
Contents
What is achondroplasia
Achondroplasia is a form of short-limbed dwarfism. The word achondroplasia literally means “without cartilage formation.” Cartilage is a tough but flexible tissue that makes up much of the skeleton during early development. However, in achondroplasia the problem is not in forming cartilage but in converting cartilage to bone (a process called ossification), particularly in the long bones of the arms and legs. Achondroplasia is the most common type of short-limbed dwarfism also called little people, a condition in which a person is very short (less than 4 feet 10 inches as an adult). The condition occurs in 1 in 15,000 to 40,000 newborns.
Achondroplasia is characterized by an unusually large head (macrocephaly), short upper arms (rhizomelic dwarfism), and short stature (adult height of approximately 4 feet). Achondroplasia does not typically cause impairment or deficiencies in mental abilities. If the bones that join the head and neck do not compress the brainstem or upper spinal cord (craniocervical junction compression), life expectancy is near normal.
Achondroplasia is similar to another skeletal disorder called hypochondroplasia, but the features of achondroplasia tend to be more severe.
Achondroplasia is genetic disorder caused by a change (mutation) in the fibroblast growth factor receptor 3 (FGFR3) gene. Achondroplasia occurs as a result of a spontaneous genetic mutation in approximately 80 percent of patients; in the remaining 20 percent achondroplasia is inherited from a parent.
All people with achondroplasia have short stature. The average height of an adult male with achondroplasia is 131 centimeters (4 feet, 4 inches), and the average height for adult females is 124 centimeters (4 feet, 1 inch). Characteristic features of achondroplasia include an average-size trunk, short arms and legs with particularly short upper arms and thighs, limited range of motion at the elbows, and an enlarged head (macrocephaly) with a prominent forehead. Fingers are typically short and the ring finger and middle finger may diverge, giving the hand a three-pronged (trident) appearance. People with achondroplasia are generally of normal intelligence.
Health problems commonly associated with achondroplasia include episodes in which breathing slows or stops for short periods (apnea), obesity, and recurrent ear infections. In childhood, individuals with the condition usually develop a pronounced and permanent sway of the lower back (lordosis) and bowed legs. Some affected people also develop abnormal front-to-back curvature of the spine (kyphosis) and back pain. A potentially serious complication of achondroplasia is spinal stenosis, which is a narrowing of the spinal canal that can pinch (compress) the upper part of the spinal cord. Spinal stenosis is associated with pain, tingling, and weakness in the legs that can cause difficulty with walking. Another uncommon but serious complication of achondroplasia is hydrocephalus, which is a buildup of fluid in the brain in affected children that can lead to increased head size and related brain abnormalities.
Achondroplasia can be diagnosed by characteristic clinical and radiographic findings in most affected individuals. In individuals in whom there is diagnostic uncertainty or atypical findings, identification of a heterozygous pathogenic variant in FGFR3 can establish the diagnosis.
Achondroplasia baby
People with achondroplasia can have a range of health problems, so it’s important to take your baby to see his health care provider for routine well-baby checkups.
At these checkups, your baby’s provider can compare your baby’s height, weight and head size to those of other babies with achondroplasia. This can help your baby’s provider spot and treat some problems early on.
People with achondroplasia often have these health problems:
- Apnea. This is when a baby stops breathing for 15 to 20 seconds or more. Babies with apnea and other breathing problems may need surgery to remove the tonsils and adenoids (lymph tissue near the throat).
- Repeat ear infections. Some babies with achondroplasia need ear tubes. These are small tubes placed in the ear that let air into the middle ear and help lower chances of ear infections. Without treatment, repeat ear infections can cause hearing loss.
- Obesity (being very overweight). Healthy eating and being active can help your child stay at a healthy weight as she grows.
- Compression of the upper end of the spinal cord. This is when the opening where the head and spine (backbone) connect is too small. The spinal cord gets squeezed (compressed), causing trouble with breathing. A small number of babies with achondroplasia die suddenly (often during sleep) from compression. If needed, surgery can widen the opening to ease pressure on the spinal cord.
- Spinal stenosis. Spinal stenosis causes the spine to narrow, putting pressure on the nerves and spinal cord. This can cause low back pain, problems with urination and weakness, tingling and pain in the legs. Symptoms usually appear when a person with achondroplasia is a teen or adult. Surgery can ease pressure on the spinal cord.
- Hydrocephalus (fluid buildup in the brain). Your baby’s provider measures your baby’s head at regular checkups to help catch hydrocephalus early. In some cases, a surgeon needs to drain the extra fluid from a baby’s brain.
- Kyphosis (a small hump in the upper back). A baby may have kyphosis due to poor muscle tone, but it usually goes away after she starts walking. Strollers or carriers that don’t give good back support can make kyphosis worse. If your child stills has kyphosis after she starts walking, she may need a back brace or surgery to correct it.
- Lordosis (inward curving of the lower back). This can develop after your child starts walking and can lead to waddling. Special exercises or physical therapy can help.
Achondroplasia causes
Achondroplasia results from specific changes (mutations) of a gene known as fibroblast growth factor receptor 3 (FGFR3). The FGFR3 gene provides instructions for making a protein that is involved in the development and maintenance of bone and brain tissue. Two specific mutations in the FGFR3 gene are responsible for almost all cases of achondroplasia. Researchers believe that these mutations cause the FGFR3 protein to be overly active, which interferes with skeletal development and leads to the disturbances in bone growth seen with this disorder.
Most babies (80 percent of achondroplasia cases) with achondroplasia are born to parents who don’t have the condition. This happens when there’s a random gene change in either the egg or sperm that join together and create a baby. Increased age of the father (advanced paternal age) may be a contributing factor in cases of sporadic achondroplasia.
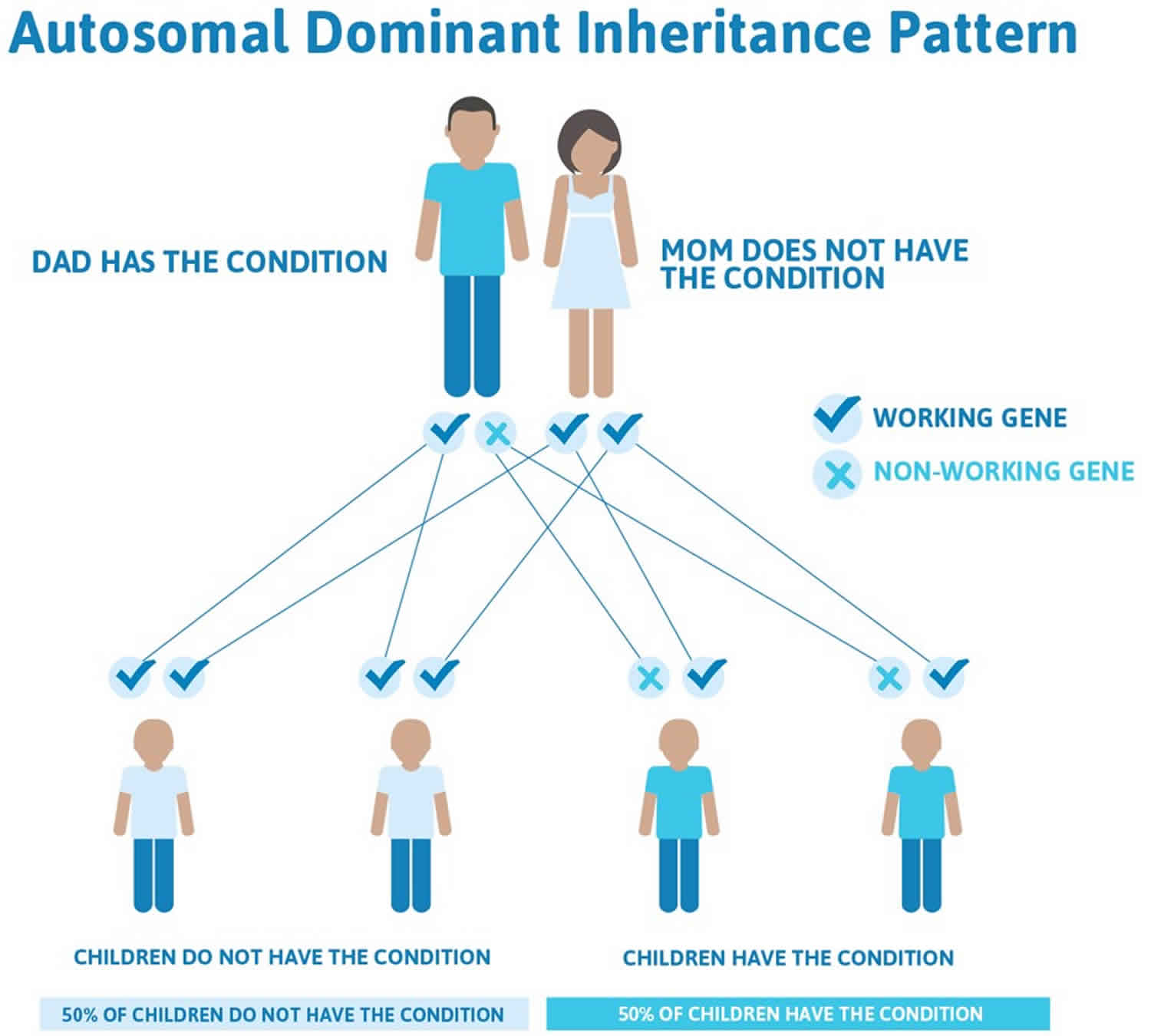
Less commonly (20 percent of achondroplasia cases), familial cases of achondroplasia follow an autosomal dominant pattern of inheritance. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary to cause a particular disorder. The abnormal gene can be inherited from either parent or can be the result of a mutated (changed) gene in the affected individual. The risk of passing the abnormal gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females.
How is achondroplasia inherited?
Achondroplasia is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Achondroplasia occurs as a result of a spontaneous genetic mutation in the FGFR3 gene in approximately 80 percent of patients; in the remaining 20 percent of people with achondroplasia have inherited an altered FGFR3 gene from one or two affected parents. Individuals who inherit two altered copies of FGFR3 gene typically have a severe form of achondroplasia that causes extreme shortening of the bones and an underdeveloped rib cage. These individuals are usually stillborn or die shortly after birth from respiratory failure.
If you or your partner has achondroplasia, you can pass it to your baby. If only one of you has the condition, there’s a 1 in 2 chance (50 percent) that your baby can have the condition.
If both you and your partner have achondroplasia, there is:
- 1 in 2 chance (50 percent) that your baby can have achondroplasia
- 1 in 4 chance (25 percent) that your baby won’t have achondroplasia
- 1 in 4 chance (25 percent) that your baby has the severe kind of achondroplasia that can lead to death
If you or your partner has achondroplasia or you’re the parent of a child with achondroplasia, talk to a genetic counselor about the condition. A genetic counselor is a person who is trained to help you understand about how genes, birth defects and other medical conditions run in families, and how they can affect your health and your baby’s health.
Genetic counseling
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Figure 1. Achondroplasia autosomal dominant inheritance pattern
Achondroplasia prevention
Genetic counseling may be helpful for prospective parents when one or both have achondroplasia. However, because achondroplasia most often develops spontaneously, prevention is not always possible.
Achondroplasia symptoms
The typical appearance of achondroplastic dwarfism can be seen at birth.
Achondroplasia baby
Infants born with achondroplasia typically have a “dome-like” (vaulted) skull, and a very broad forehead. In a small proportion there is excessive accumulation of fluid around the brain (hydrocephalus). Low muscle tone (hypotonia) in infancy is typical of achondroplasia. Acquisition of developmental motor milestones may be delayed.
A person with achondroplasia often has:
- Short height (significantly below the average height for a person of the same age and sex). The average height of an adult male with achondroplasia is 131 centimeters (4 feet, 4 inches), and the average height for adult females is 124 centimeters (4 feet, 1 inch).
- Short upper arms and thighs (compared to the forearms and lower legs)
- Large head and forehead (frontal bossing) with a flat bridge of the nose (large head-to-body size)
- Dental problems, including crowded or crooked teeth
- Broad, flat feet, short toes and short fingers
- Trident hand, a condition in which you have an extra space between the middle and ring fingers
- Decreased muscle tone. Babies with weak muscle tone may have delays in meeting developmental milestones, like sitting, standing and walking.
- Bowed legs. This is when legs curve outward between the thighs and ankles. Bowed legs can cause pain and trouble with walking. If the bowing or pain is severe, surgery can fix bowed legs.
- Narrowing of the spinal column (spinal stenosis)
- Spine curvatures called kyphosis and lordosis
Growth
Average adult height for men with achondroplasia is 131±5.6 cm; for women, 124±5.9 cm. Obesity is a major problem in achondroplasia 1). Excessive weight gain is manifest in early childhood. In adults, obesity can aggravate the morbidity associated with lumbar stenosis and contribute to nonspecific joint problems and possibly to early mortality from cardiovascular complications 2).
Development
In infancy, mild to moderate hypotonia is typical. Infants have difficulty in supporting their heads because of both hypotonia and large head size. That and differences in body habitus cause motor delays and unusual patterns of motor development such as snowplowing (using the head and feet to leverage movement) 3). Small joint hypermobility and short fingers can affect fine motor development and delay self-feeding 4).
Intelligence
Intelligence is normal unless hydrocephalus or other central nervous system complications occur. High-level executive function issues have been reported in some individuals 5).
Macrocephaly
Most children with achondroplasia are macrocephalic 6). Hydrocephalus requiring treatment, which probably occurs in 5% or fewer 7), may be caused by increased intracranial venous pressure because of stenosis of the jugular foramina 8). More recent literature suggests that in some individuals, foramen magnum stenosis may contribute to hydrocephalus – which is thus treatable by posterior fossa decompression or endoscopic third ventriculostomy 9).
Narrow craniocervical junction
Some infants with achondroplasia die in the first year of life from complications related to the craniocervical junction; population-based studies suggest that this excess risk of death may be as high as 7.5% without assessment and intervention 10). The risk appears to be secondary to central apnea associated with damage to respiratory control centers 11), and can be minimized by comprehensive evaluation of every infant with achondroplasia 12) and selective neurosurgical intervention 13). With such evaluation and management this risk may be decreased to as little as 0.3% 14). The best predictors of need for suboccipital decompression include lower-limb hyperreflexia or clonus, central hypopnea demonstrated by polysomnography, and reduced foramen magnum size, as determined by neuroimaging of the craniocervical junction. If computerized tomography (CT) is used, foraminal size can be compared with achondroplasia standards 15). Magnetic resonance imaging (MRI) examination provides direct visualization of the cord without radiation exposure, but there are no achondroplasia standards. T2-weighted MRI may show evidence of spinal cord abnormalities, which may guide operative decision making 16). In one study, all children undergoing surgical decompression of the craniocervical junction showed marked improvement of neurologic function 17). Quality of life indices determined up to 20 years after such surgery were comparable to quality of life indices in those for whom surgery was not indicated in childhood 18). A similar mechanism of injury can result in high cervical myelopathy (asymmetric or increased reflexes, weakness, persisting hypotonia, and poor balance) 19).
Restrictive pulmonary disease
In infancy a small subset of individuals with achondroplasia have restrictive pulmonary issues secondary to a small chest 20) and decreased compliance of the rib cage. Many infants show more rapid desaturations with minor respiratory events (e.g., physiologic periodic breathing or otherwise insignificant obstructive events). A small number have, as a consequence of these features, chronic hypoxemia 21). If a young infant has persistent tachypnea, failure to thrive, or evidence of respiratory failure, the polysomnogram obtained for other reasons in infants will show a low baseline oxygen saturation and/or desaturations associated with minimal respiratory irregularities. If such characteristics are recognized, referral to a pediatric pulmonologist is imperative. Treatment may include oxygen supplementation and, in a few, temporary tracheostomy. In virtually all instances, the need for a tracheostomy disappears as the child grows.
Sleep apnea
Obstructive sleep apnea is common in both older children and adults. It arises because of a combination of midface retrusion resulting in smaller airway size 22), hypertrophy of the lymphatic ring, airway malacia 23), and, perhaps, abnormal innervation of the airway musculature 24).
Clinical signs and symptoms of obstructive sleep apnea may include the following:
- Difficult morning waking
- Excessive daytime somnolence
- Respiratory pauses during sleep
- Loud snoring
- Glottal stops or gasping
- Loud sighs while sleeping
- Poor daytime concentration
- Irritability, fatigue, depression
- Bedwetting
Clinical signs and symptoms of infantile sleep apnea include the following:
- Observed apnea or exaggerated periodic breathing
- Struggling to breathe
- Poor feeding
- Coughing
- Difficulty lying flat to sleep
- Frequent awakenings
Central sleep apnea as well as obstructive sleep apnea may be present in infants. Clinical history is a poor predictor of apnea, and polysomnography should be done 25).
Middle ear dysfunction
Middle ear dysfunction is frequently a problem 26), and if inadequately treated can result in conductive hearing loss of sufficient severity to interfere with language development. More than half of children will require pressure-equalizing tube placement 27). Overall, about 40% of individuals with achondroplasia have functionally relevant hearing loss. Expressive language development is also frequently delayed 28), although the strength of the relationship between hearing loss and expressive language issues is uncertain.
Bow legs
Bowing of the lower legs is exceedingly common in those with achondroplasia. More than 90% of untreated adults have some degree of bowing 29). “Bowing” is actually a complex deformity arising from a combination of lateral bowing, internal tibial torsion, and dynamic instability of the knee 30).
Kyphosis
Kyphosis at the thoracolumbar junction is present in 90%-95% of infants with achondroplasia 31). In about 10% it does not spontaneously resolve and can result in serious neurologic sequelae 32). Preventive strategies 33) may reduce the need for surgical intervention.
Spinal stenosis
The most common medical complaint in adulthood is symptomatic spinal stenosis involving L1-L4 34). Symptoms range from intermittent, reversible, exercise-induced claudication to severe, irreversible abnormalities of leg function and of continence 35). Claudication and stenosis can both result in sensory (numbness, pain, feelings of heaviness) and motor symptoms (weakness, tripping, limited walking endurance). Vascular claudication results from engorged blood vessels after standing and walking and is fully reversible with rest. Spinal stenosis is actual impingement of the spinal cord or nerve root by the stenotic bone of the spinal canal, and symptoms are nonreversible. Symptoms localized to a particular dermatome can result from stenosis of a particular nerve root foramina.
Other orthopedic issues
- Joint laxity. Most joints are hypermobile in childhood. In general, this has minor consequences except for knee instability in a subset of individuals.
- Discoid lateral meniscus. This recently recognized structural anomaly may result in chronic knee pain in some individuals 36).
- Arthritis. Constitutive activation of FGFR-3, as in achondroplasia, may protect against development of arthritis 37).
Acanthosis nigricans
Acanthosis nigricans may be seen in about 10% of individuals with achondroplasia 38). In this population it does not reflect hyperinsulinemia or malignancy.
How do you know if your baby has achondroplasia?
Before birth, your doctor may think your baby has achondroplasia if an ultrasound shows your baby has bone problems, like shortened bones and excessive amniotic fluid surrounding the unborn infant. An ultrasound uses sound waves and a computer screen to show a picture of your baby inside the womb.
If the ultrasound shows these bone problems, your provider may recommend a prenatal test called amniocentesis (also called amnio) to confirm that your baby has achrondroplasia. In an amnio, your provider takes some amniotic fluid from around your baby in the uterus. The test checks for birth defects and genetic conditions in your baby.
Pregnant women with achondroplasia must undergo cesarean section delivery because of small pelvic size.
After birth, your baby’s provider can use X-rays, a physical exam and a blood test to check your baby for achondroplasia.
Examination of the infant after birth shows increased front-to-back head size. There may be signs of hydrocephalus (“water on the brain”).
X-rays findings can reveal achondroplasia in the newborn:
- Short, robust tubular bones
- Narrowing of the interpedicular distance of the caudal spine
- Square ilia and horizontal acetabula
- Narrow sacrosciatic notch
- Proximal femoral radiolucency
- Mild, generalized metaphyseal changes
However, if there is uncertainty, identification of the genetic variant of the FGFR3 gene by molecular genetic testing can be used to establish the diagnosis.
Clinical signs that may be used in the diagnosis of achondroplasia 39):
- Disproportionate short stature
- Macrocephaly with frontal bossing
- Backward displacement of the midface and depressed nasal bridge
- Shortening of the arms with redundant skin folds on limbs
- Limitation of elbow extension
- Shortened fingers and toes (brachydactyly)
- Trident configuration of the hands
- Bow legs
- Exaggerated inward curve of the spine (lumbar lordosis)
- Joint laxity
Achondroplasia life expectancy
People with achondroplasia seldom reach 5 feet (1.5 meters) in height. Intelligence is in the normal range.
Most babies born with achondroplasia live a normal life span, but a few may have severe bone problems that can lead to death.
Infants who receive the abnormal gene from both parents (homozygous achondroplasia) do not often live beyond a few months due respiratory insufficiency because of the small thoracic cage and neurologic deficit from cervicomedullary stenosis 40).
Increased mortality in adults with achondroplasia has been reported 41). Overall, life expectancy appeared to be decreased by about ten years 42).
Achondroplasia possible complications
Health problems that may develop include:
- Breathing problems from a small upper airway and from pressure on the area of the brain that controls breathing
- Lung problems from a small ribcage
Achondroplasia treatment
There is no specific treatment for achondroplasia. Related abnormalities, including spinal stenosis and spinal cord compression, should be treated when they cause problems.
Recommendations for managing children with achondroplasia are outlined by the American Academy of Pediatrics Committee on Genetics, which are designed to supplement guidelines for children with average stature.
As outlined in Pauli and Legare 43), the recommendations for the manifestations of achondroplasia include:
- Hydrocephalus: If signs/symptoms of increased intracranial pressure arise (accelerated head growth, bulging fontanelle, vision changes, headache), referral to a neurosurgeon is required. Computerized tomography (CT) or magnetic resonance imaging (MRI) of the brain in infancy may be done to determine the presence of hydrocephalus. Ventriculoperitoneal shunting has been the standard treatment. However, endoscopic third ventriculostomy may be beneficial in some individuals 44), implying that other mechanisms, such as obstruction of fourth ventricular exit foramina from the craniocervical stenosis, may be relevant 45).
- Craniocervical junction constriction: Predictors of the need for suboccipital decompression require evaluation by a medical professional. The best predictors of need for suboccipital decompression:
- Lower-limb hyperreflexia or clonus
- Central hypopnea demonstrated by polysomnography
- Reduced foramen magnum size, determined by CT examination of the craniocervical junction and by comparison with the norms for children with
- achondroplasia 46)
- Evidence of spinal cord compression and/or T2 signal abnormality; more recently proposed as another factor to be considered in a decision to operate 47)
- If there is clear indication of symptomatic compression, urgent referral to a pediatric neurosurgeon for decompression surgery should be initiated 48).
- Obstructive sleep apnea: Can be treated with weight reduction, surgery to remove tonsils and adenoids (adenotonsillectomy), positive airway pressure, and, rarely, surgery to create an opening in the neck (tracheostomy). Improvement in disturbed sleep and some improvement in neurologic function can result from these interventions 49). In rare instances in which the obstruction is severe enough to require tracheostomy, surgical intervention to advance the midface has been used to alleviate upper airway obstruction 50).
- Middle ear dysfunction: Ear tubes may be needed until the age of seven or eight to manage frequent middle ear infections and prevent potential hearing loss. Speech evaluation with implementation of appropriate therapies is warranted at any age if concerns arise. Routine developmental screening should be done at each well child evaluation and clinical genetics evaluation.
- Short stature: Studies on the use of growth hormone have shown initial acceleration of growth, but with lessening effect over time and little lasting benefit 51). On average, only about 3 cm of additional adult height can be expected 52).
- Extended limb lengthening using various techniques remains an option for some. Increases in height of up to 30-35 cm may be obtained 53). Complications are frequent and may be serious 54). Although some have advocated performing these procedures as early as ages six to eight years, many pediatricians, clinical geneticists, and ethicists have advocated postponing such surgery until the young person is able to participate in making an informed decision.
- At least in North America, only a tiny proportion of affected individuals elect to undergo extended limb lengthening. The Medical Advisory Board of Little People of America has published a statement regarding use of extended limb lengthening.
- Obesity: Measures to avoid obesity should begin in early childhood. Standard weight-by-height grids specific for achondroplasia should be used to monitor progress 55). It is important to note that these curves are not ideal weight-for-height curves; they were generated from thousands of data points from individuals with achondroplasia.
- Body mass index (BMI) standards have been generated for children age 16 and under 56). BMI has not been standardized for adults with achondroplasia; comparison to average-stature BMI curves will yield misleading results 57).
- Bow legs (varus deformity): Symptomatic bowing of the legs (varus deformity) requires referral to an orthopedist 58). However, asymptomatic bowing does not usually warrant surgical correction. Presence of progressive, symptomatic bowing should prompt referral to an orthopedist. Varus deformity alone, without symptoms, does not usually warrant surgical correction. Various interventions may be elected (e.g., guided growth using 8-plates, valgus-producing and derotational osteotomies). No controlled studies comparing outcomes of treatment options have been completed.
- Kyphosis: Preventive measures including prohibition of unsupported sitting in the first 12-18 months of life decrease risk of developing a fixed backwards curve in the mid-spine (kyphosis). Bracing or surgery may be necessary, depending on the degree of severity of such a deformity if preventive measures are unsuccessful.
- Kyphosis improves significantly or resolves in the majority of children upon assuming an orthograde posture and beginning to walk 59).
- In children in whom spontaneous remission does not arise after trunk strength increases and the child begins to walk, bracing is usually sufficient to prevent persistence of the thoracolumbar kyphosis 60).
- If a severe kyphosis persists, spinal surgery may be necessary to prevent neurologic complications 61).
- Spinal stenosis: If signs/symptoms of spinal stenosis arise, urgent surgical referral is appropriate. Extended and wide laminectomies 62) are usually recommended. Urgency depends on level (e.g., thoracic vs lumbar) and degree of stenosis 63).
- Immunization: All routine immunizations are necessary.
- Adaptive needs: Environmental modifications of the home and school may be necessary to accommodate for short stature. In school these may include step stools, lowered light switches, appropriate-height toilets or other means to make them accessible, lower desks, and foot support in front of chairs. All children need to be able to independently escape the building should an emergency arise. Small hands and ligamentous laxity can make fine motor activities difficult. Appropriate adaptations include the use of smaller keyboards, weighted pens, and smoother writing surfaces. Most children should have an IEP or 504 plan. Pedal extenders for driving are almost always needed. Also needed may be workplace modification such as lower desks, smaller keyboards, step stools, and toileting access.
- Agents/circumstances to avoid: Rear-facing car seats should be used as long as possible to avoid injury from motor vehicle accident. Avoid soft-back infant seats. Avoid activities in which there is risk of injury to the craniocervical junction, such as collision sports; use of a trampoline; diving from diving boards; vaulting in gymnastics; and hanging upside down from the knees or feet on playground equipment (due to risk of falling onto their head or neck).
- Socialization: Patients with achondroplasia may encounter difficulties in socialization and school adjustment. Support groups such as Little People of America (https://www.lpaonline.org/) can help assist families with these issues through peer support, personal example, and social awareness programs.
References [ + ]


{kind=link}