Contents
McKusick Kaufman syndrome
McKusick-Kaufman syndrome is a rare autosomal recessive genetic syndrome caused by mutations in the MKKS gene (also called BBS6 gene) that affects the development of the hands, feet, heart, and reproductive system 1, 2, 3, 4, 5, 6, 7, 8. McKusick-Kaufman syndrome is characterized by a combination of 3 clinical features: extra fingers and/or toes (polydactyly), congenital heart disease and genital abnormalities with hydrometrocolpos (a buildup of secretions or fluid in the vagina and uterus as a result of outlet obstruction frequently linked to imperforate hymen or vaginal agenesis where part of the vagina fails to develop) in females, and hypospadias (a genital defect where the opening is on the underside of the penis instead of at the tip), a downward-curving penis (chordee) and cryptorchidism (a condition in which one or both of the testes fail to descend into the scrotum) in males 9, 10. In female babies, the most common genital abnormality is hydrometrocolpos, where there is an accumulation of secretions within the vagina or extending up to the uterus, primarily caused by the accumulation of cervical secretions due to maternal estrogen stimulation. Hydrometrocolpos can occur if part of the vagina fails to develop (vaginal agenesis) or if a membrane blocks the opening of the vagina (imperforate hymen). The blockage allows fluid to build up in the vagina and uterus, stretching these organs and leading to a fluid-filled mass. In female infants, there is a notable presentation of a large cystic abdominal mass emerging from the pelvis caused by dilatation of the vagina and uterus. Other genital abnormalities associated with McKusick-Kaufman syndrome can include a urethral opening on the underside of the penis (hypospadias), a downward curvature of the penis (chordee), and undescended testes (cryptorchidism) in males.
Furthermore, in people with McKusick-Kaufman syndrome, the extra digits are typically on the same side of the hand or foot as the pinky or little toe also called postaxial polydactyly. The congenital heart disease in individuals with McKusick-Kaufman syndrome can include an atrial septal defect (a congenital heart defect where there is a hole in the wall or atrial septum separating the two upper chambers of the heart or atria) or a ventricular septal defect, which is a hole in the wall between the heart’s lower chambers, the ventricles, which can range from small to large.
The signs and symptoms of McKusick-Kaufman syndrome overlap significantly with those of another autosomal recessive genetic disorder called Bardet-Biedl syndrome 11. However, Bardet-Biedl syndrome has several features that are not typically seen in people with McKusick-Kaufman syndrome. Bardet-Biedl syndrome (BBS) signs and symptoms include a gradual loss of vision (retinitis pigmentosa), mental retardation, extra fingers or toes (polydactyly), kidney abnormalities, and obesity. Individuals with Bardet-Biedl syndrome typically have normal weight at birth, but gain significant weight during infancy, which can lead to long-term health issues such as heart disease and type 2 diabetes. Because some of these features are not apparent at birth, both McKusick-Kaufman syndrome and Bardet-Biedl syndrome can be difficult to tell apart in infancy and early childhood.
Both McKusick-Kaufman syndrome and Bardet-Biedl syndrome belong to a group of conditions called ciliopathies 11. Ciliopathies are inherited disorders that affect the structure or function of cilia, the microscopic, finger-like projections found on the surface of cells. Cilia are involved in signaling pathways that transmit information between cells.
McKusick-Kaufman syndrome is a rare genetic condition with only 100+ individuals with McKusick-Kaufman syndrome having been reported in the medical literature. McKusick-Kaufman syndrome was first described in 1978 in the Old Order Amish population, where it affects an estimated 1 in 10,000 people 12, 13, 14, 15, 16, 17, 18, 19, 20. However, the incidence of McKusick-Kaufman syndrome in non-Amish populations is unknown 1. In addition, McKusick-Kaufman syndrome is relatively rare compared to Bardet-Biedl syndrome outside the Amish population.
McKusick-Kaufman syndrome is caused by biallelic mutations in the MKKS gene or BBS6 gene that provides instructions for making a protein that plays an important role in early development of limbs, heart, and the reproductive system 2, 1. This protein’s structure suggests that it may belong to a family of proteins called chaperonins. Proteins must be folded into the correct shape to function properly, and chaperonins help them do that. The MKKS protein is thought to play a role in cell division and cell transport. Specifically, the MKKS protein is thought to transport molecules between the cytoplasm and the nucleus of the cell. The MKKS protein also combines with other proteins to form a complex within the cell. This complex is used to assemble the molecules involved in transporting materials that support the function of cilia. Some mutations in the MKKS gene can also cause Bardet-Biedl syndrome, a condition that is related to McKusick-Kaufman syndrome.
The MKKS protein combines with other proteins to form a structure known as the chaperonin complex. The chaperonin complex serves as a scaffold for the assembly of another molecule called the BBSome. The BBSome helps transport materials that support the function of cilia, the microscopic, finger-like projections on the surface of cells. Cilia help transmit information.
Though it is not clear exactly how mutations in the MKKS gene lead to the specific signs and symptoms of McKusick-Kaufman syndrome, two particular mutation (p.His84Tyr and p.Ala242Ser mutations) in the MKKS gene appear to impair the MKKS protein’s ability to transport SMARCC1 into the nucleus of the cell. This change likely affects the activity of certain genes that are critical during early development.
McKusick-Kaufman syndrome is inherited in an autosomal recessive pattern, which means both copies of the abnormal gene in each cell must be present in order for the disease to develop. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition also called carriers. Genes come in pairs. One gene in each pair comes from the egg (mother), and the other gene comes from the sperm (father). Recessive inheritance means both genes in a pair must be abnormal to cause disease. People with only one mutated gene in the pair are called carriers. These people are most often not affected with the condition. However, they can pass the variant gene to their children.
There is currently no published clinical diagnostic criteria for McKusick-Kaufman syndrome 3.
Diagnosis of McKusick-Kaufman syndrome should be suspected in individuals with the following features 3.
In females
- Hydrometrocolpos present in 80% to 95% of females due to either vaginal atresia, imperforate hymen, or cervical atresia which leads to the development of an abdominopelvic mass with regional compression and secondary hydronephrosis 9.
- Hydrometrocolpos in infants is dilatation of the vagina and uterus caused by the accumulation of cervical secretions as a result of maternal estrogen stimulation. Hydrometrocolpos can be caused by:
- Failure of the distal third of the vagina to develop (vaginal atresia or agenesis);
- A transverse vaginal membrane;
- Imperforate hymen; in rare cases, hydrometrocolpos and polydactyly may be associated with an imperforate hymen, but many individuals reported with these findings were described at young ages and it is thus not known if the actual diagnosis was McKusick-Kaufman syndrome or Bardet-Biedl syndrome.
- Hydrometrocolpos often presents at birth as a large, cystic abdominal mass arising out of the pelvis, which can be sufficiently large to be clinically obvious and is verified using an ultrasound scan. The mass can be large enough to cause intestinal obstruction, urinary outflow obstruction leading to dilatation of the ureter (hydroureter) and kidneys (hydronephrosis), obstruction of the inferior vena cava, and/or elevation of the diaphragm resulting in breathing difficulties.
- Hydrometrocolpos in infants is dilatation of the vagina and uterus caused by the accumulation of cervical secretions as a result of maternal estrogen stimulation. Hydrometrocolpos can be caused by:
- Postaxial polydactyly (extra digits are typically on the same side of the hand or foot as the pinky or little toe or additional digits on the ulnar side of the hand and the fibular side of the foot) is seen in 90% of cases 10
- The additional digit can be fully formed or can be a rudimentary skin tag often called a “minimus”.
- If clinical examination is insufficient, radiographs may be used to determine whether the polydactyly is postaxial or mesoaxial also known as central polydactyly (a duplication of one of the middle digits [second, third, or fourth] of a hand or foot). Mesoaxial polydactyly or central polydactyly is much less common than postaxial polydactyly.
- Congenital heart disease is seen in 10% to 20% of cases
- Developmental dysplasia of the hips and lower extremity edema are other manifestations involving the limbs.
In males
- Genital malformations most commonly hypospadias (urethral opening on the underside of the penis), cryptorchidism (undescended testes) and chordee (downward curvature of the penis)
- Postaxial polydactyly (extra digits are typically on the same side of the hand or foot as the pinky or little toe or additional digits on the ulnar side of the hand and the fibular side of the foot) is seen in 90% of cases 10
- The additional digit can be fully formed or can be a rudimentary skin tag often called a “minimus”.
- If clinical examination is insufficient, radiographs may be used to determine whether the polydactyly is postaxial or mesoaxial also known as central polydactyly (a duplication of one of the middle digits [second, third, or fourth] of a hand or foot). Mesoaxial polydactyly or central polydactyly is much less common than postaxial polydactyly.
- Congenital heart disease is seen in 10% to 20% of cases
- Developmental dysplasia of the hips and lower extremity edema are other manifestations involving the limbs.
In both males and females
- Family history typically consistent with autosomal recessive inheritance (e.g., affected siblings and/or a relationship between a father and mother who are related by blood or ‘blood relatives’ or ‘couples related by blood’). Note that absence of a known family history does not exclude the diagnosis.
- Additional less consistent findings are gastrointestinal abnormalities (28%) including imperforate anus, rectovaginal or vesicovaginal fistula, Hirschsprung’s disease, and malrotation 8.
- Abnormalities of the eyes (5%) have also been described 8.
For females without a family history and who are not part of the Amish population, the clinical diagnosis of McKusick-Kaufman syndrome can be established at birth based on clinical diagnostic criteria of hydrometrocolpos with distal vaginal agenesis or a transverse vaginal membrane and postaxial polydactyly 21, 11, 15. Genetic testing can be used in proband (a person serving as the starting point for the genetic study of a family) with suggestive clinical features of McKusick-Kaufman syndrome to support the diagnosis. However, care must be taken to ensure that the proband does not have Bardet-Biedl syndrome, another autosomal recessive genetic condition with considerable clinical overlap and age-dependent clinical features including retinitis pigmentosa, obesity, and mental retardation have not developed until children are 5 years old 15. Therefore, the clinical diagnosis of McKusick-Kaufman syndrome is not confirmed until the individual has reached five years of age without fulfilling the diagnostic criteria for Bardet-Biedl syndrome, or manifesting additional findings of an alternative diagnosis. In addition, McKusick-Kaufman syndrome is relatively rare compared to Bardet-Biedl syndrome outside the Amish population.
McKusick-Kaufman syndrome treatment includes surgical repair of the obstruction causing hydrometrocolpos and drainage of the accumulated fluid. Treatment for polydactyly and congenital heart defects and any other anomalies is standard. In the newborn with severe hydrometrocolpos, care with anesthesia in the neonatal period is appropriate, as hydrometrocolpos can cause diaphragmatic compression 22.
Figure 1. McKusick-Kaufman syndrome postaxial polydactyly
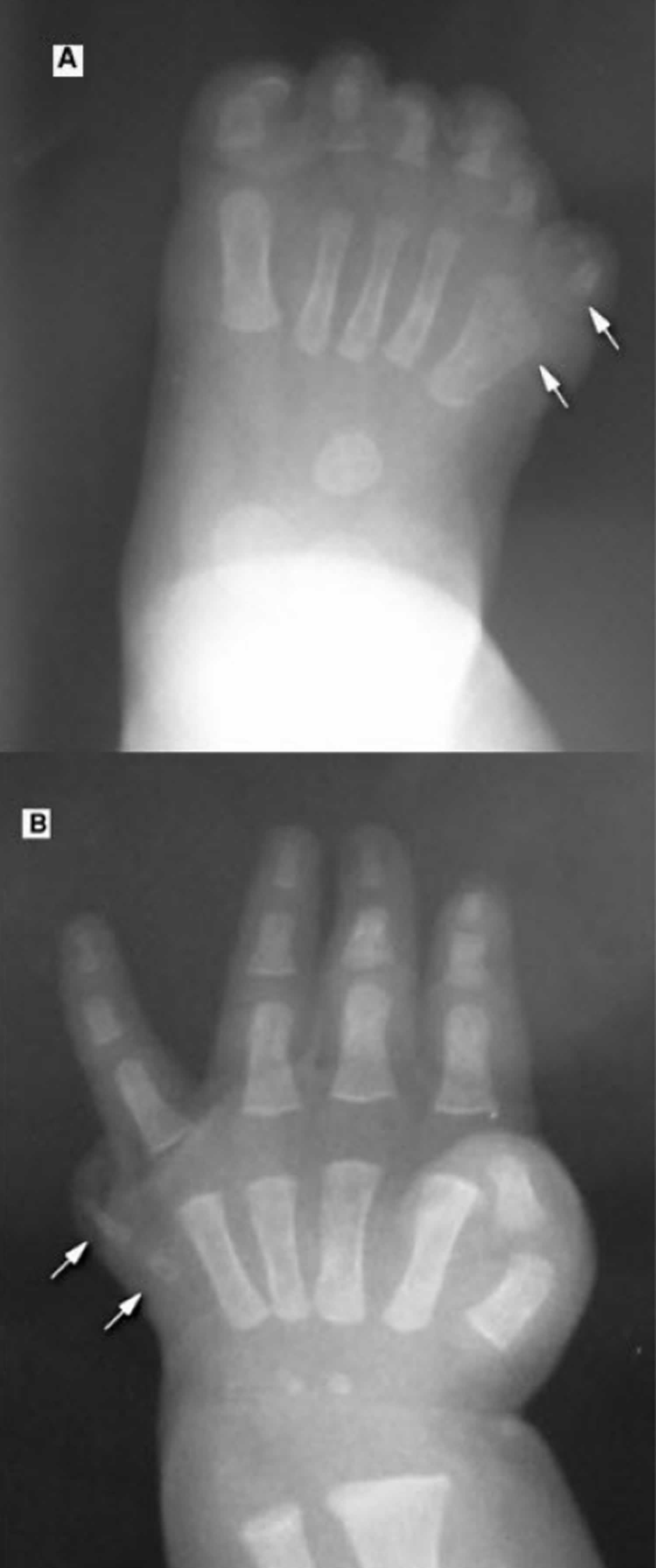
Footnotes: A 3-month-old girl with McKusick-Kaufman syndrome. Radiographs of foot (A) and hand (B) show postaxial polydactyly (arrows).
[Source 8 ]Figure 2. McKusick-Kaufman syndrome hydrometrocolpos
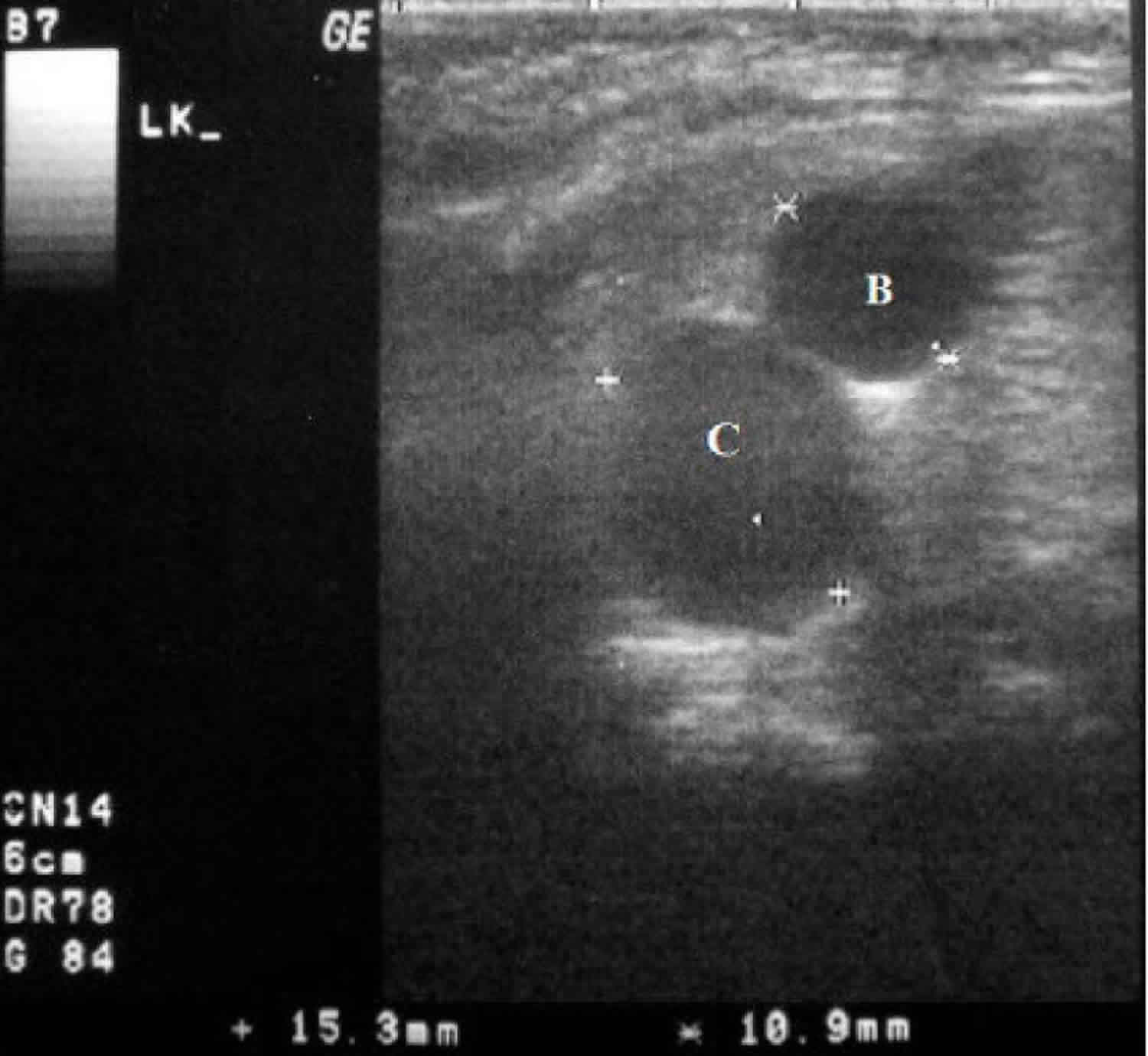
Footnotes: A 3-month-old girl with McKusick-Kaufman syndrome hydrometrocolpos. The mid-sagittal sonographic image shows a hypoechoic cystic structure measuring 15.3 mm (C) posterior to the bladder (B) with fluid-debris level in dependent part.
[Source 8 ]Figure 3. McKusick-Kaufman syndrome hydrometrocolpos MRI
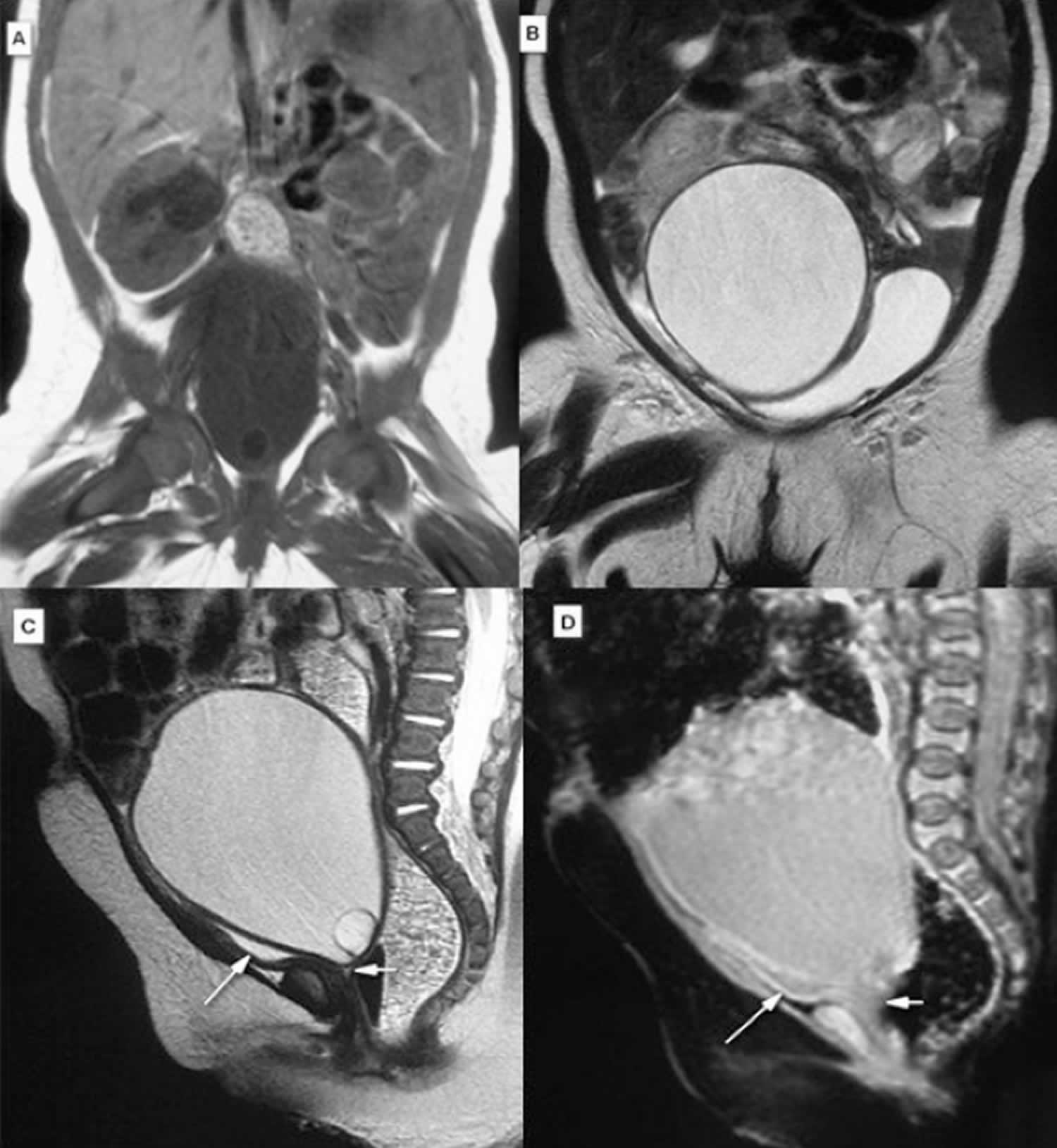
Footnotes: A 3-month-old girl with McKusick-Kaufman syndrome hydrometrocolpos. MRI of abdominopelvic cavity demonstrated a small hyposignal T1W (A) and Coronal T2W (B), sagittal T2W (C), and sagittal GRE (D) reveal hypersignal T2W triangular fluid contained structure below the larger cystic structure, anterior to the rectum, and behind the bladder, corresponding to hydrometra, atretic cervix, compressed bladder (long arrow), and atretic proximal vaginal pouch (short arrow). During cystoscopy a single small orifice at the posteroinferior wall of the bladder was detected. Approach to the uterus via this orifice was not possible; thus, exploratory laparotomy was done and the collection of fluid was drained. The laparotomy confirmed MRI and cystography findings of hydrometrocolpos, enlarged bladder, and fistula between bladder neck and atretic proximal vaginal pouch.
[Source 8 ]McKusick Kaufman syndrome cause
McKusick-Kaufman syndrome is caused by biallelic mutations in the MKKS gene or BBS6 gene that provides instructions for making a protein that plays an important role in early development of limbs, heart, and the reproductive system 2, 1. This protein’s structure suggests that it may belong to a family of proteins called chaperonins. Proteins must be folded into the correct shape to function properly, and chaperonins help them do that. The MKKS protein is thought to play a role in cell division and cell transport. Specifically, the MKKS protein is thought to transport molecules between the cytoplasm and the nucleus of the cell. The MKKS protein also combines with other proteins to form a complex within the cell. This complex is used to assemble the molecules involved in transporting materials that support the function of cilia. Some mutations in the MKKS gene can also cause Bardet-Biedl syndrome, a condition that is related to McKusick-Kaufman syndrome.
The MKKS protein combines with other proteins to form a structure known as the chaperonin complex. The chaperonin complex serves as a scaffold for the assembly of another molecule called the BBSome. The BBSome helps transport materials that support the function of cilia, the microscopic, finger-like projections on the surface of cells. Cilia help transmit information.
Though it is not clear exactly how mutations in the MKKS gene lead to the specific signs and symptoms of McKusick-Kaufman syndrome, two particular mutation (p.His84Tyr and p.Ala242Ser mutations) in the MKKS gene appear to impair the MKKS protein’s ability to transport SMARCC1 into the nucleus of the cell. This change likely affects the activity of certain genes that are critical during early development.
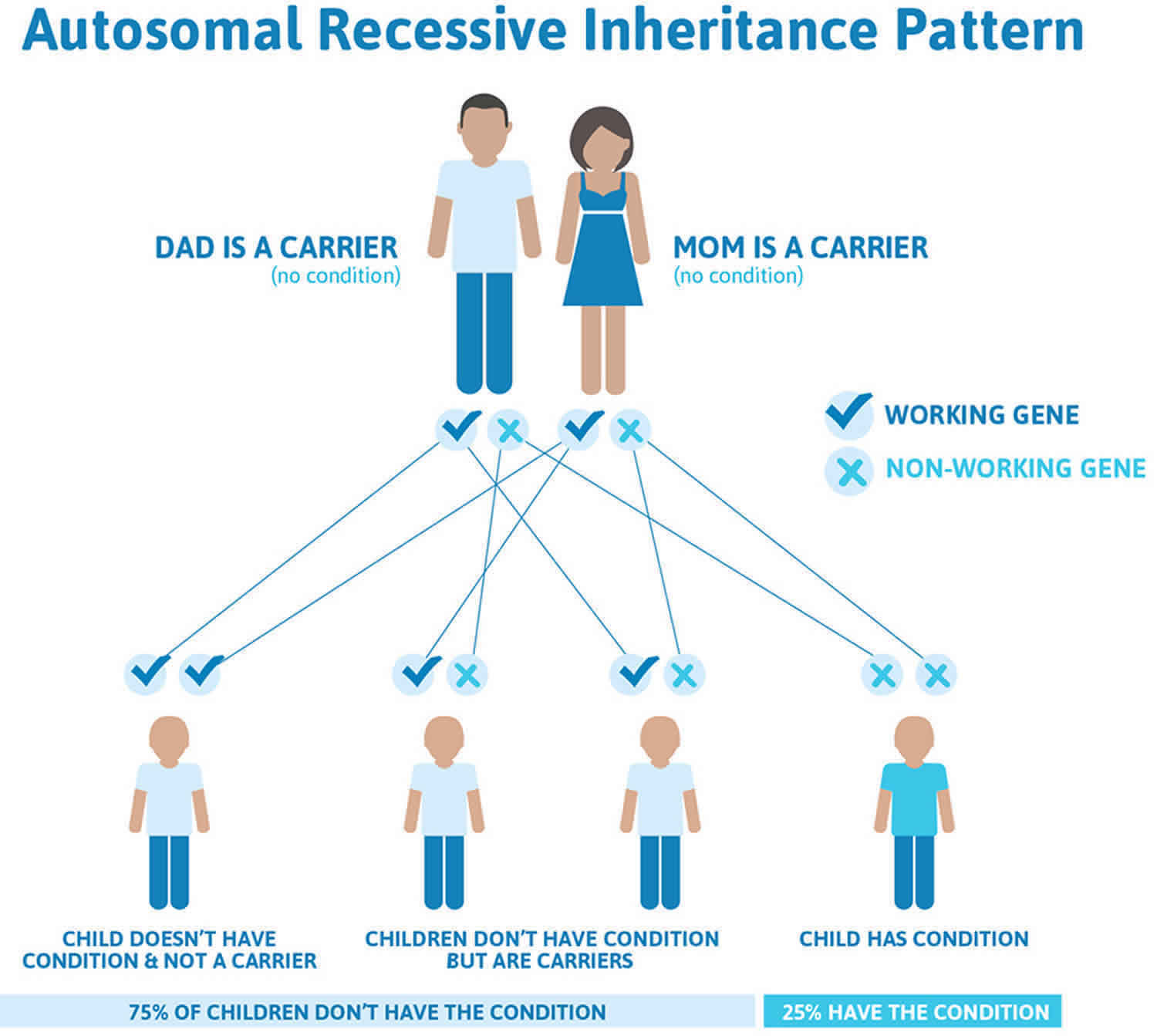
McKusick-Kaufman syndrome inheritance pattern
McKusick-Kaufman syndrome is inherited in an autosomal recessive pattern, which means both copies of the abnormal gene in each cell must be present in order for the disease to develop. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition also called carriers.
Genes come in pairs. One gene in each pair comes from the egg (mother), and the other gene comes from the sperm (father). Recessive inheritance means both genes in a pair must be abnormal to cause disease. People with only one mutated gene in the pair are called carriers. These people are most often not affected with the condition. However, they can pass the variant gene to their children.
If you are born to parents who both carry the same autosomal recessive gene, you have a 25% (1 in 4) chance of inheriting the variant gene from both parents and developing the disease. You have a 50% (1 in 2) chance of inheriting one variant gene. This would make you a carrier.
In other words, for a child born to a couple who both carry the mutated gene (but do not have signs of disease), the expected outcome for each pregnancy is:
- A 25% chance that the child is born with two normal genes (healthy)
- A 50% chance that the child is born with one normal and one variant gene (carrier, without disease)
- A 25% chance that the child is born with two variant genes (at risk for the disease)
Note: These outcomes do not mean that the children will definitely be carriers or be severely affected.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 4. McKusick-Kaufman syndrome autosomal recessive inheritance pattern
McKusick Kaufman syndrome signs and symptoms
Diagnosis of McKusick-Kaufman syndrome should be suspected in individuals with the following features 3.
In females
- Hydrometrocolpos present in 80% to 95% of females due to either vaginal atresia, imperforate hymen, or cervical atresia which leads to the development of an abdominopelvic mass with regional compression and secondary hydronephrosis 9.
- Hydrometrocolpos in infants is dilatation of the vagina and uterus caused by the accumulation of cervical secretions as a result of maternal estrogen stimulation. Hydrometrocolpos can be caused by:
- Failure of the distal third of the vagina to develop (vaginal atresia or agenesis);
- A transverse vaginal membrane;
- Imperforate hymen; in rare cases, hydrometrocolpos and polydactyly may be associated with an imperforate hymen, but many individuals reported with these findings were described at young ages and it is thus not known if the actual diagnosis was McKusick-Kaufman syndrome or Bardet-Biedl syndrome.
- Hydrometrocolpos often presents at birth as a large, cystic abdominal mass arising out of the pelvis, which can be sufficiently large to be clinically obvious and is verified using an ultrasound scan. The mass can be large enough to cause intestinal obstruction, urinary outflow obstruction leading to dilatation of the ureter (hydroureter) and kidneys (hydronephrosis), obstruction of the inferior vena cava, and/or elevation of the diaphragm resulting in breathing difficulties.
- Hydrometrocolpos in infants is dilatation of the vagina and uterus caused by the accumulation of cervical secretions as a result of maternal estrogen stimulation. Hydrometrocolpos can be caused by:
- Postaxial polydactyly (extra digits are typically on the same side of the hand or foot as the pinky or little toe or additional digits on the ulnar side of the hand and the fibular side of the foot) is seen in 90% of cases 10
- The additional digit can be fully formed or can be a rudimentary skin tag often called a “minimus”.
- If clinical examination is insufficient, radiographs may be used to determine whether the polydactyly is postaxial or mesoaxial also known as central polydactyly (a duplication of one of the middle digits [second, third, or fourth] of a hand or foot). Mesoaxial polydactyly or central polydactyly is much less common than postaxial polydactyly.
- Congenital heart disease is seen in 10% to 20% of cases
- Developmental dysplasia of the hips and lower extremity edema are other manifestations involving the limbs.
In males
- Genital malformations most commonly hypospadias (urethral opening on the underside of the penis), cryptorchidism (undescended testes) and chordee (downward curvature of the penis)
- Postaxial polydactyly (extra digits are typically on the same side of the hand or foot as the pinky or little toe or additional digits on the ulnar side of the hand and the fibular side of the foot) is seen in 90% of cases 10
- The additional digit can be fully formed or can be a rudimentary skin tag often called a “minimus”.
- If clinical examination is insufficient, radiographs may be used to determine whether the polydactyly is postaxial or mesoaxial also known as central polydactyly (a duplication of one of the middle digits [second, third, or fourth] of a hand or foot). Mesoaxial polydactyly or central polydactyly is much less common than postaxial polydactyly.
- Congenital heart disease is seen in 10% to 20% of cases
- Developmental dysplasia of the hips and lower extremity edema are other manifestations involving the limbs.
In both males and females
- Family history typically consistent with autosomal recessive inheritance (e.g., affected siblings and/or a relationship between a father and mother who are related by blood or ‘blood relatives’ or ‘couples related by blood’). Note that absence of a known family history does not exclude the diagnosis.
- Additional less consistent findings are gastrointestinal abnormalities (28%) including imperforate anus, rectovaginal or vesicovaginal fistula, Hirschsprung’s disease, and malrotation 8.
- Abnormalities of the eyes (5%) have also been described 8.
Table 1. Clinical features of individuals with McKusick-Kaufman syndrome
| Clinical Findings | Number of Individuals (%) | |
|---|---|---|
| Genitourinary malformations in females | Hydrometrocolpos | 42/44 (95%) |
| Vaginal agenesis | 26/44 (59%) | |
| Urogenital sinus | 16/44 (36%) | |
| Ectopic urethra | 8/44 (18%) | |
| No urethral opening | 6/44 (14%) | |
| No vaginal opening | 4/44 (9%) | |
| Genitourinary tract fistulae | 6/44 (14%) | |
| Postaxial polydactyly – limbs affected | Hands only | 12/42 (29%) |
| Feet only | 6/42 (14%) | |
| Hands & feet 1 | 11/42 (26%) | |
| Four-limb polydactyly 1 | 11/42 (26%) | |
| Other digital anomalies | Syndactyly | 12/49 (24%) |
| Metacarpal/tarsal anomalies | 8/49 (16%) | |
| Postaxial minimus | 6/49 (12%) | |
| Brachydactyly | 3/49 (6%) | |
| Absent phalanges | 2/49 (4%) | |
| Interstitial polydactyly | 0/48 (0%) | |
| Heptadactyly | 2/48 (4%) | |
| Cardiac malformations | Congenital heart defects comprising: atrioventricular canal defect; atrial septal defect; ventricular septal defect; complex congenital heart disease with an atrioventricular canal defect, small aorta, and hypoplastic left ventricle; tetralogy of Fallot; and a patent ductus arteriosus have been reported in individuals with an McKusick-Kaufman syndrome | 7/49 (14%) |
| Renal anomalies | Hydronephrosis | 31/49 (63%) |
| Hydroureter | 12/49 (24%) | |
| Renal cysts | 2/49 (4%) | |
| Calyceal dilatation | 7/49 (14%) | |
| Renal atrophy/hypoplasia2 | 2/49 (4%) | |
| Corticomedullary dysplasia 3 | 3/46 (6%) | |
| Nonfunctioning kidney | 2/49 (4%) | |
| Gastrointestinal malformations | Imperforate anus | 4/49 (8%) |
| Anal atresia | 1/49 (2%) | |
| Hirschsprung disease | 6/49 (12%) | |
| Anteriorly placed anus | 2/49 (4%) | |
Footnotes:
1 Four-limb polydactyly involves both hands and both feet; polydactyly of the hands and feet means that both upper and lower limbs are affected, but not every limb.
2 Renal dysplasia is a histologic diagnosis that describes abnormal differentiation of the renal parenchyma.
3 Corticomedullary dysplasia is abnormal differentiation of both the cortex and the medulla of the kidney. If focal, renal function may be preserved; if bilateral and extensive, renal failure can result.
McKusick Kaufman syndrome diagnosis
For females without a family history and who are not part of the Amish population, the clinical diagnosis of McKusick-Kaufman syndrome can be established at birth based on clinical diagnostic criteria of hydrometrocolpos with distal vaginal agenesis or a transverse vaginal membrane and postaxial polydactyly 21, 11, 15. Genetic testing can be used in proband (a person serving as the starting point for the genetic study of a family) with suggestive clinical features of McKusick-Kaufman syndrome to support the diagnosis. However, care must be taken to ensure that the proband does not have Bardet-Biedl syndrome, another autosomal recessive genetic condition with considerable clinical overlap and age-dependent clinical features including retinitis pigmentosa, obesity, and mental retardation have not developed until children are 5 years old 15. Therefore, the clinical diagnosis of McKusick-Kaufman syndrome is not confirmed until the individual has reached five years of age without fulfilling the diagnostic criteria for Bardet-Biedl syndrome, or manifesting additional findings of an alternative diagnosis. In addition, McKusick-Kaufman syndrome is relatively rare compared to Bardet-Biedl syndrome outside the Amish population.
Table 2. Recommended evaluations following initial diagnosis in individuals with McKusick-Kaufman syndrome
| System/Concern | Evaluation | Comment |
|---|---|---|
| Genitourinary malformations | Pelvic ultrasound | In females; in males, inspection of the genitalia should be performed. |
| Polydactyly & syndactyly | Skeletal radiographs to detect osseous polydactyly & syndactyly | Provide referral to surgical specialist as needed. |
| Cardiac malformation | Echocardiogram | Specialist referral as appropriate |
| Possible Bardet- Biedl syndrome. A subset of those with a diagnosis of McKusick-Kaufman syndrome as infants may with age develop findings that lead to a revised diagnosis of Bardet-Biedl syndrome. | Assessment of height, weight, & head circumference & initiation of a carefully maintained growth chart to document obesity | If obesity or short stature present, this may indicate a diagnosis of Bardet-Biedl syndrome |
| Determination of developmental status by standard screening tools to detect developmental delay | If present, this may indicate a diagnosis of Bardet-Biedl syndrome. | |
| Ophthalmologic exam & electroretinogram to evaluate for manifestations of retinal dystrophy | Specialist referral to ophthalmologist for monitoring is frequently recommended. | |
| Renal ultrasound | To detect pelvicalyceal abnormalities, renal hypoplasia, or cystic dysplasia of the kidneys | |
| Rectal biopsy in those w/severe constipation to exclude Hirschsprung disease | ||
| Genetic counseling | By genetics professionals | To inform affected persons & families re nature, mode of inheritance, & implications of McKusick-Kaufman syndrome to facilitate medical & personal decision making |
McKusick Kaufman syndrome differential diagnosis
Bardet-Biedl syndrome
Bardet-Biedl syndrome also called BBS is a rare complex genetic disorder that affects many parts of the body characterized by retinal dystrophy (a progressive eye disorder that leads to blindness, characterized by tunnel vision and night blindness), truncal obesity, polydactyly (extra fingers and/or toes), genitourinary and kidney abnormalities, learning difficulties, developmental delay, speech and language difficulties and hypogonadism 23, 24, 25, 26. Individuals with Bardet-Biedl syndrome (BBS) have a retinal degeneration similar to retinitis pigmentosa (RP). The retina is a light-sensitive layer of tissue at the back of your eyes that converts images into electrical signals for your brain. Rod and cone photoreceptors in the retina convert light into electrical signals that the brain interprets as vision. People with Bardet-Biedl syndrome (BBS) with retinal degeneration that is characterized by rod-cone dystrophy experience a gradual decline in their vision, because the photoreceptors (specialized cells in the retina that convert light into electrical signals that the brain can use for vision) degenerate. People with Bardet-Biedl syndrome (BBS) also have at least three additional non-eye features such as intellectual disability, truncal obesity (a condition where fat is disproportionately distributed onto the abdomen and chest rather than the arms and legs), polydactyly (a condition where a person has more than the normal number of fingers or toes), hypogonadism (a condition where the body’s sex glands [ovaries in women or testicles in men], produce little to no sex hormones), or kidney abnormalities as primary signs and symptoms 23, 24, 27. However, the signs and symptoms of Bardet-Biedl syndrome vary among affected individuals, even among members of the same family 28, 29.
In 1866, Laurence and Moon described four affected siblings with retinal dystrophy, obesity, and mental retardation 30. Three of them (males) also had small external genitalia and an abnormal gait 30. In 1920s, Bardet 31, 32 and Biedl 33, 34 separately reported a similar clinical features in individuals, who also had polydactyly (extra fingers and/or toes) and the condition was coined Laurence-Moon-Bardet-Biedl syndrome. The overlapping features of these cases suggested that the two disorders, Laurence-Moon-Bardet-Biedl syndrome and Bardet-Biedl syndrome (BBS) represented variable expression of a single condition 35, 36, 37 although this is controversial 38, 39. However, it is now generally considered that Bardet-Biedl syndrome (BBS) and Laurence-Moon syndrome (LMS) are distinct conditions 27.
Bardet-Biedl syndrome can result from mutations in at least 26 different BBS genes 23, 25, 40. The proteins produced from BBS genes are are known or suspected to play critical roles in the maintenance and function of cilia. Cilia are microscopic, finger-like projections that stick out from the surface of many types of cells. Cilia are involved in cell movement and many different chemical signaling pathways. Cilia are also necessary for the perception of sensory input such as sight, hearing, and smell.
Mutations in BBS genes lead to problems with the structure and function of cilia. Defects in these cell structures probably disrupt important chemical signaling pathways during development and lead to abnormalities of sensory perception. Researchers believe that defective cilia are responsible for most of the features of Bardet-Biedl syndrome.
About one-quarter of all cases of Bardet-Biedl syndrome result from mutations in the BBS1 gene. Another 20 percent of cases are caused by mutations in the BBS10 gene. The other BBS genes each account for only a small percentage of all cases of Bardet-Biedl syndrome. In about 25 percent of people with Bardet-Biedl syndrome, the cause of the disorder is unknown.
In individuals with Bardet-Biedl syndrome who have mutations in one of the BBS genes, mutations in additional genes may be involved in causing or modifying the course of the disorder. Studies suggest that these modifying genes may be known BBS genes or other genes. The additional genetic changes could help explain the variability in the signs and symptoms of Bardet-Biedl syndrome. However, this phenomenon appears to be uncommon, and it has not been found consistently in scientific studies.
Bardet-Biedl syndrome (BBS) is more common than Laurence-Moon syndrome, with a prevalence of 1 in 125,000 to 1 in 160,000 newborns in North America and Europe 37, 28, 23. Bardet-Biedl syndrome (BBS) is more common on the island of Newfoundland (off the east coast of Canada), where it affects an estimated 1 in 17,000 newborns 23. Bardet-Biedl syndrome (BBS) also occurs more frequently, due to increased marriages among consanguineous, affecting about 1 in 13,500 newborns in the Bedouin population of Kuwait; 1:6900 in Jahra district and 1:3700 in Faroe Islands 41, 42, 43, 44, 23.
Vision loss is one of the major features of Bardet-Biedl syndrome 23, 24. Most patients with Bardet-Biedl syndrome will experience the loss of light-sensing tissue at the back of the eye called the retina 27. The retina is part of the eye involved in detecting and decoding incoming images. Incoming light is focused onto the retina at the back of the eye. The retina is composed of cells called “rods and cones”. They translate incoming light into nerve impulses the brain can use. This gradual loss of the rod and cone cells on the retina is described as “retinal dystrophy”. Symptoms associated with cone-rod dystrophy may not become apparent until 7 or 8 years of age when children begin to complain of problems with night vision with an inability to see in dimly lit environments, such as a sidewalk lit only by streetlights 27. This “night blindness” may progress to variable degrees by blind spots that develop in the side (peripheral) vision. In most people, the vision becomes progressively weaker through the first and second decades of life 27. Over time, these blind spots enlarge and merge to produce tunnel vision. Affected individuals often first lose peripheral vision, and see only what is directly in front of their focus point. They see in what is termed ‘tunnel vision’ 27. Most people with Bardet-Biedl syndrome also eventually lose central vision (poor visual acuity) and become legally blind by mid-teens or early adulthood 27. In some people, the degeneration of the retina may follow a characteristic course, referred to as “retinitis pigmentosa” (RP). Retinitis pigmentosa (RP) begins with night blindness, followed by a loss of the ability to discriminate colors from one another, and finally to a progressive tunnel vision 27. Additional effects on the eye characteristic to individuals with Bardet-Biedl syndrome include: lazy eye (strabismus), clouding of the lens of the eyes (cataracts), and an increased pressure within the eyes that can result in damage to the optic nerve conducting signals to the brain (glaucoma) 27.
Truncal obesity is another characteristic feature of Bardet-Biedl syndrome 23, 24, 27. The term ‘truncal obesity’ refers to a condition where fat is disproportionately distributed onto the abdomen and chest rather than the arms and legs 27. Individuals can be described as having an apple-shape body type. Weight is usually normal at birth but weight gain is quickly evident through the first year of life in as many as 90% of people with Bardet-Biedl syndrome 27. Complications of obesity can include type 2 diabetes, high blood pressure (hypertension), and abnormally high cholesterol levels (hypercholesterolemia). Type 2 diabetes has been estimated to affect up to 45% of patients with Bardet-Biedl syndrome 27. High blood pressure (hypertension), and abnormally high cholesterol levels (hypercholesterolemia) may further complicate problems with the heart and blood vessels seen in patients with Bardet-Biedl syndrome. The heart functions as a pump for the blood, moving the blood through the vessels that bring it throughout the body. The heart relies on valves that keep the flow moving in the forward direction. With age, stiffening of the heart valves is completely normal. The stiffening is due to calcium laying down on the valves, and this process if described by the word “stenosis”. People with Bardet-Biedl syndrome may experience stenosis of their heart valves prematurely 27. People with Bardet-Biedl syndrome may also have defects of the heart’s muscular walls 27. The heart muscle is designed such that every motion is smoothly orchestrated. Defects in the heart muscle predispose people with Bardet-Biedl syndrome to heart beat abnormalities, referred to as “arrhythmias” 27.
Other major signs and symptoms of Bardet-Biedl syndrome include the presence of extra fingers or toes (polydactyly), intellectual disability or learning problems, and abnormalities of the genitalia. People with Bardet-Biedl syndrome may be born with an extra digit near the pinky or an extra toe near the fifth “little” toe (polydactyly). This finding occurs in approximately 70 percent of patients. Specifically, the presence of an extra toe is more common than that of an extra finger 27. In medical terminology, this is described as ‘postaxial polydactyly’. Fingers and toes may also show webbing, called “syndactyly”. Syndactyly is especially common between the second and third toes 27. Fingers and toes may occasionally be abnormally short in length. This characteristic is called “brachydactyly” 27. The feet may overall be short in length, of wide width and carry a flat arch.
Another major feature of Bardet-Biedl syndrome is a small size and poor function of the male gonads, termed “testicular hypogonadism”. This may manifest as a small penis, failure of the testes to descend into the scrotum termed “cryptorchidism” or a delay in the onset of puberty. Undescended testicles are a concern because they are associated with a greater risk for testicular cancer and should be surgically managed. An undescended testicle needs to be treated surgically with a procedure called orchiopexy before a child is 2 years old to increase his chance for fertility later in life. Affected females may also have complex genital and urinary tract abnormalities. Affected females may demonstrate an underdeveloped uterus, fallopian tubes, or ovaries. Menstruation cycles are often delayed from the average first age of onset and may also follow an irregular cycle. Pregnant women with Bardet-Biedl syndrome should be followed closely by obstetricians that are well trained in dealing with high-risk pregnancies.
Problems with fertility arise in both men and women. Most affected males produce reduced amounts of sex hormones (testosterone), and they are usually unable to father biological children (infertile) 23, 24.
Some individuals with Bardet-Biedl syndrome may also have abnormalities of the structure and function of the kidneys, which can be serious or life-threatening 23, 24. Kidney defects are highly variable but generally result in an accumulation of urine in the kidneys that results in inappropriate pressures within the kidneys, leading to stretching of important structures 27. The dilation resulting from this fluid accumulation is called “hydronephrosis” 27. It can be monitored by medical imaging such as ultrasound, abdominal x-ray, etc. One common risk that accompanies hydronephrosis includes bacterial infection of the kidneys. The inflammation associated with infection of the kidneys is called “pyelonephritis” 27. These complications to kidney functions can often predispose individuals with Bardet-Biedl syndrome to end-stage renal disease (ESRD) also known as kidney failure. Other clinical feature of Bardet-Biedl syndrome include the development of kidney cysts and damage to the microscopic filtration unit of the kidney 27. Kidneys are responsible for filtering the blood and damage to the filtration systems will result in urine that is dark red blood, possibly even foamy. In scenarios of kidney failure, patients may require dialysis and kidney transplantation. In the scenario that a patient requires kidney transplantation, the use of kidney-protective immunosuppressive medications have been associated with an extra increase in weight gain. This extra weight gain can further complicate any pre-existing diabetes and heart conditions.
Mild-to-moderate learning difficulties are common in individuals with Bardet-Biedl syndrome 27. Often, learning disabilities are attributed to weakened cognitive capacity. Some individuals affected with Bardet-Biedl syndrome may have true learning disabilities due to dysfunction of brain development 27. However, it is important to be sure that suspected disabilities (eg: delayed speech or reading skills) are not due to underlying vision impairment 27. Neurological impairments may manifest in poor coordination, gross and fine motor skills, and social milestones (eg: ability to play complicated games with other children). Many patients report a significant degree of clumsiness and often walk with legs in a wide-based stance. Walking heel-to-toe may be difficult 27.
Additional features of Bardet-Biedl syndrome can include impaired speech, delayed development of motor skills such as standing and walking, behavioral problems such as emotional immaturity and inappropriate outbursts, and clumsiness or poor coordination 23, 24. Distinctive facial features, dental abnormalities, unusually short or fused fingers or toes, and a partial or complete loss of the sense of smell (anosmia) have also been reported in some people with Bardet-Biedl syndrome 23, 24. They have a decreased ability to sense smells due to a change in the size to a brain center called the “olfactory bulb”. This is a relatively a mild problem but may impact safety if people are unable to sense for example, a gas leak from the stove 27.
In the absence of one of these 4 primary clinical features (i.e., vision loss, intellectual disability, obesity, polydactyly), the diagnosis of Bardet-Biedl syndrome (BBS) is made when at least two secondary features are observed, including hepatic fibrosis, diabetes mellitus, reproductive and developmental abnormalities, growth retardation, speech delays, or cardiovascular problems 23, 24.
Some individuals with Bardet-Biedl syndrome may also experience problems with their liver and digestive system. The liver is responsible for many body processes. Among them, it produces a green-brown digestive fluid that the body needs to break food down properly. Specifically, the liver conducts bile through thin ducts that can develop dilation or stricture and leak digestive fluid into the liver, where it causes damage in the form of scarring. More rarely, problems with digestive system may be due to Hirschsprung disease. Hirschsprung disease describes an absence of the nerves normally found in the colon that control the innate motion of the colon and move food along the tract.
A subgroup of affected individuals may exhibit some distinct facial features. These features including deep-set, widely-spaced eyes with downward-slanting lid folds, a flat nasal bridge with nostrils that flare forward, and a long groove (philtrum) in the center of the upper lip. Individuals may have a high-arched palate, with fewer teeth than expected The teeth may have short roots and lie crowded within the mouth.
Other Disorders with Overlapping Clinical Features
Ellis-van Creveld syndrome is a rare autosomal recessive genetic disorder that causes short-limb dwarfism (chondrodysplasia with acromelic growth restriction), a narrow chest, and is characterized by polydactyly (extra fingers and toes), malformed nails (nails dystrophy), and congenital heart disease 45, 46, 47, 48, 49. Ellis-van Creveld syndrome can also cause dental abnormalities and other skeletal issues, with the most severe health problems often stemming from congenital heart defects or breathing difficulties due to the small chest.
Pallister-Hall syndrome is an extremely rare autosomal dominant genetic disorder caused by mutations in the GLI3 gene, leading to a spectrum of congenital anomalies, including extra fingers/toes (polydactyly) or fused fingers/toes (syndactyly), a non-cancerous mass on the hypothalamus (hypothalamic hamartoma), and an underdeveloped epiglottis (bifid epiglottis) 50, 51. The severity varies, with mild cases potentially resembling isolated polydactyly and severe cases including laryngotracheal clefts that can be fatal in newborns. Overlap of McKusick-Kaufman syndrome with Pallister-Hall syndrome has been described due to hydrometrocolpos and postaxial polydactyly 52. Pallister-Hall syndrome is characterized by a spectrum of anomalies ranging from polydactyly, asymptomatic bifid epiglottis, and hypothalamic hamartoma at the mild end to laryngotracheal cleft with neonatal lethality at the severe end. Individuals with mild Pallister-Hall syndrome may be incorrectly diagnosed as having isolated postaxial polydactyly type A. Individuals with Pallister-Hall syndrome can have pituitary insufficiency and may die as neonates from undiagnosed adrenal insufficiency.
Hydrometrocolpos can be a feature of several malformation syndromes including 17:
- Mayer-Rokitansky-Küster-Hauser syndrome (MRKH syndrome) is a congenital condition in genetic females where the Müllerian ducts (which develop into the uterus and upper vagina) fail to develop properly, leading to a missing uterus and a partially or completely absent vagina, while secondary sexual characteristics are normal. The primary symptom is primary amenorrhea (failure to start menstruation by age 15), and it can be classified into Type 1 (isolated genital anomalies) or Type 2 (genital anomalies associated with kidney, skeletal, or other organ issues). Treatment often involves vaginal dilation for sexual activity and thorough psychological counseling to address issues with identity and infertility.
- Herlyn-Werner-Wunderlich syndrome also known as Obstructed Hemivagina and Ipsilateral Renal Anomaly (OHVIRA) syndrome, is a rare congenital condition characterized by a triad of abnormalities: a double uterus (didelphys uterus, a rare congenital condition where a female is born with two separate uterine cavities, each with its own cervix and sometimes a double vagina), an obstructed hemivagina, and an ipsilateral (on the same side) renal agenesis (absence of a kidney) 53, 54, 55. Herlyn-Werner-Wunderlich syndrome is a type of Müllerian duct anomaly that typically presents after puberty with symptoms like pelvic pain, painful periods, or a palpable abdominal mass due to the obstructed menstrual flow. Diagnosis is often delayed, and Magnetic Resonance Imaging (MRI) is considered the gold standard for evaluation.
- Intraabdominal teratoma, a type of germ cell tumor that may contain several different types of tissue, such as hair, muscle, and bone. Teratomas may be mature or immature, based on how normal the cells look under a microscope. Sometimes teratomas are a mix of mature and immature cells. Teratomas usually occur in the ovaries in women, the testicles in men, and the tailbone in children. They may also occur in the central nervous system (brain and spinal cord), chest, or abdomen. Teratomas may be benign (not cancer) or malignant (cancer).
- VACTERL association. VACTERL is an acronym for Vertebral defects, Anal atresia, Cardiac defects, Tracheo-esophageal fistula, Renal anomalies, and Limb abnormalities 56, 57, 58. VACTERL association presents differently in each affected person — not all patients will experience the same combination of symptoms — or the same degree of severity of symptoms. To be diagnosed with VACTERL, a baby must have anomalies in at least three of these symptoms.
- Trichorhinophalangeal syndrome (TRPS) is a rare genetic disorder with three main types (TRPS1, TRPS2, and TRPS3), all characterized by distinctive features of the hair (“tricho”), nose (“rhino”), and fingers/toes (“phalangeal”) 59, 60, 61. Key features include sparse, slow-growing hair; a characteristic pear-shaped nose; and bone deformities, especially cone-shaped finger epiphyses and hip malformations. Trichorhinophalangeal Syndrome Type 2 (TRPS2) also involves multiple cartilaginous exostoses (bony growths) and can be associated with intellectual disability and hydrometrocolpos rarely, while Trichorhinophalangeal Syndrome Type 3 (TRPS3) features severe short stature and short metacarpals 62.
- Vaginal agenesis. Rarely, vaginal agenesis can be described with chromosome aberrations 63.
McKusick Kaufman syndrome treatment
McKusick-Kaufman syndrome treatment includes surgical repair of the obstruction causing hydrometrocolpos and drainage of the accumulated fluid. Treatment for polydactyly and congenital heart defects and any other anomalies is standard. In the newborn with severe hydrometrocolpos, care with anesthesia in the neonatal period is appropriate, as hydrometrocolpos can cause diaphragmatic compression 22.
Table 3. Treatment of individuals with McKusick-Kaufman syndrome
| Manifestation/Concern | Treatment | Considerations/Other |
|---|---|---|
| Hydrometrocolpos | Prompt surgical repair of obstruction and drainage of accumulated fluid | Hydrometrocolpos can also present after neonatal period. |
| Polydactyly & syndactyly | Standard treatment | |
| Congenital heart defects | ||
| Renal anomalies | ||
| Anal anomalies & Hirschsprung disease |
Table 4. Recommended surveillance for individuals with McKusick-Kaufman syndrome
| System/Concern | Evaluation | Frequency |
|---|---|---|
| Hydrometrocolpos | Watch for later complications of surgery for hydrometrocolpos, including recurrent urinary tract infections and re-stenosis and infection of vaginal tract. | No monitoring; prompt eval of symptoms & signs of abdominal distention |
| Possible Bardet-Biedl syndrome 1 | Serial growth measurement to track height & weight to document obesity that can occur with Bardet-Biedl syndrome | Annually until at least age 5 yrs |
| Developmental assessments to detect developmental disabilities that can occur with Bardet-Biedl syndrome | ||
| Regular ophthalmologic exam & electroretinogram (if appropriate) to evaluate for visual signs & symptoms of retinitis pigmentosa | Annually after age 5 yrs | |
| If renal anomaly present, monitor renal function & blood pressure. | Frequency of follow up of renal anomalies per specialist | |
| Monitor for development of severe constipation w/referral for rectal biopsy to exclude Hirschsprung disease. | Review at well child health exams |
Footnote:
1 A subset of those with a diagnosis of McKusick-Kaufman syndrome as infants may with age develop findings that lead to revision of the diagnosis to Bardet-Biedl syndrome.
[Source 3 ]McKusick Kaufman syndrome prognosis
Studies on life span have not been performed on individuals with McKusick-Kaufman syndrome, but life span is not known to be reduced apart from the morbidity and mortality associated with hydrometrocolpos and congenital heart disease 17, 3.
- McKusick-Kaufman syndrome. https://medlineplus.gov/genetics/condition/mckusick-kaufman-syndrome[↩][↩][↩][↩]
- MKKS gene. https://medlineplus.gov/genetics/gene/mkks[↩][↩][↩]
- Slavotinek AM. McKusick-Kaufman Syndrome. 2002 Sep 10 [Updated 2020 Dec 3]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1502[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Scott CA, Marsden AN, Rebagliati MR, Zhang Q, Chamling X, Searby CC, Baye LM, Sheffield VC, Slusarski DC. Nuclear/cytoplasmic transport defects in BBS6 underlie congenital heart disease through perturbation of a chromatin remodeling protein. PLoS Genet. 2017 Jul 28;13(7):e1006936. doi: 10.1371/journal.pgen.1006936[↩]
- Slavotinek, A., Stone, E., Mykytyn, K. et al. Mutations in MKKS cause Bardet-Biedl syndrome. Nat Genet 26, 15–16 (2000). https://doi.org/10.1038/79116[↩]
- Slavotinek AM, Searby C, Al-Gazali L, Hennekam RC, Schrander-Stumpel C, Orcana-Losa M, Pardo-Reoyo S, Cantani A, Kumar D, Capellini Q, Neri G, Zackai E, Biesecker LG. Mutation analysis of the MKKS gene in McKusick-Kaufman syndrome and selected Bardet-Biedl syndrome patients. Hum Genet. 2002 Jun;110(6):561-7. doi: 10.1007/s00439-002-0733-3[↩]
- Adam A, Hellig J, Mahomed N, Lambie L. Recurrent Urinary Tract Infections in a Female Child With Polydactyly and a Pelvic Mass: Consider the McKusick-Kaufman Syndrome. Urology. 2017 May;103:224-226. doi: 10.1016/j.urology.2017.01.024[↩]
- Mostafavi SH, Hooman N, Hallaji F. McKusick-Kaufman Syndrome: Atretic Upper Vaginal Pouch; an Unusual Urogenital MR Finding. J Radiol Case Rep. 2009;3(5):1-5. doi: 10.3941/jrcr.v3i5.126[↩][↩][↩][↩][↩][↩][↩][↩]
- Chitayat D, Hahm SY, Marion RW, Sachs GS, Goldman D, Hutcheon RG, Weiss R, Cho S, Nitowsky HM. Further delineation of the McKusick-Kaufman hydrometrocolpos-polydactyly syndrome. Am J Dis Child. 1987;141:1133–1136. doi: 10.1001/archpedi.1987.04460100111042[↩][↩][↩]
- David A, Bitoun P, Lacombe D, Lambert JC, Nivelon A, Vigneron J, Verloes A. Hydrometrocolpos and polydactyly: a common neonatal presentation of Bardet-Biedl and McKusick-Kaufman syndromes. J Med Genet. 1999 Aug;36(8):599-603. https://pmc.ncbi.nlm.nih.gov/articles/instance/1762973/pdf/v036p00599.pdf[↩][↩][↩][↩][↩]
- Schaefer E, Durand M, Stoetzel C, Doray B, Viville B, Hellé S, Danse JM, Hamel C, Bitoun P, Goldenberg A, Finck S, Faivre L, Sigaudy S, Holder M, Vincent MC, Marion V, Bonneau D, Verloes A, Nisand I, Mandel JL, Dollfus H. Molecular diagnosis reveals genetic heterogeneity for the overlapping MKKS and BBS phenotypes. Eur J Med Genet. 2011 Mar-Apr;54(2):157-60. doi: 10.1016/j.ejmg.2010.10.004[↩][↩][↩][↩]
- Mckusick VA, Bauer RL, Koop CE, et al. Hydrometrocolpos as a simply inherited malformation. JAMA 1964;189:813–16. doi: 10.1001/jama.1964.03070110015003[↩]
- Kaufman RL, McAlister WH, Ho CK, Hartmann AF. Family studies in congenital heart disease VI. The association of severe obstructive left heart lesions, vertebral and renal anomalies; a second family. Birth Defects Orig Artic Ser. 1974;10(7):93-104.[↩]
- Dungy CI, Aptekar RG, Cann HM. Hereditary hydrometrocolpos with polydactyly in infancy. Pediatrics. 1971 Jan;47(1):138-41.[↩]
- Slavotinek AM, Biesecker LG. Phenotypic overlap of McKusick-Kaufman syndrome with bardet-biedl syndrome: a literature review. Am J Med Genet. 2000 Nov 27;95(3):208-15.[↩][↩][↩][↩][↩]
- Hou JW. Bardet-Biedl syndrome initially presenting as McKusick-Kaufman syndrome. J Formos Med Assoc. 2004 Aug;103(8):629-32.[↩]
- Mallmann MR, Reutter H, Mack-Detlefsen B, Gottschalk I, Geipel A, Berg C, Boemers TM, Gembruch U. Prenatal Diagnosis of Hydro(metro)colpos: A Series of 20 Cases. Fetal Diagn Ther. 2019;45(1):62-68. doi: 10.1159/000486781[↩][↩][↩]
- Cantani A, Tacconi ML, Benincori N, Picarazzi A, Ceccoli D, Gaudino S. Rare syndromes. The Kaufman-McKusick syndrome. A review of the 44 cases reported in the literature. Ann Genet. 1987;30(2):70-4.[↩]
- Cantani A, Santillo C, Cozzi F. McKusick-Kaufman syndrome: report of the 66th case complicated by a staphyloma of the left eye. Padiatr Padol. 1991;26(4):193-6.[↩]
- Hamel BC, ter Haar BG. Hydrometrocolpos, polydactylie en aangeboren hartafwijking (het McKusick-Dungy-Kaufman syndroom) [Hydrometrocolpos, polydactylia and congenital heart defect (the McKusick-Dungy-Kaufman syndrome)]. Tijdschr Kindergeneeskd. 1984 Aug;52(4):129-33. Dutch.[↩]
- Mallmann MR, Reutter H, Mack-Detlefsen B, Gottschalk I, Geipel A, Berg C, Boemers TM, Gembruch U. Prenatal Diagnosis of Hydro(metro)colpos: A Series of 20 Cases. Fetal Diagn Ther. 2019;45(1):62-68. https://doi.org/10.1159/000486781[↩][↩]
- Tekin I, Ok G, Genc A, Tok D. Anaesthetic management in McKusick-Kaufman syndrome. Paediatr Anaesth. 2003 Feb;13(2):167-70. doi: 10.1046/j.1460-9592.2003.00954.x[↩][↩]
- Bardet-Biedl syndrome. https://medlineplus.gov/genetics/condition/bardet-biedl-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Bardet-Biedl syndrome. https://rarediseases.info.nih.gov/diseases/6866/x[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- What is Bardet-Biedl Syndrome? https://bbsuk.org.uk/what-is-bardet-biedl-syndrome[↩][↩]
- Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, Heath O, McManamon PJ, O’Leary E, Pryse-Philips W. The cardinal manifestations of Bardet–Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Eng J Med. 1989;321:1002–1009. doi: 10.1056/NEJM198910123211503[↩]
- Bardet-Biedl Syndrome. https://rarediseases.org/rare-diseases/bardet-biedl-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Beales PL, Warner AM, Hitman GA, Thakker R, Flinter FA. Bardet–Biedl syndrome: A molecular and phenotypic study of 18 families. J Med Genet. 1997;34:92–98. doi: 10.1136/jmg.34.2.92[↩][↩]
- Riise R, Andreasson S, Borgstrom M, Wright AF, Tommerup N, Rosenberg T, Tornqvist K. Intrafamilial variation of the phenotype in Bardet–Biedl syndrome. Br J Ophthalmol. 1997;81:378–385. doi: 10.1136/bjo.81.5.378[↩]
- Laurence JZ, Moon RC. Four cases of “retinitis pigmentosa” occurring in the same family, and accompanied by general imperfections of development. 1866. Obes Res. 1995 Jul;3(4):400-3. doi: 10.1002/j.1550-8528.1995.tb00166.x[↩][↩]
- Bardet G. Sur un syndrome d’obesite congenitale avec polydactylie et retinite pigmentaire (contribution a l’etude des formes cliniques de l’obesite hypophysaire) These de Paris (Le Grand) 1920;470:107.[↩]
- Bardet G. On congenital obesity syndrome with polydactyly and retinitis pigmentosa (a contribution to the study of clinical forms of hypophyseal obesity. Obes Res. 1995;3:387–399. doi: 10.1002/j.1550-8528.1995.tb00165.x[↩]
- Biedl A. Ein Geschwister mit adiposogenitaler Dystrophie. Dtsch Med Wochenschr. 1922;48:1630.[↩]
- Biedl A. A pair of siblings with adiposo-genital dystrophy. Obes Res. 1995;3:404. doi: 10.1002/j.1550-8528.1995.tb00167.x[↩]
- Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, Heath O, McManamon PJ, O’Leary E, Pryse-Phillips W. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989 Oct 12;321(15):1002-9. doi: 10.1056/NEJM198910123211503[↩]
- Solis-Cohen S, Weiss E. Dystrophia adiposogenitalis, with atypical retinitis pigmentosa and mental deficiency—The Laurence–Biedl syndrome. A report of four cases in one family. Am J Med Sci. 1925;169:489–505.[↩]
- Klein D, Amman F. The syndrome of Laurence-Moon-Bardet-Biedl and allied disease in Switzerland. Clinical, genetic and epidemiological studies. J Neurol Sci. 1969;9:479–513. doi: 10.1016/0022-510x(69)90091-4[↩][↩]
- Schachat AP, Maumenee IH. Bardet–Biedl syndrome and related disorders. Arch Ophthalmol. 1982;100:285–288. doi: 10.1001/archopht.1982.01030030287011[↩]
- Laurence-Moon and Bardet-Biedl syndromes. Lancet. 1988 Nov 19;2(8621):1178. https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(88)90242-5/fulltext[↩]
- Forsyth RL, Gunay-Aygun M. Bardet-Biedl Syndrome Overview. 2003 Jul 14 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1363[↩]
- Farag TI, Teebi AS. High incidence of Bardet Biedl syndrome among the Bedouin. Clin Genet. 1989;36(6):463–464. doi: 10.1111/j.1399-0004.1989.tb03378.x[↩]
- Moore SJ, Green JS, Fan Y, et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet A. 2005;132A(4):352–360. doi: 10.1002/ajmg.a.30406[↩]
- Hjortshøj TD, Grønskov K, Brøndum-Nielsen K, Rosenberg T. A novel founder BBS1 mutation explains a unique high prevalence of Bardet-Biedl syndrome in the Faroe Islands. Br J Ophthalmol. 2009;93(3):409–413. doi: 10.1136/bjo.2007.131110[↩]
- Farag TI, Teebi AS. Bardet–Biedl and Laurence–Moon syndromes in a mixed Arab population. Clin Genet. 1988;33:78–82. doi: 10.1111/j.1399-0004.1988.tb03414.x[↩]
- Ellis-van Creveld syndrome. https://medlineplus.gov/genetics/condition/ellis-van-creveld-syndrome[↩]
- Da Silva JD, Tkachenko N, Soares AR. Ellis-van Creveld Syndrome. 2023 Oct 26. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK596643[↩]
- Kamal R, Dahiya P, Kaur S, Bhardwaj R, Chaudhary K. Ellis-van Creveld syndrome: A rare clinical entity. J Oral Maxillofac Pathol. 2013 Jan;17(1):132-5. doi: 10.4103/0973-029X.110716[↩]
- Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, King L, Francomano C, Freisinger P, Spranger S, Marino B, Dallapiccola B, Wright M, Meitinger T, Polymeropoulos MH, Goodship J. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet. 2000 Mar;24(3):283-6. doi: 10.1038/73508. Erratum in: Nat Genet 2000 May;25(1):125.[↩]
- Ruiz-Perez VL, Tompson SW, Blair HJ, Espinoza-Valdez C, Lapunzina P, Silva EO, Hamel B, Gibbs JL, Young ID, Wright MJ, Goodship JA. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am J Hum Genet. 2003 Mar;72(3):728-32. doi: 10.1086/368063[↩]
- Biesecker LG. GLI3-Related Pallister-Hall Syndrome. 2000 May 25 [Updated 2024 Feb 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1465[↩]
- Pallister-Hall Syndrome. https://rarediseases.org/rare-diseases/pallister-hall-syndrome[↩]
- Kos S, Roth K, Korinth D, Zeilinger G, Eich G. Hydrometrocolpos, postaxial polydactyly, and hypothalamic hamartoma in a patient with confirmed Pallister-Hall syndrome: a clinical overlap with McKusick-Kaufman syndrome. Pediatr Radiol. 2008 Aug;38(8):902-6. doi: 10.1007/s00247-008-0870-5[↩]
- Jhirwal M, Singh P, Sharma C, Khera P. Herlyn-Werner-Wunderlich (HWW) syndrome with kyphoscoliosis: a rare urogenital anomaly in a teenage girl. BMJ Case Rep. 2021 Mar 22;14(3):e238688. doi: 10.1136/bcr-2020-238688[↩]
- Karaca L, Pirimoglu B, Bayraktutan U, Ogul H, Oral A, Kantarci M. Herlyn-Werner-Wunderlich syndrome: a very rare urogenital anomaly in a teenage girl. J Emerg Med. 2015 Mar;48(3):e73-5. https://doi.org/10.1016/j.jemermed.2014.09.064[↩]
- Khaladkar SM, Kamal V, Kamal A, Kondapavuluri SK. The Herlyn-Werner-Wunderlich Syndrome – A Case Report with Radiological Review. Pol J Radiol. 2016 Aug 24;81:395-400. doi: 10.12659/PJR.897228[↩]
- VACTERL association. https://medlineplus.gov/genetics/condition/vacterl-association[↩]
- VACTERL Association (VATER syndrome). https://www.chop.edu/conditions-diseases/vacterl-association-vater-syndrome[↩]
- VATER Syndrome/VACTERL Association. https://www.cincinnatichildrens.org/health/v/vacterl[↩]
- Trichorhinophalangeal syndrome type I. https://medlineplus.gov/genetics/condition/trichorhinophalangeal-syndrome-type-i/[↩]
- Trichorhinophalangeal Syndrome Type I. https://rarediseases.org/rare-diseases/trichorhinophalangeal-syndrome-type-i/[↩]
- Trichorhinophalangeal Syndrome Type II. https://rarediseases.org/rare-diseases/trichorhinophalangeal-syndrome-type-ii/[↩]
- Schinzel A, Riegel M, Baumer A, Superti-Furga A, Moreira LM, Santo LD, Schiper PP, Carvalho JH, Giedion A. Long-term follow-up of four patients with Langer-Giedion syndrome: clinical course and complications. Am J Med Genet A. 2013 Sep;161A(9):2216-25. doi: 10.1002/ajmg.a.36062[↩]
- Anant M, Raj N, Yadav N, Prasad A, Kumar S, Saxena AK. Two Distinctively Rare Syndromes in a Case of Primary Amenorrhea: 18p Deletion and Mayer-Rokitansky-Kuster-Hauser Syndromes. J Pediatr Genet. 2020 Sep;9(3):193-197. doi: 10.1055/s-0039-1700577[↩]




{kind=link}