Contents
- What is pulmonary hypertension
- Pulmonary hypertension types and causes
- Pulmonary Arterial Hypertension
- How common is pulmonary arterial hypertension?
- How does pulmonary arterial hypertension affect my body?
- How serious is pulmonary arterial hypertension?
- Could pulmonary hypertension run in my family?
- Can pulmonary arterial hypertension be reversed?
- Who does pulmonary arterial hypertension affect?
- Pregnancy and pulmonary arterial hypertension
- Pulmonary arterial hypertension treatment
- Pulmonary Hypertension Due to Left Heart Disease
- Pulmonary Hypertension Due to Lung Disease and/or Hypoxia
- Pulmonary Hypertension Due to Pulmonary Artery Obstructions
- Pulmonary Hypertension Due to Unknown Causes
- Pulmonary hypertension life expectancy
- Pulmonary hypertension symptoms
- Pulmonary hypertension causes
- Pulmonary hypertension diagnosis
- Pulmonary hypertension functional classification
- Pulmonary hypertension differential diagnosis
- Pulmonary hypertension treatment
- All Types of Pulmonary Hypertension
- Pulmonary hypertension medications
- Surgery
- Treatments for pulmonary arterial hypertension
- Group 2 Pulmonary Hypertension Due to Left Heart Disease
- Group 3 Pulmonary Hypertension Due to Lung Disease
- Group 4 Pulmonary Hypertension Due to Chronic Blood Clots in the Lungs
- Group 5 Pulmonary Hypertension Due to Unknown Causes
- Pulmonary hypertension prognosis
- Living with pulmonary hypertension
What is pulmonary hypertension
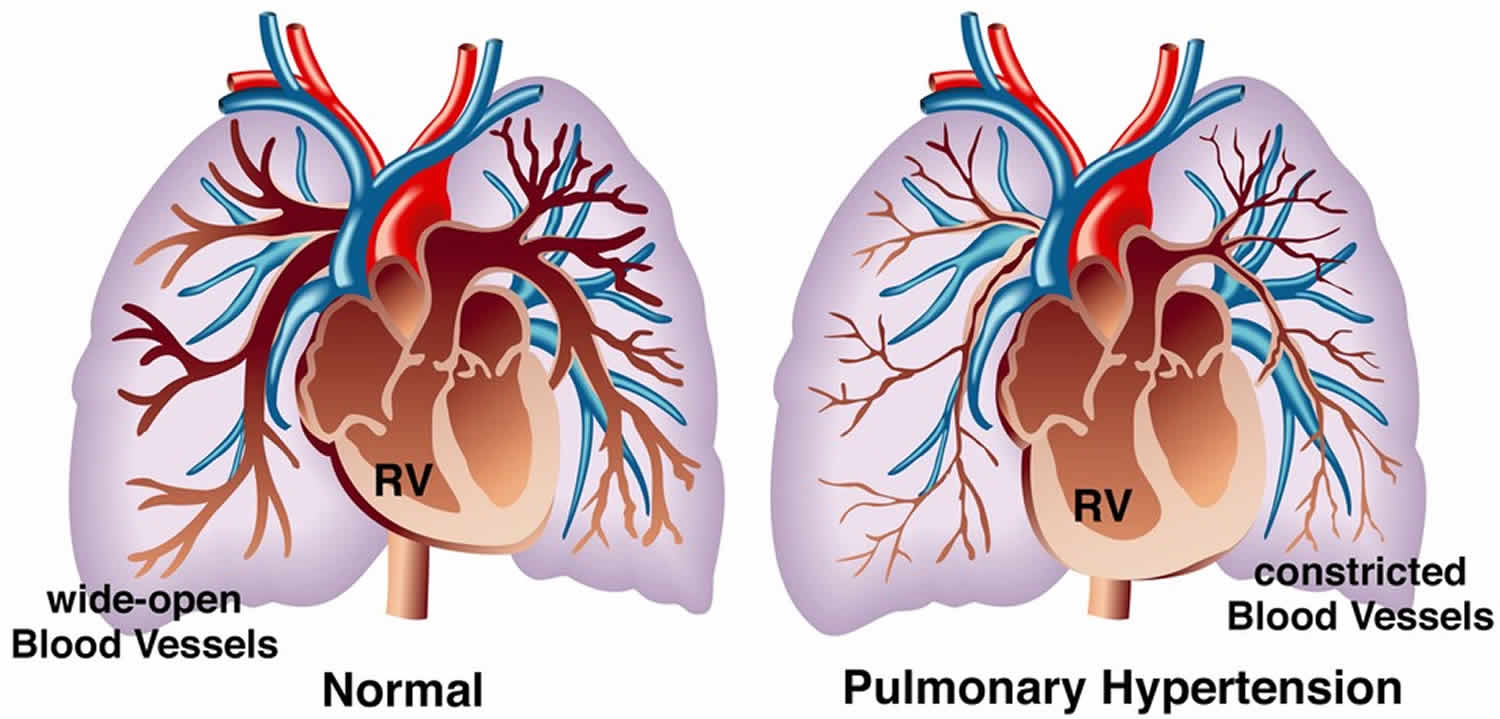
Pulmonary hypertension is a rare serious progressive (meaning it gets worse over time) condition characterized by abnormally high blood pressure (hypertension) in the blood vessels that supply your lungs (pulmonary arteries), the blood vessel that carries deoxygenated blood (venous blood) from the right side of your heart to your lungs, leading to increased resistance to blood flow and strain on the right side of your heart and if left untreated, leads to right ventricular failure and death (Figures 1 and 2) 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16. The pulmonary artery is the main blood vessel that carries blood from your heart to your lungs. Blood (deoxygenated blood or venous blood) moves from your heart to your lungs through blood vessels called pulmonary arteries. The pulmonary artery carries blood from the right side of your heart to your lungs to pick up a fresh supply of oxygen. Once in your lungs, the deoxygenated blood (venous blood) travels through many small, thin blood vessels called capillaries. There, the blood picks up more oxygen and transfers carbon dioxide to the lungs—a process called gas exchange. The oxygen-rich blood passes from your lungs back to your heart through the pulmonary veins. If the small blood vessels in your lungs become narrowed, blocked or damaged, the blood does not flow through them as well. As a result, blood can’t flow through your lungs as well as it should. This can increase the blood pressure in the pulmonary arteries and cause pulmonary hypertension. For a person at rest, normal mean pulmonary artery pressure (mPAP) is typically considered to be less than 20 mmHg or between 8-20 millimeters of mercury (8-20 mmHg). In other words, a mean pulmonary artery pressure (mPAP) of 20 mmHg is considered the upper limit of normal value 17, 18, 18, 19. In an individual with pulmonary hypertension, resting mean pulmonary arterial blood pressure (mPAP) is 20 mmHg or higher as measured by right heart catheterization, with pulmonary capillary wedge pressure (PAWP) less than 15 mm Hg and increased pulmonary vascular resistance (PVR) above 3 Wood units (240 dyne/second/cm5) at rest 20, 21, 22, 23, 24. Pulmonary artery wedge pressure (PAWP) also known as pulmonary artery occlusion pressure (PAOP) is a measurement of the pressure in the pulmonary arteries obtained by inflating a balloon-tipped catheter (often a Swan-Ganz catheter) that is “wedged” into a small branch of the pulmonary artery, pressure reading then reflects the pressure in the pulmonary veins and, therefore, the left atrium and it’s used to estimate left ventricular end-diastolic pressure (LVEDP), which is an indicator of the preload or filling volume of the left ventricle. Normally, pulmonary artery wedge pressure (PAWP) or pulmonary artery occlusion pressure (PAOP) is around 6 to 12 mmHg. An elevated pulmonary artery wedge pressure (PAWP) or pulmonary artery occlusion pressure (PAOP) greater than 18 mmHg in the context of normal oncotic pressure suggests left heart failure. Pulmonary vascular resistance (PVR) is the resistance against blood flow from the 4 pulmonary veins of the lung to the left atrium 25.
This increase in mean pulmonary artery pressure (mPAP) greater than 20 mmHg occurs when the small arteries of your lungs become abnormally narrow, thick and stiff, and can’t expand as well to allow blood through. The reduced blood flow makes it harder for the right-hand side of the heart to pump blood through the arteries. In pulmonary hypertension, the blood vessels specifically in the lungs are affected. They can become stiff, damaged or narrow, and the right side of the heart must work harder to pump blood through. If the right-hand side of your heart has to continually work harder, it can gradually become weaker. This can lead to heart failure. Pulmonary hypertension results in right ventricular pressure/volume overload leading to right ventricular heart failure and death 26.
More recently, the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology and the European Respiratory Society published guidelines further reducing the cut-off value for pulmonary vascular resistance (PVR) to 2 Woods units (160 dyne/second/cm5), based on the available data on the upper limit of pulmonary vascular resistance in healthy individuals 27. Although these lower cut-off values better reflect the normal ranges of pulmonary hemodynamic variables, they have not yet resulted in new therapeutic recommendations, and the efficacy of treatment of pulmonary arterial hypertension (PAH) in patients with a pulmonary vascular resistance (PVR) of 2-3 Woods units or mean pulmonary artery pressure (mPAP) of 21-24 mm Hg is unknown 1. The new hemodynamic definitions in the 2022 guidelines of the European Society of Cardiology-European Respiratory Society have yet to be accepted by all major pulmonary hypertension research and clinical entities in the US and internationally 1.
Pulmonary hypertension is a rare serious condition that can affect people of all ages, but it’s more common in people who have another heart or lung condition. Pulmonary hypertension can be present even if you have normal “regular” (systemic) blood pressure because the blood vessels in the lung are very different from the blood vessels in the rest of the body.
Pulmonary hypertension in general quite common; it is diagnosed in more than 2% of all patients discharged from U.S. hospitals and in up to 9% of echocardiograms performed in a community setting 28. In the UK, the observed pulmonary hypertension prevalence has doubled in the last 10 years and is currently 125 cases/million inhabitants 29. Estimates of 1% of the global population and up to 10% of individuals aged >65 years with pulmonary hypertension have been reported 30. Due to the presence of heart and lung causes of pulmonary hypertension, prevalence is higher in individuals aged >65 years 31. Globally, left heart disease is the leading cause of pulmonary hypertension 31. Lung disease, especially chronic obstructive pulmonary disease (COPD), is the second most common cause 31. In developing countries, coronary artery disease, some infectious diseases (schistosomiasis, human immunodeficiency virus [HIV]), and high altitude represent important causes of pulmonary hypertension 31. Irrespective of the underlying condition, developing pulmonary hypertension is associated with worsening symptoms and increased mortality 31.
Many common conditions and diseases are complicated by pulmonary hypertension or right ventricular failure, or both, including 1, 3, 32, 33, 34, 35:
- Problems with the smaller branches of the pulmonary arteries (pulmonary arterial hypertension or PAH)
- Conditions that affect the left side of the heart
- Shortage of oxygen in the body (hypoxia) due to heart or lung disease (such as chronic obstructive pulmonary disease [COPD]) or high altitude
- Blood clots (pulmonary embolism) that cause narrowing or a blockage in the pulmonary arteries
- Autoimmune or connective tissue diseases that damage the lungs, such as scleroderma and rheumatoid arthritis
- Birth defects of the heart (congenital heart disease)
- Portal hypertension (resulting from liver disease)
- Chronic liver disease
- End stage renal disease
- Heart failure (heart failure with reduced ejection fraction [HFrEF], heart failure with preserved ejection fraction [HFpEF])
- Heart valve disease
- HIV infection
- Schistosomiasis
- Drugs and toxins (such as appetite suppressants, cocaine, or amphetamines)
- Myeloproliferative disorders (overproduction of red blood cells or white blood cells)
- Hemoglobinopathies (abnormal oxygen-carrying proteins in red blood cells, such as found in sickle cell anemia)
- Chronic hemolysis
- Obesity with obstructive sleep apnea or sleep disordered breathing
- Thyroid disorders
- Lung disease, such as chronic obstructive pulmonary disease (COPD) or pulmonary fibrosis or any other severe chronic lung condition.
The presence of pulmonary hypertension in all of these conditions is associated with worse outcomes 34, 35.
Not all pulmonary hypertension is the same. Pulmonary hypertension is a general term used to describe high blood pressure in the lungs from any cause. There are 5 different groups of pulmonary hypertension based on different causes. These groups are defined by the World Health Organization (WHO) and are referred to as pulmonary hypertension WHO Groups 27.
The WHO classification of pulmonary hypertension is based on the mechanism or underlying cause 27:
- Group 1: Pulmonary arterial hypertension (PAH) can be idiopathic (i.e., primary pulmonary hypertension) or due to congenital left to right intracardiac shunts, portal hypertension, persistent pulmonary hypertension of the newborn, collagen vascular diseases, HIV infection.
- Group 2: Pulmonary hypertension caused by left heart disease (pulmonary venous hypertension)
- Group 3: Pulmonary hypertension caused by lung diseases, hypoxia, or both
- Group 4: Chronic thromboembolic pulmonary hypertension and pulmonary hypertension caused by pulmonary artery obstructions
- Group 5: Pulmonary hypertension caused by unclear or multifactorial mechanisms.
The majority of pulmonary hypertension diagnoses relate to left heart disease (WHO group 2) or lung disease (WHO group 3), with only a small fraction accounted for by pulmonary arterial hypertension (PAH) (WHO group 1) and chronic thromboembolic pulmonary hypertension (WHO group 4) 30. Idiopathic pulmonary hypertension (WHO group 5) in particular is rare, with an estimated incidence of approximately 1 case per million and a prevalence of 7 cases per million 28. Because current disease-specific pulmonary hypertension medications are approved only for idiopathic pulmonary hypertension (WHO group 5) and other pulmonary arterial hypertension WHO group 1 (pulmonary arterial hypertension [PAH]) conditions, it is critical for treating physicians to have a thorough understanding of the differential diagnosis and workup that is required for patients with suspected pulmonary hypertension.
Among the five pulmonary hypertension groups, pulmonary arterial hypertension (PAH) (WHO group 1) is one of the most aggressive types of pulmonary hypertension 36. Because most of the progress in the specialty has been made in the research and clinical care of pulmonary arterial hypertension (PAH) (WHO group 1). Pulmonary arterial hypertension arises spontaneously, hereditarily, or as a complication of liver cirrhosis, connective tissue disease, HIV infection, congenital heart disease, schistosomiasis, or drug and toxin use 36. Symptoms of pulmonary arterial hypertension and other types of pulmonary hypertension are non-specific, frequently leading to delays in diagnosis and treatment.
In many pulmonary hypertension cases, no cause can be identified, and in these cases the disease is referred to as idiopathic pulmonary hypertension (IPAH). Sometimes pulmonary hypertension can develop due to another medical condition, including connective tissue disease, congenital heart disease, liver disease, HIV, and others. This type of pulmonary hypertension is known as associated pulmonary arterial hypertension (APAH). Some families have a form of pulmonary hypertension that can be inherited. This is known as heritable pulmonary arterial hypertension (HPAH). Chronic thromboembolic pulmonary hypertension (CTEPH) is a form of pulmonary hypertension caused by old blood clots in the lungs (pulmonary embolism).
If left untreated, pulmonary hypertension can lead to right heart failure and death. Pulmonary hypertension can’t be cured, but treatments can reduce your symptoms and help you manage your condition. Fortunately, because of major advancements made in the past two decades, therapies that target the pulmonary arteries (pulmonary hypertension-targeted therapies) are available to help relieve symptoms, improve quality of life and slow down the progression of the disease in patients with WHO Group 1 pulmonary arterial hypertension (PAH).
Patients with WHO Groups 2 and 3 pulmonary hypertension can benefit from treating the underlying left heart and lung diseases, respectively. Patients with WHO Group 4 pulmonary hypertension can benefit from either a surgery to remove the clots or a pulmonary hypertension-targeted therapy if they are unable to have the surgery or have pulmonary hypertension remaining after the surgery.
Treating pulmonary hypertension
If you are newly diagnosed with pulmonary arterial hypertension (PAH), you should be referred to an accredited pulmonary hypertension care center for thorough evaluation. Because pulmonary arterial hypertension (PAH) is such a rare disease, it is extremely valuable to see a specialist at an accredited center to ensure you are getting the most up-to-date treatment options.
Pulmonary hypertension can’t be cured, but treatment can reduce the symptoms and help you manage your condition.
Pulmonary hypertension usually gets worse over time. Left untreated, it may cause heart failure, which can be fatal, so it’s important treatment is started as soon as possible.
If another condition is causing pulmonary hypertension, the underlying condition should be treated first. This can sometimes prevent the pulmonary arteries being permanently damaged.
Treatments for pulmonary hypertension may include anticoagulant medicines to reduce the blood’s ability to thicken (clot) and diuretics to remove excess fluid as a result of heart failure.
You may also be offered medication to widen the blood vessels.
Home oxygen treatment may also be prescribed if the level of oxygen in your blood is low. DO NOT change how much oxygen is flowing without asking your doctor. Have a backup supply of oxygen at home or with you when you go out. Keep the phone number of your oxygen supplier with you at all times.
A number of FDA-approved medications are available for the treatment of pulmonary hypertension. These medications are administered in several ways: directly into the vein (intravenously), beneath the skin (subcutaneously), orally, and by inhalation.
- Some pulmonary hypertension medications work by mimicking prostaglandin, a substance that pulmonary hypertension patients tend to be deficient in. These treatments are called are prostacyclins, and include epoprostenol (Flolan™), treprostinil (Remodulin™ and Tyvaso™), and iloprost (Ventavis™).
- Other pulmonary hypertension medications work by reversing the effects of endothelin, a substance that pulmonary hypertension patients tend to have in excess. These are called endothelin receptor antagonists, and include bosentan (Tracleer™) and ambrisentan (Letairis®).
- Still other pulmonary hypertension treatments work by allowing the lungs to produce more of their own natural vasodilators. Called PDE 5 Inhibitors, this category of treatment includes sildenafil (Revatio™) and tadalafil (Adcirca™).
- Doctors may also prescribe the anticoagulant warfarin (Coumadin™) to prevent blood clots, diuretics to reduce fluid retention, and supplemental oxygen to help patients breathe.
Other important tips to follow:
- Avoid pregnancy
- Avoid heavy physical activities and lifting
- Avoid traveling to high altitudes
- Get a yearly flu vaccine, as well as other vaccines such as the pneumonia vaccine
- Stop smoking
The Lung Association recommends patients and caregivers join:
- The Living with Lung Disease Support Community to connect with others facing this disease: https://www.inspire.com/groups/american-lung-association-lung-disease
- Pulmonary Hypertension Association: https://phassociation.org/
- Pulmonary Hypertension Association UK: http://www.phauk.org/
Figure 1. Pulmonary hypertension
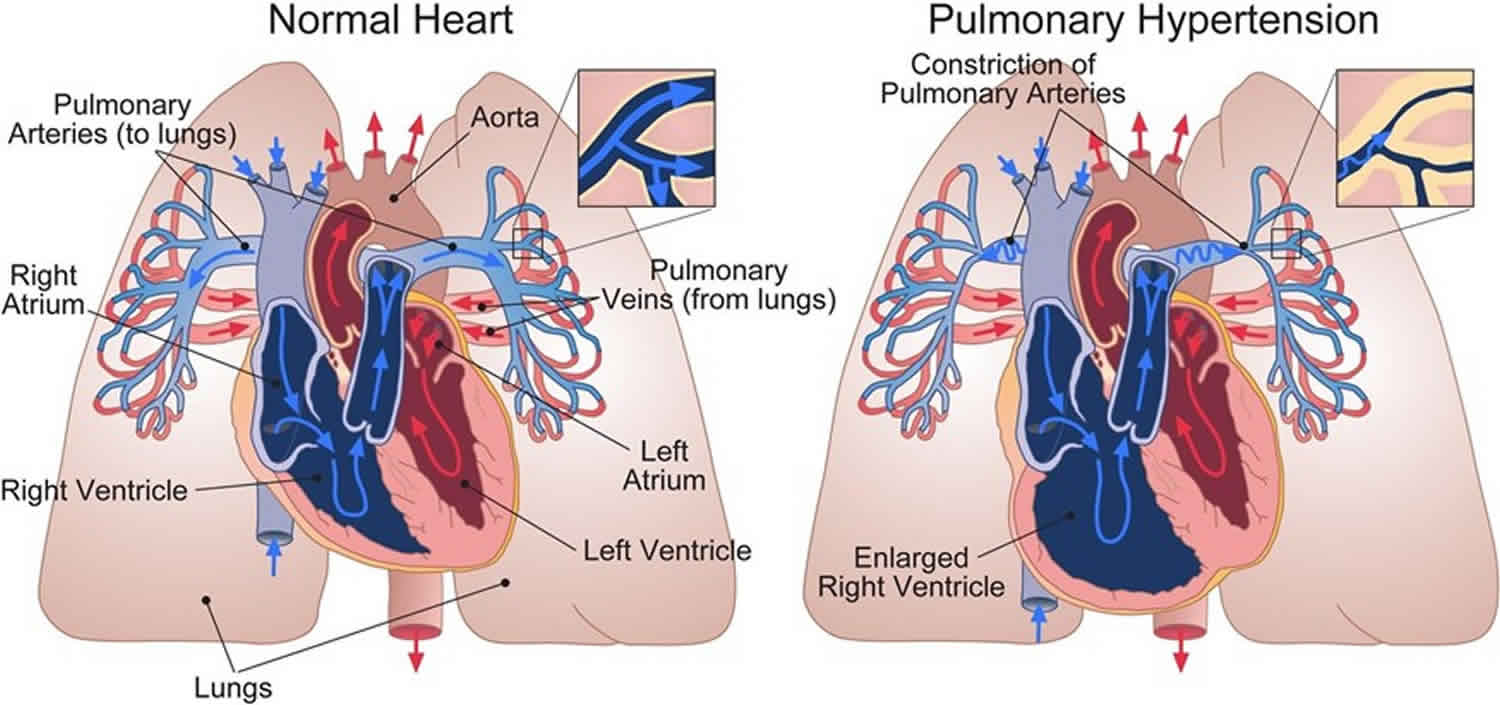
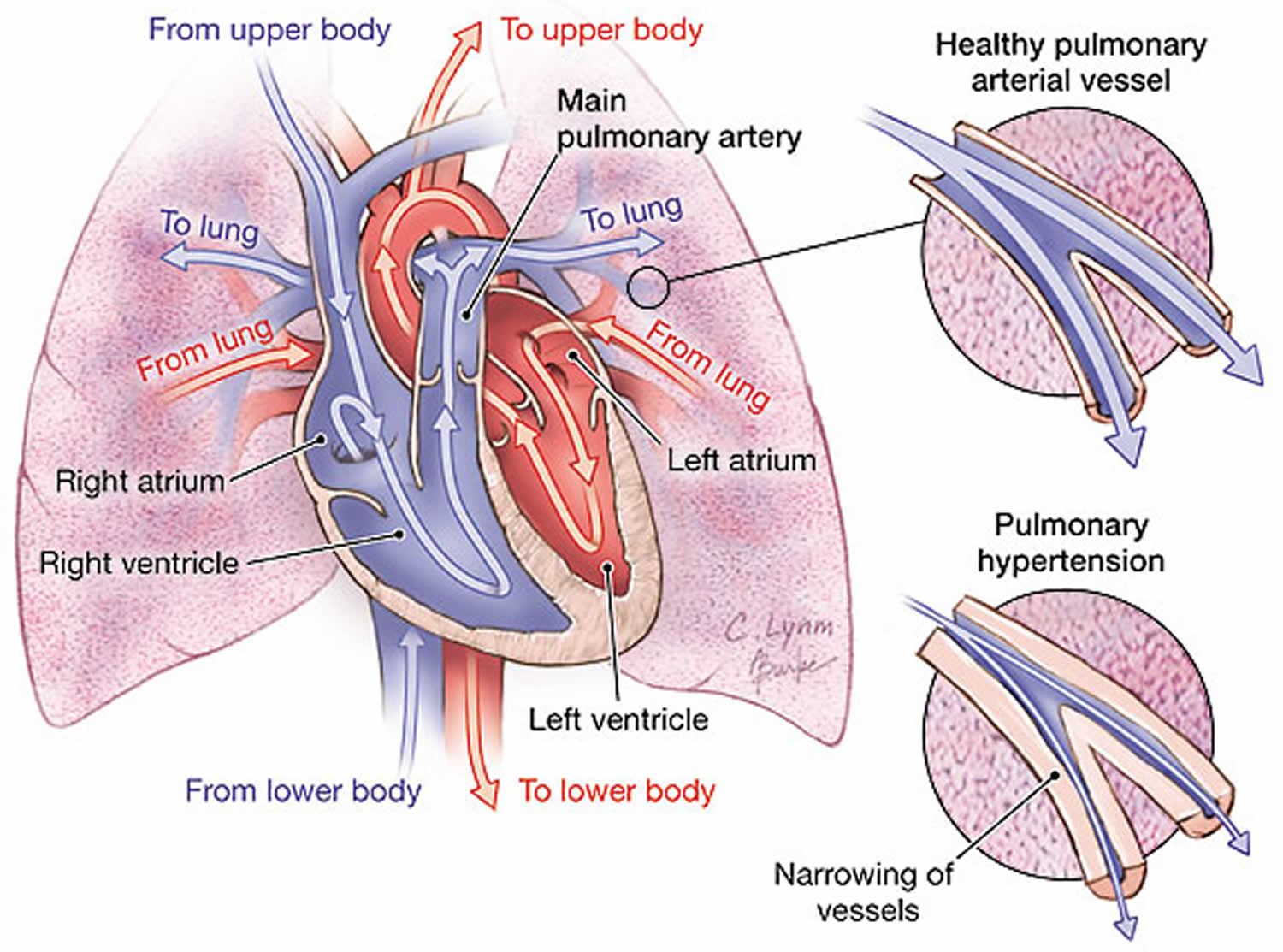
Figure 2. Pulmonary artery anatomy
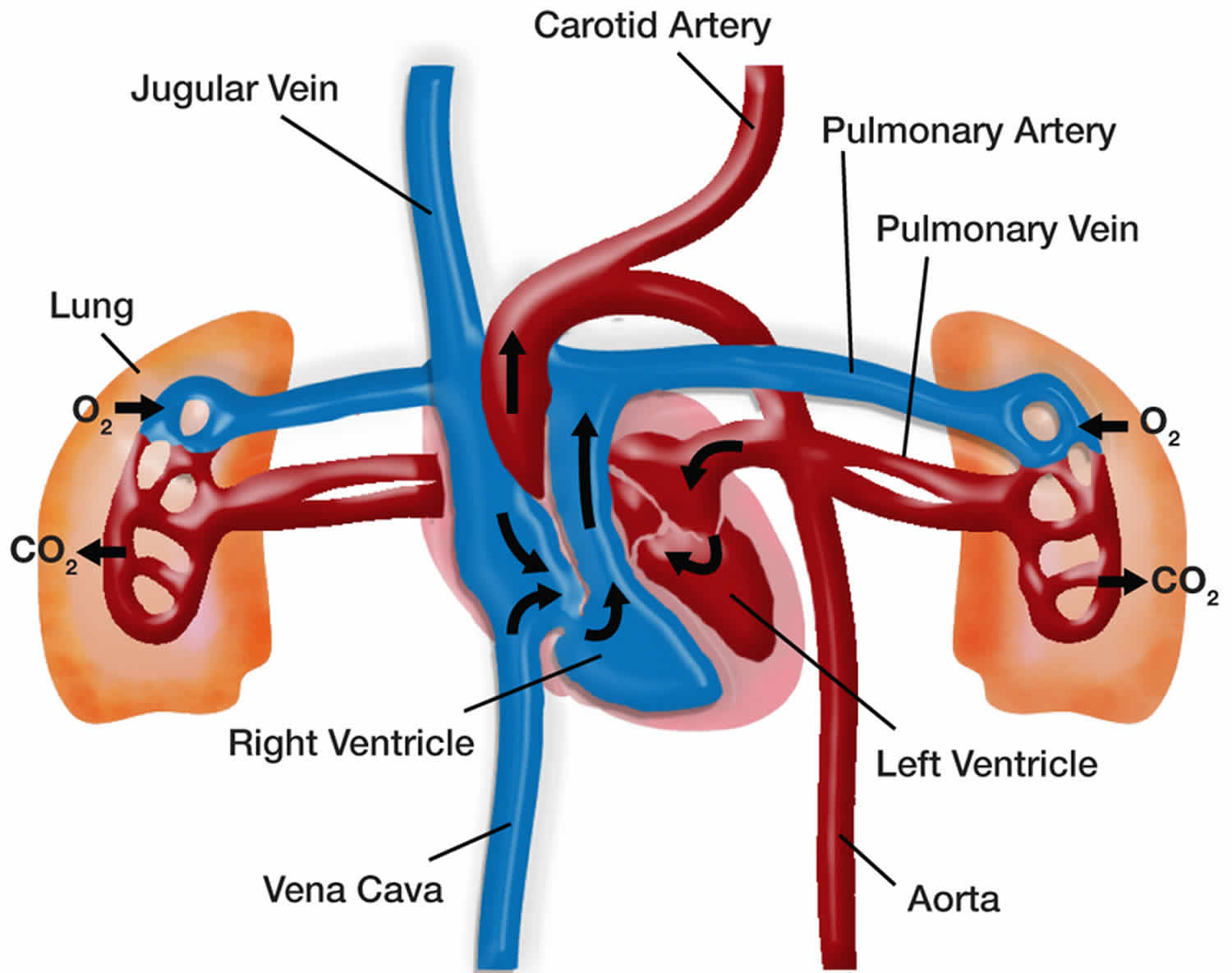
Figure 3. Pulmonary hypertension diagnostic algorithm
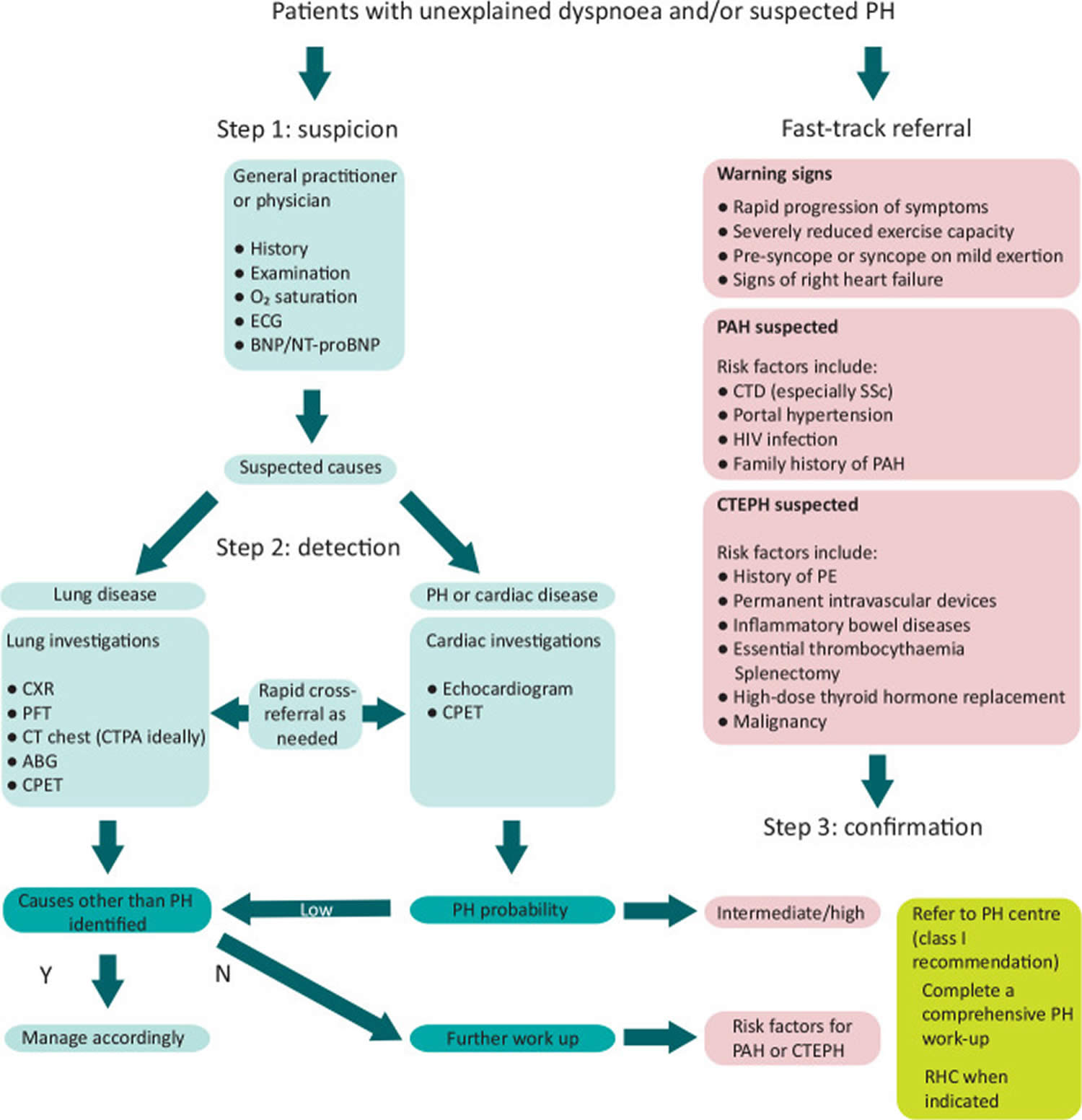
Footnotes: Diagnostic algorithm for patients with unexplained dyspnea and/or suspected pulmonary hypertension. Adapted from the 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension 27.
Abbreviations: ABG = arterial blood gas; BNP = brain natriuretic peptide; CPET = cardiopulmonary exercise testing; CT = computed tomography; CTEPH = chronic thromboembolic pulmonary hypertension; ECG = electrocardiogram; HIV = human immunodeficiency virus; N = no; NT-proBNP = N-terminal pro-brain natriuretic peptide; PAH = pulmonary arterial hypertension; PE = pulmonary embolism; PFT = pulmonary function tests; PH = pulmonary hypertension; Y = yes.
[Source 12 ]Pulmonary hypertension types and causes
| Classification* | Epidemiology | Hemodynamic characteristics† | Treatment |
|---|---|---|---|
| Group 1‡ | |||
|
|
| |
| Group 2‡ | |||
|
|
| |
| Group 3‡ | |||
|
|
| |
| Group 4‡ | |||
|
|
| |
| Group 5‡ | |||
|
|
|
|
Footnotes: World Health Organization (WHO) Pulmonary hypertension classification, epidemiology, hemodynamic characteristics, and treatments.
‡ Pulmonary hypertension is clinically divided into 5 groups:
- Group 1: Pulmonary arterial hypertension (PAH)
- Group 2: Pulmonary hypertension caused by left heart disease
- Group 3: Pulmonary hypertension caused by lung diseases, hypoxia, or both
- Group 4: Chronic thromboembolic pulmonary hypertension and pulmonary hypertension caused by pulmonary artery obstructions
- Group 5: Pulmonary hypertension caused by unclear or multifactorial mechanisms.
* According to the Sixth World Symposium on Pulmonary Hypertension.
† Defined by Sixth World Symposium on Pulmonary Hypertension, 2018. The 2022 European Respiratory Society-European Society of Cardiology guidelines suggest a cut-off for pulmonary vascular resistance of 2 Woods units.
Abbreviations: COPD = chronic obstructive pulmonary disease; COPD-PH = pulmonary hypertension related to COPD; ILD-PH = pulmonary hypertension related to interstitial lung disease; LVEF = left ventricular ejection fraction; mPAP = mean pulmonary arterial pressure; OSA = obstructive sleep apnea; PaCO2 = partial pressure of carbon dioxide in arterial blood; PAH = pulmonary arterial hypertension; PAWP = pulmonary arterial wedge pressure; PH = pulmonary hypertension; PVR = pulmonary vascular resistance; SpO2 = arterial oxygen saturation measured by pulse oximeter.
[Source 1 ]Table 1. Hemodynamic definitions of pulmonary hypertension
| Definitions | Characteristics | WHO Clinical Groups# |
|---|---|---|
| Pulmonary hypertension (PH) | Mean pulmonary arterial pressure (mPAP) >20 mmHg | |
| Pre-capillary pulmonary hypertension | Mean pulmonary arterial pressure (mPAP) >20 mmHg | 1, 3, 4 and 5 |
| Pulmonary arterial wedge pressure (PAWP) ≤15 mmHg | ||
| Pulmonary vascular resistance (PVR) ≥3 Wood Units | ||
| Isolated post-capillary pulmonary hypertension (IpcPH) | mPAP >20 mmHg | 2 and 5 |
| PAWP >15 mmHg | ||
| PVR <3 WU | ||
| Combined pre- and post-capillary pulmonary hypertension (CpcPH) | mPAP >20 mmHg | 2 and 5 |
| PAWP >15 mmHg | ||
| PVR ≥3 WU | ||
| Exercise pulmonary hypertension *** | mPAP/cardiac output slope between rest and exercise >3 mmHg/L/min |
Abbreviations: mPAP = mean pulmonary arterial pressure; PAWP = pulmonary arterial wedge pressure; PVR = pulmonary vascular resistance; WU = Wood Units. #: Group 1 = pulmonary arterial hypertension (PAH); Group 2 = pulmonary hypertension due to left heart disease; Group 3 = pulmonary hypertension due to lung diseases and/or hypoxia; Group 4 = pulmonary hypertension due to pulmonary artery obstructions; Group 5 = pulmonary hypertension with unclear and/or multifactorial mechanisms.
Footnotes: *** Exercise pulmonary hypertension, defined by an mPAP/cardiac output (CO) slope >3 mmHg/L/min between rest and exercise, has been re-introduced 48. The mPAP/cardiac output (CO) slope is strongly age dependent and its upper limit of normal ranges from 1.6–3.3 mmHg/L/min in the supine position 48. An mPAP/cardiac output (CO) slope >3 mmHg/L/min is not physiological in subjects aged <60 years and may rarely be present in healthy subjects aged >60 years 48. A pathological increase in pulmonary pressure during exercise is associated with impaired prognosis in patients with exercise dyspnea 49 and in several cardiovascular conditions 50, 51, 52, 53. Although an increased mPAP/CO slope defines an abnormal hemodynamic response to exercise, it does not allow for differentiation between pre- and post-capillary causes. The PAWP/CO slope with a threshold >2 mmHg/L/min may best differentiate between pre- and post-capillary causes of exercise pulmonary hypertension 54, 55.
[Source 13 ]Pulmonary hypertension key facts
Pulmonary hypertension is common, with multiple subtypes and causes that have individualized treatments 12.
The hemodynamic definition of pulmonary hypertension by right heart catheterization has changed to a lower mean pulmonary arterial pressure (mPAP) at rest of >20 mmHg based upon the upper normal limit in healthy individuals along with supportive prognostic data. However, there is currently no evidence for the efficacy of targeted pulmonary hypertension medications in patients with mPAP <25 mmHg and pulmonary vascular resistance (PVR) <3 Wood Units, which remains an evidence gap.
Early detection and referral to a specialist pulmonary hypertension center is crucial to institute targeted management that can improve symptoms and optimize prognosis. Physicians should be familiar with the streamlined pathway for suspicion, detection and confirmation of pulmonary hypertension, the latter step being performed at pulmonary hypertension centers. Fast-track referral pathways should be taken for clinically high-risk patients and for patient cohorts ‘at-risk’ of developing pulmonary arterial hypertension (PAH) or chronic thromboembolic pulmonary hypertension (CTEPH).
Patients with ongoing shortness of breath (dyspnea) after 3 months of anticoagulation in the setting of a history of previous thromboembolic disease require further evaluation for chronic thromboembolic pulmonary disease (CTEPD) or chronic thromboembolic pulmonary hypertension (CTEPH).
Approximately three-quarters of patients with connective tissue disease-associated PAH have scleroderma (also called systemic sclerosis – a rare, chronic autoimmune disease characterized by the hardening and thickening of the skin and internal organs due to the buildup of scar tissue or fibrosis) as the underlying connective tissue disease 56. The prevalence of PAH in patients with scleroderma ranges from 7% to 19% and carries a poor prognosis, which can be improved with early detection and initiation of targeted pulmonary hypertension therapy 57. The benefits of systematic screening for PAH in patients with scleroderma are well known but the screening methods used vary between institutions 58. The latest pulmonary hypertension guidelines recommend annual systematic screening for pulmonary hypertension in patients with scleroderma, with the DETECT algorithm recommended for screening asymptomatic adults with scleroderma of more than 3 years’ duration, forced vital capacity (FVC) ≥40%, and a transfer factor (DLCO) <60% 7. The diffusing capacity for carbon monoxide (DLCO) also known as the transfer factor for carbon monoxide (TLCO) test measures how effectively your lungs transfer oxygen from inhaled air to the blood, helping clinicians diagnose and monitor conditions such as emphysema, pulmonary fibrosis, and pulmonary hypertension 59, 60.
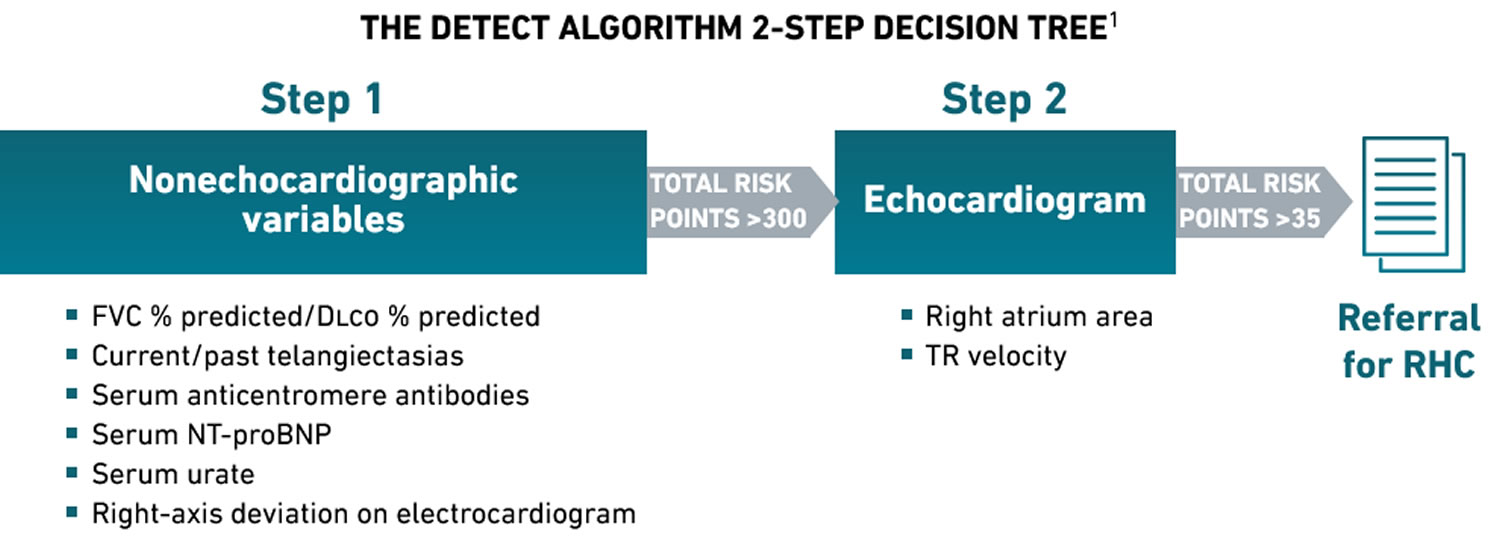
The DETECT algorithm is an evidence-based, non-invasive, multiparametric clinical screening tool that has a high sensitivity and high negative predictive value for PAH detection in scleroderma 61. Using the DETECT algorithm tool minimizes missed diagnoses, identifies milder pulmonary hypertension, and addresses resource utilisation, particularly for echocardiography.
The 2022 European Society of Cardiology/European Respiratory Society Guidelines for pulmonary hypertension gave the DETECT algorithm the highest class recommendation (Class 1) to identify asymptomatic patients with pulmonary arterial hypertension (PAH) in those with scleroderma of >3 years’ disease duration, an forced vital capacity (FVC) ≥40%, and a DLCO <60% 7.
The DETECT algorithm recommended right heart catheterization in 62% of patients (referral rate) and missed 4% of PAH patients (false negatives) 61. By comparison, using the European Society of Cardiology/European Respiratory Society Guidelines to these patients, 29% (n=24) of diagnoses were missed while requiring an right heart catheterization referral rate of 40% 61.
Figure 4. DETECT algorithm
Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is a rare serious progressive (meaning it gets worse over time) condition characterized by abnormally high blood pressure (hypertension) in the pulmonary artery, the blood vessel that carries deoxygenated blood (venous blood) from the right side of your heart to your lungs, leading to increased resistance to blood flow and strain on the right side of the heart 63, 64, 2, 27, 65, 66, 67, 68. Blood (deoxygenated blood or venous blood) moves from your heart to your lungs through blood vessels called pulmonary arteries. The pulmonary artery carries blood from the right side of your heart to your lungs to pick up a fresh supply of oxygen. Once in your lungs, the deoxygenated blood (venous blood) travels through many small, thin blood vessels called capillaries. There, the blood picks up more oxygen and transfers carbon dioxide to the lungs—a process called gas exchange. The oxygen-rich blood passes from your lungs back to your heart through the pulmonary veins. If the small blood vessels in your lungs become narrowed, blocked or damaged, the blood does not flow through them as well. As a result, blood can’t flow through your lungs as well as it should. This can increase the blood pressure in the pulmonary arteries and cause pulmonary hypertension.
Pulmonary arterial hypertension (PAH) is one form of a broader serious condition known as pulmonary hypertension. In pulmonary hypertension most of the very small arteries throughout your lungs become narrowed in diameter, which increases the resistance to blood flow through your lungs, making it harder for blood to flow through them. To overcome the increased resistance, blood pressure increases in the pulmonary artery and in the right ventricle of your heart, which is the chamber that pumps blood into the pulmonary artery. Ultimately, the increased blood pressure can damage the right ventricle of your heart, which can lead to right-sided heart failure and potentially heart failure.
Primary pulmonary hypertension (PAH) is classified in the World Health Organization’s (WHO) classification system as part of group 1. The WHO classification of pulmonary hypertension is based on the mechanism or underlying cause 27:
- Group 1: Pulmonary arterial hypertension (PAH) can be idiopathic (i.e., primary pulmonary hypertension) or due to congenital left to right intracardiac shunts, portal hypertension, persistent pulmonary hypertension of the newborn, collagen vascular diseases, HIV infection.
- Group 2: Pulmonary hypertension secondary to left heart disease (pulmonary venous hypertension)
- Group 3: Pulmonary hypertension associated with hypoxemia
- Group 4: Pulmonary hypertension due to chronic thrombotic disease, embolic disease, or both
- Group 5: Miscellaneous
In the United States, about 1,000 new cases of pulmonary arterial hypertension (PAH) are diagnosed each year 69. Pulmonary arterial hypertension (PAH) is most common in women between the ages of 30-60 70. Pulmonary arterial hypertension (PAH) is 2 to 7 times times more likely to be diagnosed in females than males. Males over age 65 who develop pulmonary arterial hypertension (PAH) are more likely to have severe cases. Pulmonary arterial hypertension (PAH) can also affect infants. This condition is known as persistent pulmonary hypertension in the neonate (PPHN) or pulmonary hypertension in newborns.
The exact cause of pulmonary arterial hypertension (PAH) is unknown. It is unlike other forms of pulmonary hypertension, where high blood pressure in the lungs is caused by underlying heart or lung disease. Researchers believe that pulmonary arterial hypertension (PAH) occurs when there is injury to the cells that line the blood vessels of your lung, which over time results in this blood vessel disease. If the cause of this change is unknown it is referred to as idiopathic pulmonary arterial hypertension (IPAH) 71. If the change is believed to be caused by a genetic mutation it is called heritable pulmonary arterial hypertension or familial pulmonary arterial hypertension. Approximately 15-20% of pulmonary arterial hypertension (PAH) patients have heritable pulmonary arterial hypertension (familial pulmonary arterial hypertension). Other conditions that are associated with the development of pulmonary arterial hypertension (PAH) include: connective tissue disorders like scleroderma, systemic lupus erythematosus (SLE), critical congenital heart disease, or Down syndrome. Researchers have also identified nongenetic factors that increase the risk of developing pulmonary arterial hypertension. These include certain drugs used as appetite suppressants and several illegal drugs, such as cocaine and methamphetamine. Pulmonary arterial hypertension is also a rare complication of certain infectious diseases such as HIV and schistosomiasis or associated with other medical conditions like cirrhosis of the liver and congenital heart diseases.
Pulmonary arterial hypertension (PAH) causes and subtypes 12:
- Idiopathic pulmonary arterial hypertension (IPAH)
- Non-responders at vasoreactivity testing
- Acute responders at vasoreactivity testing
- Heritable pulmonary arterial hypertension (HPAH) also called familial pulmonary arterial hypertension (FPAH)
- Pulmonary arterial hypertension associated with drugs and toxins 24
- Aminorex
- Fenfluramine
- Dexfenfluramine
- Benfluorex
- Methamphetamines
- Dasatinib
- Toxic rapeseed oil
- Cocaine
- Phenylpropanolamine
- L-tryptophan
- St John’s wort
- Amphetamines
- Interferon-α and -β
- Alkylating agents
- Bosutinib
- Direct-acting antiviral agents against hepatitis C virus
- Leflunomide
- Indirubin (Chinese herb Qing-Dai)
- Pulmonary arterial hypertension associated with:
- Connective tissue disease
- HIV (human immunodeficiency virus) infection
- Portal hypertension
- Congenital heart disease
- Schistosomiasis
- Pulmonary arterial hypertension (PAH) with features of venous/capillary involvement e.g., pulmonary capillary hemangiomatosis or pulmonary veno-occlusive disease
- Persistent pulmonary hypertension of the newborn (PPHN)
Pulmonary arterial hypertension (PAH) signs and symptoms occur when the increased blood pressure in the pulmonary artery cannot fully overcome the increased resistance to blood flow. As a result, the flow of oxygenated blood from your lungs to the rest of the body is insufficient. Shortness of breath during physical activity (exertional dyspnea) and fainting spells are the most common symptoms of pulmonary arterial hypertension (PAH). Some people with pulmonary arterial hypertension may experience additional symptoms, particularly as the condition worsens. Other symptoms include dizziness, swelling (edema) of the ankles or legs, fatigue, chest pain, and a rapid heart rate. If left untreated, pulmonary arterial hypertension (PAH) can lead to serious complications such as right heart failure, arrhythmias, and blood clots.
Pulmonary arterial hypertension (PAH) diagnosis involves a combination of medical history, physical exam, and tests such as echocardiogram and cardiac catheterization. Your doctor may also refer you to a pulmonologist (lung specialist) or cardiologist (heart specialist). These specialists will run specific tests to check your heart and lung function. They’ll determine what form of pulmonary hypertension you have (PAH or another form). They’ll also evaluate how far your condition has progressed.
There’s no cure for pulmonary arterial hypertension (PAH). Treatment options vary from person to person and depends on the cause, the type and severity of the pulmonary arterial hypertension. If you are newly diagnosed with pulmonary arterial hypertension (PAH), you should be referred to an accredited pulmonary hypertension care center for thorough evaluation. Because pulmonary arterial hypertension (PAH) is such a rare disease, it is extremely valuable to see a specialist at an accredited center to ensure you are getting the most up-to-date treatment options.
Pulmonary arterial hypertension (PAH) key facts
- Pulmonary arterial hypertension (PAH) is a specific type of pulmonary hypertension where the tiny blood vessels in the lungs become scarred.
- The symptoms of pulmonary arterial hypertension (PAH) are common to other diseases which makes it more difficult to diagnose.
- Pulmonary arterial hypertension (PAH) is a complex condition that will usually worsen without the right treatment.
Figure 5. Pulmonary arterial hypertension pathophysiology and current therapeutic targets
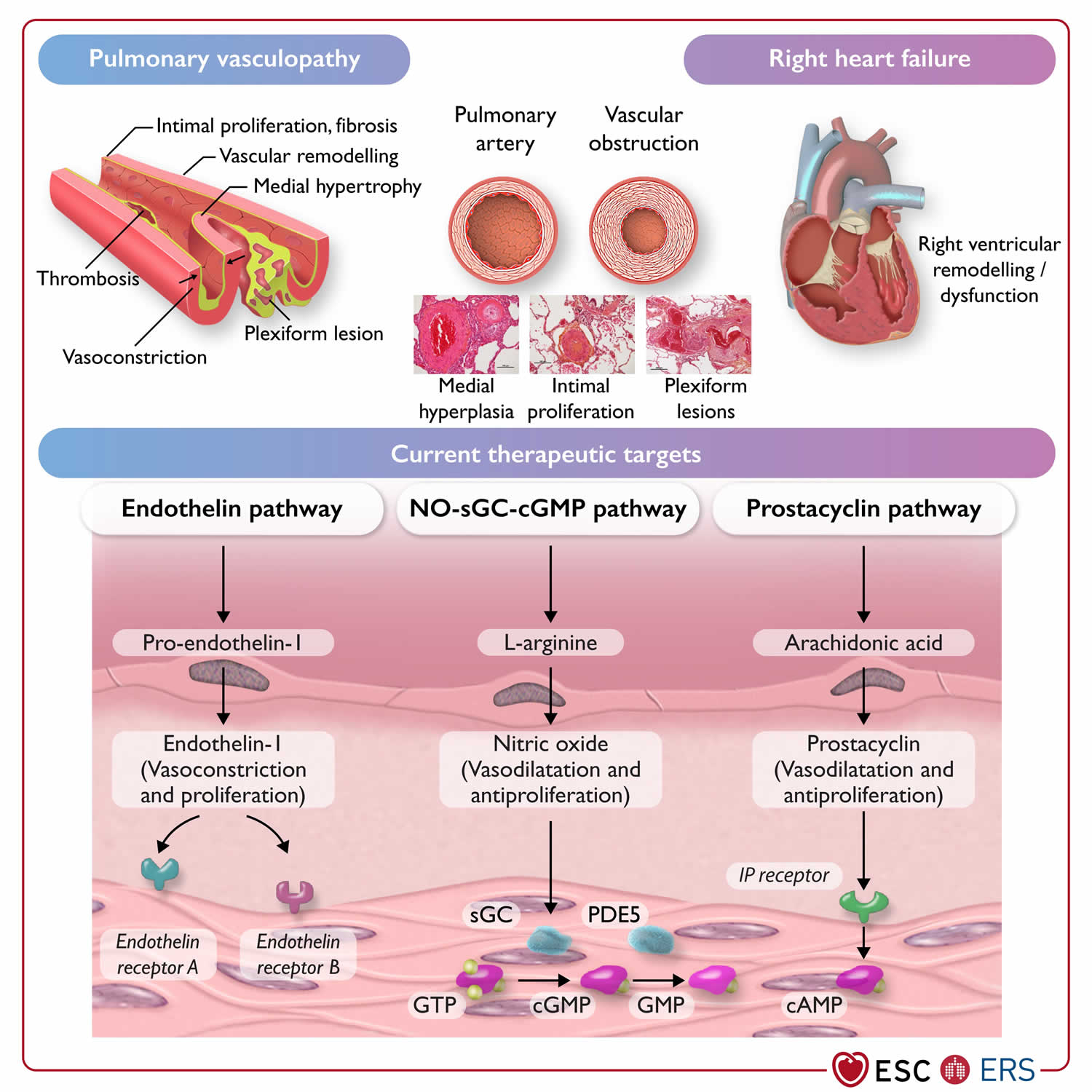
Footnotes: Pathophysiology and current therapeutic targets of pulmonary arterial hypertension (group 1).
Abbreviations: cAMP, cyclic adenosine monophosphate; (c)GMP, (cyclic) guanosine monophosphate; GTP, guanosine-5′-triphosphate; IP receptor, prostacyclin I2 receptor; NO, nitric oxide; PDE5, phosphodiesterase 5; sGC, soluble guanylate cyclase.
[Source 27 ]See your doctor if you’re having problems with:
- A fast heart rate (120 beats per minute).
- A respiratory infection or cough that’s getting worse.
- Constantly feeling dizzy or lightheaded.
- Episodes of chest pain or discomfort with physical activity.
- Extreme fatigue or decreased ability to do your normal activities.
- Nausea or lack of appetite.
- Restlessness or confusion.
- Shortness of breath that’s gotten worse, especially if you wake up feeling short of breath.
- Swelling in your ankles, legs or stomach that’s gotten worse.
- Trouble breathing with regular activities or at rest.
- Weight gain (2 pounds in one day or 5 pounds in one week).
When should I go to the emergency room?
Go to the emergency department (ER) or call your local emergency number if you have:
- A fast heart rate (120-150 beats per minute) that won’t go down.
- Fainting spells with loss of consciousness.
- Complications with your IV or infusion pump. These include infection, catheter displacement, solution leak, bleeding and IV pump malfunction.
- Shortness of breath that doesn’t go away when you rest.
- Sudden and severe chest pain.
- Sudden and severe headache.
- Sudden weakness or paralysis in your arms or legs.
How common is pulmonary arterial hypertension?
Pulmonary arterial hypertension (PAH) isn’t as common as other forms of pulmonary hypertension, including those caused by underlying heart or lung disease. Each year, about 500 to 1,000 people are diagnosed with pulmonary arterial hypertension (PAH) in the U.S 70. In Western countries, about 25 per 1 million people are living with pulmonary arterial hypertension (PAH). In a U.S. registry of 2039 patients with pulmonary artery hypertension (PAH), the average age was 51.7 (plus or minus 14.5 years) with a female-to-male ratio of 3.9 to 1. Of those patients, 46.6% (950) were classified as idiopathic PAH (IPAH). Estimates are that idiopathic PAH (IPAH) affects 1 in 1 million, usually young females who are otherwise normal. The median survival, if left untreated, is 2.8 years 72, 73.
How does pulmonary arterial hypertension affect my body?
Pulmonary arterial hypertension strains the right side of your heart, which pumps oxygen-poor blood to your lungs. This strain can lead to right-sided heart failure. Furthermore, pulmonary arterial hypertension (PAH) slows down blood flow between your heart and lungs. This means less blood can enter your lungs to gain fresh oxygen. As a result, blood flow to the rest of your body also slows down. So, your organs and tissues can’t get enough oxygen. Without treatment, pulmonary arterial hypertension (PAH) can be fatal.
How serious is pulmonary arterial hypertension?
Pulmonary arterial hypertension (PAH) is a serious condition that can be life-threatening. An early diagnosis and swift treatment can help you live longer and have a better quality of life. PAH is a rare condition, affecting about 15-50 people per million in the US. Since patients with PAH are often not diagnosed until their symptoms are severe, they may only have a few years to live unless they get proper treatment. Fortunately, advances in therapies have led to impressive improvements for patients with pulmonary arterial hypertension.
Could pulmonary hypertension run in my family?
Yes, but heritable PAH (HPAH) is relatively uncommon. Of the small percentage of people who do carry the pulmonary hypertension gene, only a small number of carriers will develop the disease. Genetic testing is available to find out if you carry the pulmonary hypertension gene.
Can pulmonary arterial hypertension be reversed?
Currently, medications can slow down pulmonary arterial hypertension (PAH) progression but not reverse the damage already done. However, researchers are working on promising new medications that could help reverse pulmonary arterial hypertension (PAH). Such medications would repair damage to the endothelial cells that line your pulmonary arteries.
Talk with your doctor to learn more about the latest research and clinical trials for pulmonary arterial hypertension (PAH) therapies.
Who does pulmonary arterial hypertension affect?
Pulmonary arterial hypertension (PAH) can affect adults at any age. It’s more common among females, who are usually diagnosed between the ages of 30 and 60. Males over age 65 who develop pulmonary arterial hypertension (PAH) are more likely to have severe cases. Pulmonary arterial hypertension (PAH) can also affect infants. This condition is known as persistent pulmonary hypertension in the neonate (PPHN) or pulmonary hypertension in newborns.
Pregnancy and pulmonary arterial hypertension
Historically, pregnancy in women with pulmonary arterial hypertension (PAH) and other forms of severe pulmonary hypertension has been associated with maternal mortality rates of up to 56% and neonatal mortality rates of up to 13% 74. With improved treatment of pulmonary arterial hypertension (PAH) and new approaches to managing women during pregnancy and the peri-partum period, maternal mortality has declined but remains high, ranging 11–25% 75, 76, 77, 78, 79. For these reasons, previous European Society of Cardiology and European Respiratory Society Guidelines for the diagnosis and treatment of pulmonary hypertension have recommended that patients with pulmonary arterial hypertension should avoid pregnancy 80, 81. However, there are reports of favourable pregnancy outcomes in women with pulmonary hypertension, including, but not limited to, women with idiopathic pulmonary arterial hypertension (IPAH) who respond to calcium channel blocker therapy 77, 78, 82, 83. Nonetheless, pregnancy remains associated with unforeseeable risks, and may accelerate pulmonary hypertension progression 84. Women with pulmonary hypertension can deteriorate at any time during or after pregnancy. Therefore, physicians have a responsibility to inform patients about the risks of pregnancy, so that women and their families can make informed decisions.
Women with poorly controlled disease, indicated by an intermediate- or high-risk profile and signs of right ventricular dysfunction, are at high risk of adverse outcomes; in the event of pregnancy, they should be carefully counseled and early termination should be advised 27. For patients with well-controlled disease, a low-risk profile, and normal or near-normal resting hemodynamics who consider becoming pregnant, individual counselling and shared decision-making are recommended 27. In such cases, alternatives such as adoption and surrogacy may also be explored. Pre-conception genetic counselling should also be considered in heritable pulmonary arterial hypertension.
Women with pulmonary hypertension who become pregnant or present during pregnancy with newly diagnosed PAH should be treated, whenever possible, in centers with a multidisciplinary team experienced in managing pulmonary hypertension in pregnancy 27. If pregnancy is continued, PAH therapy may have to be adjusted. It is recommended to stop endothelin receptor antagonists (ERAs), riociguat, and selexipag because of potential or unknown teratogenicity 85. Despite limited evidence, calcium channel blockers, Phosphodiesterase 5 inhibitors, and inhaled/i.v./subcutaneous (s.c.) prostacyclin analogues are considered safe during pregnancy 82, 86.
Pregnancy in pulmonary hypertension is a very sensitive topic and requires empathic communication. Psychological support should be offered whenever needed.
Birth control
Women with pulmonary hypertension of childbearing potential should be provided with clear contraceptive advice, considering the individual needs of the woman but recognizing that the implications of contraceptive failure are significant in pulmonary hypertension. With appropriate use, many forms of contraception, including oral contraceptives, are highly effective. In patients treated with bosentan, reduced efficacy of hormonal contraceptives should be carefully considered 87. Using hormonal implants or an intrauterine device (IUD) are alternative options with low failure rates. Surgical sterilization may be considered but is associated with peri-operative risks. Emergency post-coital hormonal contraception is safe in pulmonary hypertension.
Pulmonary arterial hypertension treatment
Treatment options vary from person to person and depends on the cause, the type and severity of the pulmonary arterial hypertension. If you are newly diagnosed with pulmonary arterial hypertension (PAH), you should be referred to an accredited pulmonary hypertension care center for thorough evaluation. Because pulmonary arterial hypertension (PAH) is such a rare disease, it is extremely valuable to see a specialist at an accredited center to ensure you are getting the most up-to-date treatment options.
The management of pulmonary arterial hypertension (PAH) is based on New York Heart Association (NYHA) Functional Classification (i.e., patient symptoms and functional status) with the goal of positive impact on the quality of life by improving symptoms and functional status.
The New York Heart Association (NYHA) Functional Classification is a system that categorizes heart failure patients based on their symptoms and physical activity limitations, ranging from Class I (no limitation) to Class IV (symptoms at rest).
Here’s a breakdown of the NYHA classifications. New York Heart Association (NYHA) classification system groups heart failure into four categories by number. You may see Roman numerals used for these category names.
- Class 1 (I) heart failure. There are no heart failure symptoms.
- Class 2 (II) heart failure. Everyday activities can be done without difficulty. But exertion causes shortness of breath or fatigue.
- Class 3 (III) heart failure. It’s difficult to complete everyday activities.
- Class 4 (IV) heart failure. Shortness of breath occurs even at rest. This category includes the most severe heart failure.
Lifestyle and home remedies
Lifestyle changes may help improve pulmonary hypertension symptoms. Try these tips:
- Eat healthy. Eat a healthy diet rich in whole grains, fruits and vegetables, lean meats, and low-fat dairy products. Try to stay away from saturated fat, trans fat and cholesterol. Limit salt.
- Stay as active as possible and manage your weight. Even mild forms activity might be too exhausting for some people who have pulmonary hypertension. For others, moderate exercise, such as walking, might be helpful — especially when done during oxygen therapy. Your health care team can help you plan an appropriate exercise program.
- Don’t smoke. If you smoke, the most important thing you can do for your heart and lungs is to stop. If you need support quitting, ask your health care team for treatment that can help. Avoid secondhand smoke too, if possible.
- Get plenty of rest. Resting can reduce tiredness related to pulmonary hypertension.
- Avoid high altitudes. High altitudes can make pulmonary hypertension worse. If you live at an altitude of 8,000 feet (2,438 meters) or higher, you might be told to consider moving to a lower altitude.
- Avoid activities that can excessively lower blood pressure. These include sitting in a hot tub or sauna or taking long hot baths or showers. Such activities lower blood pressure and can cause fainting or even death. Also, do not do activities that cause a lot of straining, such as lifting heavy objects or weights.
- Give your health care team a list of your medicines. Some medicines can make pulmonary hypertension worse or affect its treatment.
- Get regular health checkups. Many pulmonary hypertension care centers require visits every few months and regular testing such as echocardiograms and 6-minute walk testing. Your doctor may also have you complete cardiopulmonary exercise testing (CPET), a specialized type of exercise test that measures your exercise ability. Some centers will do right heart catheterizations every year to see how well treatment is working on managing pulmonary pressures and heart function. Additionally, it is essential to take your medications exactly as directed, being careful not to run out or change your schedule unless directed by your doctor.
- Get recommended vaccines. Respiratory infections can cause serious health concerns for people with pulmonary hypertension. Ask your health care team about recommend vaccines to prevent common viral infections.
- Talk to your doctor before becoming pregnant. Pregnancy can put strain on your body and for a woman with pulmonary arterial hypertension (PAH) be possibly life-threatening, so this is a subject you should discuss with your doctor prior to becoming pregnant. Pulmonary hypertension can cause serious complications to both mother and baby during pregnancy. Birth control pills can increase the risk of blood clots. Talk to your doctor about other birth control options.
Medications
If you have pulmonary arterial hypertension (PAH), you may get medicines to treat your symptoms and help you feel better. Medicines also may be used to treat or prevent complications. Pulmonary arterial hypertension (PAH) specific medications come in multiple forms: oral, inhaled and subcutaneous (meaning delivered by an injection or IV). The medicines for pulmonary arterial hypertension (PAH) work in a few ways. Some allow blood to flow more easily through the arteries of your lungs. Others help your heart and lungs work better.
Pulmonary arterial hypertension (PAH) specific medications aim to restore balance among one or more of three substances that are produced by your lungs: nitric oxide, endothelin, and prostacyclin. Although a test does not currently exist to determine which of these substances is not balanced, pulmonary arterial hypertension (PAH) medications act on these three pathways to help slow how quickly your disease worsens.
Table 2. Pulmonary arterial hypertension medications
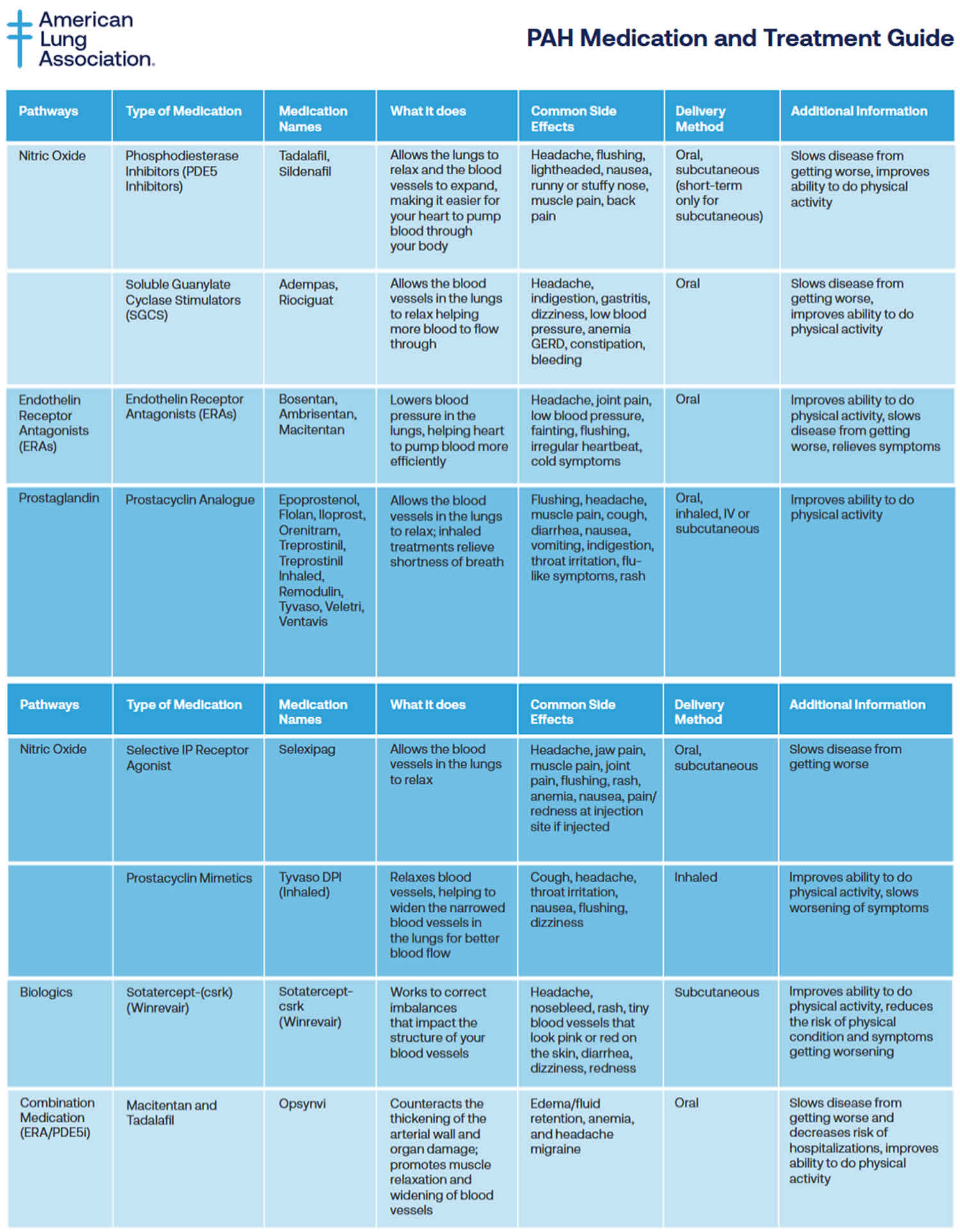
Pulmonary arterial hypertension (PAH) specific medications may include:
If you have pulmonary hypertension, you may get medicines to treat your symptoms and help you feel better. Medicines also may be used to treat or prevent complications. Treatment may include:
- Medicines to relax blood vessels (prostaglandins or prostacyclins) also called vasodilators, these medicines help open narrowed blood vessels and improve blood flow. The medicine comes in many forms. It may be breathed in, taken by mouth or given by IV. Some types are given continuously through a small pump attached to the body. Examples of vasodilators to treat pulmonary hypertension include epoprostenol (Flolan, Veletr), treprostinil (Remodulin, Tyvaso, others), Iloprost (Ventavis) and selexipag (Uptravi).
- Inhaled treatments to relieve shortness of breath and improve your ability to do physical activity. Prostacyclin analogues include Epoprostenol, Flolan, Iloprost, Orenitram, Treprostinil, Treprostinil Inhaled, Remodulin, Tyvaso, Veletri, Ventavis. Delivery Method: Oral, inhaled, IV or subcutaneous. Common Side Effects: Flushing, headache, muscle pain, cough, diarrhea, nausea, vomiting, indigestion, throat irritation, flu-like symptoms, rash.
- Selective nonprostanoid IP prostacyclin receptor agonist called Selexipag (Uptravi) targets the prostacyclin pathway by helping to prevent clotting in the arteries and by slowing the thickening of blood vessels 89. Selexipag comes as a tablet to take by mouth. It is usually taken with food twice a day. There is also injection form of Selexipag (Uptravi). Common Side Effects: Headache, jaw pain, muscle pain, joint pain, flushing, rash, anemia, nausea, pain/redness at injection site if injected.
- Tyvaso DPI (Inhaled). Tyvaso DPI is a drug-device combination therapy comprised of a small, portable, reusable, breath-powered, dry powder inhaler (DPI) for the delivery of treprostinil. Common Side Effects: Cough, headache, throat irritation, nausea, flushing, dizziness.
- Soluble guanylate cyclase (sGC) stimulators. This type of medicine relaxes the pulmonary arteries and lowers pressure in the lungs. Examples include riociguat (Adempas). Do not take these medicines if you’re pregnant.
- Medicines to widen blood vessels. Medicines called endothelin receptor antagonists (ERAs) reverse the effect of a substance in the walls of blood vessels that causes them to narrow. Such medicines include bosentan (Tracleer), macitentan (Opsumit) and ambrisentan (Letairis). They may improve energy level and symptoms. Do not take these medicines if you are pregnant.
- Medicines to increase blood flow. Medicines called phosphodiesterase 5 (PDE5) inhibitors may be used to increase blood flow through the lungs. These medicines also are used to treat erectile dysfunction. They include sildenafil (Revatio, Viagra) and tadalafil (Adcirca, Alyq, Cialis).
- Sotatercept-csrk (Winrevair) is in a class of medications called activin signaling inhibitors. Sotatercept-csrk (Winrevair) works by blocking certain substances to slow or stop the tissue changes that happen with pulmonary arterial hypertension (PAH). Sotatercept-csrk (Winrevair) can improve your ability to exercise and perform your usual activities with fewer symptoms. It can also reduce the risk of your physical condition and symptoms worsening. Sotatercept-csrk comes as a solution (liquid) to inject subcutaneously (just under the skin). It is usually given once every 3 weeks. Common Side Effects: Headache, nosebleed, rash, tiny blood vessels that look pink or red on the skin, diarrhea, dizziness, redness.
- Macitentan and Tadalafil (Opsynvi) combination works by relaxing smaller blood vessels in your lungs and increasing the supply of blood to your lungs, which reduces the workload of your heart. Macitentan and Tadalafil (Opsynvi) is only available for female patients under a restricted distribution program called the Macitentan-Containing Products REMS (Risk Evaluation and Mitigation Strategy) program. Male patients do not need to enroll in the REMS program. Macitentan is in a class of medications called endothelin receptor antagonists (ERAs). Macitentan works by stopping the action of endothelin, a natural substance that causes blood vessels to narrow and prevents normal blood flow in people who have PAH. Tadalafil is in a class of medications called phosphodiesterase (PDE) inhibitors. Tadalafil works by relaxing the blood vessels in the lungs to allow blood to flow more easily. Macitentan and tadalafil comes as a tablet to take by mouth. It is usually taken with or without food once a day. Common Side Effects: Edema/fluid retention, anemia, and headache/migraine.
- High-dose calcium channel blockers. These medicines help relax the muscles in the walls of blood vessels. They include amlodipine (Norvasc), diltiazem (Cardizem, Tiazac, others) and nifedipine (Procardia). Although calcium channel blockers can be effective, only a small number of people with pulmonary hypertension improve while taking them.
- Blood thinners also called anticoagulants, these medicines help prevent blood clots. One example is warfarin (Jantoven). Blood-thinning medicines slow the clotting process. The medicines can increase the risk of bleeding. This is especially true if you’re having surgery or a procedure that enters the body or creates an opening in the skin. Talk to your doctor about your risk.
- Digoxin (Lanoxin) also called digitalis, helps the heart squeeze better to pump blood. It also tends to slow the heartbeat. Digoxin reduces heart failure symptoms in people with heart failure with reduced ejection fraction (HFrEF). It may be more likely to be given to someone with a heart rhythm disorder, such as atrial fibrillation. The level of digoxin in the body must be checked using a blood test. If too much digoxin builds up in your blood, side effects may occur, including loss of appetite, nausea, vomiting and headaches. The heart rhythm can also become too fast or too slow. Always report any side effects of Digoxin to your doctor right away.
- Water pills also called diuretics. These medicines help the kidneys remove excess fluid from the body. This reduces the amount of work the heart has to do. Diuretics also may be used to reduce fluid buildup in the lungs, legs and belly area. Some diuretics make the body lose potassium and magnesium. Your doctor may recommend supplements to treat this. If you’re taking a diuretic, you may have regular blood tests to check your potassium and magnesium levels.
- Oxygen therapy. Breathing pure oxygen is sometimes recommended as a treatment for pulmonary hypertension. This treatment may be suggested if you live at a high altitude or have sleep apnea. Some people with pulmonary hypertension need oxygen therapy all the time.
Calcium channel blockers and the vasoactive substance are mainly used for idiopathic pulmonary hypertension (IPAH). Many new agents have been introduced, and their effectiveness can be measured by a “6-minute walk test” 90, 91, 92. Oral, high-dose calcium channel blockers (diltiazem, nifedipine) are the first-line treatment but used only in those with vasoreactivity testing positive for acute vasodilator response with short-acting pulmonary vasodilators such as adenosine, nitric oxide, or epoprostenol. The criteria for testing positive is a fall in pulmonary artery pressure to more than 10 mmHg with an increase or no change in cardiac output. Although first-line useful only in 5% of patients with idiopathic pulmonary hypertension (IPAH) and should not be used in non-responders to vasoreactivity test due to the risk of harm rather than any improvement 64. Vasoactive substances such as endothelin receptor antagonists, phosphodiesterase inhibitors, and prostanoids alter the mechanisms causing pulmonary artery smooth muscle proliferation and contraction 64.
For New York Heart Association (NYHA) Functional Class 2:
- Oral endothelin receptor antagonists (ambrisentan, bosentan, macitentan), macitentan, and modified bosentan have been shown to reduce morbidity and mortality in some studies.
- Phosphodiesterase type-5 inhibitors, PDE5 inhibitors (sildenafil, tadalafil) relax arterial smooth muscles and pulmonary artery vasodilation while inhibiting vascular remodeling.
- Non-parenteral prostanoids can be added.
For New York Heart Association (NYHA) Functional Class 3, Class 4, and those unresponsive to previous therapies:
Prostanoid agents (epoprostenol, treprostinil, iloprost): Continuous long-term intravenous epoprostenol infusion for which a semi-permanent central venous catheter is required is considered the most effective therapy. It has been shown to improve mortality, but a short half-life and high cost are the limitations. For those who cannot tolerate intravenous infusion, inhaled or subcutaneous prostanoids can be considered. Treprostinil can be used by various routes such as intravenous, subcutaneous, and inhalation. Oral prostanoids are still under clinical trials. Benefits include vasodilation, platelet inhibition, antiproliferative, and inotropic effects 93.
Soluble guanylate cyclase stimulators (riociguat, cinaciguat) are under clinical trials and are beneficial in pulmonary artery hypertension as they have a dual mode of action. They stimulate the receptor to mimic nitric oxide action and increase the sensitivity of guanylyl cyclase to endogenous nitric oxide. Riociguat has been shown to improve exercise capacity and decrease pulmonary vascular resistance in the studies.
Selexipag, a newer drug, is a selective IP prostacyclin receptor agonist. Monotherapy does relieve symptoms in patients with PAH but has not been shown to improve the prognosis and survival, and this has led to a shift to combination therapy which has shown improvement in survival (especially combination therapy including prostaglandins).
Combination therapy is being used now with the goal of targeting different mechanisms involved in the pathogenesis of PAH simultaneously (prostacyclin, endothelin, and nitric oxide pathways). A combination is considered better than increasing the dose of a single drug used and has better outcomes. Although the most common combination used is ERAs and PDE5 inhibitors, which have also been shown to reduce hospitalization in a study, but with newer drugs available now, other combinations can also be used.
Some observational studies suggest an improvement in survival in primary pulmonary hypertension with long-term anticoagulation. Also, based on the symptoms and with progression to heart failure, certain other drugs like diuretics, digoxin, and oxygen can be added.
Table 3. Dosing of pulmonary arterial hypertension medication in adults
| Starting dose | Target dose | |
|---|---|---|
| Calcium channel blockers | ||
| Amlodipine | 5 mg o.d. | 15–30 mg o.d.a |
| Diltiazem | 60 mg b.i.d.b | 120–360 mg b.i.d.b |
| Felodipine | 5 mg o.d. | 15–30 mg o.d.a |
| Nifedipine | 10 mg t.i.d. | 20–60 mg b.i.d. or t.i.d. |
| Endothelin receptor antagonists (oral administration) | ||
| Ambrisentan | 5 mg o.d. | 10 mg o.d. |
| Bosentan | 62.5 mg b.i.d. | 125 mg b.i.d. |
| Macitentan | 10 mg o.d. | 10 mg o.d. |
| Phosphodiesterase 5 inhibitors (oral administration) | ||
| Sildenafil | 20 mg t.i.d. | 20 mg t.i.d.c |
| Tadalafil | 20 or 40 mg o.d. | 40 mg o.d. |
| Prostacyclin analogues (oral administration) | ||
| Beraprost sodium | 20 µg t.i.d. | Maximum tolerated dose up to 40 µg t.i.d. |
| Beraprost extended release | 60 µg b.i.d. | Maximum tolerated dose up to 180 µg b.i.d. |
| Treprostinil | 0.25 mg b.i.d. or 0.125 mg t.i.d. | Maximum tolerated dose |
| Prostacyclin receptor agonist (oral administration) | ||
| Selexipag | 200 µg b.i.d. | Maximum tolerated dose up to 1600 µg b.i.d. |
| Soluble guanylate cyclase stimulator (oral administration) | ||
| Riociguat d | 1 mg t.i.d. | 2.5 mg t.i.d. |
| Prostacyclin analogues (inhaled administration) | ||
| Iloprost e | 2.5 µg 6–9 times per day | 5.0 µg 6–9 times per day |
| Treprostinil e | 18 µg 4 times per day | 54–72 µg 4 times per day |
| Prostacyclin analogues (i.v. or s.c. administration) | ||
| Epoprostenol i.v. | 2 ng/kg/min | Determined by tolerability and effectiveness; typical dose range at 1 year is 16–30 ng/kg/min, with wide individual variability |
| Treprostinil s.c. or i.v. | 1.25 ng/kg/min | Determined by tolerability and effectiveness; typical dose range at 1 year is 25–60 ng/kg/min, with wide individual variability |
Abbreviations: b.i.d. = twice daily; i.v. = intravenous; o.d., once daily; s.c. = subcutaneous; t.i.d. = three times daily.
Footnotes: Dosages are those commonly used in clinical practice. This does not exclude the use of alternative dosages.
a The daily dosages of amlodipine and felodipine can be administered in a single dose or divided into two doses.
b There are different release formulations of diltiazem, some of which should be administered o.d. or t.i.d.
c Sildenafil is approved at a dose of 20 mg t.i.d. but doses used in practice vary widely and are sometimes higher.
d In patients at risk of systemic hypotension, riociguat may be started at 0.5 mg t.i.d.
e Doses provided are for nebulizers and may differ with the use of other formulations and other inhalation devices.
Patient follow up
The optimal timing of follow-up right heart catheterization has not been determined. While some centers regularly perform invasive follow-up assessments, others perform them as clinically indicated, and there is no evidence that any of these strategies is associated with better outcomes.
Table 4. Pulmonary arterial hypertension follow-up
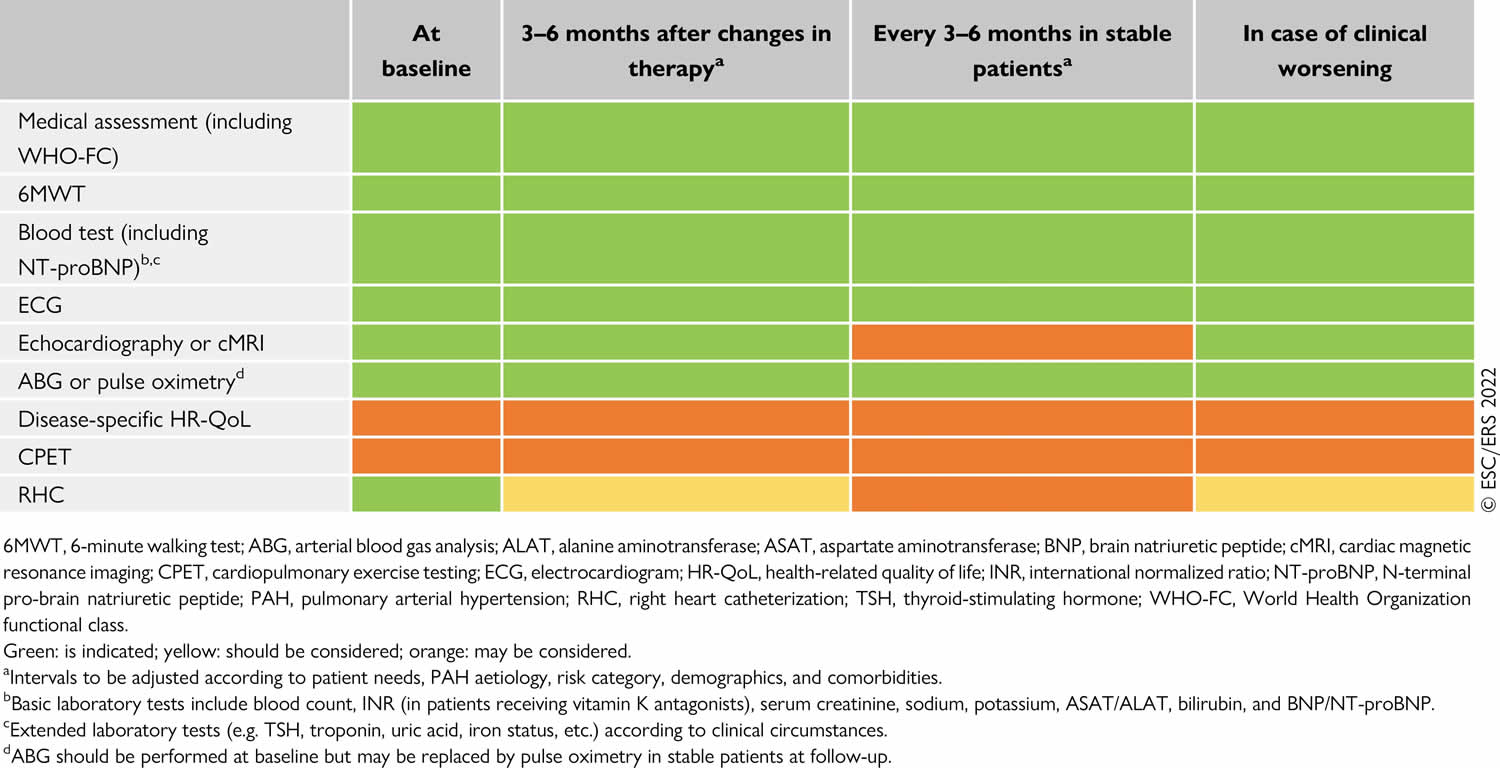
Pulmonary rehabilitation
Pulmonary rehabilitation is a supervised six – eight week exercise and education program that helps people with chronic lung diseases improve their breathing and overall well-being through exercise, education, and behavioral changes. Pulmonary rehabilitation may improve your exercise endurance, muscle strength and quality of life.
Pulmonary rehabilitation is an outpatient program and may be based in a hospital or a clinic. You may also be able to receive certain forms of pulmonary rehabilitation in your own home.
To find a pulmonary rehabilitation program in your area, visit:
Surgery and other procedures
If medicines do not help control the symptoms of pulmonary hypertension, surgery may be recommended 94. Surgeries and procedures to treat pulmonary hypertension may include:
- Atrial septostomy. This treatment may be recommended if medicines don’t control pulmonary hypertension symptoms. In an atrial septostomy, a doctor creates an opening between the upper left and right chambers of your heart. The opening reduces the pressure on the right side of the heart. Potential complications include irregular heartbeats called arrhythmias.
- Lung transplant or heart-lung transplant. Sometimes, a lung or heart-lung transplant may be needed, especially for younger people who have idiopathic pulmonary arterial hypertension. After a transplant, medicine must be taken for life to help reduce the chance of rejection.
Pulmonary Hypertension Due to Left Heart Disease
WHO Group 2 includes pulmonary hypertension due to left heart disease or post-capillary pulmonary hypertension and is defined by mean pulmonary arterial blood pressure (mPAP) >20 mmHg in the presence of an elevated pulmonary capillary wedge pressure (PAWP) >15 mmHg and normal pulmonary vascular resistance (PVR) ≤2 Woods Unit (160 dyne/second/cm5). In this group of pulmonary hypertension, the arteries and lungs are not as thick or stiff as WHO Group 1, but there are problems with how the heart squeezes or relaxes, or problems with the valves on the left side of the heart. Because of this, the left heart is unable to keep up with the blood returning from the lungs — causing a “backup” of blood which raises pressure in the lungs. WHO Group 2 is the most common form of pulmonary hypertension. In general, Group 2 pulmonary hypertension reflects the severity of left heart disease, and treatment should be directed at the underlying cause before further assessing the pulmonary hypertension itself. Targeted pulmonary hypertension therapies are not recommended in isolated pulmonary hypertension due to left heart disease, with studies showing potentially harmful outcomes 95.
Pulmonary hypertension associated with left heart disease is further subdivided into 12:
- Heart failure with preserved ejection fraction (HFpEF)
- Heart failure with reduced (≤40%) or mildly reduced (41–49%) left ventricular ejection fraction (HFrEF)
- Pulmonary hypertension associated valvular heart disease
- Pulmonary hypertension associated congenital/acquired cardiovascular conditions leading to post-capillary pulmonary hypertension.
When to refer patients with possible heart failure with preserved ejection fraction (HFpEF)-associated pulmonary hypertension to specialist centers for further evaluation by right-heart catheterization? Right-heart catheterization is recommended in cases with an intermediate probability of heart failure with preserved ejection fraction (HFpEF)-associated pulmonary hypertension when risk factors of pulmonary arterial hypertension / chronic thromboembolic pulmonary hypertension (PAH/CTEPH) are present and/or if there is evidence of right ventricular (RV) abnormality. If the probability of heart failure with preserved ejection fraction (HFpEF)-associated pulmonary hypertension is high, management should focus on the underlying left heart disease 23.
Pulmonary Hypertension Due to Lung Disease and/or Hypoxia
WHO Group 3 includes pulmonary hypertension due to chronic lung disease and/or hypoxia (low oxygen levels). These lung diseases include obstructive lung disease where the lung airways narrow and make it harder to exhale (e.g. COPD or emphysema); restrictive lung disease in which the lungs have a tough time expanding when one inhales (e.g. interstitial lung disease or pulmonary fibrosis); sleep apnea; and living in an area of high altitude for a long period of time. Arteries in the lungs tighten so that blood can only go to areas of the lungs that are receiving the most air and oxygen. This tightening leads to high blood pressure throughout the lungs. Pulmonary hypertension due to lung disease (Group 3 pulmonary hypertension) is the second commonest pulmonary hypertension subtype.
Pulmonary hypertension associated with lung diseases and/or hypoxia is further subdivided into 12:
- Pulmonary hypertension associated with obstructive lung disease or emphysema
- Pulmonary hypertension associated with restrictive lung disease
- Pulmonary hypertension associated with lung disease with mixed restrictive/obstructive pattern
- Pulmonary hypertension associated with hypoventilation syndromes
- Pulmonary hypertension associated with hypoxia without lung disease (eg high altitude)
- Pulmonary hypertension associated with developmental lung disorders.
Pulmonary Hypertension Due to Pulmonary Artery Obstructions
WHO Group 4 is due to pulmonary artery obstructions caused by chronic thromboembolic pulmonary hypertension (CTEPH) or other pulmonary artery obstructions causes including sarcomas, other malignant or non-malignant tumours, arteritis without connective tissue disease, congenital pulmonary arterial stenoses, and hydatidosis. Chronic thromboembolic pulmonary hypertension (CTEPH) can occur when the body is not able to dissolve a blood clot in the lungs. This can lead to scar tissue in the blood vessels of the lungs, which blocks normal blood flow and makes the right side of the heart work harder. This type of pulmonary hypertension is unique because it can potentially be cured through pulmonary thromboendarterectomy (PTE) surgery to remove the blood clots. However, not all chronic thromboembolic pulmonary hypertension patients are eligible for this surgery. A drug is also available for chronic thromboembolic pulmonary hypertension patients if a doctor determines that a patient is not a candidate for the pulmonary thromboendarterectomy surgery or if pulmonary hypertension remains after the surgery.
Pulmonary Hypertension Due to Unknown Causes
WHO Group 5 is where pulmonary hypertension is secondary to other diseases in ways that are not well understood. These associated conditions include, but are not limited to 12:
- Hematological disorders including inherited and acquired chronic hemolytic anaemia, sickle cell anemia and chronic myeloproliferative disorders.
- Systemic disorders including sarcoidosis, pulmonary Langerhans’s cell histiocytosis, and neurofibromatosis type 1
- Metabolic disorders including glycogen storage diseases and Gaucher’s disease
- Chronic renal failure with or without hemodialysis
- Pulmonary tumor thrombotic microangiopathy
- Fibrosing mediastinitis
- Splenectomy (spleen removal)
Pulmonary hypertension life expectancy
There’s no simple answer to this question. Every pulmonary hypertension patient is different, and new research with the potential to improve the outlook for this disease is being conducted all the time. Your journey with pulmonary hypertension depends on many factors, including the severity of your disease and how you respond to treatment. pulmonary hypertension-specific therapies are available that can prolong and improve your quality of life. Once in the care of a pulmonary hypertension specialist and on treatment, many pulmonary hypertension patients live for many years.
The outlook for pulmonary hypertension varies, depending on factors such as:
- what’s causing it
- how quickly it’s diagnosed
- how advanced your symptoms are
- whether you have another underlying health condition
The specialist in charge of your care will be able to give you more detailed information.
Having pulmonary hypertension can affect your ability to carry out everyday activities.
Pulmonary hypertension symptoms
The most common symptoms of pulmonary hypertension can also be caused by other more common medical problems, such as asthma, emphysema or chronic obstructive pulmonary disease [COPD]. Therefore, diagnosing pulmonary hypertension is difficult and requires a specialist. Physical examination signs can include visible or enlarged veins on the side of the neck, irregular heart sounds or swelling in the abdomen or legs and feet.
Symptoms are common across all types of pulmonary hypertension, however the numbers below are reported for pulmonary arterial hypertension.
Symptoms of pulmonary hypertension include 96:
- Shortness of breath 86%
- Fatigue 27%
- Chest pain (angina) 22%
- Swelling (oedema) in the legs, ankles, feet or tummy (abdomen) 21%
- Fainting or light-headedness 15%
- A racing heartbeat (palpitations) 13%
- Rarely, patients will cough up blood or have a change in their voice.
The symptoms often get worse during exercise, which can limit your ability to take part in physical activities.
If you have a type of pulmonary hypertension known as pulmonary arterial hypertension (PAH), you may not have any symptoms until the condition is quite advanced.
See your doctor if you have any symptoms of pulmonary hypertension. They’ll ask you about your symptoms and medical history, and they may carry out a physical examination.
Correctly diagnosing pulmonary hypertension can sometimes take time because its symptoms are similar to those of many other heart and lung conditions.
Tests you may have include a type of heart scan called an echocardiogram, and right heart catheterisation, where a thin, flexible tube is inserted into your pulmonary artery.
Pulmonary hypertension causes
Pulmonary hypertension is caused by changes to the pulmonary arteries, the blood vessels that carry blood from the heart to the lungs.
There are five main types of pulmonary hypertension, depending on the underlying cause. These are described below.
Pulmonary arterial hypertension
WHO Group 1 pulmonary arterial hypertension (PAH) is a specific type of pulmonary hypertension that is caused by the development of scar tissue in the tiny blood vessels of the lung. This scar tissue blocks the blood flow through the lungs and causes the pressure in those blood vessels to increase.
The walls of the arteries become thick and stiff, narrowing the space for blood to pass through and increasing pulmonary artery blood pressure.
If you have been diagnosed with pulmonary arterial hypertension (PAH), you should find a specialist who deals with this disease.
- DOCTORS WHO TREAT PULMONARY HYPERTENSION: https://phassociation.org/patients/doctorswhotreatph/
- The treatment for PAH is very complicated and depends on many factors (see treatment in the treatment section below).
How pulmonary arterial hypertension affects your body
The heart and the lungs work together to deliver nourishing oxygen-rich blood throughout the body. The left side of the heart receives blood with high oxygen content from the lungs, and pumps it through the body via arteries. Oxygen is taken up by cells of the muscles and organs, and the low oxygen content blood is collected in the veins and returned to the right side of the heart. The right side of the heart pumps this blood back to the lungs where it picks up oxygen again.
Normally, the pressure in the right side of the heart and the blood vessels of the lungs is lower than in the rest of the body. This allows the blood to collect as much oxygen as possible while in the lungs. In someone with PAH, the scarred and narrowed blood vessels in the lungs make it harder for the blood to get through. This forces the right side of the heart to pump harder, under greater pressure. If this increased pressure is not treated, it can cause the right side of the heart to become overworked and possibly fail. This problem can also reduce the ability of the blood to collect enough oxygen to keep the body functioning normally.
What causes pulmonary arterial hypertension?
Pulmonary arterial hypertension (PAH) is characterized by progressive scarring of the tiny blood vessels going to the lungs. A number of diseases and conditions can cause this scarring including: connective tissue disorders like scleroderma and lupus; exposure to certain toxins and drugs, including methamphetamine and cocaine; infections, including human immunodeficiency virus (HIV) and schistosomiasis; cirrhosis of the liver; and congenital heart abnormalities. When the cause of pulmonary arterial hypertension (PAH) is unknown, it is called idiopathic pulmonary arterial hypertension (IPAH).
WHO Group 1 pulmonary arterial hypertension (PAH) can be associated with other conditions, including:
- Connective tissue diseases – such as scleroderma, a condition that causes thickened areas of skin and problems with blood vessels; sarcoidosis, or lupus
- Congenital heart problems – such as a hole in the heart
- PAH that’s inherited (passed from parents to children through genes).
- Portal hypertension – abnormally high blood pressure inside the liver, which causes veins to become swollen
- HIV infection
- Certain medications or drugs, such as street drugs and certain diet medicines
- Connective tissue diseases. (Connective tissue helps support all parts of your body, including your skin, eyes, and heart.)
- Thyroid gland disorder
- Glycogen storage disorders – glycogen is a carbohydrate that produces short-term energy
- Pulmonary veno-occlusive disease – a rare condition that causes high blood pressure in the lungs
- Pulmonary capillary hemangiomatosis – another rare condition where tiny blood vessels (capillaries) grow within the lungs, causing blockages
- Sickle cell anemia,
- Chronic hemolytic anemia,
- Schistosomiasis. This is an infection caused by a parasite. Schistosomiasis is one of the most common causes of PAH in many parts of the world.
- Splenectomy (spleen removal)
- Certain metabolic disorders
- Drugs and toxins (appetite suppressants, cocaine, amphetamines)
- Myeloproliferative disorders (overproduction of red or white blood cells)
A small number of people develop pulmonary arterial hypertension without having any other medical condition. This is called idiopathic pulmonary arterial hypertension (IPAH). In very rare cases, PAH can be inherited – this is known as heritable pulmonary arterial hypertension (HPAH).
What are risk factors for developing pulmonary arterial hypertension?
You are more likely to get pulmonary arterial hypertension (PAH) if you are a young female, since the idiopathic form of the disease occurs more often in women of childbearing age than in men. There is an inherited form of pulmonary arterial hypertension (PAH), so a family history of the disease may put you at increased risk. Patients with diseases like lupus, scleroderma, cirrhosis of the liver, and HIV infection can develop it as well. Methamphetamine and cocaine use increase the risk for developing this disease.
How pulmonary arterial hypertension is treated
The treatment of PAH has change rapidly over the last 15 years. PAH-specific medications come in pill, inhaled, and intravenous (IV)/subcutaneous forms. The medications are often used in a variety of combinations. The treatment of pulmonary arterial hypertension is very dependent on the patient, the severity of the symptoms, the test findings, and even the support that the patient has at home. In addition to specific drugs, patients with PAH are also often treated with anticoagulants (blood thinners) and supplemental oxygen either at night, as needed or continuously. There are also patients with PAH who would benefit from a specific program of pulmonary rehabilitation. All of these potential options should be discussed between the patient and the treating physician. If the disease continues to progress in spite of treatment, your doctor may talk to you about lung transplantation.
Persistent pulmonary hypertension of the newborn
In rare cases, newborn babies can have high pressure inside their blood vessels, which means their heart can’t pump enough oxygenated blood around their body. This is known as persistent pulmonary hypertension of the newborn (PPHN). Treatment in an intensive care unit may be needed if simple measures such as keeping the baby warm and giving oxygen don’t increase oxygen levels to normal.
Before a baby is born, they do not need to use their lungs to breathe, because they receive oxygen via the umbilical cord and placenta from their mother. In the womb, the lungs are filled with amniotic fluid, and the blood vessels (arteries and veins) which take blood from the heart to the lungs are constricted, or closed. This means that the pressure inside the blood vessels of the lungs is high – you will often hear doctors talk about ‘high pulmonary pressures’.
When a baby is born, they take a big breath or cry, and their lungs fill with air instead of fluid. When the lungs fill with air, the blood vessels which take blood from the heart to the lungs open up (dilate), and this means that oxygen can be carried from the lungs, back to the heart, and pumped to the brain and the rest of the body once the umbilical cord is cut. The pressure inside the lungs and the blood vessels is now low.
If there is a problem around the time of birth which interferes with this process, the blood vessels may not open up properly so the pressure inside them remains high. This is called persistent pulmonary hypertension of the newborn (PPHN). As a result of the blood vessels not opening up, blood cannot get into the lungs to pick up oxygen and then the body will not have enough oxygen for the brain and other organs, and this can make a baby unwell.
Who does persistent pulmonary hypertension of the newborn affect?
Persistent pulmonary hypertension of the newborn occurs in about two in every 1,000 births. It usually occurs in babies that are born at term (at nine months) but can occasionally occur in premature (born too early) babies as well.
What causes persistent pulmonary hypertension of the newborn?
Doctors do not always know the cause of persistent pulmonary hypertension of the newborn, but they do know that the following may be factors:
- Meconium aspiration: This is when a baby has passed faeces (poo) while still in the womb, and because the poo becomes mixed with the amniotic fluid (the fluid inside the womb), the baby can breathe it into their lungs.
Infection - Infections such as pneumonia (lung or chest infection) and bloodstream infections can make persistent pulmonary hypertension of the newborn more likely, and there may be an increased risk of these conditions if the waters broke a long time before the baby was delivered, or if there was a group B strep infection present.
- Congenital abnormalities of the heart and lungs: A small number of babies who get persistent pulmonary hypertension of the newborn will have it because they have been born with an abnormality of their heart or lungs such as a diaphragmatic hernia, or a blocked heart valve, or lungs that are smaller than they should be.
How do doctors diagnose persistent pulmonary hypertension of the newborn?
The main feature of persistent pulmonary hypertension of the newborn is that not enough oxygen is getting to the heart, brain and other organs. This causes babies to look blue or pale and to have difficulty in breathing. Doctors, nurses and midwives will use oxygen saturation monitoring to measure the amount of oxygen in the blood, expressed as a percentage.
Monitoring involves putting a small probe which looks like a sticky plaster around the baby’s hand or foot, then displaying a number on a screen. If this number is low (below 92 per cent), and does not come up to 100 per cent easily then doctors treat problems with breathing using oxygen or breathing machines, they may diagnose persistent pulmonary hypertension of the newborn.
In some hospitals, it may be possible to do an ultrasound of the baby’s heart called an echocardiogram, which can look to see if the blood vessels in the lungs are closed or constricted (pulmonary hypertension, sometimes called ‘high pulmonary pressures’) and also rule out any abnormality of the heart which may be causing the low oxygen levels.
It is likely that doctors will also do a chest X-ray to look at the baby’s lungs, and some blood tests to look for signs of infection.
How is persistent pulmonary hypertension of the newborn treated?
Initial treatment of persistent pulmonary hypertension of the newborn will consist of simple measures such as keeping the baby warm (but not too hot) and giving oxygen, usually through small prongs (short plastic tubes) in the nostrils, or in an incubator. Doctors will usually insert a cannula or drip into the baby’s hand or foot, and use this to give some antibiotics.
As a baby is not likely to feed well while they have persistent pulmonary hypertension of the newborn, they will receive fluids containing sugar for energy through a drip. If these simple measures do not bring the oxygen levels up easily, the baby is likely to need to be moved to a neonatal intensive care unit or NICU.
Treatment of persistent pulmonary hypertension of the newborn in the intensive care unit (PICU or NICU)
Ventilation (breathing machine)
It is likely that doctors will pass a breathing tube into the baby’s airway through their mouth or nose, which is connected to a breathing machine or ventilator. This will breathe for the baby while they are unwell. The machine reduces the amount of effort and energy needed to breathe, which in turn reduces the amount of oxygen that the baby needs and it also delivers the maximum possible amount of oxygen into the lungs. When the baby is connected to the breathing machine, doctors will give them some medicines to make them sleepy so that they do not feel any discomfort as well as some medicines to stop them from moving. As the breathing tube passes into their airway, the baby will not be able to cry or make noises while they are on the machine.
High Frequency Oscillation (oscillator)
This is another type of breathing machine that doctors use to help deliver oxygen into the lungs of babies with persistent pulmonary hypertension of the newborn. This machine pushes air in and out of the lungs very quickly, so it is very noisy.
Nitric oxide
This gas is a combination of nitrogen and oxygen which is given to a baby through the breathing machine, straight into the lungs. It works by opening up the closed blood vessels so that more blood flows into the lungs, and the high pressures are reduced.
Inotropes
These are medicines that are given directly into the bloodstream via a drip to help keep the baby’s blood pressure high, as this helps the heart to pump blood into the lungs.
Extra-corporeal membrane oxygenation (ECMO)
If doctors have tried all of the treatments described above, and the baby’s oxygen saturations are still low, they may consider a treatment called extra-corporeal membrane oxygenation (ECMO), which is like a heart bypass machine during heart surgery. It takes over the work of the heart and lungs, putting oxygen directly into a baby’s blood rather than relying on the lungs to add oxygen. For this treatment a baby will be moved to the Cardiac Intensive Care Unit.
Moving forward and discharge from intensive care
Once the baby’s oxygen saturation levels are normal, the doctors and nurses will start to reduce the amount of nitric oxide delivered though the breathing machine, until it is turned off altogether. They will then gradually reduce the amount of oxygen used, and the amount of breathing work being done by the ventilator. You may hear them referring to this as ‘weaning’. During this time the doctors will reduce the medicine that keeps the baby still and sleepy so that they will become more awake and aware of their surroundings.
At the same time, the doctors will gradually reduce the inotropes (medicines being given to raise blood pressure) and might consider stopping antibiotics if a full course has been given. At this time the nurses may use a feeding tube, passed into one of the baby’s nostrils and down the throat to the stomach, to start some milk feeds. These can be expressed breast milk or formula, depending on the parents’ preference.
Once the baby is doing most of the work of breathing themselves, with only a small amount of help from the breathing machine, doctors will try to take the baby off the breathing machine altogether. Once this has happened, it is likely that they will be ready to go back to their local hospital, where the baby will finish any antibiotics that are still needed, continue oxygen treatment until it is no longer needed, and help parents to establish feeding before they are discharged home.
What is the outlook for babies who have had persistent pulmonary hypertension of the newborn?
Persistent pulmonary hypertension of the newborn is a serious condition, and if a child has been moved to the intensive care unit, that is because the baby is doing very poorly. The mortality is thought to be under 10 per cent (fewer than 1 in 10 babies affected will die).
There are also some after-effects from the lack of oxygen to the brain during the illness, and up to a quarter of babies affected will have some impairment because of their illness as they grow older. This includes difficulties such as learning problems and deafness.
Pulmonary hypertension linked to left heart disease
WHO Group 2 includes pulmonary hypertension due to left heart disease. If there are problems with the left side of the heart, the right side has to work harder to pump blood through the lungs. This increases blood pressure in the pulmonary arteries.
Problems with the left side of the heart are thought to be the most common (68 percent) cause of pulmonary hypertension. These include mitral valve problems, left ventricle problems, aortic valve conditions or long-term high blood pressure.
Pulmonary hypertension linked with lung disease or lack of oxygen
WHO Group 3 pulmonary hypertension is linked with lung diseases or lack of oxygen (hypoxia), including:
- Chronic obstructive pulmonary disease (COPD) – a number of lung conditions that affect breathing
- Interstitial lung disease – a group of lung disorders that cause scarring of the lung tissue, which makes it difficult to get enough oxygen into your body
- Conditions that affect breathing while you’re in a deep sleep – such as obstructive sleep apnoea (OSA)
Low levels of oxygen in the blood make the pulmonary arteries narrow. This squeezes the blood into a smaller space, which increases blood pressure, causing pulmonary hypertension.
Pulmonary hypertension caused by blood clots
WHO Group 4 pulmonary hypertension is also called chronic thromboembolic pulmonary hypertension (CTEPH). Pulmonary hypertension can sometimes be caused by scars from previous blood clots that narrow or block the pulmonary arteries.
A blood clot that blocks one of the blood vessels that supply your lungs is called a pulmonary embolism.
Other causes of pulmonary hypertension
WHO Group 5 is where pulmonary hypertension is secondary to other diseases in ways that are not well understood. Other, less common, causes of pulmonary hypertension include:
- Sarcoidosis – a condition that causes inflammation of different organs, including the lungs and lymph nodes
- Histiocytosis X – a rare condition that causes scarring (granulomas) and air-filled cysts, mainly in the lungs
- Compression of the blood vessels in the lungs – for example, as the result of a tumor
- Blood disorders, such as polycythemia vera and essential thrombocythemia
- Systemic disorders, such as vasculitis. Systemic disorders involve many of the body’s organs.
- Metabolic disorders, such as thyroid disease and glycogen storage disease. (In glycogen storage disease, the body’s cells don’t use a form of glucose (sugar) properly.)
- Other conditions, such as tumors that press on the pulmonary arteries and kidney disease.
Pulmonary hypertension diagnosis
Pulmonary hypertension can be difficult to diagnose in a routine medical exam because the most common symptoms of pulmonary hypertension, such as breathlessness, fatigue and dizziness, are also associated with many other heart or lungs conditions. This means there can sometimes be a delay before a correct diagnosis is made.
See your doctor if you have symptoms of pulmonary hypertension, such as breathlessness, shortness of breath and tiredness.
Initial assessment
If your doctor suspects that you have pulmonary hypertension, he or she will want to review your medical and family history, perform a physical exam and perform one or more diagnostic tests.
Your doctor will ask about:
- your symptoms and how they affect your life
- your family history – although rare, pulmonary arterial hypertension can run in families
- any medication you’re currently taking
- any other medical conditions you have
You may also have a physical examination where your doctor will listen to your heart and lungs, and check for any swelling in your legs or ankles.
Preliminary Tests
To determine if you have pulmonary hypertension and what type, your medical team will schedule specialized tests. If your medical team suspects pulmonary hypertension as a result of one or more of the following tests, they will go on to schedule a right-heart catheterization, which is required to confirm diagnosis.
Blood Tests
Blood tests check the oxygen levels in the blood, they observe liver and kidney function, and they identify whether the patient has collagen vascular disease, thyroid problems, signs of infection or HIV antibodies. One test, the brain natriuretic peptide, helps to assess the strain on the heart and may also be used to monitor response to treatment.
Chest X-Rays
Chest X-rays can reveal an enlarged right ventricle or pulmonary arteries. Chest X-rays can also show signs of emphysema or scarring (interstitial fibrosis) of the lungs.
Electrocardiogram (ECG)
An electrocardiogram checks the electrical impulses of the heart. Electrodes are attached to the patient’s skin, and a recording of these impulses is made. However, an ECG alone is not enough to indicate a pulmonary hypertension diagnosis. If your doctor performs an ECG, he or she will also perform one or more additional procedures to identify pulmonary hypertension.
Echocardiogram
In this procedure, electrodes are placed on the patient’s skin, and a sonogram (high-frequency sound waves to create an image of the heart) of the heart is taken. This painless procedure is often used to make a preliminary diagnosis by estimating the pressures in the right heart and assessing how well the heart is functioning. Other heart conditions that produce symptoms similar to pulmonary hypertension may be diagnosed with an echocardiogram. In addition, an echocardiogram may be used to monitor a patient’s condition.
Pulmonary Function Tests
These tests measure how much air your lungs can hold, how much air moves in and out of them and the lungs’ ability to exchange oxygen. These tests may be performed to potentially identify its cause.
Exercise Tolerance Test (Six-Minute Walk Test)
During this test, a patient will be asked to perform an exercise, most commonly a six-minute walk. You will walk at your normal pace for six minutes. This test most often can take place in a long hallway. The 6-Minute Walk Test objectively measures how far you can walk and to see if your oxygen levels drop when you are physically active. The purpose is to identify the patient’s exercise tolerance level. This test will monitor your body’s response to treatments for heart, lung, and other health problems.
Nuclear Scan (Ventilation/Perfusion Scan or V/Q Scan)
This diagnostic tool tests for blood clots in the lungs by producing a picture of air and blood flow to the lungs. A small dose of radioactive material is breathed in and another small dose is injected via a blood vessel into the lungs. The doctor will review the images that are produced to evaluate the health of the lungs.
The Gold Standard for pulmonary hypertension Diagnosis
Right-Heart Catheterization
If the results of initial tests point to pulmonary hypertension, your doctor will schedule a right-heart catheterization (commonly referred to as a “right heart cath”). Right-heart catheterization is one of the most accurate and useful tests to get a definitive diagnosis for pulmonary hypertension. This is the only test that directly measures the pressure inside the pulmonary arteries, and it should be done in all patients at least once to confirm a patient’s diagnosis of pulmonary hypertension. During the test, doctors insert a thin, flexible tube (catheter) through a large vein in your neck, arm or groin. They then pass the catheter up into your pulmonary artery to confirm a diagnosis by accurately measuring the blood pressure in the right side of your heart and pulmonary arteries; it’s carried out in specialist national pulmonary hypertension centers.
Vasodilator Study (Acute Vasodilator Challenge)
This test is used for patients who have already been diagnosed with pulmonary hypertension to determine how much their pulmonary blood vessels can relax over a brief period of time. Its main purpose is to screen for patients who might respond favorably to calcium channel blockers, a form of medication. The test can also help determine the patient’s prognosis. With a right heart catheter in place, the patient is given drugs that relax the pulmonary arteries. The test drug is given to the patient in higher and higher doses, pausing at each dose to see how the patient reacts. Once a significant response occurs or the side effects become intolerable, the test is considered complete.
In patients with idiopathic and hereditary pulmonary arterial hypertension, acute vasodilator testing during right heart catheterization is strongly recommended based on current guidelines.
Pulmonary hypertension functional classification
If you’re diagnosed with pulmonary hypertension, your condition will be classified depending on how severe your symptoms are and your ability to do everyday tasks. This is to help work out the best treatment for you.
World Health Organization’s (WHO) Functional Class for pulmonary hypertension may fall into one of the following groups:
- WHO Functional Class 1. Pulmonary hypertension is diagnosed, but there are no symptoms during rest or exercise. Ordinary physical activity does not cause undue shortness of breath (dyspnea) or fatigue, chest pain, or near syncope (fainting)
- WHO Functional Class 2. Patients with pulmonary hypertension with no symptoms at rest. Everyday chores or activities such as going to work or the grocery store may cause some shortness of breath (dyspnea) or fatigue, chest pain, or near syncope. There’s a slight limitation of physical activity.
- WHO Functional Class 3. Patients with pulmonary hypertension is comfortable at rest, but doing simple tasks such as bathing, dressing or preparing meals causes fatigue, shortness of breath (dyspnea) and chest pain or near syncope. The ability to do physical activity becomes very limited.
- WHO Functional Class 4. Patients with pulmonary hypertension with an inability to carry out any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnea and/or fatigue may even be present at rest. Discomfort is increased by any physical activity.
Exercise testing is used to find out the severity of pulmonary hypertension. This testing consists of either a 6-minute walk test or a cardiopulmonary exercise test. A 6-minute walk test measures the distance you can quickly walk in 6 minutes. A cardiopulmonary exercise test measures how well your lungs and heart work while you exercise on a treadmill or bicycle. During exercise testing, your doctor will rate your activity level. Your level is linked to the severity of your pulmonary hypertension. Over time, you may need more exercise tests to find out how well your treatments are working. Each time testing is done, your doctor will compare your activity level with the previous one.
The World Health Organization functional class is one of the strongest predictors of survival, both at diagnosis and follow-up, and worsening World Health Organization functional class is one of the most alarming indicators of disease progression, which should trigger further investigations to identify the cause(s) of clinical deterioration 97, 98, 99, 100, 101.
Your doctor may also use a risk calculator that considers your symptoms and test results to understand what type of treatment is needed. This is called pulmonary hypertension risk stratification.
Table 5. Pulmonary hypertension risk stratification
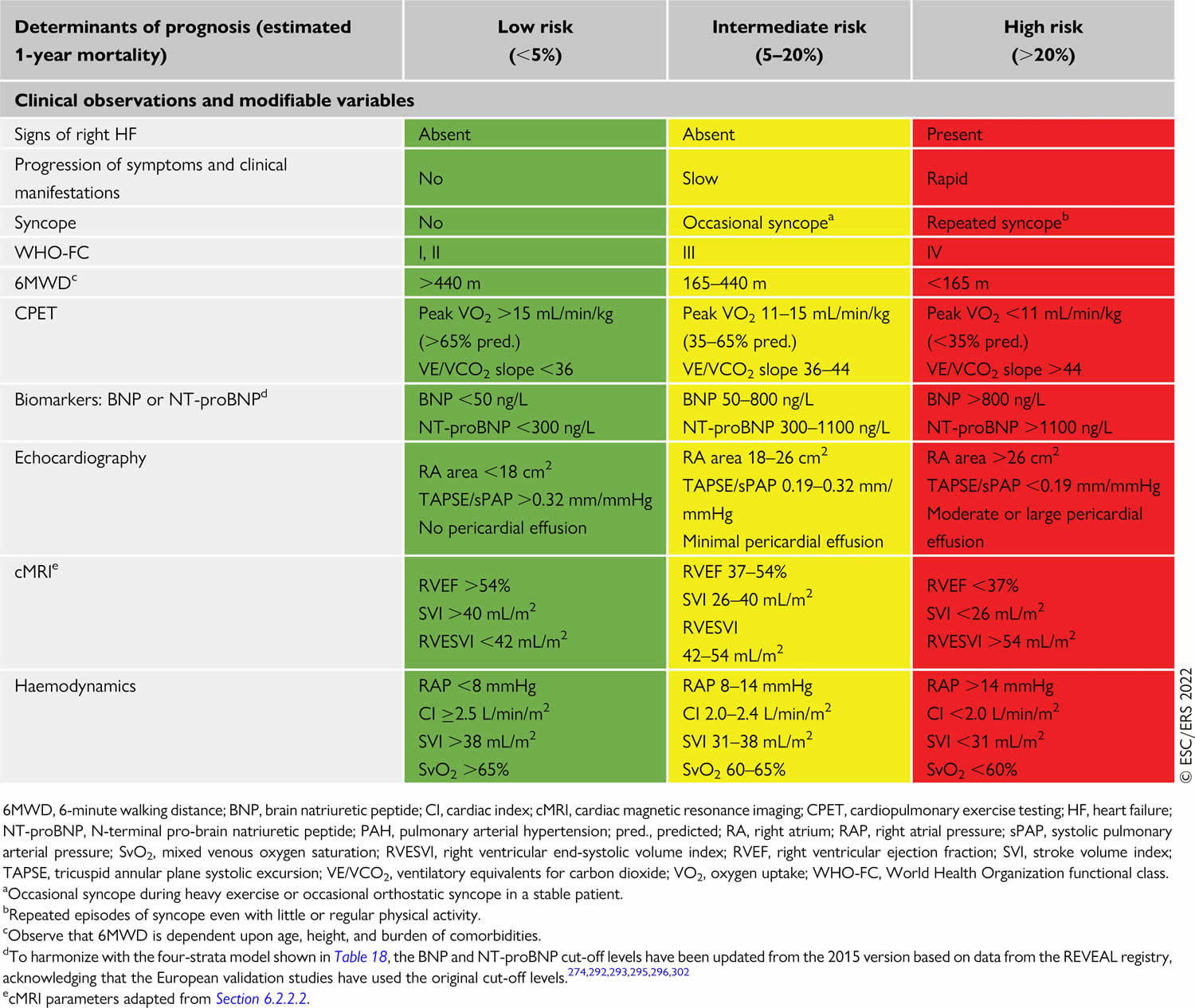
Pulmonary hypertension differential diagnosis
Pulmonary hypertension differential diagnosis is broad since the symptoms are non-specific:
- Chronic asthma
- Anemia
- Heart failure
- Chronic obstructive pulmonary disease (COPD)
- Cor pulmonale
- Dilated cardiomyopathy
- Mitral stenosis
- Connective tissue diseases
- Portal hypertension
- Obstructive sleep apnea (OSA)
Pulmonary hypertension treatment
There is currently no cure for pulmonary hypertension, however, there are treatment options available and more are on the horizon. If the cause is identified and treated early, it may be possible to prevent permanent damage to your pulmonary arteries, the blood vessels that supply your lungs.
Your doctor will take into consideration the severity of your illness (referred to as your “functional class”) and the results of your cardiac catheterization to help determine which medication is right for you. As your symptoms and pressures change, your doctor may want to adjust the type and dosage of your medication accordingly.
Pulmonary hypertension treatments include conventional medical therapies and oral, inhaled, intravenous (into the vein) and subcutaneous (into the skin) options. Depending on the severity of pulmonary hypertension, heart or lung transplant may also be an option.
Remember that each patient is different. It is essential that you talk to your own doctor about what treatment options are best for you.
All Types of Pulmonary Hypertension
Several treatments may be used for all types of pulmonary hypertension. These treatments include:
- Diuretics, also called water pills. These medicines help reduce fluid buildup in your body, including swelling in your ankles and feet.
- Blood thinners also called anticoagulants, these medicines help prevent blood clots. One example is warfarin (Jantoven). Blood-thinning medicines slow the clotting process. The medicines can increase the risk of bleeding. This is especially true if you’re having surgery or a procedure that enters the body or creates an opening in the skin. Talk to your doctor about your risk.
- Digoxin (Lanoxin) also called digitalis, helps the heart squeeze better to pump blood. It also tends to slow the heartbeat. Digoxin reduces heart failure symptoms in people with heart failure with reduced ejection fraction (HFrEF). It may be more likely to be given to someone with a heart rhythm disorder, such as atrial fibrillation. The level of digoxin in the body must be checked using a blood test. If too much digoxin builds up in your blood, side effects may occur, including loss of appetite, nausea, vomiting and headaches. The heart rhythm can also become too fast or too slow. Always report any side effects of Digoxin to your doctor right away.
- Oxygen therapy. Breathing pure oxygen is sometimes recommended as a treatment for pulmonary hypertension. This treatment may be suggested if you live at a high altitude or have sleep apnea. Some people with pulmonary hypertension need oxygen therapy all the time.
- Physical activity. Regular activity may help improve your ability to be active. Talk with your doctor about a physical activity plan that’s safe for you.
Research is ongoing for better pulmonary hypertension treatments. These treatments offer hope for the future.
Treating underlying conditions
If pulmonary hypertension is caused by another condition, such as a heart or lung problem, treatments will focus on the underlying condition.
If pulmonary hypertension is caused by blood clots that block the pulmonary arteries, you may be offered anticoagulant medicines to prevent more clots forming. You may also be offered an operation known as a pulmonary endarterectomy.
If you have pulmonary arterial hypertension (PAH), you’ll be referred to a center that specializes in treating this form of the condition (https://phassociation.org/patients/doctorswhotreatph/).
Pulmonary hypertension medications
- Calcium Channel Blockers (CCB) – Help decrease blood pressure (Only appropriate for a small minority of patients demonstrating a favorable response to vasodilator testing at the time of heart catheterization.)
- Digoxin – Assists the pumping of the heart
- Diuretics – Rids excess fluid that puts pressure on the heart
- Oxygen – Inhaled by patients via a nasal cannula or face mask
- Warfarin (Coumadin®) – “Thins” blood and prevents it from clotting
Oral Treatment Options
Endothelin Receptor Antagonists (ERAs) help prevent blood vessels from narrowing.
- Ambrisentan (Letairis®)
- Bosentan (Tracleer®)
- Bosentan (Tracleer®) for Pediatric Use
- Macitentan (Opsumit®)
Phosphodiesterase Inhibitors (PDE 5 Inhibitors) allow the lungs to produce more of its own natural vasodilators.
- Sildenafil (Revatio™)
- Sildenafil (Revatio™) for Pediatric Use
- Tadalafil (Adcirca®)
Prostacyclin Analogue allows the blood vessels in the lungs relax
- Oral Treprostinil (Orenitram®)
Selective IP Receptor Agonist targets and activates a prostacyclin receptor helping the blood vessels in the lungs relax
- Selexipag (Uptravi®)
Soluble Guanylate Cyclase (sGC) Stimulators increase the interaction of sGC with another chemical (nitric oxide) to help the blood vessels in the lungs relax.
- Riociguat (Adempas®)
Inhaled Treatment Options
Inhaled Treatment Options, such as prostacyclins, relieve shortness of breath.
- Iloprost (Ventavis®)
- Inhaled Treprostinil (Tyvaso™)
Intravenous Treatment Options
Intravenous Treatment Options open up the blood vessels and help ease symptoms of pulmonary hypertension, including chest pain and shortness of breath.
- Intravenous Treprostinil (Remodulin®)
- Epoprostenol (Flolan®)
- Room Temperature Stable Epoprostenol (Veletri®)
Subcutaneous Treatment Options
Subcutaneous Treatment Options are delivered through a portable infusion pump to open up the blood vessels and ease the symptoms of pulmonary hypertension.
- Subcutaneous Treprostinil
Surgery
Many people with pulmonary hypertension are treated with both conventional and targeted therapies, although this can be different for different people. Some people with pulmonary hypertension may need surgery. How your pulmonary hypertension is treated will depend on a number of things, for example how severe your pulmonary hypertension is, what type of pulmonary hypertension you have, etc.
Some people with pulmonary hypertension may need surgery. The three types of surgery currently used are:
- Pulmonary endarterectomy – an operation to remove old blood clots from the pulmonary arteries in the lungs in people with chronic thromboembolic pulmonary hypertension
- Balloon pulmonary angioplasty – a new procedure where a tiny balloon is guided into the arteries and inflated for a few seconds to push the blockage aside and restore blood flow to the lung; it may be considered if pulmonary endarterectomy isn’t suitable, and has been shown to lower blood pressure in the lung arteries, improve breathing, and increase the ability to exercise
- Atrial septostomy – a small hole is made in the wall between the left and right atria of the heart using a cardiac catheter, a thin, flexible tube inserted into the heart’s chambers or blood vessels; it reduces the pressure in the right side of the heart, so the heart can pump more efficiently and the blood flow to the lungs can be improved
- Transplant – in severe cases, a lung transplant or a heart-lung transplant may be needed; this type of surgery is rarely used because effective medication is available
Lung or heart-lung transplant
Lung or heart-lung transplant is a treatment option reserved for patients who are not improving on medical therapies. While transplantation can prolong survival, improve quality of life and offers a potential cure for pulmonary hypertension, it also carries risk of significant complications and many factors need to be considered before going forward.
Deciding whether transplant is right for you is a process that will involve your team of medical professionals. Most candidates experience a significant waiting time between becoming listed and receiving a transplant since there are more transplant candidates than there are donor organs.
Treatments for pulmonary arterial hypertension
There are many treatments for pulmonary arterial hypertension (PAH). Which treatment or combination of treatments you’ll be offered will depend on a number of factors, including what’s causing PAH and the severity of your symptoms.
Treatments include:
- anticoagulant medicines – such as warfarin to help prevent blood clots
- diuretics (water tablets) – to remove excess fluid from the body caused by heart failure
- oxygen treatment – this involves inhaling air that contains a higher concentration of oxygen than normal
- digoxin – derived from the foxglove plant, digoxin can improve your symptoms by strengthening your heart muscle contractions and slowing down your heart rate
There are also a number of specialist treatments for PAH that help relax the arteries in the lungs and reduce the blood pressure in the lungs.
These medicines slow the progression of PAH, and may even reverse some of the damage to the heart and lungs.
Treatments include:
- endothelin receptor antagonists – such as bosentan, ambrisentan and macitentan
- phosphodiesterase 5 inhibitors – sildenafil and tadalafil
- prostaglandins – epoprostenol, iloprost and treprostinil
- soluble guanylate cyclase stimulators – such as riociguat
- calcium channel blockers – nifedipine, diltiazem, nicardipine and amlodipine
Living with pulmonary arterial hypertension
The therapies that exist for this disease range from pills that can be taken between one to three times a day to medications that have to be given continuously through an IV line using a pump that is similar to an insulin pump used by diabetic patients. How well a patient responds to medicine is somewhat unpredictable and depends on how advanced the disease is at diagnosis as well as the underlying cause. Patients without treatment usually quickly progress to death over the course of months to years. However, with treatment, survival has improved significantly.
Typically patients will be seen in the doctor’s office at least every 3-4 months at first. In many centers, there will be regular testing such as echocardiograms and 6 minute walk testing. Some centers will obtain yearly right heart catheterizations to assess the impact of therapies on pulmonary pressures and heart function.
Special considerations for patients with PAH need to be made. For example, pregnancy should be avoided at all costs as it represents significant risk to the female patient with PAH and to the fetus. Also, many of the medications used to treat PAH may harm the fetus as it develops. Because PAH is a rare and complex disease, good care for a patient with PAH involves a close relationship with an expert in PAH and support from family and friends.
To best manage your PAH you should:
- Take your medication exactly as directed.
- Do not stop a medication without consulting with your doctor.
- Take care to never run out of medication.
- Avoid things that can put strain on the lungs and heart. This would include lifestyle choices such as smoking and the use of recreational drugs such as cocaine and amphetamines. There are also potential risks of worsening PAH during the course of normal pregnancy. These issues should be discussed between you and your doctor.
- Since PAH can lead to a form of heart failure, adhere to a low salt diet and limit fluid intake.
- Monitor your weight to recognize fluid retention. If your weight goes up, it is often helpful to let your physician or healthcare provider know, as an adjustment of medications may be needed.
- Develop strategies to help cope with fatigue and shortness of breath.
- Develop a careful exercise program with your healthcare providers.
- Get immunized against flu and pneumonia.
- Talk to your doctor about anxiety and depression, which are common in patients with PAH.
The Lung Association recommends patients and caregivers join:
- The Living with Lung Disease Support Community to connect with others facing this disease: https://www.inspire.com/groups/american-lung-association-lung-disease
- Pulmonary Hypertension Association: https://phassociation.org/
- Pulmonary Hypertension Association UK: http://www.phauk.org/
Group 2 Pulmonary Hypertension Due to Left Heart Disease
Conditions that affect the left side of the heart, such as mitral valve disease, can cause group 2 pulmonary hypertension. Treating the underlying condition will help treat pulmonary hypertension. Treatments may include lifestyle changes, medicines, and surgery.
Group 3 Pulmonary Hypertension Due to Lung Disease
Lung diseases, such as COPD (chronic obstructive pulmonary disease) and interstitial lung disease, can cause group 3 pulmonary hypertension. Certain sleep disorders, such as sleep apnea, also can cause group 3 pulmonary hypertension.
If you have this type of pulmonary hypertension, you may need oxygen therapy. This treatment raises the level of oxygen in your blood. You’ll likely get the oxygen through soft, plastic prongs that fit into your nose. Oxygen therapy can be done at home or in a hospital.
Your doctor also may recommend other treatments if you have an underlying lung disease.
Group 4 Pulmonary Hypertension Due to Chronic Blood Clots in the Lungs
Blood clots in the lungs or blood clotting disorders can cause group 4 pulmonary hypertension. If you have this type of pulmonary hypertension, your doctor will likely prescribe blood-thinning medicines. These medicines prevent clots from forming or getting larger.
Sometimes doctors use surgery to remove scarring in the pulmonary arteries due to old blood clots.
Group 5 Pulmonary Hypertension Due to Unknown Causes
Various diseases and conditions, such as thyroid disease and sarcoidosis, can cause group 5 pulmonary hypertension. An object, such as a tumor, pressing on the pulmonary arteries also can cause group 5 pulmonary hypertension.
Group 5 pulmonary hypertension is treated by treating its cause.
Pulmonary hypertension prognosis
Your life expectancy depends on many factors, including the severity of your pulmonary hypertension and how early you’re diagnosed. Talk with your doctor to learn your specific prognosis. Untreated patients have very poor survival, but some can survive into the 5th decade of life with medical treatment 102, 103. Patients who fail to respond to medications usually have the worst prognosis. Patients with persistently elevated pulmonary pressures and right heart failure usually are dead within 5 years 104, 92.
The Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL registry) PAH Risk Score Calculator is a tool clinicians may use to help prognosticate patients with pulmonary artery hypertension. The tool has validation as a predictive algorithm for 1-year survival.
You can use the PAH Risk Score Calculator here: https://pahriskcalculatorre.com/
Factors that are independently associated with decreased survival include the following:
- Men older than 60 years
- PAH associated with portal hypertension or connective tissue disorder
- Family history of PAH
- WHO Class 3 or 4 kidney insufficiency
- Resting systolic blood pressure less than 110 mmHg
- Heart rate greater than 92 beats per minute
- Six-minute walk test less than 165 m
- Brain natriuretic peptide (BNP) greater than 180 pg/ml
- Pulmonary vascular resistance greater than 32 Woods units
- Presence of pericardial effusion on echocardiogram
- TAPSE (tricuspid annular plane systolic excursion) less than 1.5 cm, percentage predicted diffusing capacity of the lung for carbon monoxide (DLCO less than 32%)
Recent United States data suggest 1-year mortality for pulmonary arterial hypertension (PAH) to be as high as 8% for intermediate-risk and 19% for high-risk patients, with 3-year mortality rates being 20% and 55% for those groups, respectively 105. These data emphasize the importance of early diagnosis and aggressive therapy.
Prognosis varies among different pulmonary arterial hypertension (PAH) subgroups based on underlying cause. The prognosis of idiopathic pulmonary hypertension (IPAH) is poor 64. The mean survival of untreated idiopathic pulmonary hypertension (IPAH) is 2 to 3 years from the diagnosis. The New York Heart Association (NYHA) functional class is an important predictor of survival, with class 4 mean survival of less than 6 months 64. The most important prognostic factor is right ventricular function which is also the cause of death in advanced idiopathic pulmonary hypertension (IPAH). Increased mortality is seen in pregnant patients with advanced idiopathic pulmonary hypertension (IPAH).
In another study, patients with scleroderma with PAH compared with patients with idiopathic pulmonary hypertension (IPAH) have a threefold higher mortality risk 106, 107, 108, 109. Furthermore, patients scleroderma with PAH stand out from those with other types of PAH, given their impaired response to traditional therapies and worse overall clinical prognosis despite exhibiting similar end-organ pathology and often presenting with milder hemodynamic impairment 106, 110. Proposed factors explaining these disparities include more pronounced inflammation, autoimmunity, the distinct nature of the underlying vasculopathy, and differing abilities of the right ventricle to adapt to the increased afterload 111, 112, 113.
The presence of pulmonary hypertension and right ventricular dysfunction in patients with left heart disease is associated with high mortality. In patients with lung disease, even nonsevere pulmonary hypertension negatively impacts survival and is associated with increased hospitalization. People with severe pulmonary hypertension have worse outcomes than those with nonsevere pulmonary hypertension 114. Chronic thromboembolic pulmonary hypertension has an excellent long-term prognosis if the disease is operable 2.
Steps you can take to improve your prognosis (outlook) include:
- Create an emergency kit. You need to have certain supplies and information with you all the time. Ask your doctor what you should include in your kit, and never leave home without it.
- Get your vaccines as your doctor recommends. It’s important to protect yourself against the flu and pneumonia.
- Keep all your follow-up appointments. Regular testing is crucial to check your lung and heart function and measure treatment progress.
- Take your medications as your doctor prescribes, and at the same time every day. Don’t make any changes to your medication routine unless your doctor tells you to do so.
Living with pulmonary hypertension
Pulmonary hypertension has no cure. However, you can work with your doctor to manage your symptoms and slow the progress of the disease.
Ongoing Care
Follow your treatment plan as your doctor advises. Call your doctor if your pulmonary hypertension symptoms worsen or change. The earlier symptoms are addressed, the easier it is to treat them.
Some symptoms, such as chest pain, may require emergency treatment. Ask your doctor when you should call him or her or seek emergency care.
Also, talk with your doctor before taking any over-the-counter medicines. Some medicines can make your pulmonary hypertension worse or interfere with the medicines you’re taking for pulmonary hypertension. Ask your doctor whether you should get a pneumonia vaccine and a yearly flu shot.
You may have a complex schedule for taking medicines. Call your doctor or nurse if you’re having problems with this schedule. Knowing the names of your medicines and how they work is helpful. Keep a list of your medicines with you. Don’t stop or change medicines unless you talk with your doctor first.
Pay careful attention to your weight. You may want to keep a daily record of your weight. You should weigh yourself at the same time each day. If you notice a rapid weight gain (2 or more pounds in 1 day or 5 or more pounds in 1 week), call your doctor. This may be a sign that your pulmonary hypertension is worsening.
Pregnancy is risky for women who have pulmonary hypertension. Consider using birth control if there is a chance you may become pregnant. Ask your doctor which birth control methods are safe for you.
Lifestyle Changes
Making lifestyle changes can help you manage your symptoms. These changes will depend on the type of pulmonary hypertension you have. Talk with your doctor about which lifestyle changes can help you.
Quit Smoking
If you smoke, quit. Smoking makes pulmonary hypertension symptoms worse. Ask your doctor about programs and products that can help you quit. Also, avoid exposure to secondhand smoke.
Follow a Healthy Diet
Following a healthy diet and maintaining a healthy weight are part of a healthy lifestyle. A healthy diet includes a variety of fruits, vegetables, and whole grains. It also includes lean meats, poultry, fish, and fat-free or low-fat milk or milk products. A healthy diet also is low in saturated fat, trans fat, cholesterol, sodium (salt), and added sugar.
Talk with your doctor about whether you need to limit the amount of salt and fluids in your diet. Ask him or her whether you also need to regulate foods that contain vitamin K. These foods can affect how well blood-thinning medicines work. Vitamin K is found in green leafy vegetables and some oils, such as canola and soybean oil.
Be Physically Active
Physical activity is an important part of a healthy lifestyle. Try to do physical activity, such as walking, regularly. This will keep your muscles strong and help you stay active. Ask your doctor how much activity is safe for you. Your doctor may tell you to limit or avoid certain activities, such as:
- Those that cause straining, such as lifting heavy objects or weights.
- Sitting in a hot tub or sauna or taking long baths. These activities can lower your blood pressure too much.
- Flying in an airplane or traveling to high-altitude areas. Your doctor may ask you to use extra oxygen during air travel.
Avoid activities that cause breathing problems, dizziness, or chest pain. If you have any of these symptoms, seek care right away.
Emotional Issues and Support
Living with pulmonary hypertension may cause fear, anxiety, depression, and stress. You may worry about your medical condition, treatment, finances, and other issues.
Talk about how you feel with your health care team. Talking to a professional counselor also can help. If you’re very depressed, your doctor may recommend medicines or other treatments that can improve your quality of life.
Joining a patient support group may help you adjust to living with pulmonary hypertension. You can see how other people who have the same symptoms have coped with them. Talk with your doctor about local support groups or check with an area medical center.
Support from family and friends also can help relieve stress and anxiety. Let your loved ones know how you feel and what they can do to help you.
The Lung Association recommends patients and caregivers join:
- The Living with Lung Disease Support Community to connect with others facing this disease: https://www.inspire.com/groups/american-lung-association-lung-disease
- Pulmonary Hypertension Association: https://phassociation.org/
- Pulmonary Hypertension Association UK: http://www.phauk.org/
- Bousseau S, Sobrano Fais R, Gu S, Frump A, Lahm T. Pathophysiology and new advances in pulmonary hypertension. BMJ Med. 2023 Mar 23;2(1):e000137. doi: 10.1136/bmjmed-2022-000137[↩][↩][↩][↩][↩]
- Oldroyd SH, Manek G, Bhardwaj A. Pulmonary Hypertension. [Updated 2024 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482463[↩][↩][↩]
- Hoeper MM, Meyer K, Rademacher J, et al. Diffusion capacity and mortality in patients with pulmonary hypertension due to heart failure with preserved ejection fraction. JACC Heart Fail 2016;4:441–9. 10.1016/j.jchf.2015.12.016[↩][↩]
- Keen J, Prisco SZ, Prins KW. Sex differences in right ventricular dysfunction: insights from the bench to bedside. Front Physiol 2020;11:623129. 10.3389/fphys.2020.623129[↩]
- Tuder RM, Archer SL, Dorfmüller P, et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 2013;62(25 Suppl):D4–12. 10.1016/j.jacc.2013.10.025[↩]
- Swisher JW, Weaver E. The Evolving Management and Treatment Options for Patients with Pulmonary Hypertension: Current Evidence and Challenges. Vasc Health Risk Manag. 2023 Mar 3;19:103-126. doi: 10.2147/VHRM.S321025[↩]
- Nazzareno Galiè, Massimiliano Palazzini, Alessandra Manes, Confirmation of survival prediction based on 2022 ESC/ERS pulmonary hypertension guidelines new haemodynamic thresholds, European Heart Journal, Volume 44, Issue 44, 21 November 2023, Pages 4692–4695, https://doi.org/10.1093/eurheartj/ehad672[↩][↩][↩]
- Karia N, Howard L, Johnson M, Kiely DG, Lordan J, McCabe C, Pepke-Zaba J, Ong R, Preiss M, Knight D, Muthurangu V, Coghlan JG. Predictors of outcomes in mild pulmonary hypertension according to 2022 ESC/ERS Guidelines: the EVIDENCE-PAH UK study. Eur Heart J. 2023 Nov 21;44(44):4678-4691. doi: 10.1093/eurheartj/ehad532[↩]
- Kovacs G, Bartolome S, Denton CP, Gatzoulis MA, Gu S, Khanna D, Badesch D, Montani D. Definition, classification and diagnosis of pulmonary hypertension. Eur Respir J. 2024 Oct 31;64(4):2401324. doi: 10.1183/13993003.01324-2024[↩]
- Kim NH, D’Armini AM, Delcroix M, Jaïs X, Jevnikar M, Madani MM, Matsubara H, Palazzini M, Wiedenroth CB, Simonneau G, Jenkins DP. Chronic thromboembolic pulmonary disease. Eur Respir J. 2024 Oct 31;64(4):2401294. doi: 10.1183/13993003.01294-2024[↩]
- Santos-Gomes J, Gandra I, Adão R, Perros F, Brás-Silva C. An Overview of Circulating Pulmonary Arterial Hypertension Biomarkers. Front Cardiovasc Med. 2022 Jul 14;9:924873. doi: 10.3389/fcvm.2022.924873[↩]
- Virsinskaite R, Karia N, Kotecha T, Schreiber BE, Coghlan JG, Knight DS. Pulmonary hypertension – the latest updates for physicians. Clin Med (Lond). 2023 Sep;23(5):449-454. doi: 10.7861/clinmed.2023-23.5.Cardio4[↩][↩][↩][↩][↩][↩][↩]
- 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. European Respiratory Journal 2023 61(1): 2200879; DOI: https://doi.org/10.1183/13993003.00879-2022[↩][↩]
- SEC Working Group for the 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension and SEC Guidelines Committe. Comments on the 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Rev Esp Cardiol (Engl Ed). 2023 May;76(5):294-300. English, Spanish. doi: 10.1016/j.rec.2022.11.001[↩]
- Boucly A, Bertoletti L, Fauvel C, Dewavrin MG, Gerges C, Grynblat J, Guignabert C, Hascoet S, Jaïs X, Jutant EM, Lamblin N, Meyrignac O, Riou M, Savale L, Tromeur C, Turquier S, Valentin S, Simonneau G, Humbert M, Sitbon O, Montani D; on behalf on the French PH Network. Evidence and unresolved questions in pulmonary hypertension: Insights from the 5th French Pulmonary Hypertension Network Meeting. Respir Med Res. 2024 Nov;86:101123. doi: 10.1016/j.resmer.2024.101123[↩]
- Price LC, Weatherald J. The new 2022 pulmonary hypertension guidelines: some small steps and some giant leaps forward for evidence-based care. Eur Respir J. 2023 Jan 6;61(1):2202150. doi: 10.1183/13993003.02150-2022[↩]
- Maron BA, Brittain EL, Choudhary G, Gladwin MT. Redefining pulmonary hypertension. Lancet Respir Med. 2018 Mar;6(3):168-170. doi: 10.1016/S2213-2600(17)30498-8[↩]
- Maron BA, Wertheim BM, Gladwin MT. Under Pressure to Clarify Pulmonary Hypertension Clinical Risk. Am J Respir Crit Care Med. 2018 Feb 15;197(4):423-426. doi: 10.1164/rccm.201711-2306ED[↩][↩]
- Condliffe R, Kovacs G. Identifying early pulmonary arterial hypertension in patients with systemic sclerosis. Eur Respir J. 2018 Apr 4;51(4):1800495. doi: 10.1183/13993003.00495-2018[↩]
- Chapter 57 – Pulmonary Hypertension in Non-Pulmonary Arterial Hypertension Patients. Vascular Medicine: A Companion to Braunwald’s Heart Disease (Second Edition) 2013, Pages 687-696. https://doi.org/10.1016/B978-1-4377-2930-6.00057-4[↩]
- Humbert M, Kovacs G, Hoeper MM, et al. ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023 Jan 6;61(1):2200879. doi: 10.1183/13993003.00879-2022[↩]
- Galiè N, McLaughlin VV, Rubin LJ, Simonneau G. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J. 2019 Jan 24;53(1):1802148. doi: 10.1183/13993003.02148-2018[↩]
- Vachiéry JL, Tedford RJ, Rosenkranz S, Palazzini M, Lang I, Guazzi M, Coghlan G, Chazova I, De Marco T. Pulmonary hypertension due to left heart disease. Eur Respir J. 2019 Jan 24;53(1):1801897. doi: 10.1183/13993003.01897-2018[↩][↩]
- Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019 Jan 24;53(1):1801913. doi: 10.1183/13993003.01913-2018[↩][↩]
- Widrich J, Shetty M. Physiology, Pulmonary Vascular Resistance. [Updated 2024 Jan 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK554380[↩]
- Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB, National Heart, Lung, and Blood Institute Working Group on Cellular and Molecular Mechanisms of Right Heart Failure. Circulation. 2006 Oct 24; 114(17):1883-91.[↩]
- Humbert M, Kovacs G, Hoeper MM, Badagliacca R, et al. ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022 Oct 11;43(38):3618-3731. doi: 10.1093/eurheartj/ehac237. Erratum in: Eur Heart J. 2023 Apr 17;44(15):1312. doi: 10.1093/eurheartj/ehad005[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- 58 – Pulmonary Hypertension. Murray and Nadel’s Textbook of Respiratory Medicine (Sixth Edition) Volume 2, 2016, Pages 1031-1049.e4 https://doi.org/10.1016/B978-1-4557-3383-5.00058-0[↩][↩]
- National Audit of Pulmonary Hypertension, 10th Annual Report. https://digital.nhs.uk/data-and-information/publications/statistical/national-pulmonary-hypertension-audit/2019#[↩]
- Hoeper MM, Humbert M, Souza R, et al. A global view of pulmonary hypertension. Lancet Respir Med 2016;4:306–22. 10.1016/S2213-2600(15)00543-3[↩][↩][↩][↩][↩]
- Hoeper MM, Humbert M, Souza R, et al. A global view of pulmonary hypertension. Lancet Respir Med 2016; 4: 306–322. doi:10.1016/S2213-2600(15)00543-3[↩][↩][↩][↩][↩]
- Maron BA, Choudhary G, Khan UA, et al. Clinical profile and underdiagnosis of pulmonary hypertension in US veteran patients. Circ Heart Fail 2013;6:906–12. 10.1161/CIRCHEARTFAILURE.112.000091[↩]
- Maron BA, Hess E, Maddox TM, et al. Association of borderline pulmonary hypertension with mortality and hospitalization in a large patient cohort: insights from the Veterans Affairs clinical assessment, reporting, and tracking program. Circulation 2016;133:1240–8. 10.1161/CIRCULATIONAHA.115.020207[↩]
- Seeger W, Adir Y, Barberà JA, et al. Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol 2013;62(25 Suppl):D109–16. 10.1016/j.jacc.2013.10.036[↩][↩]
- Vachiéry J-L, Adir Y, Barberà JA, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol 2013;62(25 Suppl):D100–8. 10.1016/j.jacc.2013.10.033[↩][↩]
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53:1801913. 10.1183/13993003.01913-2018[↩][↩]
- Ross H, Feingold ML, Train JS. Mesenteric venous thrombosis. N Engl J Med. 2002 Apr 18;346(16):1252-3; author reply 1252-3.[↩]
- Rosenkranz S, Gibbs JSR, Wachter R, et al. Left ventricular heart failure and pulmonary hypertension. Eur Heart J 2016;37:942–54. 10.1093/eurheartj/ehv512[↩]
- Kessler R, Faller M, Weitzenblum E, et al. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med 2001;164:219–24. 10.1164/ajrccm.164.2.2006129[↩]
- Blanco I, Tura-Ceide O, Peinado VI, et al. Updated perspectives on pulmonary hypertension in COPD. Int J Chron Obstruct Pulmon Dis 2020;15:1315–24. 10.2147/COPD.S211841[↩]
- Lettieri CJ, Nathan SD, Barnett SD, et al. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 2006;129:746–52. 10.1378/chest.129.3.746[↩]
- Nathan SD, Shlobin OA, Ahmad S, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration 2008;76:288–94. 10.1159/000114246[↩]
- Shorr AF, Wainright JL, Cors CS, et al. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J 2007;30:715–21. 10.1183/09031936.00107206[↩]
- Leber L, Beaudet A, Muller A. Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: identification of the most accurate estimates from a systematic literature review. Pulm Circ 2021;11:2045894020977300. 10.1177/2045894020977300[↩]
- Delcroix M, Torbicki A, Gopalan D, et al. ERS statement on chronic thromboembolic pulmonary hypertension. Eur Respir J 2021;57:2002828. 10.1183/13993003.02828-2020[↩]
- Kramm T, Wilkens H, Fuge J, et al. Incidence and characteristics of chronic thromboembolic pulmonary hypertension in Germany. Clin Res Cardiol 2018;107:548–53. 10.1007/s00392-018-1215-5[↩]
- Shlobin OA, Kouranos V, Barnett SD, et al. Physiological predictors of survival in patients with sarcoidosis-associated pulmonary hypertension: results from an international registry. Eur Respir J 2020;55:1901747. 10.1183/13993003.01747-2019[↩]
- Zeder K, Banfi C, Steinrisser-Allex G, Maron BA, Humbert M, Lewis GD, Berghold A, Olschewski H, Kovacs G. Diagnostic, prognostic and differential-diagnostic relevance of pulmonary haemodynamic parameters during exercise: a systematic review. Eur Respir J. 2022 Oct 13;60(4):2103181. doi: 10.1183/13993003.03181-2021[↩][↩][↩]
- Ho JE, Zern EK, Lau ES, et al. Exercise pulmonary hypertension predicts clinical outcomes in patients with dyspnea on effort. J Am Coll Cardiol 2020; 75: 17–26. doi:10.1016/j.jacc.2019.10.048[↩]
- Stamm A, Saxer S, Lichtblau M, et al. Exercise pulmonary haemodynamics predict outcome in patients with systemic sclerosis. Eur Respir J 2016; 48: 1658–1667. doi:10.1183/13993003.00990-2016[↩]
- Hasler ED, Muller-Mottet S, Furian M, et al. Pressure-flow during exercise catheterization predicts survival in pulmonary hypertension. Chest 2016; 150: 57–67. doi:10.1016/j.chest.2016.02.634[↩]
- Lewis GD, Murphy RM, Shah RV, et al. Pulmonary vascular response patterns during exercise in left ventricular systolic dysfunction predict exercise capacity and outcomes. Circ Heart Fail 2011; 4: 276–285. doi:10.1161/CIRCHEARTFAILURE.110.959437[↩]
- Zeder K, Avian A, Bachmaier G, et al. Exercise pulmonary resistances predict long-term survival in systemic sclerosis. Chest 2021; 159: 781–790. doi:10.1016/j.chest.2020.08.2110[↩]
- Eisman AS, Shah RV, Dhakal BP, et al. Pulmonary capillary wedge pressure patterns during exercise predict exercise capacity and incident heart failure. Circ Heart Fail 2018; 11: e004750. doi:10.1161/CIRCHEARTFAILURE.117.004750[↩]
- Bentley RF, Barker M, Esfandiari S, et al. Normal and abnormal relationships of pulmonary artery to wedge pressure during exercise. J Am Heart Assoc 2020; 9: e016339. doi:10.1161/JAHA.120.016339[↩]
- Condliffe R, Kiely DG, Peacock AJ, Corris PA, Gibbs JS, Vrapi F, Das C, Elliot CA, Johnson M, DeSoyza J, Torpy C, Goldsmith K, Hodgkins D, Hughes RJ, Pepke-Zaba J, Coghlan JG. Connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med. 2009 Jan 15;179(2):151-7. doi: 10.1164/rccm.200806-953OC[↩]
- Weatherald J, Montani D, Jevnikar M, Jaïs X, Savale L, Humbert M. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev. 2019 Jul 31;28(153):190023. doi: 10.1183/16000617.0023-2019[↩]
- Humbert M, Yaici A, de Groote P, Montani D, Sitbon O, Launay D, Gressin V, Guillevin L, Clerson P, Simonneau G, Hachulla E. Screening for pulmonary arterial hypertension in patients with systemic sclerosis: clinical characteristics at diagnosis and long-term survival. Arthritis Rheum. 2011 Nov;63(11):3522-30. doi: 10.1002/art.30541[↩]
- Enright Md P. Office-based DLCO tests help pulmonologists to make important clinical decisions. Respir Investig. 2016 Sep;54(5):305-11. doi: 10.1016/j.resinv.2016.03.006[↩]
- Modi P, Goldin J, Cascella M. Diffusing Capacity of the Lungs for Carbon Monoxide. [Updated 2024 Oct 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK556149[↩]
- Coghlan JG, Denton CP, Grünig E, Bonderman D, Distler O, Khanna D, Müller-Ladner U, Pope JE, Vonk MC, Doelberg M, Chadha-Boreham H, Heinzl H, Rosenberg DM, McLaughlin VV, Seibold JR; DETECT study group. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis. 2014 Jul;73(7):1340-9. doi: 10.1136/annrheumdis-2013-203301[↩][↩][↩]
- Screening for PAH-CTD. https://www.suspectpahctd.com/screening/[↩]
- Krowl L, Anjum F, Kaul P. Pulmonary Idiopathic Hypertension. [Updated 2023 Apr 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK519041[↩]
- Pahal P, Sharma S. Idiopathic Pulmonary Artery Hypertension. [Updated 2023 Apr 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482251[↩][↩][↩][↩][↩]
- Gaine S, Chin K, Coghlan G, Channick R, Di Scala L, Galie N, et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur Respir J. 2017;50:1602493. doi: 10.1183/13993003.02493-2016[↩]
- Kato M, Atsumi T. Pulmonary arterial hypertension associated with connective tissue diseases: a review focusing on distinctive clinical aspects. Eur J Clin Invest. 2018 doi: 10.1111/eci.12876[↩]
- Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Resp Crit Care. 2006;173:1023–1030. doi: 10.1164/rccm.200510-1668OC[↩]
- McLaughlin VV, Hoeper MM, Channick RN, Chin KM, Delcroix M, Gaine S, Ghofrani HA, Jansa P, Lang IM, Mehta S, Pulido T, Sastry BKS, Simonneau G, Sitbon O, Souza R, Torbicki A, Tapson VF, Perchenet L, Preiss R, Verweij P, Rubin LJ, Galiè N. Pulmonary Arterial Hypertension-Related Morbidity Is Prognostic for Mortality. J Am Coll Cardiol. 2018 Feb 20;71(7):752-763. doi: 10.1016/j.jacc.2017.12.010[↩]
- Pulmonary arterial hypertension. https://medlineplus.gov/genetics/condition/pulmonary-arterial-hypertension[↩]
- Learn About Pulmonary Arterial Hypertension. https://www.lung.org/lung-health-diseases/lung-disease-lookup/pulmonary-arterial-hypertension/learn-about-pulmonary-arterial-hypertension[↩][↩]
- Badesch D.B., Raskob G.E., Elliott C.G., et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137(2):376–387. doi: 10.1378/chest.09-1140[↩]
- Zanatta E, Polito P, Famoso G, Larosa M, De Zorzi E, Scarpieri E, Cozzi F, Doria A. Pulmonary arterial hypertension in connective tissue disorders: Pathophysiology and treatment. Exp Biol Med (Maywood). 2019 Feb;244(2):120-131. doi: 10.1177/1535370218824101[↩]
- Berteloot L, Proisy M, Jais JP, Lévy M, Boddaert N, Bonnet D, Raimondi F. Idiopathic, heritable and veno-occlusive pulmonary arterial hypertension in childhood: computed tomography angiography features in the initial assessment of the disease. Pediatr Radiol. 2019 May;49(5):575-585. doi: 10.1007/s00247-018-04331-y[↩]
- Weiss BM, Zemp L, Seifert B, Hess OM. Outcome of pulmonary vascular disease in pregnancy: a systematic overview from 1978 through 1996. J Am Coll Cardiol. 1998 Jun;31(7):1650-7. doi: 10.1016/s0735-1097(98)00162-4[↩]
- Bédard E, Dimopoulos K, Gatzoulis MA. Has there been any progress made on pregnancy outcomes among women with pulmonary arterial hypertension? Eur Heart J. 2009 Feb;30(3):256-65. doi: 10.1093/eurheartj/ehn597[↩]
- Duarte AG, Thomas S, Safdar Z, Torres F, Pacheco LD, Feldman J, deBoisblanc B. Management of pulmonary arterial hypertension during pregnancy: a retrospective, multicenter experience. Chest. 2013 May;143(5):1330-1336. doi: 10.1378/chest.12-0528[↩]
- Jaïs X, Olsson KM, Barbera JA, Blanco I, Torbicki A, Peacock A, Vizza CD, Macdonald P, Humbert M, Hoeper MM. Pregnancy outcomes in pulmonary arterial hypertension in the modern management era. Eur Respir J. 2012 Oct;40(4):881-5. doi: 10.1183/09031936.00141211[↩][↩]
- Kiely DG, Condliffe R, Webster V, Mills GH, Wrench I, Gandhi SV, Selby K, Armstrong IJ, Martin L, Howarth ES, Bu’lock FA, Stewart P, Elliot CA. Improved survival in pregnancy and pulmonary hypertension using a multiprofessional approach. BJOG. 2010 Apr;117(5):565-74. doi: 10.1111/j.1471-0528.2009.02492.x[↩][↩]
- Luo J, Shi H, Xu L, Su W, Li J. Pregnancy outcomes in patients with pulmonary arterial hypertension: A retrospective study. Medicine (Baltimore). 2020 Jun 5;99(23):e20285. doi: 10.1097/MD.0000000000020285[↩]
- Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015 Oct;46(4):903-75. doi: 10.1183/13993003.01032-2015 Erratum in: Eur Respir J. 2015 Dec;46(6):1855-6. doi: 10.1183/13993003.51032-2015[↩]
- Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M; ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016 Jan 1;37(1):67-119. doi: 10.1093/eurheartj/ehv317[↩]
- Kamp JC, von Kaisenberg C, Greve S, Winter L, Park DH, Fuge J, Kühn C, Hoeper MM, Olsson KM. Pregnancy in pulmonary arterial hypertension: Midterm outcomes of mothers and offspring. J Heart Lung Transplant. 2021 Mar;40(3):229-233. doi: 10.1016/j.healun.2020.12.002[↩][↩]
- Corbach N, Berlier C, Lichtblau M, Schwarz EI, Gautschi F, Groth A, Schüpbach R, Krähenmann F, Saxer S, Ulrich S. Favorable Pregnancy Outcomes in Women With Well-Controlled Pulmonary Arterial Hypertension. Front Med (Lausanne). 2021 Jul 5;8:689764. doi: 10.3389/fmed.2021.689764[↩]
- Bostock S, Sheares K, Cannon J, Taboada D, Pepke-Zaba J, Toshner M. The potential effects of pregnancy in a patient with idiopathic pulmonary arterial hypertension responding to calcium channel blockade. Eur Respir J. 2017 Dec 14;50(6):1701141. doi: 10.1183/13993003.01141-2017[↩]
- de Raaf MA, Beekhuijzen M, Guignabert C, Vonk Noordegraaf A, Bogaard HJ. Endothelin-1 receptor antagonists in fetal development and pulmonary arterial hypertension. Reprod Toxicol. 2015 Aug 15;56:45-51. doi: 10.1016/j.reprotox.2015.06.048[↩]
- Dunn L, Greer R, Flenady V, Kumar S. Sildenafil in Pregnancy: A Systematic Review of Maternal Tolerance and Obstetric and Perinatal Outcomes. Fetal Diagn Ther. 2017;41(2):81-88. doi: 10.1159/000453062[↩]
- van Giersbergen PL, Halabi A, Dingemanse J. Pharmacokinetic interaction between bosentan and the oral contraceptives norethisterone and ethinyl estradiol. Int J Clin Pharmacol Ther. 2006 Mar;44(3):113-8. doi: 10.5414/cpp44113[↩]
- PAH Medication & Treatment Guide. https://www.lung.org/getmedia/db7df4fe-93fa-439b-bd24-80856b9e3365/PAH-Medication-Guide.pdf[↩]
- https://www.janssenlabels.com/package-insert/product-monograph/prescribing-information/UPTRAVI-pi.pdf[↩]
- Behr J, Nathan SD, Harari S, Wuyts W, Kirchgaessler KU, Bengus M, Gilberg F, Wells AU. Sildenafil added to pirfenidone in patients with advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension: A Phase IIb, randomised, double-blind, placebo-controlled study – Rationale and study design. Respir Med. 2018 May;138:13-20. doi: 10.1016/j.rmed.2018.03.019[↩]
- Konstam MA, Kiernan MS, Bernstein D, Bozkurt B, Jacob M, Kapur NK, Kociol RD, Lewis EF, Mehra MR, Pagani FD, Raval AN, Ward C; American Heart Association Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; and Council on Cardiovascular Surgery and Anesthesia. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement From the American Heart Association. Circulation. 2018 May 15;137(20):e578-e622. doi: 10.1161/CIR.0000000000000560[↩]
- Kim D, Lee KM, Freiman MR, Powell WR, Klings ES, Rinne ST, Miller DR, Rose AJ, Wiener RS. Phosphodiesterase-5 Inhibitor Therapy for Pulmonary Hypertension in the United States. Actual versus Recommended Use. Ann Am Thorac Soc. 2018 Jun;15(6):693-701. doi: 10.1513/AnnalsATS.201710-762OC[↩][↩]
- Mishra A, Singh M, Kaluski E. The year since the guidelines: a concise update on recent advances in pulmonary hypertension. Minerva Cardioangiol. 2017 Feb;65(1):68-73. doi: 10.23736/S0026-4725.16.04240-7[↩]
- López-Meseguer M, Quezada CA, Ramon MA, Lázaro M, Dos L, Lara A, López R, Blanco I, Escribano P, Roman A; REHAP Investigators. Lung and heart-lung transplantation in pulmonary arterial hypertension. PLoS One. 2017 Nov 21;12(11):e0187811. doi: 10.1371/journal.pone.0187811. Erratum in: PLoS One. 2018 Jan 29;13(1):e0192100. doi: 10.1371/journal.pone.0192100[↩]
- Bermejo J, Yotti R, García-Orta R, et al. Sildenafil for Improving Outcomes after VAlvular Correction (SIOVAC) investigators. Sildenafil for improving outcomes in patients with corrected valvular heart disease and persistent pulmonary hypertension: a multicenter, double-blind, randomized clinical trial. Eur Heart J. 2018 Apr 14;39(15):1255-1264. doi: 10.1093/eurheartj/ehx700[↩]
- Brown LM, Chen H, Halpern S, et al. Delay in Recognition of Pulmonary Arterial Hypertension: Factors Identified From the REVEAL Registry. Chest. 2011;140(1):19-26. doi:10.1378/chest.10-1166. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3198486/[↩]
- Barst RJ, Chung L, Zamanian RT, Turner M, McGoon MD. Functional class improvement and 3-year survival outcomes in patients with pulmonary arterial hypertension in the REVEAL Registry. Chest. 2013 Jul;144(1):160-168. doi: 10.1378/chest.12-2417[↩]
- Nickel N, Golpon H, Greer M, Knudsen L, Olsson K, Westerkamp V, Welte T, Hoeper MM. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2012 Mar;39(3):589-96. doi: 10.1183/09031936.00092311[↩]
- Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, Rainisio M, Simonneau G. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002 Aug 21;40(4):780-8. doi: 10.1016/s0735-1097(02)02012-0[↩]
- Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010 Jul 13;122(2):164-72. doi: 10.1161/CIRCULATIONAHA.109.898122[↩]
- Humbert M, Sitbon O, Yaïci A, Montani D, O’Callaghan DS, Jaïs X, Parent F, Savale L, Natali D, Günther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G; French Pulmonary Arterial Hypertension Network. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010 Sep;36(3):549-55. doi: 10.1183/09031936.00057010[↩]
- Zamanian RT, Kudelko KT, Sung YK, Perez VJ, Liu J, Spiekerkoetter E. Current clinical management of pulmonary arterial hypertension. Circ Res. 2014 Jun 20;115(1):131-147. doi: 10.1161/CIRCRESAHA.115.303827[↩]
- Guillevin L, Armstrong I, Aldrighetti R, Howard LS, Ryftenius H, Fischer A, Lombardi S, Studer S, Ferrari P. Understanding the impact of pulmonary arterial hypertension on patients’ and carers’ lives. Eur Respir Rev. 2013 Dec;22(130):535-42. doi: 10.1183/09059180.00005713[↩]
- Keen C, Fowler-Davis S, McLean S, Manson J. Physiotherapy practice in pulmonary hypertension: physiotherapist and patient perspectives. Pulm Circ. 2018 Jul-Sep;8(3):2045894018783738. doi: 10.1177/2045894018783738[↩]
- Chang KY, Duval S, Badesch DB, Bull TM, Chakinala MM, De Marco T, Frantz RP, Hemnes A, Mathai SC, Rosenzweig EB, Ryan JJ, Thenappan T; PHAR Investigators *. Mortality in Pulmonary Arterial Hypertension in the Modern Era: Early Insights From the Pulmonary Hypertension Association Registry. J Am Heart Assoc. 2022 May 3;11(9):e024969. doi: 10.1161/JAHA.121.024969[↩]
- Fisher M.R., Mathai S.C., Champion H.C., et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006;54(9):3043–3050. doi: 10.1002/art.22069[↩][↩]
- Chung L., Liu J., Parsons L., et al. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: identifying systemic sclerosis as a unique phenotype. Chest. 2010;138(6):1383–1394. doi: 10.1378/chest.10-0260[↩]
- Ahmed S., Palevsky H.I. Pulmonary arterial hypertension related to connective tissue disease: a review. Rheum Dis Clin North Am. 2014;40(1):103–124. doi: 10.1016/j.rdc.2013.10.001[↩]
- Gashouta M.A., Humbert M., Hassoun P.M. Update in systemic sclerosis-associated pulmonary arterial hypertension. Presse Med. 2014;43(10 Pt 2):e293–e304. doi: 10.1016/j.lpm.2014.06.007[↩]
- Clements P.J., Tan M., McLaughlin V.V., et al. Investigators PAHQERIP-Q The pulmonary arterial hypertension quality enhancement research initiative: comparison of patients with idiopathic PAH to patients with systemic sclerosis-associated PAH. Ann Rheum Dis. 2012;71(2):249–252. doi: 10.1136/annrheumdis-2011-200265[↩]
- Chaisson N.F., Hassoun P.M. Systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2013;144(4):1346–1356. doi: 10.1378/chest.12-2396[↩]
- Overbeek M.J., Vonk M.C., Boonstra A., et al. Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: a distinctive vasculopathy. Eur Respir J. 2009;34(2):371–379. doi: 10.1183/09031936.00106008[↩]
- Kelemen B.W., Mathai S.C., Tedford R.J., et al. Right ventricular remodeling in idiopathic and scleroderma-associated pulmonary arterial hypertension: two distinct phenotypes. Pulm Circ. 2015;5(2):327–334. doi: 10.1086/680356[↩]
- Vizza CD, Hoeper MM, Huscher D, et al. Pulmonary Hypertension in Patients With COPD: Results From the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Chest. 2021 Aug;160(2):678-689. doi: 10.1016/j.chest.2021.02.012[↩]





{kind=link}