Contents
What is scleritis
Scleritis is a painful inflammation (swelling) of the white part of the eye, which is also known as the sclera. Scleritis is a serious eye condition that can lead to loss of vision. Scleritis is a severe, immune-mediated ocular inflammatory condition that is frequently associated with systemic immunologic disease. The tough, fibrous tissues of the sclera form a protective outer layer for the eye and make up 83 percent of the eye’s surface. Scleritis may involve the cornea, adjacent episclera and the uvea and thus can be vision-threatening. Scleritis is often associated with an underlying systemic disease in up to 50% of patients.
Scleritis is most common among women aged 30 to 50 year, and in almost half of all cases patients have underlying autoimmune disorder, such as rheumatoid arthritis, systemic lupus erythematosus (SLE), polyarteritis nodosa, granulomatosis with polyangiitis (formerly called Wegener granulomatosis), or relapsing polychondritis. A few cases are infectious in origin. About half of the cases of scleritis have no known cause.
There are two main types of scleritis classified by location: anterior and posterior scleritis. Scleritis is also classified by appearance of scleral inflammation (diffuse, nodular, or necrotizing). The most common form, anterior scleritis, is defined as scleral inflammation anterior to the extraocular recti muscles. Posterior scleritis is defined as involvement of the sclera posterior to the insertion of the rectus muscles.
Scleritis important facts
- Scleritis is severe, destructive, vision-threatening inflammation.
- Symptoms include deep, boring ache; photophobia and tearing; and focal or diffuse eye redness.
- Diagnosis is made clinically and by slit-lamp examination.
- Most patients require systemic corticosteroids and/or systemic immunosuppressive therapy, prescribed in consultation with a rheumatologist.
- Scleral grafts may be indicated for threatened perforation.
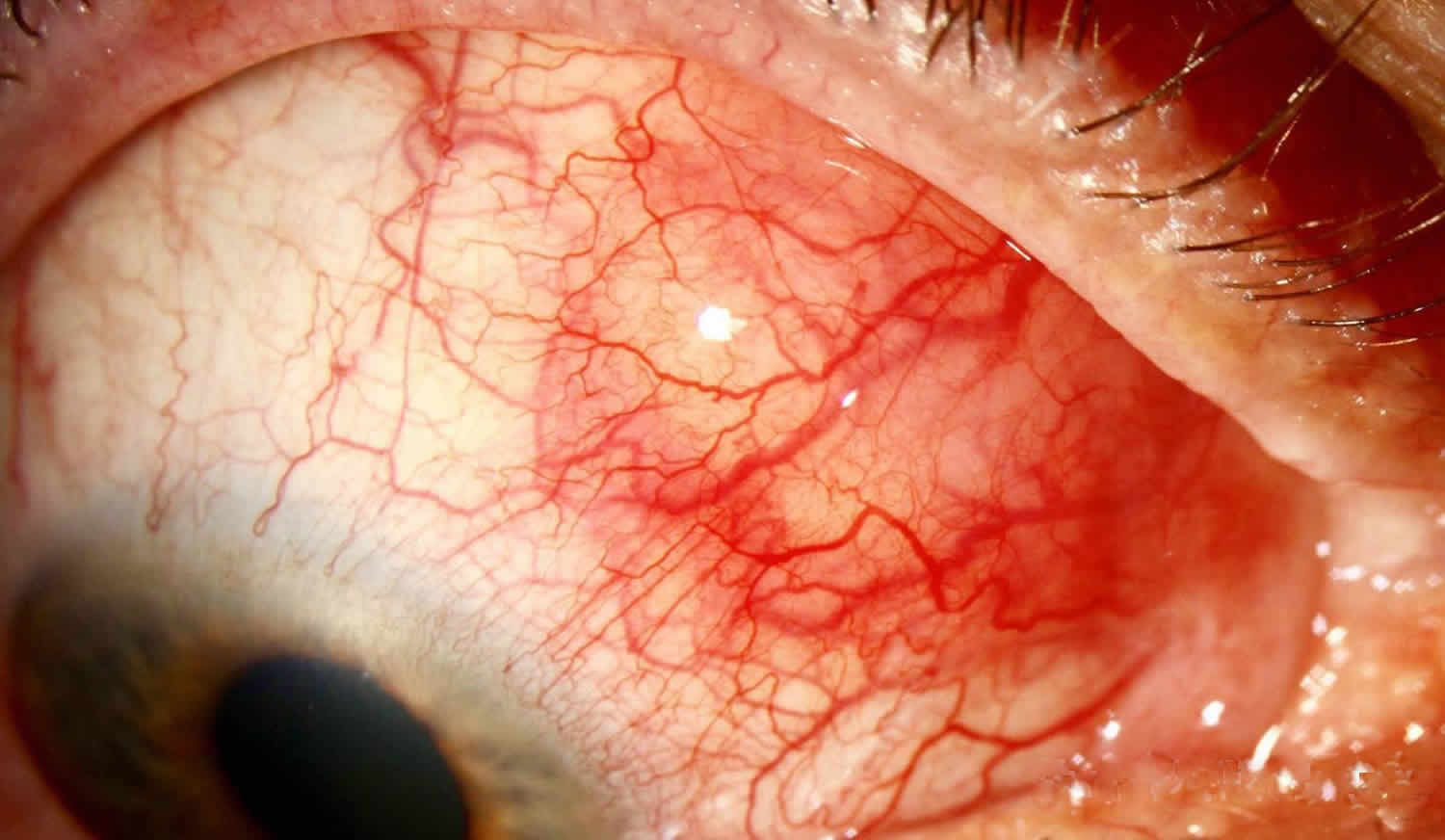
Figure 1. Scleritis
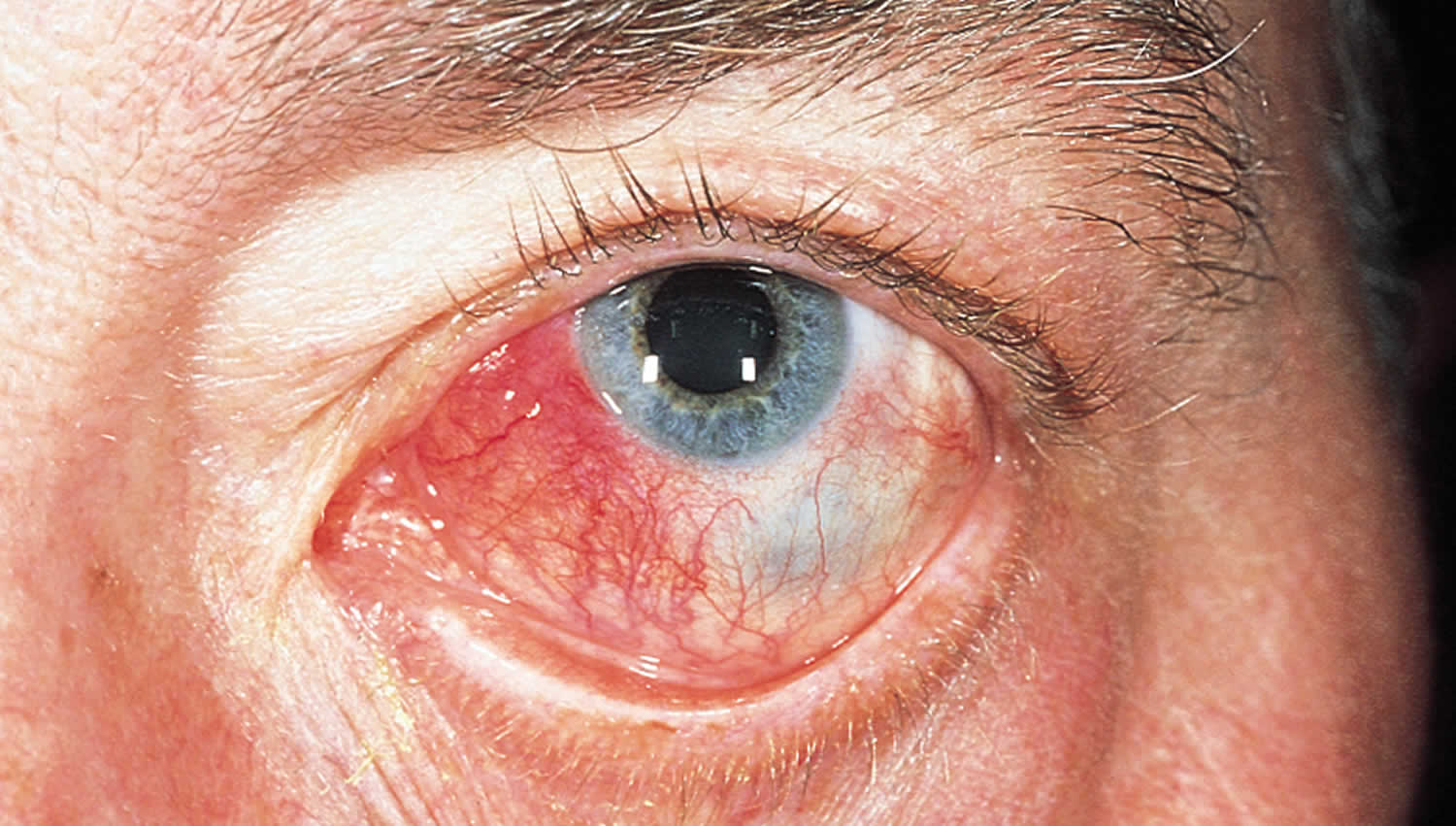
Figure 2. Nodular anterior scleritis
Footnote: Patients with nodular scleritis present with one or more firm, tender nodules. This image shows nodular thickening of the sclera with typical violaceous hue due to dilated scleral vessels. There is also overlying dilation of the conjunctival and episcleral vessels and mild chemosis. While conjunctival and episcleral involvement are often seen overlying active nodular scleritis, scleral involvement is necessary to make this diagnosis. Scleritis is often associated with underlying systemic autoimmune diseases such as rheumatoid arthritis, granulomatosis with polyangiitis (formerly called Wegener’s granulomatosis), spondyloarthritis, or systemic lupus erythematosus. Treatment is with systemic and topical non-steroidal anti-inflammatory drugs, systemic steroids, or systemic immunosuppression. Newer literature suggests that subconjunctival steroids may be effective in non-necrotizing cases.
Figure 3. Necrotizing scleritis
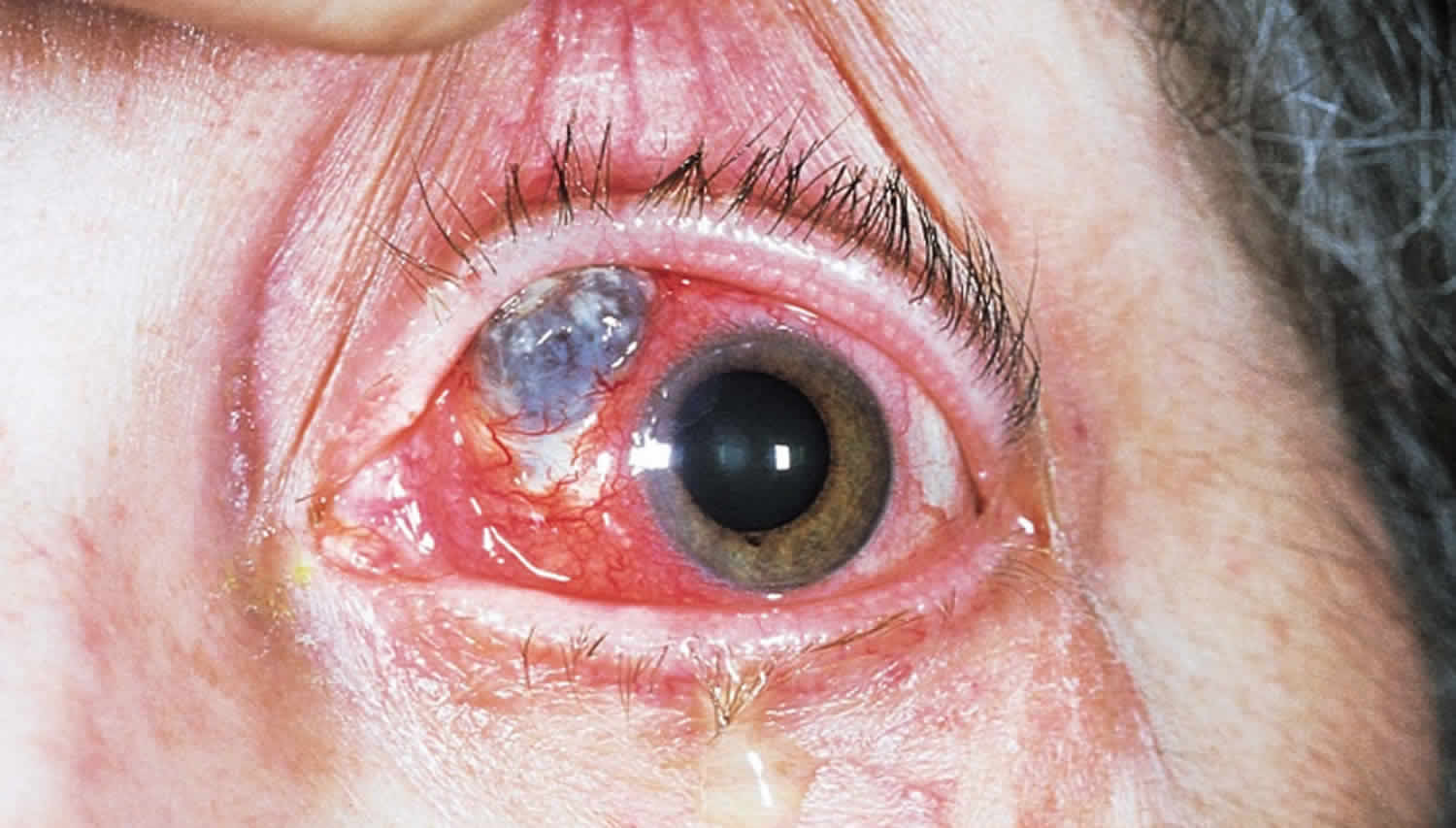
Footnote: Necrotizing scleritis is characterized by conjunctival ulceration with scleral necrosis. The blue to grayish area is the choroid, visible where there has been severe scleral loss. The smaller, white, triangular area is an avascular area that has not thinned.
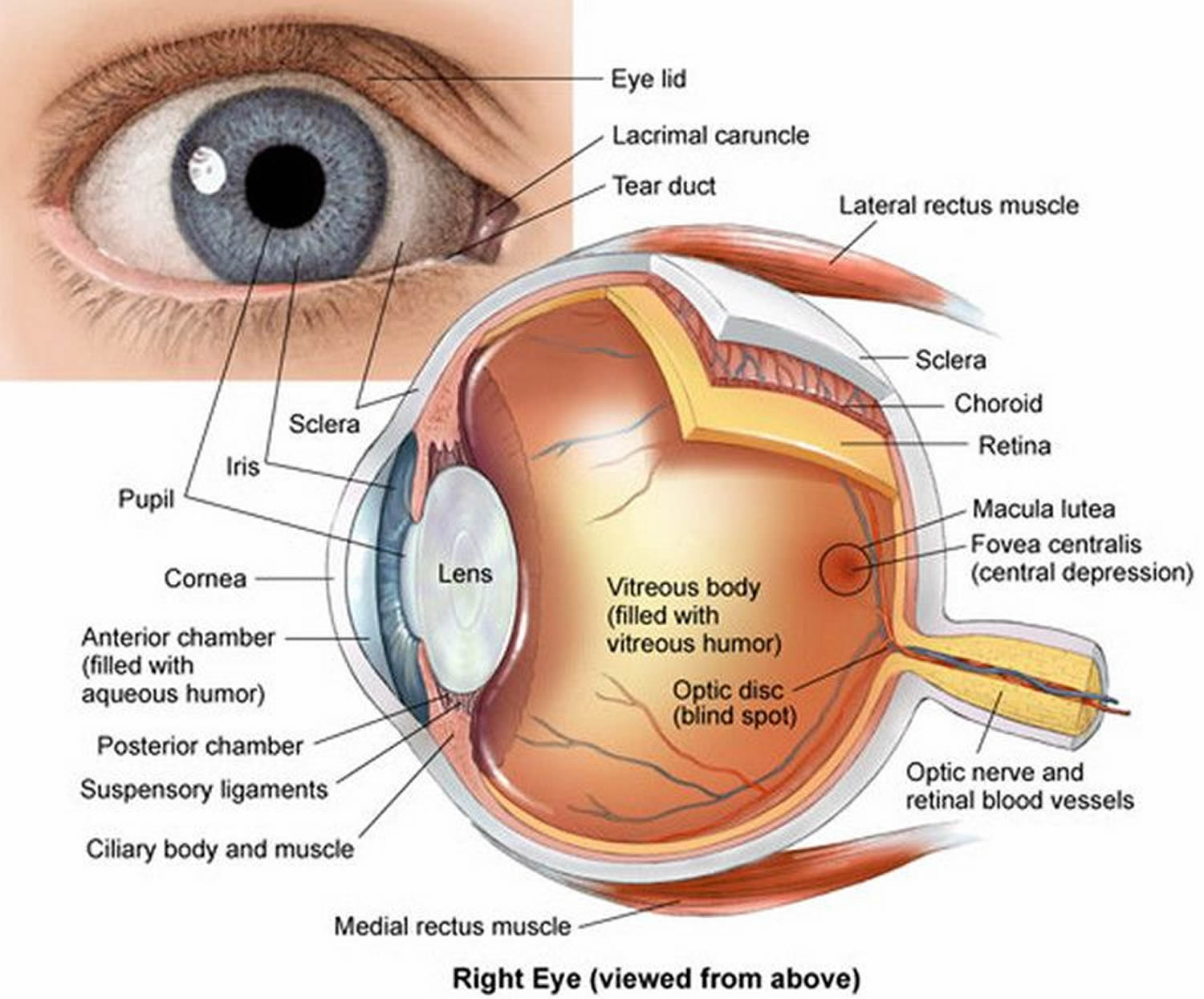
Figure 4. Eye anatomy
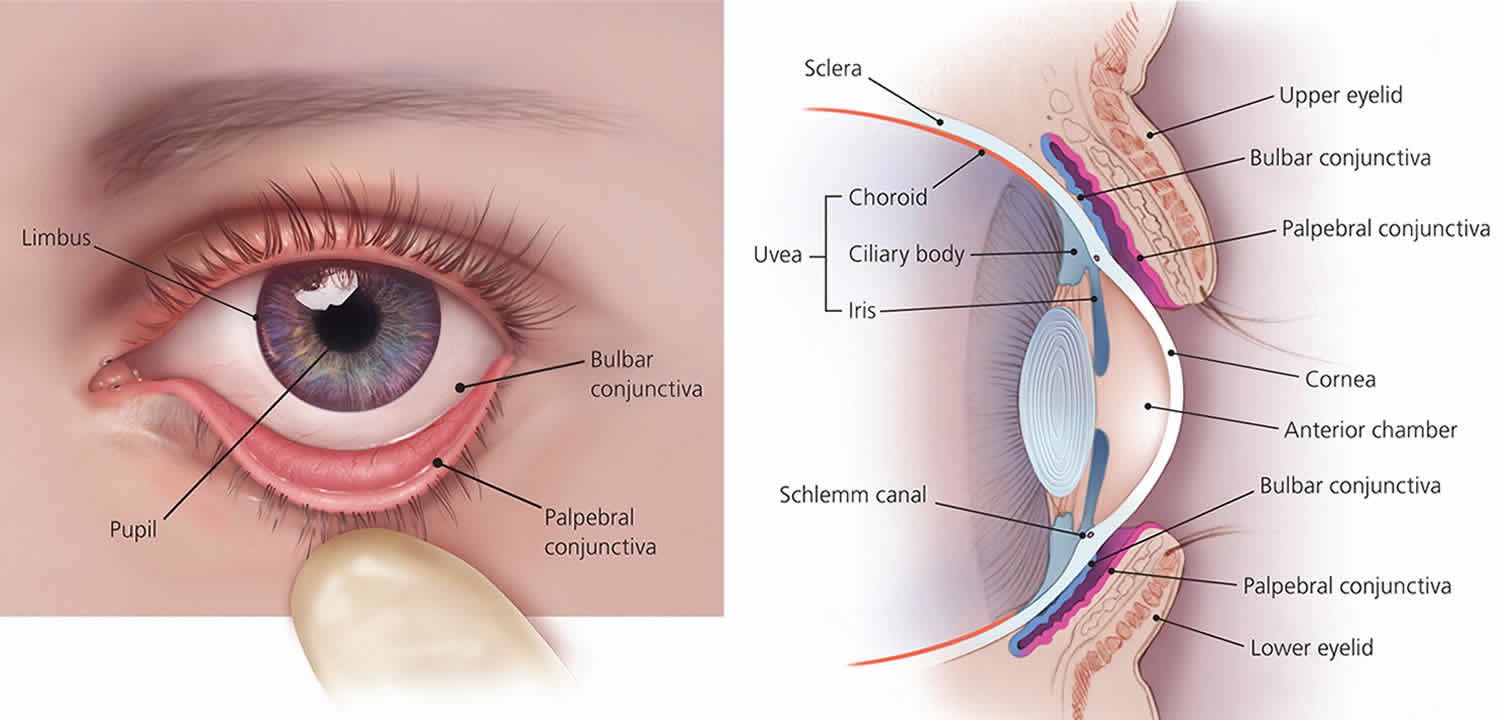
Figure 5. Structure of the human eye
Anterior scleritis
Anterior scleritis, the most common type, affects the front portion of the sclera. There are three types of anterior scleritis:
- Diffuse scleritis is the most common type and fortunately the most treatable. This type displays widespread redness and inflammation throughout the whole or a portion of the front portion of the sclera.
- Nodular scleritis, is characterized by the presence of nodules or bumps, often tender to the touch, on the surface of the eye.
- Necrotizing scleritis is the most severe form of anterior scleritis. It has the ability to destroy scleral tissues and in rare cases may lead to loss of the eye(s). This form is typically characterized by extreme pain and tenderness (although a rare form can occur without pain). A rare form of necrotizing anterior scleritis without pain can be called scleromalacia perforans. The sclera is notably white, avascular and thin. Both choroidal exposure and staphyloma formation may occur.
Posterior scleritis
Posterior scleritis, the rarer form, affects the back portion of the eye and often is not related to an underlying systemic disorder. Posterior scleritis can develop on its own or with the anterior form of scleritis. People with this form of scleritis may have pain and tenderness. Posterior scleritis can cause complications resulting in retinal detachment and angle-closure glaucoma. There is often loss of vision as well as pain upon eye movement.
The thickness of ocular coats is increased with fluid in subtenon space and around optic nerve (T sign). Inflammation of adjacent structures (e.g., dacryoadenitis) may also cause a secondary posterior scleritis with subtenon fluid 1.
The thickness of ocular coats is also increased in sympathetic ophthalmia 2, Vogt-Koyanagi-Harada syndrome, phthisis bulbi, small globe (nanophthalmos) 3, panophthalmitis, and other conditions.
Scleritis causes
Scleritis is typically associated with an autoimmune disorder or connective tissue disorders. Rheumatoid arthritis is the most common. It may also be infectious or surgically/trauma-induced. There is no known HLA association.
In other cases the cause is unknown.
Scleritis may be associated with:
- Different forms of inflammatory arthritis;
- Eye infection;
- Lupus;
- Certain connective tissue disease;
- Inflammatory bowel disease;
- Sjogren’s syndrome;
- Granulomatosis with polyangiitis;
- Scleroderma.
Scleritis may also result from trauma to the eye, or, rarely, may be caused by a fungus or a parasite.
Risk factors for scleritis
As scleritis is associated with systemic autoimmune diseases, it is more common in women. It usually occurs in the fourth to sixth decades of life. Men are more likely to have infectious scleritis than women. Patients with a history of pterygium surgery with adjunctive mitomycin C administration or beta irradiation are at higher risk of infectious scleritis due to defects in the overlying conjunctiva from calcific plaque formation and scleral necrosis. Bilateral scleritis is more often seen in patients with rheumatic disease. Two or more surgical procedures may be associated with the onset of surgically induced scleritis.
Scleritis general pathology
In scleritis, scleral edema and inflammation are present in all forms of disease. There is often a zonal granulomatous reaction that may be localized or diffuse. If localized, it may result in near total loss of scleral tissue in that region. Most commonly, the inflammation begins in one area and spreads circumferentially until the entire anterior segment is involved.
Inflammation of the sclera can involve a non-granulomatous process (lymphocytes, plasma cells, macrophages) or a granulomatous process (epitheliod cells, multinucleated giant cells) with or without associated scleral necrosis.
Scleritis pathophysiology
As there are different forms of scleritis, the pathophysiology is also varied. Scleritis associated with autoimmune disease is characterized by zonal necrosis of the sclera surrounded by granulomatous inflammation and vasculitis. Eosinophilic fibrinoid material may be found at the center of the granuloma. These eyes may exhibit vasculitis with fibrinoid necrosis and neutrophil invasion of the vessel wall.
There is an increase in inflammatory cells including T-cells of all types and macrophages. T-cells and macrophages tend to infiltrate the deep episcleral tissue with clusters of B-cells in perivascular areas. There may be cell-mediated immune response as there is increased HLA-DR expression as well as increased IL-2 receptor expression on the T-cells. Plasma cells may be involved in the production of matrix metalloproteinases and TNF-alpha. In idiopathic necrotizing scleritis, there may be small foci of scleral necrosis and mainly nongranulomatous inflammation with mainly mononuclear cells (lymphocytes, plasma cells and macrophages). Microabscesses may be found in addition to necrotizing inflammation in infectious scleritis.
Vasculitis is not prominent in non-necrotizing scleritis.
Scleritis signs and symptoms
Scleritis symptoms
Scleritis symptoms can include severe pain and tenderness of the eye. A severe pain that may involve the eye and orbit is usually present. This pain is characteristically dull and boring in nature and exacerbated by eye movements. Worsening of the pain during eye movement is due to the extraocular muscle insertions into the sclera. It may be worse at night and awakens the patient while sleeping. This pain may radiate to involve the ear, scalp, face and jaw.
Scleritis causes pain (often characterized as a deep, boring ache) severe enough to interfere with sleep and appetite. This pain can also extend to regions of the jaw, face, or head of the affected side. Inflammation (swelling) and redness of the white portion of the eye is also common. Blurred vision, tearing (lacrimation), and extreme sensitivity to light (photophobia) may occur. In some cases, partial or complete loss of vision is possible.
Hyperemic patches develop deep beneath the bulbar conjunctiva and are more violaceous than those of episcleritis or conjunctivitis. The palpebral conjunctiva is normal. The involved area may be focal (usually one quadrant of the globe) or involve the entire globe and may contain a hyperemic, edematous, raised nodule (nodular scleritis) or an avascular area (necrotizing scleritis). Posterior scleritis is less common and is less likely to cause red eye but more likely to cause blurred or decreased vision.
In severe cases of necrotizing scleritis, perforation of the globe and loss of the eye may result. Connective tissue disease occurs in 20% of patients with diffuse or nodular scleritis and in 50% of patients with necrotizing scleritis. Necrotizing scleritis in patients with connective tissue disease signals underlying systemic vasculitis.
Scleritis signs
Scleritis presents with a characteristic violet-bluish hue with scleral edema and dilatation. Other signs vary depending on the location of the scleritis and degree of involvement. In the anterior segment there may be associated keratitis with corneal infiltrates or thinning, uveitis, and trabeculitis. With posterior scleritis, there may be chorioretinal granulomas, retinal vasculitis, serous retinal detachment and optic nerve edema with or without cotton-wool spots.
Non-ocular signs are important in the evaluation of the many systemic associations of scleritis. Epistaxis, sinusitis and hemoptysis are present in granulomatosis with polyangiitis (formerly known as Wegener’s). Arthritis with skin nodules, pericarditis, and anemia are features of rheumatoid arthritis. Systemic lupus erythematous may present with a malar rash, photosensitivity, pleuritis, pericarditis and seizures. In addition to scleritis, myalgias, weight loss, fever, purpura, nephropathy and hypertension may be signs of polyarteritis nodosa.
Scleritis complications
Scleritis complications are frequent and include peripheral keratitis, uveitis, cataract and glaucoma. Central stromal keratitis may also occur in the absence of treatment. Sclerokeratitis in which peripheral cornea is opacified by fibrosis and lipid deposition with neighboring scleritis may occur particularly with herpes zoster scleritis. Sclerosing keratitis may present with crystalline deposits in the posterior corneal lamellae. Sclerokeratitis may move centrally gradually and thus opacify a large segment of the cornea. Vitritis (cells and debris in vitreous) and exudative detachments occur in posterior scleritis.
Scleritis diagnosis
It’s important to schedule a complete eye examination with an ophthalmologist the moment you experience any scleritis symptoms. Scleritis left untreated can lead to vision loss.
The onset of scleritis is gradual. Most patients develop severe boring or piercing eye pain over several days. Globe tenderness and redness may involve the whole eye or a small localized area.
Diagnosis of scleritis is made clinically and by slit-lamp examination. Smears or rarely biopsies are necessary to confirm infectious scleritis. B-scan ultrasonography and orbital magnetic resonance imaging (MRI) may be used for the detection of posterior scleritis. Ultrasonographic changes include scleral and choroidal thickening, scleral nodules, distended optic nerve sheath, fluid in Tenons capsule, or retinal detachment.
During your examination, your ophthalmologist will inspect the inside and outside of your eye using a slit lamp, which focuses an intense beam of light into the eye while he or she looks through a microscope.
As scleritis can be associated with other disorders, your ophthalmologist will want to discuss your overall health and may work with your primary care physician or other specialists to identify any underlying causes. Blood tests, magnetic resonance imaging (MRI) or other testing may be needed.
Laboratory test
As scleritis may occur in association with many systemic diseases, laboratory workup may be extensive. As mentioned earlier, the autoimmune connective tissue diseases of rheumatoid arthritis, lupus, sero-negative spondylarthropathies and vasculitides such as granulomatosis with polyangiitis and polyarteritis nodosa are most frequently seen. In addition to complete physical examination, laboratory studies should include assessment of blood pressure, renal function, and acute phase response. Laboratory tests include complete blood count (CBC) with differential, erythrocye sedimentation rate (ESR) or C-reactive protein (CRP), serum autoantibody screen (including antinuclear antibodies, anti-DNA antibodies, rheumatoid factor, antineutrophil cytoplasmic antibodies), urinalysis, syphilis serology, serum uric acid and sarcoidosis screen.
Scleritis prognosis
Visual loss is related to the severity of the scleritis. Patients with mild or moderate scleritis usually maintain excellent vision. Scleritis may be active for several months or years before going into long-term remission. Patients with necrotizing scleritis have a high incidence of visual loss and an increased mortality rate.
Of patients with scleritis, 14% lose significant visual acuity within 1 year, and 30% lose significant visual acuity within 3 year. Patients with necrotizing scleritis and underlying systemic vasculitis have a mortality rate of up to 50% in 10 year (mostly due to heart attack).
Scleritis treatment
Scleritis is a serious condition that can lead to loss of vision. Therefore, scleritis needs to be treated as soon as scleritis symptoms become evident.
Treatment varies depending on the type of scleritis. In most cases, corticosteroid pills and nonsteroid anti-inflammatory drugs (NSAIDs) can be used to reduce pain and inflammation. Eye solutions or antibiotics may be prescribed as well.
Nonsteroid anti-inflammatory drugs (NSAIDs)
Oral non-steroidal anti-inflammatory drugs (NSAIDs) are the first-line agent for mild-to-moderate scleritis. These consist of non-selective or selective cyclo-oxygenase inhibitors (COX inhibitors). Non-selective COX-inhibitors such as flurbiprofen, indomethacin and ibuprofen may be used. Indomethacin 50mg three times a day or 600mg of ibuprofen three times a day may be used. Patients using oral NSAIDS should be warned of the side effects of gastrointestinal (GI) side effects including gastric bleeding. Patients with renal compromise must be warned of renal toxicity. NSAIDS that are selective COX-2 inhibitors may have fewer gastrointestinal side effects but may have more cardiovascular side effects.
Corticosteroids
Rarely, NSAIDs are sufficient for mild cases of scleritis. Topical corticosteroids may reduce ocular inflammation but treatment is generally systemic. Usually a systemic corticosteroid (e.g., prednisone 1 to 2 mg/kg oral once/day for 7 days, then tapered off by day 10) is the initial therapy. If inflammation returns, a longer dose of oral corticosteroid, also starting at prednisone 1 to 2 mg/kg oral once/day or pulsed intravenous corticosteroid, such as methylprednisolone 1000 mg/day IV for 3 days, can be considered.
Immunomodulatory agents
If patients are unresponsive to or intolerant of systemic corticosteroids or have necrotizing scleritis and connective tissue disease, systemic immunosuppression with cyclophosphamide, methotrexate, mycophenolate mofetil, or biologic agents (eg, rituximab, adalimumab) is indicated but only in consultation with a rheumatologist.
Likewise, immunomodulatory agents should be considered in those who might otherwise be on chronic steroid use. Consultation with a rheumatologist or other internist is recommended. Patients with rheumatoid arthritis may be placed on methotrexate. Patients with granulomatosis with polyangiitis may require cyclosphosphamide or mycophenolate. Cyclosporine is nephrotoxic and thus may be used as adjunct therapy allowing for lower corticosteroid dosing. Mycophenolate mofetil may eliminate the need for corticosteroids. However, there is a risk of hematologic and hepatic toxicity.
More recently, tumor necrosis factor (TNF) alpha inhibitors such as infliximab have shown promise in the treatment of non-infectious scleritis refractory to other treatment. Treatment consists of repeated infusions as the treatment effect is short-lived. TNF-alpha inhibitors may also result in a drug-induced lupus-like syndrome as well as increased risk of lymphoproliferative disease. All patients on immunomodulatory therapy must be closely monitored for development of systemic complications with these medications.
Medical follow up
Adjustment of medications and dosages is based on the level of clinical response. Laboratory testing may be ordered regularly to follow the therapeutic levels of the medication, to monitor for systemic toxicity, or to determine treatment efficacy.
In more severe cases, surgery may be necessary to repair scleral tissues and prevent further loss of vision.
In cases where scleritis is caused by another disorder within the body, treatment of that disease is also necessary to control scleritis symptoms. Keep in mind that despite treatment, scleritis may recur. It’s important to see your ophthalmologist and other specialists regularly to properly treat scleritis.
Surgery
Scleral grafts may be indicated for threatened perforation. Clinical examination is usually sufficient for diagnosis. Formal biopsy may be performed to exclude a neoplastic or infective cause. However, one must be prepared to place a scleral reinforcement graft or other patch graft as severe thinning may result in the presentation of intraocular contents. Small corneal perforations may be treated with bandage contact lens or corneal glue until inflammation is adequately controlled, allowing for surgery.
Primary indications for surgical intervention include scleral perforation or the presence of excessive scleral thinning with a high risk of rupture. High-grade astigmatism caused by staphyloma formation may also be treated. Reinforcement of the sclera may be achieved with preserved donor sclera, periosteum or fascia lata. A lamellar or perforating keratoplasty may be necessary. Cataract surgery should only be performed when the scleritis has been in remission for 2-3 months. Small incision clear corneal surgery is preferred, and one must anticipate a return of inflammation in the postsurgical period.
Surgical follow up
Surgical biopsy of the sclera should be avoided in active disease, though if absolutely necessary, the surgeon should be prepared to bolster the affected tissue with either fresh or banked tissue (i.e., preserved pericardium, banked sclera or fascia lata). Areas with imminent scleral perforation warrant surgical intervention, though the majority of patients often have scleral thinning or staphyloma formation that do not require scleral reinforcement.
- Kumawat B, Tripathy K, Venkatesh P, et al. Lacrimal abscess mimicking a choroidal mass: an ultrawide field evaluation. Can J Ophthalmol J Can Ophtalmol 2016;51:e92-94. doi:10.1016/j.jcjo.2016.01.010[↩]
- Tripathy K, Mittal K, Chawla R. Sympathetic ophthalmia following a conjunctival flap procedure for corneal perforation. BMJ Case Rep 2016;2016. doi:10.1136/bcr-2016-214344[↩]
- Venkatesh P, Chawla R, Tripathy K, et al. Scleral resection in chronic central serous chorioretinopathy complicated by exudative retinal detachment. Eye Vis Lond Engl 2016;3:23. doi:10.1186/s40662-016-0055-5[↩]



{kind=link}