
Contents
What is Niemann Pick disease
Niemann-Pick disease is a group of rare inherited lysosomal storage disorders that affects many body systems and can affect both children and adults. Niemann-Pick diseasehas a wide range of symptoms that vary in severity. Niemann-Pick disease is divided into four main types: type A, type B, type C1, and type C2. These types are classified on the basis of genetic cause and the signs and symptoms of the condition.
Acid Sphingomyelinase Deficiency includes Niemann-Pick Disease Type A and Type B, which are caused by a lack of the enzyme acid sphingomyelinase leading to a build-up of toxic materials in the body.
Niemann-Pick Disease Type C is a devastating neurodegenerative disease caused by an accumulation of lipids (fats) in the liver, brain and spleen.
Niemann-Pick disease type A occurs more frequently among individuals of Ashkenazi (eastern and central European) Jewish descent than in the general population. The incidence within the Ashkenazi population is approximately 1 in 40,000 individuals. Niemann-Pick disease type A incidence in other populations is not known, but it is considered extremely rare and estimated to be about 0.5 to 1 per 100,000 births 1.
Combined, Niemann-Pick disease types C1 and C2 are estimated to affect 1 in 100,000 to 150,000 individuals; however, type C1 is by far the more common type, accounting for 95 percent of cases. The disease occurs more frequently in people of French-Acadian descent in Nova Scotia. In Nova Scotia, a population of affected French-Acadians were previously designated as having Niemann-Pick disease type D, however, it was shown that these individuals have mutations in the gene associated with Niemann-Pick disease type C1.
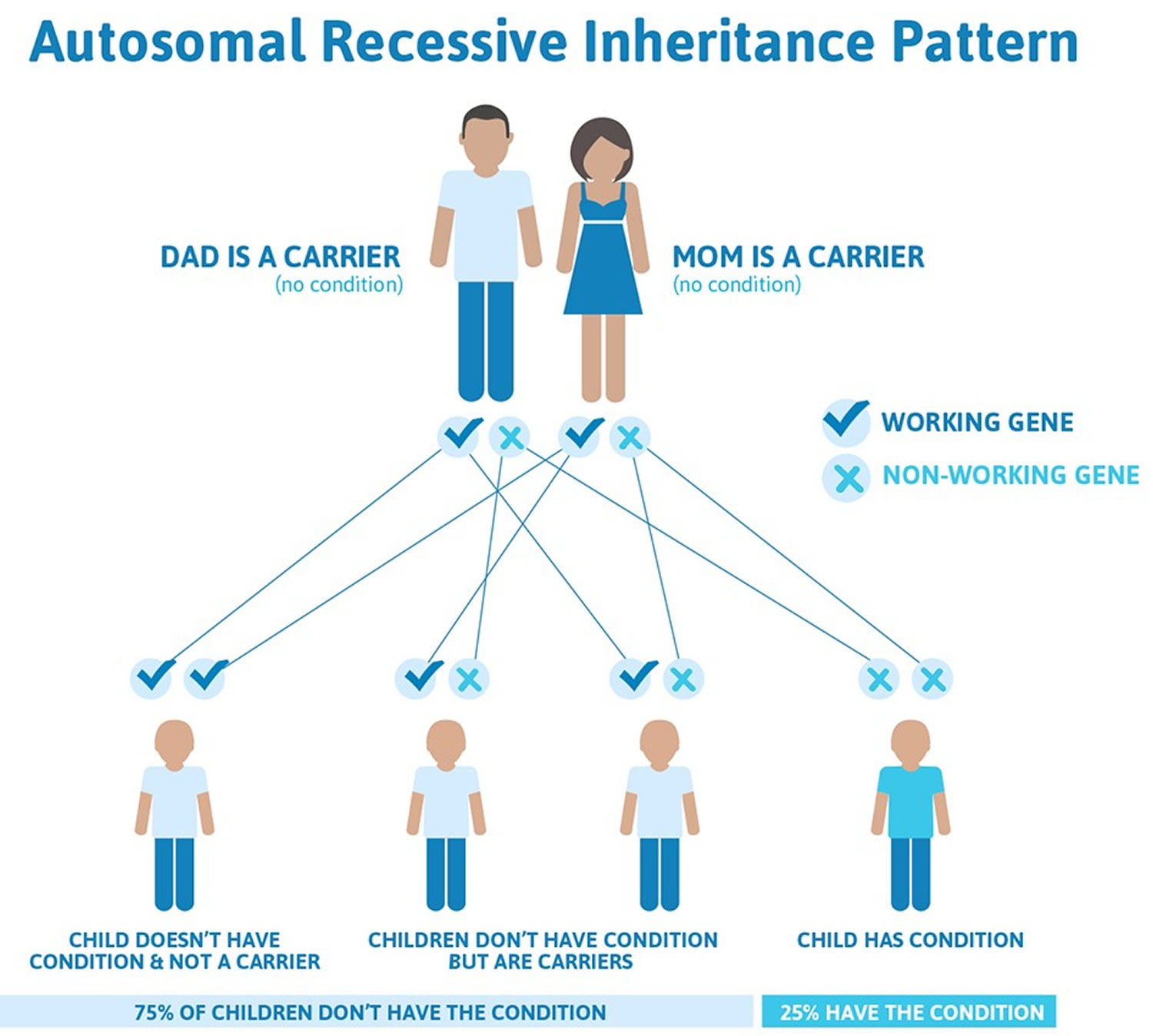
Niemann Pick disease is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Recessive genetic disorders occur when an individual inherits an abnormal (mutated) gene for the same trait from each parent. If an individual receives one normal gene and one mutated gene, the person will be a carrier for the disease, but usually will not show symptoms. For a couple who are both carriers the risk with each pregnancy for them to have an affected child is 25%, a child who is a carrier is 50%, and a child who is unaffected and is not a carrier is 25%. In recessive genetic disorders such as Niemann Pick disease the risk is the same for male and female offspring.
Figure 1. Niemann Pick disease autosomal recessive inheritance pattern
Niemann Pick disease type A
Niemann-Pick Disease Type A and Niemann-Pick Disease Type B were once thought to be separate diseases, but are now understood to be opposite ends of a spectrum of the same disease. They are both caused by a deficiency of the enzyme acid sphingomyelinase and therefore are known as Acid Sphingomyelinase Deficiency Niemann-Pick disease 2. Many variations exist within this spectrum, in terms of clinical symptoms and rate of progression.
Niemann Pick disease type A is a rare inherited lysosomal storage disorder in which harmful quantities of a fatty substance called sphingomyelin build up in the body’s cells and organs. In Niemann-Pick Disease Type B this build up mainly occurs in the liver, spleen and lungs. However, in Niemann Pick disease type A build up also occurs in the brain leading to aggressive neurological problems. A small number of patients may be described as having A/B variant, falling in the middle of the spectrum and exhibiting neurological problems which may become more apparent over time.
Niemann Pick disease is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Niemann Pick disease type A is inherited when two copies of a faulty gene (a mutation) are passed on to a child. In every pregnancy of a couple who each carry a copy of the faulty Niemann-Pick gene, there is a 1 in 4 chance (25%) that their child will have Niemann-Pick disease. This is known as autosomal recessive inheritance.
There are three common mutations that account for Niemann Pick disease type A in the Ashkenazi Jewish population, and the estimated incidence is ~1 in 40,000. The incidence in other populations is not known, but it is considered extremely rare and estimated to be about 1 in 10 million.
Sadly, in most cases of Niemann Pick disease type A life expectancy rarely exceeds three years of age.
Niemann-Pick disease type A causes
Niemann-Pick disease types A and B is caused by mutations in the SMPD1 gene. This gene provides instructions for producing an enzyme called acid sphingomyelinase. This enzyme is found in lysosomes, which are compartments within cells that break down and recycle different types of molecules. Acid sphingomyelinase is responsible for the conversion of a fat (lipid) called sphingomyelin into another type of lipid called ceramide. Mutations in SMPD1 lead to a shortage of acid sphingomyelinase, which results in reduced break down of sphingomyelin, causing this fat to accumulate in cells. This fat buildup causes cells to malfunction and eventually die. Over time, cell loss impairs function of tissues and organs including the brain, lungs, spleen, and liver in people with Niemann-Pick disease types A and B – causing cell death and the enlargement of the liver and spleen.
Niemann-Pick disease type A symptoms
Symptoms of Niemann-Pick Disease Type A develop within the first few months of life and may include a combination of:
- Feeding difficulties
- Prolonged jaundice
- Swelling of the abdomen from enlargement of the liver and spleen (usually occurs around 3-6 months of age)
- Progressive loss of early motor skills
- Failure to thrive
- A cherry red spot in the eye (not noticeable to parents)
- (Generally) a very rapid decline leading to death by two to five years of age
Infants with Niemann-Pick disease type A usually develop an enlarged liver and spleen (hepatosplenomegaly) by age 3 months and fail to gain weight and grow at the expected rate (failure to thrive). The affected children develop normally until around age 1 year when they experience a progressive loss of mental abilities and movement (psychomotor regression). Children with Niemann-Pick disease type A also develop widespread lung damage (interstitial lung disease) that can cause recurrent lung infections and eventually lead to respiratory failure. All affected children have an eye abnormality called a cherry-red spot, which can be identified with an eye examination. Children with Niemann-Pick disease type A generally do not survive past early childhood.
Niemann-Pick disease type A diagnosis
Niemann-Pick Disease Type A is diagnosed by measuring the level of the enzyme acid sphingomyelinase, in the white blood cells. This can be done by testing a small blood sample. The diagnosis is usually confirmed by DNA sequencing to identify mutations.
The enzyme deficiency at the cause of Niemann-Pick Disease Type A arises from mutations (or gene faults) in a gene on chromosome 11. An affected person will have inherited two faulty genes, one on each chromosome 11.
Niemann-Pick disease type A treatment
There are no no effective treatment or cure, for Niemann-Pick Disease Type A 3. However, patients will benefit from palliative treatments (individual medications that will help to treat the symptoms related to the condition).
Niemann Pick disease type B
Niemann-Pick Disease Type B and Niemann-Pick Disease Type A were once thought to be separate diseases, but are now understood to be opposite ends of a spectrum of the same disease. They are both caused by a deficiency of the enzyme acid sphingomyelinase and therefore are known as Acid Sphingomyelinase Deficiency Niemann-Pick disease. Many variations exist within this spectrum, in terms of clinical symptoms and rate of progression.
Niemann Pick disease type B is a very rare inherited lysosomal storage disorder in which harmful quantities of a fatty substance called sphingomyelin build up in the body’s cells and organs. In Niemann Pick disease type B this build up mainly occurs in the liver, spleen and lungs. Unlike Niemann-Pick Disease Type A, Niemann Pick disease type B does not affect the brain and, although growth may be slow, those affected will survive into adulthood with many being able to lead a full and active life. A small number of patients may be described as having A/B variant, falling in the middle of the spectrum and exhibiting neurological problems which may become more apparent over time.
It is inherited when two copies of a faulty gene (a mutation) are passed on to a child. In every pregnancy of a couple who each carry a copy of the faulty Niemann-Pick gene, there is a 1 in 4 chance (25%) that their child will have Niemann-Pick disease. This is known as autosomal recessive inheritance.
The incidence of Niemann Pick disease type B is estimated as 1 in 250,000 in the general population.
Niemann-Pick disease type B causes
Niemann-Pick Disease Type B is caused by a severe deficiency of the enzyme acid sphingomyelinase. This is required to break down a fatty substance called sphingomyelin. Failure to break down sphingomyelin causes an accumulation and enlargement of the liver and spleen.
Niemann-Pick disease type B symptoms
The first symptoms of Niemann-Pick Disease Type B are usually an enlarged liver and/or spleen in early childhood.
Symptoms can include:
- A progressive enlargement of organs
- Poor growth
- Susceptibility to respiratory infections
- Bleeding problems
- Bone pain
- Increased stress on the heart
There is usually no neurological involvement in Niemann Pick disease type B. Most patients will survive into adulthood, but not without experiencing health problems.
Niemann-Pick disease type B usually presents in mid-childhood. The signs and symptoms of this type are similar to type A, but not as severe. People with Niemann-Pick disease type B often have hepatosplenomegaly, recurrent lung infections, and a low number of platelets in the blood (thrombocytopenia). They also have short stature and slowed mineralization of bone (delayed bone age). About one-third of affected individuals have the cherry-red spot eye abnormality or neurological impairment. People with Niemann-Pick disease type B usually survive into adulthood.
Niemann-Pick disease type B diagnosis
Niemann-Pick Disease Type B is diagnosed by measuring the level of the enzyme acid sphingomyelinase in the white blood cells. This can be done by testing a small blood sample. The diagnosis is usually confirmed by DNA sequencing to identify mutations.
The enzyme deficiency at the cause of Niemann Pick disease type B arises from mutations in a gene on chromosome 11.
Niemann-Pick disease type B treatment
There is currently no specific approved treatment, or cure, for Niemann-Pick Disease Type B. However, patients will benefit from palliative treatments (individual medications that will help to treat the symptoms related to the condition). These improve the quality of life of patients who are experiencing symptoms.
There are clinical trials currently taking place investigating new therapeutic options for patients with Niemann Pick disease type B.
Niemann Pick disease type C
Niemann-Pick Type C is a rare inherited neurodegenerative disease that affects infants, children and adults. Niemann-Pick Type C is very different than Niemann Pick Type A or Niemann Pick B (Acid Sphingomyelinase Deficiency Disease). Niemann-Pick Type C patients are not able to metabolize cholesterol and other lipids properly within the cell. Consequently, excessive amounts of cholesterol accumulate within the liver and spleen and excessive amounts of other lipids accumulate in the brain. Niemann-Pick Type C causes a secondary reduction of acid sphingomyelinase activity, which led all three types to be considered forms of the same disease.
Niemann Pick disease type C belongs to a larger group of more than 50 disorders known as lysosomal storage disorders. Lysosomes are membrane-bound compartments within cells. They contain enzymes that break down large molecules such as proteins, carbohydrates and fats into their building blocks. Abnormal functioning of a transport protein leads to the accumulation of cholesterol and other fatty substances in various tissues of the body, including brain tissue.
Niemann Pick disease type C affects the processing of cholesterols produced within neurons, and not those outside of the cells. Thus, diets low in fats and cholesterols do not affect the neurological disease course. Furthermore, cholesterol-lowering drugs have not been shown to be effective at altering the course of the disease 4.
The age of onset and rate of disease progression can vary greatly from person to person; for example some children develop neurological symptoms early in childhood, whereas others may remain symptom free for a number of years.
Niemann Pick disease type C is always fatal. The majority of children with Niemann Pick disease type C die before age 20 (many die before the age of 10). Late onset of symptoms can lead to longer life spans but it is extremely rare for any person with Niemann Pick disease type C to reach age 40.
Niemann-Pick Type C is inherited when two copies of a faulty gene (a mutation) are passed on to a child. In every pregnancy of a couple who each carry a copy of the faulty Niemann-Pick gene, there is a 1 in 4 chance (25%) that their child will have Niemann-Pick disease. This is known as autosomal recessive inheritance.
Niemann Pick disease type C is divided into two subtypes, Niemann Pick disease type C1 and Niemann Pick disease type C2, as each is caused by a different gene mutation. Approximately 95 per cent of Niemann Pick disease type C cases are caused by genetic mutations in the Niemann Pick disease type C1 gene, with the other five per cent caused by mutations in the Niemann Pick disease type C2 gene.
The incidence of Niemann Pick disease type C is widely reported at 1 in 120,000, although recent evidence suggests this may be an under-estimate.
Many physicians have little experience with Niemann Pick disease type C. Thus, affected individuals and families often face a significant delay in diagnosis. Clinical experts on Niemann Pick disease type C have developed a Suspicion Index Tool to help physicians unfamiliar with the disorder to diagnose Niemann Pick disease type C 5. This tool creates a risk prediction score based on the specific manifestations present in an individual, broken down into visceral, neurological, and psychiatric categories. The original tool was effective in diagnosing individuals over the age of 4 years. Subsequently the same group derived a version that improved on diagnosing Niemann Pick disease type C in children younger than 4 years 6. Further study and refinement of the Suspicion Index Tool is necessary to determine its usefulness in clinical practice.
Niemann-Pick disease type C causes
Mutations in either the NPC1 or NPC2 gene cause Niemann-Pick disease type C 7. Approximately 95% of affected individuals have mutations in NPC1. The proteins produced from these genes are involved in the movement of lipids within cells. Mutations in these genes lead to a shortage of functional protein, which prevents movement of cholesterol and other fatty substances, leading to their accumulation in cells. Because these lipids are not in their proper location in cells, many normal cell functions that require lipids (such as cell membrane formation) are impaired. The accumulation of lipids as well as the cell dysfunction eventually leads to cell death, causing the tissue and organ damage seen in Niemann-Pick disease types C1 and C2.
Investigators have determined that the NPC1 gene is located on the long arm (q) of chromosome 18 (18q11.2). The NPC2 gene is located on the long arm of chromosome 14 (14q24.3). Chromosomes, present in the nucleus of human cells, carry the genes that contain genetic information for each individual. Each chromosome has a short arm designated “p” and a long arm designated “q.”
The exact function of the Niemann Pick disease type C1 and Niemann Pick disease type C2 proteins is not fully understood. Researchers do know that the protein products of these genes are involved in the movements (trafficking) of large molecules within cells. When Niemann Pick disease type C1 or Niemann Pick disease type C2 gene is mutated insufficient levels of functional protein products are made. This causes abnormal accumulation of cholesterol in the peripheral tissues of the body such as the liver and spleen, and accumulation of cholesterol and glycosphingolipids (complex compounds consisting of fatty material and carbohydrates) in the brain. The accumulation of these materials causes the various observable symptoms of Niemann Pick disease type C.
Niemann-Pick disease type C symptoms
Symptoms of Niemann-Pick Disease Type C vary with age of onset and from patient to patient.They may include:
- Jaundice at (or shortly after) birth
- An enlarged spleen and/or liver (hepatosplenamegaly)
- Difficulty with upward and downward eye movements (Vertical Supranuclear Gaze Palsy)
- Unsteadiness of gait, clumsiness, problems in walking (ataxia)
- Difficulty in posturing of limbs (dystonia)
- Slurred, irregular speech (dysarthria)
- Learning difficulties and progressive intellectual decline (cognitive dysfunction/dementia)
- Sudden loss of muscle tone which may lead to falls (cataplexy)
- Tremors accompanying movement and, in some cases, seizures
- Swallowing problems (dysphagia)
Individuals with Niemann Pick disease type C can have onset of symptoms at different ages that have been grouped historically as: perinatal (shortly before and after birth), early infantile (3 months to < 2 years), late infantile (2 to < 6 years), juvenile (6 to < 15 years), and adult (15 years and greater). Niemann Pick disease type C affects neurologic and psychiatric functions, as well as various internal organs (visceral). Symptoms arise at different times and follow independent progression. Visceral symptoms are more typically seen in individuals presenting at a younger age. Neurologic and psychiatric symptoms often occur slowly over time, and thus feature more prominently in individuals presenting in the later age groups.
Because Niemann Pick disease type C is a highly variable disorder, it is important to note that affected individuals will not have all of the symptoms described below and that every individual case is unique. Some children will develop severe, life-threatening complications early in life; others have a mild disease that may go undiagnosed well into adulthood. Parents should talk to their child’s physician and medical team about the specific symptoms and overall prognosis.
In perinatal Niemann Pick disease type C, the accumulation of fluid in the fetal abdomen (fetal ascites) may be present and persist after birth. These infants often have prolonged severe interruption or suppression of the flow of bile from the liver (cholestasis). Features of cholestasis include yellowing of the skin, mucous membranes and whites of the eyes (jaundice), failure to thrive, and growth deficiency. Enlargement of the liver (hepatomegaly) or spleen (splenomegaly) is present in a high percentage of affected individuals in this age group. Lipid-containing (foam) cells may accumulate in the lungs, resulting in lung disease. Liver and lung disease can progress to cause life-threatening complications during this period. Surviving individuals will develop neurological symptoms at a later age.
In the early infantile period, affected individuals may present with abnormal enlargement of the liver or spleen as the only noticeable symptom (isolated hepato-/splenomegaly), and that may remain the only symptom for many years. In other cases, additional symptoms develop including lack of muscle tone (hypotonia) often by 1 or 2 years of age. Affected individuals may also experience delays in the acquisition of skills requiring the coordination of mental and physical activities (delayed psychomotor development).
A characteristic early finding in children with Niemann Pick disease type C is impairment of the ability to look upward and downward (vertical supranuclear gaze palsy). Specifically, affected children lose their ability to rapidly move their eyes up and down. To compensate, they may blink their eyes, jerk their heads, or make abnormal movements. Eventually, vertical eye movements are lost, and side to side (horizontal) eye movements are also affected.
Hearing loss can occur in some individuals with Niemann Pick disease type C. Affected individuals may develop high frequency sensorineural hearing loss, in which transmission of sensory inputs from the auditory nerves to the brain is impaired. Up to 74% of individuals develop clinically significant hearing loss in at least one ear. Hearing loss may be the first problem seen in adults.
The classic presentation of Niemann Pick disease type C occurs during middle to late childhood with clumsiness or difficulty in drawing and writing, often noted by teachers and parents. Vertical supranuclear gaze palsy may be first reported during this time from a careful neurological exam or observations by the parents. Other neurological abnormalities may be the first apparent symptoms, specifically lack of muscle coordination (cerebellar ataxia). Children with cerebellar ataxia often have difficulties with balance and trouble with walking (unsteady gait). They may fall frequently and be considered clumsy. Affected children may also experience progressive difficulty speaking (dysarthria), resulting in slurred and eventually unintelligible speech. Children may lose previously acquired speech skills. Difficulty swallowing (dysphagia) may also develop and can become progressively worse, so that modifications such as thickening fluids or using special utensils may be recommended. Eventually a feeding tube may be required to maintain adequate nutrition. The dysphagia can lead to trouble swallowing saliva and other secretions. This may result in the inhalation of foreign materials into the airways and lungs (aspiration pneumonia).
During this time, affected individuals may also develop slowly progressive impairment of intellectually ability (cognitive impairment) that can initially be mistaken for learning disabilities. Furthermore, psychiatric disturbances and the progressive loss of memory and intellectual ability (dementia) can develop.
Additional neurologic findings can include drooling, epileptic seizures, and cataplexy. Cataplexy is characterized by a sudden loss of muscle tone and strength that can cause a sudden head drop, a weak, rubbery sensation in the legs, or in severe cases collapse. Cataplexy is often caused by strong emotions, typically laughter, in individuals with Niemann Pick disease type C (gelastic cataplexy). Dystonia, a large group of movement disorders, is also common. Dystonia is generally characterized by involuntary muscle contractions that force the body into abnormal, sometimes painful, movements and positions (postures). Some individuals may develop a tremor marked by rhythmic, jerking movements (myoclonic tremor). Sleep disturbances or irregularities such as narcolepsy or sleep apnea have also been reported.
Adolescent or adult onset of Niemann Pick disease type C may be associated with a similar neurological presentation as in childhood onset. However, the rate of progression is often much slower. Specific manifestations may vary, but can include cerebellar ataxia, dysarthria, dysphagia, cognitive impairment, and other movement disorders such as dystonia or tremor. Vertical supranuclear gaze palsy is invariably present, but can be difficult to appreciate initially. Although systemic symptoms are more common in infancy or childhood, they can also occur in individuals with adolescent or adult onset Niemann Pick disease type C. Isolated splenomegaly may be the presenting symptom in some adolescents or adults.
Psychiatric issues that have been described in individuals with adolescent onset of Niemann Pick disease type C include learning difficulties, behavioral problems, difficulty with expressive language, and attention deficit-hyperactivity disorder. Psychotic or manic episodes may occur in some affected individuals. Adults greater than 30 years of age may experience impairment of executive functions (dysexecutive syndrome) characterized by problems with complex thinking and reasoning tasks such as difficulty with organization and planning.
In some cases, older adults may first be misdiagnosed with dementia or psychiatric illness such as major depression or schizophrenia. Individuals have been described in the medical literature with other psychiatric manifestations such as obsessive-compulsive disorder, bipolar disorders, and hallucinations.
Following a long term gradual neurological decline death often results from aspiration pneumonia and subsequent respiratory failure, or intractable epilepsy not responding to medical intervention.
Niemann-Pick disease type C diagnosis
Niemann-Pick Disease Type C is difficult to diagnosis as the symptoms are non-specific to the disease and will vary from person to person. Niemann Pick disease type C is diagnosed by taking a small piece of skin (a skin biopsy) to see whether there is accumulation of fatty substances within the cells.
Diagnosis can also be made via a DNA analysis if the mutations in the affected child are known. This can be done fairly simply on a blood test but is only carried out in a few specialist centers.
Niemann-Pick disease type C treatment
There is no cure for Niemann-Pick Disease Type C, although patients benefit from palliative treatments (individual medications that will help to treat the symptoms related to the condition). Occupational therapy can be used to help with posture, speech and movement.
Treatment of Niemann Pick disease type C may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, ophthalmologists, pulmonologists, gastroenterologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Psychosocial support for the entire family is essential as well. Genetic counseling would benefit affected individuals and their families.
Current treatment is directed toward the specific symptoms apparent in each individual. Difficulty swallowing (dysphagia) should be monitored and evaluated regularly for the risk of aspiration. Swallowing difficulties may first be managed by softening solids and thickening liquids. A speech therapist can work with the individual to optimize swallowing function. Eventually, a gastronomy tube may be required to meet adequate caloric needs. With this procedure, a thin tube is placed into the stomach via a small incision in the abdomen, allowing for the direct intake of food or medicine.
Seizures often respond, at least partially, to anti-seizure medications (antiepileptics). Eventually, in an advanced stage of the disease, seizures may no longer respond to such medications (refractory seizures). Cataplexy may be treated by specific drugs including tricyclic antidepressants and central nervous system stimulants such as clomipramine, protriptyline or modafinil. Drugs that block the neurotransmitter acetylcholine (anticholinergic agents) have been effective in treating dystonia and tremor. Botulinum toxin injections can be used to treat severe dystonia. Drugs have also been used to treat various psychiatric illnesses, such as antipsychotic medications to treat psychosis and antidepressants to treat mood disorders.
Sleep abnormalities observed in Niemann Pick disease type C are diverse. Many individuals suffer from poor sleep quality due to fragmented myoclonus during slow wave sleep. Total sleep time, and time spent in different stages of sleep (REM and slow wave) may be decreased. Some individuals may suffer from insomnia which can be linked to underlying psychiatric diseases, such as anxiety or depression. When hypotonia is severe, especially in combination with enlarged adenoids and tonsils, disordered breathing with long respiratory pauses during sleep (obstructive sleep apnea) may occur. This diagnosis often requires an overnight sleep study. If the obstructive sleep apnea is severe, the patients may need a machine supplying continuous positive air pressure (CPAP) with a mask to keep the airways open during sleep. Insomnia and other sleep problems should be treated with melatonin, and if needed nocturnal sedatives.
Various services that may be beneficial to affected patients include an individualized educational plan encompassing physical therapy, speech therapy and occupational therapy. Parents, siblings, and other family members of affected individuals may find support, resources for respite care, and information on Niemann Pick disease type C through their primary pediatrician and the various professional and parent organizations.
In 2009, the European Medicines Agency approved the use of Zavesca© for the treatment of progressive neurological manifestations in adult and pediatric patients with Niemann Pick disease type C. Zavesca© has been shown to delay the progression and stabilize certain symptoms of the disease. However, this drug is not suitable for every affected individual and you are advised to discuss all medical issues with your doctor.
Studies have shown that miglustat (Zavesca®) may be able to slow the progression of neurological symptoms associated with Niemann Pick disease type C. Miglustat blocks the synthesis of glycosphingolipids, one of the substances that accumulates in the brain of individuals with Niemann Pick disease type C. The U.S. Food and Drug Administration (FDA) has not approved miglustat (Zavesca®) for the treatment of individuals with Niemann Pick disease type C, although the drug is approved for the treatment of another lysosomal storage disease known as Gaucher disease. Miglustat has been used off-label in the U.S. to treat individuals with Niemann Pick disease type C. Miglustat is available for the treatment of Niemann Pick disease type C in Australia, Canada, New Zealand, and several countries in Asia, Europe, and South America as Zavesca®, and in Japan as Brazaves®.
Currently there are limited data on patients with Niemann Pick disease type C with advanced neurological disease being commenced on miglustat. Based on the French experience of miglustat treatment in 20 children the Niemann Pick disease type C disability scores improved or stabilized in 75% of the patients with late-infantile onset disease (onset of symptoms < 5 years of age) but no patients with the early infantile onset form (onset of symptoms < 1 year of age) had a good neurological outcome 8. Only one patient out of 9 children treated before 4 years of age demonstrated stabilisation. More data are needed to determine of the efficacy of miglustat in patients below the age of 4 years.
Miglustat therapy is NOT appropriate for patients who have profound neurological disease, which, in the opinion of the attending physician, would make it difficult to assess for any improvements with therapy. Such symptoms may include but are not limited to 9:
- Profound dementia resulting in the need for 24 h care
- Inability to ambulate without a wheelchair
- Complete lack of verbal communication
- Swallowing difficulties profound enough to require tube feeding through a per-cutaneous gastrostomy.
Investigational Therapies
Several therapies have been studied or are currently being studied to assess their long-term safety and effectiveness as potential treatments for individuals with Niemann Pick disease type C. These therapies include vorinostat, 2-hydroxypropyl-β-cyclodextrin and oral arimoclomol. Clinical trials testing the safety and efficacy of intrathecal or intravenous preparations of 2-hydroxypropyl-β-cyclodextrin 10, specifically VTS270, and arimoclomol are currently in phase 2/3 clinical trials being studied for their effectiveness in treating Niemann Pick disease type C.
Niemann Pick disease life expectancy
- Most cases of Niemann Pick disease type A life expectancy rarely exceeds three years of age.
- Niemann-Pick disease type B usually presents in mid-childhood. People with Niemann-Pick disease type B usually survive into adulthood, but not without experiencing health problems.
- Niemann Pick disease type C is always fatal. The majority of children with Niemann Pick disease type C die before age 20 (many die before the age of 10). Late onset of symptoms can lead to longer life spans but it is extremely rare for any person with Niemann Pick disease type C to reach age 40.
Niemann Pick disease symptoms
Symptoms of Niemann-Pick Disease Type A develop within the first few months of life and may include a combination of:
- Feeding difficulties
- Prolonged jaundice
- Swelling of the abdomen from enlargement of the liver and spleen (usually occurs around 3-6 months of age)
- Progressive loss of early motor skills
- Failure to thrive
- A cherry red spot in the eye (not noticeable to parents)
- (Generally) a very rapid decline leading to death by two to five years of age
The first symptoms of Niemann-Pick Disease Type B are usually an enlarged liver and/or spleen in early childhood.
Symptoms can include:
- A progressive enlargement of organs
- Poor growth
- Susceptibility to respiratory infections
- Bleeding problems
- Bone pain
- Increased stress on the heart
There is usually no neurological involvement in Niemann Pick disease type B. Most patients will survive into adulthood, but not without experiencing health problems.
Symptoms of Niemann-Pick Disease Type C vary with age of onset and from patient to patient.They may include:
- Jaundice at (or shortly after) birth
- An enlarged spleen and/or liver (hepatosplenamegaly)
- Difficulty with upward and downward eye movements (Vertical Supranuclear Gaze Palsy)
- Unsteadiness of gait, clumsiness, problems in walking (ataxia)
- Difficulty in posturing of limbs (dystonia)
- Slurred, irregular speech (dysarthria)
- Learning difficulties and progressive intellectual decline (cognitive dysfunction/dementia)
- Sudden loss of muscle tone which may lead to falls (cataplexy)
- Tremors accompanying movement and, in some cases, seizures
- Swallowing problems (dysphagia)
The signs and symptoms of Niemann-Pick disease types C1 and C2 are very similar; these types differ only in their genetic cause. Niemann-Pick disease types C1 and C2 usually become apparent in childhood, although signs and symptoms can develop at any time. People with these types usually develop difficulty coordinating movements (ataxia), an inability to move the eyes vertically (vertical supranuclear gaze palsy), poor muscle tone (dystonia), severe liver disease, and interstitial lung disease. Individuals with Niemann-Pick disease types C1 and C2 have problems with speech and swallowing that worsen over time, eventually interfering with feeding. Affected individuals often experience progressive decline in intellectual function and about one-third have seizures. People with these types may survive into adulthood.
Niemann Pick disease diagnosis
Diagnosis of Niemann-Pick disease begins with a thorough physical exam, which can show an early warning sign such as an enlarged liver or spleen. Your doctor will also take a detailed medical history and discuss symptoms and family health history. Niemann-Pick disease is rare, and its symptoms can be confused with other diseases. Diagnostic techniques depend on the type of Niemann-Pick disease.
- Niemann-Pick Type A or B. Using a blood or skin sample (biopsy), experts measure how much sphingomyelinase is in white blood cells to confirm the diagnosis.
- Niemann-Pick Type C. Experts take a small sample of skin to test for Niemann-Pick to assess how the cells move and store cholesterol.
Other tests also may be done, such as:
- Magnetic resonance imaging (MRI). An MRI of the brain may show loss of brain cells. But in the early stages of Niemann-Pick, an MRI may be normal because symptoms typically occur before the loss of brain cells.
- Eye exam. An eye exam can show signs that may be an indication of Niemann-Pick disease, such as eye movement difficulties.
- Genetic testing. DNA testing of a blood sample may show the specific abnormal genes that cause Niemann-Pick types A, B and C. DNA tests can show who the carriers are for all types of Niemann-Pick disease if the mutations have been described in the first person identified in a family (the index case).
- Prenatal testing. Ultrasound can detect the enlarged liver and spleen that’s caused by type C. And amniocentesis or chorionic villus sampling may be used to confirm a diagnosis of Niemann-Pick.
Niemann Pick disease treatment
No cure exists for Niemann-Pick disease. No effective treatment is available to people with type A or B. For people with mild to moderate type C, a drug called miglustat (Zavesca) may be an option. An international study of 92 people with type C Niemann-Pick showed improved neurological symptoms after taking miglustat regularly for an average of two years.
Physical therapy is an important part of treatment to help maintain mobility as long as possible. People with Niemann-Pick disease need to see their doctors regularly, because the disease progresses and symptoms worsen.
- The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. Schuchman EH. J Inherit Metab Dis. 2007 Oct; 30(5):654-63. https://www.ncbi.nlm.nih.gov/pubmed/17632693/[↩]
- Niemann-Pick Disease Type A. http://www.npuk.org/niemann-pick-disease/asmd-npd/[↩]
- Long Y, Xu M, Li R, et al. Induced Pluripotent Stem Cells for Disease Modeling and Evaluation of Therapeutics for Niemann-Pick Disease Type A. Stem Cells Translational Medicine. 2016;5(12):1644-1655. doi:10.5966/sctm.2015-0373. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5189647/[↩]
- The effect of cholesterol-lowering agents on hepatic and plasma cholesterol in Niemann-Pick disease type C. Patterson MC, Di Bisceglie AM, Higgins JJ, Abel RB, Schiffmann R, Parker CC, Argoff CE, Grewal RP, Yu K, Pentchev PG. Neurology. 1993 Jan; 43(1):61-4. https://www.ncbi.nlm.nih.gov/pubmed/8423912/[↩]
- Wraith JE, Sedel F, Pineda M, Wijburg FA, Hendriksz CJ, Fahey M, Walterfang M, Patterson MC, Chadha-Boreham H, Kolb SA: Niemann-Pick type C Suspicion Index tool: analyses by age and association of manifestations. Journal of inherited metabolic disease 2014;37(1):93-101.[↩]
- Pineda M, Mengel E, Jahnova H, Heron B, Imrie J, Lourenco CM, van der Linden V, Karimzadeh P, Valayannopoulos V, Jesina P et al: A Suspicion Index to aid screening of early-onset Niemann-Pick disease Type C (NP-C). BMC pediatrics 2016;16:107.[↩]
- Vanier MT, Duthel S, Rodriguez-Lafrasse C, Pentchev P, Carstea ED. Genetic heterogeneity in Niemann-Pick C disease: a study using somatic cell hybridization and linkage analysis. Am J Hum Genet. 1996;58:118–125. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1914948/pdf/ajhg00014-0123.pdf[↩]
- Héron B, Valayannopoulos V, Baruteau J, et al. Miglustat therapy in the French cohort of paediatric patients with Niemann-Pick disease type C. Orphanet J Rare Dis. 2012;7:36. doi: 10.1186/1750-1172-7-36. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3465012/[↩]
- Geberhiwot T, Moro A, Dardis A, et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet Journal of Rare Diseases. 2018;13:50. doi:10.1186/s13023-018-0785-7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5889539/[↩]
- Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1-2 trial. Ory DS, Ottinger EA, Farhat NY, King KA, Jiang X, Weissfeld L, Berry-Kravis E, Davidson CD, Bianconi S, Keener LA, Rao R, Soldatos A, Sidhu R, Walters KA, Xu X, Thurm A, Solomon B, Pavan WJ, Machielse BN, Kao M, Silber SA, McKew JC, Brewer CC, Vite CH, Walkley SU, Austin CP, Porter FD. Lancet. 2017 Oct 14; 390(10104):1758-1768. http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(17)31465-4/fulltext[↩]





{kind=link}