Contents
What is dilated cardiomyopathy
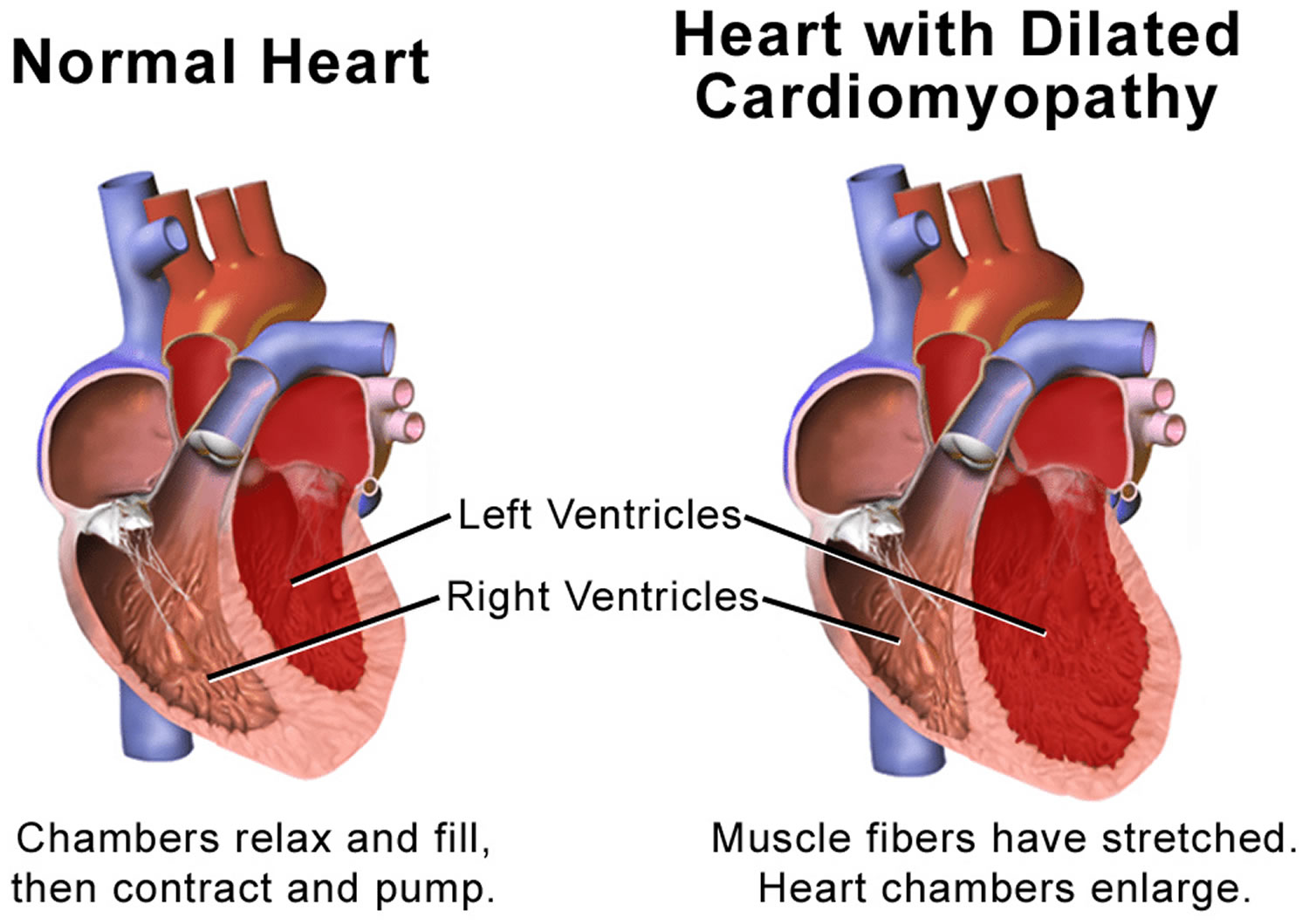
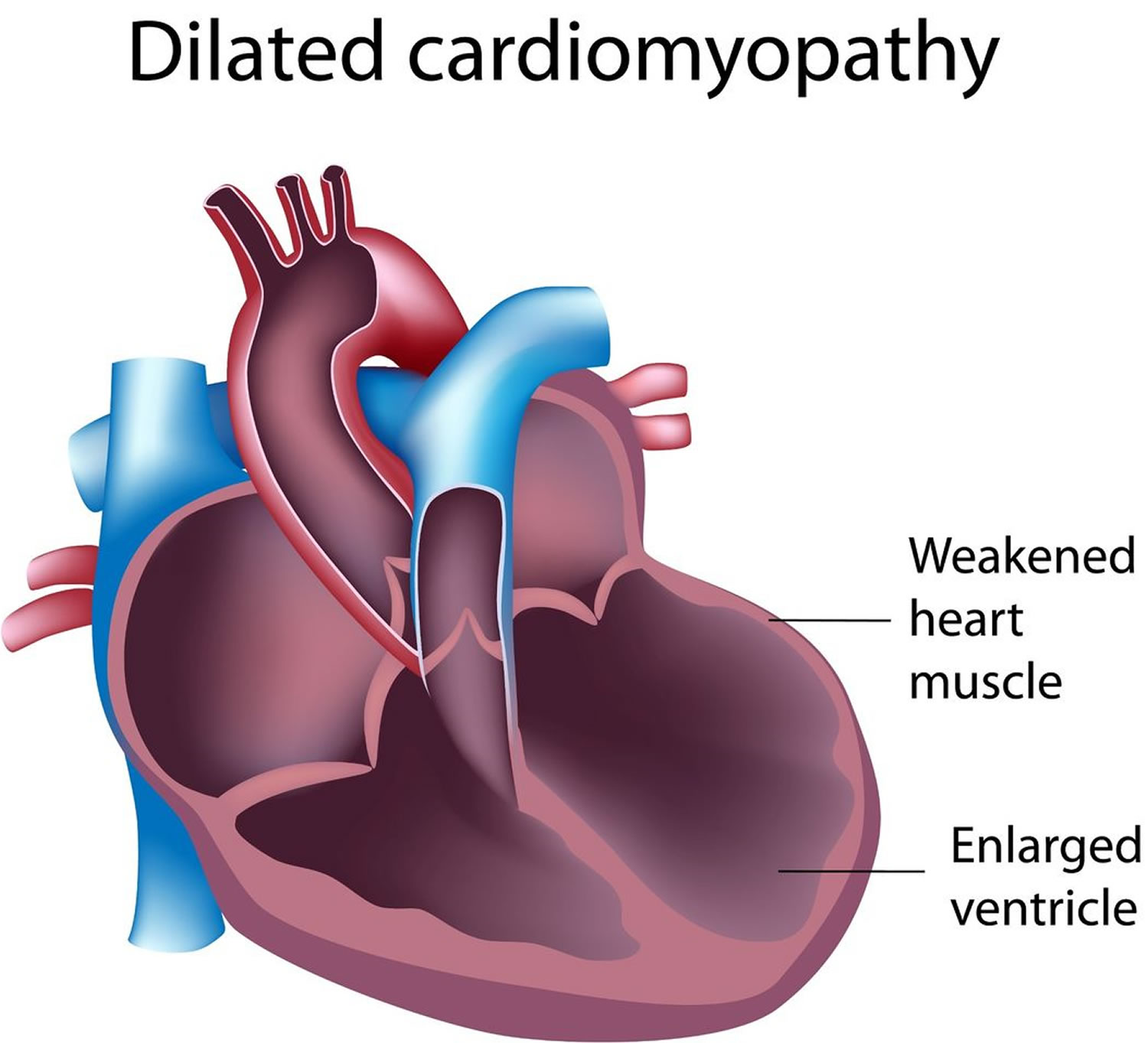
Dilated cardiomyopathy is a disease of your heart muscle where it becomes stretched or ‘enlarged’ and thin (see Figure 4 below). This means that your heart is unable to pump blood around your body efficiently. Dilated cardiomyopathy is the most common form of cardiomyopathy (heart muscle disease). Dilated cardiomyopathy usually starts in the left ventricle (the heart’s main pumping chamber) and over time can affect the right ventricle and then to the atria. The weakened chambers of the heart don’t pump effectively, causing the heart muscle to work harder. Over time, the heart loses the ability to pump blood effectively. Dilated cardiomyopathy can lead to heart failure, heart valve disease, irregular heart rate, and blood clots in the heart.
Dilated cardiomyopathy is the most common type cardiomyopathy, occurring mostly in adults 20 to 60.
In dilated cardiomyopathy the muscle walls of the heart have become stretched (dilated) and thin, so the heart can’t contract (squeeze) properly to pump blood around the body. This can lead to fluid building up in the lungs, ankles, abdomen and other organs of the body and a feeling of being breathless. This collection of symptoms is known as heart failure.
In most cases dilated cardiomyopathy develops slowly, so the heart can be quite severely affected before someone is diagnosed. In some cases, there may also be mitral regurgitation. This is when some of your blood flows in the wrong direction through the mitral valve, from the left ventricle to the left atrium.
It is estimated that 750,000 people in the United States have dilated cardiomyopathy; roughly half of these cases are familial.
Other Names for Dilated Cardiomyopathy
- Alcoholic cardiomyopathy. (A term used when overuse of alcohol causes the disease)
- Congestive cardiomyopathy
- Diabetic cardiomyopathy
- Familial dilated cardiomyopathy
- Idiopathic cardiomyopathy
- Ischemic cardiomyopathy (A term used when coronary heart disease, also called coronary artery disease or heart attack cause the disease. Not all forms of dilated cardiomyopathy are ischemic in origin.)
- Peripartum cardiomyopathy. (A term used when the disease develops in a woman shortly before or after she gives birth.)
- Primary cardiomyopathy
How serious is dilated cardiomyopathy?
If you have dilated cardiomyopathy, the left ventricle of your heart becomes dilated (stretched or ‘baggy’). As a result, the heart muscle becomes weak, thin or floppy and is unable to pump blood around the body efficiently.
If you have dilated cardiomyopathy, you’re at greater risk of heart failure, where the heart fails to pump enough blood around the body at the right pressure.
Heart failure typically causes shortness of breath, extreme tiredness and ankle swelling.
There’s also a risk of heart valve problems, an irregular heartbeat and blood clots. You’ll need to have regular appointments with your doctor so the disease can be monitored.
Human Heart
The heart is a specialized muscle that contracts regularly and continuously, pumping blood to the body and the lungs. It has four chambers – two at the top (the atria), and two at the bottom (the ventricles). See the Figures 1 to 3 below.
The heart is made up of three layers:
- the endocardium
- the myocardium
- the pericardium.
The endocardium is a thin layer on the inside of the heart, lining the chambers and valves. The myocardium is the thick, muscular layer of the heart that contracts and squeezes the blood out of the heart. It’s this layer that is affected by cardiomyopathy. The pericardium is a thin, double layer that forms a protective sac around the outside of the heart. It contains a small amount of fluid – called pericardial fluid – which acts as a lubricant when the heart is contracting.
The pumping action of the heart is caused by a flow of electricity through the heart that repeats itself in a cycle. This is normally triggered by the heart’s natural pacemaker, the SA node (sino-atrial node), which is in the right atrium (see Figure 4 below). The SA node sends out regular electrical impulses, which make the atria contract and pump blood into the ventricles. The electrical impulse then passes to the ventricles through a form of ‘junction box’ called the AV node (atrio-ventricular node). This causes the ventricles to contract and pump blood out of the heart. The blood from the right ventricle goes through the pulmonary artery to the lungs, and the blood from the left ventricle goes through the aorta and then around the body.
Figure 1. The anatomy of the heart
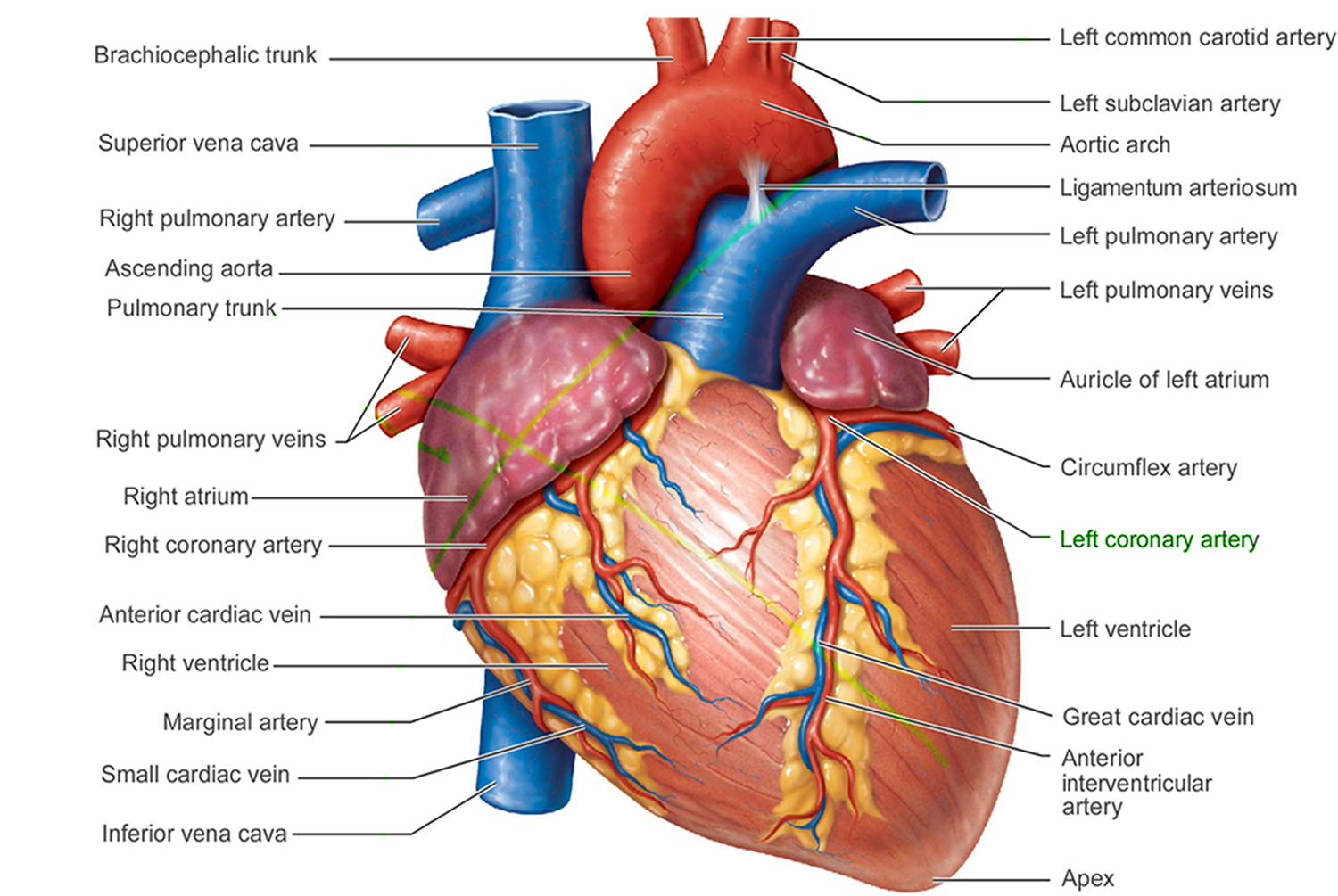
Figure 2. The anatomy of the heart chambers
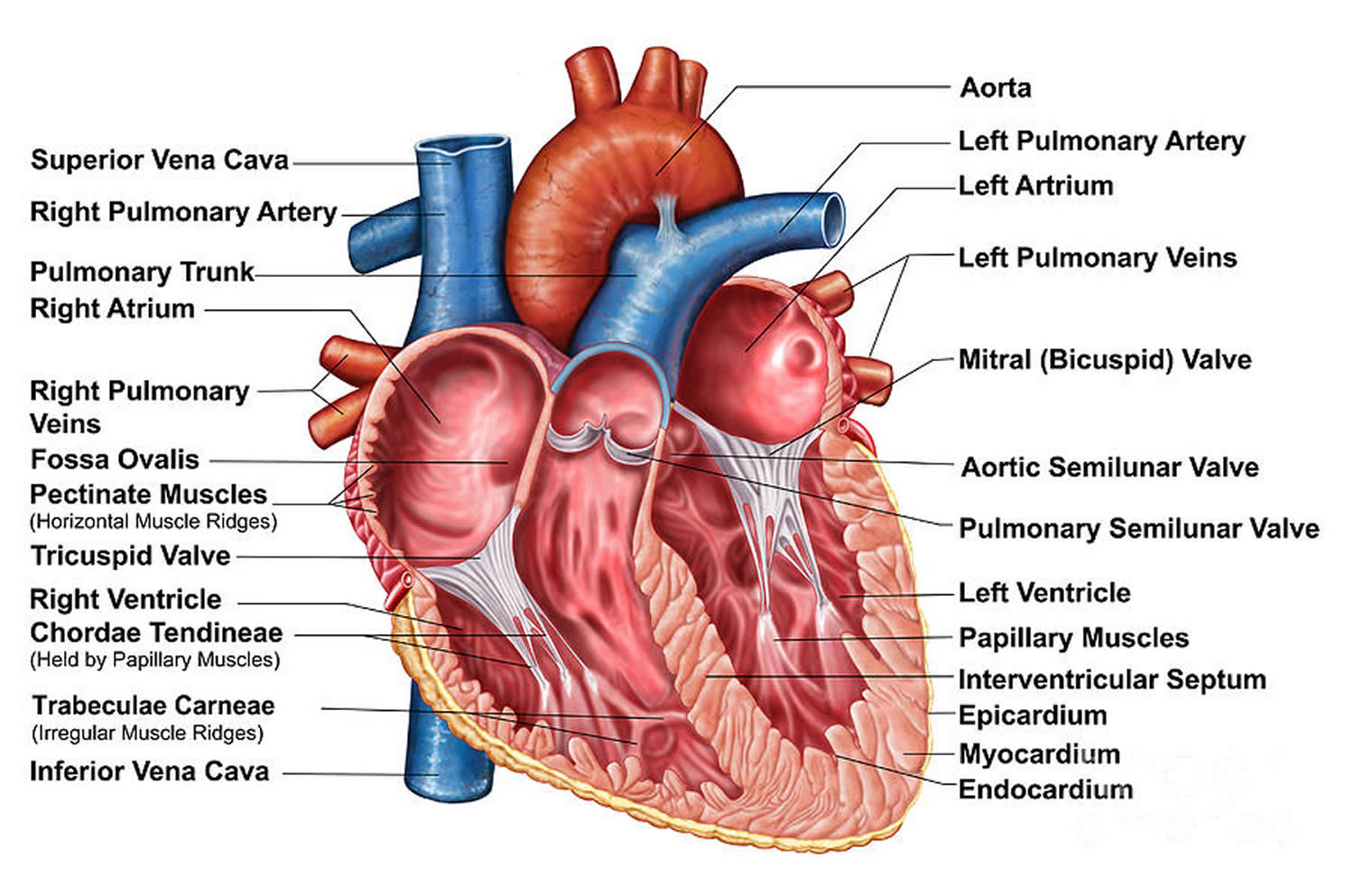 Figure 3. Normal heart blood flow
Figure 3. Normal heart blood flow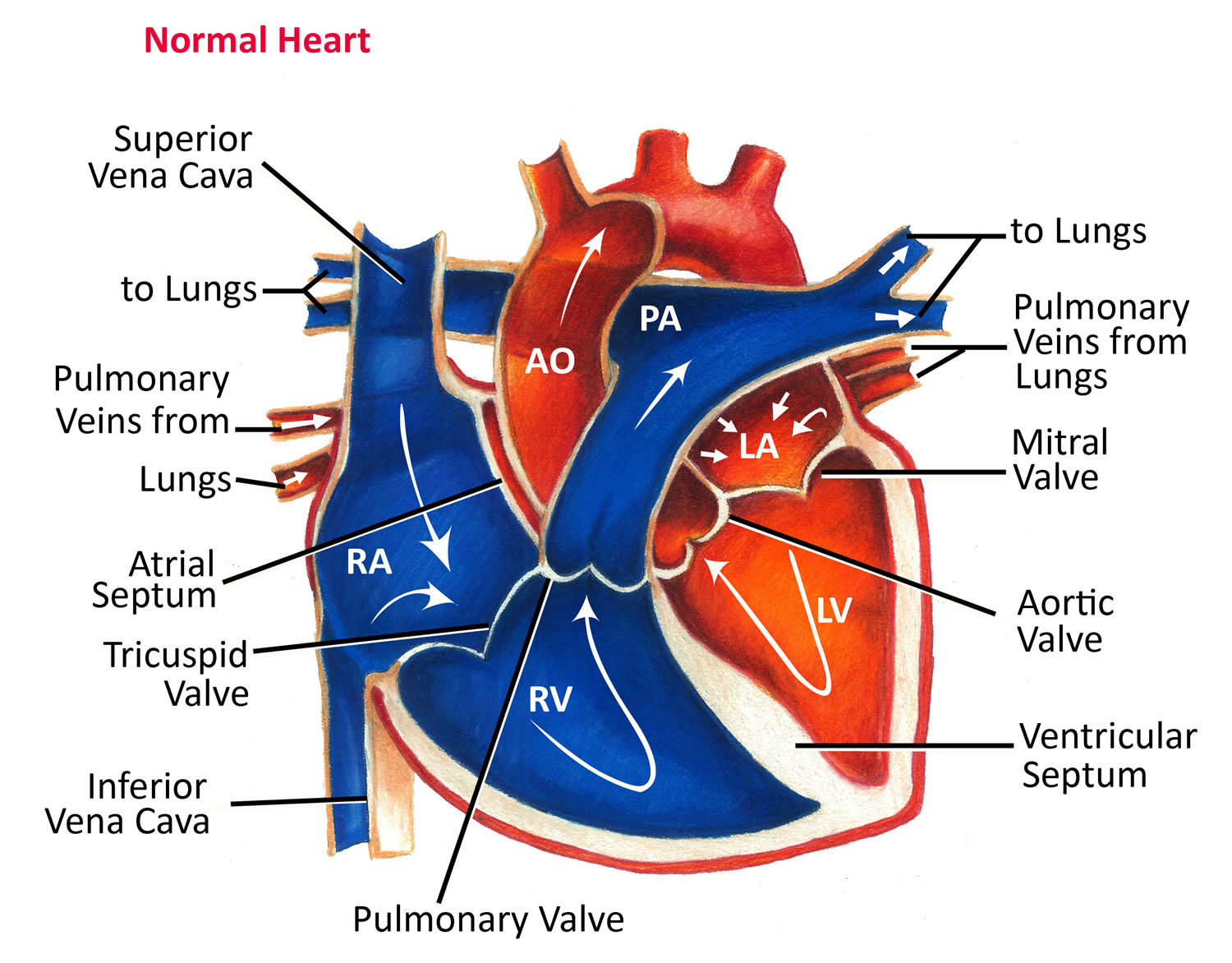 Figure 4. The heart’s electrical system
Figure 4. The heart’s electrical system
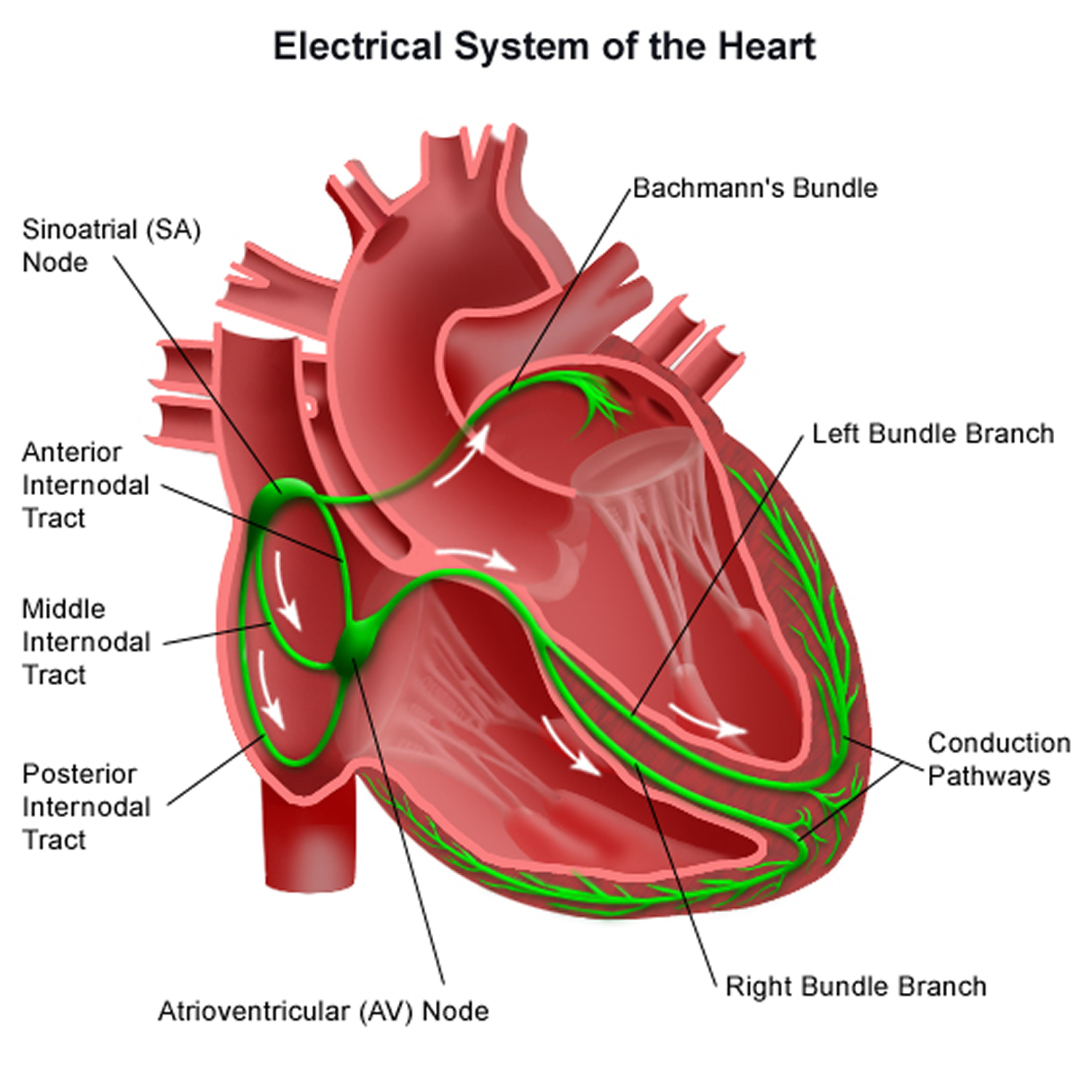 Figure 5. Dilated cardiomyopathy
Figure 5. Dilated cardiomyopathy
Dilated cardiomyopathy life expectancy
This has a poor prognosis. Fifty percent of patients die within 2 years; 25% of patients survive longer than 5 years. The two most common causes of death are progressive cardiac failure and arrhythmia.
In children with dilated cardiomyopathy, approximately 35% recover completely, 35% stabilize and the remaining may progressively worsen. Children with dilated cardiomyopathy are more prone to congestive heart failure and have a higher rate of heart transplantation compared with other forms of cardiomyopathy. However, improved medical therapy may eventually change this scenario. Current average five-year survival rates for children with dilated cardiomyopathy are 40–50%. If the cause of dilated cardiomyopathy is myocarditis, children are more likely to improve and have a better outcome than children with other causes of dilated cardiomyopathy.
Dilated cardiomyopathy causes
Dilated cardiomyopathy can affect both children and adults. In most cases, the exact reason for dilated cardiomyopathy is unknown and the condition is called ‘idiopathic dilated cardiomyopathy’. Idiopathic means that there is no known cause.
Inherited dilated cardiomyopathy is caused by a change or mutation in one or more genes. Up to one-third of the people who have dilated cardiomyopathy inherit it from their parents. If you have dilated cardiomyopathy, there is a 50 per cent chance that your child will inherit the condition.
Sometimes dilated cardiomyopathy is caused by other things, such as:
- Uncontrolled high blood pressure
- Coronary heart disease, heart attack
- Diabetes, thyroid disease, viral hepatitis and HIV
- Unhealthy lifestyle – such as a lack of vitamins and minerals in the diet, heavy alcohol drinking and recreational drug use
- Viral infection that causes inflammation of the heart muscle
- Infections, including those caused by bacteria, fungi and parasites
- Heart valve problem
- Auto-immune disease, a disease of the body tissues or vessels – such as Wegener’s granulomatosis, sarcoidosis, amyloidosis, lupus, polyarteritis nodosa, vasculitis or muscular dystrophy
- Inheriting a mutated (changed) gene that makes you more vulnerable to the disease.
- Pregnancy – cardiomyopathy can sometimes develop as complications during the last month of pregnancy or within 5 months of birth (Peripartum cardiomyopathy and Postpartum cardiomyopathy)
- Certain toxins such as cobalt
- Certain drugs (such as cocaine and amphetamines) and two medicines used to treat cancer (doxorubicin and daunorubicin)
In some people with dilated cardiomyopathy there may be more than one reason to explain their condition. For example, some people may carry a gene mutation that makes them more vulnerable to viral infections in the heart.
Viral infections
We’re all exposed to many viruses every day. Normally, your body’s immune system is very efficient at dealing with these viruses. However, sometimes a virus can affect the heart muscle of an apparently well person, even though the person has no other symptoms of having a virus. This is known as viral myocarditis.
Viral myocarditis is commonly caused by a number of different viruses. It is thought that dilated cardiomyopathy may occur when your heart muscle is badly damaged by the initial infection. Or it may be that the virus triggers your body’s own defence system (the immune system) which then attacks and damages your heart muscle.
Auto-immune disease
Your body’s immune system is responsible for defending your body against infection – for example, against viruses and bacteria. Sometimes your immune system breaks down and starts to attack your body’s own tissues. This is called auto-immune disease. Some people who are diagnosed with dilated cardiomyopathy appear to have this condition.
Exposure to toxins or certain medicines
In very rare cases, exposure to certain chemicals can cause dilated cardiomyopathy. In these cases, scientists don’t know if the person developed the condition because they already had a genetic tendency to develop it, or whether the toxins caused the cardiomyopathy.
Some anti-cancer medicines, such as anthracyclines, can cause dilated cardiomyopathy. These medicines are a very effective treatment for cancer, but they are toxic and can have harmful effects on your heart muscle, which can lead to dilated cardiomyopathy. The risk of this happening is usually related to the total dose of anthracyclines received.
If you’ve had treatment with anthracyclines, you should be seen by a cardiologist (heart specialist) at least once a year. If you know that you have a heart condition and that you will need treatment for cancer, you should discuss this with your cardiologist before you start the treatment.
In some cases, alcohol can be the cause of dilated cardiomyopathy. Over time, regularly drinking too much alcohol can weaken your heart by damaging your heart muscle. It can take years for dilated cardiomyopathy to develop and so the effects on your heart may not be seen immediately.
Risk factors for developing dilated cardiomyopathy
Dilated cardiomyopathy most commonly occurs in men, ages 20 to 50. But it can also occur in women. Other risk factors include:
- Damage to the heart muscle from a heart attack
- Family history of dilated cardiomyopathy
- Inflammation of heart muscle from immune system disorders, such as lupus
- Neuromuscular disorders, such as muscular dystrophy
Inherited dilated cardiomyopathy
Research has shown that dilated cardiomyopathy can be linked to an individual’s genetic make-up 1. Scientists have identified some gene mutations that are known to affect the development of heart muscle, and that may cause dilated cardiomyopathy. However, there are many other gene mutations that may also cause the condition. This makes genetic testing of individuals to find out if they have the condition more difficult.
Mutations in more than 30 genes have been found to cause familial dilated cardiomyopathy 2. These genes provide instructions for making proteins that are found in cardiac muscle cells called cardiomyocytes.
Many of these proteins play important roles in the contraction of the cardiac muscle through their association with cell structures called sarcomeres. Sarcomeres are the basic units of muscle contraction; they are made of proteins that generate the mechanical force needed for muscles to contract. Many other proteins associated with familial dilated cardiomyopathy make up the structural framework (the cytoskeleton) of cardiomyocytes. The remaining proteins play various roles within cardiomyocytes to ensure their proper functioning.
Mutations in one gene, TTN, account for approximately 20 percent of cases of familial dilated cardiomyopathy 2. The TTN gene provides instructions for making a protein called titin, which is found in the sarcomeres of many types of muscle cells, including cardiomyocytes. Titin provides structure, flexibility, and stability to sarcomeres. Titin also plays a role in chemical signaling and in assembling new sarcomeres. The TTN gene mutations that cause familial dilated cardiomyopathy result in the production of an abnormally short titin protein. It is unclear how the altered protein causes familial dilated cardiomyopathy, but it is likely that it impairs sarcomere function and disrupts chemical signaling.
It is unclear how mutations in the other genes cause familial dilated cardiomyopathy. It is likely that the changes impair cardiomyocyte function and reduce the ability of these cells to contract, weakening and thinning cardiac muscle.
People with familial dilated cardiomyopathy often do not have an identified mutation in any of the known associated genes. The cause of the condition in these individuals is unknown.
Familial dilated cardiomyopathy is described as nonsyndromic or isolated because it typically affects only the heart. However, dilated cardiomyopathy can also occur as part of syndromes that affect other organs and tissues in the body. These forms of the condition are described as syndromic and are caused by mutations in other genes.
Familial dilated cardiomyopathy has different inheritance patterns depending on the gene involved.
In 80 to 90 percent of cases, familial dilated cardiomyopathy is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person inherits the mutation from one affected parent. However, some people who inherit the altered gene never develop features of familial dilated cardiomyopathy. (This situation is known as reduced penetrance.) Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
In rare instances, this condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
In other rare cases, this condition is inherited in an X-linked pattern. In these cases, the gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell increases the risk of developing heart disease, but females with such a mutation may not develop familial dilated cardiomyopathy. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes familial dilated cardiomyopathy. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Alcoholic cardiomyopathy
Drinking too much alcohol can raise the levels of some fats in the blood (triglycerides).
It can also lead to high blood pressure, heart failure and an increased calorie intake. (Consuming too many calories can lead to obesity and a higher risk of developing diabetes.) Excessive alcohol consumption contributes to alcoholism, suicide and accidents.
Excessive drinking and binge drinking can lead to stroke. Other serious problems include fetal alcohol syndrome, cardiomyopathy, cardiac arrhythmia and sudden cardiac death, cancer and other diseases.
If you drink alcohol, do so in moderation. This means an average of one to two drinks per day for men and one drink per day for women. (A drink is one 12 oz. beer, 4 oz. of wine, 1.5 oz. of 80-proof spirits, or 1 oz. of 100-proof spirits.)
Drinking more alcohol increases such dangers as alcoholism, high blood pressure, obesity, stroke, breast cancer, suicide and accidents.
Also, it’s not possible to predict in which people alcoholism will become a problem. Given these and other risks, the American Heart Association cautions people NOT to start drinking if they do not already drink alcohol.
Ischemic cardiomyopathy
Coronary heart disease or coronary artery disease, is a common term for the buildup of plaque in the heart’s arteries that could lead to heart attack.
With coronary artery disease, plaque first grows within the walls of the coronary arteries until the blood flow to the heart’s muscle is limited.
This is also called ischemia. It may be chronic, narrowing of the coronary artery over time and limiting of the blood supply to part of the muscle. Or it can be acute, resulting from a sudden rupture of a plaque and formation of a thrombus or blood clot.
The traditional risk factors for coronary artery disease are high LDL “bad” cholesterol, low HDL “good” cholesterol, high blood pressure, family history, diabetes, smoking, being post-menopausal for women and being older than 45 for men. Obesity is also be a risk factor.
Coronary artery disease begins in childhood, so that by the teenage years, there is evidence that plaques that will stay with us for life are formed in most people. Preventive measures instituted early are thought to have greater lifetime benefits. Living a healthy lifestyle that incorporates good nutrition, weight management and getting plenty of physical activity can play a big role in avoiding coronary artery disease.
Major risk factors you can modify, treat or control
Tobacco smoke
Smokers’ risk of developing coronary heart disease is much higher than that of nonsmokers. Cigarette smoking is a powerful independent risk factor for sudden cardiac death in patients with coronary heart disease. Cigarette smoking also acts with other risk factors to greatly increase the risk for coronary heart disease. Exposure to other people’s smoke increases the risk of heart disease even for nonsmokers.
High blood cholesterol
As blood cholesterol rises, so does risk of coronary heart disease. When other risk factors (such as high blood pressure and tobacco smoke) are present, this risk increases even more. A person’s cholesterol level is also affected by age, sex, heredity and diet.
Here’s the lowdown on:
- Total Cholesterol: Your total cholesterol score is calculated using the following equation: HDL + LDL + 20 percent of your triglyceride level.
- Low-density-lipoprotein (LDL) cholesterol = “bad” cholesterol. A low LDL cholesterol level is considered good for your heart health. However, your LDL number should no longer be the main factor in guiding treatment to prevent heart attack and stroke, according to the latest guidelines from the American Heart Association. For patients taking statins, the guidelines say they no longer need to get LDL cholesterol levels down to a specific target number. Lifestyle factors, such as a diet high in saturated and trans fats can raise LDL cholesterol.
- High-density-lipoprotein (HDL) cholesterol = “good” cholesterol. With HDL (good) cholesterol, higher levels are typically better. Low HDL cholesterol puts you at higher risk for heart disease. People with high blood triglycerides usually also have lower HDL cholesterol. Genetic factors, type 2 diabetes, smoking, being overweight and being sedentary can all result in lower HDL cholesterol.
- Triglycerides. Triglyceride is the most common type of fat in the body. Normal triglyceride levels vary by age and sex. A high triglyceride level combined with low HDL cholesterol or high LDL cholesterol is associated with atherosclerosis, the buildup of fatty deposits in artery walls that increases the risk for heart attack and stroke.
High blood pressure
High blood pressure increases the heart’s workload, causing the heart muscle to thicken and become stiffer. This stiffening of the heart muscle is not normal, and causes the heart not to work properly. It also increases your risk of stroke, heart attack, kidney failure and congestive heart failure. When high blood pressure exists with obesity, smoking, high blood cholesterol levels or diabetes, the risk of heart attack or stroke increases even more.
Physical inactivity
An inactive lifestyle is a risk factor for coronary heart disease. Regular, moderate-to-vigorous physical activity helps reduce the risk of heart and blood vessel disease. Even moderate-intensity activities help if done regularly and long term. Physical activity can help control blood cholesterol, diabetes and obesity, as well as help lower blood pressure in some people.
Obesity and overweight
People who have excess body fat — especially if a lot of it is at the waist — are more likely to develop heart disease and stroke even if they have no other risk factors. Overweight and obese adults with risk factors for cardiovascular disease such as high blood pressure, high cholesterol, or high blood sugar can make lifestyle changes to lose weight and produce clinically meaningful reductions in triglycerides, blood glucose, HbA1c, and risk of developing Type 2 diabetes. Many people may have difficulty losing weight. But a sustained weight loss of 3 to 5% body weight may lead to clinically meaningful reductions in some risk factors, larger weight losses can benefit blood pressure, cholesterol, and blood glucose.
Diabetes mellitus
Diabetes seriously increases your risk of developing cardiovascular disease. Even when glucose levels are under control, diabetes increases the risk of heart disease and stroke, but the risks are even greater if blood sugar is not well controlled. At least 68% of people >65 years of age with diabetes die of some form of heart disease and 16% die of stroke. If you have diabetes, it’s extremely important to work with your healthcare provider to manage it and control any other risk factors you can. Persons with diabetes who are obese or overweight should make lifestyle changes (e.g., eat better, get regular physical activity, lose weight ) to help manage blood sugar.
Stress
Individual response to stress may be a contributing factor. Some scientists have noted a relationship between coronary heart disease risk and stress in a person’s life, their health behaviors and socioeconomic status. These factors may affect established risk factors. For example, people under stress may overeat, start smoking or smoke more than they otherwise would.
Diet and Nutrition
A healthy diet is one of the best weapons you have to fight cardiovascular disease. The food you eat (and the amount) can affect other controllable risk factors: cholesterol, blood pressure, diabetes and overweight. Choose nutrient-rich foods — which have vitamins, minerals, fiber and other nutrients but are lower in calories — over nutrient-poor foods. Choose a diet that emphasizes intake of vegetables, fruits, and whole grains; includes low-fat dairy products, poultry, fish, legumes, nontropical vegetable oils, and nuts; and limits intake of sweets, sugar-sweetened beverages, and red meats. And to maintain a healthy weight, coordinate your diet with your physical activity level so you’re using up as many calories as you take in.
Peripartum and postpartum cardiomyopathy
Peripartum cardiomyopathy is defined as a non-familial form of peripartum heart failure characterized as an ‘idiopathic cardiomyopathy presenting with heart failure secondary to left-ventricular systolic dysfunction towards the end of pregnancy or in the months following delivery, where no other cause of heart failure is found’ as proposed by the Working Group on peripartum cardiomyopathy of the Heart Failure Association of the European Society of Cardiology 3.
Very little is known about the incidence of peripartum cardiomyopathy. Most studies have been conducted in the USA, South Africa, or Haiti with few from the rest of the world, including Europe. The studies that have been performed were mostly single-centre case series. From the available literature, the incidence of peripartum cardiomyopathy appears to be around 1 in 2500–4000 in the USA, 1 in 1000 in South Africa, and 1 in 300 in Haiti 3. Prospective, population-based, well-conducted, epidemiological studies are required. Peripartum cardiomyopathy can occur from mid to late pregnancy or soon after delivery and is known as peripartum cardiomyopathy. It’s possible that, in these circumstances, some women may have already had dilated cardiomyopathy but it had not been diagnosed.
For most women, the dilated heart returns to normal within six to eight weeks of the delivery, provided they get the appropriate treatment. However, it’s possible that the condition could develop again in subsequent pregnancies.
If you’ve had peripartum cardiomyopathy and have not fully recovered from it, your doctor may advise you not to have any more children.
Women who have dilated cardiomyopathy or peripartum cardiomyopathy should seek specialist advice before planning another baby or if they find themselves unexpectedly pregnant. This is particularly important if you’re receiving medication for your cardiomyopathy, as this can affect the baby.
The precise mechanisms that lead to peripartum cardiomyopathy remain ill-defined, but a number of contributing factors have received attention. These include general risk factors for cardiovascular disease (such as hypertension, diabetes, and smoking) and pregnancy-related factors (such as age, number of pregnancies, number of children born, use of medication facilitating birth, and malnutrition) 4.
Prolactin, 16 kDa prolactin, and cathepsin D
Recent data suggest involvement of a cascade involving oxidative stress, the prolactin-cleaving protease cathepsin D, and the nursing-hormone prolactin, in the pathophysiology of peripartum cardiomyopathy. Oxidative stress appears to be a trigger that activates cathepsin D in cardiomyocytes and cathepsin D, subsequently, cleaves prolactin into an angiostatic and pro-apoptotic subfragment 5. Patients with acute peripartum cardiomyopathy have increased serum levels of oxidized low-density lipoprotein, indicative of enhanced systemic oxidative stress, as well as increased serum levels of activated cathepsin D, total prolactin, and the cleaved, angiostatic, 16 kDa prolactin fragment 5.
In a mouse model, the 16 kDa prolactin fragment has potentially detrimental cardiovascular actions that could play a pathophysiological role in peripartum cardiomyopathy. It inhibits endothelial cell proliferation and migration, induces endothelial cell apoptosis and disrupts already formed capillary structures.3 This form of prolactin also promotes vasoconstriction3 and impairs cardiomyocyte function 5. Consistent with the idea that 16 kDa prolactin-mediated apoptosis may contribute to the pathogenesis of peripartum cardiomyopathy, pro-apoptotic serum markers (e.g. soluble death receptor sFas/Apo-1) are increased in peripartum cardiomyopathy patients and are predictive of impaired functional status and mortality 4.
In this regard, an efficient antioxidant defence mechanism in the maternal heart, late in pregnancy and the post-partum period, seem crucial as markers of cellular oxidation rise during pregnancy, culminating in the last trimester (as part of normal pregnancy-related physiology) 6. Experimental data in a mouse model of peripartum cardiomyopathy (i.e. mice with a cardiomyocyte-restricted deletion of the signal transducer and activator of transcription-3, STAT3) suggest that defective antioxidant defence mechanisms may be responsible for the development of peripartum cardiomyopathy 5.
Furthermore, a key functional role of an activated oxidative stress–cathepsin D–16 kDa prolactin cascade in peripartum cardiomyopathy is strongly supported by the observation that suppression of the production of prolactin by the dopamine D2 receptor agonist, bromocriptine, prevented the onset of peripartum cardiomyopathy in the mouse model of peripartum cardiomyopathy 5.
Preliminary reports of the possible clinical effects of bromocriptine in patients with acute peripartum cardiomyopathy are discussed below.
Other putative pathophysiological mechanisms
Inflammation
In addition to oxidative stress, inflammation may play a role in the pathophysiology of peripartum cardiomyopathy. Serum markers of inflammation [including the soluble death receptor sFas/Apo-1, C-reactive protein, interferon gamma (IFN-γ), and IL-6] are elevated in patients with peripartum cardiomyopathy 5. This mechanism is underscored by the apparent clinical benefit of the anti-inflammatory agent pentoxifylline in a non-randomized trial in 58 patients with peripartum cardiomyopathy 7. Furthermore, that failure to improve is clinically associated with persistently elevated IFN-γ suggests that inflammatory status is important in the prognosis of patients with peripartum cardiomyopathy 8.
Viruses
Viral infection of the heart is another possible cause of peripartum inflammation, although clinical data are far from conclusive. Although some reports have implicated cardiotropic enteroviruses in peripartum cardiomyopathy 9, others have not found a higher frequency of viral infections in patients with peripartum cardiomyopathy than in those with idiopathic dilated cardiomyopathy 10. Human immunodeficiency virus infection does not seem to be implicated in peripartum cardiomyopathy 11.
Autoimmune system
In addition, autoimmune responses may play a role in the pathophysiology of peripartum cardiomyopathy. For example, serum derived from peripartum cardiomyopathy patients affects in vitro maturation of dendritic cells differently than serum from healthy post-partum women 12. High titres of auto-antibodies against selected cardiac tissue proteins have been found in the majority of women with peripartum cardiomyopathy. Circulating auto-antibodies to every type of cardiac tissue were identified in all 10 cases screened by Lamparter et al. 13. Warraich et al. 14 reported higher titres of antibodies (IgG and IgG subclasses) against cardiac myosin heavy chain in patients with peripartum cardiomyopathy compared with those with IDCM. Furthermore, these titres correlated with clinical presentation and with New York Heart Association (NYHA) functional class. In addition, the potential role of microchimerism, due to the introduction of foetal cells of haematopoietic origin into the maternal circulation, has been raised 15.
Whether or not these findings are causal in peripartum cardiomyopathy, or secondary to cardiac damage due to another mechanism, is not clear.
Genetic susceptibility to peripartum cardiomyopathy
Few data are available with which to formally evaluate any genetic contribution to susceptibility to peripartum cardiomyopathy and the studies that have been published are largely case reports rather than systematic studies. There are a number of reports in the literature of peripartum cardiomyopathy in women with mothers or sisters who had the same diagnosis. A widely cited study from the 1960s 16, identified 3 of 17 probands with peripartum cardiomyopathy who had a definite family history of the condition. Since that time, there have been several other carefully documented examples of two or three affected female first-degree relatives. Frequently, uncertainty exists about whether such cases fulfil formal peripartum cardiomyopathy diagnostic criteria (i.e. absence of pre-existing heart disease) or whether, in contrast, the affected women have an inherited dilated cardiomyopathy that only became apparent during the haemodynamic stress of pregnancy. There have been reports of women with peripartum cardiomyopathy, who have male relatives affected by dilated cardiomyopathy, arguing that at least some familial cases are examples of dilated cardiomyopathy rather than a specific peripartum cardiomyopathy 17. Recently, however, there have been two reports which more strongly support the suggestion that some cases of peripartum cardiomyopathy may in fact be part of familial dilated cardiomyopathy. In one study from the Netherlands, van Spaendonck-Zwarts et al. 18 studied 90 families with familial dilated cardiomyopathy and investigated the presence of peripartum cardiomyopathy; in addition, they also examined peripartum cardiomyopathy patients and performed cardiac screening of their first-degree relatives. Their data suggest that a subset of peripartum cardiomyopathy is an initial manifestation of familial dilated cardiomyopathy and this was corroborated by the identification of a causative mutation in one family. In another study from the USA, Morales et al.. 19 did similar observations in a large cohort study. These findings together may have important implications for cardiology screening in such families.
Nevertheless, the very high incidence in certain communities is suggestive of environmental risk factors 20, although a common genetic founder mutation cannot be excluded. Studies in immigrant populations in the USA suggest an intermediate level of risk in African-Americans and a low incidence in Hispanics (more in keeping with changing environment than genetic origins) 21.
Within populations, there clearly remains scope for variable genetic susceptibility, just as there is with other forms of heart failure. Future research could evaluate both the common variants: common disease contribution to peripartum cardiomyopathy susceptibility and, potentially, analysis of uncommon larger-effect alleles (e.g. by re-sequencing). A number of candidate gene pathways would be promising targets, e.g. genetic variants in the JAK/STAT signalling cascade; however, no associations have been detected to date.
On the basis of these results, general genetic testing is not recommended as a routine but is currently being done as part of research projects.
Dilated cardiomyopathy prevention
Healthy lifestyle habits can help you prevent or minimize the effects of dilated cardiomyopathy. If you have dilated cardiomyopathy:
- Don’t smoke.
- Don’t drink alcohol, or drink in moderation.
- Don’t use cocaine or other illegal drugs.
- Eat a healthy diet, especially a low-salt (sodium) diet.
- Maintain a healthy weight.
- Follow an exercise regimen recommended by your doctor.
- Get enough sleep and rest.
Dilated cardiomyopathy symptoms
Most people who are affected by dilated cardiomyopathy remain well. Some people have a few symptoms and others may develop problems which need more complex treatment.
In most cases dilated cardiomyopathy develops slowly, so some people can have quite severe symptoms before they are diagnosed.
The symptoms of dilated cardiomyopathy are similar to those of heart failure. Heart failure is a term used to describe a group of symptoms caused when your heart muscle becomes less efficient at pumping blood around your body.
The symptoms of heart failure include:
- Shortness of breath (dyspnea) when you’re active or lying down
- Swelling (edema) of the ankles, feet, lower back and abdomen
- Swelling of your abdomen due to fluid buildup (ascites)
- Chest pain
- Excessive tiredness
- Palpitations. This is the sensation of your own heart beating and can feel like extra or skipped beats. In some cases, palpitations may start suddenly and feel very fast, and may be accompanied by sweating or light-headedness.
- Extra or unusual sounds heard when your heart beats (heart murmurs)
- Fatigue
- Reduced ability to exercise
- Swelling (edema) in your legs, ankles and feet
Not everyone who has dilated cardiomyopathy goes on to develop all the symptoms of heart failure. The symptoms usually come on slowly, but sometimes they can come on suddenly. Once the condition has been diagnosed, in most cases dilated cardiomyopathy symptoms can be controlled with medication or other treatments.
Dilated cardiomyopathy complications
Complications from dilated cardiomyopathy include:
- Heart failure. Poor blood flow from the left ventricle can lead to heart failure. Your heart might not be able to supply your body with the blood it needs to function properly.
- Heart valve regurgitation. Enlargement of the left ventricle may make it harder for your heart valves to close, causing a backward flow of blood and making your heart pump less effectively.
- Fluid buildup (edema). Fluid can build up in the lungs, abdomen, legs and feet (edema).
- Abnormal heart rhythms (arrhythmias). Changes in your heart’s structure and changes in pressure on your heart’s chambers can contribute to the development of arrhythmias.
- Sudden cardiac arrest. Dilated cardiomyopathy can cause your heart to suddenly stop beating.
- Blood clots (emboli). Pooling of blood (stasis) in the left ventricle can lead to blood clots, which may enter the bloodstream, cut off the blood supply to vital organs, and cause stroke, heart attack or damage to other organs. Arrhythmias can also cause blood clots.
Arrhythmias
Arrhythmias are a common complication in people with dilated cardiomyopathy.
When your heart muscle becomes dilated, it stretches the cells in the heart muscle and can cause scars to develop. These abnormalities can interfere with the way that the electrical impulses pass through your heart muscle and can lead to slow, fast or erratic abnormal heart rhythms known as arrhythmias.
Arrhythmias can cause a fall in blood pressure and lead to episodes of dizziness or cause blackouts. They can sometimes slow the flow of blood through your heart and lead to an increased risk of having a stroke. If this is the case, your cardiologist will prescribe anticoagulants for you to reduce that risk.
Some arrhythmias need to be corrected by delivering a controlled electric shock. This procedure is known as a cardioversion.
In atrial fibrillation – an abnormal heart rhythm – the heart electrical impulses are sent from different places (apart from the heart’s natural pacemaker [the SA node]) in the atria in a disorganised way. This makes your heart beat uncontrollably and often very fast. It can lead to feelings of palpitations or fluttering in your chest. The condition can usually be controlled with medication.
Ventricular tachycardias are arrhythmias that affect the ventricles, the lower pumping chambers of your heart. Electrical signals in the ventricles become disorganised and take over the heartbeat independently from the SA node. This leads to a rapid heartbeat. Ventricular tachycardias can be controlled with medication, but they can sometimes lead to more life-threatening arrhythmias and the risk of sudden death.
Ventricular ectopics usually occur as single extra beats, originating in the ventricles. They should be investigated to rule out a serious arrhythmia, but they normally don’t need any further treatment. They can be found in healthy people too.
Heart block can occur in a small number of people with dilated cardiomyopathy. This is when the electrical impulse travels down to the ventricles slowly, or may even be completely blocked. This affects the way that your heart contracts, and often causes your heart to beat too slowly.
Blood clots
People with dilated cardiomyopathy have an increased risk of blood clots forming in the heart, because the blood flows through the heart more slowly than normal. The formation of clots increases the risk of having a stroke. Some people with dilated cardiomyopathy will be prescribed anticoagulants to reduce this risk.
Angina
Angina is usually caused by a narrowing of the coronary arteries, the blood vessels that supply blood to your heart muscle, and causes symptoms such as chest pain. If a coronary artery becomes completely blocked, it can cause a heart attack. However, in people with dilated cardiomyopathy, angina is usually caused by the high pressure on the dilated wall of the left ventricle. This pressure reduces the supply of blood to your heart muscle, causing chest tightness or pain. Treatment for this is usually medication to reduce the pressure and widen the arteries.
Heart murmurs
Heart murmurs are extra or unusual sounds from your heart that can be heard through a stethoscope. In people with dilated cardiomyopathy it is usually caused by a structural problem in the heart valves such as a leaking heart valves. If your doctor hears a heart murmur, they may send you for an echocardiogram to check the structure of your heart.
Is there a risk of sudden death in people with dilated cardiomyopathy?
Dilated cardiomyopathy is a common condition, and the majority of affected people remain well and have few or no symptoms. Research has shown that, with proper treatment and follow-up, most people with dilated cardiomyopathy live a normal life. However, because there is a very small risk of developing a life-threatening arrhythmia, a small proportion of people with dilated cardiomyopathy are at risk of sudden death.
Some arrhythmias can cause your heart to beat too fast and chaotically, eventually causing your heart to stop beating. This is a cardiac arrest and can lead to sudden death. This is different to a heart attack, which happens when one of the coronary arteries that supply your heart with blood becomes blocked and the heart muscle which it supplies may be starved of oxygen.
Dilated cardiomyopathy diagnosis
Your doctor will take a personal and family medical history. Then, he or she will also do a physical exam using a stethoscope to listen to your heart and lungs, and order tests. Your doctor may refer you to a heart specialist (cardiologist) for testing.
Tests your doctor might order include:
- Blood tests. These tests give your doctor information about your heart. They also may reveal if you have an infection, a metabolic disorder or toxins in your blood that can cause dilated cardiomyopathy.
- Chest X-ray. Your doctor may order a chest X-ray to check your heart and lungs for changes or abnormalities in the heart’s structure and size, and for fluid in or around your lungs.
- Electrocardiogram (ECG). An electrocardiogram — also called an ECG or EKG — records electrical signals as they travel through your heart. Your doctor can look for patterns that may be a sign of an abnormal heart rhythm or problems with the left ventricle. Your doctor may ask you to wear a portable ECG device (Holter monitor) to record your heart rhythm for a day or two.
- Echocardiogram. This primary tool for diagnosing dilated cardiomyopathy uses sound waves to produce images of the heart, allowing your doctor to see whether your left ventricle is enlarged. This test can also reveal how much blood is ejected from the heart with each beat and whether blood is flowing in the right direction.
- Exercise stress test. Your doctor may have you perform an exercise test, either walking on a treadmill or riding a stationary bike. Electrodes attached to you during the test help your doctor measure your heart rate and oxygen use.This type of test can show the severity of problems caused by dilated cardiomyopathy. If you’re unable to exercise, you may be given medication to create stress on your heart.
- CT or MRI scan. In some situations, your doctor might order one of these tests to check the size and function of your heart’s pumping chambers.
- Cardiac catheterization. For this invasive procedure, a long, narrow tube is threaded through a blood vessel in your arm, groin or neck into your heart. The test enables your doctor to see your coronary arteries on X-ray, measure pressure in your heart and collect a sample of muscle tissue to check for damage that indicates dilated cardiomyopathy.This procedure may involve having a dye injected into your coronary arteries to help your doctor study your coronary arteries (coronary angiography).
- Genetic screening or counseling. If your doctor can’t identify the cause of dilated cardiomyopathy, he or she may suggest screening of other family members to see if the disease is inherited in your family.
Dilated cardiomyopathy treatment
At present there is no cure for dilated cardiomyopathy.
If you have dilated cardiomyopathy, your doctor might recommend treatment for the underlying cause, if known. Treatment may also be suggested in order to improve blood flow and prevent further damage to your heart.
Medications
Doctors usually treat dilated cardiomyopathy with a combination of medications. Depending on your symptoms, you might need two or more of these drugs.
Drugs that have proved useful in the treatment of heart failure and dilated cardiomyopathy include:
- Angiotensin-converting enzyme (ACE) inhibitors. ACE inhibitors are a type of drug that widens or dilates blood vessels (vasodilator) to lower blood pressure, improve blood flow and decrease the heart’s workload. ACE inhibitors may improve heart function.Side effects include low blood pressure, low white blood cell count, and kidney or liver problems.
- Angiotensin II receptor blockers. These drugs have many of the beneficial effects of ACE inhibitors and may be an alternative for people who can’t tolerate ACE inhibitors. Side effects include diarrhea, muscle cramps and dizziness.
- Beta blockers. A beta blocker slows your heart rate, reduces blood pressure and may prevent some of the harmful effects of stress hormones, which are substances produced by your body that can worsen heart failure and trigger abnormal heart rhythms.Beta blockers may reduce signs and symptoms of heart failure and improve heart function. Side effects include dizziness and low blood pressure.
- Diuretics. Often called water pills, diuretics remove excess fluid and salt from your body. The drugs also decrease fluid in your lungs, so you can breathe more easily.
- Digoxin. This drug, also known as digitalis, strengthens your heart muscle contractions. It also tends to slow the heartbeat. Digoxin may reduce heart failure symptoms and improve your ability to be active.
- Blood-thinning medications. Your doctor may prescribe drugs, including aspirin or warfarin, to help prevent blood clots. Side effects include excessive bruising or bleeding.
Devices
Implantable devices used to treat dilated cardiomyopathy include:
- Biventricular pacemakers, which use electrical impulses to coordinate the actions of the left and right ventricles.
- Implantable cardioverter-defibrillators (ICDs), which monitor heart rhythm and deliver electrical shocks when needed to control abnormal, rapid heartbeats, including those that cause the heart to stop. They can also function as pacemakers.
- Left ventricular assist devices (LVADs), which are mechanical devices implanted into the abdomen or chest and attached to a weakened heart to help it pump. They usually are considered after less invasive approaches are unsuccessful.
Heart transplant
You may be a candidate for a heart transplant if medications and other treatments are no longer effective.
Living with dilated cardiomyopathy
Dilated cardiomyopathy is a common form of cardiomyopathy and research has shown that with proper treatment and follow-up, most people with the condition live a normal life.
However, because there is a very small risk of getting a life-threatening abnormal heart rhythm, a small proportion of people with dilated cardiomyopathy are at risk of sudden cardiac death. It’s important to discuss this risk with your doctor, who may offer medication or advise that you need to have an ICD fitted if you are at an increased risk.
You may have to make some small changes to manage with your symptoms, but you should be able to continue to work and drive a car for example
However, you will not be able to drive a commercial passenger vehicle and you may have to reconsider manual jobs which involve strenuous activity.
Lifestyle and home remedies
If you have dilated cardiomyopathy, these self-care strategies may help you manage your symptoms:
- Exercise. Talk to your doctor about what activities would be safe and beneficial for you. In general, competitive sports aren’t recommended because they can increase the risk of the heart stopping and causing sudden death.
- Quit smoking. Your doctor can give you advice on what methods can help you stop.
- Don’t use illegal drugs or drink alcohol excessively. Using cocaine or other illegal drugs can strain your heart. Before you drink alcohol, ask your doctor for advice.
- Maintain a healthy weight. Extra weight makes your heart work harder. Lose weight if you’re overweight or obese.
- Eat a heart-healthy diet. Eating whole grains and a variety of fruits and vegetables, and limiting salt, added sugar, and cholesterol, saturated fats and trans fats is good for your heart. Ask your doctor for a referral to a dietitian if you need help planning your diet.
- Cardiomyopathy. https://www.nhlbi.nih.gov/health-topics/cardiomyopathy[↩]
- Familial dilated cardiomyopathy. https://ghr.nlm.nih.gov/condition/familial-dilated-cardiomyopathy[↩][↩]
- Sliwa K, Hilfiker-Kleiner D, Petrie MC, Mebazaa A, Pieske B, Buchmann E, Regitz-Zagrosek V, Schaufelberger M, Tavazzi L, van Veldhuisen DJ, Watkins H, Shah AJ, Seferovic PM, Elkayam U, Pankuweit S, Papp Z, Mouquet F, McMurray JJ. Current state of knowledge on aetiology, diagnosis, management, and therapy of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Working Group on peripartum cardiomyopathy. Eur J Heart Fail 2010;12:767–778. http://onlinelibrary.wiley.com/doi/10.1093/eurjhf/hfq120/full[↩][↩]
- Peripartum cardiomyopathy. Lancet. 2006 Aug 19;368(9536):687-93. http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(06)69253-2/fulltext[↩][↩]
- A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell. 2007 Feb 9;128(3):589-600. http://www.cell.com/cell/fulltext/S0092-8674(07)00060-8[↩][↩][↩][↩][↩][↩]
- Oxidative stress and normal pregnancy. Clin Endocrinol (Oxf). 2002 Nov;57(5):609-13. https://www.ncbi.nlm.nih.gov/pubmed/12390334[↩]
- The addition of pentoxifylline to conventional therapy improves outcome in patients with peripartum cardiomyopathy. Eur J Heart Fail. 2002 Jun;4(3):305-9. https://www.ncbi.nlm.nih.gov/pubmed/12034156[↩]
- Reversal of IFN-gamma, oxLDL and prolactin serum levels correlate with clinical improvement in patients with peripartum cardiomyopathy. Eur J Heart Fail. 2008 Sep;10(9):861-8. doi: 10.1016/j.ejheart.2008.07.005. Epub 2008 Sep 2. https://www.ncbi.nlm.nih.gov/pubmed/18768352[↩]
- [Peripartum dilated cardiomyopathy. A model of multifactor disease?]. Rev Med Interne. 1993;14(10):1033. https://www.ncbi.nlm.nih.gov/pubmed/8009029[↩]
- Incidence of myocarditis in peripartum cardiomyopathy. Am J Cardiol. 1994 Sep 1;74(5):474-7. https://www.ncbi.nlm.nih.gov/pubmed/8059728[↩]
- Long-term outcome of Peripartum cardiomyopathy in a population with high seropositivity for human immunodeficiency virus. Int J Cardiol Published online ahead of print 12 September 2009[↩]
- Ellis JE, Ansari AA, Fett JD, et al. Inhibition of Progenitor Dendritic Cell Maturation by Plasma from Patients with Peripartum Cardiomyopathy: Role in Pregnancy-associated Heart Disease. Clinical and Developmental Immunology. 2005;12(4):265-273. doi:10.1080/17402520500304352. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2270740/[↩]
- Clinical and immunologic characteristics in peripartum cardiomyopathy. Int J Cardiol. 2007 May 16;118(1):14-20. Epub 2006 Aug 10. https://www.ncbi.nlm.nih.gov/pubmed/16904777[↩]
- Impact of pregnancy-related heart failure on humoral immunity: clinical relevance of G3-subclass immunoglobulins in peripartum cardiomyopathy. Am Heart J. 2005 Aug;150(2):263-9. https://www.ncbi.nlm.nih.gov/pubmed/16086928[↩]
- Is peripartum cardiomyopathy an organ-specific autoimmune disease? Autoimmun Rev. 2002 Feb;1(1-2):73-7. https://www.ncbi.nlm.nih.gov/pubmed/12849062[↩]
- Familial occurrence of postpartal heart failure. Arch Intern Med. 1963 May;111:651-5. https://jamanetwork.com/journals/jamainternalmedicine/article-abstract/568580[↩]
- [Familial cardiomyopathy. Peripartum and primary congestive cardiomyopathy in a sister and brother]. Ugeskr Laeger. 1976 Oct 11;138(42):2567-9. https://www.ncbi.nlm.nih.gov/pubmed/969012[↩]
- Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation. 2010 May 25;121(20):2169-75. doi: 10.1161/CIRCULATIONAHA.109.929646. Epub 2010 May 10. http://circ.ahajournals.org/content/121/20/2169.long[↩]
- Morales A, Painter T, Li R, et al. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation. 2010;121(20):2176-2182. doi:10.1161/CIRCULATIONAHA.109.931220. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2900861/[↩]
- Epidemiology of heart failure in sub-Saharan Africa. Expert Rev Cardiovasc Ther. 2009 Feb;7(2):169-80. doi: 10.1586/14779072.7.2.169. https://www.ncbi.nlm.nih.gov/pubmed/19210213[↩]
- Incidence, mortality, and racial differences in peripartum cardiomyopathy. Am J Cardiol. 2007 Jul 15;100(2):302-4. Epub 2007 Jun 6. https://www.ncbi.nlm.nih.gov/pubmed/17631087[↩]





{kind=link}