Contents
What is neurofibromatosis
Neurofibromatosis is one of the most common genetic disease that cause tumors to grow along your nerves (neurofibromas) and less frequently, in the brain and spinal cord, and produce other abnormalities such as skin changes and bone deformities. A neurofibroma is a type of nerve tumor that forms soft bumps on or under the skin. A neurofibroma can develop within a major or minor nerve anywhere in the body. This common type of benign nerve tumor tends to form more centrally within the nerve. Sometimes it arises from several nerve bundles (plexiform neurofibroma). The neurofibromas begin in the supporting cells that make up the nerves and the myelin sheath–the thin membrane that envelops and protects the nerves. In neurofibromatosis, the tumors are usually non-cancerous (benign), although occasionally they can be cancerous. These neurofibromas can form wherever there are nerve cells in the body.
In addition to benign tumors to growing on nerves, neurofibromatosis also affects the development of other systems and tissues including; the cardiovascular system, bones, skin, brain, eyes, respiratory system, gastrointestinal tract and hormonal system.
Neurofibromatosis affects all races, all ethnic groups and both sexes equally. Neurofibromatosis is one of the most common genetic disorders in the United States (one in every 2,500 to 3,000 births). The neurofibromatoses affects more than 100,000 Americans; this makes neurofibromatosis more prevalent than Cystic Fibrosis, hereditary Muscular Dystrophy, Huntington’s Disease and Tay Sachs combined.
Although many affected persons inherit the disorder, between 30 and 50 percent of new cases arise spontaneously through mutation (change) in an individual’s genes. Once this change has taken place, the mutant gene can be passed on to succeeding generations.
There are three types of neurofibromatosis that are each associated with unique signs and symptoms 1:
- Neurofibromatosis type 1. Symptoms of neurofibromatosis type 1, which may be evident at birth are skin changes (cafe-au-lait spots, freckling in armpit and groin area); two or more growths on the iris of the eye; bone abnormalities; optic gliomas (tumor on the optic nerve); a larger than normal head circumference, and abnormal development of the spine, a skull bone, or the tibia and tumors on the nerve tissue or under the skin. Signs and symptoms are usually present at birth and nearly always by the time the child is 10 years old. Neurofibromatosis type 1 is the most common type, affecting about 1 in 3,000 births. Neurofibromatosis type 1 is caused by a change in a gene on chromosome 17.
- Neurofibromatosis type 2 is less common and is characterized by slow-growing tumors on the vestibular branch of the right and left eighth cranial nerves, which are called vestibular schwannomas or acoustic neuromas; hearing loss; ringing in the ears; poor balance; brain and/or spinal tumors; and cataracts at a young age. It often starts in the teen years. Neurofibromatosis type 2 is a very rare form caused by a change in a gene on chromosome 22.
- Neurofibromatosis type 3 also known as Schwannomatosis causes schwannomas. The distinctive feature of schwannomatosis is the development of multiple schwannomas (tumors made up of certain cells) everywhere in the body except on the vestibular branch of the 8th cranial nerve. The dominant symptom is pain, which develops as a schwannoma enlarges or compresses nerves or adjacent tissue. Some people may develop numbness, tingling, or weakness in the fingers and toes. Neurofibromatosis type 3 is the rarest type and is similar to type 2 except it is not associated with inner ear tumors and hearing loss.
Neurofibromatosis type 1 and neurofibromatosis type 2 are distinct conditions. Neurofibromatosis type 1 cannot turn into neurofibromatosis type 2 or vice versa. It is important to have a correct diagnosis from a doctor who knows about the condition as the health management plan for each of these conditions is different.
All three types of neurofibromatosis are inherited in an autosomal dominant manner. This means that to be affected, a person only needs a change (mutation) in one copy of the responsible gene in each cell (see Figure 1). In some cases, an affected person inherits the mutation from an affected parent. Other cases may result from new (de novo) mutations in the gene. These cases occur in people with no history of the disorder in their family. A person with neurofibromatosis has a 50% chance with each pregnancy of passing along the altered gene to his or her child.
- Approximately 50% of those affected with Neurofibromatosis have a prior family history of neurofibromatosis. The other 50% of cases are the result of spontaneous genetic mutation. If an individual does not have neurofibromatosis, s/he can not pass it on to his/her children.
There is no cure for neurofibromatosis. The effects of neurofibromatosis are unpredictable and have varying manifestations and degrees of severity. Treatment is aimed at controlling symptoms and may include surgery to remove tumors, radiation therapy, and/or medicines.
Figure 1. Neurofibromatosis autosomal dominant inheritance pattern
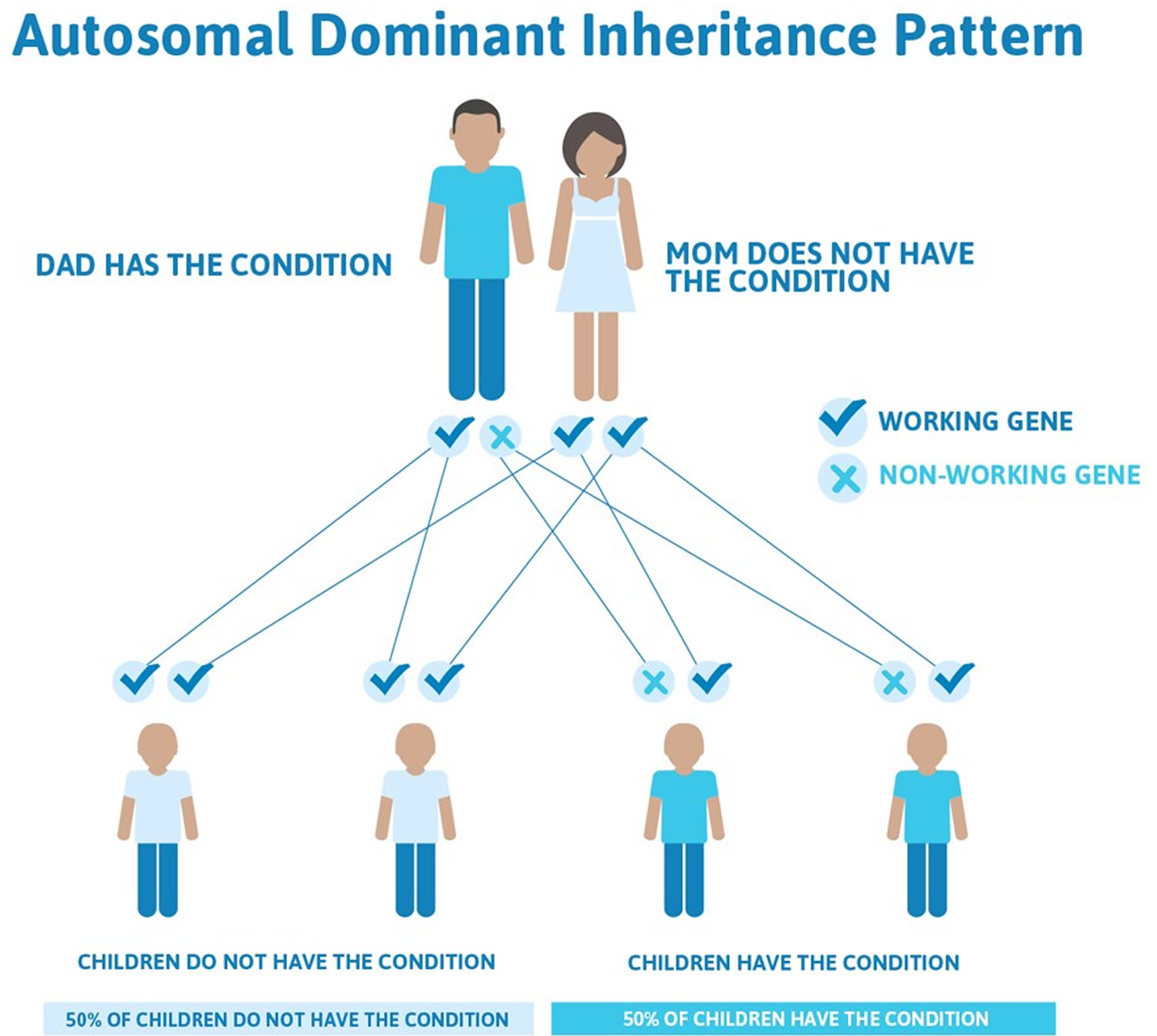
Figure 2. Structure of a neuron
Neurofibromatosis type 1
Neurofibromatosis 1 is also known as von Recklinghausen disease or peripheral neurofibromatosis. Neurofibromatosis type 1 is one of the most common genetic inherited conditions, affecting up to 1 in 2,500 individuals. It is at least as common as Cystic Fibrosis, Muscular Dystrophy and Huntington’s disease.
Neurofibromatosis 1 usually appears in childhood. Signs are often evident at birth or shortly afterward, and almost always by age 10. Signs and symptoms are often mild to moderate, but can vary in severity.
Signs and symptoms include:
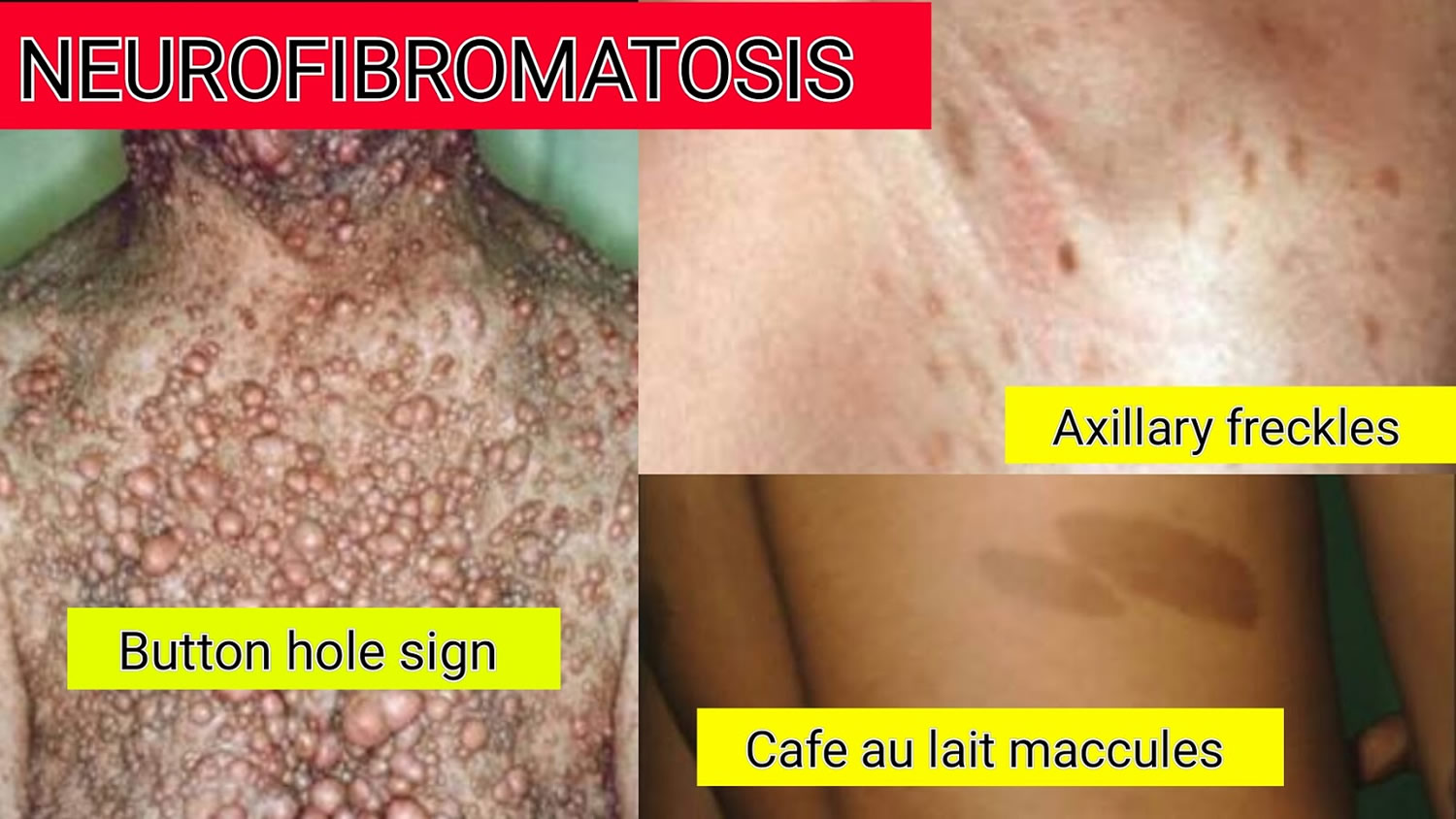
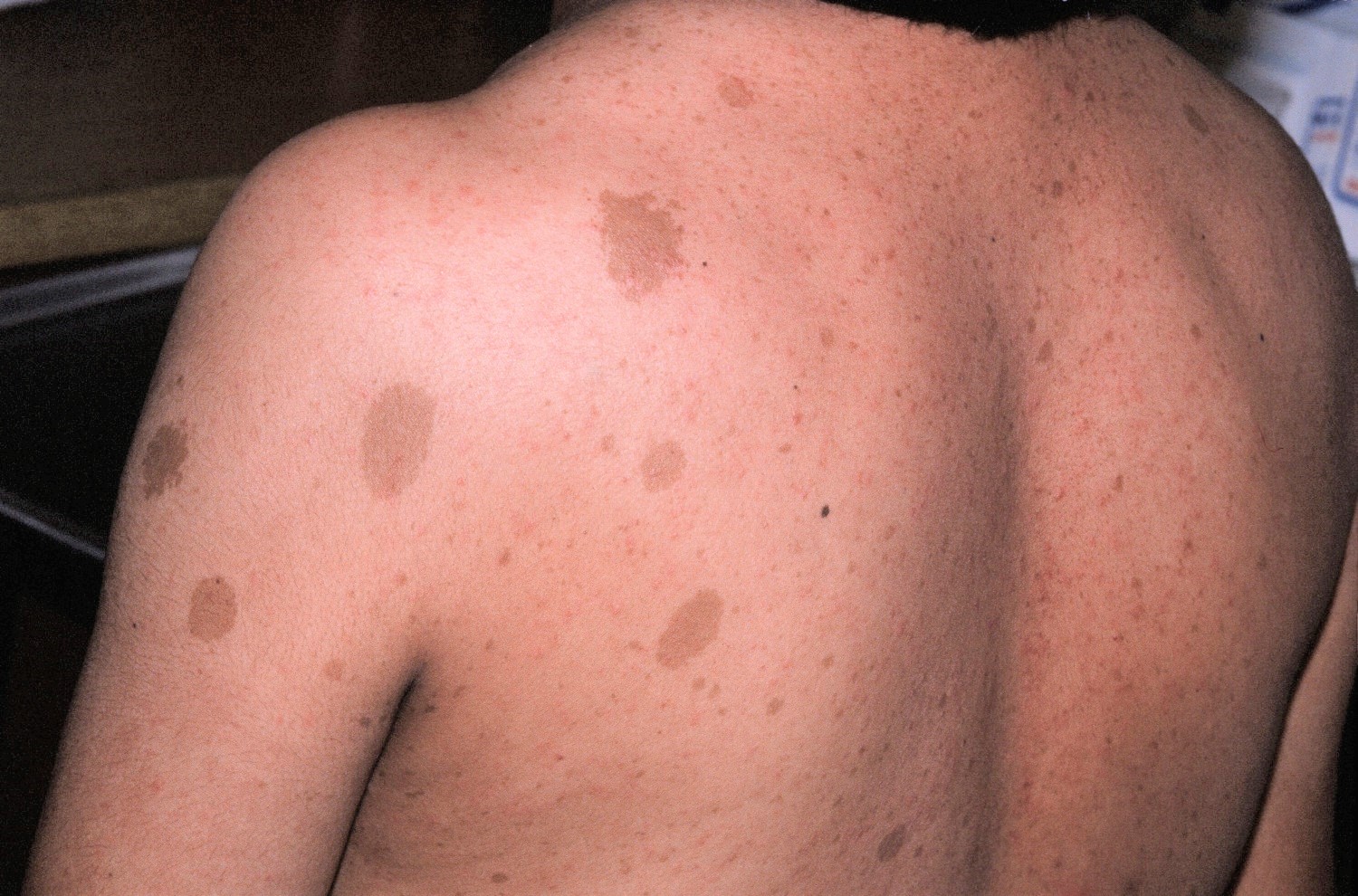
- Flat, light brown spots on the skin (cafe au lait spots). These harmless spots are common in many people. Having more than six cafe au lait spots is a strong indication of neurofibromatosis 1 (see Figure 3 below). They are usually present at birth or appear during the first years of life and then stabilize.
- Freckling in the armpits or groin area. Freckling usually appears by ages 3 to 5. Freckles are smaller than cafe au lait spots and tend to occur in clusters in skin folds.
- Tiny bumps on the iris of your eye (Lisch nodules). These harmless nodules can’t easily be seen and don’t affect your vision.
- Soft bumps on or under the skin (neurofibromas). These benign tumors usually develop in or under the skin, but can also grow inside of the body. Sometimes, a growth will involve multiple nerves (plexiform neurofibroma).
- Bone deformities. Abnormal bone growth and a deficiency in bone mineral density can cause bone deformities such as a curved spine (scoliosis) or bowed lower leg.
- Tumor on the optic nerve (optic glioma). These tumors usually appear by age 3, rarely in late childhood and adolescence, and almost never in adults.
- Learning disabilities. Impaired thinking skills are common in children with neurofibromatosis 1, but are usually mild. Often there is a specific learning disability, such as problem with reading or mathematics. Attention-deficit/hyperactivity disorder (ADHD) is also common.
- Larger than average head size. Children with neurofibromatosis 1 tend to have a larger than average head size due to increased brain volume.
- Short stature. Children with neurofibromatosis 1 often are below average in height.
Figure 3. Café-au-lait spots
Neurofibromatosis type 2
Neurofibromatosis type 2 has previously been known as Bilateral Acoustic Neurofibromatosis and Central Neurofibromatosis. Neurofibromatosis type 2 is rarer, affecting 1 in 25,000 to 1 in 40,000 individuals.
Signs and symptoms of Neurofibromatosis 2 usually result from the development of benign, slow-growing tumors (acoustic neuromas) in both ears. Also known as vestibular schwannomas, these tumors grow on the nerve that carries sound and balance information from the inner ear to the brain.
Signs and symptoms generally appear in the late teen and early adult years, and can vary in severity.
Signs and symptoms can include:
- Gradual hearing loss
- Ringing in the ears
- Poor balance
- Headaches
Sometimes Neurofibromatosis 2 can lead to the growth of schwannomas in other nerves of the body, including the cranial, spinal, visual (optic) and peripheral nerves. Signs and symptoms of these schwannomas can include:
- Numbness and weakness in the arms or legs
- Pain
- Balance difficulties
- Facial drop
- Vision problems or the development of cataracts
Neurofibromatosis type 3
Neurofibromatosis type 3 (Schwannomatosis) is the rarest type affecting around 1 in 40,000 people. This condition is distinguishable from neurofibromatosis 2 in that people affected by this condition do not develop tumours on the hearing nerves (vestibular nerves).
The features of Schwannomatosis include the formation of benign tumours (schwannomas) on cells, called Schwann cells, which surround the nerves and allow them to conduct information around the body. These tumours typically develop on cranial, spinal and peripheral nerves, but as noted above do not form on the hearing nerves. Also, unlike in neurofibromatosis2, people affected with Schwannomatosis do not go on to develop any other types of tumour.
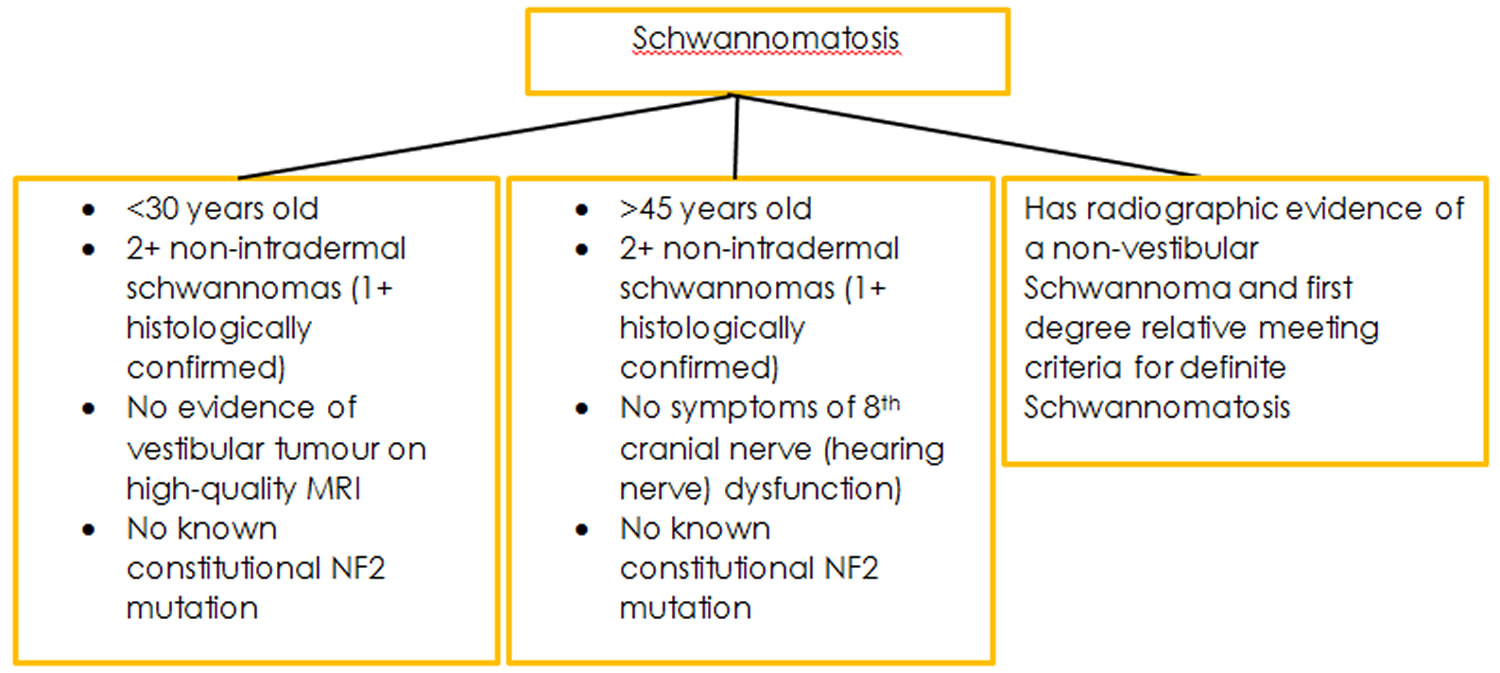
The diagnostic criteria for this condition are still evolving. This is due to the condition only recently having been characterised, and only a small number of people affected identified. As more research is conducted, and more people with the condition identified a more definitive criteria for diagnosis will form.
This rare type of neurofibromatosis 3 usually affects people after the age of 20. Schwannomatosis causes tumors to develop on skull (cranial), spinal and peripheral nerves — but not on the nerve that carries sound and balance information from the inner ear to the brain. Because tumors don’t usually grow on both hearing nerves, schwannomatosis doesn’t cause the hearing loss experienced by people with Neurofibromatosis 2.
Schwannomatosis causes chronic pain, which can be disproportionately larger than the neurological problems they have and can occur anywhere in your body. This is also often the first symptom of the condition. This pain can occur in any part of the body. As pain is often the only symptom diagnosis can often take a considerable length of time.
Other symptoms include:
- Numbness or weakness in various parts of your body
- Loss of muscle.
Neurofibromatosis causes
Neurofibromatosis is caused by genetic defects (mutations) that either are passed on by a parent or occur spontaneously at conception. The specific genes involved depend on the type of neurofibromatosis:
- Neurofibromatosis 1. The Neurofibromatosis 1 gene is located on chromosome 17. This gene normally produces a protein called neurofibromin that helps regulate cell growth. The mutated gene causes a loss of neurofibromin, which allows cells to grow uncontrolled.
- Neurofibromatosis 2. The Neurofibromatosis 2 gene is located on chromosome 22, and produces a protein call merlin. The mutated gene causes a loss of merlin, leading to uncontrolled cell growth.
- Schwannomatosis. So far, two genes are known to cause schwannomatosis. The genetics of Schwannomatosis is more complex than that of neurofibromatosis 1 and neurofibromatosis 2. While families affected by this condition usually show an autosomal dominant form of inheritance it sometimes appears as if the condition skips generations and therefore a careful family history needs to be taken down to include anyone in the family who showed symptoms of unexplained pain. Genetic testing is not currently available for Schwannomatosis as the specific genes involved are still being researched, though a candidate gene, and several potentially associated ones have been identified. A diagnosis of mosaic (or segmental) Schwannomatosis is also possible. This arises where only one area of the body is affected by the condition.
Risk factors for neurofibromatosis
The biggest risk factor for neurofibromatosis is a family history of the disorder. About half of people with neurofibromatosis 1 and neurofibromatosis 2 inherited the disease. Neurofibromatosis1 and neurofibromatosis2 that isn’t inherited results from new gene mutations.
Neurofibromatosis 1 and neurofibromatosis 2 are both autosomal dominant disorders, which means that any child of a parent with the disorder has a 50 percent chance of inheriting the genetic mutation.
The inheritance pattern for schwannomatosis is less clear. Researchers currently estimate that the risk of inheriting schwannomatosis from an affected parent is about 15 percent.
Neurofibromatosis symptoms
Neurofibromatosis is variable. Even within the same family there is no way to predict how someone will be affected. Some will be mildly affected with very few or no health problems while others can have serious life-threatening problems or problems that cause difficulty day-to-day and limits what they are able to do.
Neurofibromatosis type 1 symptoms
In neurofibromatosis type 1, these benign tumors commonly grow on the skin. A common feature of neurofibromatosis type 1 is café au lait spots, which are harmless coffee-colored skin patches. Most people have one or two of these spots, but people with neurofibromatosis1 always have six or more. In neurofibromatosis type 1, tumors can also appear on the optic nerve and on the iris in the eye (Lisch nodules) and neurofibromas on or under the skin (usually pea sized lumps). These tumors might or might not affect your vision.
Other features shown to be associated with neurofibromatosis 1 include:
- Large head
- Short stature
- Low muscle tone
- Coordination difficulties
- Scoliosis
- ADHD type behaviours
- Autism Spectrum Disorder (ASD)
- Cardiovascular (heart-related) problems
Other complications to be aware of include an increased risk to rare tumors and cancers, high blood pressure and other complications.
Neurofibromatosis type 2 symptoms
Neurofibromatosis 2 is a variable condition; there is currently no way of knowing how someone will be affected by neurofibromatosis2. Even within a family, each person will present with different symptoms or features.
In neurofibromatosis type 2, these benign tumors are more generally found within the spinal cord and brain. The most common features of neurofibromatosis type 2 are benign tumours which grow on the 8th cranial nerve in both ears, which often results in hearing loss and issues with balance. Symptoms can include hearing loss and problems with swallowing, speech, balance and eye movements.
Neurofibromatosis2 is usually diagnosed following MRI scans most commonly after presentation to a doctor with symptoms of hearing loss, ringing in the ears (tinnitus) and issues with balance. These symptoms are most commonly the result of benign tumours on the vestibular nerves in the ears. Other benign (and some malignant) tumors may also be found throughout the nervous system, typically in the brain and spine.
Tumors on both hearing nerves (bilateral vestibular schwannomas) are the main criteria for diagnosis. The presence of these tumors distinguish neurofibromatosis 2 from neurofibromatosis 1 and Schwannomatosis (neurofibromatosis type 3). Alternatively, diagnosis can be made where there is a family history of neurofibromatosis 2 along with other nervous system tumors.
While tumors on the hearing nerves (eighth cranial nerve) are the most common other tumors people with neurofibromatosis2 can develop tumors on other nerves throughout the body as well. The most commonly found tumors are called “schwannomas” because they form on cells that surround the nerves called Schwann cells, which allow the nerves to conduct information around the body. The location of the Schwann cells affected by a tumor will determine the symptoms that will arise.
There are several symptoms to be aware of and watch for in someone affected by neurofibromatosis 2. These include:
- Sudden changes to hearing
- Changes in vision
- Sudden, severe headaches
- Vomiting
- Unexplained drowsiness
- Loss of sensation, tingling or weakness in an arm or leg
- Development of pain
- Dizziness, unsteadiness or difficulties with balance
- Unexplained drowsiness
Neurofibromatosis 2 is very different to neurofibromatosis 1 in that people with neurofibromatosis 2 do not usually have a large number of skin changes as seen in neurofibromatosis 1, and most people with the condition will need operations or other treatments for brain or spinal cord tumors at some time.
Schwannomatosis (neurofibromatosis type 3)
For neurofibromatosis type 3 or Schwannomatosis, the main feature is chronic pain, which can be greater than expected from the underlying neurological problems the person affected faces.
Neurofibromatosis prognosis
In most cases, symptoms of Neurofibromatosis type 1 are mild, and individuals live normal and productive lives. In some cases, however, Neurofibromatosis type 1 can be severely debilitating and may cause cosmetic and psychological issues. The course of Neurofibromatosis type 2 varies greatly among individuals. Loss of hearing in both ears develops in most individuals with Neurofibromatosis type 2. In some cases of Neurofibromatosis type 2, the damage to nearby vital structures, such as other cranial nerves and the brain stem, can be life-threatening. Most individuals with Neurofibromatosis type 3 (Schwannomatosis) have significant pain. In some extreme cases the pain will be severe and disabling.
Living with neurofibromatosis
Neurofibromatosis can’t be cured, but any symptoms that arise can be treated and/or managed. Regular medical attention may be required throughout childhood and into adulthood, particularly if tumors become cancerous, though this is rare.
Neurofibromatosis complications
Complications of neurofibromatosis vary, even within the same family. Generally, complications result from tumor growth distorting nerve tissue or pressing on internal organs.
Neurofibromatosis 1 complications
Complications of neurofibromatosis 1 include:
- Neurological problems. Learning and thinking difficulties are the most common neurological problem associated with neurofibromatosis1. Uncommon complications include epilepsy and buildup of excess fluid in the brain.
- Concerns with appearance. Visible signs of neurofibromatosis — such as extensive cafe au lait spots, numerous neurofibromas in the facial area or large neurofibromas — can cause anxiety and emotional distress, even if they’re not medically serious.
- Skeletal problems. Some children have abnormally formed bones, which can result in bowing of legs and fractures that sometimes don’t heal. neurofibromatosis1 can cause curvature of the spine (scoliosis) that may need bracing or surgery. Neurofibromatosis 1 is also associated with decreased bone mineral density, which increases your risk of weak bones (osteoporosis). There are two types of very rare skeletal abnormalities that children with neurofibromatosis 1 can have. They generally are present at birth or soon after. The long bones in the arms and legs sometimes do not develop properly, and can appear more curved than is normal, which may not require treatment. Pseudoarthrosis, or the formation of a false joint caused by fracture, is the extreme and sometimes occurs in the long bones e.g. tibia and radius, due to the way in which the bones develop. Sphenoid wing dysplasia relates to the abnormal development of bones in the eye socket. These complications will require supervision and possibly treatment from an orthopedic surgeon (bone doctor).
- Vision problems. Occasionally in children, an optic glioma can develop, affecting vision. In over half of the 15 – 20% of children who have neurofibromatosis type 1 with these tumors do not ever have any symptoms, without careful monitoring by an ophthalmologist (eye doctor) they can go on to cause problems with eyesight. The presence of an optic glioma can sometimes confirm the diagnosis of neurofibromatosis 1. Guidelines recommend a yearly eye exam by an ophthalmologist until at least age 8 with less frequent monitoring after that point as this is when the chance of developing a symptomatic optic glioma eases off.
- Problems during times of hormonal change. Hormonal changes associated with puberty, pregnancy or menopause might cause an increase in neurofibromas. Most women with neurofibromatosis1 have healthy pregnancies but will likely need monitoring by an obstetrician familiar with the disorder.
- Cardiovascular problems. People with neurofibromatosis1 have an increased risk of high blood pressure and, rarely, blood vessel abnormalities.
- Breathing problems. Rarely, plexiform neurofibromas can put pressure on your airway.
- Plexifrom neurofibromas: Benign tumours that develop on peripheral nerves, and tend to grow over large areas involving a whole section of a nerve including its branches. They often appear as a lumpy mass under the skin. They can occur anywhere in the body, including deep within the body. The skin that overlies them can often be thickened, pigmented or hairy. This specific type of neurofibroma often develops much earlier than the skin (dermal) neurofibromas described above, and develop in approximately 30 – 50% of those diagnosed with neurofibromatosis 1. These plexiform neurofibromas need to be monitored as they can become malignant. Malignancy is most common during a person’s 20s and 30s.
- Cancer. An estimated 3 to 5 percent of people with neurofibromatosis1 develop cancerous tumors. These usually arise from neurofibromas under the skin or from plexiform neurofibromas. People with neurofibromatosis1 also have a higher risk of other forms of cancer, such as breast cancer, leukemia, brain tumors and some types of soft tissue cancer.
- Benign adrenal gland tumor (pheochromocytoma). This noncancerous tumor secretes hormones that raise your blood pressure. A pheochromocytoma is generally surgically removed.
- Learning disabilities: Approximately half of all children given a diagnosis of neurofibromatosis 1 will have some sort of learning or attention difficulties. Despite this large percentage it is rare for children with neurofibromatosis 1 to have a diagnosis of intellectual disability. Children with neurofibromatosis 1 can struggle with speech and language, concentration, perception of space, following instructions and co-ordination. Their behavior can be erratic, impulsive and inconsistent. They may not understand social cues or reasoning behind appropriate behaviours. These learning problems are not unique to neurofibromatosis 1 and are dealt with in the same way as other children without neurofibromatosis 1.
- Epilepsy: 3-7% of children with a diagnosis of neurofibromatosis 1 also have epilepsy according to research. The epileptic seizures suffered by those with neurofibromatosis1 are the same as those suffered by anyone with epilepsy/seizures in the general population and therefore are treated in the same way.
- Malignant Peripheral Nerve Sheath Tumours are cancerous tumors which can develop from plexiform neurofibromas. Anyone with a plexiform neurofibroma needs to watch for:
- Rapid growth of the plexiform
- A change in the texture of the plexiform from soft to hard
- Development of unexplained pain
- If pain begins for no apparent reason and does not go away.These cancers can also form spontaneously, and so everyone with neurofibromatosis 1 needs to watch for:
- Rapidly growing lumps under the skin
- New and painful lumps under the skin
- Unexplainable and persistent pain.In cases where a plexiform is present and when a lump grows spontaneously and any of the above changes occur it is very important to see your doctor.
- OTHER TUMORS/CANCERS: People with neurofibromatosis1 have a 1 in 10 chance (10%) of developing an neurofibromatosis 1 related cancer across their lifetime. Adults with neurofibromatosis 1 are known to have a slightly increased risk for developing glomus tumours, brain tumours, breast cancer, gastrointestinal stromal tumours (GIST), pheochromocytoma (rare, benign tumour of the adrenal glands) compared to the general population. Children also have an increased risk of a rare form of leukaemia and a tumour that grows near the bladder (rhabdomyosarcoma).
- HIGH BLOOD PRESSURE: Guidelines indicate it is very important for anyone with neurofibromatosis1 to have their blood pressure monitored on a yearly basis. While people with neurofibromatosis1 are just as likely as someone not affected by neurofibromatosis1 to have high blood pressure for unknown reasons, there are two rare conditions affecting people with neurofibromatosis1 that makes monitoring of blood pressure especially important. As noted above, pheochromocytoma, a tumour that grows on the adrenal glands above the kidneys, if present causes an increase in blood pressure. Similarly, a narrowing in the arteries that lead to the kidneys (renal artery stenosis) results in increased blood pressure.
The above is not a definitive list of associated features and complications, but rather an outline of the more common ones associated with neurofibromatosis 1.
Neurofibromatosis 2 complications
Complications of neurofibromatosis 2 include:
- Partial or total deafness
- Facial nerve damage
- Vision problems
- Small benign skin tumors (skin schwannomas)
- Weakness or numbness in the extremities
- Multiple benign brain tumors or spinal tumors requiring frequent surgeries (meningiomas)
Schwannomatosis complications
The pain caused by schwannomatosis can be debilitating and may require surgical treatment or management by a pain specialist.
Neurofibromatosis diagnosis
Neurofibromatosis is usually diagnosed in childhood. If a doctor sees several signs indicating someone has neurofibromatosis, they might refer them to a specialist for further testing. Prenatal genetic testing is possible if neurofibromatosis is known to run in the family.
Your doctor will start with a physical examination and review of your medical history and family medical history.
Neurofibromatosis 1 often can be diagnosed based on physical examination. Your doctor may use a special lamp to check your skin for cafe au lait spots. A physical examination and family history are also important for a diagnosis of neurofibromatosis 2.
Your doctor also might recommend:
- Eye exam. An eye doctor can detect Lisch nodules and cataracts.
- Ear exam. Tests such as audiometry, electronystagmography and brainstem auditory evoked response can help assess hearing and balance problems in people with NF2.
- Imaging tests. X-rays, CT scans or MRIs can help identify bone abnormalities, tumors in the brain or spinal cord, and very small tumors. An MRI might be used to help identify optic gliomas. Imaging tests are also often used to monitor NF2 and schwannomatosis.
- Genetic tests. Tests to identify neurofibromatosis 1and neurofibromatosis 2 are available and can be done prenatally. Ask your doctor about genetic counseling. Genetic tests for schwannomatosis are limited.
For a diagnosis of neurofibromatosis 1, you must have at least two signs of the condition. If your child has only one sign and no family history of neurofibromatosis 1, your doctor will likely monitor the child for the development of any additional signs. A diagnosis of neurofibromatosis 1 is usually made by age 4.
Genetic testing may help establish the diagnosis.
Neurofibromatosis type 1
To receive a diagnosis of neurofibromatosis 1 according to the National Institute of Health statement a person must have at least 2 of the following:
- A first degree relative with neurofibromatosis type 1
- Six or more café au lait patches (brown “birth marks) >0.5 cm in children and >1.5 cm in adults
- Axillary (armpit) or groin freckling
- 2 or more neurofibromas (pea sized lumps on the skin) of any type OR 1 plexiform neurofibroma (larger areas on the skin that appear swollen)
- 2 or more Lisch nodules (benign pigmented tumors also known as iris hamartomas)
- Optic pathway glioma
- Bony dysplasia (enlargement) of the sphenoid wing (part of a bone in the skull) or
- Thinning of the long bone cortex (one of the three layers of bone) with or without pseudoarthrosis (a false joint caused through naturally unmendable fracture) of the long bones.
Along with the features required for diagnosis there are other features and complications that may arise for a person with neurofibromatosis type 1:
- Epilepsy
- Scoliosis – curvature of the spine
- Learning disabilities
- Large head
- Early or late onset of puberty
- Short stature
- High blood pressure (can be caused by a narrowing in the arteries to the kidneys and occasionally due to a tumor in the adrenal glands
- Somewhat increased risk for rare malignancies occurring in the brain, nerves or spinal cord compared with the general population
- Brain tumors
- Stroke (cerebrovascular occlusion)
- Itchy skin
Neurofibromatosis type 2
To receive a diagnosis of neurofibromatosis 2 according to the National Institute of Health statement a person must have:
- A first degree relative with neurofibromatosis type 2
- Bilateral Vestibular Schwannomas
If you have a first degree relative with neurofibromatosis type 2, you may only need to have:
- Unilateral Vestibular Schwannomas AND
- Any 2 of:
- Meningioma
- Glioma
- Neurofibroma
- Schwannoma
- Posterior subcapsular opacities (cataracts)
There are now also alternative diagnostic criteria for neurofibromatosis type 2, The Manchester diagnostic criteria takes into account people who do not have a family history of neurofibromatosis 2:
EITHER
- Unilateral Vestibular Schwannoma AND
- Any 2 of:
- Meningioma
- Glioma
- Neurofibroma
- Schwannoma
- Posterior subcapsular opacities (cataracts)
OR
- 2 or more Meniongiomas AND
- Any 2 of:
- Glioma
- Neurofibroma
- Schwannoma
- Posterior subcapsular opacities (cataracts)
- Or Unilateral Vestibular Schwannoma
Schwannomatosis (neurofibromatosis type 3)
Because Schwannomatosis is rare and the genetic cause of the condition remains unknown the diagnostic criteria for this condition are still up for consideration. However, in 2005 researchers based at the Massachusetts General Hospital published diagnostic criteria based upon what is currently known.
Figure 4. Schwannomatosis diagnostic criteria
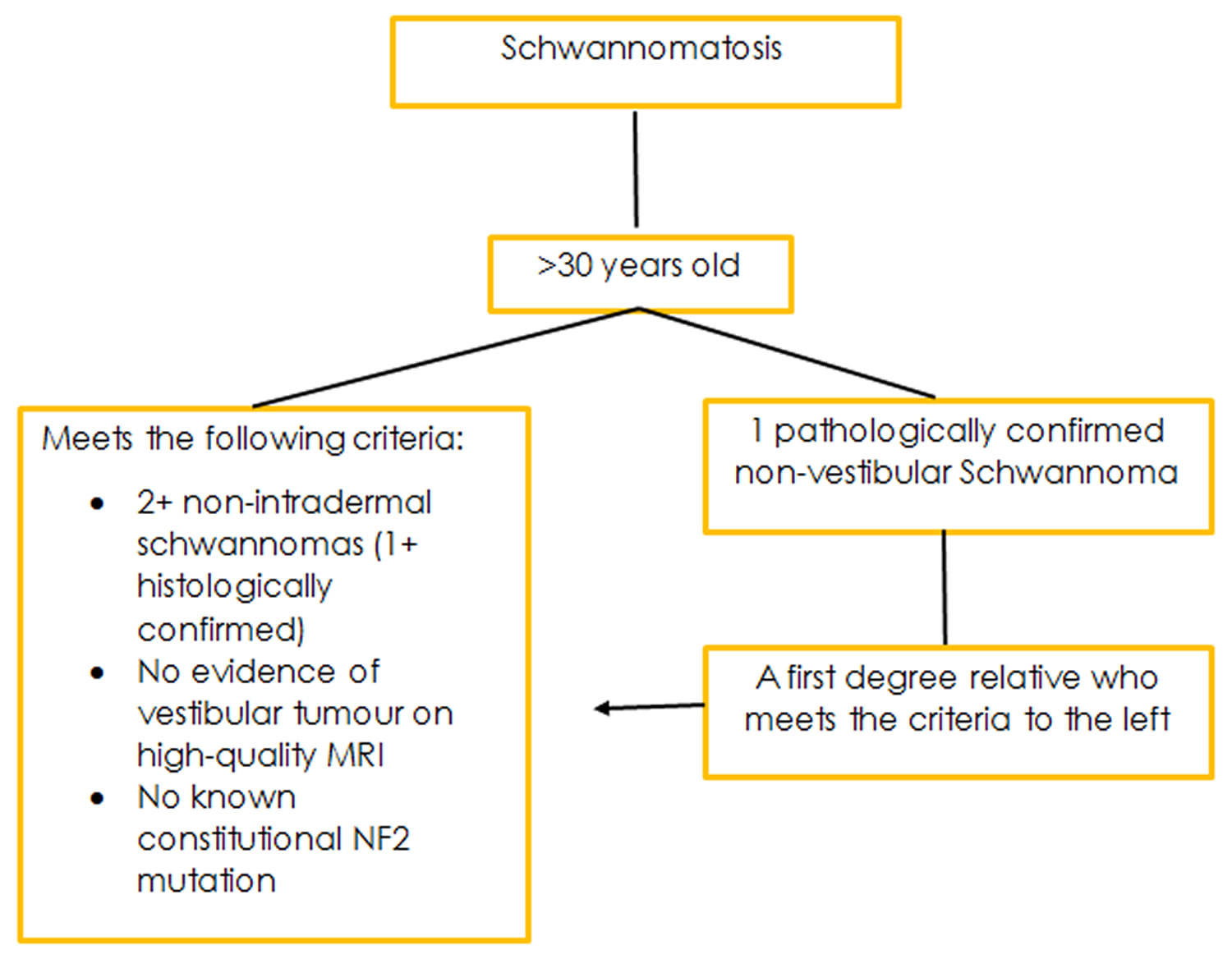
Figure 5. Schwannomatosis additional diagnostic criteria
Neurofibromatosis treatment
Neurofibromatosis can’t be cured, but treatments are available for your signs and symptoms. Generally, the sooner you or your child is under the care of a doctor trained in treating neurofibromatosis, the better the outcome.
Monitoring
If your child has neurofibromatosis 1, your doctor is likely to recommend yearly age-appropriate checkups to:
- Assess your child’s skin for new neurofibromas or changes in existing ones
- Check for signs of high blood pressure
- Evaluate your child’s growth and development — including height, weight and head circumference — according to growth charts available for children with neurofibromatosis 1
- Check for signs of early puberty
- Evaluate your child for any skeletal changes and abnormalities
- Assess your child’s learning development and progress in school
- Obtain a complete eye examination
Contact your doctor promptly if you notice any changes in signs or symptoms between visits. It’s important to rule out the possibility of a cancerous tumor and to obtain appropriate treatment at an early stage.
Treatment may include surgery, focused radiation, or chemotherapy. Surgery to remove neurofibromatosis type 2 tumors completely is one option. Surgery for vestibular schwannomas does not restore hearing and usually reduces hearing. Sometimes surgery is not performed until functional hearing is lost completely. Surgery may result in damage to the facial nerve and some degree of facial paralysis. Focused radiation of vestibular schwannoma carries of a lower risk of facial paralysis than open surgery, but is more effective o shrinking small to moderate tumors than larger tumors. Chemotherapy with a drug that targets the blood vessels of vestibular schwannoma can reduce the size of the tumor and improves hearing, but some tumors do not respond at all and sometimes respond only temporarily. Bone malformations can often be corrected surgically, and surgery can also correct cataracts and retinal abnormalities. Pain usually subsides when tumors are removed completely.
Surgery and other procedures
Your doctor might recommend surgery or other procedures to treat severe symptoms or complications of neurofibromatosis.
- Surgery to remove tumors. Symptoms can be relieved by removing all or part of tumors that are compressing nearby tissue or damaging organs. If you have neurofibromatosis 2 and have experienced hearing loss, brainstem compression or tumor growth, your doctor might recommend surgery to remove acoustic neuromas that are causing your problems. Complete removal of schwannomas in people with schwannomatosis can ease pain substantially.
- Stereotactic radiosurgery. This procedure delivers radiation precisely to your tumor and doesn’t require an incision. Stereotactic radiosurgery might be an option to remove acoustic neuromas if you have neurofibromatosis 2. Stereotactic radiosurgery can help preserve your hearing.
- Auditory brainstem implants and cochlear implants. These devices might help improve your hearing if you have neurofibromatosis 2 and hearing loss.
Cancer treatment
Malignant tumors and other cancers associated with neurofibromatosis are treated with standard cancer therapies, such as surgery, chemotherapy and radiation therapy. Early diagnosis and treatment are the most important factors resulting in good outcome.
Pain medications
Managing pain is an important part of treatment for schwannomatosis. Your doctor might recommend:
- Gabapentin (Neurotin) or pregabalin (Lyrica) for nerve pain
- Tricyclic antidepressants such as amitriptyline
- Serotonin and norepinephrine reuptake inhibitors such as duloxetine (Cymbalta)
- Epilepsy medications such as topiramate (Topamax) or carbamazepine (Carbatrol, Tegretol)
Coping and support
Caring for a child with a chronic condition such as neurofibromatosis can be a challenge. It may help to keep in mind that many children with neurofibromatosis grow up to live healthy lives with few, if any, complications.
To help you cope:
- Find a primary care doctor you can trust and who can coordinate your child’s care with other specialists. The Children’s Tumor Foundation 2 has an online tool to help you find a neurofibromatosis specialist in your area.
- Join a support group for parents who care for children with neurofibromatosis, ADHD, special needs or chronic illnesses in general.
- Accept help for daily needs such as cooking, cleaning, caring for your other children or simply giving you a needed break.
- Seek academic support for children with learning disabilities.





{kind=link}