Contents
Spinal cord tumor
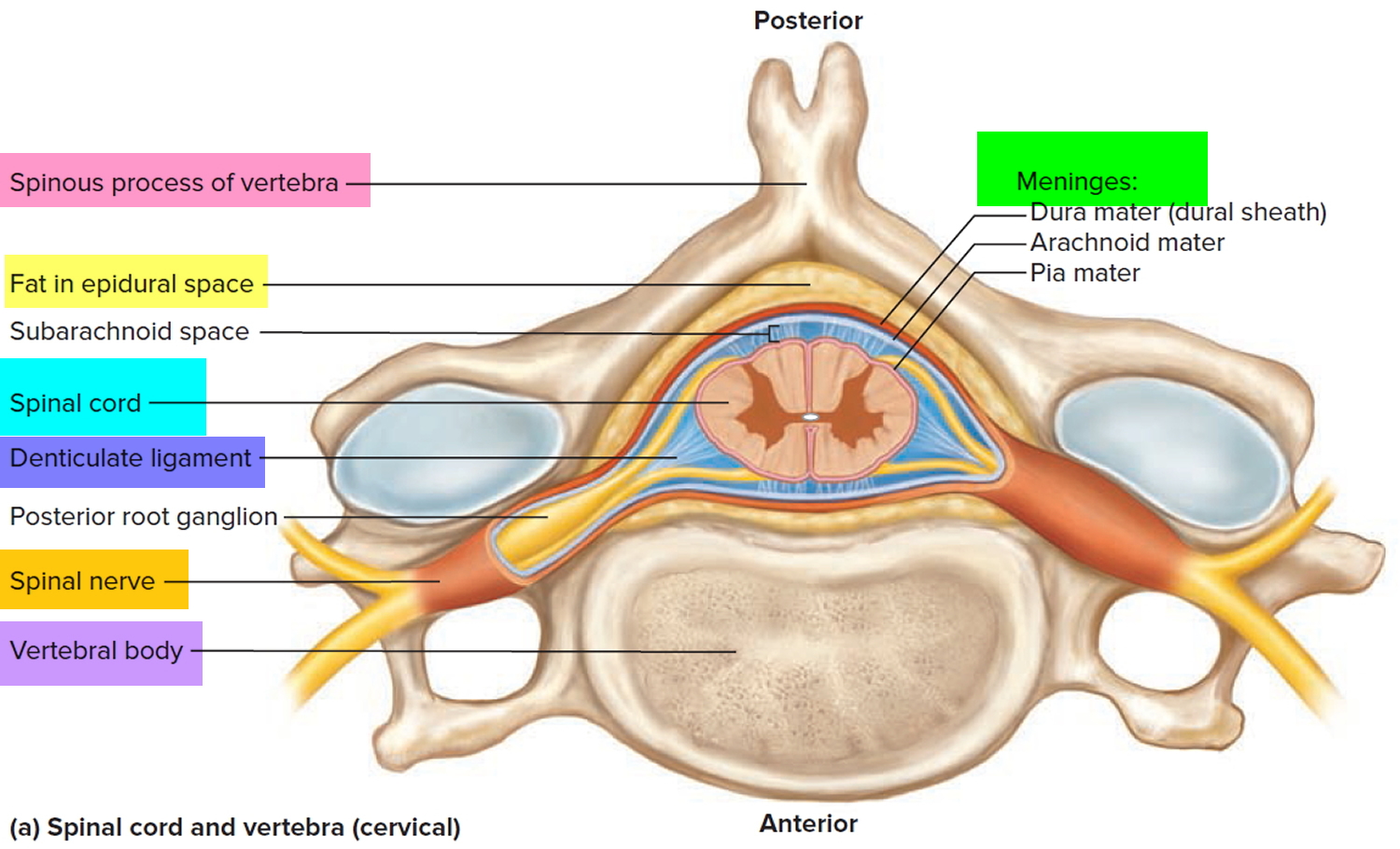
Tumors of the spinal cord are abnormal growths of tissue that develops within your spinal canal or within the bones of your spine. It may be cancerous or noncancerous 1. The spinal cord and the brain are the primary components of the central nervous system (CNS). Benign tumors are noncancerous, and malignant tumors are cancerous. The spinal cord is housed within rigid, bony quarters (i.e., spinal column), so any abnormal growth, whether benign or malignant, can place pressure on sensitive tissues and impair function (see Figure 2).
Figure 1. Spinal cord
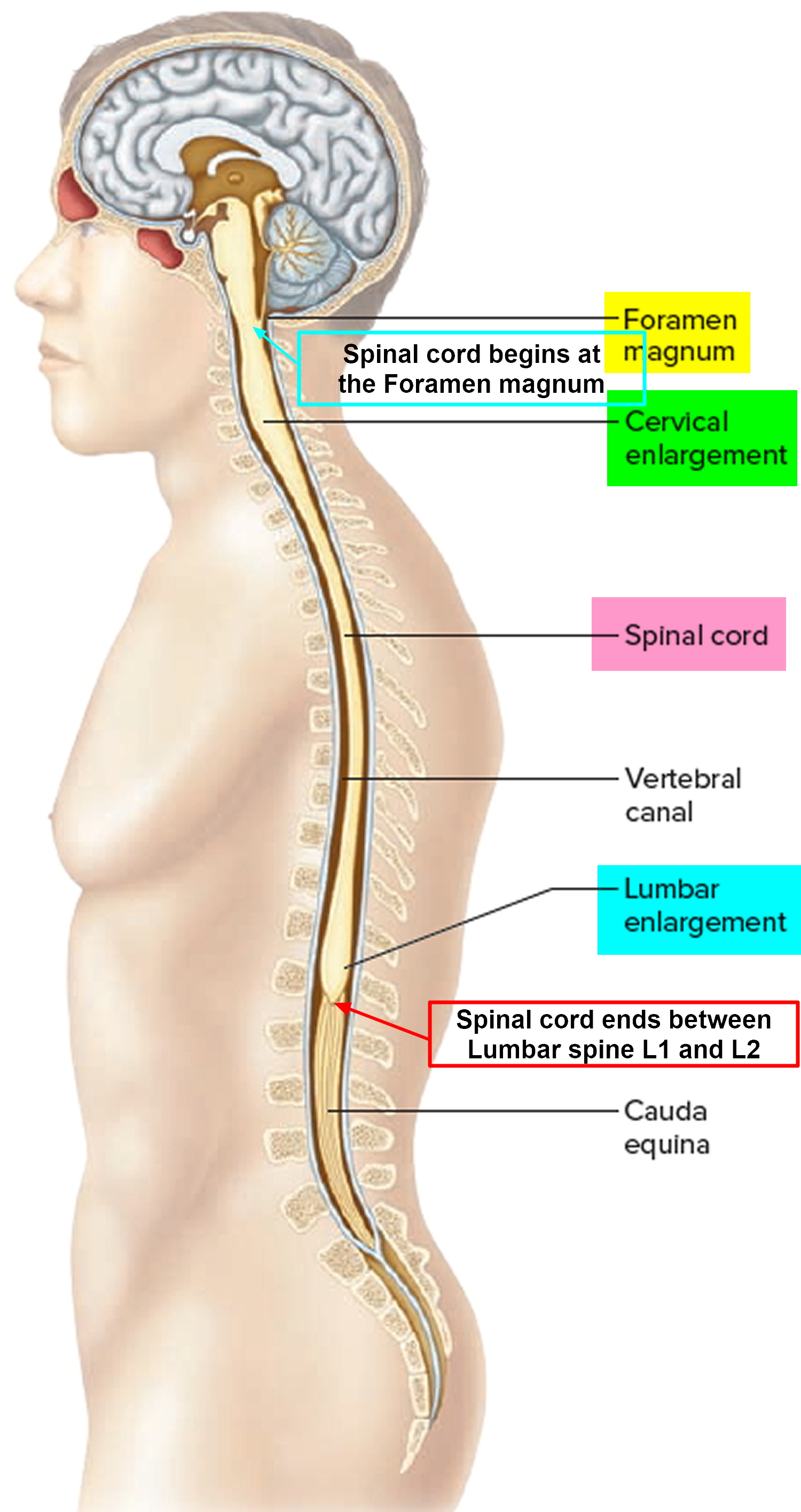
Figure 2. Spinal cord anatomy
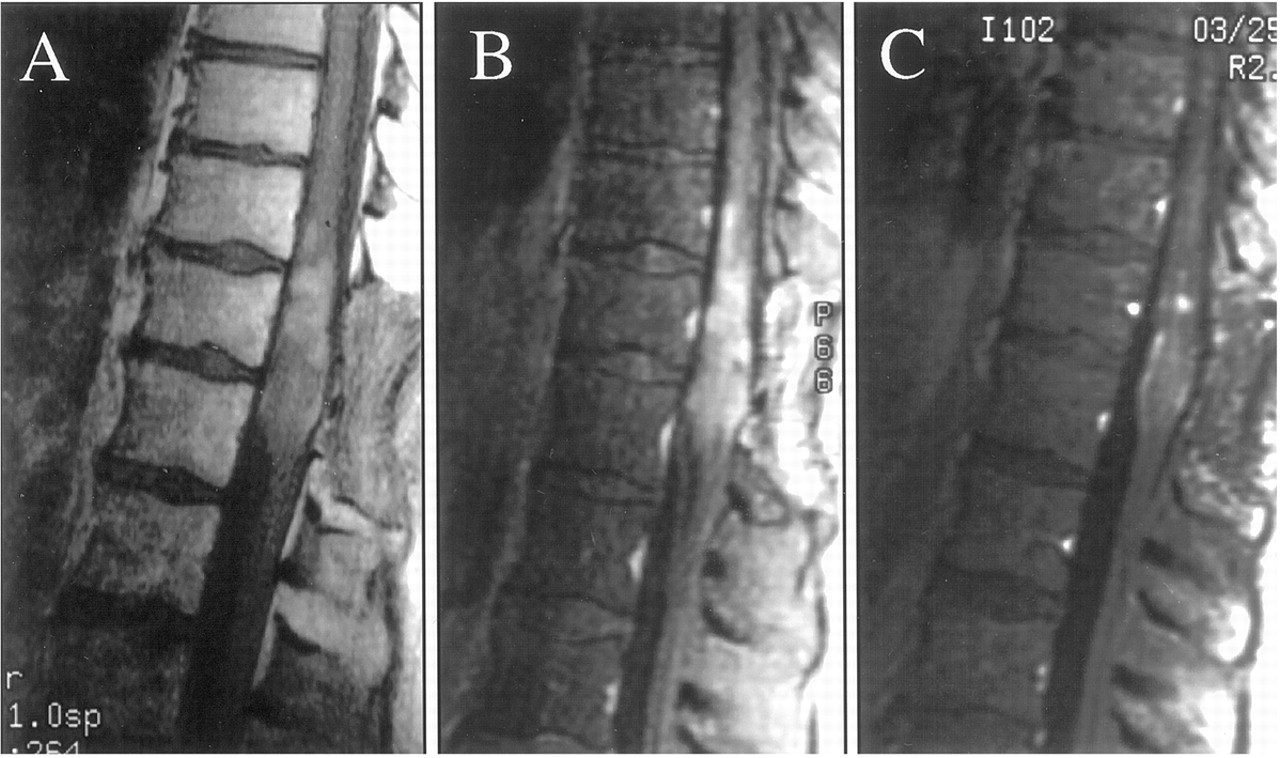
Tumors that affect the bones of the spine (vertebrae) are known as vertebral tumors.
Tumors that begin within the spinal cord itself are called spinal cord tumors.
Tumors that originate in the spinal cord are called primary tumors. Most primary tumors are caused by out-of-control growth among cells that surround and support neuron, specific genetic disease (such as neurofibromatosis type 1 and tuberous sclerosis), or from exposure to radiation or cancer-causing chemicals.
Metastatic, or secondary, tumors in the spinal cord are caused by cancer cells that break away from a primary tumor located in another region of the body. Tumors can place pressure on sensitive tissues and impair function.
Spinal cord tumor symptoms and signs
Depending on the location and type of spinal tumor, different signs and symptoms can develop, especially as a tumor grows and affects your spinal cord, surrounding nerves or blood vessels. Signs and symptoms of tumors affecting the spinal cord may include:
- Back pain, sometimes radiating to other parts of your body
- Loss of sensation, especially in your arms or legs
- Difficulty walking, sometimes leading to falls
- Decreased sensitivity to pain, heat and cold
- Loss of bowel or bladder function
- Muscle weakness that may occur in varying degrees and in different parts of your body, depending on which nerves or part of the spinal cord is compressed
Back pain is a common early symptom of both noncancerous and cancerous spinal tumors. Pain may also spread beyond your back to your hips, legs, feet or arms and may become more severe over time in spite of treatment.
Spinal tumors progress at different rates. In general, cancerous spinal tumors grow more quickly, and noncancerous spinal tumors tend to develop very slowly.
Causes of spinal cord tumor
It’s not clear why most spinal tumors develop. Experts suspect that defective genes play a role. But it’s usually not known whether such genetic defects are inherited, occur spontaneously or are caused by something in the environment, such as exposure to certain chemicals. In some cases, however, spinal cord tumors are linked to known inherited syndromes, such as neurofibromatosis 2 and von Hippel-Lindau disease.
Risk factors of spinal cord tumor
Spinal cord tumors are more common in people who have:
- Neurofibromatosis 2. In this hereditary disorder, noncancerous tumors develop on or near the nerves related to hearing, which may lead to progressive hearing loss in one or both ears. Some people with neurofibromatosis 2 also develop spinal canal tumors, frequently multiple and of several different types.
- Von Hippel-Lindau disease. This rare, multisystem disorder is associated with noncancerous blood vessel tumors (hemangioblastomas) in the brain, retina and spinal cord and with other types of tumors in the kidneys or adrenal glands.
- A prior history of cancer. Any type of cancer can travel to the spine, but the cancers that may be more likely to affect the spine include breast, lung, prostate and multiple myeloma.
Complications of Spinal cord tumors
Both noncancerous and cancerous spinal tumors can compress the spinal cord and nerves, leading to a loss of movement or sensation at and below the level of the tumor and sometimes to changes in bowel and bladder function. Nerve damage may be permanent.
However, if a spinal tumor is caught early and treated aggressively, it may be possible to prevent further loss of function and, with aggressive rehabilitation, regain nerve function. Depending on its location, a tumor that compresses the spinal cord itself may be life-threatening.
Diagnosis of Spinal cord tumors
Spinal tumors sometimes may be overlooked because they’re not common and their symptoms resemble those of more common conditions. For that reason, it’s especially important that your doctor know your complete medical history and perform both general physical and neurological exams.
If your doctor suspects a spinal tumor, one or more of the following tests can help confirm the diagnosis and pinpoint the tumor’s location:
Spinal magnetic resonance imaging (MRI). MRI uses a powerful magnet and radio waves to produce images of your spine. MRI accurately shows the spinal cord and nerves and yields better pictures of bone tumors than computerized tomography (CT) scans do. A contrast agent that helps to highlight certain tissues and structures may be injected into a vein in your hand or forearm during the test.
Some people may feel claustrophobic inside the MRI scanner or find the loud thumping sound it makes disturbing. But you’re usually given earplugs to help with the noise, and some scanners are equipped with televisions or headphones. If you’re very anxious, ask about a mild sedative to help calm you. In certain situations, a general anesthetic may be necessary.
Computerized tomography (CT). This test uses a narrow beam of radiation to produce detailed images of your spine. Sometimes it may be combined with an injected contrast dye to make abnormal changes in the spinal canal or spinal cord easier to see.
Biopsy. The only way to determine the precise nature of a spinal or vertebral tumor is to examine a small tissue sample (biopsy) under a microscope. The biopsy results will help determine treatment options.
How the sample is obtained depends on your overall health and the location of the tumor. Doctors may use a fine needle to withdraw a small amount of tissue, or the sample may be obtained during surgery. These procedures can be associated with significant risks and should only be performed at a center that specializes in spine tumors.
Treatment of Spinal cord tumors
Ideally, the goal of spinal tumor treatment is to eliminate the tumor completely, but this goal may be complicated by the risk of permanent damage to the spinal cord and surrounding nerves. Doctors also must take into account your age and overall health. The type of tumor and whether it arises from the structures of the spine or spinal canal or has spread to your spine from elsewhere in your body also must be considered in determining a treatment plan.
Treatment options for most spinal tumors include:
Monitoring. Some spinal tumors may be discovered before they cause symptoms — often when you’re being evaluated for another condition. If small tumors are noncancerous and aren’t growing or pressing on surrounding tissues, watching them carefully may be all that’s needed.
This is especially true in older adults for whom surgery or radiation therapy may pose special risks. During observation, your doctor will likely recommend periodic CT or MRI scans to monitor the tumor.
Surgery. This is often the treatment of choice for tumors that can be removed with an acceptable risk of spinal cord or nerve injury damage.
Newer techniques and instruments allow neurosurgeons to reach tumors that were once considered inaccessible. The high-powered microscopes used in microsurgery make it easier to distinguish tumor from healthy tissue.
Doctors also can monitor the function of the spinal cord and other important nerves during surgery, thus minimizing the chance of their being injured. In some instances, very high frequency sound waves might be used during surgery to break up tumors and remove the fragments.
Unfortunately, even with the latest technological advances in surgery, not all tumors can be removed completely. When the tumor can’t be removed completely, surgery may be followed by radiation therapy or chemotherapy or both.
Recovery from spinal surgery may take weeks or longer, depending on the procedure. You may experience a temporary loss of sensation or other complications, including bleeding and damage to nerve tissue.
Radiation therapy. This may be used to eliminate the remnants of tumors that remain after surgery, to treat inoperable tumors or to treat those tumors where surgery is too risky. It may also be the first line therapy for metastatic tumors (those that travel to the spine region from other cancers of the body). Radiation may also be used to relieve pain or when surgery poses too great a risk.
Medications may help ease some of the side effects of radiation, such as nausea and vomiting.
Depending on the type of tumor you have, your radiation therapy team may be able to modify your treatment to help prevent damage to surrounding tissue from the radiation and improve the treatment’s effectiveness. Modifications may range from simply changing the dosage of radiation to using sophisticated techniques such as 3-D conformal radiation therapy.
A specialized type of radiation therapy called proton beam therapy may be used to treat some vertebral tumors, such as chordomas and chondrosarcomas, and some childhood cancers when spinal radiation is required.
Stereotactic radiosurgery. This method of delivering radiation is capable of delivering a high dose of precisely targeted radiation. In stereotactic radiosurgery, doctors use computers to focus radiation beams on tumors with pinpoint accuracy and from multiple angles.
There are different types of technology used in radiosurgery to stereotactically deliver radiation to treat spinal tumors, such as a Gamma Knife machine.
Stereotactic radiosurgery has certain limits on the size and specific type of the tumors that can be treated, but where appropriate, it has proved quite effective, and growing research supports its use for the treatment of spinal and vertebral tumors. However, further study is needed to determine the best technique, radiation dose and schedule for stereotactic radiosurgery in the treatment of spinal tumors.
Chemotherapy. A standard treatment for many types of cancer, chemotherapy uses medications to destroy cancer cells or stop them from growing. Your doctor can determine whether chemotherapy might be beneficial for you, either alone or in combination with radiation therapy.
Side effects may include fatigue, nausea, vomiting, increased risk of infection and hair loss.
Other drugs. Because surgery and radiation therapy as well as tumors themselves can cause inflammation inside the spinal cord, doctors sometimes prescribe corticosteroids to reduce the swelling, either after surgery or during radiation treatments. Although corticosteroids reduce inflammation, they are usually used only for short periods to avoid serious side effects as muscle weakness, osteoporosis, high blood pressure, diabetes and an increased susceptibility to infection.
Acupuncture. Although there aren’t any alternative medicines that have been proved to cure cancer, acupuncture may help relieve some of your symptoms. During acupuncture treatment, a practitioner inserts tiny needles into your skin at precise points. Research shows that acupuncture may be helpful in relieving nausea and vomiting. Acupuncture may also help relieve certain types of pain in people with cancer.
Be sure to discuss the risks and benefits of complementary or alternative treatment that you’re thinking of trying with your doctor. Some treatments, such as herbal remedies, could interfere with medicines you’re taking.
Spinal cord tumor types
There are two main types of spinal cord tumors that may affect the spinal cord 2:
- Intramedullary tumors begin in the cells within the spinal cord itself, such as astrocytomas or ependymomas.
- Extramedullary tumors (outside the spinal cord tissue) develop within the supporting network of cells around the spinal cord. Although they don’t begin within the spinal cord itself, these types of tumors may affect spinal cord function by causing spinal cord compression and other problems. Examples of extramedullary tumors that can affect the spinal cord include schwannomas, meningiomas and neurofibromas.
Tumors from other parts of the body can spread (metastasize) to the vertebrae, the supporting network around the spinal cord or, in rare cases, the spinal cord itself.
Extramedullary tumors
These tumors may be intradural or extradural. Most intradural tumors are benign, usually meningiomas and neurofibromas, which are the most common primary spinal tumors. Most extradural tumors are metastatic, usually from carcinomas of the lungs, breasts, prostate, kidneys, or thyroid or from lymphoma (eg, Hodgkin lymphoma, lymphosarcoma, reticulum cell sarcoma).
Intradural and extradural tumors cause neurologic damage by compressing the spinal cord or nerve roots. Most extradural tumors invade and destroy bone before compressing the cord.
Symptoms and Signs of Extramedullary tumors
Pain is an early symptom, especially for extradural tumors. It is progressive, unrelated to activity, and worsened by recumbency. Pain may occur in the back, radiate along the sensory distribution of a particular dermatome (radicular pain), or both. Usually, neurologic deficits referable to the spinal cord eventually develop. Common examples are spastic weakness, incontinence, and dysfunction of some or all of the sensory tracts at a particular segment of the spinal cord and below. Deficits are usually bilateral.
Many patients with extramedullary tumors present with pain, but some present with sensory deficits of the distal lower extremities, segmental neurologic deficits, symptoms of spinal cord compression, or a combination. Symptoms of spinal cord compression can worsen rapidly and result in paraplegia and loss of bowel and bladder control. Symptoms of nerve root compression are also common; they include pain and paresthesias followed by sensory loss, muscular weakness, and, if compression is chronic, muscle wasting, which occurs along the distribution of the affected roots.
Diagnosis of extramedullary tumors
- MRI
Patients with segmental neurologic deficits or suspected spinal cord compression require emergency diagnosis and treatment.
The following suggest spinal tumors:
- Progressive, unexplained, or nocturnal back or radicular pain
- Segmental neurologic deficits
- Unexplained neurologic deficits referable to the spinal cord or nerve roots
- Unexplained back pain in patients with primary tumors of the lungs, breasts, prostate, kidneys, or thyroid or with lymphoma
Diagnosis is by MRI of the affected area of the spinal cord. CT with myelography is an alternative but is less accurate.
If MRI does not show a spinal cord tumor, clinicians consider other spinal masses (eg, abscesses, arteriovenous malformations) and paravertebral tumors. Spinal x-rays, taken for other reasons, may show bone destruction, widening of the vertebral pedicles, or distortion of paraspinal tissues, especially if the tumor is metastatic.
Treatment of extramedullary tumors
- Corticosteroids
- Excision, radiation therapy, or both
For patients with neurologic deficits, corticosteroids (eg, dexamethasone 100 mg IV, then 10 mg po qid) are begun immediately to reduce spinal cord edema and preserve function. Tumors compressing the spinal cord are treated as soon as possible.
Some well-localized primary spinal cord tumors can be excised surgically. Deficits resolve in about half of these patients. If tumors cannot be surgically excised, radiation therapy is used, with or without surgical decompression. Compressive metastatic extradural tumors are usually surgically excised from the vertebral body, then treated with radiation therapy. Noncompressive metastatic extradural tumors may be treated with radiation therapy alone but may require excision if radiation therapy is ineffective.
Intramedullary tumors
The most common are gliomas (eg, ependymomas, low-grade astrocytomas). Intramedullary tumors infiltrate and destroy spinal cord parenchyma; they may extend over multiple spinal cord segments or result in a syrinx (fluid-filled cavity that develops in the spinal cord).
Astrocytoma
Astrocytoma is a type of cancer that can form in the brain or spinal cord. Astrocytoma begins in cells called astrocytes. Astrocytes are the most abundant glial cells in the CNS and constitute over 50% of the tissue in some areas of the brain and spinal cord. Glial cells (neuroglia) protect the neurons and help them function. The word glia, which means “glue,” implies one of their roles—to bind neurons together and provide a supportive framework for the nervous tissue. In the fetus, they form a scaffold that guides young migrating neurons to their destinations. Wherever a mature neuron is not in synaptic contact with another cell, it is covered with glial cells. This prevents neurons from contacting each other except at points specialized for signal transmission, thus giving precision to their conduction pathways.They cover the entire spinal cord surface and most nonsynaptic regions of the neurons in the gray matter. They are named for their many-branched, somewhat starlike shape. Astrocytes have the most diverse functions of any glia: they form a supportive framework for the nervous tissue.
Figure 1. Astrocytes
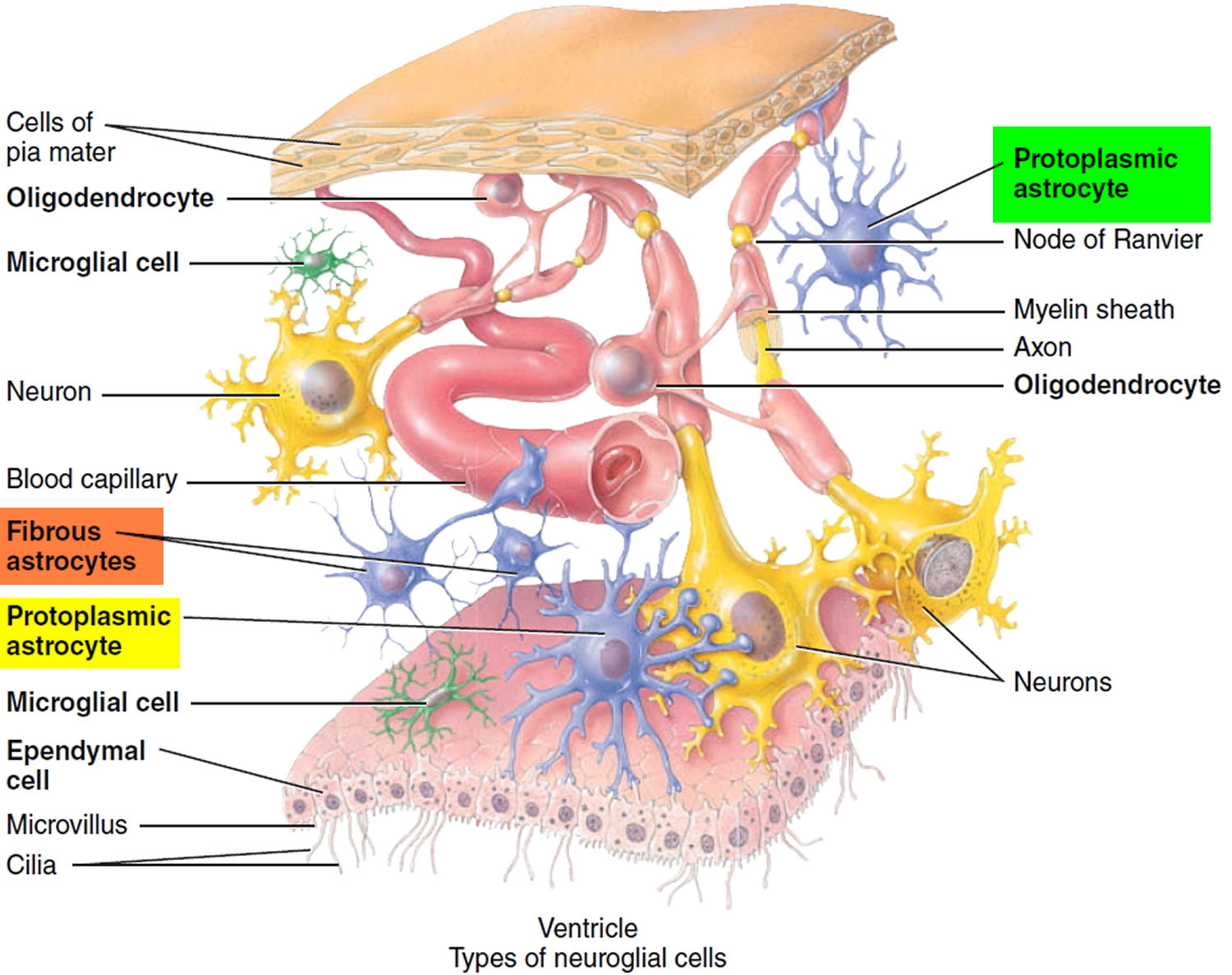
Astrocytoma signs and symptoms depend on the location of your tumor. Astrocytomas that occur in the brain can cause seizures, headaches and nausea. Astrocytomas that occur in the spinal cord can cause weakness and disability in the area affected by the growing tumor.
Astrocytoma can be a slow-growing tumor, or it can be an aggressive cancer that grows quickly. The aggressiveness (grade) of your astrocytoma determines your prognosis and treatment options.
Diagnosis of Astrocytoma
Tests and procedures used to diagnose astrocytoma include:
Neurological exam. During a neurological exam, your doctor will ask you about your signs and symptoms. He or she may check your vision, hearing, balance, coordination, strength and reflexes. Problems in one or more of these areas may provide clues about the part of your brain that could be affected by a brain tumor.
Imaging tests. Imaging tests can help your doctor determine the location and size of your brain tumor. MRI is often used to diagnose brain tumors, and it may be used along with specialized MRI imaging, such as functional MRI, perfusion MRI and magnetic resonance spectroscopy.
Other imaging tests may include CT and positron emission tomography (PET).
Removing a sample of tissue for testing (biopsy). A biopsy can be done with a needle before surgery or during surgery to remove your astrocytoma, depending on your particular situation and the location of your tumor. The sample of suspicious tissue is analyzed in a laboratory to determine the types of cells and their level of aggressiveness.
Specialized tests of the tumor cells can tell your doctor the types of mutations the cells have acquired. This gives your doctor clues about your prognosis and may guide your treatment options.
Treatment for Astrocytoma
Astrocytoma treatments include:
Surgery to remove the astrocytoma. Your brain surgeon (neursurgeon) will work to remove as much of the astrocytoma as possible. The goal is to remove all of the cancer, but sometimes the astrocytoma is located near sensitive brain tissue that makes that too risky. Even removing some of the cancer may reduce your signs and symptoms.
For some people, surgery may be the only treatment needed. For others, additional treatments may be recommended to kill any cancer cells that might remain and reduce the risk that the cancer will return.
Radiation therapy. Radiation therapy uses high-energy beams, such as X-rays or protons, to kill cancer cells. During radiation therapy, you lie on a table while a machine moves around you, directing beams to precise points in your brain.
Radiation therapy might be recommended after surgery if your cancer wasn’t removed completely or if there’s an increased risk your cancer will return. Radiation is often combined with chemotherapy for aggressive cancers. For people who can’t undergo surgery, radiation therapy and chemotherapy may be used as a primary treatment.
Chemotherapy. Chemotherapy uses drugs to kill cancer cells. Chemotherapy drugs can be taken in pill form or through a vein in your arm. In certain situations, a circular wafer of chemotherapy medicine can be placed in your brain after surgery where it slowly dissolves and releases the medication.
Chemotherapy is often used after surgery to kill any cancer cells that might remain. It can be combined with radiation therapy for aggressive cancers.
Clinical trials. Clinical trials are studies of new treatments. These studies give you a chance to try the latest treatment options, but the risk of side effects may not be known. Ask your doctor whether you might be eligible to participate in a clinical trial.
Supportive (palliative) care. Palliative care is specialized medical care that focuses on providing relief from pain and other symptoms of a serious illness. Palliative care specialists work with you, your family and your other doctors to provide an extra layer of support that complements your ongoing care. Palliative care can be used while undergoing other aggressive treatments, such as surgery, chemotherapy or radiation therapy.
Ependymomas
Ependymomas arise from the ependymal cells that line the ventricles of the brain and the center of the spinal cord 3. Ependymal cells secrete and circulate cerebrospinal fluid.
Ependymomas are soft, grayish, or red tumors which may contain cysts or mineral calcifications.
Ependymomas are relatively rare tumors in adults, accounting for 2-3% of primary brain tumors 3. However, they are the sixth most common brain tumor in children 3. About 30% of pediatric ependymomas are diagnosed in children younger than 3 years of age.
These tumors are divided into four major types:
- Subependymomas (grade I): Typically slow-growing tumors.
- Myxopapillary ependymomas (grade I): Typically slow-growing tumors.
- Ependymomas (grade II): The most common of the ependymal tumors. This type can be further divided into the following subtypes, including cellular ependymomas, papillary ependymomas, clear cell ependymomas, and tancytic ependymomas.
- Anaplastic ependymomas (grade III): Typically faster-growing tumors.
Location of ependymomas
The various types of ependymomas appear in different locations within the brain and spinal cord column. Subependymomas usually appear near a ventricle. Myxopapillary ependymomas tend to occur in the lower part of the spinal column. Ependymomas are usually located along, within, or next to the ventricular system. Anaplastic ependymomas are most commonly found in the brain in adults and in the lower back part of the skull (posterior fossa) in children. They are rarely found in the spinal cord.
Symptoms of ependymomas
Symptoms of an ependymoma are related to the location and size of the tumor. In babies, increased head size may be one of the first symptoms. Irritability, sleeplessness, and vomiting may develop as the tumor grows. In older children and adults, nausea, vomiting, and headache are the most common symptoms.
Headaches are a common symptom, and are generally worse in the morning. A tumor near the brainstem may cause one or both eyes to cross, balance problems or trouble walking.
Neck pain may result from a tumor growing near the brainstem or the upper part of the spinal cord.
Spinal cord tumors often cause leg or back pain which can be severe enough to awaken the person from sleep. Tingling sensations, numbness, or weakness in the arms or legs may also occur. Difficulty with bladder or bowel control may be caused by an ependymoma in the lower part of the spine.
Cause of ependymomas
Like many tumor types, the exact cause of ependymomas is not known.
Ependymomas diagnosis
Magnetic Resonance Imaging (MRI) scans and/or Computerized Axial Tomography (CT) scans are required for patients suspected of having a brain tumor. The MRI, which uses radio waves and a magnetic field, provides details about the location of the tumor and which parts of the brain or spinal cord are involved prior to surgery. CT scanners are sophisticated x-ray devices connected to computers. CT scans are useful for getting a quick view of the brain to see if there is a tumor and whether it has caused hydrocephalus (the buildup of cerebrospinal fluid in the brain). However, only microscopic examination of a sample of tissue obtained during surgery or biopsy confirms the exact diagnosis.
About 10–15% of ependymomas spread, or metastasize, through the cerebrospinal fluid. The tumor cells then may grow independently, in or along the spinal cord, or rarely, in other places in the brain. Infratentorial tumors are more likely to spread to the spine than supratentorial tumors.
Ependymomas almost never spread outside of the central nervous system (brain and spinal cord). A spinal MRI scan and lumbar puncture (spinal tap) will be performed to determine whether the tumor has spread to the spine and/or spinal fluid. The fluid obtained during the spinal tap will be tested for the presence of tumor cells. Your doctor will decide the appropriate time to perform these tests. The results are used to guide treatment.
Treatment for ependymomas
The first step of ependymoma treatment is to remove as much of the tumor as possible. Studies clearly show that patients whose tumor can be “grossly” removed (removing all tumor that can be seen) have the best chance of long-term survival. The amount of tumor that can be removed, however, depends on the location of the tumor. High-powered microscopes in the operating room help the surgeon see tumor located in and around the ventricles or the brainstem. Removal of all visible tumor is not always possible, especially if the tumor is attached to the brainstem or involves other important areas of the brain.
In children, ependymomas often fill the fourth ventricle and extend through its floor into the brainstem, or out to the side of the lower brainstem where the nerves that affect swallowing are located, making safe removal of those portions of tumor difficult.
An MRI of the brain should be done within a day or two of surgery to determine how much visible tumor remains, if any.
Radiation is usually recommended for older children and adults following surgery, in some cases even if the tumor was completely removed.
There are different methods of administering radiation. External beam radiation is given five days a week for six weeks. Conformal beam radiation therapy is a type of external beam radiation that contours the radiation beams to the shape of the tumor. Stereotactic radiosurgery is a way of giving a single or a few high doses of precisely focused radiation to the tumor. This is often used for ependymomas that grow back after conventional radiation. Your doctor will decide which form of radiation is best for you. Because of the long-term side effects of radiating young children, chemotherapy may be used to delay radiation therapy in the very young child. If the tumor grows despite the chemotherapy, radiation therapy may be considered.
The role of chemotherapy in treating newly diagnosed ependymomas is not clear. However, it may be used to treat tumors that have grown back after radiation therapy, or to delay radiation in infants and very young children. It is not clear which chemotherapy drugs are the most effective against ependymomas. Drugs such as cisplatin and carboplatin may cause shrinkage in about half of ependymomas, although not usually for a long time. Either standard chemotherapy, or experimental chemotherapy as part of a clinical trial, are often used for patients whose tumors regrow after radiation.
A significant number of children, particularly those who are very young at the time of treatment, experience some degree of decreased intellect and learning problems following radiation to large areas of the brain. The severity of the learning problems is correlated with the location and amount of brain radiated and inversely related to the age of the child. Radiation to just the back part of the brain does not cause as many problems as does radiation to the upper part of the brain. When the upper part of the brain is radiated, teachers and parents often report significant problems with reading, math and short-term memory. The role of special education programs to help these children develop their strengths and identify their weaknesses is of prime importance.
Older children and adults tend to have fewer problems. Poor growth may be a consequence of radiation therapy damage to the hypothalamus or pituitary gland which produce several important hormones. Replacement of these hormones is often necessary, under the supervision of a pediatric endocrinologist. Radiation to the spine at a young age may also cause short stature. Weakening of the muscles next to the spine is also possible. The long-term effects of chemotherapy in children are still being studied. Platinum-based drugs often cause hearing loss, as can radiation therapy when delivered near the ears. Both radiation and chemotherapy can increase the chance of developing a second cancer or brain tumor. Some chemotherapy can cause infertility.
The risk of incurring long term side effects due to treatment must be weighed against the outcome if these therapies are not administered. Your doctor can help you look at these concerns and understand the risks based on your child’s particular treatment plan, as well as help you balance the benefits of therapy against the potential risks.
Recurrence of ependymomas
The extent of tumor removal continues to be the strongest factor influencing recurrence and survival. Age at diagnosis, amount of tumor remaining after surgery, whether the tumor has spread, and the therapy given can all influence outcome. Researchers are also studying the biologic features of these tumors to determine which may be useful in predicting prognosis. For patients whose tumors recur after initial treatment, re-operation when possible, followed by additional therapy, can be helpful. Newer investigational treatments include various forms of chemotherapy, and re-irradiation (possibly with radiosurgery) to the areas of tumor recurrence.
Chordoma
A chordoma is a rare cancerous (malignant) primary bone tumor that usually occurs along the spine or where the skull sits atop the spine (skull base) 4. This type of tumor most often occurs at the skull base, spine or bottom of the spine (sacrum). It forms from small remnants of a coil of cells in the embryo that develops into the disks of the spinal column.
A chordoma is found in men twice as often as in women, with most tumors occurring between ages 50 and 70, although can be seen at any age.
Though commonly slow growing, a chordoma is a difficult tumor to treat because it’s near the spinal cord or other critical structures, such as the carotid artery and brain tissue 4.
Diagnosis of chordoma
Tests and procedures used to diagnose a chordoma include:
Removing a sample of cells for laboratory testing (biopsy). A biopsy is a procedure to remove a sample of suspicious cells for laboratory testing. In the lab, specially trained doctors called pathologists examine the cells under microscopes to determine whether cancer cells are present.
It is important to seek the opinion of a doctor experienced with diagnosing and treating chordomas. The person performing your biopsy will ideally consult with experienced surgeons to plan the procedure so that it can be done in a way that won’t interfere with a later operation.
Obtaining more detailed imaging. Your doctor may recommend imaging tests to help visualize your chordoma and determine whether it has spread beyond the spine or skull base. Tests may include an MRI or CT scan.
After you receive a diagnosis of chordoma, your doctor will develop a treatment plan tailored to your needs in consultation with an expert in cancer and radiation therapy (radiation oncologist) and a surgical oncologist.
Treatment for chordoma
Chordoma treatment depends on the size and its location as well as whether it has invaded nerves or other tissue. Options may include surgery, radiation therapy — including proton therapy — stereotactic radiosurgery, chemotherapy and targeted therapies. Tumors may recur after treatment.
If you decide to undergo chordoma surgery, ask about your doctor’s experience with complex cranial or spinal surgery. This type of surgery results in fewer complications when done by highly experienced, multidisciplinary surgical teams with expertise in chordomas.
Treatment for a sacral spine tumor
Surgery. The goal of surgery for a sacral spine tumor is usually to remove the entire tumor in one piece, if possible. Surgery may be difficult to perform because the tumor is near critical structures in the spinal cord.
Radiation therapy. Radiation therapy uses high-energy beams, such as X-rays or protons, to kill cancer cells. During radiation therapy, you lie on a table as a machine moves around you, directing the radiation beams to precise points on your body. Radiation therapy may be used before or after surgery or if surgery isn’t an option.
Treatment with newer types of radiation treatment, such as proton therapy, allows doctors to use higher doses of radiation while protecting healthy tissue, which may be more effective in treating a chordoma.
Radiosurgery. Stereotactic radiosurgery uses multiple beams of radiation to kill the cancer cells in a very small area. Each beam of radiation isn’t very powerful, but the point where all the beams meet — at the chordoma — receives a large dose of radiation to kill the cancer cells.
Other treatments. Sometimes chemotherapy and targeted drug therapy are used to treat a chordoma.
Treatment for skull base tumor
Treatment for a skull base tumor usually involves surgery followed by radiation therapy. The goal of surgery is to remove as much of the tumor as possible without harming nearby healthy tissue or causing undue new problems. Complete resection may not be an option if it’s near critical structures, such as the carotid artery. Endoscopic surgery as well as traditional approaches may be needed or used together to remove as much of the tumor as possible at the lowest risk possible.
Surgery is usually followed by radiation therapy to kill any remaining cancer cells and help prevent recurrence.
Neurofibroma
A neurofibroma is a type of nerve tumor that forms soft bumps on or under the skin 5. A neurofibroma can develop within a major or minor nerve anywhere in the body. This common type of benign nerve tumor tends to form more centrally within the nerve. Sometimes it arises from several nerve bundles (plexiform neurofibroma).
Symptoms are often mild or absent. If the tumor presses against nerves or grows within them, you may experience pain or numbness in the affected area.
A neurofibroma is usually noncancerous (benign) 5. Rarely, it can become cancerous (malignant) 5.
Diagnosis of neurofibroma
A neurofibroma can arise with no known cause, or it may appear in people with a genetic condition called neurofibromatosis type 1. These tumors are most often found in people ages 20 to 40 years.
Your doctor will diagnose a neurofibroma based on a physical examination, a discussion with you about your medical history, or the results of an imaging test such as a CT or MRI scan. These imaging studies can help pinpoint where the tumor is, find very small tumors, and identify what tissues are affected or nearby. Your doctor may have you undergo a PET scan to get an indication of whether it is benign. You may also have a biopsy done by a radiologist before surgery to diagnose the mass as being a neurofibroma.
Treatment for neurofibroma
Neurofibroma treatment usually isn’t needed for a single, small — less than an inch (about 2 centimeters) — tumor under the skin. Neurofibroma treatment usually involves monitoring or surgery.
Monitoring. Your doctor may recommend observation of a tumor if it’s in a place that makes removal difficult or if it’s small and causes no problems. Observation includes regular checkups and imaging tests to see if your tumor is growing.
Surgery to remove the tumor. Symptoms can be relieved by removing all or part of a neurofibroma that’s pressing on nearby tissue or damaging organs. What type of operation is performed depends on the location and size of your tumor and whether it’s intertwined with more than one nerve. The goal of surgery is to remove as much of the tumor as possible without causing further nerve damage.
After surgery, you may need physical rehabilitation. Physical therapists and occupational therapists can guide you through specific exercises that keep your muscles and joints active, prevent stiffness, and help restore your function and feeling.
Clinical trials. You may be eligible for a clinical trial testing an experimental treatment.
- Brain and Spinal Tumors Information Page. National Institute of Neurological Disorders and Stroke. https://www.ninds.nih.gov/Disorders/All-Disorders/Brain-and-Spinal-Tumors-Information-Page[↩]
- Spinal Cord Tumors. Merck Manual. https://www.merckmanuals.com/professional/neurologic-disorders/intracranial-and-spinal-tumors/spinal-cord-tumors[↩]
- Ependymoma. American Brain Tumor Association. http://www.abta.org/brain-tumor-information/types-of-tumors/ependymoma.html[↩][↩][↩]
- Chordoma. Mayo Clinic. http://www.mayoclinic.org/diseases-conditions/chordoma/cdc-20355401[↩][↩]
- Neurofibroma. Mayo Clinic. http://www.mayoclinic.org/diseases-conditions/neurofibroma/cdc-20352978[↩][↩][↩]





{kind=link}