
Contents
Canavan disease also called Canavan-Van Bogaert-Bertrand disease or spongiform leukodystrophy, is a rare, progressive, and fatal neurological hereditary disorder that begins in infancy 1). Canavan disease is part of a group of genetic diseases referred to as leukodystrophies. Leukodystrophies disrupt the growth or maintenance of the myelin sheath, which is the covering that protects nerves and promotes the efficient transmission of nerve impulses. Canavan disease is associated with the deficiency of an essential enzyme resulting in the loss of white matter in the brain, that damages the ability of nerve cells (neurons) in the brain to send and receive messages.
Neonatal/infantile Canavan disease is the most common and is usually associated with the most severe symptoms. Most infants appear healthy immediately after birth, but within 2 to 6 months, obvious deficits in gross motor development will be apparent. The infant may not be able to move, turn over, control head movements, or sitting without support. Other common features of infantile Canavan disease include weak muscle tone (hypotonia), an unusually large head size (macrocephaly), and irritability. Most infants have difficulty with feeding and swallowing difficulties, develop seizures and sleep disturbances may also develop. Some children require nasogastric feeding or permanent feeding gastrostomies 2). Joint stiffness increases, so that these children resemble individuals with cerebral palsy 3).
The mild/juvenile form of Canavan disease is less common 4). Affected individuals may have normal or mildly delayed speech or motor development early in childhood without regression. These delays may be so mild and nonspecific that they are never recognized as being caused by Canavan disease. In spite of developmental delay most of these children can be educated in typical classroom settings and may benefit from speech therapy or tutoring as needed 5). Most children with mild forms of Canavan disease have normal head size, although macrocephaly, retinitis pigmentosa, and seizures have been reported in a few individuals.
Canavan disease is caused by mutations in the ASPA gene and is inherited in an autosomal recessive pattern 6). While Canavan disease occurs in people of all ethnic backgrounds, it is most common in people of Ashkenazi (eastern and central European) Jewish heritage, and among Saudi Arabians 7). The carrier frequency of Canavan disease among the Ashkenazi varies from 1:37 to 1:57 individuals, yielding approximate prevalence rates ranging from 1:6000 to 1:14,000. Studies suggest that Canavan disease affects 1 in 6,400 to 13,500 people in the Ashkenazi Jewish population. The incidence in other populations is unknown.
Canavan disease can be identified by a simple prenatal blood test that screens for the missing enzyme or for mutations in the gene that controls aspartoacylase. Both parents must be carriers of the defective gene in order to have an affected child. When both parents are found to carry the Canavan gene mutation, there is a one in four (25 percent) chance with each pregnancy that the child will be affected with Canavan disease 8).
The life expectancy for people with Canavan disease varies. Most people with the neonatal/infantile form live only into childhood, although some survive into adolescence or beyond 9). People with the mild/juvenile form do not appear to have a shortened lifespan 10).
Canavan disease causes progressive brain atrophy. There is no cure, nor is there a standard course of treatment. Treatment is symptomatic and supportive. Supportive care is very important to ease the symptoms of the disease. Lithium and gene therapy are being studied.
Canavan disease patients have a variable average lifespan. Most people with the neonatal or infantile form survive only into childhood, although some studies report life expectancy up to adolescence or beyond. People with the mild or juvenile form do not appear to have a shortened lifespan.
Canavan disease is caused by mutations in the ASPA gene and is inherited in an autosomal recessive pattern 11). While Canavan disease occurs in people of all ethnic backgrounds, it is most common in people of Ashkenazi (eastern and central European) Jewish heritage, and among Saudi Arabians 12). The carrier frequency of Canavan disease among the Ashkenazi varies from 1:37 to 1:57 individuals, yielding approximate prevalence rates ranging from 1:6000 to 1:14,000. Studies suggest that Canavan disease affects 1 in 6,400 to 13,500 people in the Ashkenazi Jewish population. The incidence in other populations is unknown.
Children with Canavan disease may be irritable and over time experience sleep disturbances 13). While fever is not a symptom, per se, these children may develop infections which can be accompanied by fever 14). Experts strongly recommend that you discuss your child’s symptoms with your pediatrician.
Canavan disease causes progressive brain atrophy. There is no cure, nor is there a standard course of treatment. Treatment is symptomatic and supportive.[2][3] Management may include provision of adequate nutrition and hydration, treatment of infectious diseases, and protection of the airway. Physical therapy may help to minimize contractures and maximize motor abilities and seating posture. Special education programs can enhance communication skills. Seizures are treated with anti-epileptic drugs. Gastrostomy can help to maintain adequate food intake and hydration when swallowing difficulties exist 15).
- Neonatal/infantile Canavan disease. Treatment is supportive and directed to providing adequate nutrition and hydration, managing infectious diseases, and protecting the airway. Hospice care is a resource used by the families of the individuals affected by the disease. Physical therapy minimizes contractures and maximizes motor abilities and seating posture; special education programs enhance communication skills. Seizures are treated with antiepileptic drugs. Gastrostomy may be needed to maintain adequate food intake and hydration when swallowing difficulties exist.
- Mild/juvenile Canavan disease. May require speech therapy or tutoring but no special medical care.
Surveillance 16):
- Neonatal/infantile Canavan disease. Follow up every six months to evaluate developmental status and evidence of any new problems.
- Mild/juvenile Canavan disease. Annual routine follow up by a pediatric neurologist or a developmental pediatrician is indicated.
Mutations in the ASPA gene cause Canavan disease. The ASPA gene provides instructions for making an enzyme called aspartoacylase. This enzyme normally breaks down a compound called N-acetyl-L-aspartic acid (NAA), which is predominantly found in neurons in the brain, into aspartate (aspartic acid) and acetate 17). The function of N-acetyl-L-aspartic acid (NAA) is unclear. Researchers had suspected that it played a role in the production of the myelin sheath, but recent studies suggest that N-acetyl-L-aspartic acid (NAA) does not have this function. The enzyme may instead be involved in the transport of water molecules out of neurons.
Mutations in the ASPA gene reduce the function of aspartoacylase, which prevents the normal breakdown of N-acetyl-L-aspartic acid (NAA). The mutations that cause the neonatal/infantile form of Canavan disease severely impair the enzyme’s activity, allowing N-acetyl-L-aspartic acid (NAA) to build up to high levels in the brain. The mutations that cause the mild/juvenile form of the disorder have milder effects on the enzyme’s activity, leading to less accumulation of N-acetyl-L-aspartic acid (NAA). Levels of NAA are markedly increased in the patient’s plasma, urine, and cerebrospinal fluid.
An excess of NAA in the brain is associated with the signs and symptoms of Canavan disease. Studies suggest that if NAA is not broken down properly, the resulting chemical imbalance interferes with the formation of the myelin sheath as the nervous system develops. A buildup of NAA also leads to the progressive destruction of existing myelin sheaths. Nerves without this protective covering malfunction, which disrupts normal brain development. Abnormal myelination and the associated prominent swollen and vacuolated astrocytes are the fundamental hallmarks of aspartoacylase deficiency. These effects justify the name spongiform leukodystrophy. However, the precise mechanism by which accumulated NAA causes the pathogenesis of this spongiform degeneration remains uncertain.
Inheritance pattern
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
A person must inherit two copies of a abnormal ASPA gene, one from each parent, in order to be affected by the condition (25% chance). If a person inherits only one abnormal ASPA gene then they will be a carrier (50% chance). These outcomes occur randomly. They remain the same in every pregnancy and are the same for boys and girls.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing Canavan disease.
Figure 1. Canavan disease autosomal recessive inheritance pattern
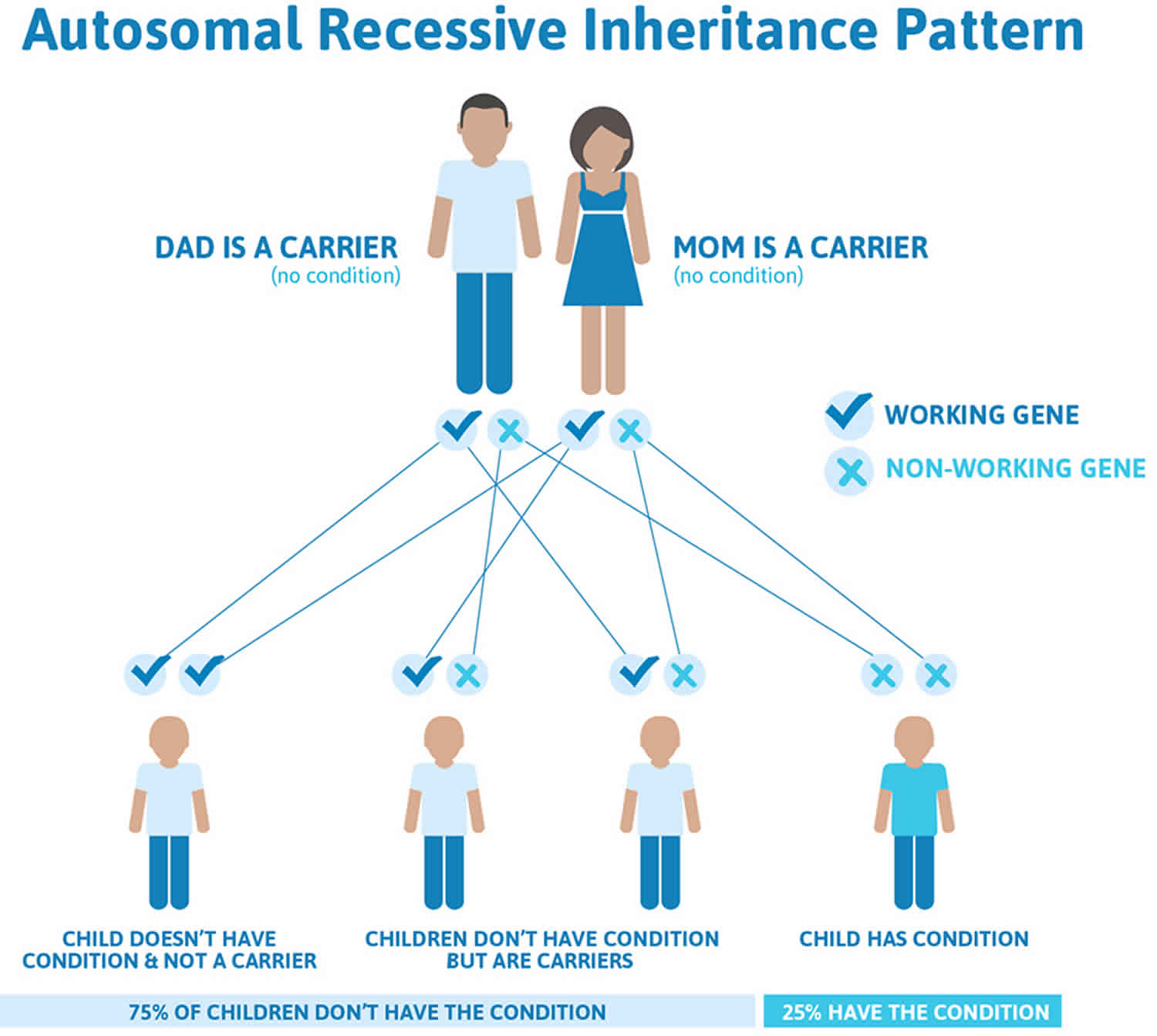
Genetic counseling is recommended for people who want to have children and have a family history of Canavan disease. Counseling should be considered if both parents are of Ashkenazi Jewish descent. For this group, DNA testing can almost always tell if the parents are carriers.
A diagnosis may be made before the baby is born (prenatal diagnosis) by testing the amniotic fluid, the fluid that surrounds the womb.
Genetic counseling
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
The symptoms and progression of Canavan disease varies from case to case. Canavan disease symptoms usually becomes apparent between 3 and 6 months of age and the initial symptoms usually include extremely poor head control, an abnormally large head (macrocepahly), and severely diminished muscle tone (hypotonia) resulting in “floppiness.” Affected infants may be generally unresponsive (apathetic), lethargic or irritable. Some infants may experience difficulty swallowing (dysphagia), which contributes to feeding difficulties. Parents tend to notice it when their child is not reaching certain developmental milestones, including head control.
Canavan disease symptoms include:
- Abnormal posture with flexed arms and straight legs
- Food material flows back into the nose
- Feeding problems
- Increasing head size
- Irritability
- Poor muscle tone, especially of the neck muscles
- A lack of head control when baby is pulled from a lying to a sitting position
- Poor visual tracking, or blindness
- Reflux with vomiting
- Seizures
- Severe intellectual disability
- Swallowing difficulties
Affected infants also show delays in reaching developmental milestones (e.g., sitting or standing unassisted) and most never walk independently. The progressive loss of abilities requiring the coordination of mental and muscular activity (psychomotor regression) and mental retardation also become apparent during infancy. Most affected infants do learn to smile, laugh, raise their heads and interact socially.
Additional symptoms that affect children with Canavan disease include seizures, sleep disorders, feeding difficulties, nasal regurgitation, backflow of acid from the stomach to the esophagus (reflux) sometimes associated with vomiting, and deterioration of the nerves of the eyes (optic nerves) that transmit impulses from the nerve-rich membrane lining the eyes (retina) to the brain (optic atrophy). Optic atrophy may cause reduced visual responsiveness. In most case, hearing is unaffected, but hearing loss can occur.
As affected infants age, hypotonia may eventually develop into spasticity, a condition characterized by involuntary muscle spasms that result in slow, stiff movements of the legs. Affected individuals may eventually exhibit uncontrolled rigid extensions and rotations of the arms, legs, fingers, and toes (decerebrate rigidity) or paralysis. Canavan disease eventually progresses to cause life-threatening complications; however, the severity and progression of the disease varies. Some individuals develop life-threatening complications in infancy; others live beyond their teen-age years.
In the last few years, a mild form of Canavan disease has been recognized, with characteristic mutations of the ASPA gene and only slightly increased NAA in the urine. These children may be only slightly delayed, can learn and go to school. The head may be somewhat enlarged, but the typical white matter changes associated with Canavan may be absent. The prognosis is certainly much better.
This disorder nay cause severe disabilities such as:
- Blindness
- Inability to walk
- Intellectual disability
- Lack of motor skills
- Difficulty feeding
- Hypotonia
- Paralysis
- Blindness
- Seizures
- Death
With Canavan disease, the central nervous system breaks down. People are likely to become disabled and the prognosis for neonatal/infantile Canavan disease is poor. For severe disease, the prognosis is grave. Survival beyond the first decade of life is not always possible 18). For mild disease, the life expectancy varies. Most patients make it through puberty and may have an average lifespan.
The life expectancy for people with Canavan disease varies. Most people with the neonatal/infantile Canavan disease live only into childhood, although some survive into adolescence or beyond 19). People with the mild/juvenile Canavan disease do not appear to have a shortened lifespan 20). Prognosis often depends upon the clinical course of the disease as well as the level of medical care provided 21).
Canavan disease should be suspected in individuals with 22):
- The triad of hypotonia, head lag, and macrocephaly after age three to five months
- Poor visual following and difficulties with suck and swallow
- Developmental delays (with regression in infantile form and without regression in mild/juvenile form)
- Leukodystrophy on neuroimaging (generalized in infantile form and localized to basal ganglia in mild/juvenile form)
- Elevated N-acetylaspartic acid (NAA) in urine using gas chromatography-mass spectrometry
A physical exam may show:
- Exaggerated reflexes
- Joint stiffness
- Loss of tissue in the optic nerve of the eye
Tests for Canavan disease include:
- Blood chemistry
- CSF chemistry
- Genetic testing for aspartoacylase gene mutations
- Head CT scan
- Head MRI scan
- Urine or blood chemistry for elevated aspartic acid
- DNA analysis
Establishing the diagnosis
The diagnosis of Canavan disease is established in a proband with typical clinical findings and elevated N-acetylaspartic acid (NAA) in urine using gas chromatography-mass spectrometry and/or biallelic pathogenic variants in ASPA identified by molecular genetic testing (see Table 1).
Note: (1) Although NAA concentration is also elevated in the blood and cerebrospinal fluid (CSF) of children with neonatal/infantile (severe) Canavan disease, elevated concentration of NAA in urine is sufficient for diagnosis of affected individuals 23). (2) Canavan disease is associated with decreased aspartoacylase enzyme activity; individuals with severe Canavan disease may have unmeasurable enzyme activity, and carriers (heterozygotes) may have enzyme activity ~50% of normal. Aspartoacylase enzyme activity may not be reliable in the diagnosis of Canavan disease because enzyme activity fluctuates with culture conditions; therefore, measurement of the urinary concentration of NAA is the preferred diagnostic method 24).
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Laboratory Studies
In symptomatic infants with compatible clinical features and neuroimaging findings suggesting aspartoacylase deficiency, the diagnosis is concluded by observing elevated levels of urine NAA. Compatible clinical features include hypotonia, poor head control, and macrocephaly. A determination can be reached more specifically by detecting deficient aspartoacylase activity in cultured skin fibroblasts. If elevated urine NAA and skin fibroblast testing are diagnostic, genetic testing is obtained only for genetic counseling.
Urine levels of NAA are increased up to 200 times that of the reference range. Gas chromatography or mass spectrometry are employed for measurement.
Prenatal Diagnosis
Prenatal diagnosis is best accomplished by measuring NAA levels in amniotic fluid. This method utilizes a stable isotope dilution coupled with gas chromatography or mass spectrometry, and also molecular analysis.
Neuroimaging
A CT scan shows a characteristic low attenuation of white matter in contrast to the relatively unaffected gray matter. This difference is mainly due to spongiform degeneration and edema of the white matter. No post contrast enhancement is usually seen.
MRI features include a relatively enlarged brain, also called megalencephaly. There is typically a diffuse bilateral white matter involvement. Unlike other leukodystrophies, subcortical U-fibers are usually involved early in the disease process. T1 shows a low signal in the white matter while T2 shows a high signal in white matter. There is no post contrast enhancement. MR spectroscopy shows markedly elevated levels of NAA, and an elevated NAA:creatine ratio. The mnemonic “CaNAAvan” is useful for remembering this information.
Table 1. Molecular Genetic Testing Used in Canavan Disease
| Gene | Test Method | Proportion of Pathogenic Variants 2 Detectable by This Method | |
|---|---|---|---|
| ASPA | Targeted testing 3 | p.Glu285Ala, p.Tyr231Ter | Ashkenazi Jewish: 98% 25) Non-Ashkenazi Jewish: 3% 26) |
| p.Ala305Glu | Ashkenazi Jewish: 1% 27) Non-Ashkenazi Jewish: 30%-60% 28) | ||
| Sequence analysis 6 | ~99% 29) | ||
| Gene-targeted deletion/duplication analysis 8 | 9 reported 30) | ||
Footnote:
3. Various molecular methods may be used to detect targeted variants.
6. Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.
8. Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods that may be used include: quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
An aspartoacylase deficiency has no effective treatment. The treatment of Canavan disease is directed toward the specific symptoms that are apparent in each individual. Management aims to maintain nutrition and hydration, protect the airways, prevent seizures, minimize the risk of contractures, and cure infections. An assessment of the patient’s nutritional and developmental status is recommended for guiding the management. Physical therapy and early intervention may help to improve posture and communication skills, respectively. If swallowing difficulties occur, feeding gastrostomy tube is typically used to maintain adequate nutrition and hydration in the presence of dysphagia to ensure proper nutrition and hydration. A gastrostomy tube can also decrease the risk of aspiration due to the patient’s lack of sound reflexes. Seizures may be treated with anti-seizure (anti-convulsant) medications.
Physical therapy and position changes are helpful to reduce the risk of contractures and ulcers. They also help improve the patient’s sitting posture. Botulinum toxin injections can be employed to treat spasticity. Special education programs and interventions can be considered.
Genetic counseling and carrier testing will benefit families in which this disease occurs.
References [ + ]


{kind=link}