
Contents
What is Waardenburg syndrome
Waardenburg syndrome is a group of genetic conditions that can cause hearing loss and changes in coloring (pigmentation) of the hair, skin, and eyes 1. Although most people with Waardenburg syndrome have normal hearing, moderate to profound hearing loss can occur in one or both ears. The hearing loss is present from birth (congenital). People with Waardenburg syndrome often have very pale blue eyes or different colored eyes, such as one blue eye and one brown eye. Sometimes one eye has segments of two different colors. Distinctive hair coloring (such as a patch of white hair or hair that prematurely turns gray) is another common sign of the condition. The features of Waardenburg syndrome vary among affected individuals, even among people in the same family.
Waardenburg syndrome is named after the investigator (PJ Waardenburg) who first precisely described the disorder in 1951 2. At least 1,400 cases have since been recorded in the medical literature. Evidence suggests that Waardenburg syndrome may have a frequency of approximately one in 40,000 births and account for about two to five percent of cases of congenital deafness 3. Waardenburg syndrome appears to affect males and females relatively equally.
Waardenburg syndrome is most often inherited as an autosomal dominant trait. This means only one parent has to pass on the faulty gene for a child to be affected.
There are four main types of Waardenburg syndrome, which are distinguished by their physical characteristics and sometimes by their genetic cause. The most common are type 1 and type 2 and they have very similar features, although people with type 1 almost always have eyes that appear widely spaced and people with type 2 do not. In addition, hearing loss occurs more often in people with type 2 than in those with type 1.
Type 3 (Klein-Waardenburg syndrome) and type 4 (Waardenburg-Shah syndrome) are rarer.
Type 3 (Klein-Waardenburg syndrome) includes abnormalities of the arms and hands in addition to hearing loss and changes in pigmentation. Type 4 (Waardenburg-Shah syndrome) has signs and symptoms of both Waardenburg syndrome and Hirschsprung disease, an intestinal disorder that causes severe constipation or blockage of the intestine 1.
The multiple types of Waardenburg syndrome result from defects in different genes. Most people with Waardenburg syndrome have a parent with the disease, but the symptoms in the parent can be quite different from those in the child.
Waardenburg syndrome affects an estimated 1 in 40,000 people 1. Waardenburg syndrome accounts for 2 to 5 percent of all cases of congenital hearing loss.
There is no specific treatment for Waardenburg syndrome. Symptoms will be treated as needed. Special diets and medicines to keep the bowel moving are prescribed to those people who have constipation. Hearing should be checked closely. Once hearing problems are corrected, most people with Waardenburg syndrome should be able to lead a normal life. Those with rarer forms of the syndrome may have other complications.
Possible complications
Complications may include:
- Constipation severe enough to require part of large bowel to be removed
- Hearing loss
- Self-esteem problems, or other problems related to appearance
- Slight decreased intellectual functioning (possible, unusual)
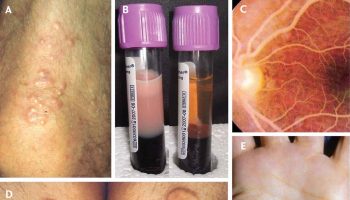
Figure 1. Waardenburg syndrome eyes
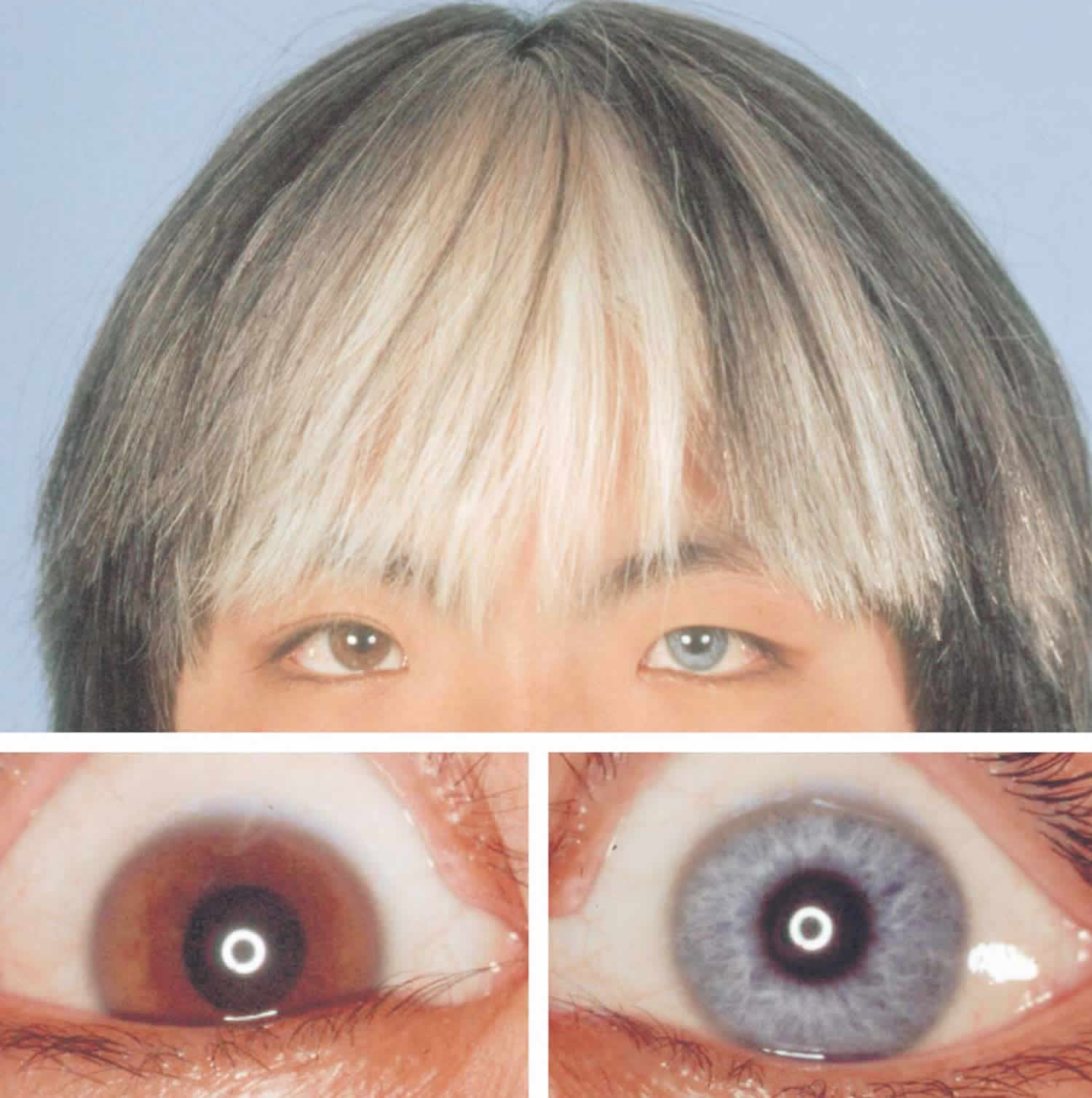
Footnote: Hair and anterior segment of the eyes of patient with Waardenburg syndrome type 2. Clusters of white hair “white locks” can be seen in the frontal hair. The hypopigmented part of the right iris appears blue and the remaining portion is brown. Left iris is blue.
[Source 4]Waardenburg syndrome type 1
Waardenburg syndrome type 1 is characterized by eyes that appear widely spaced, congenital hearing loss, and patchy pigment disturbances of the iris, hair and skin 5. Mutations in the PAX3 gene cause the symptoms observed in this condition. Treatment is symptomatic and supportive 5. Waardenburg syndrome type 1 is inherited in an autosomal dominant manner 5.
80%-99% of people with Waardenburg syndrome type 1 have these symptoms:
- Abnormality of vision
- Congenital sensorineural hearing impairment
- Heterochromia iridis (different colored eyes)
- Hypopigmented skin patches
- Lacrimation abnormality (abnormality of tear production)
Waardenburg syndrome type 1 cause
Waardenburg syndrome type 1 is caused by mutations in the PAX3 gene. Researchers believe that mutations in the PAX3 gene destroy the ability of the PAX3 protein to bind to DNA and regulate the activity of other genes. As a result, pigment cells (melanocytes) do not develop in certain areas of the skin, hair, eyes, and inner ear, which leads to the hearing loss and patchy loss of pigmentation that are characteristic features of Waardenburg syndrome. In addition, these mutations disrupt the development of certain craniofacial bones, causing the widely spaced eyes that are unique to Waardenburg syndrome type 1 6.
Waardenburg syndrome type 2
Waardenburg syndrome type 2 is characterized by varying degrees of deafness and pigmentation (coloring) abnormalities of the eyes, hair and/or skin. Waardenburg syndrome type 2 differs from type 1 and some other types of Waardenburg syndrome by the absence of dystopia canthorum (lateral displacement of the inner canthi of the eyes) 7. Sensorineural hearing loss occurs in the majority of people with Waardenburg syndrome type 2, and heterochromia iridis (differences in eye coloring) occurs in about half. Progressive hearing loss has also been reported in some people with Waardenburg syndrome type 2 8.
Waardenburg syndrome type 2 may be caused by changes (mutations) in any of several genes, but in many cases the genetic cause is unknown. While inheritance is usually autosomal dominant, sometimes Waardenburg syndrome type 2 is not inherited, occurring for the first time in someone with no family history of the condition. Treatment may include the use of hearing aids and/or cosmetic products or treatments (if desired) for skin hypopigmentation 7.
Waardenburg syndrome type 2 can be further divided into subtypes based on the genetic cause, when the responsible gene can be identified.
Waardenburg syndrome type 2 is characterized by varying degrees of deafness and pigmentation (coloring) anomalies of the eyes, hair and skin, but without dystopia canthorum (lateral displacement of the inner canthi of the eyes). Sensorineural hearing loss is present in the majority of people with Waardenburg syndrome2 (almost 80%), and heterochromia iridum (differences in eye coloring) is present in almost half 7. Progressive hearing loss has also been reported in some people with Waardenburg syndrome type 2 8. In some people, particularly those with Waardenburg syndrome type 2E, signs of Kallmann syndrome are present 7.
Is hearing loss in people with Waardenburg syndrome type 2 likely to be progressive?
Progressive hearing loss has been reported in people with Waardenburg syndrome type 2. Although some studies reporting progressive hearing loss in people with Waardenburg syndrome did not clearly describe the subtype of Waardenburg syndrome present, it is believed that progressive hearing loss is limited to those with type 2 9. Experts are not aware of data describing the proportion of people with type 2 who have progressive versus non-progressive hearing loss.
Waardenburg syndrome type 2 inheritance
Waardenburg syndrome type 2 (Waardenburg syndrome2) is usually inherited in an autosomal dominant manner 7. This means that having a change (mutation) in only one copy of the responsible gene in each cell is enough to cause features of the condition.
In some cases, an affected person inherits the mutated gene from an affected parent. In other cases, the mutation occurs for the first time in a person with no family history of the condition. This is called a de novo mutation. When a person with a mutation that causes an autosomal dominant condition has children, each child has a 50% (1 in 2) chance to inherit that mutation.
Waardenburg syndrome type 2D is the only subtype reportedly inherited in an autosomal recessive manner 10. This means that to be affected, a person must have a mutation in both copies of the responsible gene in each cell. Affected people inherit one mutated copy of the gene from each parent, who is referred to as a carrier. Carriers of an autosomal recessive condition typically do not have any signs or symptoms (they are unaffected). When 2 carriers of an autosomal recessive condition have children, each child has a:
- 25% (1 in 4) chance to be affected
- 50% (1 in 2) chance to be an unaffected carrier like each parent
- 25% chance to be unaffected and not be a carrier
Identifying the gene responsible for Waardenburg syndrome type 2 is necessary to determine the subtype that is present in a person or family.
Waardenburg syndrome type 2 diagnosis
Waardenburg syndrome type 2 is diagnosed by the presence of 2 of the 5 major criteria needed for a general diagnosis of Waardenburg syndrome, with no dystopia canthorum (lateral displacement of the inner canthi of the eyes) 11.
In contrast to the general diagnosis of Waardenburg syndrome type 2, made based on symptoms, the diagnosis of a subtype of Waardenburg syndrome type 2 relies on identifying the genetic cause of Waardenburg syndrome type 2. For some subtypes, the general location (locus) of the responsible gene is known, but the specific responsible gene has not yet been identified. There are currently 5 subtypes of Waardenburg syndrome type 2:
- Type 2A is caused by a change (mutation) in the MITF gene on chromosome 3 12
- Type 2B is associated with a locus on chromosome 1 13
- Type 2C is associated with a locus on chromosome 8 14
- Type 2D is caused by mutations is the SNAI2 gene on chromosome 8 10
- Type 2E is caused by mutations in the SOX10 gene on chromosome 22 15
Waardenburg syndrome type 3
Waardenburg syndrome type 3 (Klein-Waardenburg syndrome) is a very rare subtype of Waardenburg syndrome that is characterized by limb anomalies (predominantly involving upper limbs, with hypoplasia of the musculoskeletal system, flexion contractures, fusion of the carpal bones, syndactylia) in association with congenital sensorineural hearing loss, minor defects in structures arising from neural crest, resulting in pigmentation anomalies of eyes (hypopigmentation abnormalities of irides), hair, and skin and minor facial dysmorphism in combination with dystopia canthorum 16.
Waardenburg syndrome type 3 (Klein-Waardenburg syndrome) is caused by heterozygous or homozygous mutations in the PAX3 (2q36.1) gene.
Incidence is unknown, but Waardenburg syndrome type 3 (Klein-Waardenburg syndrome) is the rarest form of all Waardenburg syndrome types.
Waardenburg syndrome type 3 diagnosis
Diagnosis is made through criteria of the association of Waardenburg syndrome1 manifestations with limb anomalies. PAX3 gene analysis confirms diagnosis resulting in an abnormality of melanocytes of skin, ears and hair.
The defects are more severe in homozygous than heterozygous forms. Genetic counseling is recommended.
Antenatal diagnosis
Antenatal diagnosis is possible for affected parents.
Waardenburg syndrome type 3 management and treatment
Hearing aids to counter hearing loss, effective therapy to improve language, communication, and cognitive skill and limbs physiotherapy are recommended. Associated manifestations are treated as appropriate (e.g. skin and eyes protection from the sun).
Waardenburg syndrome type 3 prognosis
Due to the very few number of cases described so far, information on prognosis and quality of life is limited. Disease progression is variable, with symptoms being more severe in homozygous than in heterozygous forms.
Waardenburg syndrome type 4
Waardenburg syndrome type 4, also known as Waardenburg-Shah syndrome, is characterized by hearing loss; changes in coloring (pigmentation) of the hair, skin, and eyes; and Hirschsprung disease, an intestinal disorder that causes severe constipation or blockage of the intestine.
Waardenburg syndrome type 4 (Waardenburg-Shah syndrome) is further divided into types 4A, 4B, and 4C based on their genetic cause.
- Type 4A is caused by mutations in the EDNRB gene, mutations in EDN3 cause 4B, and mutations in SOX10 cause type 4C.
Waardenburg syndrome type 4 (Waardenburg-Shah syndrome) is usually inherited in an autosomal dominant fashion; however, some cases of Waardenburg syndrome type 4 (Waardenburg-Shah syndrome) appear to have an autosomal recessive pattern of inheritance 17.
80%-99% of people with Waardenburg syndrome type 4 have these symptoms:
- Abnormal macular morphology
- Abnormality of vision
- Aganglionic megacolon (enlarged colon lacking nerve cells)
- Constipation
- Hearing impairment or deafness
Waardenburg syndrome symptoms
Waardenburg syndrome symptoms may include:
- Cleft lip (rare)
- Constipation
- Deafness (more common in type 2 disease)
- Extremely pale blue eyes or eye colors that don’t match (heterochromia)
- Pale color skin, hair, and eyes (partial albinism)
- Difficulty completely straightening joints
- Possible slight decrease in intellectual function
- Wide-set eyes (in type 1)
- White patch of hair or early graying of the hair
Less common types of this disease may cause problems with the arms or intestines.
Primary features of Waardenburg syndrome may include distinctive facial abnormalities; unusually diminished pigmentation (hypopigmentation) of the hair, the skin, and/or the irides or the iris of both eyes (partial albinism); and/or deafness that is present at birth (congenital). However, as mentioned earlier, associated symptoms and findings may be extremely variable, including among affected members of the same family (kindred). For example, while some affected individuals may have only one characteristic feature, others may have several abnormalities associated with the disorder.
In some individuals with Waardenburg syndrome, there is abnormal sideways (lateral) displacement of the inner angles (canthi) of the eyes formed by the junction of the upper and lower eyelids (dystopia canthorum). In addition, the condition may be associated with unusually low (inferior) openings to the tear (lacrimal) ducts and an increased susceptibility to infections of the lacrimal sacs (dacryocystitis). (Each inner canthus opens into a small space that contains the opening to a lacrimal duct.) Due to dystopia canthorum, affected individuals may have an abnormally wide, high nasal bridge and underdeveloped nasal “wings” (hypoplastic nasal alae), resulting in narrow nostrils. In addition, in some cases, the eyebroWaardenburg syndrome may be unusually bushy and/or may grow together (synophrys). Rarely, affected individuals also have widely spaced eyes (ocular hypertelorism). As mentioned previously, researchers have described different forms of Waardenburg syndrome based upon certain symptoms and specific genetic findings. Waardenburg syndrome type 2 is distinguished from Waardenburg syndrome type 1 by the absence of dystopia canthorum.
In some individuals with Waardenburg syndrome, additional facial abnormalities may be present. These may include an unusually rounded nasal tip that may be slightly upturned; abnormal “smoothness” of the vertical groove of the upper lip (philtrum); full lips; and/or mild protrusion of the lower jaw (mandibular prognathism). There have also been a few reports in which the disorder has been associated with incomplete closure of the roof of the mouth (cleft palate) and/or an abnormal groove in the upper lip (cleft lip).
Waardenburg syndrome is often associated with pigmentary abnormalities due to deficiency of the pigment melanin. Some with the disorder have a white forelock (poliosis) at birth that tends to disappear with age or patches of white hair other than a forelock. (There have also been cases in which a black rather than a white forelock is present.) The eyebrows, eyelashes, and scalp hair may become prematurely gray or white (beginning as early as mid-childhood, adolescence, or early adulthood). In addition, some affected individuals have irregular patchy skin regions that lack pigmentation (leukoderma or vitiligo), particularly on the face and arms. Waardenburg syndrome may also be associated with underdevelopment (hypoplasia) of the connective tissue fibers that comprise most of the colored region (iris) of both eyes (irides). As a result, affected individuals may have unusually pale blue eyes or differences in the pigmentation of the two irides or within different areas of the same iris (heterochromia irides). For example, the iris of one eye may be blue while the other has a different color or one or both irides may seem unusually “mottled” in appearance. Some reports suggest that heterochromia irides may be more frequent in Waardenburg syndrome type 2 while the presence of a white forelock and depigmented skin patches are more common in those with Waardenburg syndrome type 1.
Some individuals with Waardenburg syndrome are also affected by congenital deafness. Such hearing impairment appears to result from abnormalities or absence of the organ of Corti, a structure within the hollow, coiled passage of the inner ear (cochlea). The organ of Corti contains minute hair cells that convert sound vibrations into nerve impulses, which are then transmitted via the auditory nerve (vestibulocochlear nerve) to the brain. Abnormalities of the organ of Corti may prevent the transmission of such nerve impulses, resulting in hearing impairment (known as sensorineural or cochlear deafness). In most affected individuals with Waardenburg syndrome, congenital sensorineural deafness affects both ears (bilateral). However, in rare cases, only one side may be affected (unilateral). Evidence suggests that congenital sensorineural deafness is more frequently associated with Waardenburg syndrome type 2 than Waardenburg syndrome type 1.
In some cases, characteristic facial, eye (ocular), and hearing (auditory) features of Waardenburg syndrome may occur in association with bilateral malformations of the arms and hands (upper limbs). This form of the disorder, which has been described as a severe presentation of Waardenburg syndrome1, is sometimes referred to as Waardenburg syndrome type 3 (Klein-Waardenburg syndrome) or Waardenburg syndrome with upper limb anomalies. Bilateral defects may include underdevelopment (hypoplasia) and abnormal shortness of the upper limbs; abnormal bending of certain joints of the fingers in fixed positions (flexion contractures); fusion of wrist (carpal) bones; and/or webbing or fusion (syndactyly) of certain fingers. In some cases, other skeletal abnormalities may be present, such as abnormal elevation of the shoulder blades (Sprengel deformity).
A fourth form of Waardenburg syndrome has also been described in which primary features of Waardenburg syndrome occur in association with Hirschsprung disease. This form of the disorder may be referred to as Waardenburg syndrome type 4 (Waardenburg-Shah syndrome) or Waardenburg-Hirschsprung disease. Hirschsprung disease (also known as aganglionic megacolon) is a gastrointestinal (GI) disorder characterized by absence of certain nerve cell bodies (ganglia) in the smooth muscle wall within a region of the large intestine (i.e., colon). As a result, there is absence or impairment of the involuntary, rhythmic contractions that propel food through the GI tract (peristalsis). Associated symptoms and findings may include an abnormal accumulation of feces within the colon; widening of the colon above the affected segment (megacolon); abdominal bloating (distension); vomiting; lack of appetite (anorexia); failure to grow and gain weight at the expected rate (failure to thrive); and/or other abnormalities.
Rare cases of Waardenburg syndrome type 4 have been described in which affected individuals have also had neurologic symptoms due to abnormalities of the brain and spinal cord (central nervous system). In such instances, additional findings have included growth restriction; abnormally diminished muscle tone (hypotonia); flexion or extension of certain joints in various fixed postures (arthrogryposis); and/or other abnormalities.
Waardenburg syndrome causes
Mutations in the EDN3, EDNRB, MITF, PAX3, SNAI2, and SOX10 genes can cause Waardenburg syndrome 1. These genes are involved in the formation and development of several types of cells, including pigment-producing cells called melanocytes. Melanocytes make a pigment called melanin, which contributes to skin, hair, and eye color and plays an essential role in the normal function of the inner ear. Mutations in any of these genes disrupt the normal development of melanocytes, leading to abnormal pigmentation of the skin, hair, and eyes and problems with hearing.
Waardenburg syndrome types 1 and 3 are caused by mutations in the PAX3 gene. Mutations in the MITF or SNAI2 gene can cause Waardenburg syndrome type 2.
Mutations in the SOX10, EDN3, or EDNRB gene can cause Waardenburg syndrome type 4. In addition to melanocyte development, these genes are important for the development of nerve cells in the large intestine. Mutations in one of these genes result in hearing loss, changes in pigmentation, and intestinal problems related to Hirschsprung disease.
In some cases, the genetic cause of Waardenburg syndrome has not been identified.
Waardenburg syndrome inheritance pattern
Waardenburg syndrome is usually inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person has one parent with the condition. A small percentage of cases result from new mutations in the gene; these cases occur in people with no history of the disorder in their family.
Some cases of Waardenburg syndrome type 2 and type 4 appear to have an autosomal recessive pattern of inheritance, which means both copies of the gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition.
Genetic counseling may help you understand the risks of passing Waardenburg syndrome on to any children you have.
- To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself. Online directories are provided by the American College of Medical Genetics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) and the National Society of Genetic Counselors (https://www.findageneticcounselor.com/).
Figure 2. Waardenburg syndrome autosomal dominant inheritance pattern
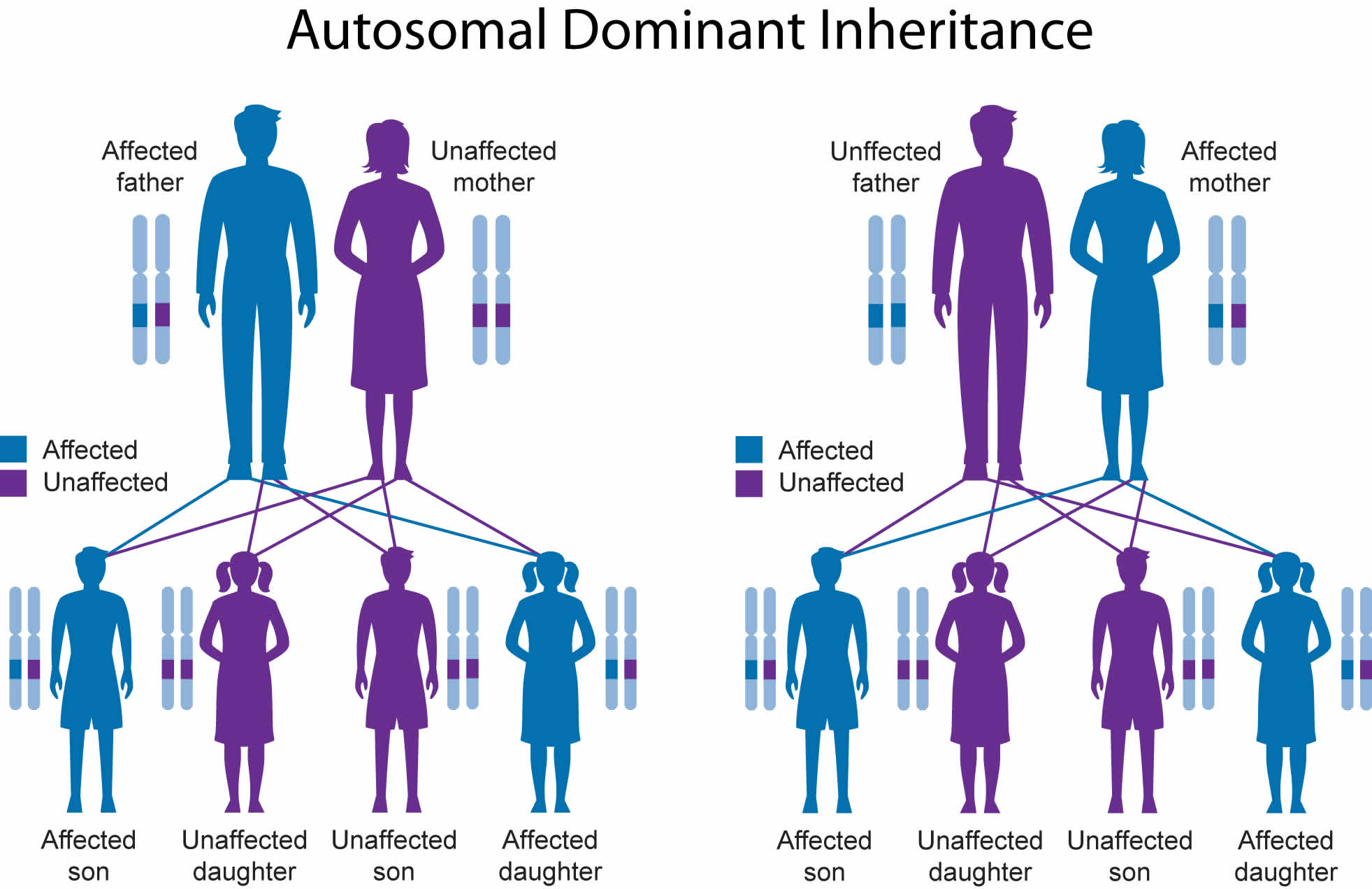
Figure 3. Waardenburg syndrome autosomal recessive inheritance pattern

Waardenburg syndrome diagnosis
Waardenburg syndrome may be diagnosed at birth or early childhood (or, in some cases, at a later age) based upon a thorough clinical evaluation, identification of characteristic physical findings, a complete patient and family history, and various specialized studies. For example, in those with suspected Waardenburg syndrome, diagnostic evaluation may include use of a caliper to measure the distances between the inner angles of the eyes (inner canthi), the outer angles of the eyes (outer canthi), and the pupils (interpupillary distances). (A caliper is an instrument with two hinged, movable, curved arms used to measure thickness or diameter.) Researchers indicate that obtaining and evaluating a composite of these measurements (i.e., using a predefined biometric index known as the “W-index”) may sometimes be helpful in confirming the presence or absence of dystopia canthorum, a finding that may suggest Waardenburg syndrome1.
Additional diagnostic studies may be conducted to help detect or characterize certain abnormalities potentially associated with Waardenburg syndrome. Such studies may include examination with an illuminated microscope to visualize internal structures of the eyes (slit-lamp examination); specialized hearing (auditory) tests; and/or advanced imaging techniques, such as to evaluate inner ear abnormalities, skeletal defects (e.g., seen in Waardenburg syndrome3), Hirschsprung disease (e.g., seen in Waardenburg syndrome4), etc. For example, researchers indicate that computed tomography (CT) scanning may help to characterize inner ear defects responsible for congenital sensorineural deafness. (During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of internal structures.) In selected cases, diagnostic evaluation may also include the removal (biopsy) and microscopic examination of certain tissue samples, such as rectal biopsies to help confirm Hirschsprung disease. In some instances, additional diagnostic studies may also be recommended.
A diagnosis of Waardenburg syndrome is made based on the presence of signs and symptoms. In 1992, the Waardenburg Consortium proposed diagnostic criteria, which includes both major and minor criteria. A diagnosis of Waardenburg syndrome type 1 (the most common type) needs 2 major, or 1 major and 2 minor of the following criteria 18:
Major criteria:
- Congenital sensorineural hearing loss (present from birth)
- Iris pigmentary (coloration) abnormality, such as heterochromia iridis (complete, partial, or segmental); pale blue eyes (isohypochromia iridis); or pigmentary abnormalities of the fundus (part of the eye opposite the pupil)
- Abnormalities of hair pigmentation, such as white forelock (lock of hair above the forehead), or loss of hair color
- Dystopia canthorum – lateral displacement of inner angles (canthi) of the eyes (in Waardenburg syndrome types 1 and 3 only)
- Having a 1st degree relative with Waardenburg syndrome
Minor criteria:
- Leukoderma (white patches of skin) present from birth
- Synophrys (connected eyebrows, or “unibrow”) or medial eyebrow flare
- Broad or high nasal bridge (uppermost part of the nose)
- Hypoplasia (incomplete development) of the nostrils
- Premature gray hair (under age 30)
Waardenburg syndrome type 2 has features similar to type 1, but the inner canthi of the eyes are normal (no dystopia canthorum present).
Waardenburg syndrome type 3 also has similar features to Waardenburg syndrome type 1, but is additionally characterized by musculoskeletal abnormalities such as muscle hypoplasia; flexion contractures (inability to straighten joints); or syndactyly (webbed or fused fingers or toes).
Waardenburg syndrome type 4 has similar features to Waardenburg syndrome type 2, but with Hirschsprung disease (a condition resulting from missing nerve cells in the muscles of part or all of the large intestine).
Waardenburg syndrome treatment
The treatment of Waardenburg syndrome is directed toward the specific symptoms that are apparent in each individual. Such treatment may require the coordinated efforts of a team of medical professionals, such as physicians who specialize in skin disorders (dermatologists); eye specialists (ophthalmologists); hearing specialists; physicians who diagnose and treat disorders of the skeleton, joints, muscles, and related tissues (orthopedists); physicians who specialize in diseases of the digestive tract (gastroenterologists); speech-language pathologists; physical therapists; and/or other health care professionals.
Early recognition of sensorineural deafness may play an important role in ensuring prompt intervention and appropriate supportive management. In some instances, physicians may recommend treatment with a cochlear implant, a device in which electrodes implanted in the inner ear stimulate the auditory nerve to send impulses to the brain. In addition, early, special instruction may be recommended to assist in the development of speech and certain methods (e.g., sign language, lip reading, the use of communication devices, etc.) that may aid communication.
Because individuals with pigmentary abnormalities of the skin may be prone to sunburns and a risk of skin cancer, physicians may recommend avoiding direct sunlight, using sunscreen with a high sun protection factor (SPF 50+), wearing sunglasses and coverings that help to protect against the sun (e.g., hats, long sleeves, pants, etc.), and following other appropriate measures. For those with diminished pigmentation of the irides, lateral displacement of the inner angles of the eyes (dystopia canthorum), and/or other associated ocular abnormalities, ophthalmologists may also recommend certain supportive measures. These may include the use of specially tinted glasses or contact lenses (e.g., to help reduce possible sensitivity to light), measures to help prevent or treat infection, or other preventive or therapeutic steps.
In individuals with upper limb abnormalities, treatment may include physical therapy and various orthopedic techniques, potentially including surgical measures. In addition, surgery may sometimes be recommended to help treat other abnormalities that may be associated with the disorder. The specific surgical procedures performed will depend upon the severity and location of the anatomical abnormalities, their associated symptoms, and other factors.
For example, for affected individuals with Hirschsprung disease, treatment may require removal of the affected intestinal region and surgical “rejoining” of healthy intestinal areas. In some instances, before surgical correction of the condition, treatment may require the creation of an artificial outlet for the colon through an opening in the abdominal wall (i.e., a temporary colostomy).
Additional supportive services that may be beneficial for some affected individuals include special education and/or other medical, social, or occupational services. Genetic counseling will also be of benefit for affected individuals and their families. Other treatment for this disorder is symptomatic and supportive.
- Waardenburg syndrome. https://ghr.nlm.nih.gov/condition/waardenburg-syndrome[↩][↩][↩][↩]
- Waardenburg Syndrome. https://rarediseases.org/rare-diseases/waardenburg-syndrome/[↩]
- Clinical aspects of hereditary hearing loss. Kochhar A, Hildebrand MS, Smith RJ. Genet Med. 2007 Jul; 9(7):393-408. https://www.ncbi.nlm.nih.gov/pubmed/17666886/[↩]
- Ohno, Naonori et al. “Clinical findings in Japanese patients with Waardenburg syndrome type 2.” Japanese journal of ophthalmology 47 1 (2003): 77-84. https://pdfs.semanticscholar.org/0b4c/d44fe900cd494d72daa9857dbb8c69ae9b5e.pdf[↩]
- Milunsky JM. Waardenburg Syndrome Type I. 2001 Jul 30 [Updated 2017 May 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1531[↩][↩][↩]
- PAX3. Genetics Home Reference (GHR). April 2006; http://ghr.nlm.nih.gov/gene/PAX3[↩]
- Véronique Pingault. Waardenburg syndrome type 2. Orphanet. October, 2015; http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=895[↩][↩][↩][↩][↩]
- Koyama H, Kashio A, Sakata A, et al. The Hearing Outcomes of Cochlear Implantation in Waardenburg Syndrome. BioMed Research International. 2016; https://www.hindawi.com/journals/bmri/2016/2854736/[↩][↩]
- Koyama H, Kashio A, Sakata A, et al. The Hearing Outcomes of Cochlear Implantation in Waardenburg Syndrome. BioMed Research International. 2016 https://www.hindawi.com/journals/bmri/2016/2854736[↩]
- Carol A. Bocchini. WAARDENBURG SYNDROME, TYPE 2D; WS2D. OMIM. http://omim.org/entry/608890[↩][↩]
- Chen Y, Yang F, Zheng H, Zhou J, Zhu G, Hu P4, Wu W. Clinical and genetic investigation of families with type II Waardenburg syndrome. Mol Med Rep. March, 2016; 13(3):1983-1988.[↩]
- Cassandra L. Kniffin. WAARDENBURG SYNDROME, TYPE 2A; WS2A. OMIM. http://omim.org/entry/193510[↩]
- Cassandra L. Kniffin. WAARDENBURG SYNDROME, TYPE 2B; WS2B. OMIM. http://www.omim.org/entry/600193?search=waardenburg%20type%202b&highlight=type%202b%20waardenburg[↩]
- Cassandra L. Kniffin. WAARDENBURG SYNDROME, TYPE 2C; WS2C. OMIM. http://omim.org/entry/606662[↩]
- Cassandra L. Kniffin. WAARDENBURG SYNDROME, TYPE 2E; WS2E. OMIM. http://omim.org/entry/611584[↩]
- Waardenburg syndrome type 3. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=896[↩]
- Waardenburg syndrome. Genetics Home Reference (GHR). http://ghr.nlm.nih.gov/condition/waardenburg-syndrome[↩]
- Milunsky JM. Waardenburg Syndrome Type I. 2001 Jul 30 [Updated 2017 May 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1531/[↩]

{kind=link}