Contents
What is ataxia
Ataxia is a term for a group of disorders that affect muscle control or coordination of voluntary movements, balance and speech.
Ataxia is usually caused by damage to a part of the brain known as the cerebellum, but it can also be caused by damage to the spinal cord or other nerves.
This damage can be part of an underlying condition such as MS, or can be caused by a head injury, lack of oxygen to the brain, or long-term, excessive alcohol consumption.
Hereditary ataxia is caused by a faulty gene passed on by family members, who may or may not be affected.
Any part of the body can be affected, but people with ataxia often have difficulties with:
- balance and walking
- picking up objects
- difficulties with speech
- swallowing
- tasks that require a high degree of control, such as writing and eating
- vision and eye movement
The exact symptoms and their severity vary depending on the type of ataxia a person has.
Persistent ataxia usually results from damage to the part of your brain that controls muscle coordination (cerebellum). Many conditions can cause ataxia, including alcohol abuse, certain medications, stroke, tumor, cerebral palsy, brain degeneration and multiple sclerosis. Inherited defective genes also can cause the condition.
Treatment for ataxia depends on the cause. Adaptive devices, such as walkers or canes, might help you maintain your independence. Physical therapy, occupational therapy, speech therapy and regular aerobic exercise also might help.
Ataxia prognosis
The outlook for ataxia can vary considerably and largely depends on the type of ataxia you have. Some types may remain relatively stable or even improve with time, but most will get progressively worse over many years.
Life expectancy is generally shorter than normal for people with hereditary ataxia, although some people can live well into their 50s, 60s or beyond. In more severe cases, the condition can be fatal in childhood or early adulthood.
For acquired ataxia, the outlook depends on the underlying cause. Some cases may improve or stay the same, while other cases may get gradually worse over time and reduce life expectancy.
If you aren’t aware of having a condition that causes ataxia, such as multiple sclerosis, see your doctor as soon as possible if you:
Lose balance
Lose muscle coordination in a hand, arm or leg
Have difficulty walking
Slur your speech
Have difficulty swallowing
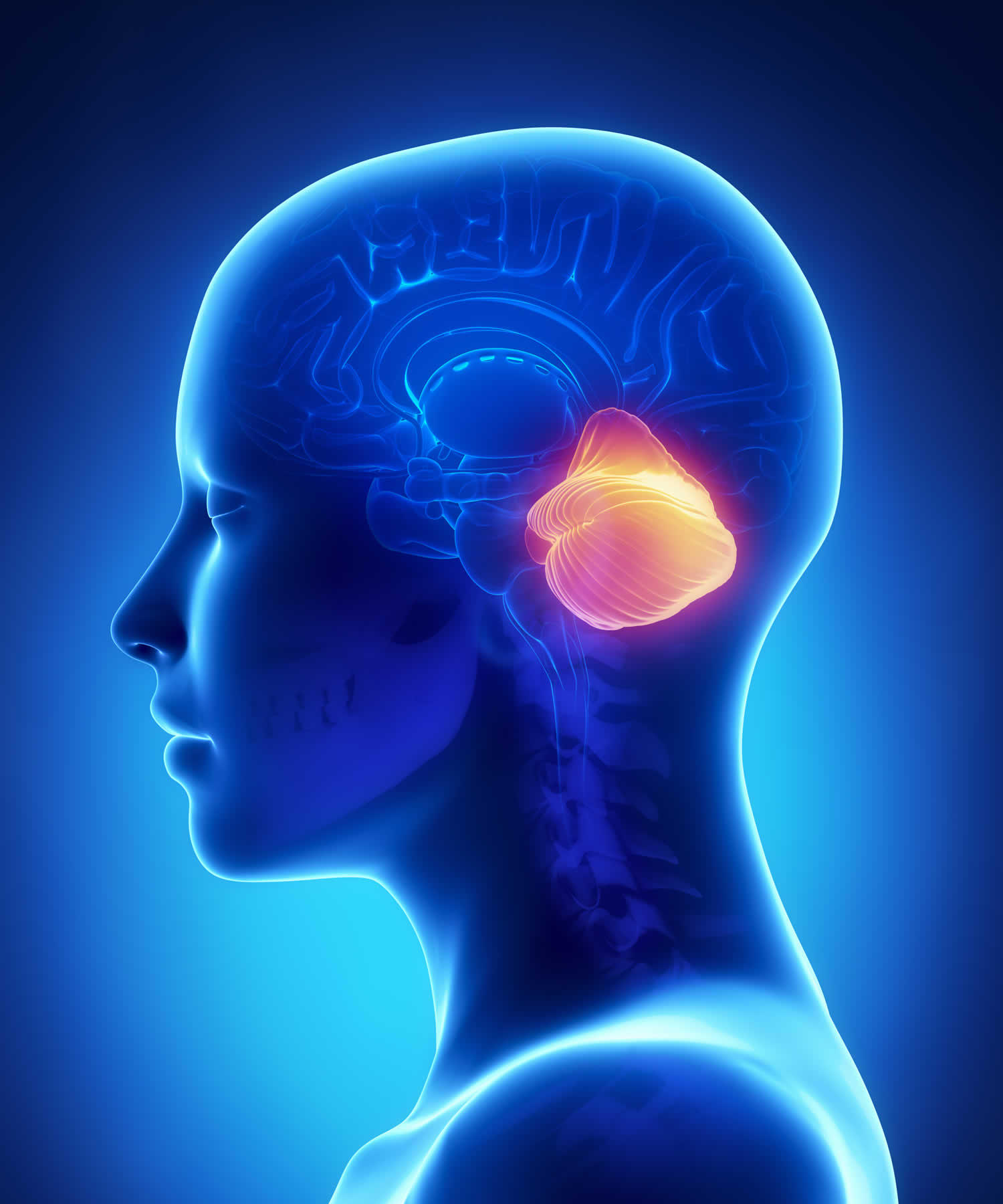
Figure 1. Human brain
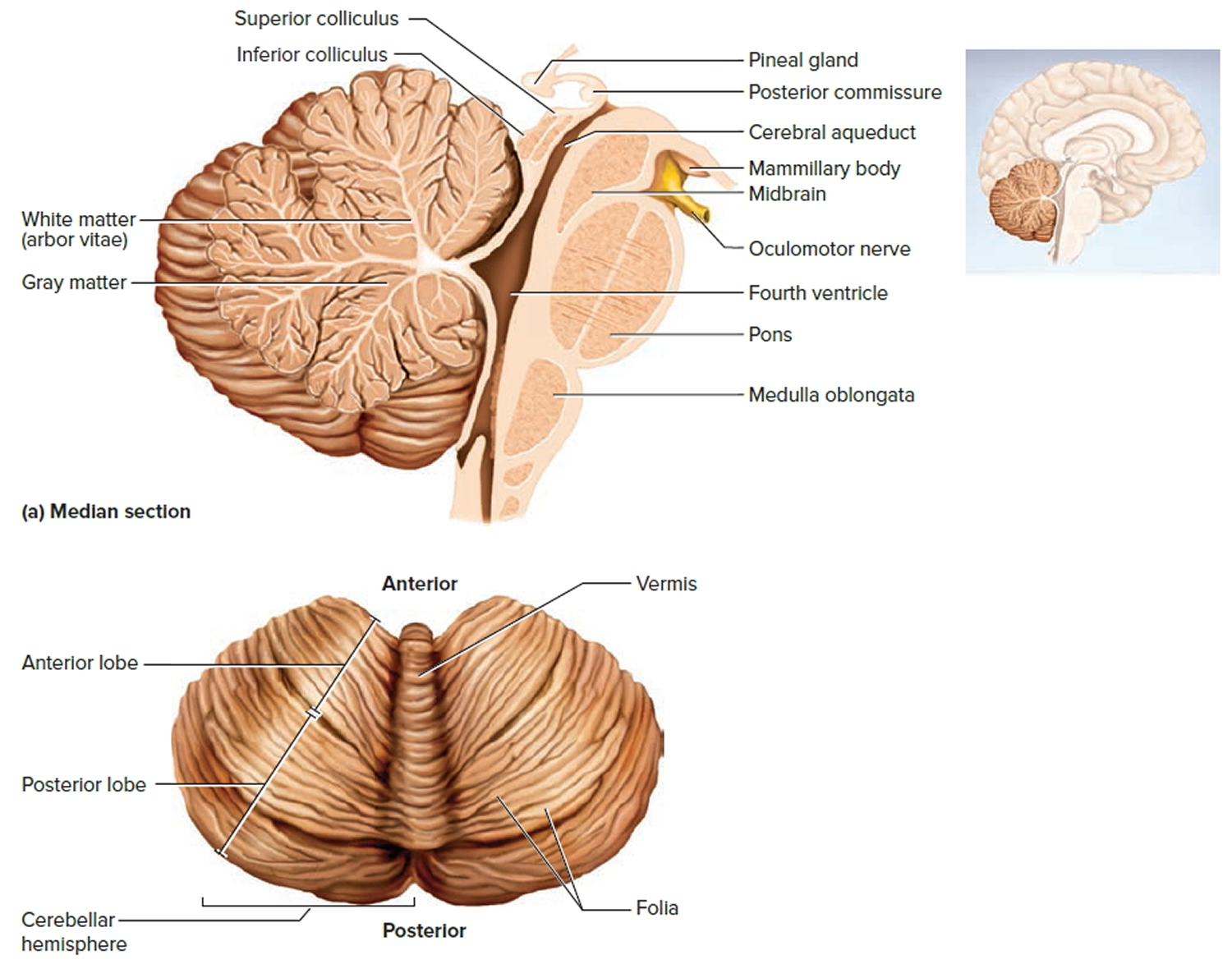
Figure 2. Cerebellum of brain
Types of ataxia
Some types of ataxia affect children from an early age, while other types may not develop until much later in adulthood.
Depending on the type of ataxia, the symptoms may stay the same, get progressively worse, or slowly improve.
Some of the main types of ataxia are described below.
There are many different types of ataxia, which can be divided into three broad categories:
- Acquired ataxia – where symptoms develop as the result of trauma, a stroke, multiple sclerosis (MS), a brain tumour, nutritional deficiencies, or other problems that damage the brain or nervous system
- Hereditary ataxia – where symptoms develop slowly over many years and are caused by faulty genes that a person inherits from their parents; the most common type is Friedreich’s ataxia
- Idiopathic late-onset cerebellar ataxia – where the brain is progressively damaged over time for reasons that are unclear.
Friedreich’s ataxia
Friedreich’s ataxia is the most common type of hereditary ataxia (caused by genes you’ve inherited). It’s thought to affect at least 1 in every 50,000 people.
Friedreich’s ataxia is a rare inherited disease that causes nervous system damage and movement problems. It usually begins in childhood and leads to impaired muscle coordination (ataxia) that worsens over time. The disorder is named after Nicholaus Friedreich, a German doctor who first described the condition in the 1860s.
Friedreich’s ataxia symptoms usually first develop before the age of 25, although it can develop in people much older than this.
In Friedreich’s ataxia the spinal cord and peripheral nerves degenerate, becoming thinner 1. The cerebellum, part of the brain that coordinates balance and movement, also degenerates to a lesser extent. This damage results in awkward, unsteady movements and impaired sensory functions. The disorder also causes problems in the heart and spine, and some people with the condition develop diabetes. The disorder does not affect thinking and reasoning abilities (cognitive functions).
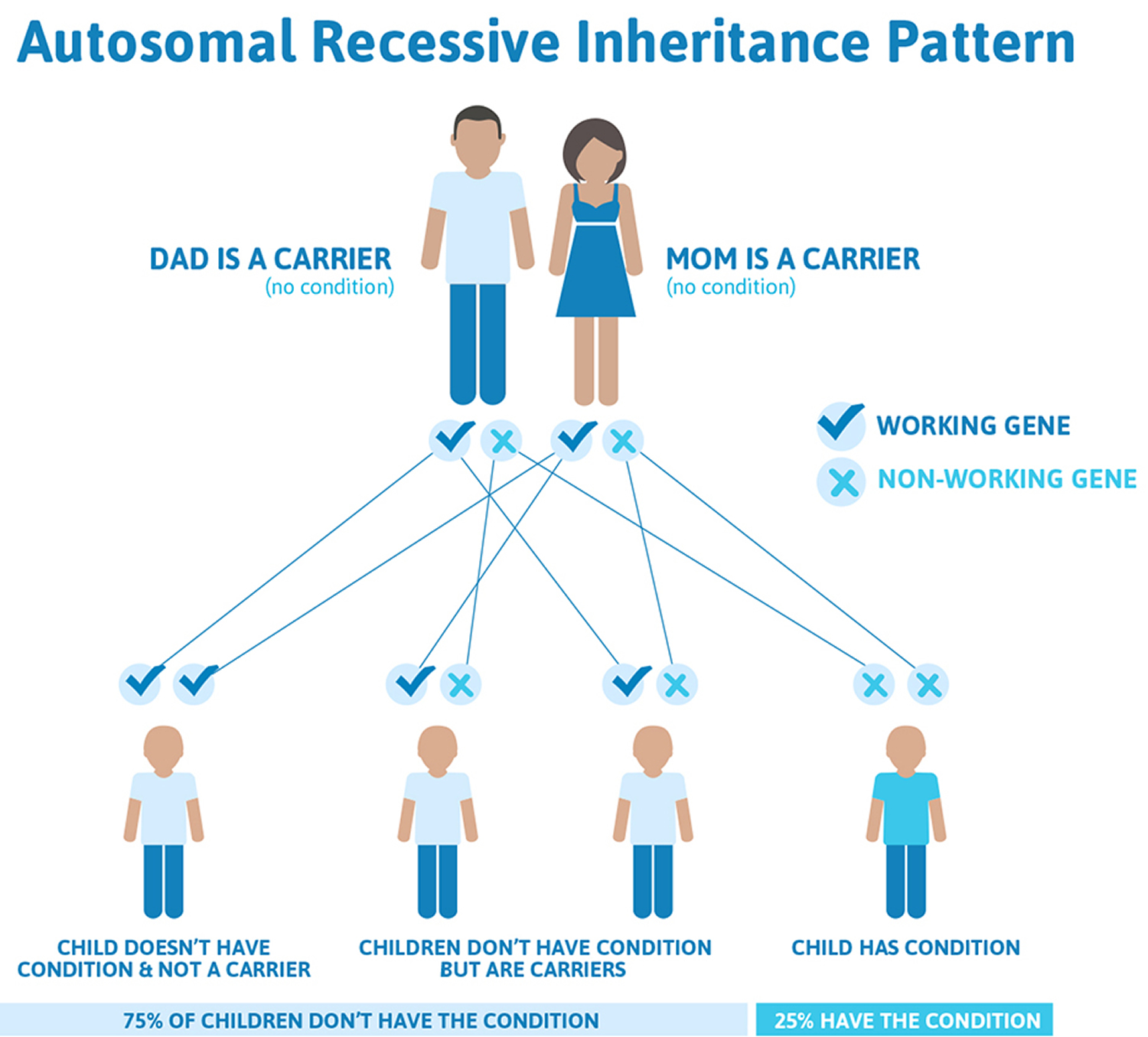
Friedreich’s ataxia is caused by a defect (mutation) in a gene labeled FXN. The disorder is recessive, meaning it occurs only in someone who inherits two defective copies of the gene, one from each parent. Although rare, Friedreich’s ataxia is the most common form of hereditary ataxia, affecting about 1 in every 50,000 people in the United States. Both male and female children can inherit the disorder.
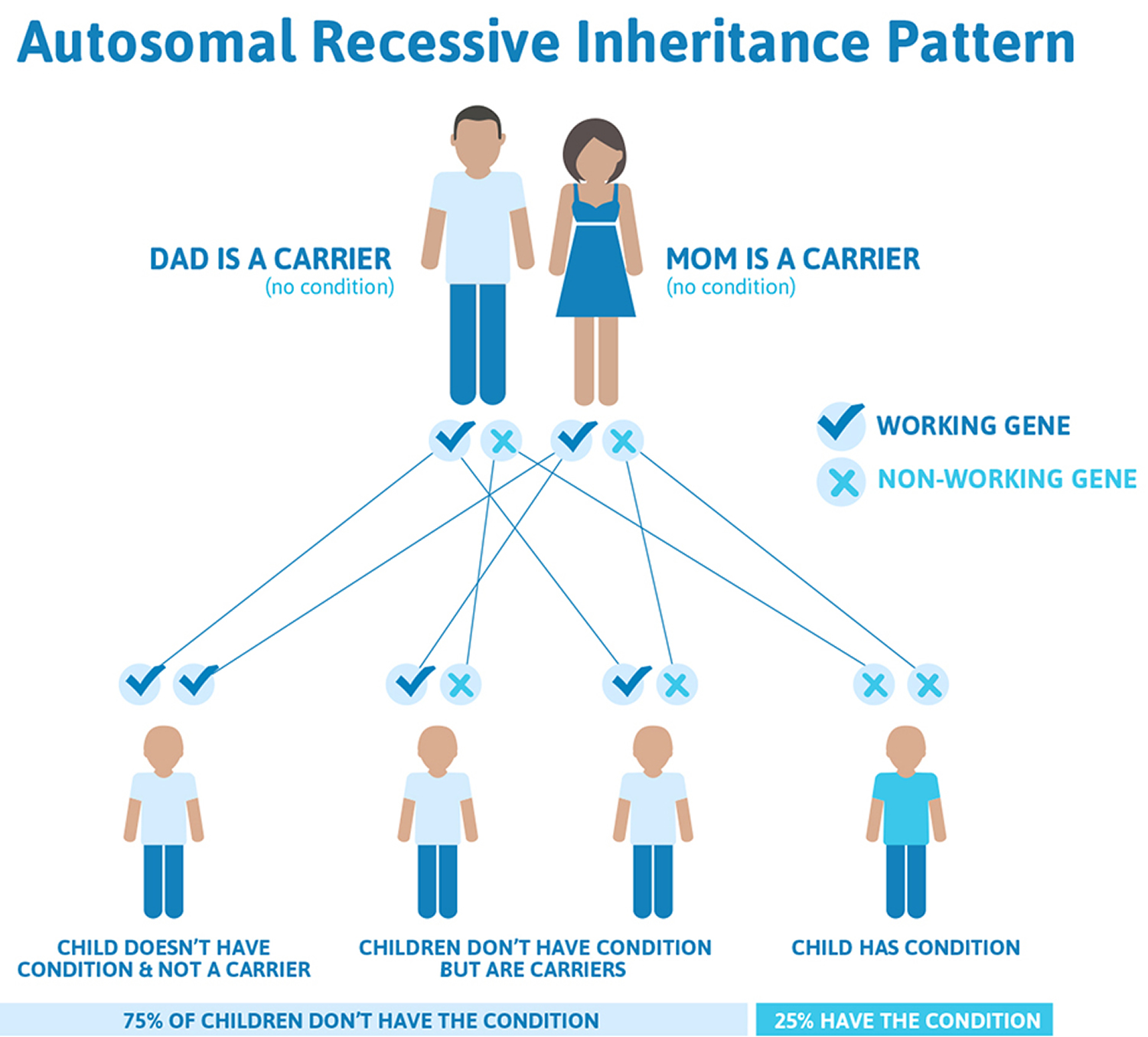
Friedreich’s ataxia inheritance
Friedreich’s ataxia is an autosomal recessive disease, meaning individuals only develop symptoms if they inherit two copies of the defective FXN gene, one from their father and one from their mother. A person who has only one abnormal copy of the gene is called a carrier. A carrier will not develop the disease but could pass the gene mutation on to his or her children. If both parents are carriers, their children will have a 1 in 4 chance of having the disease and a 1 in 2 chance of inheriting one abnormal gene that they, in turn, could pass on to their children. About one in 90 Americans of European ancestry carries an abnormal FXN gene.
In 1996, an international research team identified the Friedreich’s ataxia gene on chromosome 9. The FXN gene codes for production of a protein called “frataxin.” In the normal version of the gene, a sequence of DNA (labeled “GAA”) is repeated between 7 and 22 times. In the defective FXN gene, the repeat occurs over and over again—hundreds, even up to a thousand times.
This abnormal pattern, called a triplet repeat expansion, has been implicated as the cause of several dominantly inherited diseases, but Friedreich’s ataxia is the only known recessive genetic disorder caused by the problem. Almost all people with Friedreich’s ataxia have two copies of this mutant form of FXN, but it is not found in all cases of the disease. About two percent of affected individuals have other defects in the FXN gene that are responsible for causing the disease.
The triplet repeat expansion greatly disrupts the normal production of frataxin. Frataxin is found in the energy-producing parts of the cell called mitochondria. Research suggests that without a normal level of frataxin, certain cells in the body (especially peripheral nerve, spinal cord, brain and heart muscle cells) cannot effectively produce energy and have been hypothesized to have a buildup of toxic byproducts leading to what is called “oxidative stress.” It also may lead to increased levels of iron in the mitochondria. When the excess iron reacts with oxygen, free radicals can be produced. Although free radicals are essential molecules in the body’s metabolism, they can also destroy cells and harm the body. Research continues on this subject.
Figure 3. Friedreich’s ataxia autosomal recessive inheritance pattern
Friedreich’s ataxia signs and symptoms
Symptoms typically begin between the ages of 5 and 15 years, although they sometimes appear in adulthood and on rare occasions as late as age 75. The first symptom to appear is usually gait ataxia, or difficulty walking. The ataxia gradually worsens and slowly spreads to the arms and the trunk. There is often loss of sensation in the extremities, which may spread to other parts of the body. Other features include loss of tendon reflexes, especially in the knees and ankles. Most people with Friedreich’s ataxia develop scoliosis (a curving of the spine to one side), which often requires surgical intervention for treatment.
Signs and symptoms of Friedreich’s ataxia can include:
- problems with balance and co-ordination, often causing wobbliness, clumsiness and frequent falls
- increasingly slurred, slow and unclear speech (dysarthria)
- increasing weakness in the legs – many people find walking difficult and need to use a wheelchair after around 10 to 20 years
- difficulty swallowing (dysphagia)
- abnormal curvature of the spine (scoliosis)
- total or partial vision loss and hearing loss
- diabetes
- thickening of the heart muscles (hypertrophic cardiomyopathy), which can cause chest pain, breathlessness and an irregular heartbeat
- loss of sensation in the hands and feet (peripheral neuropathy)
Dysarthria (slowness and slurring of speech) develops and can get progressively worse. Many individuals with later stages of Friedreich’s ataxia develop hearing and vision loss.
Other symptoms that may occur include chest pain, shortness of breath, and heart palpitations. These symptoms are the result of various forms of heart disease that often accompany Friedreich’s ataxia, such as hypertrophic cardiomyopathy (enlargement of the heart), myocardial fibrosis (formation of fiber-like material in the muscles of the heart), and cardiac failure. Heart rhythm abnormalities such as tachycardia (fast heart rate) and heart block (impaired conduction of cardiac impulses within the heart) are also common.
About 20 percent of people with Friedreich’s ataxia develop carbohydrate intolerance and 10 percent develop diabetes. Most individuals with Friedreich’s ataxia tire very easily and find that they require more rest and take a longer time to recover from common illnesses such as colds and flu.
The symptoms of Friedreich’s ataxia usually get gradually worse over many years, however the rate of progression varies from person to person. Generally, within 10 to 20 years after the appearance of the first symptoms, the person is confined to a wheelchair, and in later stages of the disease individuals may become completely incapacitated. People with the condition tend to have a shorter life expectancy than normal. Many people live until at least their 30s, and some can live into their 60s or beyond.
Friedreich’s ataxia can shorten life expectancy, and heart disease is the most common cause of death. However, some people with less severe features of Friedreich’s ataxia live into their sixties, seventies, or older.
Friedreich’s ataxia diagnosis
A diagnosis of Friedreich’s ataxia requires a careful clinical examination, which includes a medical history and a thorough physical exam, in particular looking for balance difficulty, loss of proprioception (joint sensation), absence of reflexes, and signs of neurological problems. Genetic testing now provides a conclusive diagnosis. Other tests that may aid in the diagnosis or management of the disorder include:
- electromyogram (EMG), which measures the electrical activity of muscle cells,
- nerve conduction studies, which measure the speed with which nerves transmit impulses,
- electrocardiogram (ECG), which gives a graphic presentation of the electrical activity or beat pattern of the heart,
- echocardiogram, which records the position and motion of the heart muscle,
- blood tests to check for elevated glucose levels and vitamin E levels, and
- magnetic resonance imaging (MRI) or computed tomography (CT) scans, tests which provide brain and spinal cord images that are useful for ruling out other neurological conditions.
Genetic testing is essential for proper clinical diagnosis, and can aid in prenatal diagnosis and determining a person’s carrier status. Genetic counselors can help explain how Friedreich’s ataxia is inherited. Psychological counseling and support groups for people with genetic diseases may also help affected individuals and their families cope with the disease.
A primary care physician can screen people for complications such as heart disease, diabetes and scoliosis, and can refer individuals to specialists such as cardiologists, physical therapists, and speech therapists to help deal with some of the other associated problems.
Support and information for families is also available through a number of private organizations. These groups can offer ways to network and communicate with others affected by Friedreich’s ataxia. They can also provide access to patient registries, clinical trials information, and other useful resources.
Friedreich’s ataxia treatment
As with many degenerative diseases of the nervous system, there is currently no cure or effective treatment for Friedreich’s ataxia. However, many of the symptoms and accompanying complications can be treated to help individuals maintain optimal functioning as long as possible. Doctors can prescribe treatments for diabetes, if present; some of the heart problems can be treated with medication as well. Orthopedic problems such as foot deformities and scoliosis can be corrected with braces or surgery. Physical therapy may prolong use of the arms and legs. Advances in understanding the genetics of Friedreich’s ataxia are leading to breakthroughs in treatment. Research has moved forward to the point where clinical trials of proposed treatments are presently occurring for Friedreich’s ataxia.
Ataxia telangiectasia
Ataxia-telangiectasia is a rare inherited disorder that affects the nervous system, immune system, and other body systems. Ataxia-telangiectasia is a rare, recessive genetic disorder of childhood that occurs in between 1 out of 40,000 and 1 out of 100,000 persons worldwide. The ailment is progressive. This disorder is characterized by progressive difficulty with coordinating movements (ataxia) beginning in early childhood, usually before age 5. Affected children typically develop difficulty walking, problems with balance and hand coordination, involuntary jerking movements (chorea), muscle twitches (myoclonus), and disturbances in nerve function (neuropathy). The movement problems typically cause people to require wheelchair assistance by adolescence and the disease is generally fatal to patients by the time they reach their twenties. People with this disorder also have slurred speech and trouble moving their eyes to look side-to-side (oculomotor apraxia). Small clusters of enlarged blood vessels called telangiectases, which occur in the eyes and on the surface of the skin, are also characteristic of this condition.
The primary features of classic ataxia-telangiectasia:
- Progressive gait and truncal ataxia with onset between ages one and four years
- Progressively slurred speech
- Oculomotor apraxia (inability to follow an object across visual fields)
- Choreoathetosis (writhing movements)
- Oculocutaneous telangiectasia (usually evident by age 6 years)
- Frequent infections (with accompanying evidence of serum and cellular immunodeficiencies)
- Hypersensitivity to ionizing radiation with increased susceptibility to cancer (usually leukemia or lymphoma)
Other features:
- Premature aging with strands of gray hair
- Endocrine abnormalities including insulin-resistant diabetes mellitus and premature ovarian failure (i.e., normal menarche followed by irregular menses and loss of ovarian function before age 40 years) 2
People with ataxia-telangiectasia often have a weakened immune system, and many develop recurrent sinus and respiratory infections and chronic lung infections. Patients usually have immune system abnormalities and are very sensitive to the effects of radiation treatments.
They also have an increased risk of developing cancer, particularly cancer of blood-forming cells (leukemia) and cancer of immune system cells (lymphoma). Affected individuals are very sensitive to the effects of radiation exposure, including medical x-rays. The life expectancy of people with ataxia-telangiectasia varies greatly, but affected individuals typically live into early adulthood.
In the United States, where recurrent infections typical of the disorder are usually controlled by antibiotics, patients are at high risk of developing and dying of cancer, particularly leukemias and lymphomas.
Affected individuals tend to have high amounts of a protein called alpha-fetoprotein (AFP) in their blood. The level of this protein is normally increased in the bloodstream of pregnant women, but it is unknown why individuals with ataxia-telangiectasia have elevated AFP or what effects it has in these individuals.
Ataxia-telangiectasia causes
Mutations in the ATM (for ataxia telangiectasia, mutated) gene cause ataxia-telangiectasia. The ATM (for ataxia telangiectasia, mutated) gene provides instructions for making a protein that helps control cell division and is involved in DNA repair. This protein plays an important role in the normal development and activity of several body systems, including the nervous system and immune system. The ATM protein assists cells in recognizing damaged or broken DNA strands and coordinates DNA repair by activating enzymes that fix the broken strands. Efficient repair of damaged DNA strands helps maintain the stability of the cell’s genetic information.
Mutations in the ATM gene reduce or eliminate the function of the ATM protein. Without this protein, cells become unstable and die. Cells in the part of the brain involved in coordinating movements (the cerebellum) are particularly affected by loss of the ATM protein. The loss of these brain cells causes some of the movement problems characteristic of ataxia-telangiectasia. Mutations in the ATM gene also prevent cells from responding correctly to DNA damage, which allows breaks in DNA strands to accumulate and can lead to the formation of cancerous tumors.
Ataxia-telangiectasia is inherited in an autosomal recessive pattern, which means both copies of the ATM gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition.
About 1 percent of the United States population carries one mutated copy and one normal copy of the ATM gene in each cell. These individuals are called carriers. Although ATM mutation carriers do not have ataxia-telangiectasia, they are more likely than people without an ATM mutation to develop cancer; female carriers are particularly at risk for developing breast cancer. Carriers of a mutation in the ATM gene also may have an increased risk of heart disease.
Figure 4. Ataxia telangiectasia autosomal recessive inheritance pattern
Ataxia-telangiectasia signs and symptoms
The first signs of the disease, which include delayed development of motor skills, poor balance, and slurred speech, usually occur during the first decade of life. Telangiectasias (tiny, red “spider” veins), which appear in the corners of the eyes or on the surface of the ears and cheeks, are characteristic of the disease, but are not always present and generally do not appear in the first years of life.
About 20-40 percent of those with ataxia-telangiectasia develop cancer, most frequently acute lymphocytic leukemia or lymphoma. Many individuals with ataxia-telangiectasia have a weakened immune system, making them susceptible to recurrent respiratory infections. Other features of the disease may include mild diabetes mellitus, premature graying of the hair, difficulty swallowing, and delayed physical and sexual development. Children with ataxia-telangiectasia usually have normal or above normal intelligence.
Signs and symptoms of ataxia-telangiectasia can include:
- difficulty walking – most children need to use a wheelchair by 10 years of age
- increasingly slurred, slow and unclear speech (dysarthria)
- difficulty swallowing (dysphagia)
- small spider-like clusters of red blood vessels in the corner of their eyes and on their cheeks (telangiectasias)
- very slow eye movements, which may mean the person has to move their head a lot to compensate for this
- a weakened immune system – children with ataxia-telangiectasia are more vulnerable to infections, particularly infections of the sinuses, lungs and airways, such as pneumonia
- an increased risk of cancer, particularly acute lymphoblastic leukaemia or lymphoma – up to 40% of people with ataxia-telangiectasia will develop cancer
The symptoms of ataxia-telangiectasia tend to get worse quite quickly. People with the condition usually live until the age of 19 to 25, although some may live into their 50s.
Neurologic
The most obvious characteristic of classic ataxia-telangiectasia is progressive cerebellar ataxia. Shortly after learning to walk, children with ataxia-telangiectasia begin to stagger. Although the neurologic status of some children appears to improve from age two to four years, ataxia subsequently progresses. (The transient improvement is probably attributable to the rapid learning curve of young children.)
The ataxia begins as purely truncal but within several years involves peripheral coordination as well. Writing and drawing are affected by age five years.
By age ten years, most children become confined to a wheelchair.
Slurred speech and oculomotor apraxia are noted early. Both horizontal and vertical saccadic eye movements are affected 3. Drooling is common.
Choreoathetosis is found in almost all individuals with ataxia-telangiectasia. Myoclonic jerking and intention tremors are present in about 25%.
All teenagers with classic ataxia-telangiectasia need help with dressing, eating, washing, and toileting.
Muscle strength is normal at first but wanes with disuse, especially in the legs. Contractures in the fingers and toes are common in older individuals. Deep tendon reflexes are decreased or absent in older individuals; plantar reflexes are upgoing or absent
Intelligence is typically normal; however, learning difficulties are common. Slow motor and verbal responses make it difficult for individuals to complete ‘timed’ IQ tests. Many American and British individuals with classic ataxia-telangiectasia have finished high school with good grades; some have finished college or university, often with the assistance of aides and attentive parents and sibs.
Dystonia and adult-onset spinal muscular atrophy have also been observed.
Immunodeficiency, present in 60%-80% of individuals with classic ataxia-telangiectasia, is variable and does not correlate well with the frequency, severity, or spectrum of infections. The most consistent immunodeficiency reported in classic ataxia-telangiectasia is poor antibody response to pneumococcal polysaccharide vaccines 4. Serum concentration of the immunoglobulins IgA, IgE, and IgG2 is often reduced. NK lymphocyte levels are occasionally elevated, most notably in individuals from Costa Rica 5.
The immunodeficiency is not progressive, and some evidence suggests that T-cell lymphopenia may normalize after age 20 years 6.
Of note, immune status was more seriously impaired in 80 individuals with ataxia-telangiectasia and two null ATM alleles: both T- and B-cell lymphopenia and more frequent recurrent sinopulmonary infections were observed [Staples et al 2008]. Children with lymphopenia tend to have the most deleterious pathogenic variants, lowest (enzymatic) ATM kinase levels, most severe phenotypes, most frequent sinopulmonary infections, and poorest prognosis [Verhagen et al 2012].
Infection. In contrast to the spectrum of infection observed in most immunodeficiency disorders, the spectrum of infection in classic ataxia-telangiectasia does not include opportunistic infections. The frequency and severity of infections correlate more with ATM kinase levels and general nutritional status than with immune status.
Some individuals with classic ataxia-telangiectasia develop chronic bronchiectasis.
While individuals with frequent infections may occasionally benefit from prophylactic antibiotics and/or intravenous immunoglobulin (IVIG) replacement therapy 4, longevity has increased substantially even in those not receiving IVIG.
Pulmonary. In older individuals, pulmonary failure, with or without identifiable infections, is a major cause of failing health and death 7. Life-threatening lymphocytic infiltration of the lung has been reported 8.
Other findings. Liver enzyme levels are often elevated in ataxia-telangiectasia, without apparent liver pathology. (Thus, liver biopsy is usually not revealing.)
Cancer. The risk for malignancy in individuals with classic ataxia-telangiectasia is 38%.
Leukemia and lymphoma account for about 85% of malignancies. Younger children tend to have acute lymphocytic leukemia (ALL) of T-cell origin and older children are likely to have an aggressive T-cell leukemia. Lymphomas are usually B-cell types.
As individuals with classic ataxia-telangiectasia are living longer, other cancers and tumors including ovarian cancer, breast cancer, gastric cancer, melanoma, leiomyomas, and sarcomas have also been observed.
Ataxia-telangiectasia prognosis
Ataxia-telangiectasia is presently incurable and unrelenting. If they are lucky enough not to develop cancer, most ataxia-telangiectasia children are dependent on wheelchairs by the age of ten, not because their muscles are too weak, but because they cannot control them. Later, ataxia-telangiectasia patients usually die from respiratory failure or cancer by their early or mid-twenties. A few ataxia-telangiectasia patients live into their forties, but they are extremely rare.
Life expectancy. Over the past 25 years, for reasons that are unclear, the life expectancy of individuals with ataxia-telangiectasia has increased considerably. Most affected individuals now live beyond age 25 years. Some have survived into their 50s 9.
Ataxia-telangiectasia diagnosis
Ataxia-telangiectasia should be suspected in children who have the following clinical, MRI (magnetic resonance imaging), and preliminary laboratory findings.
Clinical findings. Progressive cerebellar dysfunction between ages one and four years manifests as:
- Gait and truncal ataxia;
- Head tilting;
- Slurred speech;
- Oculomotor apraxia and abnormal ocular saccades. “If oculomotor apraxia cannot be clearly documented in a cooperative patient, the diagnosis of A-T should be viewed with suspicion”.
Note: The diagnosis of ataxia-telangiectasia is most difficult in very young children: they do not yet exhibit all the characteristic features of ataxia-telangiectasia and are typically unable to cooperate during neurologic examination.
MRI (magnetic resonance imaging). The classic cerebellar findings are atrophy of the frontal and posterior vermis and both hemispheres. Note: Although a small cerebellum is not always apparent on MRI in young children, diffusion-weighted MRI allowed quantitation of cerebellar corticomotor pathway pathology in children as young as age three years, suggesting that this imaging may be useful in early confirmation of the diagnosis of ataxia-telangiectasia when the necessary equipment and expertise are available 10.
Preliminary laboratory findings
Newborn screening for severe combined immunodeficiency identifies reduced T-cell receptor excision circle (TREC) levels. This method of newborn screening most likely identifies the estimated 50% of children with ataxia-telangiectasia who have lymphopenia; however, it may be less sensitive in older children with ataxia-telangiectasia (in whom T cell lymphopenias are less severe) 11.
Serum concentration of alpha-fetoprotein (AFP) is elevated above10 ng/mL in about 95% of individuals with ataxia-telangiectasia.
Note: (1) Serum AFP concentration may remain above normal in unaffected children until age 24 months. (2) Persistent elevation of AFP does not necessarily indicate ongoing cerebellar damage or correlate with prognosis.
Chromosome analysis. A 7;14 chromosome translocation is identified in 5%-15% of cells in routine chromosome studies of peripheral blood of individuals with ataxia-telangiectasia. The break points are commonly at 14q11 (the T-cell receptor-alpha locus) and at 14q32 (the B- cell immunoglobulin heavy chain receptor [IGH] locus).
Establishing the Diagnosis
The diagnosis of ataxia-telangiectasia is established in a proband either by molecular genetic testing to document the presence of biallelic (homozygous or compound heterozygous) ATM pathogenic variants or (when available) by immunoblotting to test for absent or reduced ATM protein.
Molecular genetic testing approaches can include single-gene testing or use of a multi-gene panel.
- Single-gene testing. Sequence analysis of ATM is performed first, followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found.
- Targeted analysis for the following ATM pathogenic variants in specific populations can be performed first when appropriate:
- Amish: c.1564_1565delAG
- North African Jewish: c.103C>T (p.Arg35Ter)
- Sardinian: c.3894dupT
- A multi-gene panel that includes ATM and other genes of interest, such as PP2A, may also be considered. Note: The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and over time.
Immunoblotting for ATM protein in a lymphoblastoid cell line is more than 95% sensitive and more than 98% specific for diagnosing ataxia-telangiectasia 12. This testing may be performed to help interpret sequence variants of uncertain significance or confirm a diagnosis when only one ATM pathogenic variant is identified.
Of individuals with ataxia-telangiectasia:
- About 90% have no detectable ATM protein (i.e., <15% of control levels);
- About 10% have trace amounts to 15% of control levels of ATM protein;
- About 1% have a near-normal amount of ATM protein that lacks ATM serine/threonine kinase activity (so-called “kinase-dead” protein) 13.
Note: The presence of more than 15% ATM protein suggests one of the following:
- Another diagnosis
- An ATM pathogenic missense variant
- A leaky splicing ATM variant
- An ATM pathogenic variant resulting in “kinase-dead” protein
- Possibly, a pathogenic variant in a gene encoding an ancillary ATM-activating phosphatase like PP2A. However, to the authors’ knowledge, PP2A deficiency results mainly from somatic pathogenic PP2A variants in tumor cells or PP2A inhibitors. To date, a person with PP2A deficiency has not been described.
Ataxia-telangiectasia treatment
There is no cure for ataxia-telangiectasia at this time and there is currently no way to slow the progression of the disease. At this time, treatments are directed only toward partially alleviating some symptoms as they appear. Because ataxia-telangiectasia is a rare, “orphan” disease, very little research data is available on pharmaceutical therapies that may aid these children. Physical, occupational and speech therapy are used to help maintain flexibility, gamma-globulin injections help supplement the immune systems of ataxia-telangiectasia patients, and high-dose vitamin regimes are being undertaken with some moderate results.
The cloning and sequencing of the gene named ATM (for ataxia telangiectasia, mutated) has opened several avenues of research to develop better treatment, including: (1) gene therapy; (2) the design of drugs to correct the function of the altered protein; and (3) direct replacement of the functional protein. Physical, occupational, and speech therapy are used to help maintain flexibility, gamma-globulin injections help supplement the immune systems of ataxia-telangiectasia patients, and high-dose vitamin regimens are being researched with some moderate results.
Research shows that a protein kinase called ATM reacts to DNA damage by chemically modifying and triggering accumulation of a molecular or tumor suppressor called p53. This tumor suppressor is defective in about half of all human cancers and is the master control switch for a process that normally prevents cells from dividing. In ataxia-telangiectasia patients, the ATM protein is missing or defective. This delays the accumulation of p53, allowing cells to replicate without repair of their DNA and thereby increasing the risk of cancer.
Neurologic. Supportive therapy can minimize drooling, choreoathetosis, myoclonus/tremor, and ataxia. However, individual responses to specific medications (e.g., amantadine and 4-aminopyridine) and to treatments used for myoclonus vary 14. Thus, it is recommended that treatment options be discussed with an experienced neurologist.
Early and continued physical therapy can minimize the risk for contractures (which appear in almost all individuals with time and often lead to other problems such as pressure sores and pain) and scoliosis (which can, for example, be the consequence of prolonged sitting in a wheelchair – particularly the tendency to lean on the same elbow).
Although steroids are reported to temporarily improve the neurologic symptoms of ataxia-telangiectasia in children, the symptoms reappear within days of their discontinuation 15.
Immunodeficiency. IVIG replacement therapy should be considered for individuals with frequent and severe infections and very low IgG levels 4.
Pulmonary. The European Respiratory Society 16 has prepared extensive guidelines for the multidisciplinary respiratory management of ataxia-telangiectasia, emphasizing the need for monitoring of immune function, recurrent infection, pulmonary function, swallowing, nutrition, and scoliosis, all of which can contribute to increased respiratory morbidity and mortality in persons with ataxia-telangiectasia.
Cancer. Because cells from individuals with ataxia-telangiectasia are 30% more sensitive to ionizing radiation than cells from controls, conventional doses of ionizing radiation are potentially lethal in individuals with ataxia-telangiectasia. Thus, the use of radiotherapy and some radiomimetic chemotherapeutic agents should be administered carefully and monitored closely 17.
Doses of some chemotherapeutic agents are often reduced by 25%-50% and longer recovery periods between treatments are considered to allow for the slower DNA repair that occurs in ataxia-telangiectasia 17.
Prevention of Secondary Complications
Pulmonary and nutritional complications of dysphagia are common. Often, gastrostomy tube feedings are recommended to manage these comorbidities. Children with disorders with predictable progression (like ataxia-telangiectasia) and impaired swallowing may benefit from early rather than late placement of a feeding tube 18.
Anesthesia carries unique potential risks in persons with ataxia-telangiectasia because of impaired coordination of swallowing, increased risk of aspiration, reduced respiratory capacity, and propensity to infections 19. In a recent review, 24% of patients required supplemental oxygen (maximum duration 24 hours) post anesthesia; mild postoperative hypothermia was also relatively common 20.
Surveillance
The European Respiratory Society 16 has prepared extensive guidelines for the multidisciplinary respiratory management of ataxia-telangiectasia, emphasizing the need for monitoring of immune function, recurrent infection, pulmonary function, swallowing, nutrition, and scoliosis, all of which could contribute to increased respiratory morbidity and mortality in ataxia-telangiectasia.
Parents should be counseled to monitor for – and report to a physician – the early warning signs of malignancy (which can occur at any age) including weight loss, bruising, and localized pain or swelling. Periodic complete blood counts are warranted.
Immune status needs to be monitored if severe recurrent infections occur or immunomodulatory therapy is in progress.
Spinocerebellar ataxia
Spinocerebellar ataxias (also called SCA’s) are a group of hereditary ataxias that often don’t begin until adulthood, affecting people from the age of 25 up to 80, depending on the type of spinocerebellar ataxia. Occasionally, some types of spinocerebellar ataxia begin in childhood.
The Spinocerebellar ataxias are caused by mutations in different genes. For example, spinocerebellar ataxia 1 is linked to a default in the gene SCA1.
Through research, more than 100 types of spinocerebellar ataxias have been discovered since 1965. One of the common genetic defects is a an expansion of a CAG triplet repeat. In this way, it is similar to fragile-X syndrome, Huntington disease and myotonic dystrophy, all of which exhibit a triplet repeat expansion of a gene. In the case of spinocerebellar ataxia I, the gene is SCA1, found on chromosome 6. The protein product of the gene – called ataxin-1 – varies in size, depending on the size of the CAG triplet repeat.
Persons with spinocerebellar ataxia experience a degeneration of the spinal cord and the cerebellum. The cerebellum is concerned with coordination of movements, so the “wasting away” of this critical control center results in a loss of muscle coordination. Atrophy in the spine can bring spasticity.
The symptoms vary depending on the type of spinocerebellar ataxia. They can include:
- problems with balance and co-ordination – many people find walking difficult and need to use a wheelchair after a few years
- increasingly slurred, slow and unclear speech (dysarthria)
- difficulty swallowing (dysphagia)
- muscle stiffness and cramps
- loss of sensation in the hands and feet (peripheral neuropathy)
- memory loss and difficulties with spoken language
- slow eye movement, which means people have to move their head to compensate
- reduced bladder control (urinary urgency or incontinence)
Episodic ataxia
Episodic ataxia is characterized as bouts or attacks of ataxia symptoms. The episodic ataxias are a relatively rare group of conditions which, as their name suggests, tend to affect people in bouts or attacks poor coordination and balance (ataxia). During these episodes, many people also experience dizziness (vertigo), nausea and vomiting, migraine headaches, blurred or double vision, slurred speech, and ringing in the ears (tinnitus). Seizures, muscle weakness, and paralysis affecting one side of the body (hemiplegia) may also occur during attacks. Additionally, some affected individuals have a muscle abnormality called myokymia during or between episodes. This abnormality can cause muscle cramping, stiffness, and continuous, fine muscle twitching that appears as rippling under the skin.
During an episode, someone with episodic ataxia may experience:
- problems with balance and co-ordination
- slurred, slow and unclear speech (dysarthria)
- muscle spasms
- involuntary eye movements (nystagmus)
- vertigo, migraines and tinnitus
Episodic ataxia usually first develops during the teenage years to adulthood. The episodes can last from several minutes to hours and are usually the result of certain triggers, such as emotional stress, caffeine, alcohol, certain medications, physical activity, illness, sudden movement, stress or exercise. The frequency of attacks ranges from several per day to one or two per year. Between episodes, some affected individuals continue to experience ataxia, which may worsen over time, as well as involuntary eye movements called nystagmus.
The symptoms of episodic ataxia may disappear as a person gets older, although sometimes the condition gets gradually worse over time. Medication can often help control attacks, and life expectancy is usually normal.
Researchers have identified at least seven types of episodic ataxia, designated type 1 through type 7. The types are distinguished by their pattern of signs and symptoms, age of onset, length of attacks, and, when known, genetic cause.
Episodic ataxia is uncommon, affecting less than 1 in 100,000 people. Only types 1 and 2 have been identified in more than one family.
Episodic ataxia type 2 is most common form of the condition and the most well understood, and they are called episodic ataxia type 1 and 2. Both of these occur in families and are inherited in what is known as an autosomal dominant manner. This means that an affected individual has a 50% chance of passing the gene on to their children.
Figure 5. Episodic ataxia autosomal dominant inheritance pattern
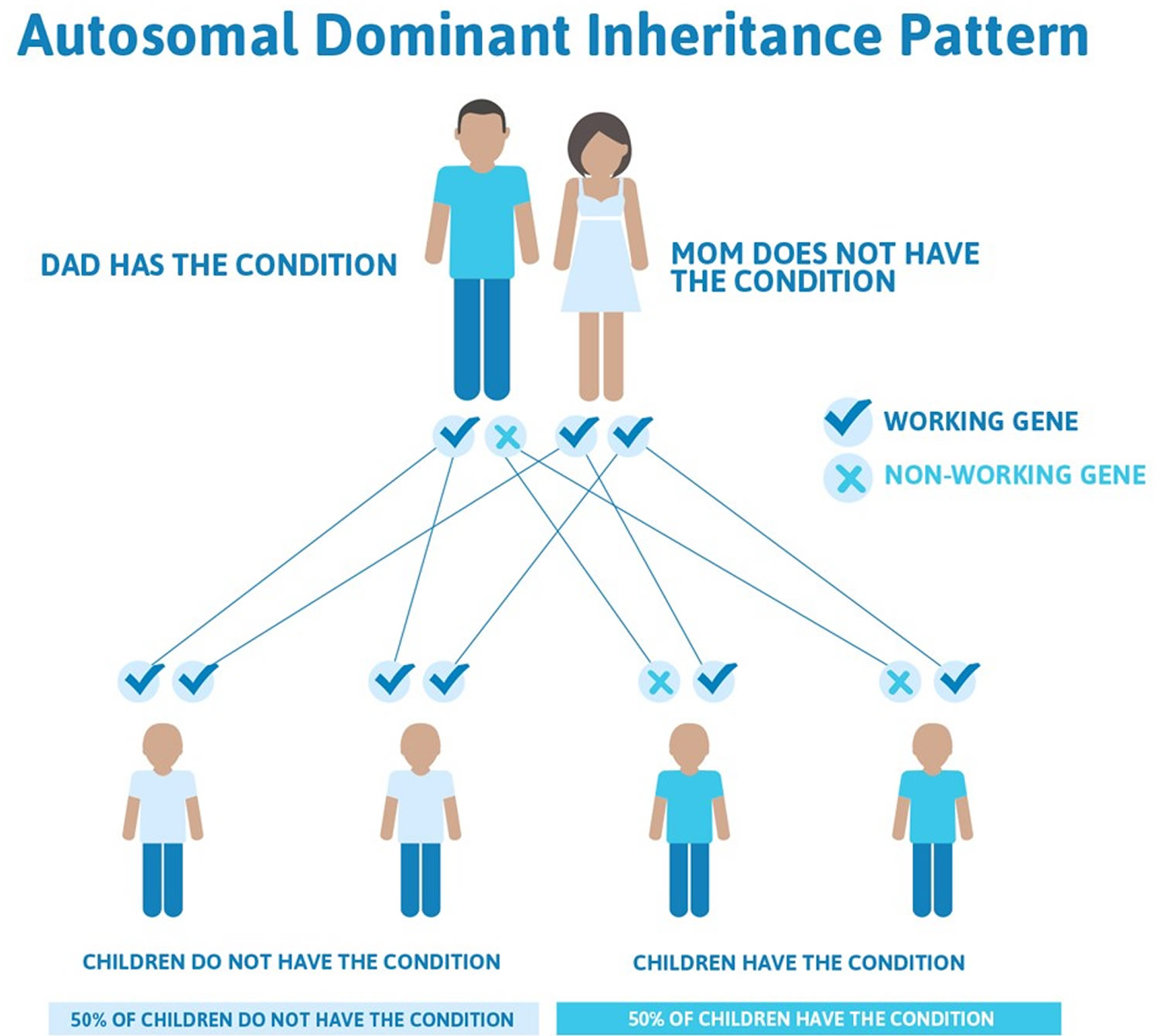
Episodic ataxia can be caused by mutations in several genes that play important roles in the nervous system. Three of these genes, KCNA1, CACNA1A, and CACNB4, provide instructions for making proteins that are involved in the transport of charged atoms (ions) across cell membranes. The movement of these ions is critical for normal signaling between nerve cells (neurons) in the brain and other parts of the nervous system. Mutations in the KCNA1, CACNA1A, and CACNB4 genes are responsible for episodic ataxia types 1, 2, and 5, respectively.
Mutations in the SLC1A3 gene have been found to cause episodic ataxia type 6. This gene provides instructions for making a protein that transports a brain chemical (neurotransmitter) called glutamate. Neurotransmitters, including glutamate, allow neurons to communicate by relaying chemical signals from one neuron to another.
Researchers believe that mutations in the KCNA1, CACNA1A, CACNB4, and SLC1A3 genes alter the transport of ions and glutamate in the brain, which causes certain neurons to become overexcited and disrupts normal communication between these cells. Although changes in chemical signaling in the brain underlie the recurrent attacks seen in people with episodic ataxia, it is unclear how mutations in these genes cause the specific features of the disorder.
The genetic causes of episodic ataxia types 3, 4, and 7 have not been identified. Researchers are looking for additional genes that can cause episodic ataxia.
Episodic ataxia type 2
It involves people having episodes of ataxia that last hours to days. Exertion and stress often trigger the episodes. Common symptoms are vertigo, nausea and vomiting, nystagmus (involuntary eye movements) and some people have migraine. It is a rare disorder, representing less than 1% of the inherited ataxia syndromes. Acetozolamide often dramatically helps with the symptoms during the episodes of ataxia.
Not all people respond well to acetozolamide and some people may be allergic to it. If acetazolamide is ineffective or causes side effects there are other drugs that can be tried. As stress often triggers attacks, stress management techniques (e.g. meditation) can be helpful in controlling symptoms. Alcohol and caffeine should be avoided, and regular but modest exercise should be encouraged. In some cases patients with this form of ataxia develop a slowly progressive ataxia, this most frequently coincides with the attacks lessening in frequency. Episodic ataxia type 2 is caused by mutations in a calcium channel found in cells mainly in the cerebellum. Calcium channels normally are important for maintaining the correct amount of calcium in cells which is important for the cells to perform their normal functions. It is important to see a neurologist, who will monitor the condition, on a regular basis.
Episodic ataxia type 1
Episodic ataxia type 1 is very rare – in a relatively recent publication only a few families had been identified. It is characterized by short attacks (seconds or minutes) of incoordination and dysarthria (speech problem) that generally last minutes. Myokymia (twitching of muscles) is present in most people. It can be twitching of muscles in hand, fingers, sometimes the tongue. Myokymia is frequently accentuated during attacks. The severity of the symptoms varies greatly between individuals, both in terms of the severity and frequency of attacks and also the amount of myokymia. Most people do not have symptoms between attacks but others may have slight ataxia and tremor between attacks. Attacks can occur spontaneously or they can be triggered by rapid sudden movements, or if one is startled. Anxiety, and fatigue also increase the susceptibility to an attack. Basically, episodic ataxia type 1 is a non-progressive disorder, but some elderly people show slight permanent ataxia and tremor. Once a diagnosis is confirmed there are a number of drugs that can be tried to help with the symptoms. Episodic ataxia type 1 is caused by mutations in potassium channels in cells that may lead to problems with transmitting nervous impulses. It is important to see a neurologist, who will monitor the condition, on a regular basis.
Gluten ataxia
Gluten ataxia is caused by a sensitivity to the protein gluten found in wheat, barley and rye products. Gluten ataxia is one of the most common forms of sporadic idiopathic ataxia (where ataxia is found in people with no family history of ataxia and no known cause).
When gluten is eaten by a person with gluten sensitivity their own body’s immune system produces antibodies. These antibodies can attack the balance center of the brain, resulting in ataxia symptoms. Sometimes the peripheral nerves located outside the spinal cord, which supply muscles and are also responsible for sensation, can also be affected. This can lead to a condition called peripheral neuropathy. It can result in numbness, tingling or pain in the hands and feet. However, symptoms will vary in every person.
If gluten sensitivity is the cause of the ataxia then the only treatment for this condition is a strict gluten free diet. Gluten ataxia is diagnosed by checking your blood for the presence of antibodies.
It can take up to six months and even as long as a year for the antibodies to completely disappear from your body and so you must stick to the gluten free diet at all times. If you do consume gluten at any time, further antibodies will be produced, resulting in more damage to the balance center.
Other conditions that also require a strict gluten free diet are celiac disease, a condition that affects the gut and dermatitis herpetiformis, which is a skin condition. Sometimes all these conditions can be present in the same person.
Gluten ataxia diagnosis
If the blood tests show that you have antibodies to gluten it is usual for your doctor to refer you for a test called a gastroscopy and duodenal biopsy. This is a test that allows the gut doctor to look directly at the lining of the esophagus (gullet), the stomach and around the first bend of the small intestine – the duodenum.
The gastroscope is a long flexible tube (thinner than your little finger) with a bright light on the end. It is important that you are not on a gluten free diet when you have this done.
Using the gastroscope your doctor can see a picture of the lining of your stomach on a monitor. A biopsy is taken from the first part of the small bowel and this is examined for any evidence of inflammation (seen in celiac disease). Not everyone who has gluten ataxia will also have inflammation of the bowel.
When your doctor have your results he/she will let you know if a gluten free diet is recommended and he/she will refer you to be seen by a dietitian. Once you have seen the dietitian you can start the gluten free diet. It is very important that you are 100% strict with the diet.
Will my ataxia get better on the gluten free diet?
The aim of the diet is to stabilize the disease process and prevent further deterioration of the ataxia symptoms. Some people find that their symptoms improve whilst for others this is not quite so obvious. By repeating the blood tests and checking that the antibodies have disappeared along with the brain scan results your doctor is in a better position to show that the ataxia is stabilizing.
Gluten free is the only treatment for this condition. Most people adjust over time and there are organizations that can help along the way. If you are struggling or have any questions see your doctor or dietician.
Acute cerebellar ataxia
Acute cerebellar ataxia is sudden, uncoordinated muscle movement due to disease or injury to the cerebellum. This is the area in the brain that controls muscle movement.
Acute cerebellar ataxia causes
Acute cerebellar ataxia in children, particularly younger than age 3, may occur several weeks after an illness caused by a virus.
Viral infections that may cause this include chickenpox, Coxsackie disease, Epstein-Barr, and echovirus.
Other causes of acute cerebellar ataxia include:
- Abscess of the cerebellum
- Alcohol, medicines, and insecticides
- Bleeding into the cerebellum
- Multiple sclerosis
- Strokes of the cerebellum
- Vaccination
Outlook (Prognosis) for acute cerebellar ataxia
People whose condition was caused by a recent viral infection should make a full recovery without treatment in a few months. Strokes, bleeding, or infections may cause permanent symptoms.
Acute cerebellar ataxia possible complications
In rare cases, movement or behavioral disorders may persist.
Acute cerebellar ataxia symptoms
Ataxia may affect movement of the middle part of the body from the neck to the hip area (the trunk) or the arms and legs (limbs).
When the person is sitting, the body may move side-to-side, back-to-front, or both. Then the body quickly moves back to an upright position.
When a person with ataxia of the arms reaches for an object, the hand may sway back and forth.
Common symptoms of ataxia include:
- Clumsy speech pattern (dysarthria)
- Repetitive eye movements (nystagmus)
- Uncoordinated eye movements
- Walking problems (unsteady gait)
Acute cerebellar ataxia diagnosis
The health care provider will ask if the person has recently been sick and will try to rule out any other causes of the problem. Brain and nervous system examination will be done to identify the areas of the nervous system that are most affected.
The following tests may be ordered:
- CT scan of the head
- MRI scan of the head
- Spinal tap
Acute cerebellar ataxia treatment
Treatment depends on the cause:
- If the acute cerebellar ataxia is due to bleeding, surgery may be needed.
- For a stroke, medicine to thin the blood can be given.
- Infections may need to be treated with antibiotics or antivirals.
- Steroids may be needed for swelling (inflammation) of the cerebellum (such as from multiple sclerosis).
- Cerebellar ataxia caused by a recent viral infection may not need treatment.
Optic ataxia
Optic ataxia was first described by Rudolph Bálint in 1909 as a neurological symptom resulting in gross mis-reaching to targets in the peripheral visual field 21. Initially, optic ataxia was only seen as part of a triad of symptoms, which forms the basis of the Bálint-Holmes syndrome. Two additional symptoms are usually observed in a Bálint syndrome, namely oculomotor apraxia and simultanagnosia 22. Garcin et al. 23 were the first to report that optic ataxia can also appear as a distinct disorder in isolation. The defining characteristic of optic ataxia is the contrast between the occurrence of spatial errors in reaching movements to targets in the visual periphery and unimpaired movements to targets in the central visual field. This feature together with hand- and field-effects in unilateral cases supported the conclusion that optic ataxia represents a visuomotor coordination deficit that could neither be explained by a motor deficit nor by a sensory deficit alone 24. In contrast to their visuomotor impairments, patients with optic ataxia seem to be able to detect and localize targets in their complete surroundings and reliably perceive shape, size and orientation of targets in the peripheral visual field 25. Performing a lesion analysis with sixteen optic ataxia patients, Karnath and Perenin 26 found the lateral and medial parieto-occipital junction to be specifically affected in optic ataxia.
Other types of ataxia
There are also a number of other types of ataxia that tend to have similar symptoms to those mentioned above. These include:
- Idiopathic ataxia is the name we give a type of ataxia for which the cause is not yet known. Almost 50% of people with the condition have idiopathic ataxia, showing just how much more work there is to do.
- Acquired ataxia – this can affect people of any age and usually develops very quickly over the course of a few days, or sometimes hours; it may improve over time, stay the same or get slowly worse. Ataxia can be acquired during someone’s lifetime, and caused by something other than a genetic malfunction. Strokes, gluten intolereance, alcohol, vitamin-E deficiency and trauma to the head can all cause ataxia. Some of these types can be reversed, depending on the cause; a vitamin-E deficiency can be reduced by attention given to diet, for example, that will reduce ataxic symptoms and in some cases, eliminate them all together.
- Idiopathic late-onset cerebellar ataxia – this usually begins at around 50 years of age and gets slowly worse over time
- Ataxia with vitamin E deficiency – a similar condition to Friedreich’s ataxia caused by problems with the body’s ability to use vitamin E in the diet; it’s often possible to control the symptoms with vitamin E supplements.
Ataxia causes
Ataxia is usually caused by damage, degeneration or loss of nerve cells to a part of the brain known as the cerebellum, but it can also be caused by damage to the spinal cord or other nerves. Your cerebellum comprises two pingpong-ball-sized portions of folded tissue situated at the base of your brain near your brainstem.
The spinal cord is a long bundle of nerves that runs down the spine and connects the brain to all other parts of the body.
The right side of your cerebellum controls coordination on the right side of your body; the left side of your cerebellum controls coordination on the left.
The cerebellum is located at the base of the brain and is responsible for controlling:
- walking and sitting balance
- limb co-ordination
- eye movements
- speech
Damage can occur as a result of injury or illness (acquired ataxia) or because the cerebellum or spinal cord degenerates because of an inherited faulty gene (hereditary ataxia).
Sometimes there’s no clear reason why the cerebellum and spinal cord become damaged. This is the case for people with idiopathic late-onset cerebellar ataxia (ILOA).
Diseases that damage the spinal cord and peripheral nerves that connect your cerebellum to your muscles also can cause ataxia. Ataxia causes include:
- Head trauma. Damage to your brain or spinal cord from a blow to your head, such as might occur in a car accident can cause acute cerebellar ataxia, which comes on suddenly.
- Stroke. When the blood supply to a part of your brain is interrupted or severely reduced, depriving brain tissue of oxygen and nutrients, brain cells die.
- Cerebral palsy. This is a general term for a group of disorders caused by damage to a child’s brain during early development — before, during or shortly after birth — that affects the child’s ability to coordinate body movements.
- Autoimmune diseases. Multiple sclerosis, sarcoidosis, celiac disease and other autoimmune conditions can cause ataxia.
- Infections. Ataxia can be an uncommon complication of chickenpox and other viral infections. It might appear in the healing stages of the infection and last for days or weeks. Normally, the ataxia resolves over time.
- Paraneoplastic syndromes. These are rare, degenerative disorders triggered by your immune system’s response to a cancerous tumor (neoplasm), most commonly from lung, ovarian, breast or lymphatic cancer. Ataxia can appear months or years before the cancer is diagnosed.
- Tumor. A growth on the brain, cancerous (malignant) or noncancerous (benign), can damage the cerebellum.
- Toxic reaction. Ataxia is a potential side effect of certain medications, especially barbiturates, such as phenobarbital; sedatives, such as benzodiazepines; and some types of chemotherapy. These are important to identify because the effects are often reversible. Also, some medications you take can cause problems as you age, so you might need to reduce your dose or discontinue the medication. Alcohol and drug intoxication; heavy metal poisoning, such as from lead or mercury; and solvent poisoning, such as from paint thinner, also can cause ataxia.
- Vitamin E, vitamin B-12 or thiamine deficiency. Not getting enough of these nutrients,because of the inability to absorb enough, alcohol abuse or other reasons, can lead to ataxia.
For some adults who develop sporadic ataxia, no specific cause can be found. Sporadic ataxia can take a number of forms, including multiple system atrophy, a progressive, degenerative disorder.
Hereditary ataxias
Some types of ataxia and some conditions that cause ataxia are hereditary. If you have one of these conditions, you were born with a defect in a certain gene that makes abnormal proteins.
The abnormal proteins hamper the function of nerve cells, primarily in your cerebellum and spinal cord, and cause them to degenerate. As the disease progresses, coordination problems worsen.
You can inherit a genetic ataxia from either a dominant gene from one parent (autosomal dominant disorder) or a recessive gene from each parent (autosomal recessive disorder). In the latter case, it’s possible neither parent has the disorder (silent mutation), so there might be no obvious family history.
Different gene defects cause different types of ataxia, most of which are progressive. Each type causes poor coordination, but each has specific signs and symptoms.
Autosomal dominant ataxias
These include:
- Spinocerebellar ataxias. Researchers have labeled more than 35 autosomal dominant ataxia genes, and the number continues to grow. Cerebellar ataxia and cerebellar degeneration are common to all types, but other signs and symptoms, as well as age of onset, differ depending on the specific gene mutation.
- Episodic ataxia (EA). There are seven recognized types of ataxia that are episodic rather than progressive — EA1 through EA7. EA1 and EA2 are the most common. EA1 involves brief ataxic episodes that may last seconds or minutes. The episodes are triggered by stress, being startled or sudden movement, and often are associated with muscle twitching. EA2 involves longer episodes, usually lasting from 30 minutes to six hours, that also are triggered by stress. You might have dizziness (vertigo), fatigue and muscle weakness during your episodes. In some cases, symptoms resolve in later life.Episodic ataxia doesn’t shorten life span, and symptoms might respond to medication.
Autosomal recessive ataxias
These include:
- Friedreich’s ataxia. This common hereditary ataxia involves damage to your cerebellum, spinal cord and peripheral nerves. Peripheral nerves carry signals from your brain and spinal cord to your muscles. In most cases, signs and symptoms appear well before age 25.
The rate of disease progression varies. The first indication generally is difficulty walking (gait ataxia). The condition typically progresses to the arms and trunk. Muscles weaken and waste away over time, causing deformities, particularly in your feet, lower legs and hands.
Other signs and symptoms that might develop as the disease progresses include slow, slurred speech (dysarthria); fatigue; rapid, involuntary eye movements (nystagmus); spinal curvature (scoliosis); hearing loss; and heart disease, including heart enlargement (cardiomyopathy) and heart failure. Early treatment of heart problems can improve quality of life and survival.
- Ataxia-telangiectasia. This rare, progressive childhood disease causes degeneration in the brain and other body systems. The disease also causes immune system breakdown (immunodeficiency disease), which increases susceptibility to other diseases, including infections and tumors. It affects various organs.
Telangiectasias are tiny red “spider” veins that might appear in the corners of your child’s eyes or on the ears and cheeks. Delayed motor skill development, poor balance and slurred speech are typically the first indications of the disease. Recurrent sinus and respiratory infections are common.
Children with ataxia-telangiectasia are at high risk of developing cancer, particularly leukemia or lymphoma. Most people with the disease need a wheelchair by their teens and die before age 30, usually of cancer or lung (pulmonary) disease.
- Congenital cerebellar ataxia. This type of ataxia results from damage to the cerebellum that’s present at birth.
- Wilson’s disease. People with this condition accumulate copper in their brains, livers and other organs, which can cause neurological problems, including ataxia. Early identification of this disorder can lead to treatment that will slow progression.
Ataxia symptoms
Ataxia can develop over time or come on suddenly. A sign of a number of neurological disorders, ataxia can cause:
- Poor coordination
- Unsteady walk and a tendency to stumble
- Difficulty with fine motor tasks, such as eating, writing or buttoning a shirt
- Change in speech
- Involuntary back-and-forth eye movements (nystagmus)
- Difficulty swallowing.
Ataxia diagnosis
If you have ataxia, your doctor will look for a treatable cause. Besides conducting a physical exam and a neurological exam, including checking your memory and concentration, vision, hearing, balance, coordination, and reflexes, your doctor might request laboratory tests, including:
- Imaging studies. A CT scan or MRI of your brain might help determine potential causes. An MRI can sometimes show shrinkage of the cerebellum and other brain structures in people with ataxia. It may also show other treatable findings, such as a blood clot or benign tumor, that could be pressing on your cerebellum.
- Lumbar puncture (spinal tap). A needle is inserted into your lower back (lumbar region) between two lumbar bones (vertebrae) to remove a sample of cerebrospinal fluid. The fluid, which surrounds and protects your brain and spinal cord, is sent to a laboratory for testing.
- Genetic testing. Your doctor might recommend genetic testing to determine whether you or your child has the gene mutation that causes one of the hereditary ataxic conditions. Gene tests are available for many but not all of the hereditary ataxias.
Ataxia treatment
The treatment for ataxia can vary depending on exact what type of ataxia you have.
It’s sometimes possible to treat the underlying cause of the condition so it improves or stops getting worse, but in most cases this isn’t possible and you’ll have treatment to relieve your symptoms.
Your treatment plan
You’ll usually be cared for by a group of healthcare professionals called a multidisciplinary team, who will work with you to come up with a care plan. Your multidisciplinary team will probably include a neurologist, physiotherapist and specialist nurse, among others.
Your care plan will play an important part in the management of your condition. Your physical, social and psychological needs will be assessed, and the plan will outline how these needs can best be met. The plan will also address any future needs you may have.
You’ll normally have regular appointments with your multidisciplinary team or doctor to review your progress. In some cases, you may be seen in a specialist ataxia centre.
Treating the symptoms
Treatments for the various symptoms of ataxia are discussed in the following sections, although you may not experience all of the problems described.
Speech and language therapy
A speech and language therapist will be able to help with two of the most common symptoms of ataxia – slurred speech (dysarthria) and swallowing problems (dysphagia).
The therapist will be able to advise you about how to make your voice sound clearer. For example, they may suggest:
- changing your posture to improve the quality of your voice
- carrying out exercises to strengthen the muscles used when speaking
- speaking more slowly to emphasise each word
- using breathing techniques to improve your speech
If your speech gets worse, you may want to consider using speaking aids such as a laptop computer connected to a voice synthesiser. Your therapist will be able to advise you about the equipment available.
To treat dysphagia, your therapist will be able to teach you exercises to stimulate the nerves used to trigger your swallowing reflex and strengthen the muscles used when swallowing.
You may also be referred to a dietitian for dietary advice. For example, your diet may need to include food that’s easier to swallow.
Occupational therapy
The aim of occupational therapy is to teach you how to adapt to your gradual loss of mobility and develop new skills you can use to carry out daily activities.
An occupational therapist may be able to teach you how to use a wheelchair and other mobility devices. They can also advise you about modifications you can make to your house, such as installing guide rails or a stair lift, to help make your life easier.
Physiotherapy
If you have ataxia, physiotherapy can help you maintain the use of your arms and legs, and prevent your muscles weakening or getting stuck in one position (contractures).
A physiotherapist will be able to teach you a number of physical exercises you can do every day to help strengthen and stretch your muscles. They may also be able to recommend walking aids to help you get around.
Muscle problems
If you’re experiencing muscle spasms, cramps and stiffness, muscle relaxant medication such as baclofen or tizanidine may be used to control these symptoms.
If these aren’t effective, an injection of botulinum toxin (Botox) may be given. This works by blocking the signals from your brain to the affected muscles. The effects of the injection will usually last for up to three months.
Bladder problems
Bladder problems, such as urinary urgency or, more rarely, urinary incontinence, sometimes affect people with ataxia.
In some cases, bladder problems can be controlled using a number of self care techniques, such as limiting fluid intake during the day, planning regular trips to the toilet, and avoiding drinks known to stimulate urine production, such as caffeine and alcohol.
Some people may also require a type of medication known as antimuscarinic. This will help relax the bladder, reducing the frequent urge to urinate. Occasional injections of botulinum toxin into the bladder may also help.
Others may find it difficult to empty their bladder completely when they go to the toilet. This can lead to small amounts of urine leaking out later on. In such cases, it may be necessary to insert a small tube known as a urinary catheter into the bladder to help drain the urine.
Eye problems
Eye problems are common in some cases of ataxia. Oscillopsia is an eye problem caused by involuntary movement of the eyes from side to side or up and down. It can cause visual disruption, making tasks such as reading difficult. This can sometimes be treated using medication such as gabapentin to control the muscles that move the eyes.
Some people with ataxia experience double vision, where you see two images of a single object. It may be possible to treat this by attaching a wedge-shaped piece of glass or plastic called a prism to your glasses.
Erectile dysfunction
As a result of underlying nerve damage, some men with ataxia will experience difficulty getting or maintaining an erection (erectile dysfunction).
This can often be treated using a group of medications known as phosphodiesterase-5 (PDE-5) inhibitors, such as sildenafil (sold as Viagra). These help increase blood flow to the penis.
Fatigue
Many people with neurological conditions such as ataxia report feeling extremely tired and lethargic (lacking in energy). It’s thought this is partly caused by disturbed sleep and the physical efforts of having to cope with the loss of co-ordination.
A physiotherapist may be able to help you increase your stamina levels, and an occupational therapist can advise you about how to adapt your daily activities to help you cope with fatigue better.
Nerve pain
Damage to the nerve endings can result in nerve pain. The medical term for nerve pain is neuropathic pain, which is often experienced as a burning, aching or shooting pain, or sometimes tingling, in certain parts of the body.
Traditional painkillers such as paracetamol or ibuprofen aren’t usually effective in treating neuropathic pain, so you may be prescribed a number of medications, such as amitriptyline, gabapentin or pregabalin.
Cardiomyopathy
Cardiomyopathy (damage to the heart muscle) is a common problem in some types of ataxia. This can be serious as it can place strain on the heart, affect the normal blood flow through the heart, and cause heartbeat irregularities (arrhythmias).
If you develop cardiomyopathy, you’ll receive regular check-ups from a cardiologist (a heart specialist). You may need to take medication to treat any problems as they develop.
Depression
Living with a long-term condition such as ataxia can be stressful and can often cause intense feelings of anxiety. In some cases, this can trigger the onset of depression.
Signs that you may be depressed include feeling down or hopeless during the past month and no longer taking pleasure in the things you enjoy.
You should contact your doctor or multidisciplinary team for advice if you think you may be depressed. There are several treatments for depression, such as antidepressants and talking therapies such as cognitive behavioural therapy (CBT).
Treating the underlying cause
In a few cases of ataxia, it may be possible to improve the condition or stop it getting worse by treating the underlying cause.
For example:
- ataxia with vitamin E deficiency can often be controlled or improved with vitamin E supplements
- episodic ataxia can often be controlled with a medication called acetazolamide and by avoiding triggers such as stress, alcohol and caffeine
- acquired ataxia can sometimes be treated depending on the specific cause – for example, antibiotic or antiviral medication may help if it’s caused by an infection
If acquired ataxia is caused by serious underlying brain damage, such as damage from a stroke or a severe head injury, it may not be possible to improve the condition. If this is the case, the treatments mentioned above can be used to control your symptoms.
Coping and support
The challenges you face when living with ataxia or having a child with the condition might make you feel alone or lead to depression and anxiety. Talking to a counselor or therapist might help. Or you might find encouragement and understanding in a support group, either for ataxia or for your underlying condition, such as cancer or multiple sclerosis.
Although support groups aren’t for everyone, they can be good sources of information. Group members often know about the latest treatments and tend to share their own experiences. If you’re interested, your doctor might be able to recommend a group in your area.
- Friedreich’s Ataxia Fact Sheet. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Friedreichs-Ataxia-Fact-Sheet[↩]
- Gatti R, Perlman S. Ataxia-Telangiectasia. 1999 Mar 19 [Updated 2016 Oct 27]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26468/[↩]
- Yakusheva TA, Shaikh AG, Green AM, Blazquez PM, Dickman JD, Angelaki DE. Purkinje cells in posterior cerebellar vermis encode motion in an inertial reference frame. Neuron. 2007;54:973–85. https://www.ncbi.nlm.nih.gov/pubmed/17582336[↩]
- Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr. 2004;144:505–11. https://www.ncbi.nlm.nih.gov/pubmed/15069401[↩][↩][↩]
- Regueiro JR, Porras O, Lavin M, Gatti RA. Ataxia-telangiectasia: a primary immunodeficiency revisited. Immunology & Allergy Clinics. 2000;20:177–206.[↩]
- Staples ER, McDermott ERM, Reiman A, Byrd PJ, Ritchie S, Taylor AMR, Davies EG. Immunodeficiency in ataxia telangiectasia is correlated strongly with the presence of two null mutations in the ataxia telangiectasia mutated gene. Clin Exp Immunol. 2008;153:214–20. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2492895/[↩]
- Lockman JL, Iskander AJ, Bembea M, Crawford TO, Lederman HM, McGrath-Morrow S, Easley RB. Anesthetic and perioperative risk in the patient with ataxia-telangiectasia. Paediatr Anaesth. 2012;22:256–62. https://www.ncbi.nlm.nih.gov/pubmed/22098343 [↩]
- Tangsinmankong N, Wayne AS, Howenstine MS, Washington KR, Langston C, Gatti RA, Good RA, Nelson RP Jr. Lymphocytic interstitial pneumonitis, elevated IgM concentration, and hepatosplenomegaly in ataxia-telangiectasia. J Pediatr. 2001;138:939–41. https://www.ncbi.nlm.nih.gov/pubmed/11391347[↩]
- Dörk T, Bendix-Waltes R, Wegner RD, Stumm M. Slow progression of ataxia-telangiectasia with double missense and in frame splice mutations. Am J Med Genet A. 2004;126A:272–7. https://www.ncbi.nlm.nih.gov/pubmed/15054841[↩]
- Sahama I, Sinclair K, Pannek K, Lavin M, Rose S. Radiological imaging in ataxia telangiectasia: a review. Cerebellum. 2014b;13:521–30. https://www.ncbi.nlm.nih.gov/pubmed/24683014[↩]
- Mallott J, Kwan A, Church J, Gonzalez-Espinosa D, Lorey F, Tang LF, Sunderam U, Rana S, Srinivasan R, Brenner SE, Puck J. Newborn screening for SCID identifies patients with ataxia telangiectasia. J Clin Immunol. 2013 Apr;33(3):540–9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3591536/[↩]
- Chun HH, Sun X, Nahas SA, Teraoka S, Lai CH, Concannon P, Gatti RA. Improved diagnostic testing for ataxia-telangiectasia by immunoblotting of nuclear lysates for ATM protein expression. Mol Genet Metab. 2003;80:437–43. https://www.ncbi.nlm.nih.gov/pubmed/14654357[↩]
- Stewart GS, Last JI, Stankovic T, Haites N, Kidd AM, Byrd PJ, Taylor AM. Residual ataxia telangiectasia mutated protein function in cells from ataxia telangiectasia patients, with 5762ins137 and 7271T–>G mutations, showing a less severe phenotype. J Biol Chem. 2001;276:30133–41. https://www.ncbi.nlm.nih.gov/pubmed/11382771[↩]
- van Egmond ME, Elting JW, Kuiper A, Zutt R, Heineman KR, Brouwer OF, Sival DA, Willemsen MA, Tijssen MA, de Koning TJ. Myoclonus in childhood-onset neurogenetic disorders: The importance of early identification and treatment. Eur J Paediatr Neurol. 2015;19:726–9. https://www.ncbi.nlm.nih.gov/pubmed/26232052[↩]
- Gatti RA, Perlman S. A proposed bailout for A-T patients? Eur J Neurol. 2009;16:653–5. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3242728/[↩]
- Bhatt JM, Bush A, van Gerven M, Nissenkorn A, Renke M, Yarlett L, Taylor M, Tonia T, Warris A, Zielen S, Zinna S, Merkus PJ., European Respiratory Society. ERS statement on the multidisciplinary respiratory management of ataxia telangiectasia. Eur Respir Rev. 2015;24:565–81. http://err.ersjournals.com/content/24/138/565.long[↩][↩]
- Schütte P, Möricke A, Zimmermann M, Bleckmann K, Reismüller B, Attarbaschi A, Mann G, Bodmer N, Niggli F, Schrappe M, Stanulla M, Kratz CP. Preexisting conditions in pediatric ALL patients: Spectrum, frequency and clinical impact. Eur J Med Genet. 2016;59:143–51. https://www.ncbi.nlm.nih.gov/pubmed/26732628[↩][↩]
- Lefton-Greif MA, Crawford TO, Winkelstein JA, Loughlin GM, Koerner CB, Zahurak M, Lederman HM. Oropharyngeal dysphagia and aspiration in patients with ataxia-telangiectasia. J Pediatr. 2000;136:225–31. https://www.ncbi.nlm.nih.gov/pubmed/10657830[↩]
- McGrath-Morrow S, Lefton-Greif M, Rosquist K, Crawford T, Kelly A, Zeitlin P, Carson KA, Lederman HM. Pulmonary function in adolescents with ataxia telangiectasia. Pediatr Pulmonol. 2008;43:59–66. https://www.ncbi.nlm.nih.gov/pubmed/18041755[↩]
- Lockman JL, Iskander AJ, Bembea M, Crawford TO, Lederman HM, McGrath-Morrow S, Easley RB. Anesthetic and perioperative risk in the patient with ataxia-telangiectasia. Paediatr Anaesth. 2012;22:256–62. https://www.ncbi.nlm.nih.gov/pubmed/22098343[↩]
- Balint R. (1909). Seelenlähmung des “Schauens” optische Ataxie räumliche Störung der Aufmerksamkeit. Monatsschr. Psychiatr. Neurol. 25, 5–81[↩]
- Psychoanatomical substrates of Bálint’s syndrome. Rizzo M, Vecera SP. J Neurol Neurosurg Psychiatry. 2002 Feb; 72(2):162-78. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1737727/pdf/v072p00162.pdf[↩]
- Optic ataxia localized in 2 left homonymous visual hemifields [clinical study with film presentation]. Garcin R, Rondot P, de Recondo J. Rev Neurol, Paris. 1967 Jun; 116(6):707-14.[↩]
- Optic ataxia: a specific disruption in visuomotor mechanisms. I. Different aspects of the deficit in reaching for objects. Perenin MT, Vighetto A. Brain. 1988 Jun; 111 ( Pt 3)():643-74. https://www.ncbi.nlm.nih.gov/pubmed/3382915/[↩]
- Optic ataxia and the function of the dorsal stream: contributions to perception and action. Pisella L, Sergio L, Blangero A, Torchin H, Vighetto A, Rossetti Y. Neuropsychologia. 2009 Dec; 47(14):3033-44. https://www.ncbi.nlm.nih.gov/pubmed/19563817/[↩]
- Cortical control of visually guided reaching: evidence from patients with optic ataxia. Karnath HO, Perenin MT. Cereb Cortex. 2005 Oct; 15(10):1561-9. https://www.ncbi.nlm.nih.gov/pubmed/5619766/[↩]





{kind=link}