion of lymph node sampling after a positive sentinel lymph node biopsy 72. In both instances, the rate was around 20%.
Prostate Cancer
There are few studies of lymphedema after prostate cancer therapy. A small cross-sectional survey of men who underwent radical prostatectomy reported that 19 of 54 respondents (35.2%) had bilateral lower-limb lymphedema 73. Of note, 25 respondents reported that they had received manual lymphatic drainage therapy. Men who did not experience regression experienced more distress related to physical and mental functioning than those who did. An elevated BMI and poor general health were risk factors for lymphedema.
Sarcoma
One study measured patient demographics, surgical outcomes data, functional outcomes, and lymphedema severity with a validated scale for 289 patients who underwent limb preservation surgery of an extremity sarcoma between 2000 and 2007 74. The mean time from surgery was 35 months (range, 12–60 months). Eighty-three patients had some degree of lymphedema, including 58 with mild but definite swelling, 22 with moderate swelling, and 3 with considerable swelling. No patients had grade 4 or very severe swelling with shiny skin and skin cracking. Univariate analysis demonstrated that radiation therapy, tumor size, and tumor depth correlated with severity. The location of the sarcoma (upper or lower extremity), lymph node dissection (yes or no), and BMI did not correlate with severity. Multivariable analysis demonstrated that tumor size was the only independent predictor.
Cancer treatment as a cause of lymphedema
Lymphedema and surgery for cancer
Some surgeries to treat cancer for example, most breast cancer surgeries include removing one or more lymph nodes. Removing lymph nodes is like closing lanes on a highway. Cars can’t get through as easily and traffic slows down. Lymph fluid – like the cars – starts to back up and causes swelling in the body parts that those lymph nodes drain fluid from.
Surgery for breast cancer often involves removing lymph nodes from the armpit area (axillary lymph node dissection), which is why some people get lymphedema in the hand or arm after breast surgery. The more lymph nodes removed, the higher your risk for lymphedema. A sentinel node biopsy usually removes 2 or 3 lymph nodes. An axillary node dissection usually removes between 5 and 30 lymph nodes from the armpit (axilla).
Surgery for other types of cancer might include the removal of lymph nodes in other parts of the body, such as the pelvic area or groin. The risk of lymphedema will depend on the location and number of lymph nodes that must be removed.
Lymphedema and radiation therapy for cancer
Radiation therapy can also cause lymphedema. Radiation can damage or scar nearby lymph nodes. Damaged lymph nodes don’t work well, allowing fluid to back up and cause swelling.
Lymphedema not caused by cancer or cancer treatment
Tumors and enlarged lymph nodes can also cause lymphedema if they are pressing on and blocking the flow of lymph fluid.
Non-cancer causes of lymphedema:
- Infections that damage tissue or cause scarring
- Other health conditions, such as heart or vascular disease, arthritis, and eczema
- Gene changes or mutations that involve the lymph system
- Injury or trauma to a certain part of the body
Lymphedema Stages
If you have lymphedema, your cancer doctor (oncologist) might describe it as being stage 0, 1, 2, or 3.
- Stage 0: No visible swelling, but slight symptoms such as feeling the area is heavy, full, or tight. Reversible.
- Stage 1 (mild): Visible swelling. Area might also feel heavy, full, or tight. If arms or legs are involved, the swelling improves when the arm or leg is raised. Usually reversible.
- Stage 2 (moderate): More swelling than stage 1. If arms or legs involved, swelling doesn’t get better when the arm or leg is raised. Worse symptoms than stage 1. May be reversible if treated early.
- Stage 3 (severe): Extreme swelling that often limits self-care or everyday activities. Skin can be very dry, thickened, or discolored. May have fluid leaking or blisters. Usually not reversible.
Lymphangiosarcoma Pathophysiology
The pathogenic mechanism by which lymphedema may induce angiosarcoma has been the subject of controversy. Stewart and Treves found a high incidence of third cancers in patients with postmastectomy lymphangiosarcoma 1. Therefore, they speculated that a systemic carcinogenic factor was the main causative factor in the pathogenesis of lymphangiosarcomas.
In 1979, Schreiber and others postulated the concept of local immunodeficiency in the presence of lymphedema 75. This theory is supported by experimental evidence. In 1960, Stark and associates demonstrated that homograft skin transplanted to lymphedematous arms survive much longer than those transplanted to healthy arms 76. Therefore, lymphedema may cause some degree of local immunodeficiency and lead to oncogenesis.
The possibility that radiation therapy has an important role in the induction of lymphangiosarcoma is also postulated 77. Sternby et al 78 reported that in their study, the patient with the shortest interval between radical mastectomy and the onset of the tumor (8 months) received both preoperative radiation therapy of the breast and involved axillary lymph nodes followed by fractionated radiation. Others suggest that irradiation is not an essential factor in the pathogenesis of lymphangiosarcoma. Finally, irradiation may be an indirect cause of lymphangiosarcomas because it may cause axillary node sclerosis and thereby accelerate and aggravate the edema.
Clinical data from Swedish women with previous breast cancer who developed angiosarcomas or lymphangiosarcomas on the thoracic wall or upper limb between 1958 and 2008 showed 31 angiosarcomas developed at a median age of 71 years 79. The 14 women treated for breast cancer with radical mastectomy and radiotherapy from 1949-1988 developed angiosarcomas in edematous arms after a median 11 years, whereas 17 females treated by segmental resection, antihormonal treatment, and radiotherapy from 1980-2005 developed angiosarcomas in the irradiated field on the thoracic wall after a median 7.3 years 79.
A hemangiogenic and lymphangiogenic origin of lymphangiosarcoma has been documented 49.
A better understanding of the pathogenesis of angiosarcomas may result in better targeting of effective treatment. Further efforts to influence Vascular Endothelial Growth Factor (VEGF), which is a potent angiogenic factor and has also been shown in angiosarcomas, may improve treatment outcomes in the future 80, 81. Several studies also have shed light on the role of the immune system in patients with angiosarcoma, and these reports show that patients may benefit from anti-PD-1 (programmed death) therapy 82, 83. As for a possible preventive action, the use of lymphatic supermicrosurgery for the treatment of chronic lymphedema, such as vascularised lymph node transfer, or lymphovenous anastomosis, can probably further decrease the incidence of lymphangiosarcoma 84, 85.
Lymphangiosarcoma signs and symptoms
Lymphangiosarcoma can initially appear as a “spreading bruise” or a raised purple-red papule (small raised bump on the skin), eventually developing tissue infiltration, swelling (edema), tumor fungation, ulceration, and even bleeding due to increasing tumor size. The second most common location is in a lymphedematous upper limb secondary to radical mastectomy, known as the Stewart Treves tumor. Median size lesions range from 3 to 6 cm, while untreated lymphangiosarcomas can grow to 20 cm or larger. Although originally described in patients after a radical mastectomy, lymphangiosarcoma can occur in the following:
- The setting of congenital or hereditary lymphatic malformations, for example, Turner syndrome, Noonan syndrome, Milroy disease, lymphedema praecox, and lymphedema tarda
- Chronic infections
- Chronic venous stasis
- Morbid obesity
- Malignant obstruction
- Surgical procedures that disrupt the lymphatic flow
Patients most commonly present complaining of pain or discomfort. The range of time for the progression of chronic lymphedema to develop lymphangiosarcoma is between 4 to more than 50 years.
Lymphangiosarcoma diagnosis
Patients with lymphangiosarcoma require referral to medical and surgical oncologist.
CT scan or MRI of the head and neck will help to establish the extent of bone and soft tissue involvement as well as aid in lymph node evaluation.
- Magnetic resonance imaging (MRI) is recommended to evaluate the local extent of angiosarcomas 33. However, its true value is in question because of poor results in delineating the margin of the tumor. It may be low in signal intensity on T2-weighting and short-tau inversion recovery (STIR) imaging, reflecting the densely cellular, fibrous stroma and sparsely vascularized tumor histology 34, 35. Additional administration of intravenous contrast medium may reveal significant enhancement of the tumorous lesions. However, in patients with chronic lymphedema, nodules detected by MRI within the lymphedema should be evaluated for Stewart-Treves syndrome 36, 37.
- Chest CT scanning should be performed to rule out metastatic disease to the lungs before the patient undergoes extensive surgery.
- Chest X-ray can help in identifying pulmonary metastases and pleural effusion.
- A positron-emission tomographic (PET) scan may document the extent of subcutaneous spread and aid in planning surgical management 38. Fluorodeoxyglucose (FDG) PET/CT scanning may delineate tumor spread, including metastases, and detect the possible malignant transformation in patients with chronic lymphedema 39.
Biopsy
Analysis of a biopsy specimen is essential to the diagnosis of lymphangiosarcoma. Fine-needle aspiration (FNA) is inadequate for diagnosis of Stewart-Treves syndrome.
Histologic Findings
Histologically, lymphangiosarcomas in Stewart-Treves syndrome are indistinguishable from lymphangiosarcomas in nonlymphedematous sites. Post-lymphedema lymphangiosarcomas are characterized by proliferating vascular channels, which dissect the dermal collagen and, often, the obliterate appendages. Tumor endothelial cells lining these channels show marked hyperchromatism and pleomorphism. Mitoses are commonly seen in these tumor cells. The vascular endothelial cells appear round or oval, and they are protuberant and often project into the lumen. Erythrocytes can be seen inside these vascular channels. The overlying epidermis may be hyperkeratotic and acanthotic, or it may be atrophic. Prominent proliferation of reticular fibers can be seen in association with lymphangiosarcoma.
At electron microscopic examination, lymphangiosarcoma cells are surrounded by a complete basal lamina. In some tumor cells, pinocytosis, intercellular junctions, and cytoplasmic intermediate filaments are observed. In addition, Weibel-Palade bodies and erythrophagocytosis are often present. These ultrastructural findings suggest a vascular endothelial origin rather than a lymphatic endothelial origin.
The following immunohistologic and ultrastructural findings can be used to confirm that lymphangiosarcoma originates from blood vessels:
- Antibodies against factor VIII–related antigen are markers for endothelial cells. Although malignant endothelial cells may not always show positive staining with this marker, a more sensitive endothelial marker, lectin Ulex europaeus-I, is more likely to react with hemangiosarcoma tumor cells. However, the specificity of this marker is reduced in people with blood group O because normal epithelial cells and carcinomas also bind this lectin in these individuals.
- CD34 antigen is a marker of vascular endothelial cells and does not react with the lymphatic endothelium.
- Antikeratin antibodies show no evidence of keratin in this malignancy; this finding confirms that the tumor cells are nonepithelial in origin.
- Positive staining for laminin, CD31, collagen IV, and vimentin can aid in diagnosing the tumors as lymphangiosarcomas.
Lymphangiosarcoma Stages
After someone is diagnosed with a lymphangiosarcoma, doctors will try to figure out if it has spread, and if so, how far. This process is called staging. The stage of a cancer describes how much cancer is in the body. It helps determine how serious the cancer is and how best to treat it. Doctors also use a cancer’s stage when talking about survival statistics. Lymphangiosarcoma tends to metastasize by lymphatic or hematogenous pathways, and 20% to 45% of patients have metastatic disease at presentation. The lungs are the most common site of metastasis and can present as pleural disease, hemorrhagic pleural effusion, or pneumothorax. Other common sites include the liver, bone, soft-tissue, and lymph nodes.
Lymphangiosarcoma staging is based on the International Union Against Cancer and American Joint Committee on Cancer (AJCC) system using the TNM system, which is based on 4 key pieces of information:
- The extent of the tumor (T): How large is the cancer?
- The spread to nearby lymph nodes (N): Has the cancer spread to nearby lymph nodes?
- The spread (metastasis) to distant sites (M): Has the cancer spread to distant organs such as the lungs?
- The grade (G) of the cancer: How much do the sarcoma cells look like normal cells?
The stages of lymphangiosarcomas range from stages I (1) through IV (4). As a rule, the lower the number, the less the cancer has spread. A higher number, such as stage IV, means cancer has spread more. And within a stage, an earlier letter means a lower stage. Although each person’s cancer experience is unique, cancers with similar stages tend to have a similar outlook and are often treated in much the same way.
In 1959, McConnell and Haslam divided the course of development of lymphangiosarcoma into 3 stages 86. This staging system lacks universal application.
- Stage 1 (prolonged lymphedema):
- This stage is characterized by extensive edema that causes the degeneration of fat and collagen mainly in the deep part of the dermis.
- Edema separates the collagen bands, creating a misperception of an increased amount of fibrous tissue in the area.
- Stage 2 (premalignant angiomatosis):
- This stage involves multiple foci of small, proliferating channels in the dermis and subdermis. These vessels are lined by hyperplastic endothelial cells, as well as normal, flattened cells.
- The areas of angiomatosis vary in size, ranging from 100 µm to a couple of centimeters in diameter.
- Superficial areas can be seen as bruises or vesicles, whereas deeper areas are seen as areas of induration and hemorrhage.
- Early lesions show little evidence of malignancy, but more advanced lesions reveal early malignant transformation with an increased number of mitotic figures and pleomorphic cells.
- Stage 3 (frankly malignant angiosarcoma):
- These aggressive tumors develop from areas of premalignant angiomatosis.
- The histologic features of this malignancy are described above in stage 2.
The grade is partly used to determine the stage of a sarcoma. The staging system divides sarcomas into 3 grades (1 to 3). The grade of a sarcoma helps predict how rapidly it will grow and spread. It’s useful in predicting a patient’s outlook and helps determine treatment options.
The grade of a sarcoma is determined using a system known as the French or FNCLCC system, and is based on 3 factors:
- Differentiation: Cancer cells are given a score of 1 to 3, with 1 being assigned when they look a lot like normal cells and 3 being used when the cancer cells look very abnormal. Certain types of sarcoma are given a higher score automatically.
- Mitotic count: How many cancer cells are seen dividing under the microscope; given a score from 1 to 3 (a lower score means fewer cells were seen dividing)
- Tumor necrosis: How much of the tumor is made up of dying tissue; given a score from 0 to 2 (a lower score means there was less dying tissue present).
Each factor is given a score, and the scores are added to determine the grade of the tumor. Sarcomas that have cells that look more normal and have fewer cells dividing are generally placed in a low-grade category. Low-grade tumors tend to be slow growing, slower to spread, and often have a better outlook (prognosis) than higher-grade tumors. Certain types of sarcoma are automatically given higher differentiation scores. This affects the overall score so much that they are never considered low grade. Examples of these include synovial sarcomas and embryonal sarcomas. Here’s what the grade numbers mean:
- GX: The grade cannot be assessed (because of incomplete information).
- Grade 1 (G1): Total score of 2 or 3
- Grade 2 (G2): Total score of 4 or 5
- Grade 3 (G3): Total score of 6, 7 or 8
Table 1. Trunk and Extremities Sarcoma Stages
| AJCC stage | Stage grouping | Trunk and Extremities Sarcoma Stage description* |
|---|---|---|
| IA | T1 N0 M0 G1 or GX | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| IB | T2, T3, T4 N0 M0 G1 or GX | The cancer is: Larger than 5 cm but not more than 10 cm (T2) OR Larger than 10cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| II | T1 N0 M0 G2 or G3 | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIA | T2 N0 M0 G2 or G3 | The cancer is larger than 5 cm (2 inches) but not more than 10 cm (T2). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIB | T3 or T4 N0 M0 G2 or G3 | The cancer is: Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IV | Any T N1 M0 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has not spread to distant sites (M0). It can be any grade. |
| OR | ||
| Any T Any N M1 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has spread to distant sites such as the lungs (M1). It can be any grade. | |
Footnotes: * The following categories are not listed in the table above:
- TX: Main tumor cannot be assessed due to lack of information.
- T0: No evidence of a primary tumor.
- NX: Regional lymph nodes cannot be assessed due to lack of information.
Table 2. Retroperitoneum Sarcoma Stages
| AJCC stage | Stage grouping | Retroperitoneum Sarcoma Stage description* |
|---|---|---|
| IA | T1 N0 M0 G1 or GX | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| IB | T2, T3, T4 N0 M0 G1 or GX | The cancer is: Larger than 5 cm but not more than 10 cm OR Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 1 (G1) or the grade cannot be assessed (GX). |
| II | T1 N0 M0 G2 or G3 | The cancer is 5 cm (2 inches) or smaller (T1). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIA | T2 N0 M0 G2 or G3 | The cancer is larger than 5 cm (2 inches) but not more than 10 cm (T2). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| IIIB | T3 or T4 N0 M0 G2 or G3 | The cancer is: Larger than 10 cm but not more than 15 cm (T3) OR Larger than 15 cm (T4). It has not spread to nearby lymph nodes (N0) or to distant sites (M0). The cancer is grade 2 (G2) or grade 3 (G3). |
| OR | ||
| Any T N1 M0 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has not spread to distant sites (M0). It can be any grade. | |
| IV | Any T Any N M1 Any G | The cancer is any size (Any T) AND it has spread to nearby lymph nodes (N1). It has spread to distant sites such as the lungs (M1). It can be any grade. |
Footnotes: * The following categories are not listed in the table above:
- TX: Main tumor cannot be assessed due to lack of information.
- T0: No evidence of a primary tumor.
- NX: Regional lymph nodes cannot be assessed due to lack of information.
Lymphangiosarcoma Differential Diagnoses
Lymphangiosarcoma differential diagnoses include:
- Acquired Angioedema Due to C1 Inhibitor Deficiency
- Anaplastic Kaposi sarcoma 88, 89
- Angioendotheliomatosis
- Angiolymphoid Hyperplasia With Eosinophilia
- Melanoma
- Dermatologic Manifestations of Kaposi Sarcoma
- Dermatologic Manifestations of Metastatic Carcinomas
- Hereditary Angioedema
- Lymphangiectasia
- Lymphangioma
- Lymphocytoma Cutis
- Telangiectatic metastatic breast disease to the skin
- Hemangioendotheliomas
- Hemangiopericytomas
- Some authors have suggested a distinction between Stewart-Treves syndrome and cutaneous postradiation angiosarcoma 90
- Other lymphedema-related neoplasms 91
Lymphangiosarcoma treatment
There are not many treatment options due to lack of randomized trials. Initial surgical resection with wide margins has been shown to offer patients the best chance of survival 40. All efforts should be made to achieve negative margins even if it calls for repeat resections. The main problem with obtaining wide margins is that most lesions are relatively extensive at the time of diagnosis. Even when margins are found to be negative by histologic studies, the recurrence rate and risk of metastatic disease remain increased. For this reason, a multispecialty approached is often warranted.
Multimodal therapy including hyperthermic isolated limb perfusion with tumor necrosis factor-alpha (TNF-alpha) and melphalan, combined with radical resection of the affected skin and subcutaneous tissue including the fascia, with large safety margins, may provide enhanced survival 42.
In 2000, Grobmyer and associates 43 found no statistical significant difference in the survival rates of patients treated with chemotherapy compared with those treated with radiation therapy. Although long-term survivors after either radiation therapy or systemic chemotherapy have been reported 44, 45, the overall results have been discouraging. A questionable response to weekly paclitaxel has been described 46. As a result of these findings, these treatment options are reserved for patients with inoperable, advanced disease or those who refuse surgery.
Intra-arterial mitoxantrone or paclitaxel was used for angiosarcoma of the lower limb associated with chronic lymphedema (Stewart-Treves syndrome) in a patient with cervical cancer 47.
Complications from metastatic disease, such as pleural effusions, may require hospitalization of the patient. A CT scan may identify bilateral pulmonary involvement. Stewart-Treves syndrome patients may need further inpatient care for pain control. In 1994, Furue et al 48 demonstrated that immunotherapy may be beneficial as palliative treatment for pleural effusions caused by metastatic angiosarcoma.
Expression of VEGF-C makes angiosarcoma a good potential candidate for targeted antilymphangiogenic therapy 49.
Stewart-Treves syndrome occurring in the abdominal wall was successfully treated with eribulin mesylate, a structurally modified analog of halichondrin B, originally isolated from the marine sponge Halichondria okadai 50.
Roy et al 92 report cases of five lymphangiosarcoma patients treated in their center. In four of them, forequarter amputation was performed and they were alive and disease-free at 3, 16, 23 and 135 months 92. One patient declined amputation, and isolated limb perfusion using melphalan and TNF-alpha was performed followed by débridement of residual tumor, but finally palliative forequarter amputation was performed because of recurrent disease 92. She died from metastatic oesophageal carcinoma. On the other hand, Farzaliyev et al 93 describe two cases in which they performed the radical subfascial skin excision of the affected arm followed by mesh skin graft transplantation from both thighs. In both cases the axillary hyperthermic isolated limb perfusion (HILP) with TNF-alpha and high-dose melphalan was used (in one case as a non-curative, in the other as a neoadjuvant treatment method) 93. A complete pathological response was observed after neoadjuvant treatment (chemotherapy treatment given before surgery), no response was observed after non-curative treatment. The first patient had no evidence of disease 18 months after surgery, the latter patient died 18 months after the first presentation of disease. McKeown et al 94 report the case of a patient with lymphangiosarcoma developing 15 years after the initial diagnosis of breast cancer, who refused a surgical procedure and was treated with doxorubicin. She also died in 18 months 94. Regarding other treatment modalities, there is no difference in overall survival when using radiation therapy or chemotherapy.
Surgery
Early amputation or wide local excision provides the best chance of long-term survival in patients with Stewart-Treves syndrome 41, 95. Some authorities favor radical ablative surgery with an early diagnosis, in order to confer a reasonable prognosis with this rare but aggressive disease 96, 95.
The most common approach in patients with lymphangiosarcoma is amputation of the limb or forequarter surgery rather than wide local surgical excision. Forequarter surgery also called Forequarter Amputation is a radical surgical procedure to remove the entire upper limb, including the shoulder girdle (scapula and clavicle), typically for extensive malignant tumors of the shoulder, arm, and axilla 97, 98.. Even in cases with early surgical treatment, the prognosis is disappointing, with a high rate of local recurrence and metastasis. Metastatic disease should exclude surgical treatment unless surgery is useful for symptomatic improvement.
Chemotherapy, immunotherapy, and/or radiation therapy can be used as adjuvants (add-ons) to surgery
Radiation therapy
Due to high risk of local recurrence, adjuvant (add-on) radiotherapy with large doses and wide treatment fields is most-often recommended, except in cases where lymphangiosarcomas are radiation-induced. Radiotherapy may be palliative, but unfortunately, does not improve survival.
Chemotherapy
Chemotherapy with single-agent doxorubicin or paclitaxel is the treatment of choice for advanced regional or metastatic disease. Promising studies have shown that bevacizumab acts as a tumor stabilizer and may aid in decreasing the progression rate of the disease. Isolated reports have shown successful outcomes with thalidomide therapy. Even in cases of early surgical intervention, prognosis remains poor, with a high rate of local recurrence and metastasis.
Among available chemotherapy, 5-fluorouracil, methotrexate, bleomycin, actinomycin D, vincristine, doxorubicin, metronomic cyclophosphamide with prednisolone, paclitaxel, ifosfamide and dacarbazine can be used therapeutically 16, 20, 54, 93, 99. Some authors favor the use of paclitaxel as a first-line treatment of the advanced or metastatic angiosarcoma. Paclitaxel with or without bevacizumab, VEGF inhibitor, was tested in a randomized phase 2 trial 100. The study found that both paclitaxel with and without bevacizumab were supported as active treatment regimens, although they did not find a benefit of adding bevacizumab. However, bevacizumab as a monotherapy is an attractive option for a second-line treatment 101. Pazopanib, a multityrosine kinase inhibitor, also showed promising results and should also be considered as one of the second-line treatment options 102. The use of neoadjuvant hyperthermic isolated limb perfusion (HILP) has also the potential to improve local control 93, 92, 103.
Lymphangiosarcoma prognosis
Lymphangiosarcoma prognosis is poor with a high number of local and distant recurrences 104, 105. Most patients die of metastatic disease within 2 years 16, 20, 54.
Good prognostic factors have been found to be age (younger than 50 years), localized tumor stage, and anatomical site (trunk). The reason for increased survival in patients with lymphangiosarcomas of the trunk is unclear. Lymphangiosarcomas are extremely aggressive tumors with a high local recurrence rate and a tendency to metastasize early to many areas 106. Early diagnosis and treatment by radical ablative surgery or possibly other approaches may afford an improved prognosis; patients at risk should be carefully monitored 107, 108.
Long-term survivors are the exceptions. The 5-year survival rate reported by Sordillo et al in 1981 was 13.6% 109. In 1987, Hultberg found that patients with Stewart-Treves syndrome (lymphangiosarcoma following breast cancer surgery) had a mean survival of 20 months after tumor onset 110. Untreated patients have an average survival of 5-8 months 105. A more recent analysis showed the overall 5-year survival was 16% 79.
Metastatic lymphangiosarcoma to the lungs and chest wall are the most common cause of death in patients with Stewart-Treves syndrome. Metastases to the liver and bones can also occur. Lymphangiomas are associated with a high rate of local recurrence and metastasis, even after aggressive surgical treatment.
Lymphangiosarcoma survival rates
The 5-year survival rate reported by Sordillo et al in 1981 was 13.6% 109. In 1987, Hultberg found that patients with Stewart-Treves syndrome (lymphangiosarcoma following breast cancer surgery) had a mean survival of 20 months after tumor onset 110. Untreated patients have an average survival of 5-8 months 105. Overall survival is the length of time from either the date of diagnosis or the start of treatment for a disease, such as cancer, that patients diagnosed with the disease are still alive. Lymphangiosarcoma overall survival ranges between 18 and 31 months 111, 93, 94. A more recent analysis showed the overall 5-year survival was 16% 79. In a recent institutional review, patients with lymphangiosarcoma were found to have a 3-year and 5-year survival of 55% and 35%, respectively 2, 112, 113. Median survival has been noted at just 12 months 114. Untreated patients usually live only 5–8 months after diagnosis 19.
- Stewart FW, Treves N. Lymphangiosarcoma in post-mastectomy lymphedema. Cancer. 1948;1:64–81. doi: 10.1002/1097-0142(194805)1:1<64::aid-cncr2820010105>3.0.co;2-w[↩][↩][↩][↩]
- Murgia RD, Gross GP. Stewart-Treves Syndrome. [Updated 2023 Aug 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507833[↩][↩]
- Vojtíšek R, Sukovská E, Kylarová M, Kacerovská D, Baxa J, Divišová B, Fínek J. Stewart-Treves syndrome: Case report and literature review. Rep Pract Oncol Radiother. 2020 Nov-Dec;25(6):934-938. doi: 10.1016/j.rpor.2020.09.006[↩]
- Sepah YJ, Umer M, Qureshi A, Khan S. Lymphangiosarcoma of the arm presenting with lymphedema in a woman 16 years after mastectomy: a case report. Cases J. 2009 Sep 1;2:6887. doi: 10.4076/1757-1626-2-6887[↩][↩][↩][↩][↩]
- Gottlieb R, Serang R, Chi D, Menco H. Stewart-Treves syndrome. Radiol Case Rep. 2015 Dec 7;7(4):693. doi: 10.2484/rcr.v7i4.693[↩][↩]
- Woodward AH, Ivins JC, Soule EH. Lymphangiosarcoma arising in chronic lymphedematous extremities. Cancer. 1972 Aug;30(2):562-72. doi: 10.1002/1097-0142(197208)30:2<562::aid-cncr2820300237>3.0.co;2-v[↩]
- Mark R.J., Poen J.C., Tran L.M., Fu Y.S., Juillard G.F. Angiosarcoma. A report of 67 patients and a review of the literature. Cancer. 1996;77(11):2400–2406. doi: 10.1002/(SICI)1097-0142(19960601)77:11<2400::AID-CNCR32>3.0.CO;2-Z[↩]
- Stewart F.W., Treves N. Lymphangiosarcoma in postmastectomy lymphedema; a report of six cases in elephantiasis chirurgica. Cancer. 1948;1(1):64–81. doi: 10.1002/1097-0142(194805)1:1<64::aid-cncr2820010105>3.0.co;2-w[↩]
- Kaufmann T, Chu F, Kaufman R. Post-mastectomy lymphangiosarcoma (Stewart-Treves syndrome): report of two long-term survivals. Br J Radiol. 1991;64(765):857–860. doi: 10.1259/0007-1285-64-765-857[↩]
- Ocana A, Delgado C, Rodriguez CA, Bellido L, Izquierdo N, Martin R, Cruz JJ. Case 3. Upper limb lymphangiosarcoma following breast cancer therapy. J Clin Oncol. 2006;24(9):1477–1478. doi: 10.1200/JCO.2005.01.8473[↩][↩][↩][↩][↩]
- Fineberg S., Rosen P.P. Cutaneous angiosarcoma and atypical vascular lesions of the skin and breast after radiation therapy for breast carcinoma. Am J Clin Pathol. 1994;102(6):757–763. doi: 10.1093/ajcp/102.6.757[↩]
- Vojtísek R, Kinkor Z, Fínek J. Sekundární angiosarkomy po konzervativn lécebe nádorů prsu [Secondary angiosarcomas after conservation treatment for breast cancers]. Klin Onkol. 2011;24(5):382-8. Czech.[↩]
- Danese CA, Grishman E, Dreiling DA. Malignant vascular tumors of the lymphedematous extremity. Ann Surg. 1967;166(2):245–253. doi: 10.1097/00000658-196708000-00012[↩][↩][↩][↩][↩][↩]
- Echenique-Elizondo M, Tuneu-Valls A, Zubizarreta J, Lobo C. Stewart-Treves syndrome. Cir Esp. 2005;78(6):382–384. doi: 10.1016/S0009-739X(05)70959-3[↩]
- Stewart NJ, Pritchard DJ, Nascimento AG, Kang YK. Lymphangiosarcoma following mastectomy. Clin Orthop Relat Res. 1995 Nov;(320):135-41.[↩]
- Sharma A., Schwartz R.A. Stewart-Treves syndrome: Pathogenesis and management. J Am Acad Dermatol. 2012;67(6):1342–1348. doi: 10.1016/j.jaad.2012.04.028[↩][↩][↩][↩]
- Sordillo P.P., Chapman R., Hajdu S.I., Magill G.B., Golbey R.B. Lymphangiosarcoma. Cancer. 1981;48(7):1674–1679. doi: 10.1002/1097-0142(19811001)48:7<1674::aid-cncr2820480733>3.0.co;2-h[↩]
- Mack TM. Sarcomas and other malignancies of soft tissue, retroperitoneum, peritoneum, pleura, heart, mediastinum and spleen. Cancer. 1995;75:211–245. doi: 10.1002/1097-0142(19950101)75:1+<211::aid-cncr2820751309>3.0.co;2-x[↩]
- Berebichez-Fridman R., Deutsch Y.E., Joyal T.M. Stewart-Treves syndrome: A case report and review of the literature. Case Rep Oncol. 2016;9(1):205–211. doi: 10.1159/000445427[↩][↩][↩]
- McHaffie D.R., Kozak K.R., Warner T.F., Cho C.S., Heiner J.P., Attia S. Stewart-Treves syndrome of the lower extremity. J Clin Oncol. 2010;28(21):e351–2. doi: 10.1200/JCO.2009.26.0406[↩][↩][↩]
- Shavit E., Alavi A., Limacher J.J., Sibbald R.G. Angiosarcoma complicating lower leg elephantiasis in a male patient: An unusual clinical complication, case report and literature review. SAGE Open Med Case Rep. 2018;6:1–5. doi: 10.1177/2050313X18796343[↩]
- Soran A, D’Angelo G, Begovic M, Ardic F, Harlak A, Samuel Wieand H, Vogel VG, Johnson RR. Breast cancer-related lymphedema-what are the significant predictors and how they affect the severity of lymphedema? Breast J. 2006;12(6):536–543. doi: 10.1111/j.1524-4741.2006.00342.x[↩]
- Thomas-MacLean R, Miedema B, Tatemichi SR. Breast cancer-related lymphedema: women’s experiences with an underestimated condition. Can Fam Physician. 2005 Feb;51(2):246-7. https://pmc.ncbi.nlm.nih.gov/articles/PMC1472970[↩]
- Petrek JA, Pressman PI, Smith RA. Lymphedema: current issues in research and management. CA Cancer J Clin. 2000;50(5):292–307. doi: 10.3322/canjclin.50.5.292[↩]
- Beaulac SM, McNair LA, Scott TE, LaMorte WW, Kavanah MT. Lymphedema and quality of life in survivors of early-stage breast cancer. Arch Surg. 2002;137(11):1253–1257. doi: 10.1001/archsurg.137.11.1253[↩]
- Wagner MJ, Ravi V, Schaub SK, et al. Incidence and Presenting Characteristics of Angiosarcoma in the US, 2001-2020. JAMA Netw Open. 2024 Apr 1;7(4):e246235. doi: 10.1001/jamanetworkopen.2024.6235[↩]
- Beckjord EB, Reynolds KA, van Londen GJ, Burns R, Singh R, Arvey SR, Nutt SA, Rechis R. Population-level trends in posttreatment cancer survivors’ concerns and associated receipt of care: results from the 2006 and 2010 LIVESTRONG surveys. J Psychosoc Oncol. 2014;32(2):125-51. doi: 10.1080/07347332.2013.874004[↩][↩]
- Zhang X, McLaughlin EM, Krok-Schoen JL, Naughton M, Bernardo BM, Cheville A, Allison M, Stefanick M, Bea JW, Paskett ED. Association of Lower Extremity Lymphedema With Physical Functioning and Activities of Daily Living Among Older Survivors of Colorectal, Endometrial, and Ovarian Cancer. JAMA Netw Open. 2022 Mar 1;5(3):e221671. doi: 10.1001/jamanetworkopen.2022.1671[↩][↩][↩]
- Paskett ED, Le-Rademacher J, Oliveri JM, Liu H, Seisler DK, Sloan JA, Armer JM, Naughton MJ, Hock K, Schwartz M, Unzeitig G, Melnik M, Yee LD, Fleming GF, Taylor JR, Loprinzi C. A randomized study to prevent lymphedema in women treated for breast cancer: CALGB 70305 (Alliance). Cancer. 2021 Jan 15;127(2):291-299. doi: 10.1002/cncr.33183[↩]
- Woodward AH, Ivins JC, Soule EH. Lymphangiosarcoma arising in chronic lymphedematous extremities. Cancer. 1972;30(2):562–572. doi: 10.1002/1097-0142(197208)30:2<562::aid-cncr2820300237>3.0.co;2-v[↩][↩]
- Prudden JF, Wolarsky ER. Lymphangiosarcoma of the thigh. Case report. Arch Surg. 1967;94(3):376–379. doi: 10.1001/archsurg.1967.01330090070018[↩][↩][↩][↩]
- Rosenbaum E, Antonescu CR, Smith S, et al. Clinical, genomic, and transcriptomic correlates of response to immune checkpoint blockade-based therapy in a cohort of patients with angiosarcoma treated at a single center. J Immunother Cancer. 2022 Apr;10(4):e004149. doi: 10.1136/jitc-2021-004149[↩][↩]
- Li B, Li J, Hao K, Jin Y, Ma J, Du X. Magnetic resonance findings of Stewart-Treves Syndrome in primary limb lymphedema compared with pathology: A retrospective single-center study. Front Oncol. 2023 Feb 9;13:953524. doi: 10.3389/fonc.2023.953524[↩][↩]
- Nakazono T, Kudo S, Matsuo Y, Matsubayashi R, Ehara S, Narisawa H, Yonemitsu N. Angiosarcoma associated with chronic lymphedema (Stewart-Treves syndrome) of the leg: MR imaging. Skeletal Radiol. 2000 Jul;29(7):413-6. doi: 10.1007/s002560000225[↩][↩]
- Schindera ST, Streit M, Kaelin U, Stauffer E, Steinbach L, Anderson SE. Stewart-Treves syndrome: MR imaging of a postmastectomy upper-limb chronic lymphedema with angiosarcoma. Skeletal Radiol. 2005 Mar;34(3):156-60. doi: 10.1007/s00256-004-0807-5[↩][↩]
- Li B, Wang Z. Stewart-Treves syndrome: Magnetic resonance imaging data compared with pathological results from a single center. Oncol Lett. 2018 Jan;15(1):1113-1118. doi: 10.3892/ol.2017.7363[↩][↩]
- Dyroff S, Layfield LJ, Crim J. Angiosarcoma arising in massive localized lymphedema. Skeletal Radiol. 2020 May;49(5):815-818. doi: 10.1007/s00256-020-03373-4[↩][↩]
- Dawlatly SL, Dramis A, Sumathi VP, Grimer RJ. Stewart-Treves syndrome and the use of positron emission tomographic scanning. Ann Vasc Surg. 2011 Jul;25(5):699.e1-3. doi: 10.1016/j.avsg.2010.12.027[↩][↩]
- Chen YR, Hsieh TC, Yen KY, Kao CH. Distant metastases in a young woman with Stewart-Treves syndrome demonstrated by an FDG-PET/CT scan. Clin Nucl Med. 2014 Nov;39(11):975-6. doi: 10.1097/RLU.0000000000000349[↩][↩]
- Florou V, Wilky BA. Current and Future Directions for Angiosarcoma Therapy. Curr Treat Options Oncol. 2018 Mar 8;19(3):14. doi: 10.1007/s11864-018-0531-3[↩][↩]
- di Meo N, Drabeni M, Gatti A, Trevisan G. A Stewart-Treves syndrome of the lower limb. Dermatol Online J. 2012 Jun 15;18(6):14. https://doi.org/10.5070/D38d1463nq[↩][↩][↩]
- Farzaliyev F, Hamacher R, Steinau Professor HU, Bertram S, Podleska LE. Secondary angiosarcoma: A fatal complication of chronic lymphedema. J Surg Oncol. 2020 Jan;121(1):85-90. doi: 10.1002/jso.25598[↩][↩]
- Grobmyer SR, Daly JM, Glotzbach RE, Grobmyer AJ 3rd. Role of surgery in the management of postmastectomy extremity angiosarcoma (Stewart-Treves syndrome). J Surg Oncol. 2000 Mar;73(3):182-8. doi: 10.1002/(sici)1096-9098(200003)73:3<182::aid-jso14>3.0.co;2-n[↩][↩]
- Breidenbach M, Rein D, Schmidt T, Heindel W, Kolhagen H, Mallmann P, Kurbacher CM. Intra-arterial mitoxantrone and paclitaxel in a patient with Stewart-Treves syndrome: selection of chemotherapy by an ex vivo ATP-based chemosensitivity assay. Anticancer Drugs. 2000 Apr;11(4):269-73. doi: 10.1097/00001813-200004000-00007[↩][↩]
- Goetze S, Schmook T, Audring H, Ziegenbein C, Worm M, Schulze P. [Successful treatment of Stewart-Treves syndrome with liposomal doxorubicin]. J Dtsch Dermatol Ges. 2004 Jan. 2(1):49-52.[↩][↩]
- Paydas S, Yapar Z, Gonlusen G. Stewart-Treves syndrome: questionable response to weekly paclitaxel. Chemotherapy. 2010;56(1):66-8. doi: 10.1159/000282285[↩][↩]
- Fujisawa Y, Ito M, Mori K, Okada S, Nakamura Y, Kawachi Y, Otsuka F. Intra-arterial mitoxantrone/paclitaxel in angiosarcoma of the lower limb associated with chronic lymphedema (Stewart-Treves syndrome) in a patient with cervical cancer. Eur J Dermatol. 2011 Jan-Feb;21(1):119-20. doi: 10.1684/ejd.2010.1179[↩][↩]
- Furue M, Yamada N, Takahashi T, Kikuchi K, Tsuchida T, Ishibashi Y, Kobori O, Ihara A, Kitayama J, Minami M. Immunotherapy for Stewart-Treves syndrome. Usefulness of intrapleural administration of tumor-infiltrating lymphocytes against massive pleural effusion caused by metastatic angiosarcoma. J Am Acad Dermatol. 1994 May;30(5 Pt 2):899-903. https://www.jaad.org/article/S0190-9622(94)70109-1/abstract[↩][↩]
- Stanczyk M, Gewartowska M, Swierkowski M, Grala B, Maruszynski M. Stewart-Treves syndrome angiosarcoma expresses phenotypes of both blood and lymphatic capillaries. Chin Med J (Engl). 2013 Jan;126(2):231-7.[↩][↩][↩][↩]
- Imura Y, Nagata S, Wakamatsu T, Tanaka T, Tamiya H, Naka N, Takenaka S. A case of Stewart-Treves syndrome occurring in the abdominal wall successfully treated with eribulin: A case report. Mol Clin Oncol. 2020 Nov;13(5):49. doi: 10.3892/mco.2020.2119[↩][↩]
- Horsley JS, Styblo T: Lymphedema in the postmastectomy patient. In: Bland KI, Copeland EM, eds.: The Breast: Comprehensive Management of Benign and Malignant Diseases. Saunders, 1991, pp 701-6.[↩]
- Lymphedema (PDQ®)–Health Professional Version. https://www.cancer.gov/about-cancer/treatment/side-effects/lymphedema/lymphedema-hp-pdq[↩][↩][↩][↩]
- Ruocco V., Schwartz R.A., Ruocco E. Lymphedema: an immunologically vulnerable site for development of neoplasms. J Am Acad Dermatol. 2002;47(1):124–127. doi: 10.1067/mjd.2002.120909[↩]
- Young R.J., Brown N.J., Reed M.W., Hughes D., Woll P.J. Angiosarcoma. Lancet Oncol. 2010;11(10):983–991. doi: 10.1016/S1470-2045(10)70023-1[↩][↩][↩]
- Cozen W., Bernstein L., Wang F., Press M.F., Mack T.M. The risk of angiosarcoma following primary breast cancer. Br J Cancer. 1999;81(3):532–536. doi: 10.1038/sj.bjc.6690726[↩]
- Lee R., Saardi K.M., Schwartz R.A. Lymphedema-related angiogenic tumors and other malignancies. Clin Dermatol. 2014;32(5):616–620. doi: 10.1016/j.clindermatol.2014.04.008[↩]
- Ridner SH. Quality of life and a symptom cluster associated with breast cancer treatment-related lymphedema. Support Care Cancer. 2005 Nov;13(11):904-11. doi: 10.1007/s00520-005-0810-y[↩]
- Dunberger G, Lindquist H, Waldenström AC, Nyberg T, Steineck G, Åvall-Lundqvist E. Lower limb lymphedema in gynecological cancer survivors–effect on daily life functioning. Support Care Cancer. 2013 Nov;21(11):3063-70. doi: 10.1007/s00520-013-1879-3[↩]
- Pyszel A, Malyszczak K, Pyszel K, Andrzejak R, Szuba A. Disability, psychological distress and quality of life in breast cancer survivors with arm lymphedema. Lymphology. 2006 Dec;39(4):185-92.[↩]
- Gjorup CA, Groenvold M, Hendel HW, Dahlstroem K, Drzewiecki KT, Klausen TW, Hölmich LR. Health-related quality of life in melanoma patients: Impact of melanoma-related limb lymphoedema. Eur J Cancer. 2017 Nov;85:122-132. doi: 10.1016/j.ejca.2017.07.052[↩][↩][↩]
- McLaughlin SA, Brunelle CL, Taghian A. Breast Cancer-Related Lymphedema: Risk Factors, Screening, Management, and the Impact of Locoregional Treatment. J Clin Oncol. 2020 Jul 10;38(20):2341-2350. doi: 10.1200/JCO.19.02896[↩][↩]
- Montagna G, Zhang J, Sevilimedu V, Charyn J, Abbate K, Gomez EA, Mehrara B, Morrow M, Barrio AV. Risk Factors and Racial and Ethnic Disparities in Patients With Breast Cancer-Related Lymphedema. JAMA Oncol. 2022 Aug 1;8(8):1195-1200. doi: 10.1001/jamaoncol.2022.1628[↩][↩][↩][↩]
- DiSipio T, Rye S, Newman B, Hayes S. Incidence of unilateral arm lymphoedema after breast cancer: a systematic review and meta-analysis. Lancet Oncol. 2013 May;14(6):500-15. doi: 10.1016/S1470-2045(13)70076-7[↩][↩][↩]
- Giuliano AE, Ballman KV, McCall L, Beitsch PD, Brennan MB, Kelemen PR, Ollila DW, Hansen NM, Whitworth PW, Blumencranz PW, Leitch AM, Saha S, Hunt KK, Morrow M. Effect of Axillary Dissection vs No Axillary Dissection on 10-Year Overall Survival Among Women With Invasive Breast Cancer and Sentinel Node Metastasis: The ACOSOG Z0011 (Alliance) Randomized Clinical Trial. JAMA. 2017 Sep 12;318(10):918-926. doi: 10.1001/jama.2017.11470[↩]
- Armer JM, Ballman KV, McCall L, Ostby PL, Zagar E, Kuerer HM, Hunt KK, Boughey JC. Factors Associated With Lymphedema in Women With Node-Positive Breast Cancer Treated With Neoadjuvant Chemotherapy and Axillary Dissection. JAMA Surg. 2019 Sep 1;154(9):800-809. doi: 10.1001/jamasurg.2019.1742[↩][↩]
- Koelmeyer LA, Gaitatzis K, Dietrich MS, Shah CS, Boyages J, McLaughlin SA, Taback B, Stolldorf DP, Elder E, Hughes TM, French JR, Ngui N, Hsu JM, Moore A, Ridner SH. Risk factors for breast cancer-related lymphedema in patients undergoing 3 years of prospective surveillance with intervention. Cancer. 2022 Sep 15;128(18):3408-3415. doi: 10.1002/cncr.34377[↩][↩][↩]
- Beesley V, Janda M, Eakin E, Obermair A, Battistutta D. Lymphedema after gynecological cancer treatment : prevalence, correlates, and supportive care needs. Cancer. 2007 Jun 15;109(12):2607-14. doi: 10.1002/cncr.22684[↩]
- Ryan M, Stainton MC, Slaytor EK, Jaconelli C, Watts S, Mackenzie P. Aetiology and prevalence of lower limb lymphoedema following treatment for gynaecological cancer. Aust N Z J Obstet Gynaecol. 2003 Apr;43(2):148-51. doi: 10.1046/j.0004-8666.2003.00040.x[↩]
- Carter J, Huang HQ, Armer J, Carlson JW, Lockwood S, Nolte S, Stewart BR, Kauderer J, Hutson A, Walker JL, Fleury AC, Bonebrake A, Soper JT, Mathews C, Zivanovic O, Richards WE, Tan A, Alberts DS, Barakat RR, Wenzel L. GOG 244 – The LymphEdema and Gynecologic cancer (LEG) study: The association between the gynecologic cancer lymphedema questionnaire (GCLQ) and lymphedema of the lower extremity (LLE). Gynecol Oncol. 2019 Dec;155(3):452-460. doi: 10.1016/j.ygyno.2019.09.027[↩][↩][↩]
- Jeans C, Brown B, Ward EC, Vertigan AE, Pigott AE, Nixon JL, Wratten C. Comparing the prevalence, location, and severity of head and neck lymphedema after postoperative radiotherapy for oral cavity cancers and definitive chemoradiotherapy for oropharyngeal, laryngeal, and hypopharyngeal cancers. Head Neck. 2020 Nov;42(11):3364-3374. doi: 10.1002/hed.26394[↩][↩][↩][↩]
- Deban M, Vallance P, Jost E, McKinnon JG, Temple-Oberle C. Higher Rate of Lymphedema with Inguinal versus Axillary Complete Lymph Node Dissection for Melanoma: A Potential Target for Immediate Lymphatic Reconstruction? Curr Oncol. 2022 Aug 11;29(8):5655-5663. doi: 10.3390/curroncol29080446[↩]
- Moody JA, Botham SJ, Dahill KE, Wallace DL, Hardwicke JT. Complications following completion lymphadenectomy versus therapeutic lymphadenectomy for melanoma – A systematic review of the literature. Eur J Surg Oncol. 2017 Sep;43(9):1760-1767. doi: 10.1016/j.ejso.2017.07.003[↩]
- Neuberger M, Schmidt L, Wessels F, Linke M, Müller C, Westhoff N, Nuhn P, von Hardenberg J. Onset and burden of lower limb lymphedema after radical prostatectomy: a cross-sectional study. Support Care Cancer. 2022 Feb;30(2):1303-1313. doi: 10.1007/s00520-021-06520-2[↩]
- Friedmann D, Wunder JS, Ferguson P, O’Sullivan B, Roberge D, Catton C, Freeman C, Saran N, Turcotte RE. Incidence and Severity of Lymphoedema following Limb Salvage of Extremity Soft Tissue Sarcoma. Sarcoma. 2011;2011:289673. doi: 10.1155/2011/289673[↩]
- Schreiber H, Barry FM, Russell WC, Macon WL 4th, Ponsky JL, Pories WJ. Stewart-Treves syndrome. A lethal complication of postmastectomy lymphedema and regional immune deficiency. Arch Surg. 1979 Jan;114(1):82-5. doi: 10.1001/archsurg.1979.01370250084018[↩]
- Stark RB, Dwyer EM, De Forest M. Effect of surgical ablation of regional lymph nodes on survival of homografts. Ann NY Acad Sci. 1960. 87:140-148.[↩]
- Cabral ANF, Rocha RH, Amaral ACVD, Medeiros KB, Nogueira PSE, Diniz LM. Cutaneous angiosarcoma: report of three different and typical cases admitted in a unique dermatology clinic. An Bras Dermatol. 2017 Mar-Apr;92(2):235-238. doi: 10.1590/abd1806-4841.20175326[↩]
- Sternby NH, Gynning I, Hogeman KE. Postmastectomy angiosarcoma. Acta Chir Scand. 1961. 121:420-432.[↩]
- Styring E, Fernebro J, Jönsson PE, Ehinger A, Engellau J, Rissler P, Rydholm A, Nilbert M, Vult von Steyern F. Changing clinical presentation of angiosarcomas after breast cancer: from late tumors in edematous arms to earlier tumors on the thoracic wall. Breast Cancer Res Treat. 2010 Aug;122(3):883-7. doi: 10.1007/s10549-009-0703-8[↩][↩][↩][↩]
- Zietz C., Rössle M., Haas C. MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas. Am J Pathol. 1998;153(5):1425–1433. doi: 10.1016/S0002-9440(10)65729-X[↩]
- McLaughlin Er, Brown Lf, Weiss Sw, Mulliken Jb, Perez-Atayde A., Arbiser Jl. VEGF and its receptors are expressed in a pediatric angiosarcoma in a patient with Aicardi’s syndrome. J Invest Dermatol. 2000;114(6):1209–1210. doi: 10.1046/j.1523-1747.2000.00005-3.x[↩]
- Ishida Y., Otsuka A., Kabashima K. Cutaneous angiosarcoma: Update on biology and latest treatment. Curr Opin Oncol. 2018;30(2):107–112. doi: 10.1097/CCO.0000000000000427[↩]
- Shimizu A., Kaira K., Okubo Y. Positive PD-L1 expression predicts worse outcome in cutaneous angiosarcoma. J Glob Oncol. 2016;3(4):360–369. doi: 10.1200/JGO.2016.005843[↩]
- Chang D.W., Suami H., Skoracki R. A prospective analysis of 100 consecutive lymphovenous bypass cases for treatment of extremity lymphedema. Plast Reconstr Surg. 2013;132(5):1305–1314. doi: 10.1097/PRS.0b013e3182a4d626[↩]
- Engel H., Lin C.Y., Huang J.J., Cheng M.H. Outcomes of lymphedema microsurgery for breast cancer-related lymphedema with or without microvascular breast reconstruction. Ann Surg. 2018;268(6):1076–1083. doi: 10.1097/SLA.0000000000002322[↩]
- McConnell AH, Haslam P. Angiosarcoma in post-mastectomy lymphedema: report of five cases and review of the literature. Br J Surg. 1959. 46:322-32. doi: 10.1002/bjs.18004619804[↩]
- Soft Tissue Sarcoma Stages. https://www.cancer.org/cancer/types/soft-tissue-sarcoma/detection-diagnosis-staging/staging.html[↩][↩]
- Schwartz RA, Kardashian JF, McNutt NS, Crain WR, Welch KL, Choy SH. Cutaneous angiosarcoma resembling anaplastic Kaposi’s sarcoma in a homosexual man. Cancer. 1983 Feb 15;51(4):721-6. doi: 10.1002/1097-0142(19830215)51:4<721::aid-cncr2820510428>3.0.co;2-2[↩]
- Salameire D, Templier I, Charles J, Pinel N, Morand P, Leccia MT, Lantuejoul S. An “anaplastic” Kaposi’s sarcoma mimicking a Stewart-Treves syndrome. A case report and a review of literature. Am J Dermatopathol. 2008 Jun;30(3):265-8. doi: 10.1097/DAD.0b013e318169fd5f[↩]
- Kunkel T, Mylonas I, Mayr D, Friese K, Sommer HL. Recurrence of secondary angiosarcoma in a patient with post-radiated breast for breast cancer. Arch Gynecol Obstet. 2008 Nov;278(5):497-501. doi: 10.1007/s00404-008-0605-8[↩]
- Lee R, Saardi KM, Schwartz RA. Lymphedema-related angiogenic tumors and other malignancies. Clin Dermatol. 2014 Sep-Oct;32(5):616-20. doi: 10.1016/j.clindermatol.2014.04.008[↩]
- Roy P., Clark M.A., Thomas J.M. Stewart-Treves syndrome–treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004;30(9):982–986. doi: 10.1016/j.ejso.2004.07.027[↩][↩][↩][↩]
- Farzaliyev F., Hamacher R., Steinau Professor H.U., Bertram S., Podleska L.E. Secondary angiosarcoma: A fatal complication of chronic lymphedema. J Surg Oncol. 2020;121(1):85–90. doi: 10.1002/jso.25598[↩][↩][↩][↩][↩]
- McKeown D.G., Boland P.J. Stewart–Treves syndrome: A case report. Ann R Coll Surg Engl. 2013;95(5):e80–e82. doi: 10.1308/003588413X13629960046110[↩][↩][↩]
- Stanczyk M, Gewartowska M. Stewart-Treves syndrome. Indian J Med Res. 2014 Jan;139(1):179. https://pmc.ncbi.nlm.nih.gov/articles/PMC3994737[↩][↩]
- Roy P, Clark MA, Thomas JM. Stewart-Treves syndrome–treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004 Nov;30(9):982-6. doi: 10.1016/j.ejso.2004.07.027[↩]
- Dimas V, Kargel J, Bauer J, Chang P. Forequarter amputation for malignant tumours of the upper extremity: Case report, techniques and indications. Can J Plast Surg. 2007 Summer;15(2):83-5. doi: 10.1177/229255030701500204[↩]
- Forequarter Amputation. https://www.sciencedirect.com/topics/medicine-and-dentistry/forequarter-amputation[↩]
- Shon W., Ida C.M., Boland-Froemming J.M., Rose P.S., Folpe A. Cutaneous angiosarcoma arising in massive localized lymphedema of the morbidly obese: A report of five cases and review of the literature. J Cutan Pathol. 2011;38(7):560–564. doi: 10.1111/j.1600-0560.2011.01703.x[↩]
- Ray-Coquard I.L., Domont J., Tresch-Bruneel E. Paclitaxel given once per week with or without bevacizumab in patients with advanced angiosarcoma: A randomized phase II trial. J Clin Oncol. 2015;33(25):2797–2802. doi: 10.1200/JCO.2015.60.8505[↩]
- Agulnik M., Yarber J.L., Okuno S.H. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24(1):257–263. doi: 10.1093/annonc/mds237[↩]
- Kollár A., Jones R.L., Stacchiotti S. Pazopanib in advanced vascular sarcomas: An EORTC Soft tissue and Bone Sarcoma Group (STBSG) retrospective analysis. Acta Oncol (Madr) 2017;56(1):88–92. doi: 10.1080/0284186X.2016.1234068[↩]
- Stevenson M.G., Hoekstra H.J., Song W., Suurmeijer A.J.H., Been L.B. Histopathological tumor response following neoadjuvant hyperthermic isolated limb perfusion in extremity soft tissue sarcomas: Evaluation of the EORTC-STBSG response score. Eur J Surg Oncol. 2018;44(9):1406–1411. doi: 10.1016/j.ejso.2018.05.011[↩]
- Naka N., Ohsawa M., Tomita Y. Prognostic factors in angiosarcoma: A multivariate analysis of 55 cases. J Surg Oncol. 1996;61(3):170–176. doi: 10.1002/(SICI)1096-9098(199603)61:3<170::AID-JSO2>3.0.CO;2-8[↩]
- Stewart-Treves Syndrome. https://emedicine.medscape.com/article/1102114-overview#a2[↩][↩][↩]
- Hsu A, Matera R, Dizon DS. Stewart-Treves syndrome: a rapidly fatal complication of breast cancer treatment. Lancet Oncol. 2020 Oct;21(10):e495. doi: 10.1016/S1470-2045(20)30274-6[↩]
- Felmerer G, Dowlatshahi AS, Stark GB, Földi E, Földi M, Ahls MG, Ströbel P, Aung T. Lymphangiosarcoma: Is Stewart-Treves Syndrome a Preventable Condition? Lymphat Res Biol. 2016 Mar;14(1):35-9. doi: 10.1089/lrb.2015.0006[↩]
- Campana LG, Valpione S, Tosi A, Rastrelli M, Rossi CR, Aliberti C. Angiosarcoma on Lymphedema (Stewart-Treves Syndrome): A 12-Year Follow-up after Isolated Limb Perfusion, Limb Infusion, and Electrochemotherapy. J Vasc Interv Radiol. 2016 Mar;27(3):444-6. doi: 10.1016/j.jvir.2015.11.060[↩]
- Sordillo PP, Chapman R, Hajdu SI, Magill GB, Golbey RB. Lymphangiosarcoma. Cancer. 1981 Oct 1;48(7):1674-9. doi: 10.1002/1097-0142(19811001)48:7<1674::aid-cncr2820480733>3.0.co;2-h[↩][↩]
- Hultberg BM. Angiosarcomas in chronically lymphedematous extremities. Two cases of Stewart-Treves syndrome. Am J Dermatopathol. 1987 Oct;9(5):406-12. doi: 10.1097/00000372-198710000-00006[↩][↩]
- Chopra S., Ors F., Bergin D. MRI of angiosarcoma associated with chronic lymphoedema: Stewart Treves syndrome. Br J Radiol. 2007;80(960):e310–3. doi: 10.1259/bjr/19441948[↩]
- D’Angelo SP, Munhoz RR, Kuk D, Landa J, Hartley EW, Bonafede M, Dickson MA, Gounder M, Keohan ML, Crago AM, Antonescu CR, Tap WD. Outcomes of Systemic Therapy for Patients with Metastatic Angiosarcoma. Oncology. 2015;89(4):205-14. doi: 10.1159/000381917[↩]
- Fury MG, Antonescu CR, Van Zee KJ, Brennan MF, Maki RG. A 14-year retrospective review of angiosarcoma: clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer J. 2005 May-Jun;11(3):241-7. doi: 10.1097/00130404-200505000-00011. Erratum in: Cancer J. 2005 Jul-Aug;11(4):354.[↩]
- Benton A, Liu B, Gartenhaus LE, Hanna JA. Genomic landscape and preclinical models of angiosarcoma. Mol Oncol. 2025 Apr;19(4):965-983. doi: 10.1002/1878-0261.13744[↩]





{kind=link}