
Contents
- What is muscular dystrophy
- Muscular dystrophy types
- Muscular dystrophy causes
- Muscular dystrophy prognosis
- Muscular dystrophy life expectancy
- Muscular dystrophy complications
- Muscular dystrophy symptoms
- Muscular dystrophy diagnosis
- Muscular dystrophy treatment
What is muscular dystrophy
The muscular dystrophies are a group of hereditary diseases that cause progressive weakness and degeneration of the skeletal muscles characterized by the presence of progressive skeletal muscle weakness and loss of muscle mass. In muscular dystrophy, abnormal genes (mutations) interfere with the production of proteins needed to form healthy muscle. There is usually a defect or deficiency in essential muscle proteins which is accompanied by the death of muscle cells and tissues. Cardiac and smooth muscle may be affected in some types of muscular dystrophy.
There are many different kinds of muscular dystrophy. Symptoms of the most common variety begin in childhood, mostly in boys. Other types don’t surface until adulthood.
The most common types of muscular dystrophy are:
- Duchenne muscular dystrophy, the most common childhood form of muscular dystrophy. It is an X-linked disease, which means that the gene is passed from mothers to their sons.
- Becker muscular dystrophy, with a genetic defect very similar to that in Duchenne muscular dystrophy, but not as severe. Duchenne and Becker muscular dystrophies together affect 1 in 3,500 to 5,000 newborn males worldwide. Between 400 and 600 boys in the United States are born with these conditions each year.
- Myotonic dystrophy. Patients have myotonia (delayed relaxation of muscles after contraction), for example, after grasping a doorknob. There are several different types within this group. They occur in both men and women and affect about 1 in 8000 people 1.
There’s no cure for muscular dystrophy.
The broad aim of treatment for muscular dystrophy is to lessen the impairments of the disease and to deter functional limitations and disability. The treatment is usually on an outpatient basis, with the involvement of neurologists, orthopaediacs, physical and occupational therapists, social workers and orthotists.
Treatment is aimed at:
- Preventing contractures: active and passive stretching, physical therapy, positioning and splinting
- Maintaining muscle strength
- Promoting cardiopulmonary endurance
- Maintaining mobility and ambulation: the use of bracing and walking aids
- Monitoring pulmonary and cardiac function
- Vaccinating against influenza and pneumococcal infections when wheel chair bound and if on steroids
Surgical intervention may be necessary in some cases. It is essential to keep an appropriate exercise and diet regime as weight control is essential. The cardiac problems that occur with Emery-Dreifuss muscular dystrophy and myotonic muscular dystrophy may require a pacemaker. Genetic counseling may be required as well as family and individual counseling therapy.
Muscular dystrophy types
The major types of muscular dystrophy include:
- Becker’s muscular dystrophy (BMD) – usually noticeable at adolescence
- Congenital muscular dystrophy (CMD) – usually noticeable at birth
- Bethlem CMD
- Fukuyama CMD
- Muscle-eye-brain diseases (MEBs)
- Rigid spine syndromes
- Ullrich CMD
- Walker-Warburg syndromes (WWS)
- Duchenne muscular dystrophy (DMD)
- Emery-Dreifuss muscular dystrophy (EDMD)
- Facioscapulohumeral muscular dystrphy (FSHD)
- Limb-girdle muscular dystrophies (LGMD)
- Myotonic dystrophy (DM)
- Oculopharyngeal muscular dystrophy (OPMD)
Becker muscular dystrophy
Becker muscular dystrophy (Becker muscular dystrophy) is one of nine types of muscular dystrophy, a group of genetic, degenerative diseases primarily affecting voluntary muscles. It is named after German doctor Peter Emil Becker, who first described this variant of Duchenne muscular dystrophy in the 1950s. Becker muscular dystrophy is similar to Duchenne muscular dystrophy but allows the voluntary muscles to function better than they do in Duchenne muscular dystrophy. The heart muscle, however, can be affected similarly to the way it is in Duchenne muscular dystrophy.
Becker muscular dystrophy causes
In 1986, researchers identified the gene that, when flawed (mutation) — causes Duchenne muscular dystrophy and Becker muscular dystrophy. In 1987, the protein associated with this gene was identified and named dystrophin. Dystrophin plays a role in keeping muscle cells intact; lack of dystrophin causes cells to be fragile and easily damaged.
Mutations in the DMD gene cause the Duchenne and Becker forms of muscular dystrophy. The DMD gene provides instructions for making a protein called dystrophin. This protein is located primarily in skeletal and cardiac muscle, where it helps stabilize and protect muscle fibers. Dystrophin may also play a role in chemical signaling within cells.
Mutations in the DMD gene alter the structure or function of dystrophin or prevent any functional dystrophin from being produced. Muscle cells without enough of this protein become damaged as muscles repeatedly contract and relax with use. The damaged fibers weaken and die over time, leading to the muscle weakness and heart problems characteristic of Duchenne and Becker muscular dystrophies. Mutations that lead to an abnormal version of dystrophin that retains some function usually cause Becker muscular dystrophy, while mutations that prevent the production of any functional dystrophin tend to cause Duchenne muscular dystrophy.
Because Duchenne and Becker muscular dystrophies result from faulty or missing dystrophin, these conditions are classified as dystrophinopathies.
Becker muscular dystrophy occurs when the dystrophin protein that’s made from a particular gene on the X chromosome is only partially functional.
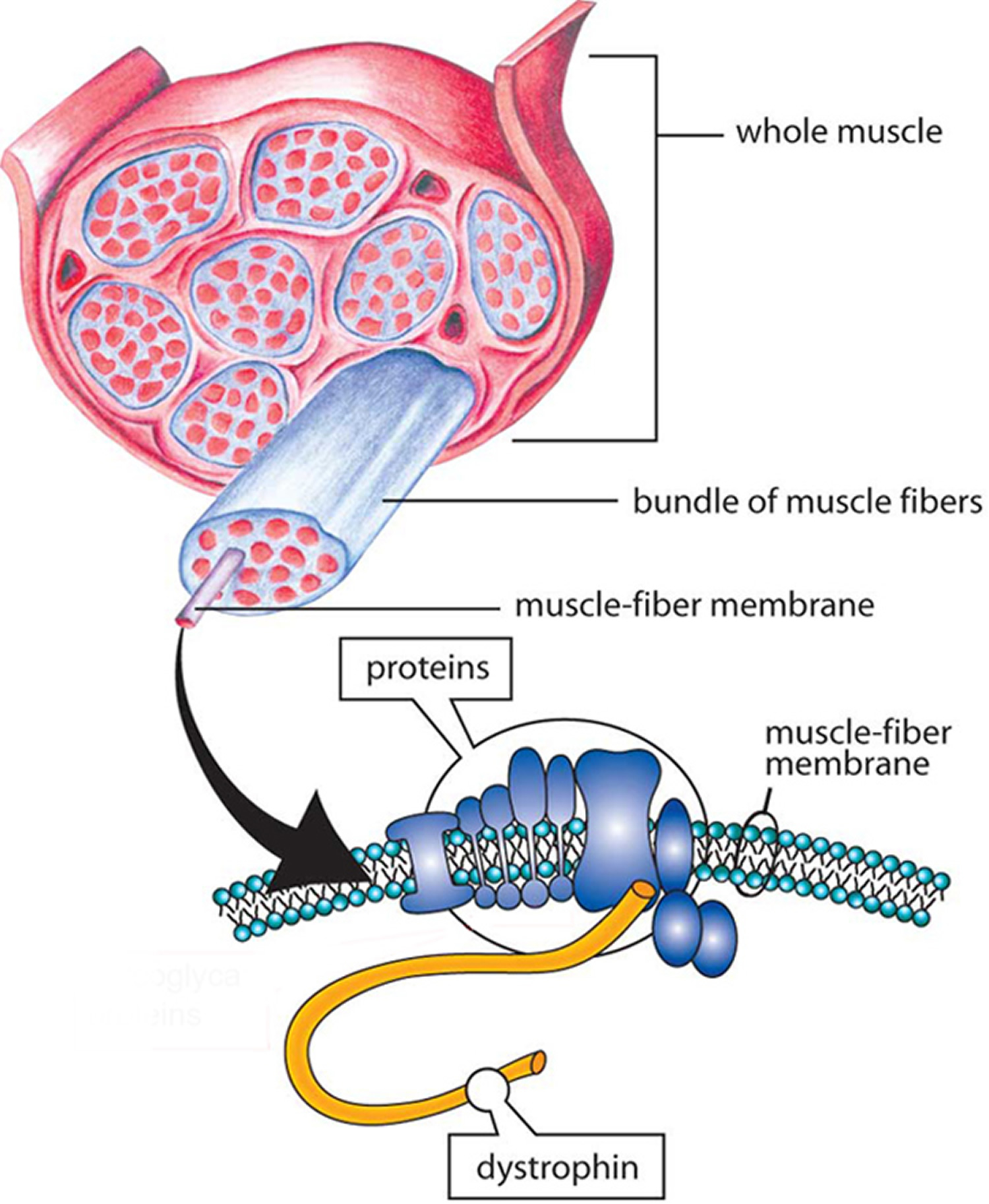
The dystrophin protein transfers the force of muscle contraction from the inside of the muscle cell outward to the cell membrane. Because it connects the center of the muscle cell to the periphery, the dystrophin protein is extremely long. One end is specialized for linking to the muscle interior, and the other end for linking to a variety of proteins at the cell membrane. The long middle section, called the rod domain, is taken up by a series of repeating units called spectrin repeats. The repeated spectrin units in the middle of the protein play an important role in linking the two ends, but the protein can still function (albeit not perfectly) with fewer of them than normal. Mutations that cause Becker muscular dystrophy decrease the number of these repeats, leading to muscle weakness.
In addition to its force-transfer role, dystrophin provides the scaffold for holding numerous molecules in place near the cell membrane. Loss of dystrophin displaces these molecules, with consequent disruptions in their functions.
While Duchenne muscular dystrophy mutations cause virtually no functional dystrophin to be made, people with Becker muscular dystrophy make dystrophin that is partially functional. They make a shortened form of the protein, which protects the muscles of those with Becker from degenerating as completely or as quickly as those of people with Duchenne muscular dystrophy.
Becker muscular dystrophy is inherited in an X-linked pattern. That means the gene that sometimes contains a mutation causing these diseases is on the X chromosome. Every boy inherits an X chromosome from his mother and a Y chromosome from his father, which is what makes him male. Girls get two X chromosomes, one from each parent. Each son born to a woman with a dystrophin mutation on one of her two X chromosomes has a 50 percent chance of inheriting the flawed gene and having Becker muscular dystrophy. Each of her daughters has a 50 percent chance of inheriting the mutation and being a carrier.
Becker muscular dystrophy primarily affects boys and men, who inherit the disease through their mothers. Women can be carriers but usually exhibit no symptoms, but can have a child with the mutation or the disease. Becker muscular dystrophy carriers are at risk for cardiomyopathy.
Figure 1. Becker muscular dystrophy X-linked inheritance pattern
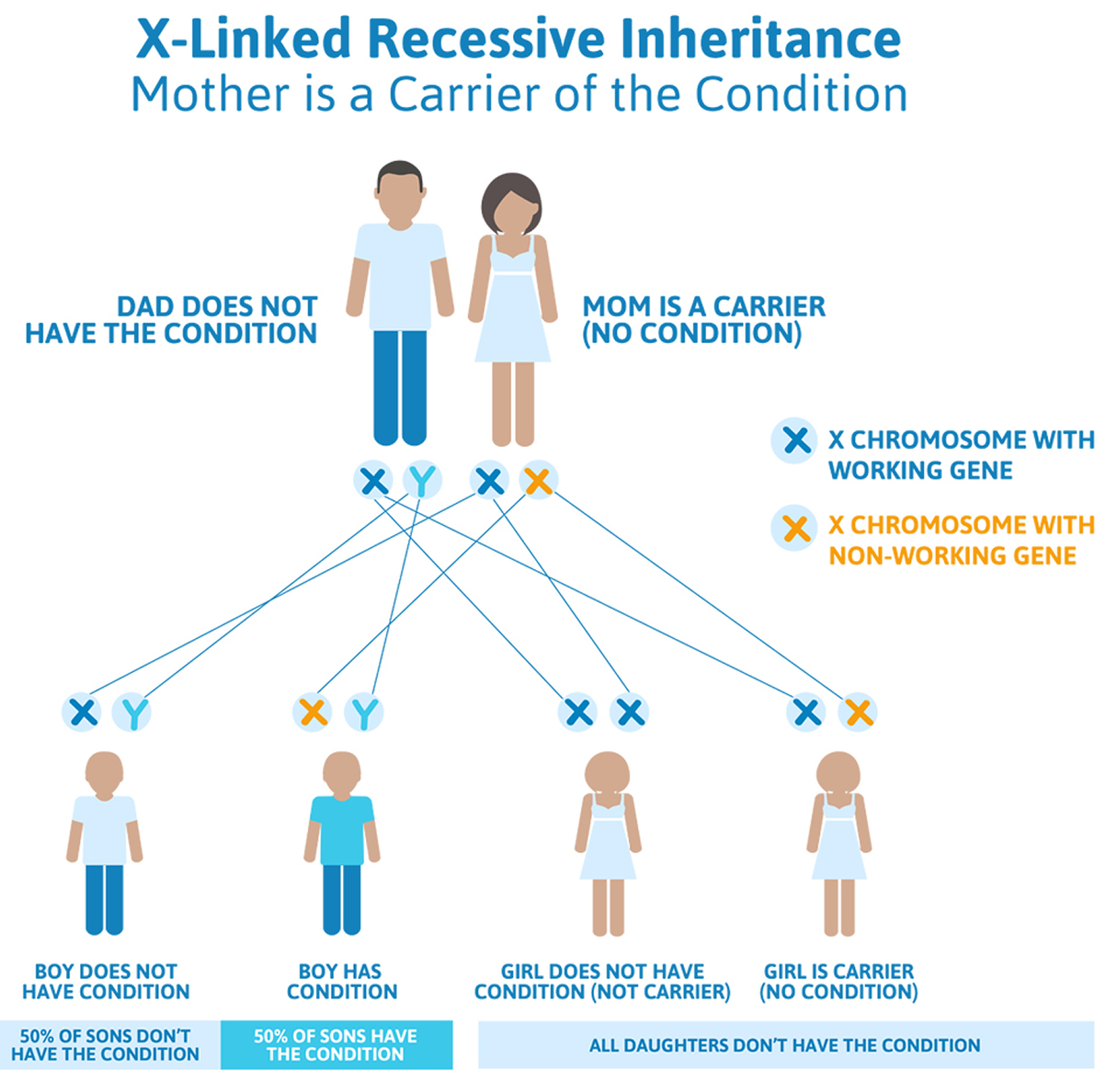
Figure 2. Becker muscular dystrophy (dystrophin)
If a mother gives birth to a son with Becker muscular dystrophy, there’s always the possibility that more than one of her egg cells has a dystrophin gene mutation, putting her at higher-than-average risk for passing the mutation to another child. Once the new mutation has been passed to a son or daughter, he or she can pass it to the next generation.
A man with Becker muscular dystrophy can’t pass the flawed gene to his sons because he gives a son a Y chromosome, not an X. But he’ll certainly pass it to his daughters, because each daughter inherits her father’s only X chromosome. They’ll then be carriers, and each of their sons will have a 50 percent chance of developing the disease, and so on.
Why don’t girls usually get Becker muscular dystrophy?
When a girl inherits a flawed dystrophin gene from one parent, she usually also gets a healthy dystrophin gene from her other parent, giving her enough of the protein to protect her from Becker muscular dystrophy. Males who inherit the mutation get Becker muscular dystrophy because they have no second dystrophin gene to make up for the faulty one.
However, although girls don’t usually get the full effects of Becker muscular dystrophy, some females with the gene flaw are somewhat affected. A minority of females with the mutation are manifesting carriers, who usually have a mild form of the disorder.
For these women, the dystrophin deficiency may result in weaker muscles that fatigue easily in the back, legs and arms. Some may even need a wheelchair or other mobility aids. Manifesting carriers may have heart problems, which can show up as shortness of breath or inability to do moderate exercise. The heart problems, if untreated, can be quite serious, even life-threatening.
A female relative of someone with Becker muscular dystrophy can get a full range of diagnostic tests to determine her carrier status. If she’s found to be a carrier, regular strength evaluations and close cardiac monitoring can help her manage any symptoms that may arise.
Carriers usually have no disease symptoms
Becker muscular dystrophy life expectancy
Most people with Becker muscular dystrophy survive well into mid- to late adulthood. If the cardiac aspects of the disease are minimal, or if they are adequately controlled through medical intervention, a normal or nearly normal life span can be expected.
Becker muscular dystrophy symptoms
Becker muscular dystrophy’s onset is usually in late childhood or adolescence, and the course is slower and less predictable than that of Duchenne muscular dystrophy. The pattern of muscle loss in Becker muscular dystrophy usually begins with the hips and pelvic area, the thighs and the shoulders. To compensate for weakening muscles, the person may walk with a waddling gait, walk on his toes or stick out the abdomen. Calves are often enlarged. There can be significant heart involvement.
The rate of muscle degeneration varies a great deal from one person to another. Some men require wheelchairs by their 30s or later, while some manage for many years with minor aids, such as canes.
Pain and sensation
Because muscular dystrophy doesn’t affect nerves directly, touch and other senses remain normal, as does control over the smooth, or involuntary, muscles of the bladder and bowel, and sexual functions.
Muscle deterioration in Becker muscular dystrophy usually isn’t painful in itself. Some people report muscle cramps at times; these usually can be treated with over-the-counter pain relievers.
The heart
Like muscles in the limbs, heart muscles also can be weakened by lack of dystrophin. People with Becker muscular dystrophy often develop cardiomyopathy — heart muscle weakness — because of a deficiency of dystrophin. The muscle layer (myocardium) of the heart deteriorates, just as the skeletal muscles do.
Damage done by Becker muscular dystrophy to the heart can become life-threatening as early as the teen years, and some people with Becker muscular dystrophy have mild skeletal muscle involvement but severe cardiac problems. For these reasons, everyone with Becker muscular dystrophy should be monitored by a cardiologist.
Breathing and coughing
Respiratory muscles often stay strong in Becker muscular dystrophy for many years, but eventually, they may become weaker than is optimal for breathing and coughing (to clear secretions from the respiratory tract).
Learning
Doctors believe that dystrophin abnormalities in the brain may cause subtle cognitive and behavioral deficits. The learning problems seen in some people with Becker muscular dystrophy seem to occur in three general areas: attention focusing, verbal learning and memory, and emotional interaction. For more on coping with intellectual effects.
Becker muscular dystrophy diagnosis
The diagnosis of Becker muscular dystrophy may be made during childhood, typically after the age of about 7. Sometimes, however, it isn’t made until adolescence or even adulthood, possibly when a young man finds he can’t keep up in physical education classes or military training.
In diagnosing any form of muscular dystrophy, a doctor usually begins by taking a patient and family history, and performing a physical examination. Much can be learned from these, including the pattern of weakness. The history and physical go a long way toward making the diagnosis, even before any complicated diagnostic tests are done.
The doctor also wants to determine whether the patient’s weakness results from a problem in the muscles themselves or in the nerves that control them. Problems in the muscle-controlling nerves, or motor neurons (originating in the spinal cord and brain and reaching out to all the muscles), can cause weakness that looks like a muscle problem but really isn’t.
Other diseases have some of the same symptoms as Becker muscular dystrophy, and it has sometimes been misdiagnosed as Duchenne muscular dystrophy (DMD) or limb-girdle muscular dystrophy (LGMD). For this reason, it’s important to go through a careful diagnostic process, usually involving genetic (DNA) testing, before making an assumption that the disorder is Becker muscular dystrophy.
Early in the diagnostic process doctors often order a special blood test called a CK (creatine kinase) level. Creatine kinase is an enzyme that leaks out of damaged muscle. When elevated creatine kinase levels are found in a blood sample, it usually means muscle is being destroyed by some abnormal process, such as muscular dystrophy or inflammation. Therefore, a high creatine kinase level suggests that the muscles themselves are the likely cause of the weakness, but it doesn’t tell exactly what the muscle disorder might be.
DNA testing of the dystrophin gene to diagnose Becker muscular dystrophy is now widely available and is usually done from a blood sample. In many cases, the DNA test alone can tell families and doctors with a fairly high degree of certainty whether the disease course is more likely to be Becker muscular dystrophy or Duchenne muscular dystrophy (DMD).
Female relatives of men and boys with Becker muscular dystrophy can undergo DNA testing to see if they are carriers of the disease. If they are, they can give birth to children who are themselves carriers or who will develop Becker muscular dystrophy.
In some cases, to be more certain about the disease and its course, a doctor may suggest a muscle biopsy in which a small sample of muscle is taken for special examination. Muscle biopsies also may be taken as part of a research study.
Becker muscular dystrophy treatment
Most therapies are supportive in nature.
People with Becker muscular dystrophy may have unexpected adverse reactions to certain types of anesthesia. It’s important that the surgical team know about that the patient has Becker muscular dystrophy so that complications can be avoided or quickly treated.
Braces, scooters and wheelchairs
Braces, also called orthoses, can support just the ankle and foot or extend over the knee. Ankle-foot orthoses are sometimes prescribed for night wear to keep feet from pointing downward and keep the Achilles tendon stretched. Orthoses also are known as orthotics.
Some people with Becker muscular dystrophy ultimately require wheelchairs or scooters. Although some look at these devices as symbols of disability, most users find they’re actually more mobile, energetic and independent when using a wheelchair than when trying to walk on very weak legs. Scooters and wheelchairs are especially valuable when covering long distances.
Cardiac care
Cardiomyopathy, which means deterioration of the heart muscle, is common in Becker muscular dystrophy. The American Academy of Pediatrics recommends that those with Becker muscular dystrophy have cardiac evaluations at least every other year beginning at age 10.
Carriers of Becker muscular dystrophy also are at higher-than-average risk of developing cardiomyopathy. The American Academy of Pediatrics suggests that carriers should undergo a complete cardiac evaluation in late adolescence or early adulthood, or sooner if symptoms occur, and should be evaluated every five years starting at age 25 to 30.
Some people with Becker muscular dystrophy who have cardiomyopathy but generally good health have been successfully treated with heart transplants.
Contractures
As muscle deteriorates, a person with muscular dystrophy often develops fixations of the joints, known as contractures. If not treated, these can become severe, causing discomfort and restricting mobility and flexibility. The impact of Becker muscular dystrophy can be significantly minimized by keeping the body as flexible, upright and mobile as possible.
There are several ways to minimize and postpone contractures. Range-of-motion exercises, performed on a regular schedule, help delay contractures by keeping tendons from shortening prematurely. It’s important that a physical therapist demonstrate the correct way to do range-of-motion exercises.
Braces on the lower legs help keep the limbs stretched and flexible, delaying the onset of contractures.
When contractures have advanced, surgery may be performed to relieve them. A tendon release procedure, also called heel cord surgery, can treat ankle and other contractures while the child is still walking.
Diet
No special dietary restrictions or additions are known to help in Becker muscular dystrophy. Most doctors recommend a diet similar to that for any growing boy, but with a few modifications.
A combination of immobility and weak abdominal muscles can lead to severe constipation, so the diet should be high in fluid and fiber, with fresh fruits and vegetables dominant.
For boys and men who use power wheelchairs, aren’t very active or who take prednisone, excessive weight gain can occur. Caloric intake should be restricted to keep weight down, as obesity puts greater stress on already weakened skeletal muscles and the heart. Doctors have found that a low-calorie diet doesn’t have any harmful effect on the muscles.
Those on prednisone and those with cardiomyopathy may require a sodium-restricted diet.
Exercise
Exercise can help build skeletal muscle, keep the cardiovascular system healthy and contribute to feeling better. But in muscular dystrophy, too much exercise could damage muscle. Consult with your doctor about how much exercise is best. A person with Becker muscular dystrophy can exercise moderately but shouldn’t go to the point of exhaustion.
Some experts recommend swimming and water exercises (aquatic therapy) as a good way to keep muscles as toned as possible without causing undue stress on them. The buoyancy of the water helps protect against certain kinds of muscle strain and injury.
Before undertaking any exercise program, make sure to have a cardiac evaluation.
Learning disabilities
Dystrophin deficiency can cause some cognitive problems in some people. Children and adults with Becker muscular dystrophy who are suspected of having a learning disability can be evaluated by a neuropsychologist through a school system’s special education department.
If a learning disability is diagnosed, educational and psychological interventions can begin right away. The specialist may prescribe exercises and techniques that can help improve these deficits, and schools can provide special help with learning.
Medications
Medications that lessen the workload on the heart are sometimes prescribed for Becker muscular dystrophy. There’s some evidence that treatment with angiotensin converting enzyme (ACE) inhibitors and beta blockers can slow the course of cardiac muscle deterioration in Becker muscular dystrophy if the medications are started as soon as abnormalities on an echocardiogram (imaging of the heart) appear, but before symptoms occur.
Medications belonging to a group known as corticosteroids have been found effective in slowing the course of Duchenne muscular dystrophy. Data for or against the use of corticosteroids in Becker muscular dystrophy are lacking. However, some physicians prescribe corticosteroids for severe Becker muscular dystrophy in much the same way as they would for Duchenne muscular dystrophy (DMD), if the patient or family wants to try this type of medication.
Prednisone is by far the most commonly prescribed corticosteroid for Duchenne muscular dystrophy/Becker muscular dystrophy in the United States. When taking at relatively high doses for long periods of time, it can have significant side effects, such as weight gain, decreased bone density, behavioral abnormalities, cataracts and growth retardation.
Physical and occupational therapy
A physical therapy program is usually part of the treatment for Becker muscular dystrophy.
The primary goals of physical therapy are to allow greater motion in the joints and to prevent contractures and scoliosis (spinal curvature). Occupational therapy focuses on specific activities and functions, such as work tasks, recreation, driving, dressing or using a computer.
Respiratory care
In some people with Becker muscular dystrophy, particularly as they age, breathing muscles can weaken, resulting in less than optimal breathing, particularly during sleep. This can be treated by a noninvasive strategy known as bilevel positive airway pressure. Coughing muscles also can become weak, allowing mucus to build up in the respiratory tract, which can lead to obstruction and infection. A device known as a CoughAssist can help with this problem.
Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is a genetic disorder characterized by progressive muscle degeneration and weakness. Duchenne muscular dystrophy is caused by an absence of dystrophin, a protein that helps keep muscle cells intact. Symptom onset is in early childhood, usually between ages 3 and 5. The disease primarily affects boys, but in rare cases it can affect girls.
Duchenne muscular dystrophy has an X-linked recessive inheritance pattern and is passed on by the mother, who is referred to as a carrier.
Duchenne muscular dystrophy causes
Duchenne muscular dystrophy occurs because the mutated gene fails to produce virtually any functional dystrophin. On the other hand, individuals with Becker muscular dystrophy genetic mutations make dystrophin that is partially functional, which protects their muscles from degenerating as badly or as quickly as in Duchenne muscular dystrophy.
Mutations in the DMD gene cause the Duchenne and Becker forms of muscular dystrophy. The DMD gene provides instructions for making a protein called dystrophin. This protein is located primarily in skeletal and cardiac muscle, where it helps stabilize and protect muscle fibers. Dystrophin may also play a role in chemical signaling within cells.
Mutations in the DMD gene alter the structure or function of dystrophin or prevent any functional dystrophin from being produced. Muscle cells without enough of this protein become damaged as muscles repeatedly contract and relax with use. The damaged fibers weaken and die over time, leading to the muscle weakness and heart problems characteristic of Duchenne and Becker muscular dystrophies. Mutations that lead to an abnormal version of dystrophin that retains some function usually cause Becker muscular dystrophy, while mutations that prevent the production of any functional dystrophin tend to cause Duchenne muscular dystrophy.
Because Duchenne and Becker muscular dystrophies result from faulty or missing dystrophin, these conditions are classified as dystrophinopathies.
Lack of dystrophin causes muscle damage and progressive weakness, beginning in early childhood.
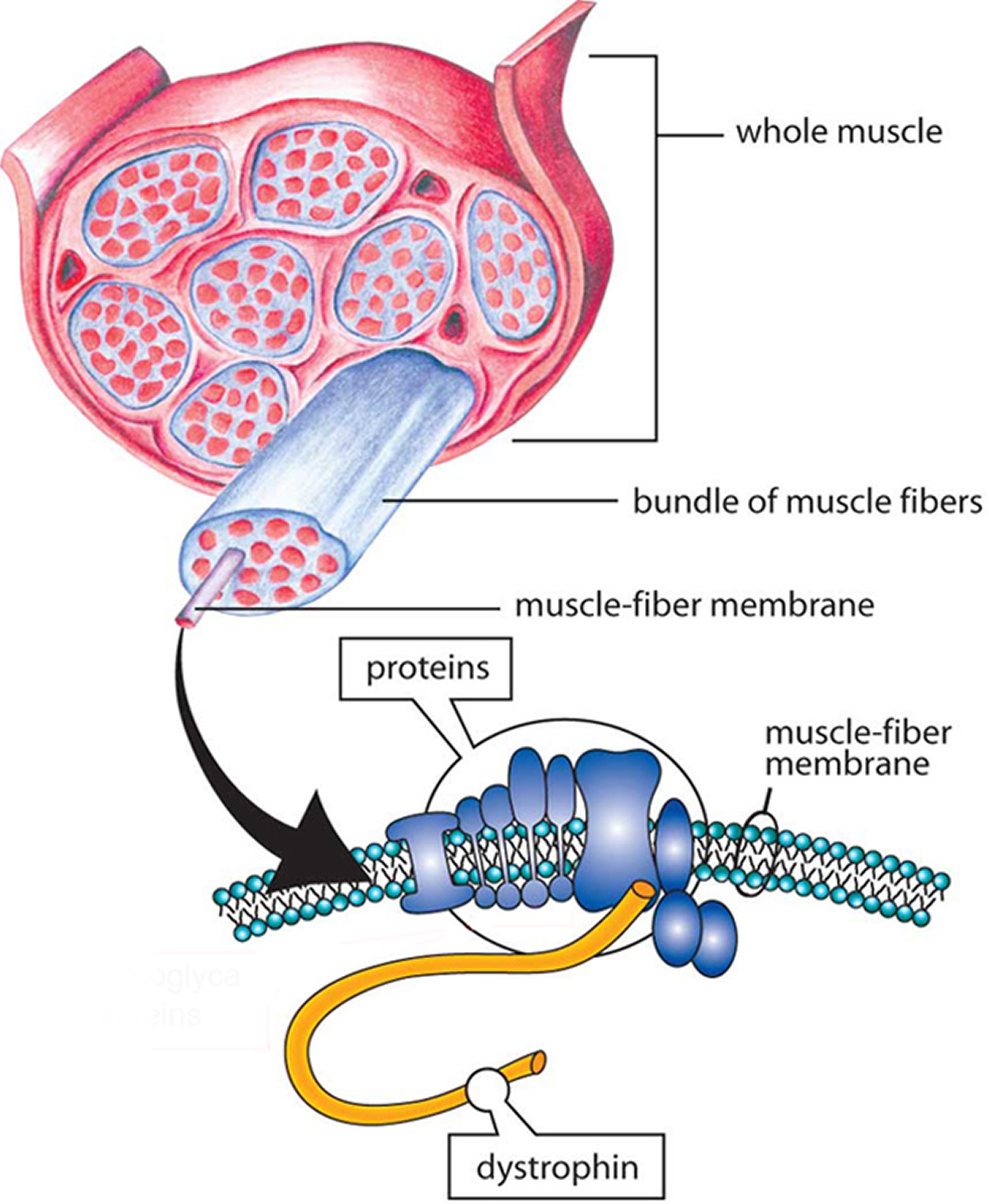
The dystrophin protein transfers the force of muscle contraction from the inside of the muscle cell outward to the cell membrane. Because it connects the center of the muscle cell to the periphery, the dystrophin protein is extremely long. One end is specialized for linking to the muscle interior and the other end is specialized for linking to a variety of proteins at the cell membrane. The long middle section, called the rod domain, is taken up by a series of repeating units called spectrin repeats.
The repeated spectrin units in the middle of the protein play an important role in linking the two ends but studies have shown that the exact number of these units is not critical for the function of the protein as a whole. Many cases of Duchenne muscular dystrophy are caused by mutations in the part of the gene that encodes this middle section. Production of the entire protein stops when the mutation is encountered.
The absence of dystrophin sets in motion a cascade of deleterious effects. Fibrous tissue begins to form in the muscle and the body’s immune system increases inflammation. In addition to its force-transfer role, dystrophin provides the scaffold for holding numerous molecules in place near the cell membrane. Loss of dystrophin displaces these molecules, with consequent disruptions in their functions.
Duchenne muscular dystrophy is inherited in an X-linked pattern, because the gene that can carry a Duchenne muscular dystrophy-causing mutation is on the X chromosome. Every boy inherits an X chromosome from his mother and a Y chromosome from his father, which is what makes him male. Girls get two X chromosomes, one from each parent.
Each son born to a woman with a dystrophin mutation on one of her two X chromosomes has a 50 percent chance of inheriting the flawed gene and having Duchenne muscular dystrophy. Each of her daughters has a 50 percent chance of inheriting the mutation and being a carrier. Carriers may not have any disease symptoms but can have a child with the mutation or the disease. Duchenne muscular dystrophy carriers are at risk for cardiomyopathy.
Although Duchenne muscular dystrophy often runs in a family, it’s possible for a family with no history of Duchenne muscular dystrophy to suddenly have a son with the disease. There are two possible explanations:
- The genetic mutation leading to Duchenne muscular dystrophy may have existed in the females of a family for some generations without anyone knowing it. Perhaps no male children were born with the disease, or, even if a boy in an earlier generation was affected, relatives may not have known what disease he had.
- The second possibility is that the child with Duchenne muscular dystrophy has a new genetic mutation that arose in one of his mother’s egg cells. Since this mutation isn’t in the mother’s blood cells, it’s impossible to detect by standard carrier testing.
If a mother gives birth to a child with Duchenne muscular dystrophy, there’s always the possibility that more than one of her egg cells has a dystrophin gene mutation, putting her at higher than average risk for passing the mutation to another child. And once the new mutation has been passed to a son or daughter, he or she can pass it to the next generation.
A man with Duchenne muscular dystrophy can’t pass the flawed gene to his sons because he gives a son a Y chromosome, not an X. But he’ll certainly pass it to his daughters, because each daughter inherits her father’s only X chromosome. They’ll then be carriers, and each of their sons will have a 50 percent chance of developing the disease and so on.
Figure 2. Duchenne muscular dystrophy X-linked recessive inheritance pattern
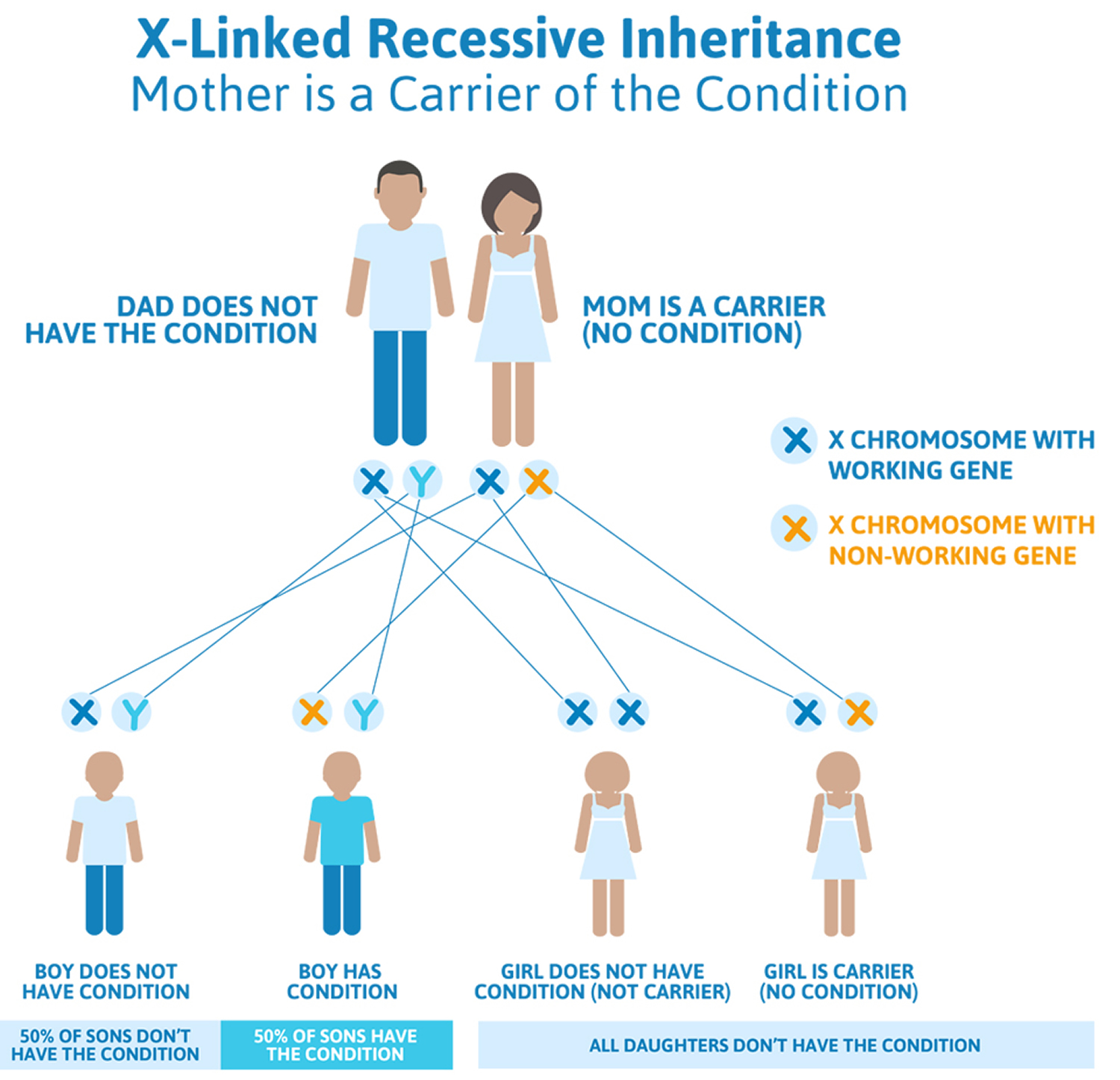
Figure 4. Duchenne muscular dystrophy
Why don’t girls usually get Duchenne muscular dystrophy?
When a girl inherits a flawed dystrophin gene from one parent, she usually also gets a healthy dystrophin gene from her other parent, giving her enough of the protein to protect her from the disease. Males who inherit the mutation get the disease because they have no second dystrophin gene to make up for the faulty one.
Early in the embryonic development of a female, either the X chromosome from the mother (maternal X) or the one from the father (paternal X) is inactivated in each cell. The choice of which chromosome to inactivate is random. In each cell, there’s a 50 percent chance that either the maternal or paternal X chromosome will be inactivated, with the other left active.
Most of the time, it doesn’t matter how many inactivated maternal and paternal X chromosomes a female has. But when there’s a mutation in an X-chromosome gene, such as in the gene for dystrophin, it matters a lot.
If a girl or woman has to rely on too many X chromosomes with the dystrophin gene mutation (meaning the Xs with the functional dystrophin genes are mostly inactivated), she’s likely to develop symptoms of Duchenne muscular dystrophy or Becker muscular dystrophy.
Usually, however, girls don’t experience the full effects of Duchenne muscular dystrophy the way boys do, although they still have symptoms of muscle weakness. A minority of females with the mutation, called manifesting carriers, have some signs and symptoms of Duchenne muscular dystrophy.
For these women, the dystrophin deficiency may result in weaker muscles in the back, legs and arms that fatigue easily. Manifesting carriers may have heart problems, which can show up as shortness of breath or inability to do moderate exercise. The heart problems, if untreated, can be quite serious, even life-threatening.
In very rare instances, a girl may lack a second X chromosome entirely, or her second X may have sustained serious damage. In these cases, she makes little or no dystrophin (depending on the type of dystrophin mutation), and she develops Duchenne muscular dystrophy or Becker muscular dystrophy just as a boy would.
A female relative of a boy with Duchenne muscular dystrophy can get a full range of diagnostic tests to determine her carrier status. If she is found to be a Duchenne muscular dystrophy carrier, regular strength evaluations and close cardiac monitoring can help her manage any symptoms that may arise.
Duchenne muscular dystrophy symptoms
Children with Duchenne muscular dystrophy are often late walkers.
In toddlers, parents may notice enlarged calf muscles. This enlargement is known as pseudohypertrophy, or “false enlargement,” because the muscle tissue is abnormal and may contain scar tissue.
A preschooler with Duchenne muscular dystrophy may seem clumsy and fall often. Parents also may notice that children have trouble climbing stairs, getting up from the floor (Gowers sign, using their hands to push on their legs to get up) or running. Intellectual impairment may also be present.
Figure 5. Duchenne muscular dystrophy
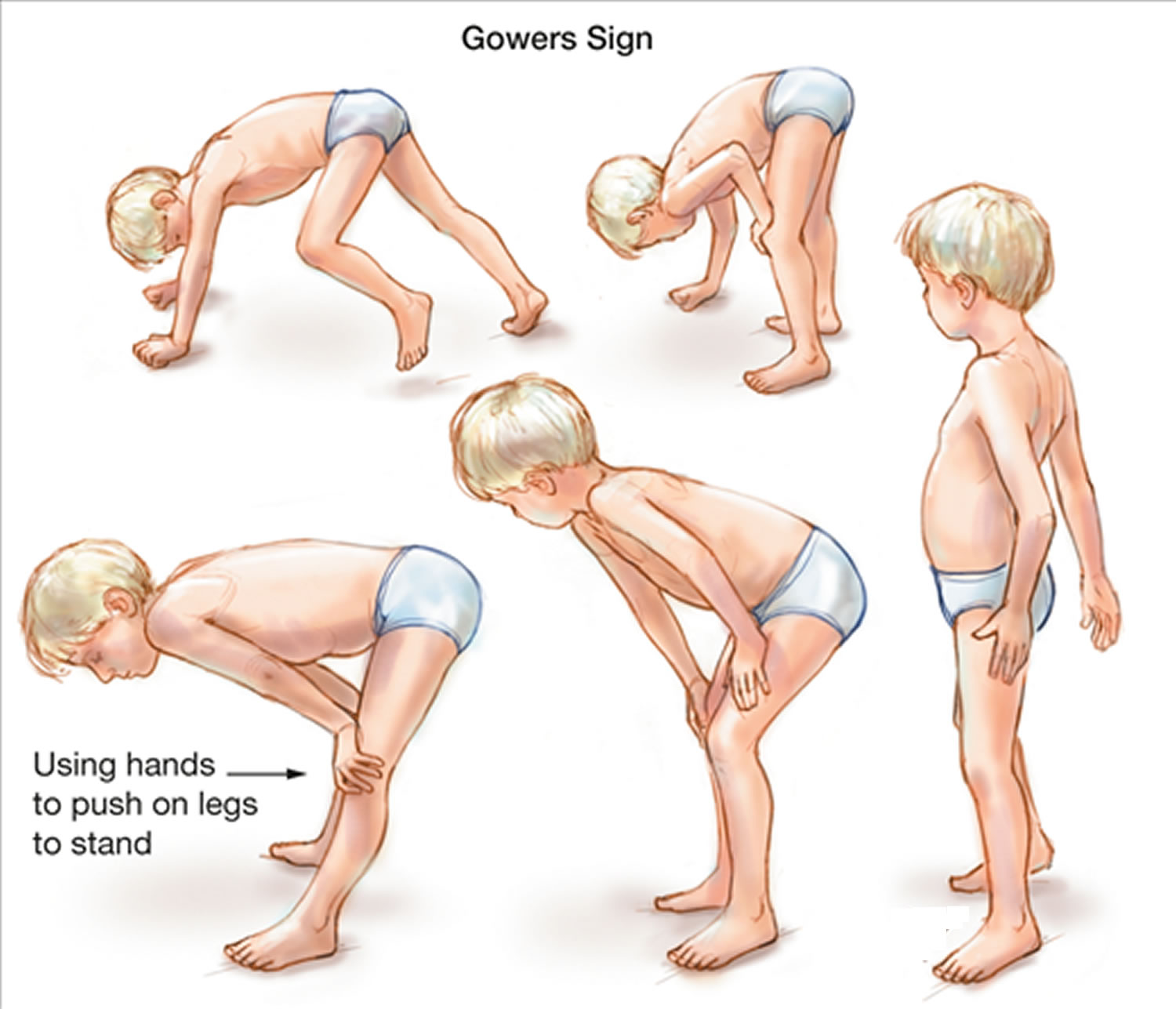
By school age, children may walk on their toes or the balls of their feet with a slightly waddling gait, and fall frequently. To try to keep their balance, they may stick out their bellies and pull back their shoulders. Children also have difficulty raising their arms.
Many children with Duchenne muscular dystrophy begin using a wheelchair sometime between ages 7 and 12. Transition to a wheelchair usually is a gradual process; at first, the chair may be required only to conserve the child’s energy when covering long distances. (Children often experience renewed independence once they fully transition to a power wheelchair.)
In the teen years, activities involving the arms, legs or trunk may require assistance or mechanical support.Pain and sensation
Pain and sensation
The muscle deterioration in Duchenne muscular dystrophy isn’t usually painful in itself. Some people report muscle cramps at times; these usually can be treated with over-the-counter pain relievers.
Because muscular dystrophy doesn’t affect nerves directly, touch and other senses are normal, as is control over the smooth, or involuntary, muscles of the bladder and bowel, and sexual functions.
The heart
Lack of dystrophin can weaken the muscle layer in the heart (myocardium), resulting in a condition calledcardiomyopathy. Over time, sometimes as early as the teen years, the damage done by Duchenne muscular dystrophy to the heart can become life-threatening. The heart should be monitored closely, usually by a pediatric cardiologist.
Respiratory function
Beginning at about 10 years of age, the diaphragm and other muscles that operate the lungs may weaken, making the lungs less effective at moving air in and out. Although the child may not complain of shortness of breath, problems that indicate poor respiratory function include headaches, mental dullness, difficulty concentrating or staying awake, and nightmares.
Weakened respiratory muscles make it difficult to cough, leading to increased risk of serious respiratory infection. A simple cold can quickly progress to pneumonia. It’s important to get flu shots, and when infections occur, to get prompt treatment.
Learning
About a third of boys with Duchenne muscular dystrophy have some degree of learning disability, although few have serious mental retardation. Doctors believe that dystrophin abnormalities in the brain may have subtle effects on cognition and behavior. Learning problems in Duchenne muscular dystrophy occur in three general areas: attention focusing, verbal learning and memory, and emotional interaction.
Children suspected of having a learning disability can be evaluated by a developmental or pediatric neuropsychologist through the school system’s special education department.
If a learning disability is diagnosed, educational and psychological interventions can begin right away. The specialist may prescribe exercises and techniques that can help improve these areas, and the school also can provide special help with learning.
Duchenne muscular dystrophy diagnosis
In diagnosing any form of muscular dystrophy, a doctor usually begins by taking a patient and family history, and performing a physical examination. Much can be learned from these, including the pattern of weakness. The history and physical go a long way toward making the diagnosis, even before any complicated diagnostic tests are done.
Early in the diagnostic process, doctors often order a blood test called a CK (creatine kinase) level. Creatine kinase is an enzyme that leaks out of damaged muscle. When elevated creatine kinase levels are found in a blood sample, it usually means muscle is being destroyed by some abnormal process, such as a muscular dystrophy or inflammation. A very high creatine kinase level suggests that the muscles themselves (and not the nerves that control them) are the likely cause of the weakness, although it doesn’t tell exactly what the muscle disorder might be.
Genetic testing
Genetic testing involves analyzing the DNA of any cells (usually blood cells are used) to see whether there is a mutation in the dystrophin gene, and if so, exactly where it occurs. Such DNA testing for dystrophin mutations is widely available in the United States.
Female relatives of men and boys with Duchenne muscular dystrophy can undergo DNA testing to see if they are carriers of the disease. Women who are Duchenne muscular dystrophy carriers can pass on the disease to their sons and their carrier status to their daughters. In a minority of cases, girls and women who are Duchenne muscular dystrophy carriers may themselves show symptoms of Duchenne muscular dystrophy, such as muscle weakness and heart problems. These symptoms may not show up until adulthood.
Several experimental drugs currently in development to treat Duchenne muscular dystrophy require knowledge of the person’s precise genetic mutation, so genetic testing has become important not only for diagnosis but possibly for future treatments.
Muscle biopsy
To obtain more information, the doctor may order a muscle biopsy, the surgical removal of a small sample of muscle from the patient. By examining this sample, doctors can tell a great deal about what’s actually happening inside the muscles.
Modern techniques can use the biopsy to distinguish muscular dystrophies from inflammatory and other disorders, and also to distinguish among different forms of muscular dystrophy. For instance, the amount of functional dystrophin protein found in a muscle biopsy sample sheds light on whether the disease course is likely to be Duchenne muscular dystrophy (with no dystrophin present) or the milder Becker muscular dystrophy (with some partially functional dystrophin present).
Duchenne muscular dystrophy treatment
People with Duchenne muscular dystrophy may have unexpected adverse reactions to certain types of anesthesia. It’s important that the surgical team know about the patient’s Duchenne muscular dystrophy so that complications can be avoided or quickly treated.
Braces, standing frames and wheelchairs
Braces, also called orthoses, support the ankle and foot, or may extend up over the knee. Ankle-foot orthoses (AFOs) are sometimes prescribed for night wear to keep the foot from pointing downward and keep the Achilles tendon stretched while the child is sleeping.
Standing for a few hours each day, even with minimal weight bearing, promotes better circulation, healthier bones and a straight spine. A standing walker or standing frame can assist people with Duchenne muscular dystrophy to stand. Some wheelchairs will raise the user into a standing position.
Sooner or later, a wheelchair is needed in Duchenne muscular dystrophy, typically by about age 12. Unless there’s an injury, such as a broken leg, wheelchair use usually is gradual. Many at first use wheelchairs for long distances, such as at school or the mall, and continue to walk at home.
Although the child and parents may dread the wheelchair as a symbol of disability, most find that when they start to use one, they are actually more mobile, energetic and independent than when trying to walk.
Other mobility and positioning aids can help parents and caregivers. Among the simplest aid is a transfer board for helping the person move in and out of the wheelchair. Mechanical lifts, shower chairs and electronic beds also can be useful.
Cardiac care
The American Academy of Pediatrics recommends that people with Duchenne muscular dystrophy have a complete cardiac evaluation by a specialist beginning in early childhood and again at least every other year until age 10. After that, the evaluations should be done every year or at the onset of symptoms of heart weakness, such as fluid retention or shortness of breath.
Female carriers of Duchenne muscular dystrophy are at higher-than-average risk of developing cardiomyopathy. The academy suggests that carriers should undergo a complete cardiac evaluation in late adolescence or early adulthood, or sooner if symptoms occur, and that they should be evaluated every five years starting at age 25 to 30.
There’s some preliminary evidence that treatment with angiotensin converting enzyme (ACE) inhibitors and beta blockers can slow the course of cardiac muscle deterioration in Duchenne muscular dystrophy if the medications are started as soon as abnormalities on an echocardiogram (ultrasound imaging of the heart) appear but before symptoms occur.
Contractures
The impact of Duchenne muscular dystrophy can be minimized significantly by keeping the body as flexible, upright and mobile as possible. There are several ways to do this.
As muscle deteriorates, a person with muscular dystrophy often develops fixations of the joints, known ascontractures. If not treated, these will become severe, causing discomfort and restricting mobility and flexibility. Contractures can affect the knees, hips, feet, elbows, wrists and fingers.
However, there are many ways to minimize and postpone contractures. Range-of-motion exercises, performed on a regular schedule, help delay contractures by keeping tendons from shortening prematurely. It’s important that a physical therapist show you how to do range-of-motion exercises correctly.
Braces on the lower legs also can help keep the limbs stretched and flexible, delaying the onset of contractures.
When contractures have advanced, surgery may be performed to relieve them. A tendon release procedure, also called heel cord surgery, is often done to treat ankle and other contractures while the child is still walking. Usually the boy will need to wear lower leg braces after this.
Diet
No special dietary restrictions or additions are known to help in Duchenne muscular dystrophy. Most doctors recommend a diet similar to that for any growing boy but with a few modifications.
A combination of immobility and weak abdominal muscles can lead to severe constipation, so the diet should be high in fluid and fiber, with fresh fruits and vegetables dominant.
For boys who use power wheelchairs, take prednisone or who aren’t very active, excessive weight gain can become a problem. For these boys, caloric intake should probably be somewhat restricted to keep weight down. Obesity puts greater stress on already weakened skeletal muscles and the heart. Doctors have found that a low-calorie diet doesn’t have any harmful effect on the muscles.
Those on prednisone and those with heart problems may need a sodium-restricted diet.
Exercise
Exercise can help build skeletal muscle, keep the cardiovascular system healthy, and contribute to feeling better. But in muscular dystrophy, too much exercise could damage muscle. Consult with your doctor about how much exercise is best. A person with Duchenne muscular dystrophy can exercise moderately but shouldn’t go to the point of exhaustion.
Many experts recommend swimming and water exercises (aquatic therapy) as a good way to keep muscles as toned as possible without causing undue stress on them. The buoyancy of the water helps protect against certain kinds of muscle strain and injury.
Before undertaking any exercise program, make sure to have a cardiac evaluation.
Learning disabilities
Children with Duchenne muscular dystrophy who are suspected of having a learning disability can be evaluated by a developmental or pediatric neuropsychologist through the school system’s special education department.
If a learning disability is diagnosed, educational and psychological interventions can begin right away. The specialist may prescribe exercises and techniques, and the school also can provide special help with learning.
Medications
Medications that lessen the workload on the heart are sometimes prescribed for Duchenne muscular dystrophy.
Medications belonging to a group known as corticosteroids have been found to be effective in slowing the course of Duchenne muscular dystrophy. The corticosteroids prednisone and deflazacort are beneficial in the treatment of Duchenne muscular dystrophy.
The FDA on Feb. 9, 2017, approved deflazacort (brand name Emflaza) to treat Duchenne muscular dystrophy 2. EMFLAZA (deflazacort) is a corticosteroid drug for the treatment of Duchenne muscular dystrophy in patients 5 years of age and older.
- EMFLAZA is taken by mouth once a day in the form of a pill or liquid at the recommended dose 3.
- EMFLAZA possible side effects:
- EMFLAZA, like other corticosteroid drugs may cause some serious side effects such as hormonal suppression, infections, heart and kidney problems, stomach or gut rupture, changes in mood and behavior, weakening of the bones, and some eye disorders. The most common side effects are Cushingoid appearance (hormonal condition causing facial puffiness), weight gain, increased appetite, upper respiratory tract infection, cough, frequent urination, excessive hairiness, and increased fat around the waist.
Several high-quality studies of these medications in Duchenne muscular dystrophy showed a significant increase in strength, timed muscle function (such as the time it took to climb stairs) and pulmonary function.
Chronic use of corticosteroids is part of the standard of care for Duchenne muscular dystrophy, but such treatment can lead to side effects and rapid withdrawal of corticosteroids can result in life-threatening complications.
Physical and occupational therapy
A physical therapy program is usually part of the treatment for Duchenne muscular dystrophy. The primary goals of physical therapy are to allow greater motion in the joints and to prevent contractures and scoliosis.
While physical therapy emphasizes mobility and, where possible, strengthening of large muscle groups, occupational therapy focuses on specific activities and functions. Occupational therapy can help with tasks for work, recreation or daily living, such as dressing or using a computer.
Respiratory care
As the muscles that assist in breathing get weaker, the bronchial system must be kept free of secretions, either by using a cough assist device or by manual assisted coughing with the help of a caregiver. A respiratory therapist or pulmonologist can be consulted for the needed information. At some point, assisted ventilation may be needed to help provide sufficient air flow into and out of the lungs.
The first step in using assisted ventilation is usually a noninvasive device, meaning one that doesn’t require any surgical procedures. The person receives air under pressure through a mask, nosepiece or mouthpiece. Noninvasive ventilation usually is required only part time, often only during sleep.
If round-the-clock ventilatory support becomes necessary, it’s possible to use noninvasive ventilation full time, under the care of a doctor knowledgeable in this practice. Some young men choose to switch to an invasive system, which means that a surgical opening called a tracheostomy is performed, allowing air to be delivered directly into the trachea (windpipe).
Spinal curvatures
In young men with Duchenne muscular dystrophy, the spine can be gradually pulled into a curved shape. The spine may curve from side to side (scoliosis) or forward in a “hunchback” shape (kyphosis).
Scoliosis usually appears after a boy has started using a wheelchair full time. The “swayback” curvature that’s sometimes seen in those who are still walking is called lordosis.
Severe scoliosis can interfere with sitting, sleeping and even breathing, so measures should be taken to try to prevent it.
Exercises to keep the back as straight as possible and advice about sitting and sleeping positions can be obtained from a physical therapist.
Spine-straightening surgery involves inserting metal rods with hooks into the spine.
Surgery for youngsters with Duchenne muscular dystrophy is usually performed in adolescence.
Congenital muscular dystrophy
Congenital muscular dystrophy refers to a group of muscular dystrophies that become apparent at or near birth. Congenital muscular dystrophy results in overall muscle weakness with possible joint stiffness or looseness. Depending on the type, congenital muscular dystrophy may involve spinal curvature, respiratory insufficiency, intellectual disabilities, learning disabilities, eye defects or seizures.
Congenital myotonic dystrophy affects about 1 per 10,000 births
Congenital muscular dystrophy types
At least 33 different types of congenital muscular dystrophy are now recognized.
1)Congenital muscular dystrophy with adducted (drawn inward) thumbs, ophthalmoplegia (paralyzed eye muscles) and intellectual disability
Description: rare form of congenital muscular dystrophy with inward-drawn thumbs, contractures (permanent shortening) of the toe joints, weakness, lack of muscle tone, delayed walking, paralysis of eye muscles and intellectual disability
Molecular basis: unknown
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
congenital muscular dystrophy with cardiomyopathy
Description: weakness beginning within first year; delayed motor milestones; slowly progressive; walking achieved in adolescence; contractures of the joints, neck and spine; progressive cardiomyopathy (cardiac muscle deterioration) beginning ages 5-12; cardiac rhythm abnormalities
Molecular basis: mutations in titin gene, causing deficiency of titin protein; protein normally plays a role in muscle assembly and force transmission in skeletal and cardiac muscles
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
2) Congenital muscular dystrophy with central nervous system atrophy and absence of large myelinated fibers in peripheral nervous system
Description: onset in newborn period; weakness, lack of muscle tone, poor motor function; respiratory failure in some; diminished size of major parts of the brain; joint contractures
Molecular basis: unknown
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
congenital muscular dystrophy with cerebellar atrophy (diminished size of the cerebellum, a part of the brain involved in motor control)
Description: nonprogresssive form of congenital muscular dystrophy with onset by 7 months, weakness, lack of muscle tone, delayed motor milestones, lack of coordination of movements, difficulty speaking, involuntary eye movements and intellectual disability
Molecular basis: unknown
Inheritance pattern: possibly recessive (requires mutations in both copies of a gene to produce symptoms)
3) Congenital muscular dystrophy with desmin inclusions (abnormal accumulations of the muscle protein desmin in some muscle fibers)
Description: onset of progressive weakness and low muscle tone at birth or during early infancy; small muscles; cardiac abnormalities in some; spinal curvatures at 8-14 years; joint contractures; respiratory impairment
Molecular basis: mutations in SEPN1 gene, causing deficiency of SEPN1 protein; protein is thought to play a role in early development or regeneration of muscle tissue
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
congenital muscular dystrophy with integrin alpha 7 mutations
Description: early-onset low muscle tone, weakness; may walk at age 2-3; respiratory involvement with disease progression
Molecular basis: mutations in the integrin-alpha 7 gene, causing a deficiency of the integrin alpha 7 beta 1 protein; protein normally provides a link between muscle fibers and the surrounding matrix
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
4) Congenital muscular dystrophy with joint hyperlaxity (abnormally flexible joints)
Description: weakness, poor muscle tone and contractures from birth; slowly progressive; walking at 1-3 years; wheelchair later, between teens and 30s; reduced respiratory capacity that does not progress; contractures in some joints and abnormal flexibility in others; spinal curvature possible; normal intelligence
Molecular basis: thought to be due to mutations in the integrin alpha 9 gene, causing a deficiency of the integrin alpha 9 protein; protein normally plays a role in how cells stick to each other and to their surroundings
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
5) Congenital muscular dystrophy with familial junctional epidermolysis bullosa
Description: onset of weakness or poor muscle tone, with skin blistering, at birth; skin blisters with injury and heat; slowly progressive; many need wheelchair by age 10; elbow contractures; respiratory impairment; cardiomyopathy; diminished brain size; treatment with 3,4-diaminopyridine, which increases signal transmission from nerve to muscle, may be helpful
Molecular basis: mutations in the gene for the plectin protein, causing a deficiency of this protein; protein is thought to provide mechanical strength to cells and tissues
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
6) Congenital muscular dystrophy with muscle hypertrophy (enlargement of muscles); also called MDC1C
Description: low muscle tone and weakness starting in first weeks of life; may sit unassisted but walking not achieved; some muscles enlarged, especially calf muscles; other muscles small, especially in shoulder area; joint contractures in some; cognitive function usually normal; mild intellectual disability or speech problems can occur
Molecular basis: mutations in gene for fukutin-related protein (FKRP), leading to FKRP deficiency; protein normally helps glycosylate (sugar-coat) a protein called alpha-dystroglycan
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
7) Congenital muscular dystrophy with muscle hypertrophy and respiratory failure; also called MDC1B
Description: early-onset weakness with involvement of the diaphragm and respiratory failure; walking at 1.5 to 2.5 years; weakness does not appear to progress; generalized muscle enlargement; contractures in ankles; spinal rigidity in about 50 percent; normal intelligence
Molecular basis: mutations in unknown gene on chromosome 1
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
8) Congenital muscular dystrophy with muscle hypertrophy and severe intellectual disability; also called MDC1D
Description: onset around 5 months, with low muscle tone and weakness; some muscles enlarged; global developmental delay; profound intellectual disability; contractures of ankles and elbows
Molecular basis: mutations in LARGE gene, leading to deficiency of LARGE protein; protein thought to play a role in sugar-coating (glycosylation) of alpha-dystroglycan protein
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
9) Congenital muscular dystrophy with myasthenic syndrome
Description: rare form of congenital muscular dystrophy with onset by time of birth; weakness, lack of muscle tone, small muscles; slowly progressive; respiratory involvement possible; most survivors able to walk as children and adults; normal intelligence
Molecular basis: DOK7 gene mutation leading to deficiency of DOK7 protein; protein normally plays a role in forming the connections between nerves and muscles
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
10) Congenital muscular dystrophy with (early) spinal rigidity
Description: onset birth to 1 year or during first decade of life; early-onset poor muscle tone, weakness; respiratory capacity often reduced; small muscles; early improvement, followed by stabilization or slow decline; spinal rigidity beginning ages 3-7, with limited ability to flex the neck and spine; spinal curvature beginning ages 4-12 and progressing; joint contractures; minor cardiac abnormalities, if any; normal intelligence
Molecular basis: mutations in SEPN1 gene, causing deficiency of SEPN1 protein; protein is thought to play a role in early development or regeneration of muscle tissue
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
11) Congenital muscular dystrophy with spinal rigidity and lamin A/C abnormality
Description: weakness within first year; respiratory involvement; rigid spine, curved spine, curved feet; cardiac rhythm abnormalities in some; premature aging in some; abnormalities of fatty tissue in some
Molecular basis: mutation in lamin A/C gene, causing an abnormality in the lamin A or C proteins; these normally form part of a membrane that surrounds the cell nucleus
Inheritance pattern: dominant (requiring a mutation in only one copy of a gene to produce symptoms)
12) Congenital muscular dystrophy with spinal rigidity and selenoprotein deficiency
Description: early-onset weakness; developmental delay; reduced respiratory capacity; fatigue; skin abnormalities; hearing loss; straight, rigid spine
Molecular basis: mutations in SBP2 gene, causing deficiency of SBP2 protein; protein normally involved in the production of selenoproteins
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
13) Congenital muscular dystrophy with structural abnormalities of mitochondria (energy-producing subunits of cells)
Description: poor muscle tone, weakness from birth, with late walking; loss of muscle tissue; cardiomyopathy; intellectual disability; mitochondria (seen in muscle biopsy samples) are enlarged and have an abnormal structure
Molecular basis: mutations in choline kinase beta gene, which leads to deficiency of choline kinase beta protein; protein normally helps make a key substance in muscle and brain
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
14) Fukuyama congenital muscular dystrophy; also called MDDGA4
Description: common in Japan; rare in Western countries; spectrum of severity; weakness and low muscle tone within first year; some achieve walking; joint contractures; spinal curvatures; seizures in 50 percent; intellectual disability; eye involvement
Molecular basis: mutations in fukutin gene, causing a deficiency of fukutin protein; protein normally helps sugar-coat (glycosylate) the alpha-dystroglycan protein in muscle and brain tissue
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
15) Merosin-deficient congenital muscular dystrophy; also called MDC1A
Description: early-onset weakness and low muscle tone; spectrum of severity; some learn to walk at age 2-3 years; spinal curvature; contractures; respiratory impairment; intelligence often normal; seizures in about 20 percent
Molecular basis: mutations in laminin alpha 2 gene, leading to deficiency of laminin alpha 2 protein; leads to deficiency of laminin 211 protein, also known as merosin; protein normally helps connect muscle fiber with surrounding matrix
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
16) Merosin-positive congenital muscular dystrophy; this is an old term referring to a variety of congenital muscular dystrophy types in which merosin is normal
Description: examples are congenital muscular dystrophy with early spinal rigidity; congenital muscular dystrophy with muscle hypertrophy; congenital muscular dystrophy with muscle hypertrophy and respiratory failure; congenital muscular dystrophy with myasthenic syndrome; and Ullrich congenital muscular dystrophy; see individual listings for different types
Molecular basis: variety of gene mutations, causing variety of protein defects that do not affect merosin protein
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
17) Santavuori muscle-eye-brain disease
Description: low muscle tone at birth; slow development; intellectual disability; eye abnormalities
Molecular basis: Mutations in POMGnT1 gene, causing deficiency of POMGnT1 protein; protein normally helps sugar-coat (glycosylate) the alpha-dystroglycan protein
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
18) Ullrich congenital muscular dystrophy
Description: early-onset weakness, poor muscle tone; severity varies; some joints have contractures; some joints have hyperlaxity (excessive flexibility); spinal rigidity, curvature; respiratory impairment; soft skin; normal cardiac function; normal intelligence
Molecular basis: mutations in COLGA1, COL6A2 or COL6A3 genes, causing deficiency of or abnormalities in collagen 6 protein; protein normally has an anchoring function in many tissues, including the matrix surrounding muscle fibers
Inheritance pattern: dominant (requiring a mutation in only one copy of a gene to produce symptoms) or recessive (requires mutations in both copies of a gene to produce symptoms)
19) Walker-Warburg syndrome: MDDGA type
Description: early-onset weakness with brain and eye abnormalities; intellectual disability
Molecular basis: mutations in B3GNT1 gene, causing deficiency of the B3GNT1 protein; protein normally helps sugar-coat (glycosylate) alpha-dystroglycan
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
20) Walker-Warburg syndrome: MDDGA1 type
Description: early-onset weakness with brain and eye abnormalities; intellectual disability
Molecular basis: mutations in POMT1 gene, causing deficiency of POMT1 protein; protein normally helps sugar-coat (glycosylate) alpha-dystroglycan
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
21) Walker-Warburg syndrome: MDDGA2 type
Description: early-onset weakness with brain and eye abnormalities; intellectual disability
Molecular basis: mutations in POMT2 gene, causing deficiency of POMT2 protein; protein normally helps sugar-coat (glycosylate) alpha-dystroglycan
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
22) Walker-Warburg syndrome: MDDGA3 type; same as Santavuori muscle-eye-brain disease
23) Walker-Warburg syndrome: MDDGA4 type; same as Fukuyama congenital muscular dystrophy
24) Walker-Warburg syndrome: MDDGB5 type; same as congenital muscular dystrophy with muscle hypertrophy (MDC1C)
25) Walker-Warburg syndrome: MDDGA6 type; same as congenital muscular dystrophy with muscle hypertrophy and severe intellectual disability (MDC1D)
26) Walker-Warburg syndrome: MDDGA7 type
Description: early-onset weakness with brain and eye abnormalities; intellectual disability
Molecular basis: mutations in ISPD gene, causing deficiency of the ISPD protein; protein normally helps sugar-coat (glycosylate) alpha-dystroglycan
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
27) Walker-Warburg syndrome: MDDGA8 type
Description: early-onset weakness with brain and eye abnormalities; intellectual disability
Molecular basis: mutations in GTDC2 gene, causing deficiency of the GTDC2 protein; protein may help sugar-coat (glycosylate) alpha-dystroglycan
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
28) Walker-Warburg syndrome: MDDGA10 type
Description: early-onset weakness with brain and eye abnormalities; intellectual disability
Molecular basis: mutations in TMEM5 gene, causing deficiency of the TMEM5 protein; protein may help sugar-coat (glycosylate) alpha-dystroglycan
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
29) Walker-Warburg syndrome: MDDGA11 type
Description: early-onset weakness with brain and eye abnormalities; intellectual disability
Molecular basis: mutations in B3GALNT2 gene, causing deficiency of the B3GALNT2 protein; protein normally helps sugar-coat (glycosylate) alpha-dystroglycan
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
30) Walker-Warburg syndrome: MDDGA12 type
Description: early-onset weakness with brain and eye abnormalities; intellectual disability
Molecular basis: Mutations in SGK196 gene, causing deficiency of SGK196 protein; protein normally may help sugar-coat (glycosylate) alpha-dystroglycan
Inheritance pattern: recessive (requires mutations in both copies of a gene to produce symptoms)
Congenital muscular dystrophy causes
Muscle cells are embedded in a web-like structure known as the extracellular matrix (ECM). The extracellular matrix is a complex mix of molecules, including many kinds of proteins linked to carbohydrates (called glycoproteins). Glycoproteins have multiple roles in the extracellular matrix, including transmitting the force of muscle contraction to the skeleton, connecting individual muscle cells to each other, transmitting signals among cells, and promoting development and repair. Many congenital muscular dystrophys are due to mutations in glycoproteins themselves, or in the enzymes that create them. One group of congenital muscular dystrophys are due to mutations in genes that help glycosylate (add sugar molecules to) the protein alpha-dystroglycan, a key glycoprotein of the extracellular matrix.
Loss of glycoproteins due to mutation interferes with normal muscle function. A consequence of most congenital muscular dystrophy-causing mutations is increased susceptibility to cellular injury from normal contraction, and/or a reduction in the ability to repair damage.
It isn’t known why the congenital muscular dystrophys cause muscle weakness earlier than other types of muscular dystrophy. One possibility is that the muscle proteins affected in congenital muscular dystrophy are required early in the development of an infant’s muscle, while muscle proteins linked to other muscular dystrophies don’t become important until the muscles begin to get a lot of use as a child grows.
It’s important to note that just because the muscle weakness in congenital muscular dystrophy starts earlier, congenital muscular dystrophy isn’t automatically more severe than other forms of muscular dystrophy. The degree and rate of progression of muscle weakness varies with different forms of congenital muscular dystrophy and from one child to the next.
In the mid-1990s, researchers found that a deficiency of a protein then called merosin and now more often called laminin 211 was the underlying cause of at least some cases of congenital muscular dystrophy. Merosin normally anchors muscle cells to a structure that encases them (like the skin on a hot dog) called the basal lamina.
Doctors began to classify congenital muscular dystrophy as either “merosin-deficient” or “merosin-positive.” The gene for merosin is on chromosome 6.
As the 20th century ended, researchers began to suspect that Ullrich’s disease, now known as Ullrich congenital muscular dystrophy, was caused by a lack of collagen 6, a ropelike protein located in the area where laminin 211 is found.
Collagen 6, which helps support the muscle fiber, probably affects muscle cells via its connection to laminin 211. Laminin 211, in turn, connects to muscle cells via either of two other proteins: integrin or dystroglycan.
Dystroglycan links the outer surface of muscle cells with structures outside them via branches, made of sugar molecules, that protrude from its surface and stick to laminin.
The branch structure helps explain why mutations in so many diverse genes all appear to cause congenital muscular dystrophy. Each of these proteins contributes in a different way to the process of “sugar-coating” (glycosylating) dystroglycan. Several forms of congenital muscular dystrophy — such as Fukuyama congenital muscular dystrophy, Santavuori muscle-eye-brain disease and Walker-Warburg syndrome — arise from defects in these glycosylation proteins.
Figure 6 below shows the physical relationships among these proteins.
Figure 6. Congenital muscular dystrophy
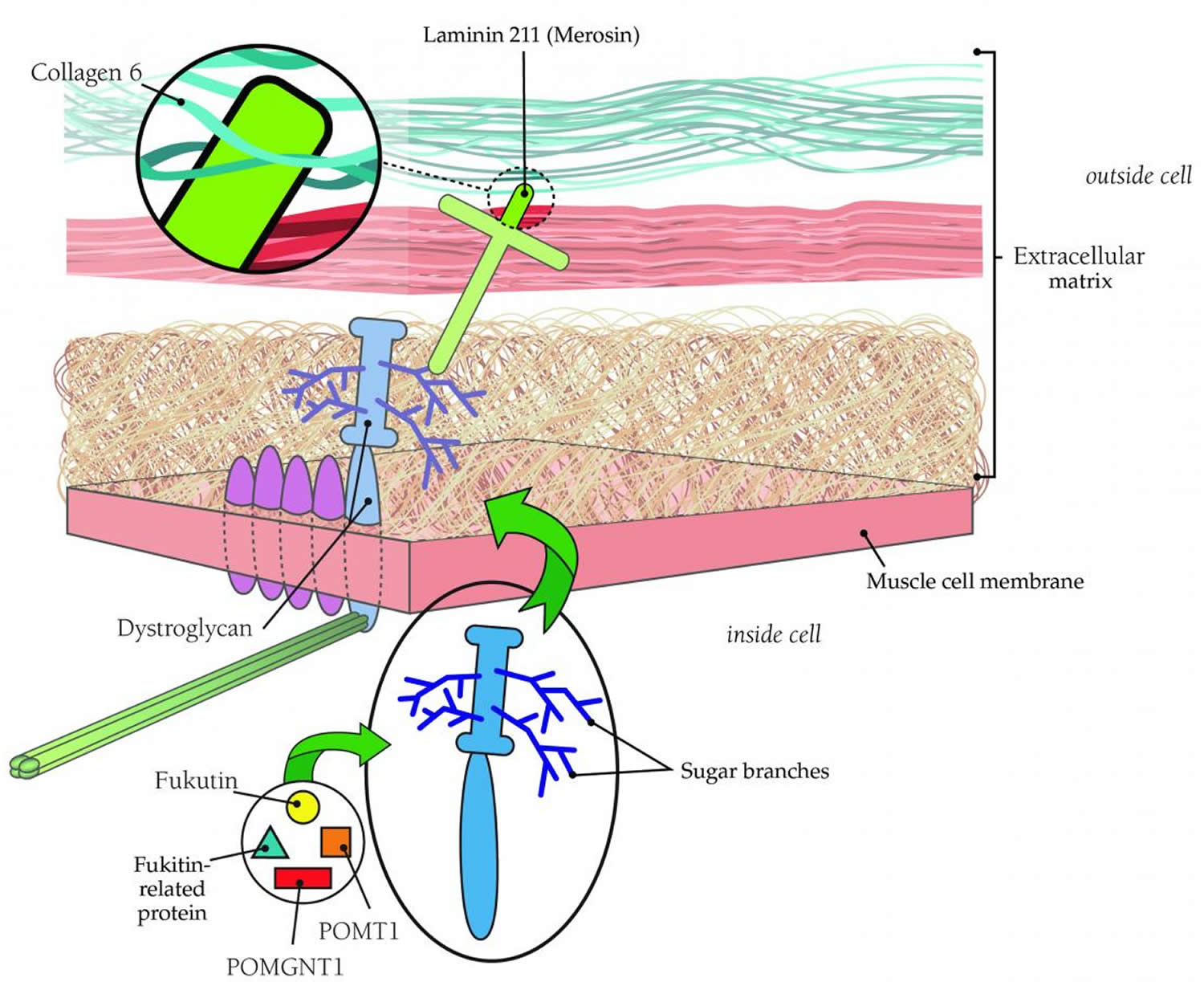
The congenital muscular dystrophies are caused by genetic defects that affect important muscle proteins. Most forms of congenital muscular dystrophy are inherited in an autosomal recessive pattern.
In brief, if a disease is recessive, two copies of the defective gene (one from each parent) are required to produce the disease. Each parent would be a carrier of the gene flaw but wouldn’t usually have the disease.
If a disease is dominant, then only one copy of the genetic defect is needed to cause the disease. Anyone with the gene flaw will have disease symptoms and can pass the disorder to children.
Figure 7. Congenital muscular dystrophy autosomal recessive inheritance pattern
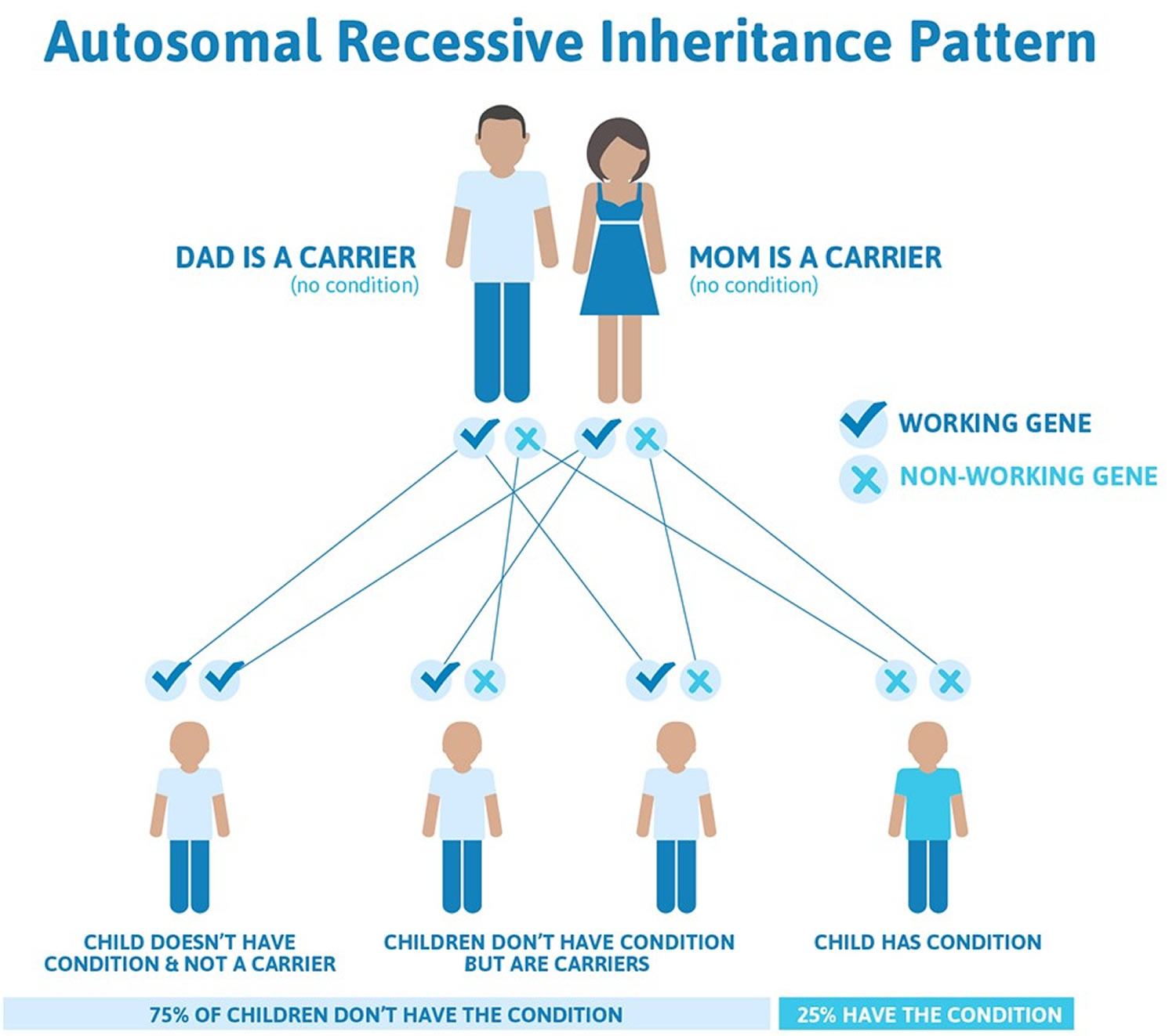
Congenital muscular dystrophy signs and symptoms
Most children with congenital muscular dystrophy exhibit some progressive muscle weakness, although they can have different symptoms, degrees of severity and rates of progression.
This weakness, usually first identified as hypotonia, or lack of muscle tone, can make an infant seem “floppy.” Later, infants and toddlers may be slow to meet motor milestones such as rolling over, sitting up or walking, or may not meet some milestones at all.
Some of the rarer forms of congenital muscular dystrophy are also accompanied by significant learning disabilities, or mental retardation.
Congenital muscular dystrophy diagnosis
A diagnosis of congenital muscular dystrophy can be confusing because for many years the term was used as a “catch-all” name to describe conditions that looked like other muscular dystrophies, but started much earlier or followed different patterns of inheritance.
In recent years, doctors have agreed that there are several categories of “true” congenital muscular dystrophy, caused by specific gene mutations, and they’re distinct from other muscular dystrophies. It’s possible that some people who received diagnoses of congenital muscular dystrophy many years ago may actually have some other known form of muscular dystrophy with an unusually early onset.
In diagnosing any form of muscular dystrophy, a doctor usually begins by taking a patient and family history and performing a physical examination. Much can be learned from these, including the pattern of weakness. The history and physical go a long way toward making the diagnosis, even before any complicated diagnostic tests are done.
The doctor also wants to determine whether the patient’s weakness results from a problem in the muscles themselves or in the nerves that control them. Problems with muscle-controlling nerves, or motor nerves, originating in the spinal cord and reaching out to all the muscles, can cause weakness that looks like a muscle problem but really isn’t.
Usually, the origin of the weakness can be pinpointed by a physical exam. Occasionally, special tests called nerve conduction studies and electromyography (EMG) are done.
In these tests, electricity and very fine pins are used to stimulate and assess the muscles or nerves individually to see where the problem lies. Electromyography is uncomfortable but not usually very painful.
Early in the diagnostic process doctors often order a special blood test called a CK (creatine kinase) level. Creatine kinase is an enzyme that leaks out of damaged muscle. When elevated creatine kinase levels are found in a blood sample, it usually means muscle is being destroyed by some abnormal process, such as a muscular dystrophy or inflammation. Therefore, a high creatine kinase level often suggests that the muscles themselves are the likely cause of the weakness, but it doesn’t tell exactly what the muscle disorder might be.
To determine which disorder is causing creatine kinase elevation, a doctor may order a muscle biopsy, the surgical removal of a small sample of muscle from the patient. By examining this sample, doctors can tell a great deal about what’s actually happening inside the muscles. Modern techniques can use the biopsy to distinguish muscular dystrophies from infections, inflammatory disorders and other problems.
Other tests on the biopsy sample can provide information about which muscle proteins are present in the muscle cells, and whether they’re present in the normal amounts and in the right locations. This can help narrow down which type of congenital muscular dystrophy is present.
Genetic (DNA) tests, generally using a blood sample, can analyze the person’s genes for particular defects that cause congenital muscular dystrophy, help predict the likely course of a disease and help families assess the risk of passing on the disease to the next generation.
Congenital muscular dystrophy treatment
The treatment plan includes a multidisciplinary team of pulmonologists, cardiologists, ophthalmologists, physiotherapists, orthopedists, possibly others, and ideally, a palliative care specialist to optimize quality of life.
A follow-up visit with a genetic counselor may be in order, but since 50 percent of children with congenital muscular dystrophy may not have a specific genetic diagnosis, supportive care should take place regardless of whether or not a specific genetic diagnosis is made.
Cardiac (heart) care
Some types of congenital muscular dystrophy, such as merosin-deficient congenital muscular dystrophy, are associated with severe cardiac complications. Since undetected heart problems can worsen over time, the guidelines recommend that everyone undergo cardiac screening at the time a congenital muscular dystrophy diagnosis is made, or as soon as possible thereafter.
Cardiac investigations should be systematically performed during follow-up examinations, and the frequency of which is dependent on the type of congenital muscular dystrophy and the level of cardiac involvement. Cardiac symptoms sometimes are atypical, especially in younger patients, and can start late in the course of the disease.
Since severe heart arrhythmia can lead to sudden death, implantation of a defibrillator should be considered.
Gastrointestinal, nutritional and oral care
Feeding and swallowing difficulties are significant problems in some types of congenital muscular dystrophy. Individuals with this problem should be observed and evaluated by a qualified specialist, using a video-fluoroscopic swallow assessment, if possible.
Recommendations for the treatment and management of feeding problems include adaptations to positioning and seating, supports for self-feeding, safe swallowing techniques and food texture modification.
If these recommendations are insufficient, gastrostomy tube feeding should be considered.
Muscle weakness and facial malformation can lead to speech problems in some people with congenital muscular dystrophy. There is no evidence that oral motor therapy and exercises help improve speech, but they may help resolve feeding problems.
Neurological issues
Problems related to congenital brain malformation, which occurs in some forms of congenital muscular dystrophy, include mental retardation, behavioral and learning problems, autistic features, emotional problems, seizures and vision problems. The Standard of Care discusses various medications for seizures and recommends that neurologists act as advocates to obtain the best possible professional and community supports and services for the child with congenital muscular dystrophy-related mental and emotional issues.
Specially adapted computers also can help children with vision problems.
Orthopedics and rehabilitation
Orthopedic symptoms, such as joint contractures, scoliosis, foot and spine deformities, rigid spine, hip dislocation and joint hypermobility are some of the most common aspects of congenital muscular dystrophy.
A conservative and preventive approach to orthopedic symptoms is recommended. Regular stretching, maintaining proper positioning and environmental supports such as braces and orthotics are generally favored over surgical interventions.
Although spinal surgery has been shown to improve the quality of life of older children with progressive spinal deformity, great care should be taken to minimize the risks of surgical intervention; postoperative, multidisciplinary care is essential.
Palliative care
Palliative care seeks to incorporate the emotional, spiritual, developmental and physical aspects of caring for a person with a life-threatening disease. It is a comprehensive and multidisciplinary model that benefits patients, caregivers and practitioners as they seek to maximize the life span and well-being of the person with congenital muscular dystrophy.
Problems that can be addressed through palliative care include fatigue, pain, depression, anger, anxiety, and other mental and emotional difficulties.
Established inpatient and outpatient palliative care resources should be offered, although other members of the medical team also may act as palliative care specialists.
Unclear diagnoses and uncertain prognoses are common features of congenital muscular dystrophy, requiring well-coordinated multidisciplinary care and strong patient-provider relationships throughout the changing course of the disease.
Respiratory care
All types of congenital muscular dystrophy can lead to the development of respiratory failure, and in some types, breathing problems may be severe from birth.
A proactive approach is favored because breathing problems can be present before they become noticeable. Weak crying, ineffective cough, choking on feedings, weight loss and repeated infections all can be signs of respiratory distress, even though, because of motor weakness, typical signs like breathlessness may not be present.
The guidelines address how to accurately assess and monitor breathing problems in children and adults with congenital muscular dystrophy and recommend that noninvasive ventilation be offered, particularly at night, before respiratory distress becomes acute.
Aggressive treatment of acute respiratory tract infections is particularly important, as these infections are the most common cause of hospital admissions and death in people with congenital muscular dystrophy.
Facioscapulohumeral muscular dystrophy
Facioscapulohumeral muscular dystrophy is a genetic muscle disorder in which the muscles of the face, shoulder blades and upper arms are among the most affected.
The long name comes from facies, the Latin word and medical term for face; scapula, the Latin word and anatomical term for shoulder blade; and humerus, the Latin word for upper arm and the anatomical term for the bone that goes from the shoulder to the elbow.
The term muscular dystrophy means progressive muscle degeneration, with increasing weakness and atrophy (loss of bulk) of muscles. In Facioscapulohumeral muscular dystrophy, weakness first and most seriously affects the face, shoulders and upper arms, but the disease usually also causes weakness in other muscles.
Facioscapulohumeral muscular dystrophy causes
Facioscapulohumeral muscular dystrophy may be inherited through either the father or the mother, or it may occur without a family history. It is almost always associated with a genetic flaw (mutation) that leads to a shorter than usual segment of DNA on chromosome 4. The segment isn’t part of any particular gene, but it nevertheless seems to interfere with the correct processing of genetic material.
A small number of people have a disorder that looks exactly like Facioscapulohumeral muscular dystrophy but don’t have the short segment on chromosome 4. The genetic cause of their disorder has yet to be identified.
The region of your chromosomes that causes Facioscapulohumeral muscular dystrophy contains a section with multiple identical units of DNA called D4Z4 repeats. Each repeat contains a copy of a gene called DUX4. This gene is used during fetal development, but in adulthood, the DNA in this region is normally “condensed,” or packed tightly together, which prevents the cellular machinery from reading the DUX4 gene. As a result, no protein is made from it once fetal development is completed.
In Facioscapulohumeral muscular dystrophy, multiple D4Z4 units are lost, which de-condenses the DNA and reactivates the DUX4 gene, allowing aberrant production of the DUX4 protein. Many Facioscapulohumeral muscular dystrophy researchers now believe that elevation of DUX4 protein causes the symptoms of Facioscapulohumeral muscular dystrophy.
In about 5 percent of people with Facioscapulohumeral muscular dystrophy, the D4Z4 region is of normal length. In these individuals, mutations in a different gene, called SMCHD1, cause the DNA in the vicinity of the D4Z4 repeats to be opened up, allowing the DUX4 gene to be read and protein to be produced.
Facioscapulohumeral muscular dystrophy is inherited in an autosomal dominant pattern, meaning it only takes one such mutation (from one parent) to cause the disorder. This altered piece of DNA also can occur spontaneously in a child as he or she develops in the womb.
Facioscapulohumeral muscular dystrophy can affect either males or females. In a small number of people with Facioscapulohumeral muscular dystrophy, the usual chromosome 4 mutation can’t be identified. In most affected people, it can be, with genetic testing.
Facioscapulohumeral muscular dystrophy is one of many genetic disorders in which germ line mosaicism is believed to occur. Germ line refers to egg or sperm cells. In this phenomenon, some sperm or egg cells in a parent carry a particular mutation.
In families with more than one child with Facioscapulohumeral muscular dystrophy but no previous family history, it’s likely that one parent has germ line mosaicism and that affected children were conceived with egg or sperm cells carrying the Facioscapulohumeral muscular dystrophy mutation. In these situations, the parents have no symptoms, and, if their blood cells are tested, they don’t show the mutation.
For help in understanding your family’s specific situation and planning for future children, it’s best to meet with a genetic counselor.
Figure 8. Facioscapulohumeral muscular dystrophy autosomal dominant inheritance pattern
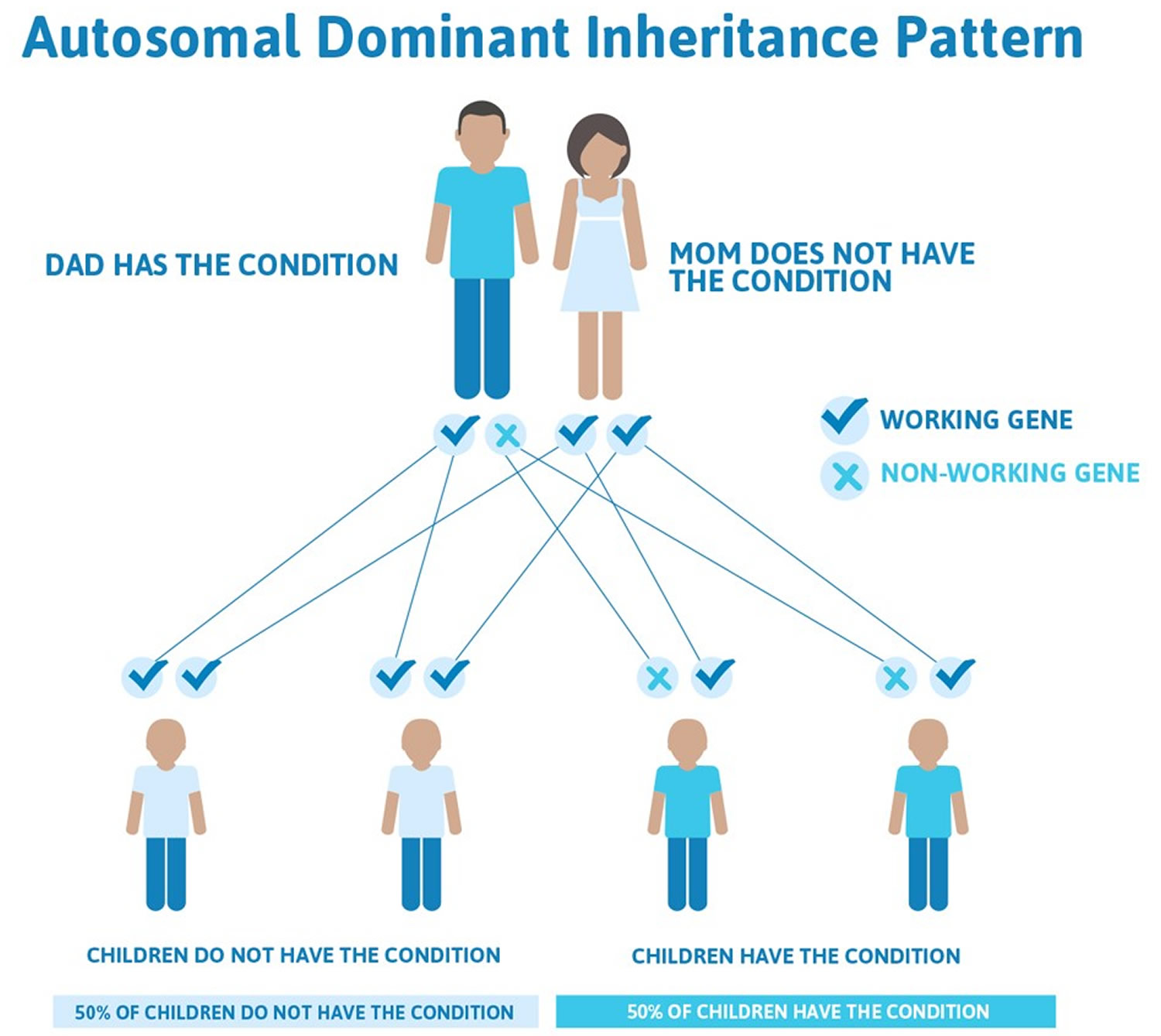
Facioscapulohumeral muscular dystrophy symptoms
Facioscapulohumeral muscular dystrophy usually begins before age 20, with weakness and atrophy of the muscles around the eyes and mouth, shoulders, upper arms and lower legs. Later, weakness can spread to abdominal muscles and sometimes hip muscles.
Some experts divide Facioscapulohumeral muscular dystrophy into adult-onset and infantile-onset forms. The adult-onset (which includes Facioscapulohumeral muscular dystrophy that begins in adolescence) is far more common.
In either type of Facioscapulohumeral muscular dystrophy, facial weakness can start in childhood. Occasionally, other Facioscapulohumeral muscular dystrophy symptoms appear in early childhood. Infantile-onset Facioscapulohumeral muscular dystrophy generally runs a more pronounced course with regard to muscle weakness and sometimes also affects hearing and vision. Preliminary evidence suggests that the infantile-onset form is associated with a larger piece of missing DNA.
About one in three people with facioscapulohumeral muscular dystrophy are unaware of any symptoms until well into adulthood. Others develop problems in early childhood. The condition tends to progress slowly.
Signs in your child may include:
- sleeping with their eyes slightly open
- an inability to squeeze their eyes tightly shut
- an inability to purse their lips – for example, to blow up balloons
Teenagers or adults may have shoulder aches, rounded shoulders or thin upper arms. As the condition progresses, it usually affects the muscles in the:
- face (facio)
- shoulders (scapula)
- upper arms (humeral)
- upper back
- calves
Around half of all people with facioscapulohumeral muscular dystrophy develop weakness in their leg muscles, and one or two in every 10 people with the condition will eventually need a wheelchair.
Facioscapulohumeral muscular dystrophy can develop unevenly, so the muscles on one side of the body may be affected more than the other. As the condition progresses slowly, it doesn’t usually shorten life expectancy.
Facioscapulohumeral muscular dystrophy diagnosis
Today, the most reliable way to diagnose facioscapulohumeral muscular dystrophy is with a test for a tiny missing section of DNA on chromosome 4. This test, which is performed on blood cells, is considered highly accurate for facioscapulohumeral muscular dystrophy, even though no specific gene has been identified as being associated with the disorder.
In people who have a family history of the disease and are showing signs of it, a DNA test is generally all that need be done to confirm whether facioscapulohumeral muscular dystrophy is likely to develop.
In many cases, however, people with no family history are suspected of having either facioscapulohumeral muscular dystrophy or some other neuromuscular disorder. In these situations, less expensive and less specific tests than the facioscapulohumeral muscular dystrophy DNA test may be done first.
One test is a creatine kinase level. This test, also performed on a blood sample, measures the amount of an enzyme known as creatine kinase in the blood. When muscle cells breakdown, as they do in muscular dystrophies and some other disorders, the creatine kinase level is elevated. Creatine kinase was formerly called creatine phosphokinase, or CPK.
Another type of diagnostic test is the electromyogram, or EMG, which measures the electrical activity in the muscles.
A nerve conduction velocity test may also be done. This involves measuring how fast signals travel from one part of a nerve to another.
Another diagnostic procedure sometimes undertaken is the muscle biopsy. In this procedure, a small piece of muscle is taken, under local anesthesia, usually from the arm or leg. Biopsy samples reveal cellular and molecular abnormalities that suggest certain muscle disorders and rule out others.
Muscle biopsies are less often performed today than in the past, especially when there’s a DNA test for the disease the doctor suspects is causing the symptoms — as there is for facioscapulohumeral muscular dystrophy. Muscle biopsy samples, however, are extremely valuable to researchers seeking to understand the relationship between the DNA results and what actually happens inside the muscle.
Confusion with other disorders
Facioscapulohumeral muscular dystrophy can be confused with polymyositis, which is neither a genetic disease nor a muscular dystrophy. It also can be confused with certain conditions of the nervous system that aren’t muscle disorders.
Facioscapulohumeral muscular dystrophy treatment
Medical treatments for Facioscapulohumeral muscular dystrophy are relatively few, and none are specific to the disease. There’s no treatment that can halt or reverse the effects of Facioscapulohumeral muscular dystrophy, but there are treatments and devices to help alleviate many of the symptoms.
Anti-inflammatory drugs known as nonsteroidal anti-inflammatories, or NSAIDs, are often prescribed to improve comfort and mobility. These are the same drugs taken by many people with arthritis and other inflammatory conditions.
Surgery
Surgical procedures to stabilize the shoulder blades (scapulae) by attaching them to the ribs have helped some people with Facioscapulohumeral muscular dystrophy.
In this procedure, the scapulae are fixed to the ribs so that they don’t move. The patient gains some leverage with the arm on the side that’s had the operation, since the scapulae no longer slide around.
Although this type of surgery may actually decrease the arm’s range of motion (since the shoulder blade can no longer rotate normally), the ability of the arm to function may be better, since the arm’s leverage point is now stable.
It’s important to go to a surgeon who fully understands Facioscapulohumeral muscular dystrophy and has had experience with this exact type of surgery.
Orthoses
Physical therapists often recommend devices such as back supports, corsets, girdles and special bras for people with Facioscapulohumeral muscular dystrophy. These supports help to compensate for weakening muscles in the upper and lower back.
Lower leg braces, known as ankle-foot orthoses, or AFOs, can compensate for weakening muscles in the lower leg that cause tripping and falling. These may be recommended by the physician or physical therapist and can be purchased as off-the-shelf or custom-made models. Some people find a lightweight, high-top shoe can be as helpful as an AFO in supporting the foot, at least in the early stages of weakness.
Physical therapists advise that those with Facioscapulohumeral muscular dystrophy shouldn’t resist using these types of devices for fear their muscles will get “lazy.” A supportive corset or AFO can help with mobility and endurance, they say, and supporting muscle in a normal position can help you use your remaining strength more effectively.
Massage or warm, moist heat (for example, from hot packs you can put in a microwave) are also good for the discomfort associated with Facioscapulohumeral muscular dystrophy.
Exercise
Since the precise underlying defect that causes muscle loss in Facioscapulohumeral muscular dystrophy isn’t yet understood, it’s hard to make precise recommendations about exercise.
However, physical therapists who have observed people with Facioscapulohumeral muscular dystrophy for many years say that moderate exercise appears to do no harm and may even be helpful, at least for muscles that haven’t severely weakened.
Therapists advise that exercise shouldn’t cause muscle cramping, significant muscle pain or extreme fatigue. An exercise program for someone with Facioscapulohumeral muscular dystrophy should be directed by a professional, such as a physical or occupational therapist, who has experience with neuromuscular disorders. The program should emphasize exercising muscles that are still relatively strong and resting those that have weakened. This can be accomplished with careful positioning and adaptation of standard exercise regimens.
Diet
There’s no specific diet known to help in Facioscapulohumeral muscular dystrophy or any other muscular dystrophy.
Myotonic muscular dystrophy
As with other types of muscular dystrophy, myotonic dystrophy involves progressive muscle weakness and muscle wasting. However, it’s often the smaller muscles that are affected first, such as those in the face, jaw and neck.
Myotonic dystrophy can appear at any time between birth and old age. It affects the same number of men and women.
Myotonic dystrophy is divided into two types:
- Type 1 myotonic dystrophy (myotonic dystrophy1) occurs when a gene on chromosome 19 called myotonic dystrophyPK contains an abnormally expanded section.
- Type 2 myotonic dystrophy (myotonic dystrophy2) is caused by an abnormally expanded section in a gene on chromosome 3 called ZNF9. myotonic dystrophy2 was originally called PROMM, for proximal myotonic myopathy, a term that has remained in use but is somewhat less common than the term myotonic dystrophy2.
The expanded sections of DNA in these two genes appear to have many complex effects on various cellular processes.
Myotonic dystrophy symptoms
myotonic dystrophy causes weakness of the voluntary muscles, although the degree of weakness and the muscles most affected vary greatly according to the type of myotonic dystrophy and the age of the person with the disorder.
Myotonia, the inability to relax muscles at will, is another feature of myotonic dystrophy. For example, it may be difficult for someone with myotonic dystrophy to let go of someone’s hand after shaking it.
As the disease progresses, the heart can develop an abnormal rhythm and the heart muscle can weaken. The muscles used for breathing can weaken, causing inadequate breathing, particularly during sleep.
In addition, in type 1 myotonic dystrophy, the involuntary muscles, such as those of the gastrointestinal tract, can be affected. Difficulty swallowing, constipation and gallstones can occur. In females, the muscles of the uterus can behave abnormally, leading to complications in pregnancy and labor.
The development of cataracts (opaque spots in the lenses of the eyes) relatively early in life is another characteristic of myotonic dystrophy, in both type 1 and type 2.
Overall intelligence is usually normal in people with myotonic dystrophy, but learning disabilities and an apathetic demeanor are common in the type 1 form. In congenital myotonic dystrophy 1, which affects children from the time of birth, there can be serious impairment of cognitive functioning. These children also may have problems with speech, hearing and vision.
Generally, the earlier myotonic dystrophy1 begins, the more profound the symptoms tend to be.
In general, myotonic dystrophy 2 has a better overall prognosis than myotonic dystrophy1. The symptoms are often relatively mild and progress slowly. myotonic dystrophy 2 rarely occurs during childhood, and there is no known congenital-onset form.
What is the progression of myotonic dystrophy?
The progression of myotonic dystrophy varies greatly among individuals, but in general, symptoms progress slowly.
The most common type of myotonic dystrophy 1 — the adult-onset form — begins in adolescence or young adulthood, often with weakness in the muscles of the face, neck, fingers and ankles. The weakness is slowly progressive for these and eventually other muscles.
When myotonic dystrophy 1 begins earlier in life than adolescence — the congenital-onset and juvenile-onset forms of the disease — it may be quite different in progression from the adult-onset type. Children with congenital-onset myotonic dystrophy1, once they survive the crucial neonatal period of respiratory muscle weakness with the help of assisted ventilation, usually show improvements in motor and breathing functions over the first year or so. They may have cognitive impairment, delayed speech, difficulty eating and drinking and various other developmental delays. Most will learn to walk. As adolescence approaches, children begin to show symptoms of the adult-onset form of myotonic dystrophy1 and follow its usual progression.
The childhood-onset form of myotonic dystrophy 1 — beginning after infancy but before adolescence — is more often characterized by cognitive and behavioral abnormalities than by physical disabilities. Eventually, muscle symptoms develop, to varying degrees.
Myotonic dystrophy 2 is, in general, a milder disease than type 1. It does not appear to have a congenital-onset form and rarely begins in childhood.
In contrast to type 1 myotonic dystrophy, the muscles affected first in myotonic dystrophy 2 are the proximal muscles — those close to the center of the body — particularly those around the hips. However, some finger weakness may be seen early as well. The disorder progresses slowly, but mobility may be impaired early because of weakness of the large, weight-bearing muscles.
Myotonic dystrophy 2 is quite rare, except in Germany and in people of German descent. Not as much is known about myotonic dystrophy 2 as about myotonic dystrophy 1.
Myotonic dystrophy treatment
Congenital-Onset myotonic dystrophy 1
Medical Management
Babies born with congenital-onset myotonic dystrophy 1 have the most complex medical challenges seen in myotonic dystrophy . Although the prognosis for these children has improved, the disease still has profound consequences and can be life-threatening, especially in the early months.
Breathing difficulties
The muscles needed for breathing are very weak in congenital-onset myotonic dystrophy 1, and a baby born with this disorder is likely to need a ventilator for an uncertain period of time. The breathing muscles do generally become stronger over time.
Cognitive impairment
Cognitive impairment and even mental retardation are common in congenital-onset myotonic dystrophy 1, often requiring early intervention during the preschool years and special education later.
Feeding difficulties
Weakness in the muscles needed for sucking may impair the baby’s ability to feed. If the weakness is interfering with nutrition and hydration, a tube can be inserted either down the nose into the stomach (nasogastric tube) or directly into the stomach from outside (gastrostomy tube) until the muscles become strong enough for the child to eat and drink by mouth.
Muscle abnormalities
Babies with congenital-onset myotonic dystrophy 1 are born “floppy,” with weak muscles and poor muscle tone. The medical term for this is hypotonia.
Motor milestones, such as sitting, standing and walking, are likely to be delayed, but ultimately are achieved by most children.
Muscle weakness in the feet may cause them to be fixed in a downward-pointing, inward-turning position. This condition is known as clubfoot or talipes equinovarus. This condition may have to be treated by casting or surgery before a child can walk.
Adult-Onset myotonic dystrophy 1/myotonic dystrophy 2 and Juvenile-Onset myotonic dystrophy 1
Medical Management
This section addresses medical management of the many symptoms of adult-onset myotonic dystrophy 1 and myotonic dystrophy 2, as well as juvenile-onset myotonic dystrophy 1. These three forms of myotonic dystrophy share similar medical management strategies.
Anesthesia warning
An unusually high rate of complications and even deaths associated with general anesthesia given during surgery have been reported in people with myotonic dystrophy 1. This can occur even if the myotonic dystrophy is mild. In fact, mild cases can be particularly dangerous because the surgeon, anesthesiologist and patient may be less likely to pay attention to the myotonic dystrophy when planning surgery.
Surgery usually can be safely undertaken with careful monitoring of cardiac and respiratory functions before, during and after the procedure. Be sure to tell the entire medical team, especially those responsible for the anesthesia, that you or your family member has myotonic dystrophy . If at all possible, have the anesthesiologist and the neurologist communicate long before the surgery.
Adverse reactions to anesthesia do not seem as serious in myotonic dystrophy 2. However, caution is advised.
Breathing and coughing muscle weakness
In myotonic dystrophy 1, breathing muscle weakness can be an important factor in the disease course. It does not seem common in myotonic dystrophy 2.
A good way to treat respiratory muscle weakness is to pump air into the lungs during the night with a small, portable “breathing booster” known as a bilevel positive airway pressure device (BiPAP is the trademarked name of the device made by Philips Respironics). It’s usually used with a face mask that can be easily put on and taken off. This kind of breathing assistance also can be used during the day, although usually that’s not necessary.
Cough assistance machines (CoughAssist is a brand name) and assisted cough techniques can help people clear out secretions, especially when a person with myotonic dystrophy 1 has a cold or chest infection.
Cataracts
Cataracts — opaque spots on the lens of the eye — are common in both myotonic dystrophy 1 and myotonic dystrophy 2. If they interfere with vision, they can be removed surgically. Caution with anesthesia and pain medication is necessary, and the surgical team should be aware of and familiar with the patient’s myotonic dystrophy .
Cognitive and behavioral abnormalities
Cognitive and behavioral abnormalities can exist at any point in myotonic dystrophy 1 or myotonic dystrophy 2, although they’re more common in myotonic dystrophy 1 and particularly when myotonic dystrophy 1 begins in childhood.
In general, these abnormalities manifest as an “avoidant” or apathetic personality, and difficulties with planning ahead, making decisions and processing visual and spatial information.
An evaluation by a neuropsychologist, special education strategies and counseling can be helpful, as can medications that increase alertness and attention (for example, modafinil), depending on the person and the details of his or her individual situation.
Daytime sleepiness
Daytime sleepiness, which is more common in myotonic dystrophy 1 but also occurs in myotonic dystrophy 2, can sometimes be helped with medication. One drug that can be used is methylphenidate (Ritalin). Newer drugs are modafinil (Provigil) and armodafinil (Nuvigil). These drugs may work on the brain’s sleep-wake cycle. Treating excessive sleepiness can make life more enjoyable for the person with myotonic dystrophy and his or her family.
Another approach that can be tried is to coax the body into a better rhythm of sleeping and waking by going to bed and getting up at the same time every day. Consult with a respiratory specialist familiar with muscular dystrophy to determine if breathing is compromised during sleep.
Gastrointestinal dysfunction
Dysfunction of the muscles of the throat or esophagus can occur in myotonic dystrophy 1, impairing swallowing.
It’s important to watch for swallowing problems, such as a tendency to choke on food or drinks, and be sure to mention them to the doctor. A swallowing specialist can help people learn to swallow more safely and, if necessary, how to change the consistencies of foods and liquids so they can be swallowed more easily.
Vomiting can be very dangerous for a person with myotonic dystrophy whose swallowing muscles are weak. A head-down position is crucial to prevent inhaling the vomit — a possibly fatal event.
Difficulty swallowing does not seem to be a common feature of myotonic dystrophy 2.
In myotonic dystrophy 1, the intestines may not move digested food along as well as they should, so constipation can be a chronic problem. A doctor can help set up a more effective bowel schedule and, if necessary, recommend laxatives, suppositories or enemas to help manage this condition.
Constipation does not seem to be a common factor in myotonic dystrophy 2.
The gallbladder, a hollow sac on the right side of the upper abdomen, likewise can be sluggish in myotonic dystrophy 1, leading to the formation of gallstones. These may cause persistent pain in the upper abdomen. Surgery to remove the gallbladder can be performed if necessary.
Gallbladder dysfunction does not seem to be a feature of myotonic dystrophy 2.
Heart abnormalities
Not everyone with myotonic dystrophy needs treatment for heart problems, but everyone should be checked for them on a regular basis. The problem seems more common in myotonic dystrophy 1 than in myotonic dystrophy 2, although both types of myotonic dystrophy can affect the heart.
The most common type of heart problem in myotonic dystrophy is an abnormal heart rhythm (arrhythmia) called a conduction disturbance. When a conduction disturbance is present, signals do not move through the heart in the normal way. This can be very serious, even causing sudden death. Therefore, it’s imperative that people with myotonic dystrophy have regular electrocardiograms (EKGs, also known as ECGs).
Some people with myotonic dystrophy develop an abnormal heart rhythm known as atrial fibrillation, in which the top part of the heart beats extremely fast, causing turbulent blood flow that can lead to clots and strokes.
Various electronic devices — pacemakers and implantable defibrillators — can be used to treat abnormal heart rhythms. Sometimes, medications also are prescribed. These include such drugs as beta blockers and anti-arrhythmic drugs.
Sometimes, especially late in the disease course, the heart muscle itself can weaken, causing a type of disorder known as cardiomyopathy. Medications can be prescribed to lessen the stress on the heart in this disorder. These are known as beta blockers and ACE inhibitors.
Insulin resistance
A phenomenon known as insulin resistance — meaning the insulin produced by the body isn’t utilized as well as it should be — can cause high blood sugar and sometimes even diabetes. Insulin resistance is common in people with myotonic dystrophy 1 and is thought to affect approximately 20 percent of those with myotonic dystrophy 2. The phenomenon often doesn’t cause any trouble but should be monitored by a physician. If it does become problematic, insulin or other medications that lower blood sugar can be prescribed.
Myotonia
Myotonia — inability to relax muscles at will — occurs in both myotonic dystrophy 1 and myotonic dystrophy 2. Grip myotonia — not being able to release one’s grip after, for example, shaking hands or holding a steering wheel — can be the main thing people notice. If myotonia is bothersome, it can be treated by drugs, such as mexiletine (Mexitil).
Pain
Pain in the skeletal muscles is a common feature of myotonic dystrophy 2 and is less common in myotonic dystrophy 1. The pain does not appear to be related to myotonia or to exercise. However, cold temperatures make it worse. Painful stiffness can occur, particularly in the legs.
A doctor may suggest an over-the-counter pain remedy or even a prescription pain medication in some circumstances. Some people find warm baths, heating pads or massage to be helpful.
Pain in the involuntary muscles — for instance, of the gastrointestinal tract or uterus — may be more common in myotonic dystrophy 1. The underlying cause of the pain, such as constipation, should be identified and treated where possible.
Some pain in the involuntary muscles may respond to heat or massage, and some may require pain medication, under a doctor’s supervision.
A doctor should always be told that myotonic dystrophy is present, as this may make a difference in how the pain should be treated. (For instance, in myotonic dystrophy 1, it’s important to avoid pain medications that affect breathing.)
Pregnancy and childbirth
In both myotonic dystrophy 1 and myotonic dystrophy 2, pregnancy can be complicated by a mother’s heart abnormalities, requiring special care and attention, particularly during labor and delivery.
In myotonic dystrophy 1, uterine and vaginal muscles may be weak, posing additional problems for pregnancy and delivery and making a surgical delivery more likely.
Reactions to anesthesia and pain medications can be unpredictable and need the attention of someone familiar with these disorders.
In addition, a mother who has mild myotonic dystrophy 1 may give birth to a child with congenital-onset myotonic dystrophy 1 who will need a neonatal intensive care unit and specialists familiar with this condition.
Skeletal muscle weakness
In myotonic dystrophy 1, weakness of the skeletal muscles is concentrated in the face, tongue, neck, forearms, hands and feet, especially at the beginning. Later on, other muscles can be affected, such as those of the thighs, and the breathing muscles.
Some people can compensate for weak foot muscles by picking up the foot from the knee and walking with a “marching” step. Eventually, though, many people with myotonic dystrophy 1 find that a cane or walker is helpful to compensate for foot and leg weakness.
A lower leg brace, called an ankle-foot orthosis or AFO, may be needed. Some people with myotonic dystrophy 1 use a wheelchair or a power scooter for convenience when covering long distances. For more, read Putting Your Best Foot Forward.
Various devices that hold the hand in a good position for using a keyboard or writing or drawing can help compensate for weak wrist and hand muscles.
In myotonic dystrophy 2, by contrast, the weakness begins in the large muscles close to the center of the body. Early in the disease course, there is weakness of the hips and thighs. The upper arms and shoulders are often involved early, and the forearms and fingers can be affected early as well.
The facial muscles usually remain strong, weakening in only a small percentage of people with myotonic dystrophy 2. The lower legs and feet tend to remain strong as well, although the calf muscles can become enlarged.
Walking aids, such as walkers, canes, and even scooters and wheelchairs, can be helpful.
Limb girdle muscular dystrophy
Limb-girdle muscular dystrophy (LGMD) is a diverse group of disorders with many subtypes categorized by disease gene and inheritance. Limb-girdle muscular dystrophy usually manifests in the proximal muscles around the hips and shoulders. (The proximal muscles are those closest to the center of the body; distal muscles are farther away from the center — for example, in the hands and feet).
The shoulder girdle is the bony structure that surrounds the shoulder area, and the pelvic girdle is the bony structure surrounding the hips. Collectively, these are called the limb girdles, and it is the observed weakness and atrophy (wasting) of the muscles connected to the limb girdles that has given this group of disorders its name.
Prevalence of limb-girdle muscular dystrophy is about 1 in 20,000 males are more affected than females. Depending on the type of muscular dystrophy, it can be first apparent from birth right through to adolescence.
Subtypes of Limb-girdle muscular dystrophy (LGMD)
Here is a list of limb-girdle muscular dystrophy subtypes. Type 1 LGMDs are dominantly inherited, requiring only one mutation for symptoms to result. Type 2 LGMDs are recessively inherited, requiring two mutations — one from each parent — for symptoms to appear. Sometimes, LGMDs are referred to by their names, not their numbers, and some types have not been assigned numbers.
Some limb-girdle muscular dystrophy subtype names:
Calpainopathy (CAPN3 mutations; recessive; LGMD2A)
Desmin myopathy (DES mutation; dominant; LGMD1E / DES mutation; recessive; LGMD2R)
Dysferlinopathy (DYSF mutations; recessive; LGMD2B)
Sarcoglycanopathies (SGCG, SGCA, SGCB, SGCD mutations; recessive; LGMD2C, LGMD2D, LGMD2E, LGMD2F)
ZASP-related myopathy (ZASP mutation; dominant; a form of myofibrillar myopathy)
Dominant LGMD subtype numbers:
LGMD1A (MYOT mutation)
LGMD1B (LMNA mutation)
LGMD1C (CAV3 mutation)
LGMD1D (DNAJB6 mutation)
LGMD1E (DES mutation)
LGMD1F (TNP03 mutation)
Recessive LGMD subtype numbers:
LGMD2A (CAPN3 mutations)
LGMD2B (DYSF mutations)
LGMD2C, also called SCARMD1 (SGCG mutations)
LGMD2D, also called SCARMD2 (SGCA mutations)
LGMD2E (SGCB mutations)
LGMD2F (SGCD mutations)
LGMD2G (TCAP mutations)
LGMD2H (TRIM32 mutations)
LGMD2I (FKRP mutations)
LGMD2J (TTN mutations)
LGMD2K (POMT1 mutations)
LGMD2L (ANO5 mutations)
LGMD2M (FKTN mutations)
LGMD2N (POMT2 mutations)
LGMD2O (POMGnT1 mutations)
LGMD2P (DAG1 mutations)
LGMD2Q (PLEC1 mutations)
LGMD2R (DES mutations)
LGMD2S (TRAPPC11 mutations)
LGMD2T (GMPPB mutations)
LGMD2U (ISPD mutations)
LGMD2V (GAA mutations)
LGMD2W (LIMS2 mutations)
LGMD2X (BVES mutations)
LGMD2Y (TOR1A1P1 mutations)
Limb-girdle muscular dystrophy symptoms
The unifying features of the Limb-girdle muscular dystrophies are the weakness and atrophy of the limb-girdle muscles. However, the age at which symptoms appear, and the speed and severity of disease progression, can vary.
Individuals may first notice a problem when they begin to walk with a “waddling” gait because of weakness of the hip and leg muscles. They may have trouble getting out of chairs, rising from a toilet seat or climbing stairs. As this weakness progresses, the person may require the use of assistive mobility devices.
Weakness in the shoulder area may make reaching over the head, holding the arms outstretched or carrying heavy objects difficult. It may become increasingly hard to keep the arms above the head for such activities as combing one’s hair or arranging things on a high shelf. Some people find it harder to type on a computer or other keyboard and may even have trouble feeding themselves.
Some of the various limb-girdle muscular dystrophy subtypes also are characterized by additional symptoms. For example, the heart can be affected in some types of limb-girdle muscular dystrophy, with weakness of the heart muscle (cardiomyopathy) and/or abnormal transmission of signals that regulate the heartbeat (conduction abnormalities or arrhythmias).
Some disease subtypes also involve the muscles used for breathing, and for that reason, respiratory function, along with cardiac function, should be monitored regularly.
Other symptoms may be present in some of the different subtypes of limb-girdle muscular dystrophy, including but not limited to: joint stiffness, muscle cramps, enlargement of calf muscles and involvement of distal muscles of the body such as those controlling the hands and feet.
Limb-girdle muscular dystrophy causes
Genes are the codes, or recipes, that cells use to manufacture the various proteins needed by the body. The genes associated with limb-girdle muscular dystrophy normally encode proteins that play vital roles in muscle function, regulation and repair. When one of these genes contains a mutation (a flaw, such as missing or incorrect information) cells cannot produce the proteins needed for healthy muscles.
There are two major groups of limb-girdle muscular dystrophys. Called limb-girdle muscular dystrophy1 and limb-girdle muscular dystrophy 2, these two groups are classified by the respective inheritance patterns: autosomal dominant and autosomal recessive. If one copy of the abnormal gene is sufficient to cause the disease, it is said to be autosomal dominant; if two copies are needed then the inheritance pattern is autosomal recessive.
Dozens of different genes, when mutated, have been shown to cause specific limb-girdle muscular dystrophy1 and limb-girdle muscular dystrophy2 subtypes. In these cases, the proteins associated with these genes are nonfunctional or deficient, and muscles are unable to function normally. Gradually, the muscles become weak enough that people experience the symptoms of limb-girdle muscular dystrophy.
In addition to the known limb-girdle muscular dystrophy1 and limb-girdle muscular dystrophy2 subtypes linked to specific genes, there are many cases of limb-girdle muscular dystrophy for which the causative gene is not yet known (and the person is not identified to have a subtype-specific form of limb-girdle muscular dystrophy). Scientists are actively working to understand the causes of these unidentified subtypes of limb-girdle muscular dystrophy, because the more we understand about all the different causes of limb-girdle muscular dystrophy and the diverse ways that muscle can be compromised, the better chance we have of finding effective therapies to intervene in the pathological process.
What is the progression of limb-girdle muscular dystrophy?
At this time, progression in each type of limb-girdle muscular dystrophy can’t be predicted with certainty, although knowing the underlying genetic mutation can be helpful. Some forms of the disorder progress to loss of walking ability within a few years and cause serious disability, while others progress very slowly over many years and cause minimal disability.
limb-girdle muscular dystrophy can begin in childhood, adolescence, young adulthood or even later. Both genders are affected equally.
When limb-girdle muscular dystrophy begins in childhood, some physicians say, the progression is usually faster and the disease more disabling. When the disorder begins in adolescence or adulthood, they say, it’s generally not as severe and progresses more slowly.
Limb-girdle muscular dystrophy treatment
Assistive devices
Simple devices like a cane or a long-handled reacher can make things easier as weakness progresses.
A power wheelchair or scooter becomes convenient when weakness in the pelvic girdle and upper legs causes frequent falls. People whose limb-girdle muscular dystrophy has reached this stage often find that a great deal of their independence returns, and they’re much less fatigued when they begin using this type of mobility equipment.
Heart, respiration and diet
The heart can be affected in limb-girdle muscular dystrophy, but this doesn’t occur as often as it does in some other forms of muscular dystrophy. Heart problems can take two forms — weakness of the heart muscle (cardiomyopathy) and abnormal transmission of signals that regulate the heartbeat (conduction abnormalities or arrhythmias). The heart should be monitored for these complications. When necessary, medications or devices (such as pacemakers) can be used to treat them.
Respiratory (breathing) function can decline over time, and this, too, should be monitored regularly. There are devices that can help sustain respiratory function.
No special dietary restrictions or additions are known to directly affect the course of limb-girdle muscular dystrophy. A doctor may advise a weight reduction or weight stabilization diet for some people, because being markedly overweight puts greater stress on already weakened muscles.
Therapy and exercise
Physical and occupational therapy programs are usually part of the treatment for limb-girdle muscular dystrophy. Occupational therapy focuses on specific activities and functions, particularly use of the hands, while physical therapy emphasizes mobility and (where possible) strengthening of large muscle groups.
The primary goals of physical therapy are to allow greater motion in the joints and to prevent contractures (freezing of the joints). These problems can arise when movement is limited, and it’s important for the patient’s comfort and function to avoid them.
In occupational therapy, the focus is on improving abilities related to your job, recreation or daily living. For example, arm supports can make tasks such as using a computer or fixing your hair less tiring.
Doctors and therapists have somewhat different opinions on the relative value or danger of various exercise regimens in people with muscular dystrophy. In limb-girdle muscular dystrophy, certain kinds of stress-causing exercises may actually hasten muscle damage.
Some experts recommend swimming and water exercises as a good way to keep muscles as toned as possible without causing undue stress on them. The buoyancy of the water helps protect against certain kinds of muscle strain and injury. Before undertaking an exercise program, make sure you’ve had a cardiac evaluation, and don’t swim alone.
Oculopharyngeal muscular dystrophy
In oculopharyngeal muscular dystrophy, symptoms aren’t usually apparent until a person is around 50 or 60 years old. It affects the muscles in the eyes (ocular) and the throat (pharyngeal).
Symptoms of oculopharyngeal muscular dystrophy can include:
- droopy eyelids
- difficulty swallowing
- progressive restriction of eye movement as the eye muscles become affected
- limb weakness around the shoulders and hips
As the eyelids droop, they can cover the eyes and impair vision. It’s also possible to develop double vision.
Dysphagia can eventually make it difficult to swallow solid foods, liquids and even small amounts of saliva. This can lead to chest infections if food and drink is accidentally swallowed the “wrong way” into the lungs. However, with treatment to manage the symptoms, a person’s life expectancy isn’t usually altered.
Emery-Dreifuss muscular dystrophy
People with Emery-Dreifuss muscular dystrophy often begin to develop symptoms during childhood or adolescence.
In the early stages, people with the condition usually develop muscle contractures (where the muscles and tendons become shortened and tightened, limiting the range of movement at nearby joints).
Areas commonly affected by muscle contractures include the arms, neck and feet. This means that people with Emery-Dreifuss muscular dystrophy may have difficulty straightening their elbows or bending their neck forward, for example.
Like all types of muscular dystrophy, Emery-Dreifuss muscular dystrophy also causes progressive muscle weakness, usually beginning in the shoulders, upper arms and lower legs. This can make it difficult to lift heavy objects or raise your arms above your head, and you may have an increased tendency to trip over things.
Later on, the hip and thigh muscles become weaker, making activities such as walking up stairs difficult. People with Emery-Dreifuss muscular dystrophy will often eventually require a wheelchair, as they become unable to walk.
Emery-Dreifuss muscular dystrophy can also affect the heart’s electrical signals, causing heart block. This can result in people with the condition developing an abnormally slow heartbeat and palpitations, which can lead to episodes of lightheadedness or fainting. The slow heartbeat can often be treated successfully with an implanted pacemaker.
Due to the risk of serious heart and respiratory problems, someone with Emery-Dreifuss muscular dystrophy will often have a shortened life expectancy. However, most people with the condition live until at least middle age.
Muscular dystrophy causes
In most cases, muscular dystrophy (muscular dystrophy) runs in families. It usually develops after inheriting a faulty gene from one or both parents. But some occur spontaneously in the mother’s egg or the developing embryo and can be passed on to the next generation.
Muscular dystrophy is caused by mutations (alterations) in the genes responsible for healthy muscle structure and function. The mutations mean that the cells that should maintain your muscles can no longer fulfil this role, leading to muscle weakness and progressive disability.
Inheriting muscular dystrophy
You have two copies of every gene (with the exception of the sex chromosomes). You inherit one copy from one parent, and the other copy from the other parent. If one or both of your parents has a mutated gene that causes muscular dystrophy, it can be passed on to you.
Depending on the specific type of muscular dystrophy, the condition can be a:
- recessive inherited disorder
- dominant inherited disorder
- sex-linked (X-linked) disorder
In a few cases, the genetic mutation that causes muscular dystrophy can also develop as a new event in the family. This is known as a spontaneous mutation. Examples include:
A recessive inherited disorder
If you have a recessive inherited disorder, it means you’ve inherited an altered version of the gene that causes the condition from both of your parents (both your copies of the gene are altered).
If a child only inherits an altered version of the gene from one parent, they’ll become a carrier of the condition. This means they’re not affected, but there’s a chance that any children they have will be if their partner is also a carrier.
If both parents carry an altered version of the gene that causes the condition, there’s a:
- 1 in 4 chance their child will have muscular dystrophy
- 1 in 4 chance their child will be healthy, but carry the mother’s faulty gene
- 1 in 4 chance their child will be healthy, but carry the father’s faulty gene
- 1 in 4 chance their child will be healthy (won’t inherit any mutated genes)
Some types of limb-girdle muscular dystrophy are inherited in this way.
A dominant inherited disorder
A dominant inherited disorder means you only need to inherit the mutated gene from one parent to be affected.
This means that if you have a child with an unaffected partner, there’s still a 50% chance of your child developing the condition.
Types of muscular dystrophy inherited in this way include myotonic dystrophy, facioscapulohumeral muscular dystrophy, oculopharyngeal muscular dystrophy and some types of limb-girdle muscular dystrophy.
A sex-linked (X-linked) disorder
Chromosomes are long, threadlike structures of DNA. A male has one X and one Y sex chromosome, and a female has two X chromosomes.
A sex-linked disorder is caused by a mutation in a gene on the X chromosome. As males only have one copy of each gene on the X chromosome, they’ll be affected if one of those genes is mutated.
As females have two copies of the X chromosome, they’re less likely to develop an X-linked condition, because the normal copy of the chromosome can usually cover for (mask) the altered version.
Females can still be affected by X-linked disorders, but the condition is usually less severe than when the gene alteration is present in an affected male.
Types of muscular dystrophy inherited in this way include Duchenne muscular dystrophy and Becker muscular dystrophy, which is why these conditions are more common and more severe in males.
Spontaneous gene mutations
Spontaneous gene mutations can occasionally cause muscular dystrophy. This is where the genes mutate for no apparent reason, changing the way the cells function. Spontaneous gene mutations can cause muscular dystrophy to develop in people who don’t have a family history of the condition.
Another way a child with no family history can be affected is when the condition is recessive. The gene mutations may have been present on both sides of the family for many generations, but may not have affected anyone until a child inherited a copy of the altered gene from both parents.
Risk factors for muscular dystrophy
Muscular dystrophy occurs in both sexes and in all ages and races. However, the most common variety, Duchenne, usually occurs in young boys. People with a family history of muscular dystrophy are at higher risk of developing the disease or passing it on to their children.
Muscular dystrophy prognosis
Muscular dystrophies are progressive diseases. Children with Duchenne muscular dystrophy often require a wheelchair by their teenage years, as do children with Becker muscular dystrophy in their late adolescence.
Because muscular dystrophies affect muscles everywhere in the body, including the lungs and heart, patients may need specialized care, such as a pacemaker. Each of the forms has unique features that require attention. For example, myotonic dystrophy can lead to impotence and high cholesterol levels.
Corticosteroids may improve muscle strength and function in Duchenne dystrophy. In patients with myotonic dystrophy, other medications improve myotonia. However, muscular dystrophies cannot be cured and weakness is progressive. Many people with either Duchenne or Becker muscular dystrophy may die in their early to late 20s.
Muscular dystrophy life expectancy
The natural history of muscular dystrophy depends on the type. Some cases may be mild and very slowly progressive, with a near normal lifespan, while other cases may have more marked progression of muscle weakness, functional disability and loss of ambulation. In Duchenne muscular dystrophy and some cases of Becker muscular dystrophy there is progressive weakness, contractures and the inability to walk. A kyphosis (back deformity) may develop which can lead to respiratory difficulty and hence an early death (Duchenne muscular dystrophy ~16 years, Becker muscular dystrophy ~42 years). With some other types of muscular dystrophy there is slow progression and thus a near normal lifespan. Some complications that may arise from muscular dystrophy (depending on the type) include cardiac arrhythmias, hypertension, dysphagia, malignant hyperthermia, respiratory problems, cataracts and hearing loss.
Life expectancy for people with myotonic dystrophy can vary considerably. Many people have a normal life expectancy, but people with the more severe congenital form (present from birth) may die while still a newborn baby, or only survive for a few years. Some people who first develop symptoms as a child or teenager may also have a shortened life expectancy. Most deaths related to myotonic dystrophy are related to pneumonia, breathing problems or heart problems.
Facioscapulohumeral muscular dystrophy can develop unevenly, so the muscles on one side of the body may be affected more than the other. As the condition progresses slowly, it doesn’t usually shorten life expectancy.
Muscular dystrophy complications
The complications of progressive muscle weakness include:
- Trouble walking. Some people with muscular dystrophy eventually need to use a wheelchair.
- Shortening of muscles or tendons around joints (contractures). Contractures can further limit mobility.
- Breathing problems. Progressive weakness can affect the muscles associated with breathing. People with muscular dystrophy may eventually need to use a breathing assistance device (ventilator), initially at night but possibly also during the day.
- Curved spine (scoliosis). Weakened muscles may be unable to hold the spine straight.
- Heart problems. Muscular dystrophy can reduce the efficiency of the heart muscle.
- Swallowing problems. If the muscles involved with swallowing are affected, nutritional problems and aspiration pneumonia may develop. Feeding tubes may be an option.
Muscular dystrophy symptoms
The main sign of muscular dystrophy is progressive muscle weakness. Specific signs and symptoms begin at different ages and in different muscle groups, depending on the type of muscular dystrophy.
Duchenne type muscular dystrophy
This is the most common form of muscular dystrophy. Although girls can be carriers and mildly affected, it’s much more common in boys.
In Duchenne muscular dystrophy, young boys start walking very late and have difficulties arising from the floor (Gowers sign, using their hands to push on their legs to get up). Intellectual impairment may also be present.
Signs and symptoms typically appear in early childhood and may include:
- Frequent falls
- Difficulty rising from a lying or sitting up position
- Trouble running and jumping
- Waddling gait
- Walking on the toes
- Large calf muscles
- Muscle pain and stiffness
- Learning disabilities
Becker muscular dystrophy
Signs and symptoms are similar to those of Duchenne muscular dystrophy, but tend to be milder and progress more slowly. Symptoms generally begin in the teens but may not occur until the mid-20s or even later.
In Becker muscular dystrophy, weakness may be limited to the quadriceps (muscles in the front of the thigh).
Other types of muscular dystrophy
Some types of muscular dystrophy are defined by a specific feature or by where in the body symptoms first begin. Examples include:
- Myotonic. Also known as Steinert’s disease, this form is characterized by an inability to relax muscles at will following contractions. Myotonic muscular dystrophy is the most common form of adult-onset muscular dystrophy. Facial and neck muscles are usually the first to be affected.
- Infants with type 1 myotonic dystrophy can have severe trouble breathing and swallowing and may not survive the neonatal (newborn) period. Adults with type 1 myotonic dystrophy may develop cataracts, hand and lower leg weakness and wasting (loss of muscle mass), and grip myotonia.
- Adults with later-onset type 2 myotonic dystrophy can have stiffness and myotonia in the thighs and hands, neck weakness, and hip weakness with trouble climbing stairs.
- Facioscapulohumeral (FSHD). Muscle weakness typically begins in the face and shoulders. The shoulder blades might stick out like wings when a person with facioscapulohumeral (FSHD) raises his or her arms. Onset usually occurs in the teenage years but may begin in childhood or as late as age 40.
- Congenital. This type affects boys and girls and is apparent at birth or before age 2. Some forms progress slowly and cause only mild disability, while others progress rapidly and cause severe impairment.
- Limb-girdle. Hip and shoulder muscles are usually the first affected. People with this type of muscular dystrophy may have difficulty lifting the front part of the foot and as a result may trip frequently. Onset usually begins in childhood or the teenage years.
Muscular dystrophy diagnosis
Your doctor is likely to start with a medical history and physical examination.
After that, your doctor may recommend:
- Enzyme tests. Damaged muscles release enzymes, such as creatine kinase (CK), into your blood. In a person who hasn’t had a traumatic injury, high blood levels of creatine kinase (CK) suggest a muscle disease — such as muscular dystrophy.
- Electromyography. An electrode needle is inserted into the muscle to be tested. Electrical activity is measured as you relax and as you gently tighten the muscle. Changes in the pattern of electrical activity can confirm a muscle disease.
- Genetic testing. Blood samples can be examined for mutations in some of the genes that cause different types of muscular dystrophy.
- Muscle biopsy. A small piece of muscle can be removed through an incision or with a hollow needle. Analysis (biopsy) of the tissue sample can distinguish muscular dystrophies from other muscle diseases.
- Heart-monitoring tests (electrocardiography and echocardiogram). These tests are used to check heart function, especially in people diagnosed with myotonic muscular dystrophy.
- Lung-monitoring tests. These tests are used to check lung function.
Muscular dystrophy treatment
There’s no cure for any form of muscular dystrophy. But treatment can help prevent or reduce problems in the joints and spine to allow people with muscular dystrophy to remain mobile as long as possible. Treatment options include medications, physical and occupational therapy, and surgical and other procedures.
Medications
Your doctor may recommend:
- Eteplirsen (Exondys 51), the first medication to be approved by the Food and Drug Administration specifically to treat Duchenne muscular dystrophy. It was approved conditionally in 2016 and will continue to be evaluated during an additional two years of use. Although the medication appears safe, it’s not clear how effective the drug is. It’s definitely not a cure for DMD, but it may increase muscle strength in some people treated with the drug. Eteplirsen acts on specific gene variants that affect approximately one in seven people with DMD.
- Corticosteroids, such as prednisone, which can help muscle strength and delay the progression of certain types of muscular dystrophy. But prolonged use of these types of drugs can cause weight gain and weakened bones, increasing fracture risk.
- Heart medications, such as angiotensin-converting enzyme (ACE) inhibitors or beta blockers, if muscular dystrophy damages the heart.
Creatine supplements
Recent research has also shown that a creatine supplement can improve muscle strength in some people with muscular dystrophy, while causing few side effects.
Creatine is a substance normally found in the body that helps supply energy to muscle and nerve cells. It’s often available as a supplement from pharmacies and health food stores.
If you have muscular dystrophy and decide to take creatine supplements, make sure you mention this to your doctors.
Therapy
Several types of therapy and assistive devices can improve the quality and sometimes the length of life in people who have muscular dystrophy. Examples include:
- Range-of-motion and stretching exercises. Muscular dystrophy can restrict the flexibility and mobility of joints. Limbs often draw inward and become fixed in that position. Range-of-motion exercises can help to keep joints as flexible as possible.
- Exercise. Low-impact aerobic exercise, such as walking and swimming, can help maintain strength, mobility and general health. Some types of strengthening exercises also might be helpful. But it’s important to talk to your doctor first because some types of exercise might be harmful.
- Braces. Braces can help keep muscles and tendons stretched and flexible, slowing the progression of contractures. Braces can also aid mobility and function by providing support for weakened muscles.
- Mobility aids. Canes, walkers and wheelchairs can help maintain mobility and independence.
- Breathing assistance. As respiratory muscles weaken, a sleep apnea device may help improve oxygen delivery during the night. Some people with severe muscular dystrophy may need to use a machine that forces air in and out of their lungs (ventilator).
Corrective surgery
In some severe cases of muscular dystrophy, surgery may be necessary to correct physical problems that can occur as a result of the condition.
For example, if your child has Duchenne muscular dystrophy, there’s a chance they’ll develop scoliosis. Surgery can correct the scoliosis or prevent it getting worse, although there haven’t been any trials to evaluate its effectiveness.
Other kinds of surgery may be used to treat specific symptoms:
- droopy eyelids can be lifted away from the eyes to improve vision
- tight joints caused by tendon contractures can be loosened to improve movement by lengthening or releasing the tendons
- weak shoulder muscles may be improved by surgically fixing the shoulder blades to the back of the ribs (scapular fixation) – however, there haven’t been any trials to evaluate the effectiveness of this treatment
If you or your child may benefit from having surgery, you’ll be referred to a specialist to discuss the procedure and the risks involved.
Treating swallowing problems
People with some types of muscular dystrophy find swallowing increasingly difficult as the condition progresses. This is known as dysphagia and it can increase your risk of choking or developing a chest infection, if food and liquid get into the lungs.
Depending on the severity of your swallowing problems, there are a number of treatments that can be used. For example, a dietitian may help you alter the consistency of your food and you may be taught some exercises by a speech and language therapist, to improve your swallowing.
If necessary, surgery can also be used to treat swallowing problems. This may involve a minor procedure to cut one of the muscles in your throat, or a small balloon may be inflated in your gullet (esophagus) to expand it.
If muscular dystrophy progresses to a point where you’re unable to get enough nutrition by swallowing, a feeding tube (gastrostomy or PEG) may need to be surgically implanted into your stomach through your abdomen (tummy).
Treating heart complications
Some types of muscular dystrophy can affect the heart muscles and the muscles used for breathing. When the condition has progressed to this stage, it can become life-threatening.
It’s important that your heart function is assessed regularly once muscular dystrophy has been diagnosed. For Duchenne and Becker muscular dystrophy, an electrocardiogram (ECG) examination of heart rhythm will be carried out at regular intervals, and you may also have an echocardiogram from time to time. A magnetic resonance imaging (MRI) scan may also be used to check for heart problems.
If any damage to your heart is detected, you may be referred to a cardiologist (heart specialist) for further tests and possibly more frequent monitoring.
You may be prescribed medication to treat your heart problems, such as ACE inhibitors to relax your arteries and make it easier for your heart to pump blood around your body, or beta-blockers to control irregular heartbeats (arrhythmias or dysrhythmias).
In some cases of myotonic or Emery-Dreifuss muscular dystrophy, a pacemaker may be fitted to correct an irregular heartbeat. A pacemaker is a small, battery operated device that can be implanted into your chest to regulate your heartbeat.
Mobility and breathing assistance
As muscular dystrophy progresses, it weakens your muscles and you gradually begin to lose mobility and strength. These physical problems can be helped with the following:
- low-impact exercise, such as swimming
- physiotherapy can be useful for maintaining muscle strength, preserving flexibility and preventing stiff joints
- physical aids, such as a wheelchair, leg braces or crutches, which can help you stand and stay mobile
- occupational therapy can help maximize or improve your independence by using different techniques, changing your environment and providing any necessary assistive equipment
Once the chest muscles become too weak to control breathing properly, you may need machines to assist with your breathing and coughing, particularly while sleeping.
Preventing respiratory infections
Respiratory infections may become a problem in later stages of muscular dystrophy. So, it’s important to be vaccinated for pneumonia and to keep up to date with influenza shots. Try to avoid contact with children or adults who have an obvious infection.
Home remedies
Dietary changes haven’t been shown to slow the progression of muscular dystrophy. But proper nutrition is essential because limited mobility can contribute to obesity, dehydration and constipation. A high-fiber, high-protein, low-calorie diet may help.
Coping and support
A diagnosis of muscular dystrophy can be extremely challenging. To help you cope:
- Find someone to talk with. You may feel comfortable discussing your feelings with a friend or family member, or you might prefer meeting with a formal support group (The Muscular Dystrophy Association https://www.mda.org/).
- Learn to discuss your child’s condition. If your child has muscular dystrophy, ask your doctor about the most appropriate ways to discuss this progressive condition with your child.
- Muscular Dystrophy. JAMA. 2011;306(22):2526. doi:10.1001/jama.2011.1794 https://jamanetwork.com/journals/jama/fullarticle/1104723[↩]
- Drug Trials Snapshots: EMFLAZA. https://www.fda.gov/drugs/informationondrugs/ucm544351.htm[↩]
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208684s000,208685s000lbl.pdf[↩]





{kind=link}