Contents
What is myelofibrosis
Myelofibrosis is a progressive, chronic disease in which the bone marrow is replaced by fibrous tissue and blood is made in organs such as the liver and the spleen, instead of in the bone marrow. Myelofibrosis is a rare bone marrow cancer where it is one of a related group of blood cancers known as “myeloproliferative neoplasms” in which bone marrow cells that produce the blood cells becomes fibrous as part of a chronic malignant (cancerous) process in which the proliferation of certain blood cell clones leads to the formation of fibrous tissue. The result is extensive scarring in your bone marrow, leading to severe anemia, weakness, body wasting (loss of body mass or size), fatigue and often an enlarged liver and spleen. With increased fibrosis of the bone marrow – other organs (spleen, liver) become a source of blood cell synthesis – consequently enlargement of the liver and spleen is the other common finding in myelofibrosis.
Myelofibrosis gets its name from the disease’s characteristics. The prefix “myelo-” in the word “myelofibrosis” means a relationship to the marrow. The presence of scar tissue gives the disease the “fibrosis” part of its name. myelofibrosis is also known by several other names, including agnogenic myeloid metaplasia and idiopathic myelofibrosis.
Myelofibrosis is an uncommon type of chronic leukemia — a cancer that affects the blood-forming tissues in the body and can occur on its own (primary myelofibrosis or idiopathic myelofibrosis) or can occur secondary to other bone marrow conditions such as myeloproliferative neoplasms that can progress to myelofibrosis include polycythemia vera, chronic myeloid leukemia and essential thrombocythemia.
The natural history of idiopathic myelofibrosis is of progressive bone marrow insufficiency with transfusion-dependent anemia and increasing organomegaly. Patients also become prone to deep infections – e.g. of the lungs. About 10% of patients develop an aggressive form of acute leukemia.
Myelofibrosis secondary to other bone marrow diseases is more common than idiopathic myelofibrosis (primary myelofibrosis).
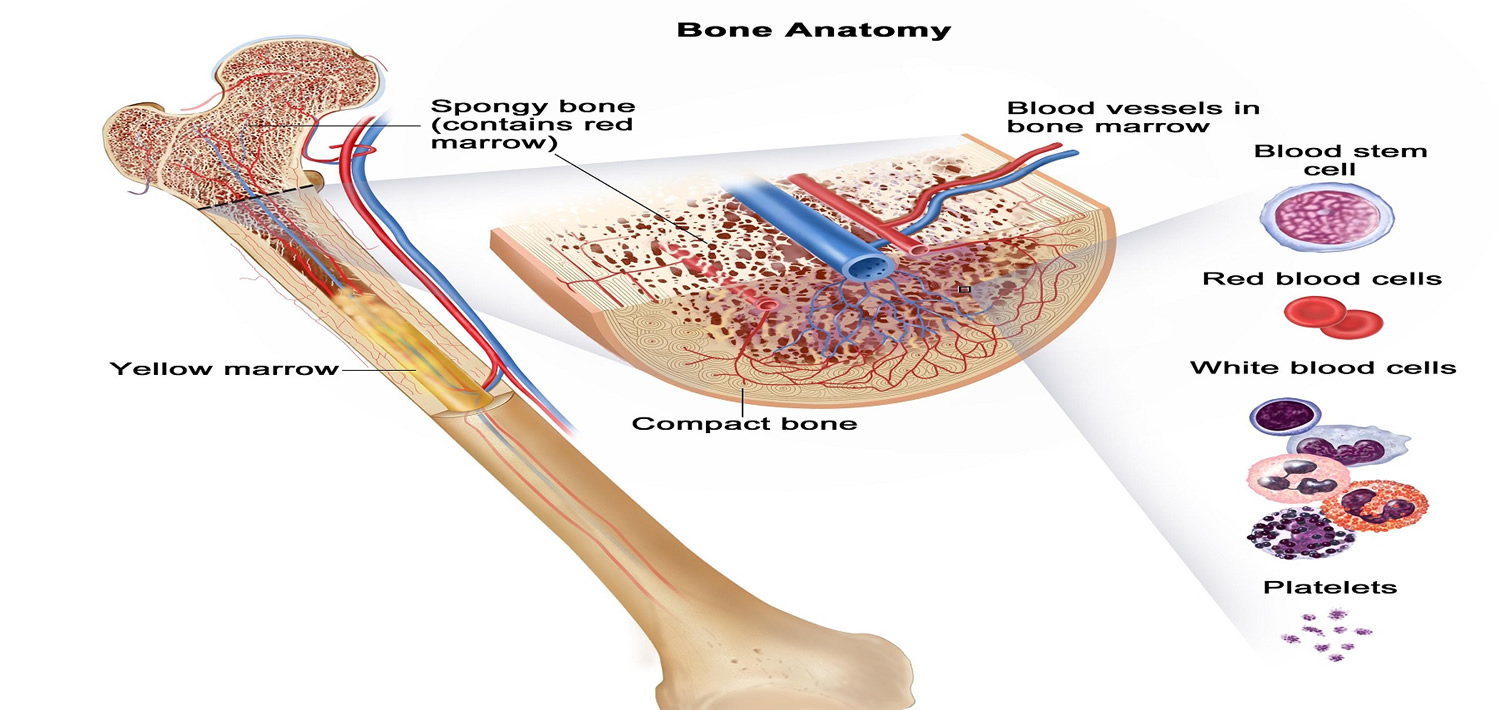
The bone marrow is the site of hematopoiesis – i.e. the organ where the blood cells are formed. It is found in the center of certain long bones (e.g. the femurs or thigh bones), pelvic bones, ribs, sternum etc (see Figure 2).
Many people with myelofibrosis may get progressively worse, and some may eventually develop a more serious form of leukemia, requiring treatment. Yet it’s also possible to have myelofibrosis and live symptom-free for years. In both cases, patients do need to be monitored regularly. The treatment goal for most patients with myelofibrosis is to relieve symptoms, reduce an enlarged spleen, improve blood cell counts (i.e., anemia), and reduce the risk of complications.
Figure 1. Blood composition
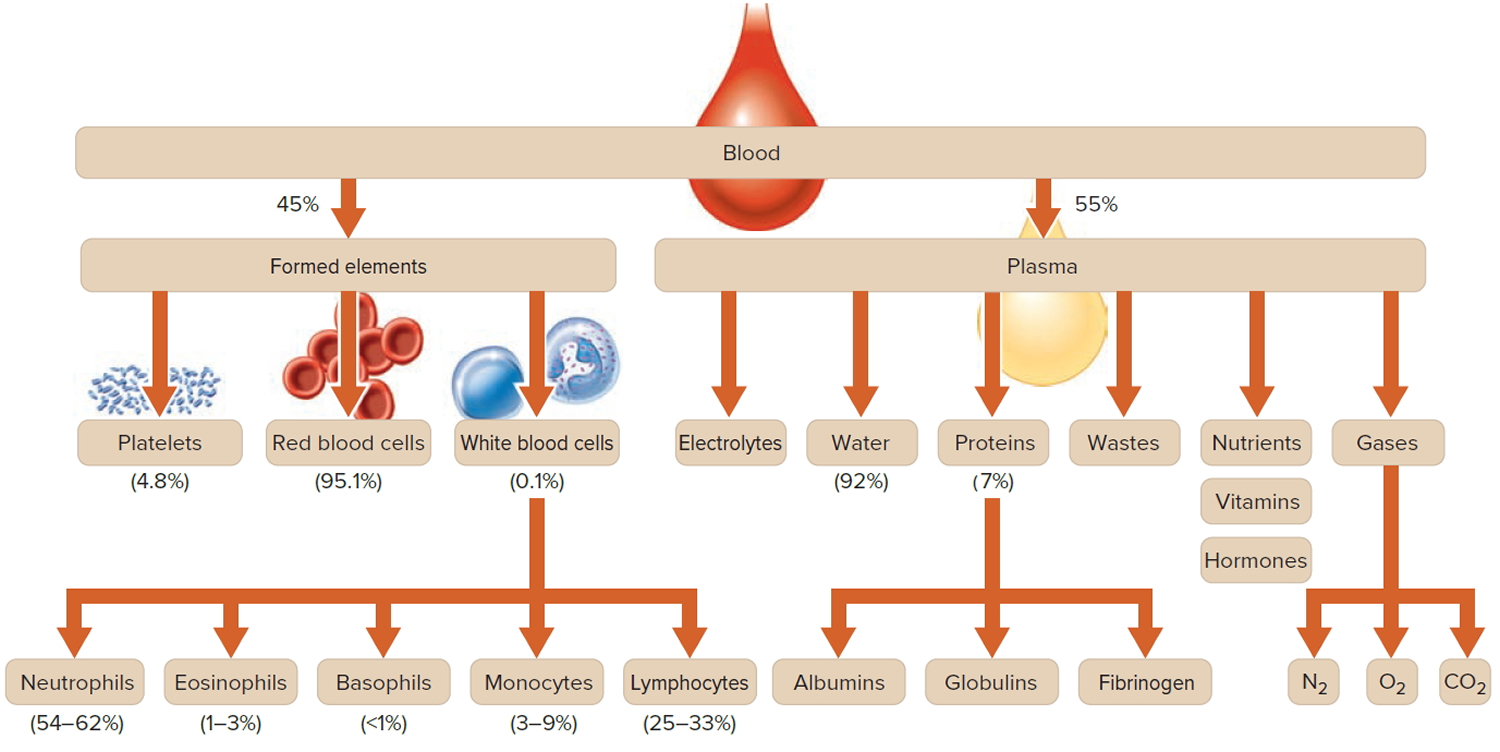
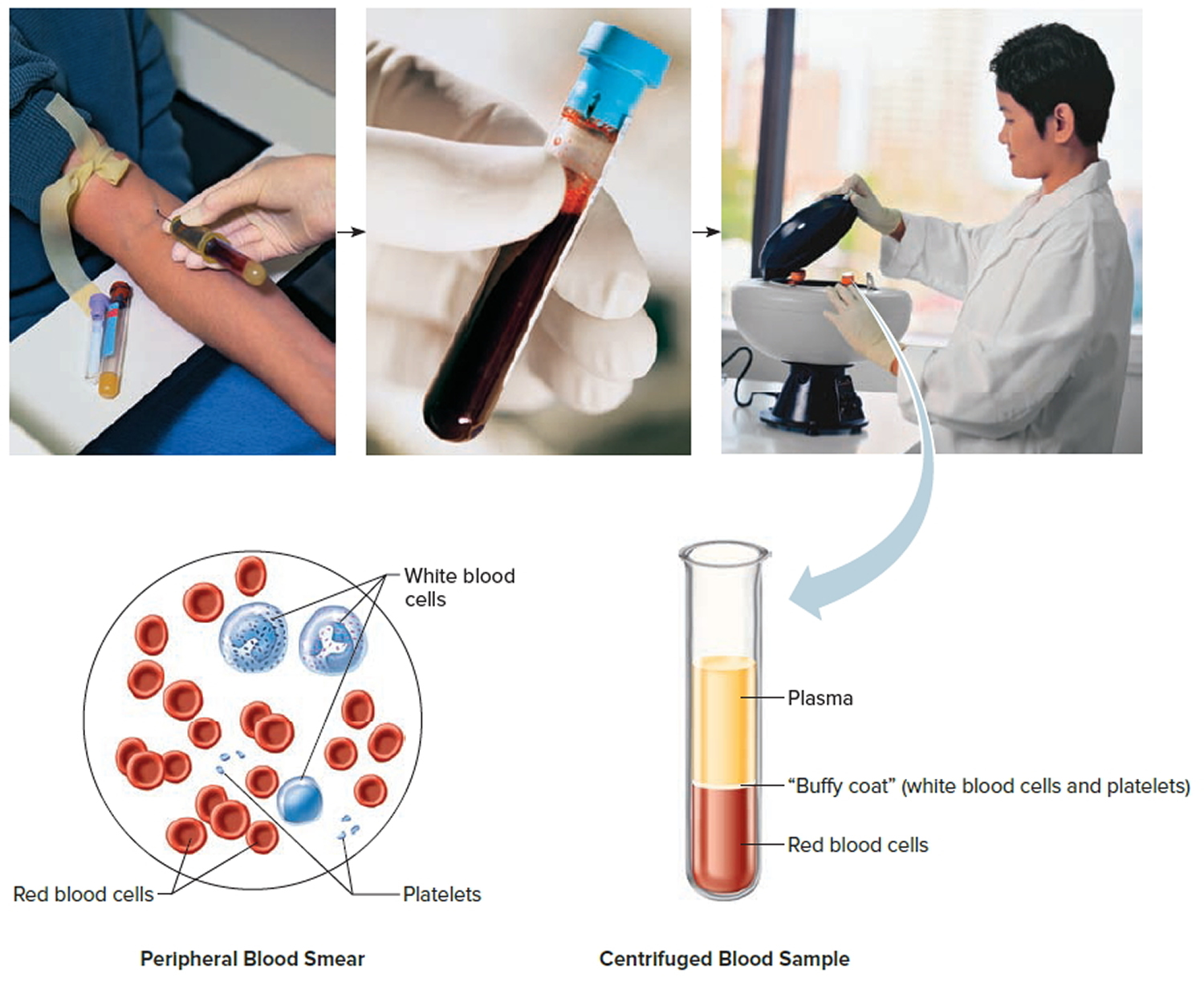
Figure 2. Bone marrow anatomy
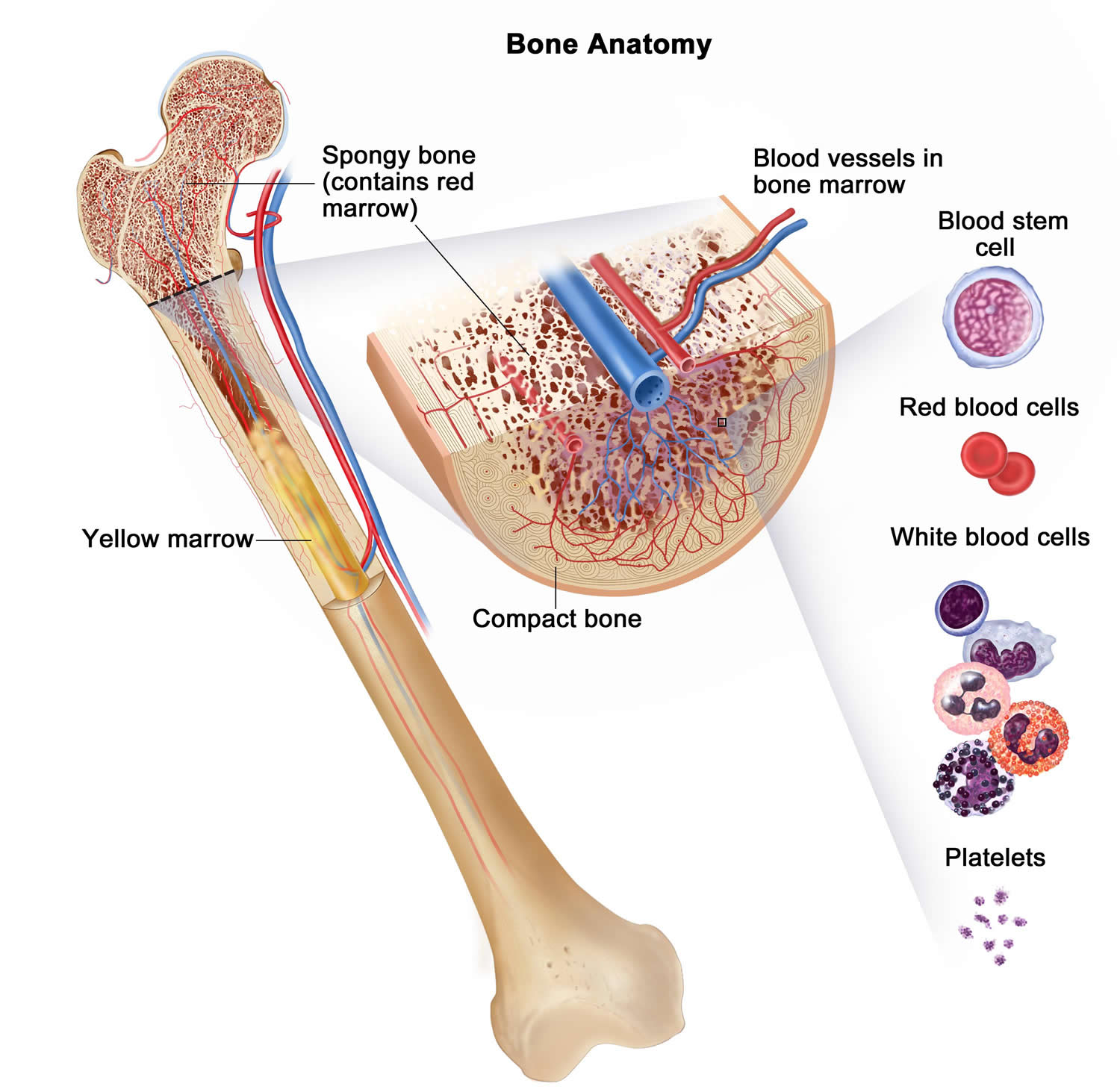
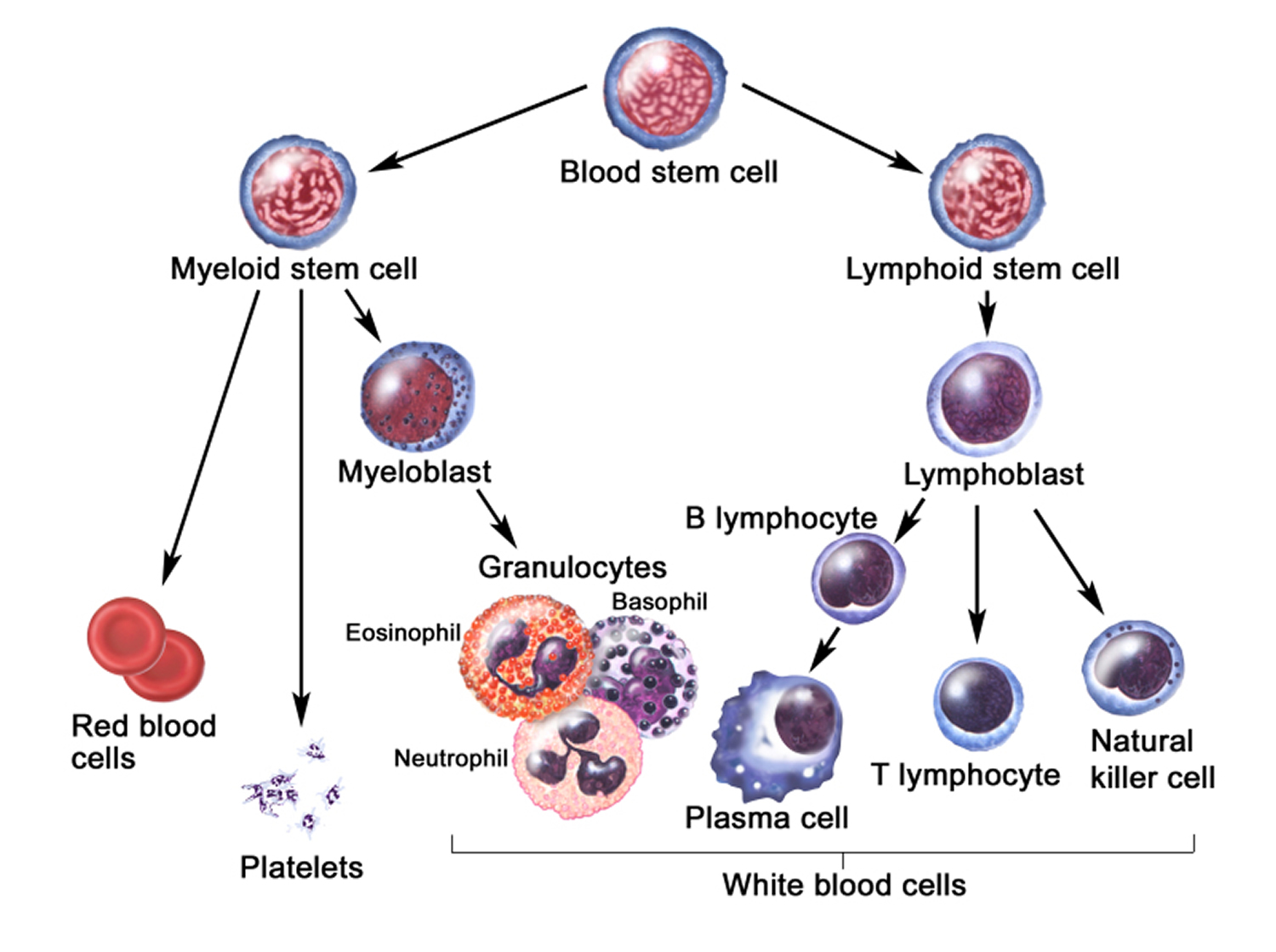
Figure 3. Blood cell development. A blood stem cell goes through several steps to become a red blood cell, platelet, or white blood cell
Primary myelofibrosis
Primary myelofibrosis is also called chronic idiopathic myelofibrosis, agnogenic myeloid metaplasia, myelosclerosis with myeloid metaplasia, and idiopathic myelofibrosis. Primary myelofibrosis is a Philadelphia-negative myeloproliferative neoplasm characterized by abnormal proliferation of megakaryocytes, deposition of fibrous connective tissues in the bone marrow, abnormal stem cell trafficking, and extramedullary hematopoiesis (myeloid metaplasia) 1. Besides varying degrees of bone marrow reticulin fibrosis, primary myelofibrosis is characterized by anemia, splenomegaly, a variety of constitutional symptoms, extramedullary hematopoiesis, a higher rate of transformation to acute myeloid leukemia (AML) than other “classic” myeloproliferative neoplasm and short survival 2. Primary myelofibrosis is an uncommon but aggressive myeloproliferative neoplasm, with an estimated prevalence of 4–6 per 100,000 individuals living in the US in 2010 3. In an international study of 1054 patients with primary myelofibrosis, the overall median survival was found to be 5.8 years, but considerable variability was observed 4. This study identified age >65 years, presence of constitutional symptoms, hemoglobin level <10 g/dL, leukocyte count >25 × 109/L, and circulating blast cells 1% or greater as independent predictors of shortened survival at diagnosis. The use of these parameters led to the definition of the International Prognostic Scoring System (IPSS), which identifies 4 prognostic groups with substantially different survival in primary myelofibrosis 4.
In a European multi-national study 5 looking at the survival of primary myelofibrosis patients diagnosed between 1980 and 1995 and 1996 and 2007, median survival increased from 4.5 to 6.5 years between the two periods studied, but there was no improvement for patients with intermediate-2 or high risk disease according to the International Prognostic Scoring System (IPSS) 6. A Swedish Cancer Registry study 7 of over 9000 myeloproliferative neoplasm patients found no improvement in survival of myeloproliferative neoplasm patients after the year 2000. Activating mutations in Janus kinase 2 (JAK2), the thrombopoietin receptor, MPL and the endoplasmic reticulum chaperone protein, calreticulin (CALR) are found in approximately 50–60%, 5–10% and 20–30% of patients with primary myelofibrosis, respectively; overall survival and leukemia-free survival are best in CALR-mutated and worst in so called “triple negative” patients 8. However, activated JAK-signal transducer and activator of transcription (STAT) signaling is universal across Philadelphia chromosome-negative (Ph−) myeloproliferative neoplasm 9, and the JAK1/2 inhibitor, ruxolitinib, is effective in higher risk patients with Pmyelofibrosis and post-polycythemia vera/essential thrombocythemia myelofibrosis (post-PV/ET myelofibrosis) without regard to the mutational status of JAK2, myelofibrosis subtype (primary myelofibrosis or post-polycythemia vera/essential thrombocythemia myelofibrosis), age, IPSS risk, performance status, baseline hemoglobin level, spleen size or symptom burden 10. Demonstration of superiority over placebo and best available therapy, respectively, in the COmyelofibrosisORT I 11 and II trials 12 established this agent as the cornerstone of drug therapy in myelofibrosis 13. Current guidelines recommend consideration of allogeneic stem cell transplantation (ASCT) for all patients with IPSS intermediate-2 and high risk disease, as well as for selected patients with intermediate-1 risk disease who have other high-risk features such as refractory, transfusion-dependent anemia, >2% circulating blasts or adverse cytogenetics 14. Efforts are in place to incorporate information on presence/absence of specific driver mutations into prognostic scoring systems, along with information on mutations in the epigenetic regulators ASXL1, EZH2 and IDH1/2, as well as the spliceosome component, SRSF2, which have also been identified as being prognostically adverse in primary myelofibrosis 15.
Myelofibrosis stages
The 2016 revision of the World Health Organization (WHO) classification of myeloproliferative neoplasms defines 2 stages of primary myelofibrosis 16:
- Prefibrotic/early (pre-primary myelofibrosis) and
- Overt fibrotic (overt primary myelofibrosis) phase.
The World Health Organization (WHO) classification of primary myelofibrosis criteria 16 mainly rely on bone marrow morphology (with megakaryocyte proliferation and atypia in both diseases, and increased age-adjusted cellularity with granulocyte proliferation and often decreased erythropoiesis in pre-primary myelofibrosis) and fibrosis grade (grade 0-1 indicates pre-primary myelofibrosis and grade 2-3 overt primary myelofibrosis); accordingly, grade 1 fibrosis is included in the pre-primary myelofibrosis category. This contrasts with the 2008 WHO classification where criteria did not explicitly define the grade of fibrosis 17, thereby resulting in variable proportions of patients with initial fibrosis included among overt primary myelofibrosis. Finally, peripheral blood leukoerythroblastosis constitutes a minor diagnostic criterion of overt primary myelofibrosis in the 2016 WHO classification, whereas anemia, leukocytosis, increased lactate dehydrogenase (LDH) and palpable splenomegaly may be present in both diseases 16.
In this “real-life” study 18, scientists studied the clinical and molecular features of patients belonging to these categories of primary myelofibrosis. The diagnosis of 661 primary myelofibrosis patients with a bone marrow biopsy at presentation was revised according to modern criteria; clinical information and annotation of somatic mutations in both driver and selected nondriver myeloid genes were available for all patients. Compared with pre-primary myelofibrosis, overt primary myelofibrosis was enriched in patients with anemia, thrombocytopenia, leukopenia, higher blast count, symptoms, large splenomegaly, and unfavorable karyotype. The different types of driver mutations were similarly distributed between the 2 categories, whereas selected mutations comprising the high mutation risk category (any mutations in ASXL1, SRSF2, IDH1/2, EZH2) were more represented in overt primary myelofibrosis. More patients with overt primary myelofibrosis were in higher International Prognostic Scoring System (IPSS) risk categories at diagnosis, and the frequency increased during follow-up, suggesting greater propensity to disease progression compared with pre-primary myelofibrosis. Median survival was significantly shortened in overt primary myelofibrosis (7.2 vs 17.6 years), with triple negativity for driver mutations and presence of high mutation risk mutations representing independent predictors of unfavorable outcome. The findings of this “real-life” study indicate that adherence to 2016 WHO criteria allows for identification of 2 distinct categories of patients with primary myelofibrosis where increased grades of fibrosis are associated with more pronounced disease manifestations, adverse mutation profile, and worse outcome, overall suggesting they might represent a phenotypic continuum. Current results also suggest that accurate differentiation between pre-primary myelofibrosis and overt primary myelofibrosis is required for meaningful interpretation of results of clinical trials with novel therapeutic agents.
Myelofibrosis complications
Complications that may result from myelofibrosis include:
- Increased pressure on blood flowing into your liver (Portal Hypertension). Normally, blood flow from the spleen enters your liver through a large blood vessel called the portal vein. Increased blood flow from an enlarged spleen can lead to high blood pressure in the portal vein (portal hypertension). This in turn can force excess blood into smaller veins in your stomach and esophagus, potentially causing these veins to rupture and bleed.
- Pain. A severely enlarged spleen can cause abdominal pain and back pain.
- Growths in other areas of your body. Formation of blood cells outside the bone marrow (extramedullary hematopoiesis) may create clumps (tumors) of developing blood cells in other areas of your body. These tumors may cause problems such as bleeding in your gastrointestinal system, coughing or spitting up of blood, compression of your spinal cord, or seizures.
- Bleeding complications. As the disease progresses, your platelet count tends to drop below normal (thrombocytopenia) and platelet function becomes impaired. An insufficient number of platelets can lead to easy bleeding — an issue that you and your doctor will want to discuss if you’re contemplating any type of surgical procedure.
- Painful bones and joints. Myelofibrosis can lead to hardening of your bone marrow and inflammation of the connective tissue that is found around the bones. This may cause bone and joint pain.
- Gout. Because myelofibrosis increases the body’s production of uric acid, a byproduct of the breakdown of purines—a substance found naturally in the body—needlelike deposits of uric acid can form in the joints, causing joint pain and inflammation (gout).
- Acute Myeloid Leukemia. In about 15 to 20 percent of patients with myelofibrosis, myelofibrosis will transform to Acute Myeloid Leukemia, a type of blood and bone marrow cancer that progresses rapidly.
Myelofibrosis prognosis
The prognosis (the likely outcome of the disease) varies widely in patients with myelofibrosis (myelofibrosis). Each patient’s prognostic risk factors are evaluated differently. A very large proportion of symptom-free patients remain stable for years without needing treatment.
Important prognostic factors include anemia, thrombocytopaenia (low platelets), age, unexplained fever, weight loss, night sweats, and certain gene abnormalities on bone marrow testing.
Myelofibrosis survival rate
While the median survival for people with myelofibrosis is about 5 years, people younger than 55 and with good prognostic factors have a median survival of 11 years. However, some people may survive for decades following a diagnosis. It is important to know that outcome data can show how groups of people with myelofibrosis responded to treatment, but cannot determine how any one person will respond. For these reasons, patients are advised to discuss survival information with their doctors.
Although there is no staging system for myelofibrosis, the International Prognostic Scoring System (IPSS), uses the following five risk factors for estimating survival from the time of diagnosis:
- Age – 65 years or older
- Anemia – Hemoglobin level lower than 10 grams per deciliter (g/dL)
- Symptoms – Such as fever, night sweats or weight loss
- Leukocytosis – An elevated white cell count of above 25,000/µl
- Circulating blood blast cells – At least 1 percent.
According to the International Prognostic Scoring System (IPSS), patients without any of these adverse features, excluding age, have a median survival of more than ten years. The presence of any three of the above adverse features reduces the median survival to two years. However, recent long-term studies of ruxolitinib (Jakafi®) suggest that it can improve survival times in patients with myelofibrosis for several years.
Myelofibrosis life expectancy
Patients may survive for 10 years or more, though the range is from 1 to 15. Acceleration of the disease progression occurs in 10-20% of cases from transformation to acute myeloblastic leukaemia. The most common causes of death are cardiovascular disease, infection and gastrointestinal bleeding.
Myelofibrosis causes
The DNA (genetic material) of a single hematopoietic (blood-forming) stem cell is damaged. This is called an “acquired mutation.” Myelofibrosis occurs when blood stem cells develop a genetic mutation. Blood stem cells have the ability to replicate and divide into the multiple specialized cells that make up your blood — red blood cells, white blood cells and platelets (see Figure 3 above). Several specific gene mutations have been identified in people with myelofibrosis.
About 90 percent of people with myelofibrosis have a mutation (a change in their DNA) in one of three genes (JAK2, CALR or MPL). All three of these gene mutations cause abnormal signaling in the JAK pathway, which regulates blood cell production.
- About 50 percent of patients have the“V617F JAK2” mutation found in the Janus kinase 2 (JAK2) gene
- About 25 percent have a mutation in the calreticulin (CALR) gene,
- Between 5 and 10 percent of myelofibrosis patients have a myeloproliferative leukemia (MPL) gene mutation.
- In addition, over last several years, mutations in many other genes, such as ASXL1, EZH2, IDH1/2, SRSF2 and TET2 have been found in 5 to 20 percent of patients with myelofibrosis; these mutations may occur in addition to JAK2, CALR, or MPL mutations, and one person may have several of them at the same time.
Knowing whether the JAK2 gene or others are associated with your myelofibrosis helps determine your prognosis and your treatment.
It’s not clear what causes the genetic mutation in blood stem cells. Exposure to petrochemicals, such as benzene and toluene, and ionizing radiation may raise the risk of developing the cancer. But only a small proportion of people exposed to these chemicals develop myelofibrosis. A theory about why myelofibrosis develops in some people is that they have inherited genes that limit their ability to detoxify the causative agents. In most cases, myelofibrosis is not an inherited disease, but there are some rare cases of familial clustering of the myeloproliferative neoplasms, including myelofibrosis. There is no known prevention.
As the mutated blood stem cells replicate and divide, they pass along the mutation to the new cells. As more and more of these mutated cells are created, they begin to have serious effects on blood production.
Eventually, this abnormal cell production overtakes the bone marrow’s ability to produce enough normal blood cells, including:
- Red blood cells, which carry oxygen to the tissues
- White blood cells, which fight infection
- Platelets, which help blood to clot.
The end result is usually a lack of red blood cells, which causes the anemia characteristic of myelofibrosis and an overabundance of white blood cells with varying levels of platelets. In people with myelofibrosis, the normally spongy bone marrow becomes scarred. The result can be severe anemia, weakness, bone pain, fatigue and increased risk of infection or bleeding.
The abnormal growth of blood-forming cells can also take place outside of the bone marrow, called “extramedullary hematopoiesis,” in such organs as the liver, spleen, lungs, lymph nodes and spinal cord, causing swelling.
An important constant feature of myelofibrosis is the overproduction of platelet-forming cells, called “megakaryocytes,” in the marrow. This causes too many platelets to be released into the blood and chemicals called “cytokines” to be released into the marrow. The cytokines stimulate the development of scar tissue in the marrow, called fibrosis. Paradoxically, the number of megakaryocytes can become so abnormal that platelet production decreases in some patients.
Platelets are small blood cells (comprising about one-tenth the volume of red cells) that stick to the site of a blood vessel injury and form a plug to seal off the injured blood vessel to stop bleeding. Normally, new platelets are made to replace used platelets in the body.
Risk factors for myelofibrosis
Although the cause of myelofibrosis often isn’t known, certain factors are known to increase your risk:
- Age. Myelofibrosis can affect anyone, but it’s most often diagnosed in people older than 50.
- Another blood cell disorder. A small portion of people with myelofibrosis develop the condition as a complication of essential thrombocythemia or polycythemia vera.
- Exposure to certain chemicals. Myelofibrosis has been linked to exposure to industrial chemicals such as toluene and benzene.
- Exposure to radiation. People exposed to high levels of radiation, such as survivors of atomic bomb attacks, have an increased risk of myelofibrosis. Some people who received a radioactive contrast material called Thorotrast, used until the 1950s, have developed myelofibrosis.
Myelofibrosis symptoms
Myelofibrosis usually develops slowly. In its very early stages, many people don’t experience signs or symptoms. About one-third of people with myelofibrosis (myelofibrosis) have no symptoms at the time of diagnosis. Patients often learn they have myelofibrosis after a routine physical exam and blood test.
As disruption of normal blood cell production increases, signs and symptoms may include:
- Feeling tired, weak or shortness of breath, usually due to a low red blood cell count (anemia)
- Pain or fullness below your ribs on the left side, due to an enlarged spleen (splenomegaly)
- Enlarged liver (on the right side of the abdomen below ribs)
- Pale skin
- Frequent infections, due to a low white blood cell count (neutropenia)
- Weight loss
- Easy bruising, as a result of a low platelet count (thrombocytopenia)
- Easy bleeding, as a result of a low platelet count (thrombocytopenia)
- Excessive sweating during sleep (night sweats)
- Fever
- Bone or joint pain
See your doctor if you’re troubled by any of the above signs or symptoms.
Myelofibrosis diagnosis
A diagnosis of myelofibrosis is made based on the World Health Organization (WHO) criteria. There is no one test that can diagnose a person as having myelofibrosis; the diagnosis is based on findings from a bone marrow biopsy exam, blood cell counts and chemistry, and physical exam.
Physical exam
Your doctor will perform a physical exam. This includes a check of vital signs, such as pulse and blood pressure, as well as checks of your lymph nodes, spleen and abdomen.
In people who have no symptoms, myelofibrosis may be suspected when a routine medical checkup reveals an enlarged spleen and abnormal blood test results.
Tests and procedures used to diagnose myelofibrosis include:
Blood Tests
The results of a blood test (a complete blood count, or CBC) that suggest a diagnosis of myelofibrosis often include:
- A decrease below the normal range in the number of red blood cells (anemia)
- An increase in the normal range in the number of white blood cells
- An increase above the normal range in platelet counts (for about one-third of patients)
- A mild to moderate decrease below the normal range in platelet counts (for about one-third of patients).
In addition to blood cell counts, blood tests may also show:
- Teardrop-shaped red cells and immature red cells and white cells in the blood (seen by microscopic examination of the blood cells)
- Giant platelets, abnormal platelet formation and circulating dwarf megakaryocytes (bone marrow cells responsible for the production of platelets)
- Elevated serum levels of uric acid, lactic dehydrogenase (LDH), alkaline phosphatase and bilirubin
- Decreased serum levels of albumin, total cholesterol and high-density lipoprotein (HDL).
Bone Marrow Tests
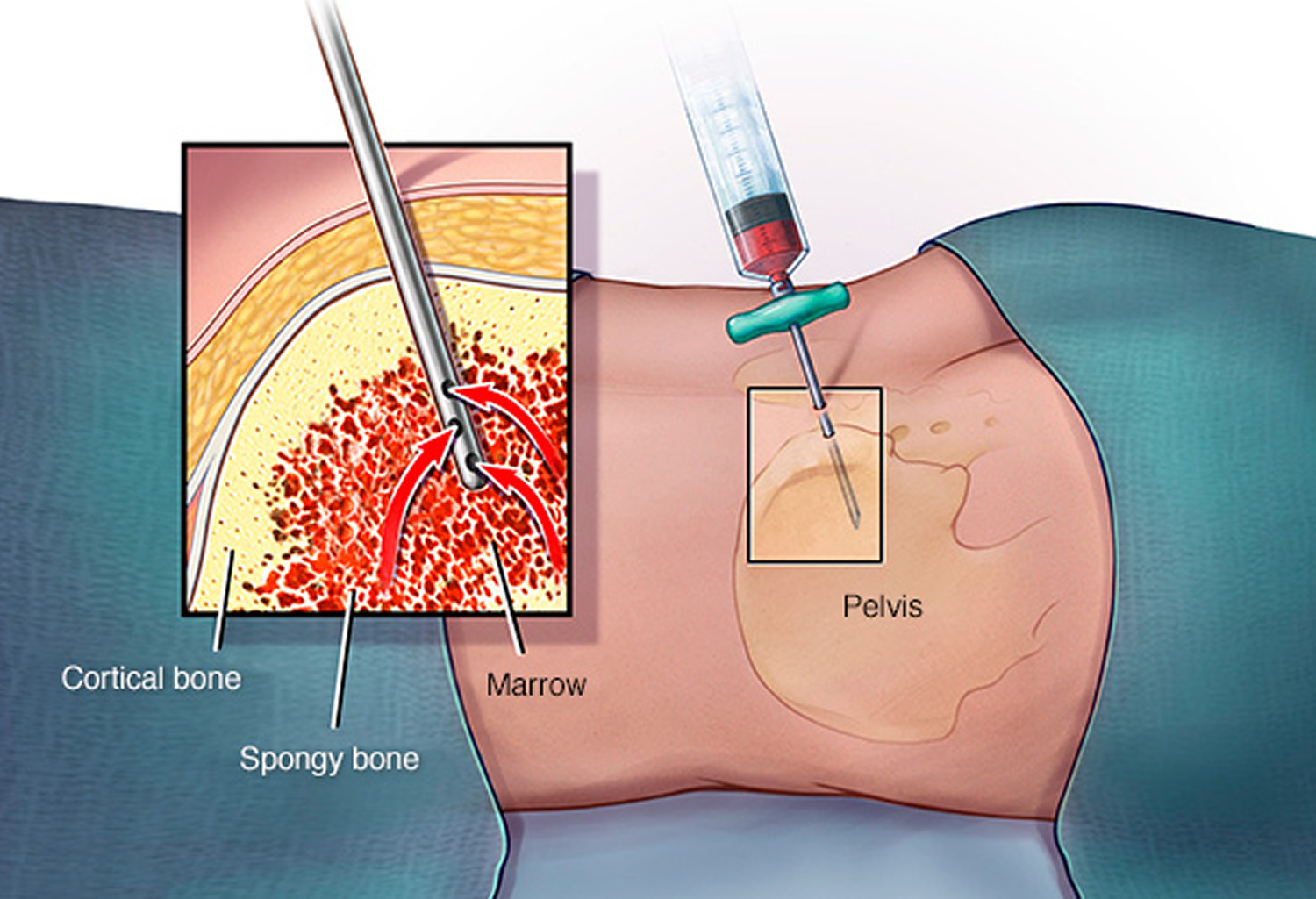
Your doctor tests your bone marrow to help confirm a diagnosis. Bone marrow testing involves two steps usually performed at the same time in a doctor’s office or a hospital:
- A bone marrow aspiration to remove a liquid marrow sample
- A bone marrow biopsy to remove a small amount of bone filled with marrow
Bone marrow biopsy and aspiration can confirm a diagnosis of myelofibrosis. In a bone marrow biopsy, a needle is used to draw a sample of hardened bone marrow from your hipbone. During the same procedure, another type of needle may be used to withdraw a sample of the liquid portion of your bone marrow. The samples are studied in a laboratory to determine the numbers and types of cells found.
Studying genetic components of the bone marrow cells can show mutations in the cells that may help eliminate other types of bone marrow disorders.
Karyotyping
Your blood or bone marrow sample may be used for a lab test called a karyotype. In this test, a microscope is used to examine the size, shape and number of chromosomes in a sampling of cells. The results of the karyotype may be helpful in making certain treatment decisions.
Gene Tests
A sample of blood or bone marrow may be analyzed in a laboratory to look for gene mutations, such as the JAK2, CALR, or MPL mutations, present in 90 percent of myelofibrosis patients.
Imaging tests
Ultrasound tests may be used to determine the size of the spleen. Magnetic resonance imaging (MRI) tests may be used to identify changes in the bone marrow that indicate myelofibrosis.
Figure 4. Bone marrow biopsy
Myelofibrosis treatment
Myelofibrosis is a chronic disease. It’s important that your doctor is experienced in treating myeloproliferative neoplasms or works in consultation with a hematology-oncologist who has experience treating myeloproliferative neoplasm patients.
Treatment consists of general supportive measures such as:
- Blood transfusion, folic acid, iron, and pyridoxine for anemia – but this does not respond to most measures
- Analgesics for pain. Allopurinol can be used for hyperuricemia to prevent gouty complications.
- Drugs such as hydroxycarbamide (most common drug used) and busulfan are used to reduce metabolic activity and high WBC (white blood cell) count and platelet levels.
- Chemotherapy (e.g. with hydroxand radiotherapy are used to reduce splenic size.
- If the spleen becomes very large and painful, transfusion requirements are high and it may be advisable to perform splenectomy.
- Stem cell transplantation
- Your doctor may suggest that you participate in a clinical trial. Clinical trials can involve therapy with new drugs and new drug combinations.
In addition, there are supportive therapies that may be used to help manage a patient’s anemia (and low blood cell count in general), enlarged spleen or other myelofibrosis-related systemic symptoms. Some of those therapies include iron, folic acid and vitamin B12 replacement and blood transfusions.
Immediate treatment may not be necessary
If you aren’t experiencing symptoms and don’t show signs of anemia, an enlarged spleen or other complications, treatment usually isn’t necessary. Instead, your doctor is likely to monitor your health closely through regular checkups and exams, watching for any signs of disease progression. Some people remain symptom-free for years.
For patients with symptoms, the goals of myelofibrosis treatment are to:
- Relieve symptoms
- Reduce an enlarged spleen
- Improve blood cell counts (i.e., anemia)
- Reduce the risk of complications.
As you develop a treatment plan with your doctor, be sure to discuss:
- The results you can expect from treatment
- Potential side effects
- The possibility of participating in a clinical trial, where you’ll have access to advanced medical treatment that may be more beneficial to you than standard treatment
You may find it helpful to bring a loved one with you to your doctor’s visits for support and to take notes and ask follow-up questions. It’s a good idea to prepare questions you’d like to ask when you visit your doctor. You can also record your conversations with your doctor and listen more closely when you get home.
Treatments that target gene mutations
Researchers are working to develop medications that target the JAK2 gene mutation that’s thought to be responsible for myelofibrosis.
The first of these medications approved by the Food and Drug Administration is ruxolitinib (Jakafi). Ruxolitinib and the other medications being developed and tested in clinical trials have been helpful in decreasing enlarged spleens and reducing symptoms associated with myelofibrosis.
It’s not yet clear whether these medications can help people with myelofibrosis live longer. But preliminary indications from clinical trials show promise.
Ruxolitinib works by stopping the action of all JAK-related genes in the body, including those found in both healthy and diseased cells. Because healthy cells are affected, side effects may occur, such as bleeding, infection, bruising, dizziness and headaches.
Treatments for anemia
If myelofibrosis is causing severe anemia, you may consider treatment, such as:
- Blood transfusions. If you have severe anemia, periodic blood transfusions can increase your red blood cell count and ease anemia symptoms, such as fatigue and weakness. Sometimes, medications can help improve anemia.
- Androgen therapy. Taking a synthetic version of the male hormone androgen may promote red blood cell production and may improve severe anemia in some people. Androgen therapy does have risks, including liver damage and masculinizing effects in women.
- Thalidomide and related medications. Thalidomide (Thalomid) and the related drugs lenalidomide (Revlimid) and pomalidomide (Pomalyst) may help improve blood cell counts and may also relieve an enlarged spleen. These drugs may be combined with steroid medications. Thalidomide and related drugs carry a risk of serious birth defects and require special precautions. This type of treatment is being studied in clinical trials.
Treatments to lower platelet counts
Drug therapy is used for myelofibrosis to lower platelet counts.
Your doctor may use one or more of the following treatments:
- Janus-associated kinase (JAK) inhibitor, such as ruxolitinib (Jakafi®)
- Chemotherapy, such as cladribine (Leustatin®) or hydroxyurea (Hydrea®)
- Immunomodulators (IMiDs), such as thalidomide (Thalomid®) and lenalidomide (Revlimid®)
- Interferon alfa (Intron® A, Roferon®-A)
- Androgen therapy, such as oxymetholone (Anadrol-50®) and danazol (Danocrine®)
- Recombinant erythropoietin (Epogen®, Procrit®)
- Glucocorticoids (also called “corticosteroids” or “steroids”)
- Bisphosphonates, such as zoledronic acid (Zometa®)
- Anagrelide hydrochloride (Agrylin®)
Treatments for enlarged spleen
If an enlarged spleen is causing complications, your doctor may recommend treatment. Your options may include:
- Chemotherapy. Chemotherapy drugs may reduce the size of an enlarged spleen and relieve related symptoms, such as pain.
- Radiation therapy. Radiation uses high-powered beams, such as X-rays, to kill cells. Radiation may be useful for a small number of patients to treat an enlarged spleen, bone pain and tumors outside the marrow. Radiation therapy can help reduce the size of the spleen, when surgical removal isn’t an option.
- Surgical removal of the spleen (splenectomy). If the size of your spleen becomes so large that it causes you pain and begins to cause harmful complications — and if you don’t respond to other forms of therapy — you may benefit from having your spleen surgically removed. Risks include infection, excessive bleeding and blood clot formation leading to stroke or pulmonary embolism. After the procedure, some people experience liver enlargement and an abnormal increase in platelet count.
- Targeted drug therapy. Ruxolitinib, which targets the gene mutation found in most cases of myelofibrosis, may be used to reduce symptoms of an enlarged spleen.
Stem cell transplant
Allogeneic stem cell transplantation — stem cell transplantation from a suitable donor — is the only treatment that has the potential to cure myelofibrosis. But it also has a high risk of life-threatening side effects.
Many people with myelofibrosis, because of age, stability of the disease or other health problems, don’t qualify for this treatment.
Prior to a stem cell transplant, also called a bone marrow transplant, you receive very high doses of chemotherapy or radiation therapy to destroy your diseased bone marrow. Then, healthy hematopoietic (blood-forming) stem cells from a compatible donor (a sibling or unrelated person whose stem cells “match” the patient’s) are infused into the myelofibrosis patient. The transplanted healthy cells travel to the patient’s bone marrow, replacing the defective stem cells. The new cells grow and provide a supply of red blood cells, white blood cells (including immune cells) and platelets.
Most patients with myelofibrosis are older and often have other health conditions that may impair organ function. Older individuals are also more likely to have other medical problems, develop complications from the treatment and have decreased tolerance for the cumulative effects of the intensive chemotherapy and for the radiation treatments needed before the transplant. However, these are generalizations. Allogeneic stem cell transplantation can be used in older people when medically appropriate. Whether or not a patient is a candidate for transplantation is determined by medical indications and the availability of a donor. There is no specific age cutoff for stem cell transplantation.
After the procedure, there’s a risk that the new stem cells will react against your body’s healthy tissues, causing potentially fatal damage (graft-versus-host disease). Other risks include organ or blood vessel damage, cataracts, and the development of a different cancer later on.
Doctors are studying a reduced-intensity transplant, also called a nonmyeloablative allogeneic stem cell transplantation or minitransplant. Reduced-intensity transplants use lower doses of pretransplant chemotherapy and radiation. Nonmyeloablative allogeneic stem cell transplantation is being used to treat some patients with leukemia, lymphoma or myeloma. Compared to a standard allogeneic stem cell transplantation, a reduced-intensity transplant delivers lower doses of chemotherapy drugs and/or radiation to the patient in preparation for the transplant. The success of reduced-intensity transplantation is a result of the graft-versus-tumor effect of the donor stem cells, rather than of high doses of chemotherapy. This approach may benefit older and sicker patients and other selected patients. Reduced-intensity transplants are now done with results that are increasingly encouraging for myelofibrosis patients.
Current Myelofibrosis Research and Clinical Trials
Some examples of treatments being studied include:
- JAK Inhibitors target abnormal JAK pathway signaling, which is present in all myelofibrosis patients, largely due to either JAK2, CALR or MPL mutations. Several JAK inhibitors are now in clinical trials and are showing effectiveness in reducing spleen size and symptoms such as night sweats and fatigue and possibly improving anemia. These new treatments include momelotinib (CYT387), pacritinib (SB1518) and NS-018.
- Histone deacetylase (HDAC) inhibitors play an important role in the regulation of gene expression. A clinical study of panobinostat (Farydak®), which is approved by the FDA for the treatment of multiple myeloma, in combination with ruxolitinib in patients with myelofibrosis is ongoing. Pracinostat is another HDAC inhibitor that is being studied in combination with ruxolitinib.
- Antifibrotic agents interfere with the process of tissue repair and fibrosis. PRM-151 is an antifibrotic therapy that is being tested in myelofibrosis. Lysin oxidase like-2 antibody is another antifibrotic medication that is being studied in a clinical trial.
- Therapies that target other pathways that may be abnormally activated in myelofibrosis are also being tested. LCL-161 is an oral therapy that blocks the activity of inhibitor of apoptosis (IAP) proteins, which promote cell survival. SL-401 is a therapy that targets the IL-3 receptor, which is found on the surface of myelofibrosis cells.
- Immune checkpoints inhibitors (nivolumab, ipilmumab) are a new class of drugs that harness the body’s immune system to fight cancer. Nivolumab (Opdivo®), which is approved by the FDA for the treatment of melanoma and non-small cell lung cancer, is now being tested as a therapy for myelofibrosis.
- Sotatercept (ACE-011) is a therapy that stimulates the production of red blood cells and is being tested to treat anemia in myelofibrosis.
- Imetelstat is a telomerase inhibitor, which affects ability of dividing cell to repair loss of DNA that happens during cell division. It is being studied in myelofibrosis to possibly improve bone marrow and normalize blood cell count.
Supportive (palliative) care
Palliative care is specialized medical care that focuses on providing relief from pain and other symptoms of a serious illness. Palliative care specialists work with you, your family and your other doctors to provide an extra layer of support that complements your ongoing care. Palliative care can be used while undergoing other aggressive treatments, such as surgery, chemotherapy or radiation therapy.
When palliative care is used along with all of the other appropriate treatments, people with cancer may feel better and live longer.
Palliative care is provided by a team of doctors, nurses and other specially trained professionals. Palliative care teams aim to improve the quality of life for people with cancer and their families. This form of care is offered alongside curative or other treatments you may be receiving.
Coping and support
Living with myelofibrosis may involve coping with pain, discomfort, uncertainty and the side effects of long-term treatments. The following steps may help ease the challenge and make you feel more comfortable and in charge of your health:
- Learn about your condition. Myelofibrosis is fairly uncommon. To help you find accurate and trustworthy information, ask your doctor to direct you toward appropriate sources. Based on these sources, find out as much as you can about myelofibrosis.
- Get support. Take this opportunity to lean on family and friends. It can be tough to talk about your diagnosis, and you’ll likely get a range of reactions when you share the news. But talking about your diagnosis and passing along information about your condition can help. So can the offers of help that often result. You may also benefit from joining a support group, either in your community or on the internet. A support group of people with the same or a similar diagnosis, such as a myeloproliferative disorder or another rare disease, can be a source of useful information, practical tips and encouragement.
- Explore ways to cope with the disease. If you have myelofibrosis, you may face frequent blood work and medical appointments and regular bone marrow exams. Some days, you may feel sick even if you don’t look sick. And some days, you may just be sick of being sick. Try to find some activities that help, whether it’s yoga, exercise, social outings or adopting a more flexible work schedule. Talk to a counselor, therapist or oncology social worker if you need help dealing with the emotional challenges of this disease.
- Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2008.[↩]
- Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med. 2000 Apr 27;342(17):1255–65. https://www.ncbi.nlm.nih.gov/pubmed/10781623[↩]
- Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the united states. Leuk Lymphoma. 2014 Mar;55(3):595–600. https://www.ncbi.nlm.nih.gov/pubmed/23768070[↩]
- Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901 http://www.bloodjournal.org/content/113/13/2895.long[↩][↩]
- Cervantes F, Dupriez B, Passamonti F, Vannucchi AM, Morra E, Reilly JT, et al. Improving survival trends in primary myelofibrosis: An international study. J Clin Oncol. 2012 Aug 20;30(24):2981–7. https://www.ncbi.nlm.nih.gov/pubmed/22826273[↩]
- Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, et al. New prognostic scoring system for primary myelofibrosis based on a study of the international working group for myelofibrosis research and treatment. Blood. 2009 Mar 26;113(13):2895–901. Description of the IPSS, the most widely used prognostic scoring system for PMF at diagnosis. http://www.bloodjournal.org/content/113/13/2895.long[↩]
- Hultcrantz M, Kristinsson SY, Andersson TM, Landgren O, Eloranta S, Derolf AR, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in sweden from 1973 to 2008: A population-based study. J Clin Oncol. 2012 Aug 20;30(24):2995–3001 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3417050[↩]
- Rumi E, Pietra D, Pascutto C, Guglielmelli P, Martinez-Trillos A, Casetti I, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. 2014 Aug 14;124(7):1062–9. Important paper showing that CALR-mutated patients with PMF have the best outcomes, and triple negative patients the worst, while the outcomes of JAK2- and MPL-mutated patients are similar. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4133481/[↩]
- Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014 May 29;123(22):e123–33. Demonstration that activated JAK-STAT signaling in MPN is not dependent on the mutational status of JAK2. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4041169/[↩]
- Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. The clinical benefit of ruxolitinib across patient subgroups: Analysis of a placebo-controlled, phase III study in patients with myelofibrosis. Br J Haematol. 2013 May;161(4):508–16 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4055021/[↩]
- Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012 Mar 1;366(9):799–807. Initial report of COMFOR I, the pivotal trial of ruxolitinib in MF conducted in North America and Australia. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4822164/[↩]
- Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012 Mar 1;366(9):787–98. Initial report of COMFOR II, the pivotal trial of ruxolitinib in MF conducted in Europe. http://www.nejm.org/doi/10.1056/NEJMoa1110556[↩]
- Cervantes F. How I treat myelofibrosis. Blood. 2014 Oct 23;124(17):2635–42. http://www.bloodjournal.org/content/124/17/2635.long[↩]
- Kroger NM, Deeg JH, Olavarria E, Niederwieser D, Bacigalupo A, Barbui T, et al. Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: A consensus process by an EBMT/ELN international working group. Leukemia. 2015 Aug 21 https://www.ncbi.nlm.nih.gov/pubmed/26293647[↩]
- Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013 Sep;27(9):1861–9. The original report of the prognostic importance of non-phenotypic driver mutations in PMF https://www.ncbi.nlm.nih.gov/pubmed/23619563[↩]
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. http://www.bloodjournal.org/content/127/20/2391[↩][↩][↩]
- Swerdlow SH, Campo E, Harris NL. In: Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, eds. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008.[↩]
- Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood 2017 129:3227-3236; doi: https://doi.org/10.1182/blood-2017-01-761999[↩]





{kind=link}