
Contents
What is Tay Sachs disease
Tay-Sachs disease is a rare inherited neurodegenerative disorder caused by the absence of beta-hexosaminidase (HexA) enzyme that results in excessive accumulation of certain fats (lipids) known as gangliosides in the brain and nerve cells. This abnormal accumulation of gangliosides progressively destroys nerve cells (neurons) in the brain and spinal cord. A cure for Tay-Sachs does not yet exist but there are many strategies for managing life with Tay-Sachs disease.
Tay Sachs disease facts
Tay-Sachs disease is very rare in the general population. The genetic mutations that cause this disease are more common in people of Ashkenazi (eastern and central European) Jewish heritage than in those with other backgrounds. Approximately one in 30 Ashkenazi Jewish people carries the altered gene for Tay-Sachs disease. The mutations responsible for this disease are also more common in individuals of Italian, Irish Catholic, and non-Jewish French Canadian descent, especially those living in the Cajun community of Louisiana and the southeastern Quebec. Tay-Sachs disease affects males and females in equal numbers.
Tay-Sachs disease is named for Warren Tay (1843-1927), a British ophthalmologist who in 1881 described a patient with a cherry-red spot on the retina of the eye. Tay-Sachs disease is also named for Bernard Sachs (1858-1944), a New York neurologist whose work several years later provided the first description of the cellular changes in Tay-Sachs disease. Dr. Sachs also recognized the familial nature of the disorder, and, by observing numerous cases, he noted that most babies with Tay-Sachs disease at that time were of Eastern European Jewish origin. Today, Tay-Sachs occurs among people of all backgrounds.
In this specific Jewish population, about one in 3,600 live births is affected. In the general population, the carrier rate for the altered gene is approximately 1 in 250-300 people.
Tay-Sachs disease is categorized as a lysosomal storage disease. Lysosomes are the major digestive units in cells. Enzymes within lysosomes break down or “digest” nutrients, including certain complex carbohydrates and fats. When an enzyme like hexosaminidase A, which are needed to breakdown certain substances like fats, are missing or ineffective, they build up in the lysosomal. This is called abnormal “storage”. When too much fatty material builds up in the lysosome, it becomes toxic destroying the cell and damaging surrounding tissue.
The most common form of Tay-Sachs disease becomes apparent in infancy. Infants with this disorder typically appear normal until the age of 3 to 6 months, when their development slows and muscles used for movement weaken. They also develop an exaggerated startle reaction to loud noises and are listlessness. Affected infants lose previously acquired skills (i.e., psychomotor regression), such as motor skills like turning over, sitting, and crawling and severely diminished muscle tone (hypotonia). Infants with hypotonia may be described as “floppy”. As the disease progresses, affected infants and children may develop cherry-red spots within the middle layer of the eyes, gradual loss of vision, and hearing loss, increasing muscle stiffness and restricted movements (spasticity), eventual paralysis, uncontrolled electrical disturbances in the brain (seizures), and deterioration of cognitive processes (dementia).
The classical form of Tay-Sachs disease occurs during infancy. This is the most common form and is usually fatal during early childhood.
Other forms of Tay-Sachs disease are very rare and they are the Juvenile (Subacute) Tay-Sachs Disease and Late-Onset Tay-Sachs Disease (adult forms of Tay-Sachs disease). Signs and symptoms can appear in childhood, adolescence, or adulthood and are usually milder than those seen with the classical Infantile Tay-Sachs disease. Characteristic features include muscle weakness, loss of muscle coordination (ataxia) and other problems with movement, speech problems, and mental illness. These signs and symptoms vary widely among people with late-onset forms of Tay-Sachs disease.
Late-onset Tay-Sachs disease occur less often than the infantile form. However, rare disorders like late-onset Tay-Sachs disease often go unrecognized. These disorders are under-diagnosed, making it difficult to determine the true frequency of such disorders in the general population.
Children with the juvenile form, also called the subacute form, develop symptoms later than those with the infantile form, and they usually live until later in childhood or adolescence. The adult form, also called late-onset Tay-Sachs disease, may occur anytime from adolescence to the mid-30s. The symptoms and severity can vary from one person to another. Some people may fall in between the juvenile and adult forms.
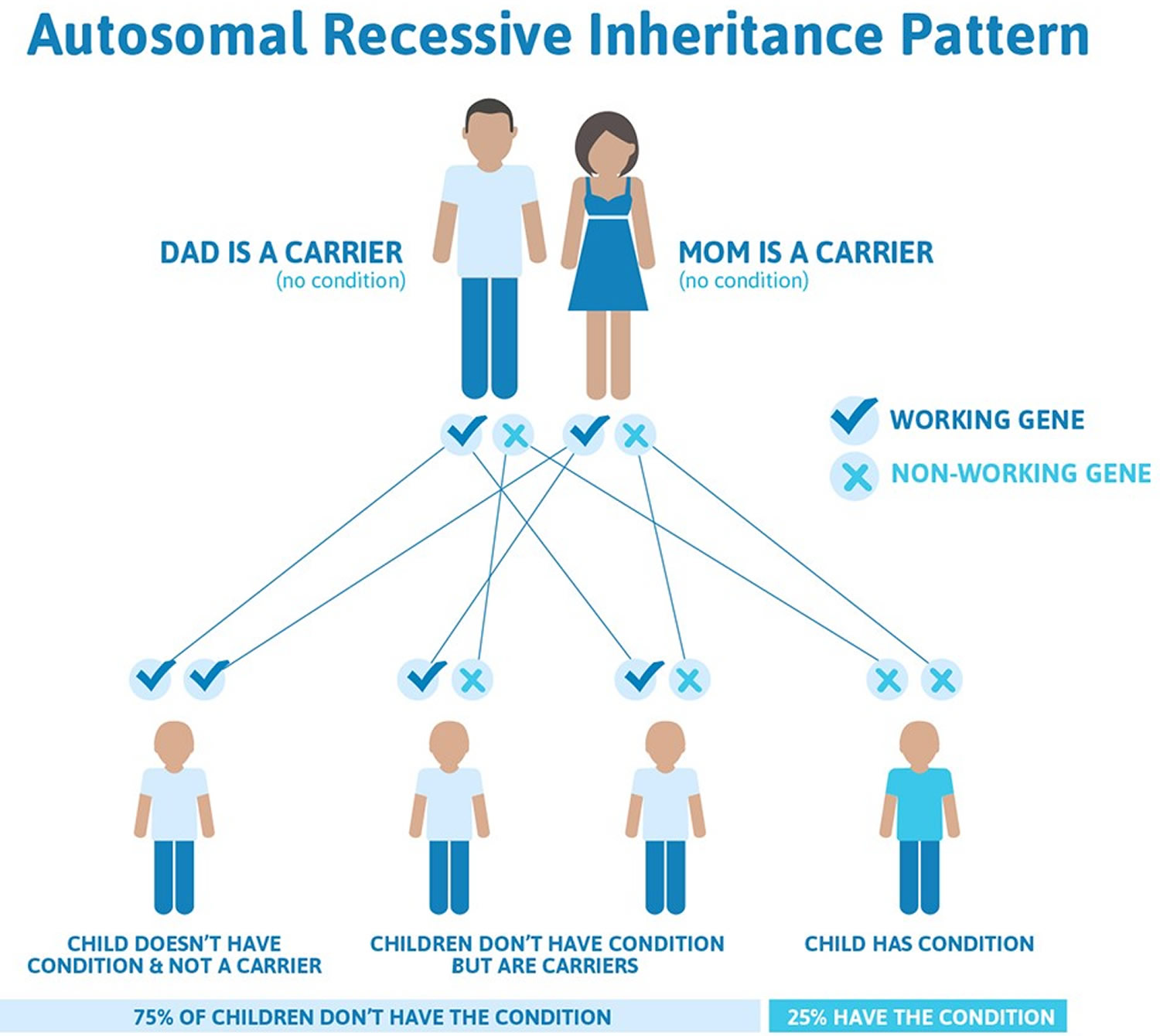
Tay-Sachs disease is inherited in an autosomal recessive manner (see Figure 1 below). The disorder results from changes (mutations) of a gene known as the HEXA gene, which regulates production of the hexosaminidase A enzyme. The HEXA gene has been mapped to the long arm (q) of chromosome 15 (15q23-q24). There is no cure for Tay-Sachs disease, and the treatment is aimed at relieving the specific symptoms that occur.
Another name for Tay-Sachs disease is GM2 gangliosidosis type 1. There are two other, related disorders, called Sandhoff disease and hexosaminidase activator deficiency that are indistinguishable from Tay-Sachs disease based on symptoms and can only be differentiated through testing to determine the underlying cause. These two disorders are also cause reduced activity of hexosaminidase, but are caused by changes in different genes. Collectively, these three disorders are known as GM2 gangliosidoses.
Other names of Tay Sachs Disease:
- B Variant GM2-Gangliosidosis
- GM2 Gangliosidosis, Type 1
- Hexoaminidase Alpha-Subunit Deficiency (Variant B)
- Hexosaminidase A deficiency
- HEXA deficiency
- Sphingolipidosis, Tay-Sachs
- TSD
Tay Sachs disease types
There are 3 forms of Tay-Sachs disease:
The form is determined by the age of the individual when symptoms first appear. Only one form of Tay-Sachs occurs in a family. If a child has Infantile, older siblings are not at risk to develop Juvenile or Late Onset Tay-Sachs later in life.
- Classic Infantile Tay-Sachs Disease -Symptoms appear around 6 months of age.
- Juvenile (Subacute) Tay-Sachs Disease -Symptoms typically appear between ages 2 and 5, but can occur anytime during childhood.
- Late-Onset Tay-Sachs Disease -Symptoms typically appear in adolescence or early adulthood, but can appear later.
Classic Infantile Tay-Sachs disease
- The First Signs – A baby with classic Infantile Tay-Sachs appears normal at birth and typically continues to develop normally for the first six months of age. Around 6 months of age, development slows. Parents may notice a reduction in vision and tracking and the baby does not outgrow normal startle response.
- A Gradual Loss of Skills – Infantile Tay-Sachs children gradually regress, losing skills one by one. Over time they are unable to crawl, turn over, sit or reach out. Other symptoms include loss of coordination, progressive inability to swallow and difficulty breathing.
- By Age 2 and beyond – Most children experience recurrent seizures by age 2 and eventually lose muscle function, mental function and sight, becoming mostly non-responsive to their environment.
Diagnosis
Tay-Sachs disease is diagnosed through a blood test to check the level of Hexosaminidase A (HexA). A follow-up DNA test may be recommended. Any doctor can order the Tay-Sachs HexA blood test. Often, diagnosis is made by a neurologist or geneticist.
Babies affected by the infantile forms of Tay-Sachs, Sandhoff, GM-1 and similar related allied diseases are frequently diagnosed by the cherry-red spot on the retina of the eye. Initially many parents notice developmental delays but pediatricians often dismiss these concerns by stating “every baby develops differently” and “the baby will catch up.” Often at about 10-14 months of age, children may start to exhibit trouble tracking and/or focusing with their eyes, so parents schedule an appointment for an eye exam. The cherry-red spot is quickly seen and an initial diagnosis of Tay-Sachs or similar devastating disease is made.
Diagnosis can also be made by a neurologist or geneticists and the completion of a metabolic evaluation.
Management
There is no treatment or cure for Tay-Sachs disease but there are ways to manage symptoms. These range from life-extending interventions like a feeding tube to comfort measures like massage to promote relaxation.
Respiratory health and seizure management are the two main symptom management challenges in Infantile Tay-Sachs disease.
Juvenile (Subacute) Tay-Sachs Disease
- First signs – Early symptoms of Juvenile Tay-Sachs include lack of coordination or clumsiness and muscle weakness such as struggling with stairs. A child may also exhibit slurred speech, swallowing difficulties and muscle cramps.
- Gradual Loss of skills – Over time, children with Juvenile Tay-Sachs slowly decline, losing their ability to walk, eat on their own and communicate. Children are prone to respiratory infections and often experience recurrent bouts of pneumonia. Many have seizures.
- Range of Severity – Juvenile Tay-Sachs has a broad range of severity. In most cases, the earlier the first signs are observed, the more quickly the disease will progress. For example, a child with first symptoms at age 2 will decline faster than a child with first symptoms at age 5.
Diagnosis
Tay-Sachs disease is diagnosed through a blood test to check the level of Hexosaminidase A (HexA). A follow-up DNA test may be recommended. Any doctor can order the Tay-Sachs HexA blood test. Often, diagnosis is made by a neurologist or geneticist.
Diagnosis can also be made by a neurologist or geneticists and the completion of a metabolic evaluation.
Juvenile Tay-Sachs does not always exhibit the cherry-red spot, which can make the road to diagnosis long and challenging. Unfortunately many healthcare providers are not aware of the rare juvenile forms of these diseases and dismiss the initial diagnosis due to the age of the child.
Management
There is no treatment or cure for Tay-Sachs disease but there are ways to manage symptoms. These range from life-extending interventions like a feeding tube to comfort measures like massage to promote relaxation. Each new diagnosis of Tay-Sachs is a unique journey.
Progressive loss of ambulatory skills followed by respiratory health and seizure management are the main symptom management issues in Juvenile Tay-Sachs disease.
Late Onset Tay-Sachs disease
- First signs – Early symptoms of Late Onset Tay-Sachs (LOTS) include clumsiness and muscle weakness in the legs. Once diagnosed, adults often reflect back to their childhood and realize they experienced symptoms much earlier such as not being athletic, speech difficulties and/or a stutter as a child or teenager. Mental health symptoms may present first which can lead to an especially long road to diagnosis. About 40% of affected adults experience mental health symptoms such as bi-polar or psychotic episodes.
- Gradual Loss of skills – Over time adults with Late Onset Tay-Sachs slowly decline. Adults frequently require more mobility assistance, i.e. cane to walker to wheelchair. Many experience speech and swallowing difficulties but few require a feeding tube.
Diagnosis
Late Onset Tay-Sachs may be hard to diagnose. Some adults go five or more years before learning their true diagnosis. In some cases it may be misdiagnosed as Multiple Sclerosis or ALS.
Adults affected by the adult form of Tay-Sachs do not exhibit the tell-tale cherry-red spot, which can contribute to a challenging and long road to diagnosis. Unfortunately many healthcare providers are not aware of the rare adult forms of these diseases and dismiss the initial diagnosis due to the age of the patient.
Adults that display mental health symptoms before physical symptoms often experience the longest road to diagnosis.
Tay-Sachs disease is diagnosed through a blood test to check the level of Hexosaminidase A (HexA). A follow-up DNA test may be recommended. Any doctor can order the Tay-Sachs HexA blood test. Often, a diagnosis is confirmed by a neurologist and/or geneticist, and after the completion of a metabolic evaluation.
Many affected adults express mixed emotions when finally receiving their diagnosis. After years of not knowing the cause of their progressive symptoms, they may experience a certain amount of relief when an accurate diagnosis is determined. At the same time, Tay-Sachs is a difficult diagnosis to receive and there can be a sense of regret and frustration.
Living with Late Onset Tay-Sachs
There is no treatment or cure for Tay-Sachs disease but there are ways to manage.
Mobility, speech and mental health are the primary symptom management issues of Late Onset Tay-Sachs. These symptoms frequently lead to other challenges related to employment, housing and communication.
Late Onset Tay-Sachs is a challenging and debilitating disorder but doesn’t always shorten life span like the childhood forms of Tay-Sachs.
Tay Sachs disease cause
What causes Tay Sachs disease
Mutations in the HEXA gene cause Tay-Sachs disease. More than 100 different mutations of the HEXA gene have been identified in individuals with the disease. Inheriting two mutated copies of the HEXA gene (homozygotes) causes deficiency of the beta-hexosaminidase A enzyme. The HEXA gene provides instructions for making part of an enzyme called beta-hexosaminidase A, which plays a critical role in the brain and spinal cord. This enzyme is located in lysosomes, which are structures in cells that break down toxic substances and act as recycling centers. Within lysosomes, beta-hexosaminidase A helps break down a fatty substance called GM2 ganglioside.
Mutations in the HEXA gene disrupt the activity of beta-hexosaminidase A, which prevents the enzyme from breaking down GM2 ganglioside. As a result, this substance accumulates to toxic levels, particularly in neurons in the brain and spinal cord. Progressive damage caused by the buildup of GM2 ganglioside leads to the destruction of these neurons, which causes the signs and symptoms of Tay-Sachs disease.
Because Tay-Sachs disease impairs the function of a lysosomal enzyme and involves the buildup of GM2 ganglioside, this condition is sometimes referred to as a lysosomal storage disorder or a GM2-gangliosidosis.
In infantile Tay-Sachs disease, there is an almost complete lack of hexosaminidase A. In late-onset Tay-Sachs disease, there is deficiency of hexosaminidase A enzyme activity. Because there is some enzyme activity, the disorder is less severe and progresses much slower than infantile Tay-Sachs disease. The exact amount of enzyme activity in late-onset Tay-Sachs disease varies greatly from one person to another. Consequently, the age of onset, severity, specific symptoms, and rate of progression of late-onset Tay-Sachs disease also vary greatly from one person to another.
The changes in the HEXA gene that cause Tay-Sachs disease are inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
If both parents are carriers of faulty HEXA genes there is a:
- 1 in 4 chance that a child will be healthy and not a carrier
- 1 in 4 chance that a child will inherit Tay-Sachs disease
- 1 in 4 chance that a child will be healthy, but a carrier.
If one parent is a carrier, there is 1 in 2 chance of a child also becoming a carrier.
Researchers have determined that the gene for Tay-Sachs disease is located on the long arm (q) of chromosome 15 (15q23-q24). Chromosomes are located in the nucleus of human cells and carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes numbered from 1 through 22 are called autosomes and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 11p13” refers to band 13 on the short arm of chromosome 11. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
Figure 1. Tay Sachs disease autosomal recessive inheritance pattern
Risk factors for Tay-Sachs disease
Risk factors for Tay-Sachs disease include having ancestors from:
- Eastern and Central European Jewish communities (Ashkenazi Jews)
- Certain French Canadian communities in Quebec
- Old Order Amish community in Pennsylvania
- Cajun community of Louisiana.
Tay Sachs disease symptoms
Tay-Sachs disease is broken down into the classic or infantile form, the juvenile form, and the adult or late-onset form. In individuals with infantile Tay-Sachs disease, symptoms typically first appear between three and five months of age. In individuals with the late-onset form, symptoms may become apparent anytime from adolescence through the mid-30s.
Symptoms of Infantile Tay-Sachs disease usually start when a child is 3 to 6 months old.
The main symptoms include:
- being overly startled by noises and movement
- being very slow to reach milestones like learning to crawl, and losing skills they have already learnt
- floppiness and weakness, which keeps getting worse until they’re unable to move (paralysis)
- difficulty swallowing
- loss of vision or hearing
- muscle stiffness
- seizures (fits)
- “Cherry-red” spots in the eyes
- movement problems
The condition is usually fatal by around 3 to 5 years of age, often due to complications of a lung infection (pneumonia).
Rarer types of Tay-Sachs disease start later in childhood (juvenile Tay-Sachs disease) or early adulthood (late-onset Tay-Sachs disease). The late-onset type doesn’t always shorten life expectancy.
Infantile Tay-Sachs Disease
The infantile form of Tay-Sachs disease is characterized by an almost complete lack of hexosaminidase A enzyme activity. The disorder often progresses rapidly, resulting in significant mental and physical deterioration.
Infants may appear completely unaffected at birth. Initial symptoms, which usually develop between 3 and 6 months, can include mild muscle weakness, twitching or jerking of muscles (myoclonic jerks), and an exaggerated startle response, such as when there is a sudden or unexpected noise. The startle response may be partly due to an increased sensitivity to sound (acoustic hypersensitivity).
Between six and 10 months, affected infants may fail to gain new motor skills. They may no longer make eye contact and there may be unusual eye movements. They may be listlessness and irritable. As affected infants age, they may experience slow growth, progressive muscle weakness, diminished muscle tone (hypotonia), and diminished mental functioning. Affected infants may also exhibit gradual loss of vision, involuntary muscle spasms that result in slow, stiff movements (spasticity), and the loss of previously acquired skills (i.e., psychomotor regression) such as crawling or sitting up.
A characteristic symptom of Tay-Sachs disease is the development of cherry red spots in the eyes. This condition occurs when the macular cells of the eye deteriorate, exposing the underlying choroid. The choroid is the middle layer of the eye that consists of blood vessels that supply blood to the retina. This characteristic finding occurs is approximately 90% of individuals with Tay-Sachs disease.
As affected infants age, more serious complications may develop, including seizures; difficulty swallowing; progressive hearing loss; confusion, disorientation, and/or deterioration of intellectual abilities (dementia); paralysis; and continued loss of vision, which can be rapid and lead to blindness. Eventually, infants may become unresponsive to their environment and surroundings. By three to five years of age, life-threatening complications may occur such as respiratory failure.
Juvenile (Subacute) Tay-Sachs Disease
The onset of this form can be anywhere between two and 10 years of age. Often, one of the first signs is clumsiness and problems with coordination. This occurs because affected children have issues controlling their body’s movements (ataxia). Behavioral problems a progressive loss of speech, life skills, and intellectual abilities also develop. Children may or may not develop a cherry-red spot in the eyes. Degeneration of the nerve that carries impulses from the eye to the brain to form images (optic atrophy) may occur. Some children may have retinitis pigmentosa, a large group of vision disorders that cause progressive degeneration of the retina, the light-sensitive membrane that coats the inside of the eyes. Children will become less responsive to their environment and surroundings. Life-threatening complications usually occur around 15 years of age.
Late-Onset Tay-Sachs Disease
The symptoms associated with late-onset Tay-Sachs disease vary greatly from one person to another. Affected individuals will not have all the symptoms listed below. The disorder progresses much slower than the infantile form. The variability of late onset Tay-Sachs may even be seen in members of the same family. One person may have symptoms in their 20s, while another reaches theirs 60s or 70s with only minor problems with their muscles.
Initial symptoms associated with late-onset Tay-Sachs disease may include clumsiness, mood alterations, and progressive muscle weakness and wasting (amyotrophy). As affected individuals age, they may exhibit tremors, muscle twitching (fasciculations), seizures, slurred speech (dysarthria), an inability to coordinate voluntary movements (ataxia), difficulty swallowing (dysphagia), and a condition known as dystonia. Dystonia is a group of disorders characterized by involuntary muscle contractions that may force certain body parts into unusual, and sometimes painful, movements and positions. Some individuals may have involuntary muscle spasms that result in slow, stiff movements (spasticity).
As late-onset Tay-Sachs disease progresses, affected individuals may experience problems with walking, running, and other similar activities. In severe instances, affected individuals may eventually need assistive devices such as braces or a wheelchair.
In some instances, affected individuals may experience mental deterioration, memory problems, and behavioral changes including short attention spans and personality changes. In approximately 40% of people, psychiatric episodes including loss of contact with reality, paranoia, hallucinations, bipolar episodes, and depression may be present.
Related Disorders
Symptoms of the following disorders can be similar to those of Tay-Sachs disease. Comparisons may be useful for a differential diagnosis:
- Sandhoff disease is a rare genetic disorder resulting in the progressive deterioration of the central nervous system (neurodegenerative disorder). A deficiency of the enzymes hexosaminidase A and B results in the accumulation of certain fats (lipids) in the brain and other organs of the body. This disorder is categorized as a lysosomal storage disease. The most common form affects infants, usually beginning between three and six months of age. Symptoms in infants may include feeding problems, general weakness, and an exaggerated startle reflex in response to sudden loud noise. Motor delays, loss of previously acquired skills, and intellectual impairment are progressive. Children can experience hearing loss, vision loss, and seizures. There are very rare forms where symptoms begin later in childhood, adolescence or adulthood. Sandhoff disease is a severe form of Tay-Sachs disease and is not limited to any specific ethnic group. Sandhoff disease is caused by changes (mutations) in the hexosaminidase B (HEXB) gene and is inherited as an autosomal recessive trait.
- Leigh syndrome is a rare genetic neurometabolic disorder. It is characterized by the degeneration of the central nervous system (i.e., brain, spinal cord, and optic nerve). The symptoms of Leigh syndrome usually begin between the ages of three months and two years, but some patients do not exhibit signs and symptoms until several years later. Symptoms are associated with progressive neurological deterioration and may include loss of previously acquired motor skills, loss of appetite, vomiting, irritability, and/or seizure activity. As Leigh syndrome progresses, symptoms may also include generalized weakness, lack of muscle tone (hypotonia), and episodes of lactic acidosis, which may lead to impairment of respiratory and kidney function. Several different genetically determined enzyme defects can cause the syndrome, initially described more than 60 years ago. Most individuals with Leigh syndrome have defects of mitochondrial energy production, such as deficiency of an enzyme of the mitochondrial respiratory chain complex or the pyruvate dehydrogenase complex. In most people, Leigh syndrome is inherited as an autosomal recessive trait. However, X-linked recessive and maternal inheritance, due to a mitochondrial DNA mutation, are additional modes of transmission.
- The neuronal ceroid lipofuscinoses are a group of progressive degenerative neurometabolic diseases. These diseases share certain similar symptoms and are distinguished in part by the age at which such symptoms appear. Two forms occur during infancy: classic infantile CLN1 disease (Santavuori disease) and CLN2 disease (late infantile type). The neuronal ceroid lipofuscinoses are characterized by abnormal accumulation of certain fatty, granular substances (i.e., pigmented lipids [lipopigments] ceroid and lipofuscin) within nerve cells (neurons) of the brain as well as other tissues of the body that may result in progressive deterioration (atrophy) of certain areas of the brain, neurological impairment, and other characteristic symptoms and physical findings.
Tay-Sachs disease is classified as a lysosomal storage disease. Several other lysosomal storage diseases can share symptoms similar to those seen in Tay-Sachs disease. Lysosomal storage diseases are inherited metabolic diseases that are characterized by an abnormal build-up of various toxic materials in the body’s cells as a result of enzyme deficiencies. There are nearly 50 of these disorders altogether, and they may affect different parts of the body, including the skeleton, brain, skin, heart, and central nervous system. New lysosomal storage disorders continue to be identified. While clinical trials are in progress on possible treatments for some of these diseases, there is currently no approved treatment for many lysosomal storage diseases.
Symptoms of the following disorders can be similar to those of late-onset Tay-Sachs disease. Comparisons may be useful for a differential diagnosis:
- Adult neuronal ceroid lipofuscinosis (ANCL) is a general term for several rare genetic disorders that belong to a group of progressive, degenerative neurometabolic disorders known as the neuronal ceroid lipofuscinoses (neuronal ceroid lipofuscinoses). These disorders share certain similar symptoms and are distinguished in part by the age at which such symptoms appear. Onset of adult neuronal ceroid lipofuscinosis is usually around the age of 30, but these disorders can occur during the teen-aged years or in people more than 50 years old. The neuronal ceroid lipofuscinoses as a group are characterized by abnormal accumulation of certain fatty, granular substances (i.e., pigmented lipids [lipopigments] ceroid and lipofuscin) within nerve cells (neurons) of the brain as well as other tissues of the body. This is accompanied by progressive deterioration (atrophy) of certain areas of the brain, neurological impairment, and other characteristic symptoms and physical findings. Adult neuronal ceroid lipofuscinosis is sometimes called Kufs disease. Historically, Kufs disease was broken down into Type A or Type B. The Aneuronal ceroid lipofuscinoses are caused by changes (mutations) in different genes and can have different signs and symptoms.
- Amyotrophic lateral sclerosis (ALS) is characterized by slight muscle weakness, clumsy hand movements, and/or difficulty performing tasks that require delicate movements of the fingers and/or hands. Muscular weakness in the legs may cause tripping and falling. People with ALS may have difficulty swallowing (dysphagia), and speech may be slowed. Additional symptoms of this disorder include progressive weakness of the lips and impairment and/or loss of function of the tongue, mouth, and/or voice box (bulbar symptoms). Leg cramps may occur during the night, most frequently in the calf and/or thigh muscles. ALS may progress quickly or slowly, and gradually additional muscles become involved. Individuals with ALS may also exhibit uncontrolled twitching of muscles (fasciculations), stiffness in the legs, and/or coughing. As the ability to move becomes progressively impaired, people with this disease are at increased risk for respiratory failure. The exact cause of ALS is unknown.
Tay-Sachs disease diagnosis
To confirm that your baby has Tay-Sachs disease, your doctor will ask you about the child’s symptoms and any hereditary family disorders. While performing a careful eye exam of your child, the doctor may see a cherry-red spot in the back of the child’s eyes, which is a sign of the disease. You may need to see a pediatric neurologist and an ophthalmologist for nervous system and eye examinations.
The diagnosis of Tay-Sachs disease may be confirmed by a thorough clinical evaluation and specialized tests, such as blood tests that measure the levels of hexosaminidase A in the body. Hexosaminidase A is reduced in people with Tay-Sachs disease, and absent or nearly absent in the infantile form.
Molecular genetic testing can confirm a diagnosis of Tay-Sachs disease. Molecular genetic testing can detect mutations in the HEXA gene known to cause the disorder, but is available only as a diagnostic service at specialized laboratories.
In some instances, it is possible that a diagnosis of Tay-Sachs disease may be suspected before birth (prenatally) based upon specialized tests, such as amniocentesis and chorionic villus sampling (CVS). During amniocentesis, a sample of fluid that surrounds the developing fetus is removed, while chorionic villus sampling involves the removal of tissue samples from a portion of the placenta. These samples are studied to determine whether hexosaminidase A is present or, as in people with Tay-Sachs disease, absent or present in greatly reduced levels. This is called an enzyme assay. Prenatal diagnosis is also possible through molecular genetic testing of tissue samples obtained through chorionic villus sampling or amniocentesis if the specific disease-causing mutation in the HEXA gene is known in the family.
Blood tests can determine whether individuals are carriers for Tay-Sachs disease (i.e., they have one copy of the disease gene). Relatives of individuals with Tay-Sachs disease should be tested to determine whether they are carriers of the disease gene. Couples who are planning to have a child and have any Jewish ancestry (not just Ashkenazi) are encouraged to undergo carrier screening before proceeding with a pregnancy.
Tay Sachs disease treatment
There is no cure for Tay-Sachs disease, but some treatments can help in managing symptoms. The goal of treatment is support and comfort tailored to each individuals.
Treatment may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, speech pathologists, specialists who asses and treat hearing problems (audiologists), eye specialists, and other health care professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may be of benefit for affected individuals and their families. Psychosocial support is recommended for the entire family.
Because of the potential of feeding difficulties, infants should be monitored for nutritional status and proper hydration. Nutritional support and supplementation may be necessary and, sometimes, the insertion of a feeding tube may be necessary. In addition to nutritional support, a feeding tube may be necessary to help prevent food, liquid or other foreign material from accidently going into the lungs (aspiration).
Supportive treatments include:
- Medication. To reduce your child’s symptoms, a number of prescription medications are available, including anti-seizure medications.
- Respiratory care. Children who have Tay-Sachs disease are at high risk of lung infections that cause breathing problems and frequently accumulate mucus in their lungs. Your child may need the mucus using chest physiotherapy (CPT) to help remove mucus from the lungs.
- Feeding tubes. Your child may have trouble swallowing, or develop respiratory problems by inhaling food or liquid into the lungs while eating. To prevent those problems, your doctor may recommend an assistive feeding device such as a gastrostomy tube, which is inserted through your child’s nose and goes to your child’s stomach. Or, a doctor trained in stomach surgery may surgically insert an esophagogastrostomy tube.
- Physical therapy. As the disease progresses, your child may benefit from physical therapy to help keep joints flexible and maintain as much ability to move (range of motion) as possible. Physical therapy can delay joint stiffness and reduce or delay the loss of function and pain that can result from shortened muscles.
Potential future treatments
Gene therapy or enzyme replacement therapy research may eventually lead to a cure or treatment to slow the progression of Tay-Sachs disease.
Enzyme replacement therapy involves replacing a missing enzyme in individuals who are deficient or lack a particular enzyme. Synthetic versions of missing enzymes have been developed and used to treat individuals with other lysosomal storage diseases including Hurler syndrome, Fabry syndrome, and Gaucher disease. However, ERT has not proven successful in people with Tay-Sachs disease. One issue is the inability to find a way for the replacement enzyme to cross the blood-brain barrier, a protective networks of blood vessels and cells that allow some materials to enter the brain, while keeping other materials out.
Gene therapy is also being studied as another possible approach to therapy for some lysosomal storage disorders. In gene therapy, the defective gene present in a patient is replaced with a normal gene to enable the production of active enzyme and prevent the development and progression of the disease in question. Given the permanent transfer of the normal gene, which can produce active enzyme at all sites of disease, this form of therapy is theoretically most likely to lead to a “cure.” However, at this time, there are still technical difficulties to resolve before gene therapy can succeed.
Chaperone therapy is also being studied for Tay-Sachs disease. This type of therapy involves very small molecules that attach to newly-created hexosaminidase A enzymes, before the mutated enzymes are broken down, and guides them to the lysosome, where the enzymes can perform their normal function. Such a molecule can also cross the blood-brain barrier. This therapy is only in initial stages of study, and more research will necessary to determine its long-term safety and effectiveness.
A drug called pyrimethamine has been tried as a treatment for Tay-Sachs disease. Affected individuals who took the medication showed increased activity of hexosaminidase A. However, this increased activity did not lead to any noticeable improvement in neurological or psychiatric symptoms. More research is necessary to determine whether pyrimethamine has any role in the treatment of Tay-Sachs disease.
Information on current clinical trials is posted on the Internet at https://www.clinicaltrials.gov/.





{kind=link}