
Contents
What is phenylketonuria
Phenylketonuria (commonly known as PKU) is an inherited disorder of deficiency of hepatic phenylalanine hydroxylase activity needed to convert the essential amino acid phenylalanine to tyrosine, resulting in increasing levels of phenylalanine in the blood. Phenylalanine is a building block of proteins (an amino acid) that is obtained through the diet. Phenylalanine is found in all proteins and in some artificial sweeteners. If phenylketonuria is not treated, phenylalanine can build up to harmful levels in the body, causing intellectual disability and other serious health problems. A dangerous buildup of phenylalanine can develop when a person with phenylketonuria eats protein-rich foods, such as milk, cheese, nuts or meat, and even grains such as bread and pasta, or eats aspartame, an artificial sweetener. This buildup of phenylalanine results in damage to nerve cells in the brain.
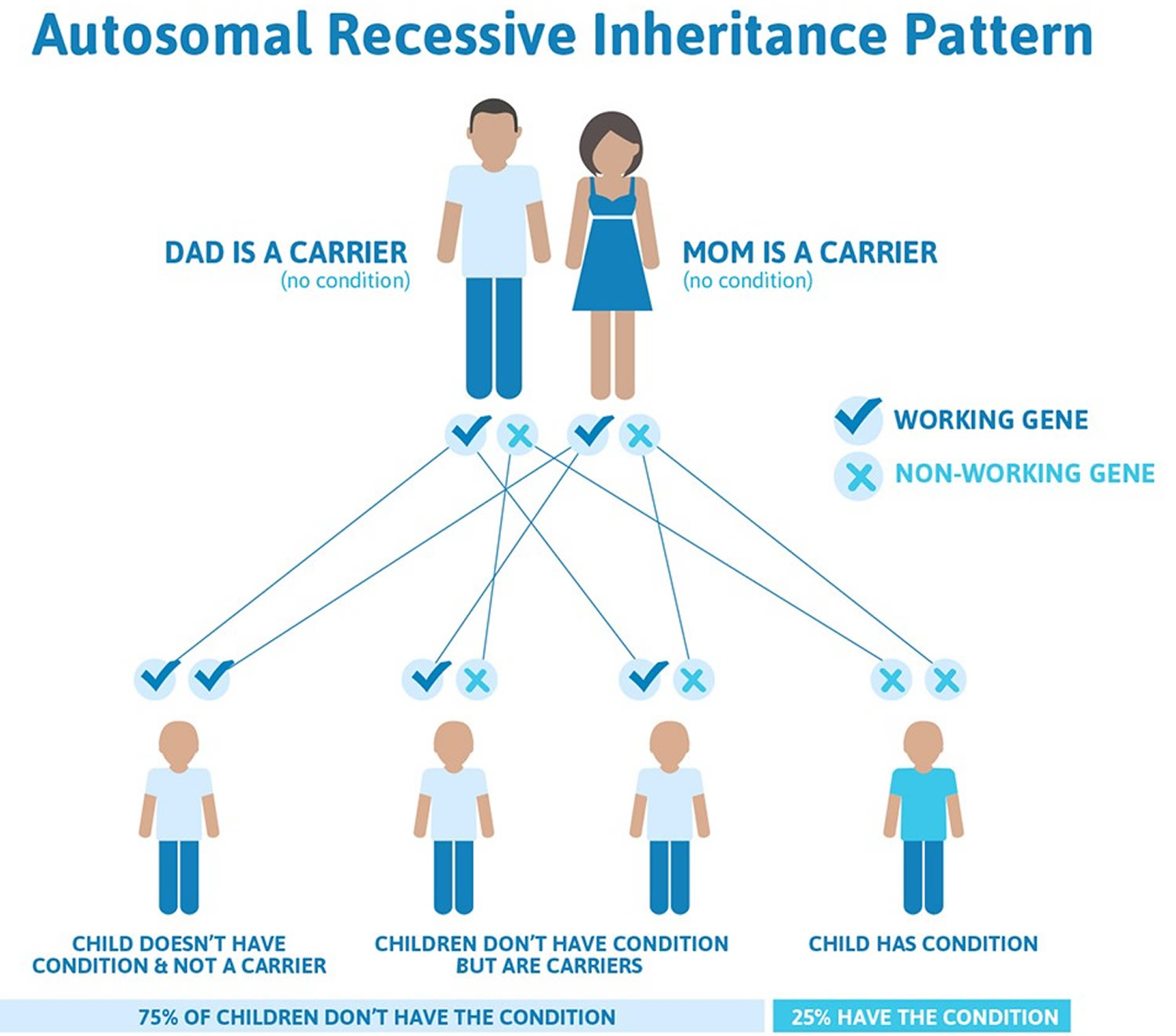
Phenylketonuria is inherited (autosomal recessive inheritance pattern). This means for a child to inherit phenylketonuria, both the mother and father must have and pass on the defective gene. A gene is a part of your body’s cells that stores instructions for the way your body grows and works. Genes come in pairs—you get one of each pair from each parent. Your baby has to inherit a gene change for phenylketonuria from both parents to have phenylketonuria (see Figure 1 below). If your baby inherits the gene from just one parent, your baby has the gene change for phenylketonuria (phenylketonuria carrier), but she/he doesn’t have phenylketonuria. When this happens, your baby is called a phenylketonuria carrier. A phenylketonuria carrier has the gene change but doesn’t have phenylketonuria. Most often, phenylketonuria is passed to children by two parents who are carriers of the disorder, but don’t know it.
The occurrence of phenylketonuria varies among ethnic groups and geographic regions worldwide. In the United States, phenylketonuria occurs in 1 in 10,000 to 15,000 newborns. Phenylketonuria happens in all ethnic groups. But it’s more common in people who are Native American and Northern European than those who are African-American, Ashkenazi Jewish or Japanese. Most cases of phenylketonuria are detected shortly after birth by newborn screening, and treatment is started promptly. As a result, the severe signs and symptoms of classic phenylketonuria are rarely seen.
The signs and symptoms of phenylketonuria (PKU) vary from mild to severe. The most severe form of this disorder is known as classic phenylketonuria. Infants with classic phenylketonuria appear normal until they are a few months old. Without treatment, these children develop permanent intellectual disability. Seizures, delayed development, behavioral problems, and psychiatric disorders are also common. Untreated individuals may have a musty or mouse-like odor as a side effect of excess phenylalanine in the body. Children with classic phenylketonuria tend to have lighter skin and hair than unaffected family members and are also likely to have skin disorders such as eczema.
Less severe forms of this condition, sometimes called variant phenylketonuria and non-phenylketonuria hyperphenylalaninemia, have a smaller risk of brain damage. People with very mild cases may not require treatment with a low-phenylalanine diet.
Babies born to mothers who have phenylketonuria and uncontrolled phenylalanine levels (women who no longer follow a low-phenylalanine diet) have a significant risk of intellectual disability because they are exposed to very high levels of phenylalanine before birth. These infants may also have a low birth weight and grow more slowly than other children. Other characteristic medical problems include heart defects or other heart problems, an abnormally small head size (microcephaly), and behavioral problems. Women with phenylketonuria and uncontrolled phenylalanine levels also have an increased risk of pregnancy loss.
Improvements in behavior and functioning have been documented in previously untreated individuals with phenylketonuria who have been placed on diet therapy 1. In children for whom therapy was delayed, improvement in IQ may occur after nutrition therapy is initiated 1. Choice of treatment modality should be based on feasibility and effectiveness for the individual patient. Among the concerns are comorbidities, contraindications due to prescribed medications, and limited access to care.
The main treatment for phenylketonuria includes:
- A lifetime diet with very limited intake of protein, because foods with protein contain phenylalanine
- Taking a phenylketonuria formula — a special nutritional supplement — for life to make sure you get enough essential protein (without phenylalanine) and nutrients that are crucial for growth and general health
A safe amount of phenylalanine differs for each person with phenylketonuria and can vary over time. In general, the idea is to consume only the amount of phenylalanine that’s necessary for normal growth and body processes, but no more. Your doctor can determine a safe amount through:
- Regular review of diet records, growth charts and blood levels of phenylalanine
- Frequent blood tests that monitor phenylalanine levels as they change over time, especially during childhood growth spurts and pregnancy
- Other tests that assess growth, development and health
Your doctor may refer you to a registered dietitian who can help you learn about the phenylketonuria diet, make adjustments to your diet when needed, and offer suggestions on ways to manage phenylketonuria diet challenges.
Foods to AVOID
Because the amount of phenylalanine that a person with PKU can safely eat is so low, it’s crucial to avoid all high-protein foods, such as:
- Milk
- Eggs
- Cheese
- Nuts
- Soybeans
- Beans
- Chicken
- Beef
- Pork
- Fish
Potatoes, grains and other vegetables that have protein will likely be limited.
Children and adults also need to avoid certain other foods and beverages, including many diet sodas and other drinks that contain aspartame (NutraSweet, Equal). Aspartame is an artificial sweetener made with phenylalanine.
Some medications may contain aspartame and some vitamins or other supplements may contain amino acids or skim milk powder. Check with your pharmacist about the contents of over-the-counter products or prescription medications.
Talk with your doctor or registered dietitian to learn more about your specific dietary needs.
Other names for phenylketonuria
- deficiency disease, phenylalanine hydroxylase
- Folling disease
- Folling’s disease
- PAH deficiency
- Phenylalanine Hydroxylase Deficiency
- phenylalanine hydroxylase deficiency
- phenylalanine hydroxylase deficiency disease
- PKU
Phenylketonuria test
All babies have a newborn screening test for phenylketonuria. Newborn screening checks for serious but rare conditions at birth. It includes blood, hearing and heart screening. With newborn screening, phenylketonuria can be found and treated early so babies can grow up healthy.
A phenylketonuria test is done a day or two after your baby’s birth. The test is done after your baby is 24 hours old and after your baby has ingested some protein in the diet to ensure accurate results.
- A nurse or lab technician collects a few drops of blood from your baby’s heel or the bend in your baby’s arm. The blood is collected on a special paper and sent to a lab for testing.
- A laboratory tests the blood sample for certain metabolic disorders, including phenylketonuria.
- If you don’t deliver your baby in a hospital or are discharged soon after the birth, you may need to schedule a newborn screening with your pediatrician or family doctor.
If this test indicates your baby may have phenylketonuria:
- Your baby may have additional tests to confirm the diagnosis, including more blood tests and urine tests
- You and your baby may have genetic testing to identify gene mutations
If newborn screening results aren’t normal, it simply means your baby needs more testing. Your baby’s provider can recommend another kind of test, called a diagnostic test. This test can check to see if your baby has phenylketonuria or if there is some other cause for abnormal test results.
If your baby is tested before he’s a full day old, it’s possible for the test to miss phenylketonuria. Some experts recommend that if your baby was tested within the first 24 hours of life, he should be tested again at 1 to 2 weeks of age.
Maternal phenylketonuria
Maternal phenylketonuria means that a woman who has phenylketonuria is pregnant. About 3,000 women of childbearing age in the United States have phenylketonuria. High maternal blood phenylalanine is associated with poor outcomes in offspring, including low birth weight, microcephaly, congenital heart defects, and intellectual disability. Maintaining maternal blood phenylalanine between 120 and 360 μmol/l before and during pregnancy results in the best outcome for offspring 2. However, 30% of clinics surveyed recommend 120–240 μmol/l as the target range for maternal blood phenylalanine 3.
When a pregnant woman’s phenylalanine levels get too high, they can cause serious problems in her baby, including 2:
- Intellectual disabilities
- Microcephaly. Microcephaly is a birth defect in which a baby’s head is smaller than expected, compared to babies of the same sex and age.
- Heart defects and other heart problems
- Low birthweight
- Delayed development
- Facial abnormalities
- Abnormally small head
- Behavioral problems
The good news is that most pregnant women who have phenylketonuria can have healthy babies if they follow their phenylketonuria meal plan. This is a special meal plan that is low in phenylalanine.
Phenylketonuria meal plans are different for each person and depend on how much phenylalanine your body can take. Health care providers at a medical center or clinic that has a special program to treat phenylketonuria can help you create a phenylketonuria meal plan. These providers can monitor your pregnancy and help you change your meal plan to help keep your blood phenylalanine at the right level. Ask your health care provider for information on a medical center or clinic that treats phenylketonuria.
Your health care provider may order ultrasound tests during your pregnancy to check on your baby’s growth.
Be sure to follow your phenylketonuria meal plan at least 3 months before getting pregnant and throughout pregnancy. If you just found out you’re pregnant, go back to your phenylketonuria meal plan right away. You can get weekly blood tests during pregnancy to make sure your phenylalanine levels aren’t too high.
Can I find out during pregnancy if my baby has phenylketonuria or is a phenylketonuria carrier?
Yes. If you or your partner has phenylketonuria or is a phenylketonuria carrier, you can have a prenatal test to find out if your baby has phenylketonuria or is a carrier. You can have either of these tests:
- Chorionic villus sampling (also called CVS). This test checks tissue from the placenta for birth defects and genetic conditions. You can get CVS at 10 to 13 weeks of pregnancy.
- Amniocentesis (also called amnio). This test checks amniotic fluid from the amniotic sac around your baby for birth defects and genetic conditions. You can get this test at 15 to 20 weeks of pregnancy.
Talk to your provider or genetic counselor if you’re thinking of having either of these tests.
How is maternal phenylketonuria treated in pregnancy?
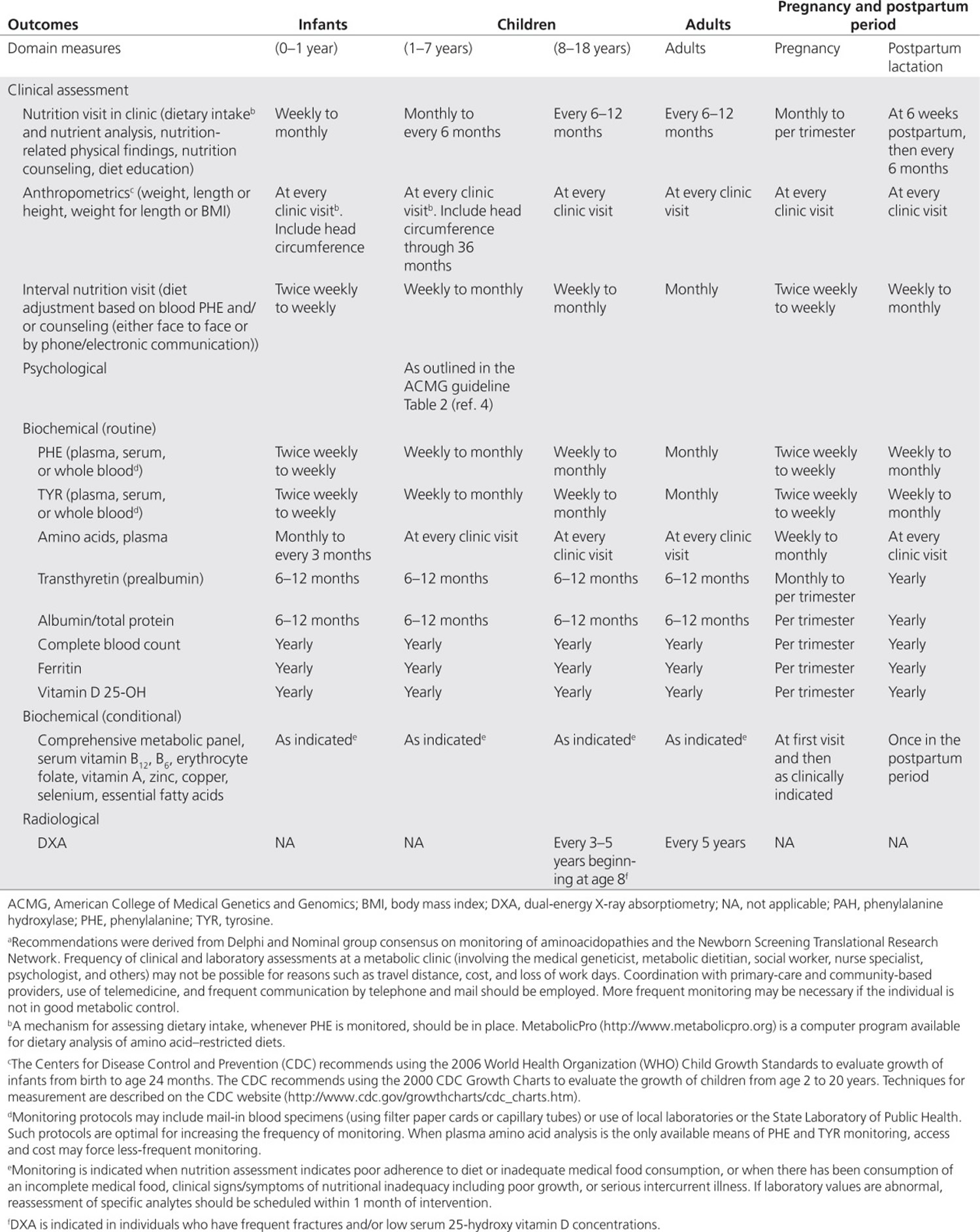
Most studies have not evaluated the effect of nutrition (beyond maternal blood phenylalanine) on outcomes. In the Maternal PKU Collaborative Study, maternal protein intake and energy were significantly and negatively correlated with blood phenylalanine throughout the entire pregnancy 4. Women with low protein intake secondary to inadequate consumption of medical food (see below under phenylketonuria treatment and phenylketonuria diet) also had low overall nutrient intake and a higher incidence of congenital anomalies in their offspring 5. Adequate energy and fat intake, as well as adequate weight gain, have all been associated with better pregnancy outcomes 4. Control of nausea and vomiting, especially in early pregnancy, is key to preventing catabolic weight loss and elevated phenylalanine levels. Neither tyrosine intake nor blood tyrosine has been reported to be associated with outcomes in maternal phenylketonuria 6. Fat and essential fatty acid intakes may be very low in individuals with phenylketonuria when their medical food contains no fat. DHA supplementation of 200–300 mg/day should be provided to all pregnant women with phenylketonuria 3. Specific monitoring recommendations during pregnancy for women with phenylketonuria are outlined in Table 2 (see Table 2 Monitoring the nutritional management of individuals with phenylalanine hydroxylase deficiency deficiency below). The use of adjunctive therapies in managing blood phenylalanine during pregnancy is a matter of debate. Large neutral amino acid monotherapy is not recommended, because it does not result in adequate control of blood phenylalanine. Because sapropterin can lower blood phenylalanine, its use should be considered on a case-by-case basis 3. Women are encouraged to continue the diet in the postpartum period and can breast-feed their non–PAH-deficient infant regardless of maternal blood phenylalanine levels.
Recommendations
- Maintain blood phenylalanine between 120 and 360 μmol/l before conception and throughout pregnancy.
- Monitor dietary intake of pregnant women with phenylketonuria to ensure nutrient adequacy.
- Consider sapropterin use on a case-by-case basis for pregnant women who have difficulty adhering to the diet.
- Do not recommend large neutral amino acids for use in pregnant women with phenylketonuria.
Phenylketonuria complications
Untreated phenylketonuria can lead to complications in infants, children and adults with the disorder. When mothers with phenylketonuria have high blood phenylalanine levels during pregnancy, fetal birth defects or miscarriage can occur.
Untreated phenylketonuria can lead to:
- Irreversible brain damage and marked intellectual disability beginning within the first few months of life
- Neurological problems such as seizures and tremors
- Behavioral, emotional and social problems in older children and adults
- Major health and developmental problems
Phenylketonuria causes
What causes phenylketonuria
Mutations in the PAH gene cause phenylketonuria. The PAH gene provides instructions for making an enzyme called phenylalanine hydroxylase. This enzyme converts the amino acid phenylalanine to other important compounds in the body. If gene mutations reduce the activity of phenylalanine hydroxylase, phenylalanine from the diet is not processed effectively. As a result, this amino acid can build up to toxic levels in the blood and other tissues. Because nerve cells in the brain are particularly sensitive to phenylalanine levels, excessive amounts of this substance can cause brain damage.
Classic phenylketonuria, the most severe form of the disorder, occurs when phenylalanine hydroxylase activity is severely reduced or absent. People with untreated classic phenylketonuria have levels of phenylalanine high enough to cause severe brain damage and other serious health problems. Mutations in the PAH gene that allow the enzyme to retain some activity result in milder versions of this condition, such as variant phenylketonuria or non-phenylketonuria hyperphenylalaninemia.
Changes in other genes may influence the severity of phenylketonuria, but little is known about these additional genetic factors.
Phenylketonuria (PKU) is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
If you and your partner are both phenylketonuria carriers, there’s a:
- 3-in-4 chance (75 percent) that your baby won’t have phenylketonuria
- 1-in-2 chance (50 percent) that your baby won’t have phenylketonuria but will be a phenylketonuria carrier
- 1-in-4 chance (25 percent) that your baby will have phenylketonuria
- 1-in-4 chance (25 percent) that your baby will not have phenylketonuria and not be a carrier
A genetic counselor can help you understand your chances of passing phenylketonuria to your baby. A genetic counselor is a person who is trained to help you understand about how genes, birth defects and other medical conditions run in families, and how they can affect your health and your baby’s health.
Figure 1. Phenylketonuria autosomal recessive inheritance pattern
How can you find out if you’re a phenylketonuria carrier?
You may not know if you’re a phenylketonuria carrier. If you’re a carrier, you don’t have any signs or symptoms of phenylketonuria and you do not have phenylketonuria.
There are two types of tests that can tell you if you’re a phenylketonuria carrier. Both are safe to take during pregnancy. Your partner can have the tests, too.
- A blood test
- A swab of cells inside your mouth. This means your provider rubs a cotton swab against the inside of your cheek to get some cells.
You and your partner may want to be tested if phenylketonuria runs in either of your families. To help you find out, take your family health history. This is a record of any health conditions and treatments that you, your partner and everyone in both of your families have had. Use this family health history form and share it with your health care provider.
Phenylketonuria prevention
If you have phenylketonuria and are considering getting pregnant:
- Follow a low-phenylalanine diet. Women with phenylketonuria can prevent birth defects by sticking to or returning to a low-phenylalanine diet before becoming pregnant. If you have phenylketonuria, talk to your doctor before you start trying to conceive.
- Consider genetic counseling. If you have phenylketonuria, a close relative with phenylketonuria or a child with phenylketonuria, you may also benefit from genetic counseling before becoming pregnant. A doctor who specializes in medical genetics (geneticist) can help you better understand how phenylketonuria is passed through your family tree. He or she can also help determine your risk of having a child with phenylketonuria and assist with family planning.
Phenylketonuria symptoms
Babies born with phenylketonuria seem normal for the first few months of life. But by age 3 to 6 months, they begin to lose interest in their surroundings. By age 1 year, children are developmentally delayed and their skin has less pigmentation than someone without the condition. If people with phenylketonuria do not restrict the phenylalanine in their diet, they develop severe intellectual and developmental disabilities.
Babies born with phenylketonuria begin to have signs and symptoms of the illness at about 6 months of age. These include:
- Jerky movements in arms and legs
- Lighter skin and eyes (Babies with phenylketonuria can’t properly make melanin, the pigment in the body that’s responsible for skin and hair color.)
- A musty odor in urine, breath, or skin that is a result of the extra phenylalanine in the body
- Seizures, shaking, or jerking movements in the arms and legs
- Skin rashes, like eczema
- Small head size, called microcephaly
- Taking longer than expected to sit, crawl or walk
- Losing interest in surroundings
- Delays in mental and social skills
- Intellectual disabilities
- Behavior problems, like being hyperactive.
- Social problems
- Stunted or slow growth
- Psychiatric disorders.
Phenylketonuria severity varies
The severity of phenylketonuria depends on the type.
- Classic phenylketonuria. The most severe form of the disorder is called classic phenylketonuria. The enzyme needed to convert phenylalanine is missing or severely reduced, resulting in high levels of phenylalanine and severe brain damage.
- Less severe forms of phenylketonuria. In mild or moderate forms, the enzyme retains some function, so phenylalanine levels are not as high, resulting in a smaller risk of significant brain damage.
But most children with the disorder still require a special phenylketonuria diet to prevent intellectual disability and other complications.
Phenylketonuria diagnosis
Newborn blood testing identifies almost all cases of phenylketonuria. All 50 states in the United States require newborns to be screened for phenylketonuria. Many other countries also routinely screen infants for phenylketonuria.
If you have phenylketonuria or a family history of it, your doctor may recommend screening tests before pregnancy or birth. It’s possible to identify phenylketonuria carriers through a blood test.
Phenylketonuria treatment
Nutrition therapy, first introduced 6 decades ago, remains the primary treatment for phenylketonuria 7. If your baby has phenylketonuria, he may need testing as often as once a week or more often for the first year of life to check his phenylalanine levels. After that, he may have testing once or twice a month throughout childhood.
Your baby needs to follow a special meal plan that is low in phenylalanine. It’s best to start this meal plan as soon as possible, ideally within the first 7 to 10 days of life.
At first, your baby gets a special protein formula that has reduced phenylalanine. Protein is important to help your baby grow and develop. The amount of phenylalanine in the formula is controlled to meet you baby’s individual needs. Your baby also can have some breast milk. Your breast milk has phenylalanine in it, so talk to your baby’s provider to find out how much breast milk your baby can have.
Physical findings associated with poorly controlled phenylketonuria include osteopenia 8 and dermatological problems 9. Asthma, recurrent headache, eczema, neurological signs, hyperactivity, and/or lethargy have all been reported in adults with phenylketonuria who discontinued dietary treatment 10. Psychological symptoms include phobias and depression 11. The majority of physical signs and symptoms of phenylketonuria reported in the literature resolved when blood phenylalanine concentrations were reduced to the treatment range.
Providing dietary phenylalanine
Because phenylalanine is an essential amino acid, limited amounts from intact protein must be provided for anabolic processes regardless of the medical food chosen. For infants, the source of phenylalanine can be either breast milk 12 or infant formula (containing DHA/arachidonic acid). A variety of strategies for introducing breast milk have been described 12. Although there is little consensus about the best way to incorporate breast milk into the diet of an infant with phenylketonuria, there is agreement that feeding at the breast, as well as feeding expressed breast milk by bottle, results in good metabolic control 13. Later in the first year, breast milk or infant formula is slowly removed in exchange for limited amounts of intact protein from solid foods containing an equivalent amount of phenylalanine.
Tracking dietary phenylalanine
There is little consensus about the best method for tracking dietary phenylalanine intake. As long as data are available on the phenylalanine content of individual food items, the most precise method of tracking phenylalanine intake is counting milligrams of phenylalanine. An exchange system (1 exchange = 15 mg phenylalanine) is easier for some patients and families. Those with higher phenylalanine tolerance may be able to achieve good phenylalanine control by counting grams of protein. This has the advantage of allowing the use of food labels as a guide. A more liberal approach of allowing “free” consumption of fruits, vegetables, and low-phenylalanine foods (<100 mg phenylalanine/100 g) allows for similar blood phenylalanine control to that of other tracking methods 14. This system has been used by clinics in Europe with good success but is not common practice in the United States 15.
Formula for people with phenylketonuria
Because of the restricted diet, people with phenylketonuria need to get essential nutrients through a special nutritional supplement. The phenylalanine-free formula provides protein and other essential nutrients in a form that’s safe for people with phenylketonuria.
Your doctor and dietitian can help you find the right type of formula.
- Formula for babies and toddlers. Because regular infant formula and breast milk contain phenylalanine, babies with phenylketonuria instead need to consume a phenylalanine-free infant formula. A dietitian can carefully calculate the amount of breast milk or regular formula to be added to the phenylalanine-free formula. Parents introduce solid foods with low levels of phenylalanine to children with phenylketonuria on the same schedule used for other infants.
- Formula for older children and adults. Older children and adults continue to drink or eat a protein substitute formula daily, as directed by a doctor or dietitian. Your daily dose of formula is divided between your meals and snacks, instead of consumed all at once. The formula for older children and adults is not the same as the one used for infants, but it works on the same principle. It provides essential protein (amino acids) without phenylalanine and is continued for life.
The need for a nutritional supplement, especially if your child doesn’t find it appealing, and the limited food choices can make the phenylketonuria diet challenging. But families need to make a firm commitment to this lifestyle change because it’s the only way to prevent the serious health problems that people with phenylketonuria can develop.
Neutral amino acid therapy
Another possible addition to the phenylketonuria diet is a supplement called neutral amino acid therapy in powder or tablet form. This supplement may block some absorption of phenylalanine. This may be a treatment option for adults with phenylketonuria. Ask your doctor or dietitian if this supplement is appropriate for your diet.
Phenylketonuria medication
The Food and Drug Administration (FDA) has approved the drug sapropterin (Kuvan) for the treatment of phenylketonuria. It works by increasing your tolerance to phenylalanine. The drug is for use in combination with a phenylketonuria diet. But it doesn’t work for everyone with phenylketonuria.
In approving sapropterin, the FDA directed that studies continue because there are no long-term studies on the drug’s efficacy and long-term safety.
Phenylketonuria diet
When your baby is ready to eat solid foods, she can eat vegetables, fruits, some grains (like low-protein cereals, breads and pasta) and other low-phenylalanine foods.
If your baby has phenylketonuria, she should NOT eat:
- Milk, cheese, ice cream and other dairy products
- Eggs
- Meat and poultry
- Fish
- Nuts
- Beans
- Food or drinks that contain aspartame. This is an artificial sweetener that has lots of phenylalanine in it. It’s sold as NutraSweet® and Equal®.
Phenylketonuria meal plans are different for each baby and can vary over time depending on how much phenylalanine your baby can take. Health care providers at a medical center or clinic that has a special program to treat phenylketonuria can help you create a phenylketonuria meal plan for your baby. Ask your baby’s health care provider for information on a medical center or clinic that treats phenylketonuria.
Your child follows the phenylketonuria meal plan through her whole life. If she eventually gets pregnant, she follows her meal plan throughout pregnancy. Most pregnant women who have phenylketonuria can have healthy pregnancies and healthy babies.
The medicine Kuvan® (sapropterin dihydrochloride) can help some people with phenylketonuria. The medicine is more likely to work in people with mild or special forms of phenylketonuria. Children who take Kuvan® must follow a special meal plan, but it may not be as strict as one for those not taking the drug. They still need regular blood tests to check phenylalanine levels.
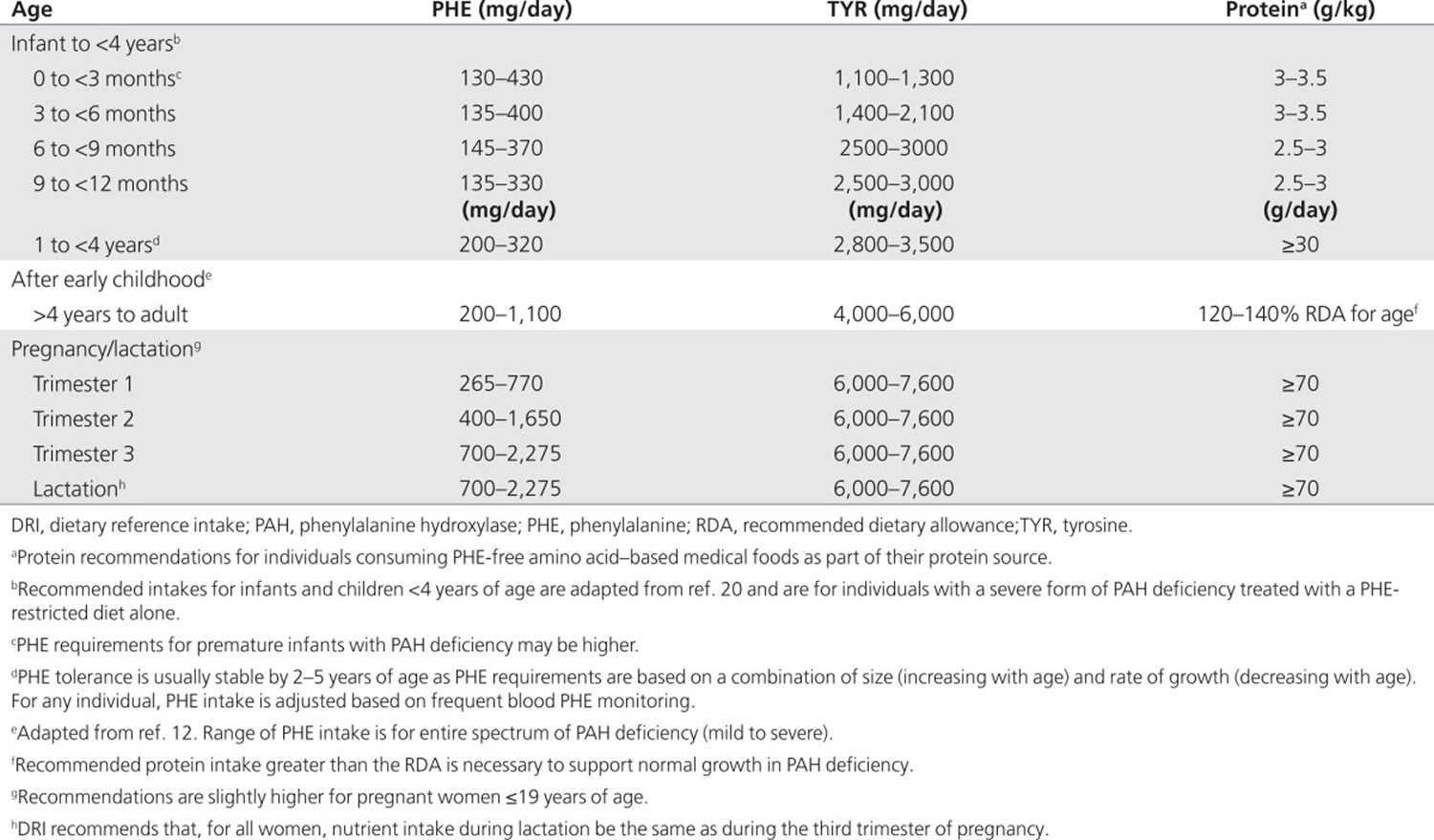
Nutrient Requirements, Sources, and Monitoring
Nutrient requirements for individuals with phenylalanine hydroxylase deficiency do not differ from those of the general population, except for phenylalanine, tyrosine, and protein (Table 1) 16. Concern about the nutritional adequacy of the diet arises because the severe restriction of foods containing intact protein necessitates lifelong reliance on semisynthetic medical foods, from which some nutrients may not be as well absorbed. In addition, nonadherence to medical food consumption, or reliance on nutritionally incomplete medical foods, increases the risk of multiple nutrient deficiencies. Therefore, monitoring of nutrient intake and laboratory indexes of nutritional adequacy is recommended (see Table 2 below).
Table 1. Recommended intakes of phenylalanine, tyrosine and protein for individuals with phenylketonuria
Protein and amino acids
Because medical food is the primary source of protein equivalents in the diet for phenylketonuria, it is a critical component of the diet throughout life. Recommendations for total protein intake exceed age- and sex-specific Dietary Reference Intakes because l-amino acids found in most medical foods are absorbed and oxidized more rapidly than amino acids in intact protein 18. Traditionally, l-amino acids (without phenylalanine) have been the source of protein equivalents in medical foods, but more recently, glycomacropeptide (GMP), an intact protein that is low in phenylalanine, has been used as a protein source in combination with limiting l-amino acids 19. l-Amino acid–based or GMP-based medical foods provide ~85% of the protein needs of individuals with a severe form of phenylalanine hydroxylase deficiency 20. Large neutral amino acids (LNAAs) can also be used as the medical food for some individuals with phenylalanine hydroxylase deficiency in conjunction with a more relaxed protein restriction.
Normal protein status, assessed by monitoring plasma amino acids and prealbumin, is achievable when total protein is provided in appropriate amounts 21. Prealbumin is an acceptable measure of protein status in individuals with phenylalanine hydroxylase deficiency 22. Mild protein insufficiency, as indicated by a prealbumin concentration of <20 mg/dl, has been associated with decreased linear growth 23.
tyrosine is conditionally essential in phenylalanine hydroxylase deficiency and must be added to the diet to maintain blood concentrations in the normal range. By itself, tyrosine supplementation does not improve neurological outcomes 24. All medical foods for the treatment of phenylketonuria are supplemented with tyrosine 25; therefore, individuals who do not adhere to their prescribed intake of medical food may have inadequate blood concentrations of tyrosine. In addition, tyrosine has a low solubility and may settle out of prepared medical foods during storage 25. Most clinicians monitor blood tyrosine routinely in individuals with phenylalanine hydroxylase deficiency 26 and low plasma tyrosine has been documented among individuals treated with diet 27. One cause could be a diurnal variation in blood tyrosine 28. Supplementation beyond that provided by medical food with tyrosine is indicated only if blood tyrosine concentrations are consistently below the normal range. Fewer than 20% of clinicians in the Inborn Errors of Metabolism survey routinely prescribe tyrosine supplementation.
Energy
Because energy expenditure varies from person to person, energy requirements must be individually assessed. Most evidence suggests that energy requirements are not increased in phenylalanine hydroxylase deficiency, and carbohydrate and fat intakes are within established recommendations 29.
Essential fatty acids
With adequate fat intake, children with phenylalanine hydroxylase deficiency are found to have normal essential fatty acid status 30. Medical foods supplemented with sources of long-chain polyunsaturated fatty acids increase blood concentrations of essential fatty acid 31. When a fat-free medical food is provided or the diet contains inadequate sources of linoleic and α-linolenic acid, essential fatty acid status should be monitored 3. Both the Inborn Errors of Metabolism survey and the Delphi survey 3 suggest that supplementation with precursor essential fatty acid, or with preformed docosahexaenoic acid (DHA), may be necessary in these individuals. In infancy, including breast milk or standard formulas containing DHA and arachidonic acid, as well as medical foods that contain DHA/arachidonic acid, can help ensure adequacy.
Micronutrients
Most medical foods available in the United States are supplemented with vitamins and minerals to provide micronutrients in amounts that meet recommendations. When a medical food does not contain adequate amounts of micronutrients or an individual’s intake is inadequate, a vitamin and mineral supplement should be included in the treatment plan. Additional biochemical monitoring (see Table 2) is indicated when there is a question about inadequate or excessive intake.
Minerals, including copper, manganese, and zinc 32, as well as selenium 33, have been reported as being deficient in the diet for phenylalanine hydroxylase deficiency. However, these minerals are now added to most medical foods. Iron deficiency (without anemia) has been reported among individuals with phenylalanine hydroxylase deficiency, and routine evaluation of iron status is recommended 34.
Vitamin B12, as well as B6, deficiency may occur if there is inadequate consumption of medical food or animal protein 35. Severe megaloblastic anemia has been reported in adolescents and adults with phenylalanine hydroxylase deficiency who are off-diet 36. Because B12 deficiency can cause neurological deficits 37, including memory loss, such symptoms could be erroneously attributed to high blood phenylalanine. Supplementation is indicated if B12 markers remain low 37. In addition, higher than normal plasma B12 levels have been reported in some on-diet individuals with phenylalanine hydroxylase deficiency 38. Both excessive and inadequate intakes of the fat-soluble vitamins A and D are possible with inappropriate medical food intake. Monitoring for clinical signs with subsequent biochemical testing is warranted.
There may be concerns with bone density that are unique to phenylketonuria. Decreased bone mineral density and bone mineral content, indicated by dual-energy X-ray absorptiometry, have been noted 39. Osteopenia, defined as bone mineral density one or more standard deviations from normal reference for age and sex, is also seen 40, but cannot be attributed to vitamin D concentrations alone. Some reports indicate that bone density is associated with adherence to nutrition therapy 8, whereas others note decreased density across groups, stratified by calcium and phosphorus intakes 39 and/or as compared with controls without phenylalanine hydroxylase deficiency 41. Aberrant findings in protein markers, correlating to bone resorption or absorption, have also been reported 42. Further research is warranted to look at the effects of phenylalanine hydroxylase deficiency itself, phenylalanine hydroxylase deficiency management, and nutrient intake on bone density.
Medical foods
Medical foods are available in a variety of flavors and in different forms (i.e., powders, ready-to-drink liquids, tablets, and bars). Convenience packaging and alternative forms of medical foods typically cost more than the powdered forms. Medical food should be consumed throughout the day and divided into at least three servings because more frequent consumption of medical foods is associated with better phenylalanine tolerance 43 and improved plasma phenylalanine concentrations 44. Although insurance coverage of medical foods is mandated in some states, there are exceptions and limitations. Therefore, access to medical food is not guaranteed in the United States, and obtaining coverage can be an arduous process 45. This is due, in part, to the fact that medical foods are not prescription medications but have a unique classification by the Food and Drug Administration that was intended to promote the development of more products for phenylketonuria and other orphan diseases 46. The classification allows a less costly regulatory process necessary to get medical foods to market. The macro- and micronutrient profiles of many medical foods for Inborn Errors of Metabolism are available at http://www.gmdi.org.
Glycomacropeptide
Glycomacropeptide is an intact whey protein low in phenylalanine, tyrosine, histidine, leucine, tryptophan, and arginine. All except phenylalanine must be added as free l-amino acids to provide an appropriate protein source for phenylketonuria 47. Although long-term efficacy and growth studies are lacking, a short-term inpatient study found no safety concerns when glycomacropeptide products replaced amino acid–based medical food 47. Various markers pointed to improved protein utilization, and lower ghrelin concentrations suggested improved satiety when glycomacropeptide products replaced each subject’s usual amino acid–based medical food as the protein source 48. Medical food products incorporating glycomacropeptide are an alternative to phenylalanine-free amino acid formulas. Because glycomacropeptide contains a small amount of phenylalanine (2–5 mg phenylalanine per gram of protein) 47, the allowance of phenylalanine from food may need to be reduced to maintain phenylalanine intake within phenylalanine tolerance limits 19.
Large neutral amino acids
The theory behind large neutral amino acids use as a medical food is described in the American College of Medical Genetics and Genomics guideline 49. Large neutral amino acids are not recommended for young children or pregnant women but should be considered for adults with phenylketonuria deficiency who are not in good metabolic control and do not adhere to other treatment options 3. Large neutral amino acids therapy has been shown to improve executive function in some adults 50. When using large neutral amino acids, 25–30% of total protein needs are provided by large neutral amino acids, with the remaining 70–75% coming from dietary protein sources 51. Protein intake and plasma amino acids should be monitored to prevent essential amino acid deficiencies. It is difficult to monitor the success of large neutral amino acid therapy because blood phenylalanine remains high, and measurement of phenylalanine concentration in the brain is impractical. Blood and urine melatonin have been suggested as surrogate markers for serotonin and may be useful in monitoring treatment.58 Serotonin has been shown to be deficient in individuals with phenylketonuria deficiency who exhibit executive function defects 52.
Modified low-protein foods
Individuals with severe forms of phenylketonuria and low phenylalanine tolerance often rely on modified low-protein foods to provide energy and variety in their diets 25. Modified low-protein foods, including low-protein breads and pasta, use the starch portion of the grain rather than the higher-protein flour 53. It is important to note that these products do not have the usual vitamin enrichment found in regular grain products. As a source of energy, these products can help to prevent weight loss, catabolism, and the resulting elevated blood phenylalanine 25. Third-party reimbursement for modified low-protein foods is not universally available 45.
Sapropterin (tetrahydrobiopterin) therapy
Sapropterin dihydrochloride (sapropterin) is the pharmaceutical form of tetrahydrobiopterin, a cofactor required for phenylalanine hydroxylase activity. Given in therapeutic doses, sapropterin appears to enhance phenylalanine hydroxylase activity in certain individuals with phenylketonuria 54. The American College of Medical Genetics and Genomics phenylketonuria guideline recommends a trial of sapropterin therapy for all individuals 49. The benefits of response fall into two categories: for individuals who are nonadherent or unable to maintain diet restriction and medical food intake, sapropterin may lower blood phenylalanine without further diet modification; and for individuals who maintain blood phenylalanine within the therapeutic range by dietary adherence, sapropterin may allow liberalization of dietary phenylalanine and less medical food intake 55.
Although sapropterin therapy may support significant dietary liberalization, it seldom allows individuals to maintain appropriate blood phenylalanine without some phenylalanine restriction and medical food. Regular monitoring of blood phenylalanine, dietary adequacy, and nutritional status continue to be essential. Individualized patient counseling includes planning and calculating the diet with a higher phenylalanine or protein allowance, choosing appropriate natural protein sources, reading labels, distributing high-phenylalanine/intact protein foods throughout the day, meeting micronutrient/vitamin requirements when the medical food prescription is decreased, and understanding the importance of consistent sapropterin dosing 56. Longer-term follow-up will be necessary to determine whether individuals with phenylketonuria remain adherent to sapropterin and dietary recommendations and if there is an impact on outcome.
Target blood phenylalanine
Blood phenylalanine has been used to monitor metabolic status and determine appropriate dietary phenylalanine intake and has been shown to be a reliable predictor of clinical outcomes 57. The companion American College of Medical Genetics and Genomics phenylalanine hydroxylase deficiency treatment guideline supports a lifelong target blood phenylalanine of 120–360 µmol/l for optimal cognitive outcome 49. Eighty percent of Genetic Metabolic Dietitians International and Southeast Regional Newborn Screening and Genetics Collaborative respondents supported 120–360 µmol/l as a goal blood phenylalanine for individuals with phenylalanine hydroxylase deficiency of all ages, yet there was recognition that the goal is difficult to achieve and may not apply in all cases 3. Regular blood phenylalanine monitoring is key to the management of phenylalanine hydroxylase deficiency. During infancy, early childhood, and pregnancy, more frequent monitoring is necessary to assess the increased dietary phenylalanine needs of anabolism (Table 1) 58. The British Medical Research Council guidelines recommend measuring phenylalanine concentrations at a standard time 58; there was a consensus that blood samples be collected at the same time of day, preferably 2–3 hours after a meal 3. It is recommended that medical food be consumed throughout the day to maintain stable concentrations of blood phenylalanine 59.
Table 2. Monitoring the nutritional management of individuals with phenylalanine hydroxylase deficiency deficiency a
Target blood tyrosine and phenylalanine:tyrosine ratio
Because both endogenous production of tyrosine and intact protein intake are greatly limited in phenylalanine hydroxylase deficiency, monitoring is necessary to ensure that supplementation is adequate to maintain the blood tyrosine in the normal range. Some clinicians routinely monitor blood phenylalanine:tyrosine ratios 26. There are reports of impairments in executive function among individuals with high phenylalanine:tyrosine ratios 60. However, at this time the clinical relevance of phenylalanine:tyrosine as a routine biomarker requires further study.
Recommendations:
- Maintain blood phenylalanine between 120 and 360 μmol/l throughout the life span for optimal outcome.
- Monitor blood phenylalanine most frequently during times of increased anabolism: infancy, childhood, and pregnancy.
- Monitor blood phenylalanine at a consistent time during the day, preferably 2–3 hours after eating.
- Maintain blood tyrosine in the normal range.
Home remedies
Some strategies to help manage phenylketonuria may include the following.
Keep track and measure correctly
If you or your child is following a low-phenylalanine diet, you’ll need to keep records of the food eaten every day to be sure you’re sticking to the specific, individualized dietary guidelines recommended by your dietitian.
To be as accurate as possible, measure food portions using standard measuring cups and spoons and a kitchen scale that reads in grams. The food amounts are compared with a food list or are used to calculate the amount of phenylalanine eaten every day. Each meal and snack includes the appropriately divided portion of your daily phenylketonuria formula.
Food diaries or computer programs are available that list the amount of phenylalanine in baby foods, solid foods, phenylketonuria formulas, and common baking and cooking ingredients.
Buy low-protein products
To add variety to your diet, buy some of the many low-protein products, such as low-protein pasta, rice, flour and bread, which are available through specialty food retailers.
These products allow people with phenylketonuria to eat some meals that more closely resemble what everyone else is eating. Like the phenylketonuria formulas, these products can be expensive, but you might consider splurging on a few favorites with the money you save on dairy and meat products.
Be creative
Talk with your dietitian to find out how you can be creative with foods to help you stay on track. For example, use seasonings and a variety of cooking methods to transform lower phenylalanine vegetables into a whole menu of different dishes. Herbs and flavorings low in phenylalanine can pack a flavorful punch. Just remember to measure and count every ingredient and adjust recipes to your individualized diet.
If you have any other health conditions, you may also need to consider those when you plan your diet. Talk with your doctor or dietitian if you have any questions.
Coping and support
Living with phenylketonuria can be challenging. These strategies may help:
- Stay informed. Knowing the facts about phenylketonuria can help you take charge of the situation. Discuss any questions with your pediatrician, family doctor, geneticist or dietitian. Read books and cookbooks specifically written for people with phenylketonuria.
- Learn from other families. Ask your doctor about local or online support groups for people dealing with phenylketonuria. Talking with others who have mastered similar challenges can be very helpful. The National phenylketonuria Alliance is an online support group for adults with phenylketonuria.
- Get help with menu planning. A registered dietitian with experience in phenylketonuria can help you devise delicious low-phenylalanine dinners. He or she may also have great ideas for holiday meals and birthdays.
- Plan ahead when you eat out. A meal at the local restaurant gives you a break from the kitchen and can be fun for the whole family. Most places offer something that fits into the phenylketonuria diet. But you may want to call ahead and ask about the menu or bring food from home.
- Find sources of financial aid. Ask your doctor or dietitian if there are programs or insurance plans that help cover the high costs of formula and low-protein foods. Also, see if your local school lunch program will accommodate special dietary needs.
- Don’t focus on food. Encourage children with phenylketonuria to focus on sports, music or favorite hobbies, not on just what they can and can’t eat. Also consider creating holiday traditions that center on special projects and activities, not just food.
- Let your child help manage his or her diet as early as possible. Toddlers can make choices about which cereal, fruit or vegetable they’d like to eat and help measure out portions. They can also help themselves to pre-measured snacks. Older children can help with menu planning, pack their own lunches and keep their own food records.
- Make your grocery list and meals with the whole family in mind. A cupboard full of restricted foods can be tempting to a child or adult with phenylketonuria, so try to focus on foods that everyone can eat. Serve stir-fried vegetables that are lower in protein. If the other family members wish, they can add peas, corn, meat and rice. Or set up a salad bar with low-protein and moderate-protein options. You can also serve the whole family a delicious low-phenylalanine soup or curry.
- Be prepared for potlucks, picnics and car trips. Plan ahead, so there’s always a phenylketonuria-friendly food option. Pack dehydrated fruit snacks, raisins and lower protein crackers for the car. Take fruit kebabs or vegetable skewers to a cookout, and make a low-phenylalanine salad for the neighborhood potluck. Other parents, friends and family members will likely be accommodating and helpful if you explain the dietary restrictions.
- Talk to teachers and other staff in your child’s school. Your child’s teachers and cafeteria staff can be a big help with the phenylketonuria diet if you take the time to explain its importance and how it works. By working with your child’s teachers, you can also plan ahead for special school events and parties so that your child always has a treat to eat.
- Maintain a positive food attitude. When children know nothing but the foods they are given, they can be surprisingly accepting of the phenylketonuria diet — especially when their parents are positive problem-solvers.
- Grosse SD. Late-treated phenylketonuria and partial reversibility of intellectual impairment. Child Dev. 2010;81:200–211. https://www.ncbi.nlm.nih.gov/pubmed/20331662[↩][↩]
- National Institutes of Health. National Institutes of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16-18, 2000 Pediatrics 2001108:972–982. https://www.ncbi.nlm.nih.gov/pubmed/11581453[↩][↩]
- GMDI Delphi Survey: Genetic Metabolic Dietitians International and Southeast Regional Newborn Screening and Genetics Collaborative. 2013.[↩][↩][↩][↩][↩][↩][↩][↩]
- Acosta PB, Matalon K, Castiglioni L, et al. Intake of major nutrients by women in the Maternal Phenylketonuria (MPKU) Study and effects on plasma phenylalanine concentrations. Am J Clin Nutr. 2001;73:792–796. https://www.ncbi.nlm.nih.gov/pubmed/11273855[↩][↩]
- Michals-Matalon K, Platt LD, Acosta P P, Azen C, Walla CA. Nutrient intake and congenital heart defects in maternal phenylketonuria. Am J Obstet Gynecol. 2002;187:441–444. https://www.ncbi.nlm.nih.gov/pubmed/12193940[↩]
- Koch R, Hanley W, Levy H, et al. The Maternal Phenylketonuria International Study: 1984-2002. Pediatrics. 2003;112 Pt 2:1523–1529. https://www.ncbi.nlm.nih.gov/pubmed/14654658[↩]
- Agency for Healthcare Research and Quality. Comparative Effectiveness of Treatment for Phenylketonuria (PKU). Comparative Effectiveness Review No. 56. (Prepared by the Vanderbilt Evidence-Based Practice Center under Contract No. 290-2007-10065-I.) AHRQ Publication No. 12-EHC035-EF. 2011 [↩]
- Porta F, Mussa A, Zanin A, Greggio NA, Burlina A, Spada M. Impact of metabolic control on bone quality in phenylketonuria and mild hyperphenylalaninemia. J Pediatr Gastroenterol Nutr. 2011;52:345–350. https://www.ncbi.nlm.nih.gov/pubmed/21336059[↩][↩]
- Belloso LM, Lowitt MH. Cutaneous findings in a 51-year-old man with phenylketonuria. J Am Acad Dermatol. 2003;49 2 Suppl Case Reports:S190–S192. https://www.ncbi.nlm.nih.gov/pubmed/12894120[↩]
- Koch R, Burton B, Hoganson G, et al. Phenylketonuria in adulthood: a collaborative study. J Inherit Metab Dis. 2002;25:333–346. https://www.ncbi.nlm.nih.gov/pubmed/12408183[↩]
- Brumm VL, Bilder D, Waisbren SE. Psychiatric symptoms and disorders in phenylketonuria. Mol Genet Metab. 2010;99 suppl 1:S59–S63. https://www.ncbi.nlm.nih.gov/pubmed/20123472[↩]
- van Rijn M, Bekhof J, Dijkstra T, Smit PG, Moddermam P, van Spronsen FJ. A different approach to breast-feeding of the infant with phenylketonuria. Eur J Pediatr. 2003;162:323–326. https://www.ncbi.nlm.nih.gov/pubmed/12692713[↩][↩]
- Kanufre VC, Starling AL, Leão E, et al. Breastfeeding in the treatment of children with phenylketonuria. J Pediatr (Rio J) 2007;83:447–452. http://www.jped.com.br/conteudo/07-83-05-447/port.pdf[↩]
- Zimmermann M, Jacobs P, Fingerhut R, et al. Positive effect of a simplified diet on blood phenylalanine control in different phenylketonuria variants, characterized by newborn BH4 loading test and PAH analysis. Mol Genet Metab. 2012;106:264–268. https://www.ncbi.nlm.nih.gov/pubmed/22607939[↩]
- GMDI Delphi Survey: Genetic Metabolic Dietitians International and Southeast Regional Newborn Screening and Genetics Collaborative. 2013[↩]
- Acosta PB. Nutrition management of patients with inherited metabolic disorders of aromatic amino acid metabolism. Acosta PB.(ed).Nutrition Management of Patients With Inherited Metabolic Disorders Jones and Bartlett Publishers; Sudbury, MA; 2010126–129.[↩]
- Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet Med. 2014 Feb; 16(2): 121–131. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3918542/[↩][↩]
- Metges CC, El-Khoury AE, Selvaraj AB, et al. Kinetics of L-[1-(13)C]leucine when ingested with free amino acids, unlabeled or intrinsically labeled casein. Am J Physiol Endocrinol Metab. 2000;278:E1000–E1009. https://www.physiology.org/doi/full/10.1152/ajpendo.2000.278.6.E1000[↩]
- van Calcar SC, Ney DM. Food products made with glycomacropeptide, a low-phenylalanine whey protein, provide a new alternative to amino acid-based medical foods for nutrition management of phenylketonuria. J Acad Nutr Diet. 2012;112:1201–1210. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3402906/[↩][↩]
- University of Washington PKU Clinic Management Guidelines 2013. http://depts.washington.edu/pku/pdfs/Management_Guidelines_Brochure_0308.pdf[↩]
- Huemer M, Huemer C, Möslinger D, Huter D, Stöckler-Ipsiroglu S. Growth and body composition in children with classical phenylketonuria: results in 34 patients and review of the literature. J Inherit Metab Dis. 2007;30:694–699. https://www.ncbi.nlm.nih.gov/pubmed/17628756[↩]
- Rocha JC, Almeida MF, Carmona C, et al. The use of prealbumin concentration as a biomarker of nutritional status in treated phenylketonuric patients. Ann Nutr Metab. 2010;56:207–211. https://www.ncbi.nlm.nih.gov/pubmed/20215742[↩]
- Arnold GL, Vladutiu CJ, Kirby RS, Blakely EM, Deluca JM. Protein insufficiency and linear growth restriction in phenylketonuria. J Pediatr. 2002;141:243–246. https://www.ncbi.nlm.nih.gov/pubmed/12183721[↩]
- Smith ML, Hanley WB, Clarke JT, et al. Randomised controlled trial of tyrosine supplementation on neuropsychological performance in phenylketonuria. Arch Dis Child. 1998;78:116–121. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1717450/pdf/v078p00116.pdf[↩]
- Macleod EL, Ney DM. Nutritional management of phenylketonuria. Ann Nestle Eng. 2010;68:58–69. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2901905/[↩][↩][↩][↩]
- Sharman R, Sullivan KA, Young RM, McGill JJ. Tyrosine monitoring in children with early and continuously treated phenylketonuria: results of an international practice survey. J Inherit Metab Dis. 2010;33 suppl 3:S417–S420. https://www.ncbi.nlm.nih.gov/pubmed/20882350[↩][↩]
- Hanley WB, Lee AW, Hanley AJ, et al. “Hypotyrosinemia” in phenylketonuria. Mol Genet Metab. 2000;69:286–294. https://www.ncbi.nlm.nih.gov/pubmed/10870846[↩]
- Kalsner LR, Rohr FJ, Strauss KA, Korson MS, Levy HL. Tyrosine supplementation in phenylketonuria: diurnal blood tyrosine levels and presumptive brain influx of tyrosine and other large neutral amino acids. J Pediatr. 2001;139:421–427. https://www.ncbi.nlm.nih.gov/pubmed/11562623[↩]
- Rocha JC, van Spronsen FJ, Almeida MF, et al. Dietary treatment in phenylketonuria does not lead to increased risk of obesity or metabolic syndrome. Mol Genet Metab. 2012;107:659–663. https://www.ncbi.nlm.nih.gov/pubmed/23137570[↩]
- Acosta PB, Yannicelli S, Singh R, et al. Intake and blood levels of fatty acids in treated patients with phenylketonuria. J Pediatr Gastroenterol Nutr. 2001;33:253–259. https://www.ncbi.nlm.nih.gov/pubmed/11593118[↩]
- Cleary MA, Feillet F, White FJ, et al. Randomised controlled trial of essential fatty acid supplementation in phenylketonuria. Eur J Clin Nutr. 2006;60:915–920. https://www.ncbi.nlm.nih.gov/pubmed/16523206[↩]
- Alexander FW, Clayton BE, Delves HT. Mineral and trace-metal balances in children receiving normal and synthetic diets. Q J Med. 1974;43:89–111. https://www.ncbi.nlm.nih.gov/pubmed/4822973[↩]
- Jochum F, Terwolbeck K, Meinhold H, Behne D, Menzel H, Lombeck I. Effects of a low selenium state in patients with phenylketonuria. Acta Paediatr. 1997;86:775–777. https://www.ncbi.nlm.nih.gov/pubmed/9240892[↩]
- Acosta PB, Yannicelli S, Singh RH, Elsas LJ, 2nd, Mofidi S, Steiner RD. Iron status of children with phenylketonuria undergoing nutrition therapy assessed by transferrin receptors. Genet Med. 2004;6:96–101. https://www.ncbi.nlm.nih.gov/pubmed/15017332[↩]
- Walter JH. Vitamin B12 deficiency and phenylketonuria. Mol Genet Metab. 2011;104 suppl:S52–S54. https://www.ncbi.nlm.nih.gov/pubmed/21824796[↩]
- Hanley WB, Feigenbaum AS, Clarke JT, Schoonheyt WE, Austin VJ. Vitamin B12 deficiency in adolescents and young adults with phenylketonuria. Eur J Pediatr. 1996;155 suppl 1:S145–S147. https://www.ncbi.nlm.nih.gov/pubmed/8828632[↩]
- Hvas AM, Nexo E, Nielsen JB. Vitamin B12 and vitamin B6 supplementation is needed among adults with phenylketonuria (PKU). J Inherit Metab Dis. 2006;29:47–53. https://www.ncbi.nlm.nih.gov/pubmed/16601867[↩][↩]
- Prince AP, Leklem JE. Vitamin B-6 status of school-aged patients with phenylketonuria. Am J Clin Nutr. 1994;60:262–268. https://www.ncbi.nlm.nih.gov/pubmed/8030605[↩]
- Allen JR, Humphries IR, Waters DL, et al. Decreased bone mineral density in children with phenylketonuria. Am J Clin Nutr. 1994;59:419–422. https://www.ncbi.nlm.nih.gov/pubmed/8310995[↩][↩]
- Pérez-Dueñas B, Cambra FJ, Vilaseca MA, Lambruschini N, Campistol J, Camacho JA. New approach to osteopenia in phenylketonuric patients. Acta Paediatr. 2002;91:899–904. https://www.ncbi.nlm.nih.gov/pubmed/12222712[↩]
- Hillman L, Schlotzhauer C, Lee D, et al. Decreased bone mineralization in children with phenylketonuria under treatment. Eur J Pediatr. 1996;155 suppl 1:S148–S152. https://www.ncbi.nlm.nih.gov/pubmed/8828633[↩]
- Millet P, Vilaseca MA, Valls C, et al. Is deoxypyridinoline a good resorption marker to detect osteopenia in phenylketonuria. Clin Biochem. 2005;38:1127–1132. https://www.ncbi.nlm.nih.gov/pubmed/16256974[↩]
- MacLeod EL, Gleason ST, van Calcar SC, Ney DM. Reassessment of phenylalanine tolerance in adults with phenylketonuria is needed as body mass changes. Mol Genet Metab. 2009;98:331–337. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2783926/[↩]
- MacDonald A, Rylance G, Hall SK, Asplin D, Booth IW. Factors affecting the variation in plasma phenylalanine in patients with phenylketonuria on diet. Arch Dis Child. 1996;74:412–417. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1511531/pdf/archdisch00613-0052.pdf[↩]
- Berry SA, Kenney MK, Harris KB, et al. Insurance coverage of medical foods for treatment of inherited metabolic disorders. Genet Med. 2013;15:978–982. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4541808/[↩][↩]
- Haffner ME. Applications of the orphan drug act to special patient populations. Drug Inf J. 1994;28:495–503.[↩]
- van Calcar SC, MacLeod EL, Gleason ST, et al. Improved nutritional management of phenylketonuria by using a diet containing glycomacropeptide compared with amino acids. Am J Clin Nutr. 2009;89:1068–1077 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2667457[↩][↩][↩]
- MacLeod EL, Clayton MK, van Calcar SC, Ney DM. Breakfast with glycomacropeptide compared with amino acids suppresses plasma ghrelin levels in individuals with phenylketonuria. Mol Genet Metab. 2010;100:303–308. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2906609/[↩]
- Vockley J, Andersson H, Antshel KM.et al. Phenylalanine hydroxylase deficiency diagnosis and management guideline Genet Med e-pub ahead of print 2 January 2014[↩][↩][↩]
- Schindeler S, Ghosh-Jerath S, Thompson S, et al. The effects of large neutral amino acid supplements in PKU: an MRS and neuropsychological study. Mol Genet Metab. 2007;91:48–54. https://www.ncbi.nlm.nih.gov/pubmed/17368065[↩]
- Ahring KK. Large neutral amino acids in daily practice. J Inherit Metab Dis. 2010;33 suppl 3:S187–S190. https://www.ncbi.nlm.nih.gov/pubmed/20300852[↩]
- Yano S, Moseley K, Azen C. Large neutral amino acid supplementation increases melatonin synthesis in phenylketonuria: a new biomarker. J Pediatr. 2013;162:999–1003. https://www.ncbi.nlm.nih.gov/pubmed/23164313[↩]
- Independence Blue Cross. Medical Foods, Low-Protein Modified Food Products, Enteral Nutrition, and Nutritional Formulas 2013[↩]
- Blau N, Erlandsen H. The metabolic and molecular bases of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Mol Genet Metab. 2004;82:101–111. https://www.ncbi.nlm.nih.gov/pubmed/15171997[↩]
- Burton BK, Grange DK, Milanowski A, et al. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis. 2007;30:700–707. https://www.ncbi.nlm.nih.gov/pubmed/17846916[↩]
- Cunningham A, Bausell H, Brown M, et al. Recommendations for the use of sapropterin in phenylketonuria. Mol Genet Metab. 2012;106:269–276. https://www.ncbi.nlm.nih.gov/pubmed/22575621[↩]
- Waisbren SE, Noel K, Fahrbach K, et al. Phenylalanine blood levels and clinical outcomes in phenylketonuria: a systematic literature review and meta-analysis. Mol Genet Metab. 2007;92:63–70. https://www.ncbi.nlm.nih.gov/pubmed/17591452[↩]
- Recommendations on the dietary management of phenylketonuria. Report of Medical Research Council Working Party on Phenylketonuria. Arch Dis Child. 1993;68:426–427. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1793880/pdf/archdisch00630-0094.pdf[↩][↩]
- MacDonald A, Rylance G, Hall SK, Asplin D, Booth IW. Factors affecting the variation in plasma phenylalanine in patients with phenylketonuria on diet. Arch Dis Child. 1996;74:412–417. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1511531/[↩]
- Luciana M, Sullivan J, Nelson CA. Associations between phenylalanine-to-tyrosine ratios and performance on tests of neuropsychological function in adolescents treated early and continuously for phenylketonuria. Child Dev. 2001;72:1637–1652. https://www.ncbi.nlm.nih.gov/pubmed/11768137[↩]





{kind=link}