



What is red yeast rice
Red yeast rice also known as Hongqu, red Koji, red fermented rice, red kojic rice, red koji rice, anka, or ang-kak, is a bright reddish purple fermented rice, which acquires its color from being cultivated with the mold Monascus purpureus (red yeast of Aspergillaceae family). Red yeast rice is a remedy belonging to Traditional Chinese Medicine, largely used nowadays as dietary supplement in Western countries to improve blood circulation by decreasing cholesterol and triglyceride levels 1. China is the world’s largest producer of red yeast rice. Red yeast rice supplements contain monacolin K (also known as Mevinolin), which is chemically identical to lovastatin, a licensed drug 2. Lovastatin is associated with various adverse effects such as myopathy and abnormal liver function test results, which can lead to serious problems if patients are not monitored and treated 3. Red yeast rice is often used by statin‐intolerant patients, although there are very few studies testing red yeast rice safety profile as compared to statins. Red yeast rice products may not be safe; some may have the same side effects as certain cholesterol-lowering drugs, and some may contain a potentially harmful contaminant. Clinical trials have reported no significant serious adverse events, but minor events include heartburn 4, flatulence 5, dizziness 4, myalgia 6 and loose stools 6.
Red yeast rice is produced by first soaking in water until the rice grains are fully saturated. The raw soaked rice can then either be directly inoculated or it can be steamed for the purpose of sterilizing and cooking the grains prior to inoculation. Inoculation is done by mixing a food fungus of the Monascus genus, mainly Monascus purpureus spores or powdered red yeast rice together with the rice that is being treated. The mix is then incubated in an environment around room temperature for 3–6 days. During this period of time, the rice should be fully cultured with Monascus purpureus, with each rice grain turning bright red in its core and reddish purple on the outside. During fermentation the naturally produced pigments give the characteristic red color to the rice and monacolins are produced. As part of the Chinese diet, Red yeast rice is used as food additive to increase the color of meat, fish and soybean products; some preparations of red yeast rice are used in food products in Chinese cuisine, including Peking duck. Moreover, it is recognized as a folk medicine for improving food digestion and blood circulation 7. The fully cultured rice is then either sold as the dried grain, or cooked and pasteurized to be sold as a wet paste, or dried and pulverized to be sold as a fine powder.
Key facts
- Some red yeast rice products contain substantial amounts of monacolin K, which is chemically identical to the active ingredient in the cholesterol-lowering drug lovastatin. Lovastatin is one of the drugs in the category known as statins. These drugs lower blood cholesterol levels by reducing the production of cholesterol by the liver. These products may lower blood cholesterol levels and can cause the same types of side effects and drug interactions as lovastatin.
- Other red yeast rice products contain little or no monacolin K. It is not known whether these products have any effect on blood cholesterol levels.
- Consumers have no way of knowing how much monacolin K is present in most red yeast rice products. The labels on these products usually state only the amount of red yeast rice that they contain, not the amount of monacolin K.
- The composition of red yeast rice products varies depending on the yeast strains and culture conditions used to manufacture them. The strains and conditions used to produce culinary red yeast rice differ from those used to produce products that are intended to lower cholesterol. Tests performed by the U.S. Food and Drug Administration (FDA) indicate that the red yeast rice sold as a food product contains only traces of monacolin K or none at all.
- The U.S. Food and Drug Administration (FDA) has determined that red yeast rice products that contain more than trace amounts of monacolin K are unapproved new drugs and cannot be sold legally as dietary supplements.
- Some red yeast rice products contain a contaminant called citrinin, which can cause kidney failure.
- Tell all your health care providers about any complementary health approaches you use. Give them a full picture of what you do to manage your health. This will help ensure coordinated and safe care.
In 1998, the FDA determined that a red yeast rice product that contained a substantial amount of monacolin K was an unapproved new drug, not a dietary supplement. On several occasions since then, the FDA has taken action against companies selling red yeast rice products that contain more than trace amounts of monacolin K, warning them that it is against the law to market these products as dietary supplements.
Despite the FDA actions, some red yeast rice products currently on the market in the United States may contain monacolin K. (Some products tested as recently as 2011 have been found to contain it in substantial amounts.) Other products may contain little or none of this component. Consumers have no way of knowing how much monacolin K is present in most red yeast rice products, and therefore have no way of knowing whether a particular product is safe, effective, or legal. The labels on these products usually state only the amount of red yeast rice that they contain, not the amounts of monacolin K or other monacolins.
Several studies have shown the lipid‐lowering effects of red yeast rice in humans, especially in comparison with placebo in patients intolerant to statins 8, 9, although only a minority of trials compared head‐to‐head red yeast rice with statins or ezetimibe 10, 11, and some quality issues (e.g., allocation concealment and blinding) have been raised 12.
If you are considering red yeast rice
- Do not use red yeast rice to replace conventional care or to postpone seeing your health care provider about a health problem.
- Do not use red yeast rice dietary supplements if you are pregnant, trying to become pregnant, or nursing a child. If you are considering giving a child a red yeast rice dietary supplement, it is especially important to consult the child’s health care provider.
- Do not take red yeast rice in addition to prescription statin drugs.
- Many Web sites, including sales sites, have information about red yeast rice. Be cautious when you evaluate information from the Web; not all of it is trustworthy.
- Federal regulations for dietary supplements are very different from—and less strict than—those for drugs.
- Tell all your health care providers about any complementary health approaches you use. Give them a full picture of what you do to manage your health. This will help ensure coordinated and safe care.
Figure 1. Red yeast rice

Red yeast rice benefits
Red yeast rice products that contain substantial amounts of monacolin K can lower blood cholesterol levels. Researchers have not reported results of any studies of red yeast rice products that contain little or no monacolin K, so whether these products have any effect on blood cholesterol is unknown.
Red yeast rice for cholesterol
In clinical trials (studies in people) of red yeast rice products that contained substantial amounts of monacolin K, the products lowered blood levels of total cholesterol and low-density lipoprotein (LDL) cholesterol (the so-called bad cholesterol that is linked to increased heart disease risk). It is important to emphasize that all of these clinical trials used products that contained substantial amounts of monacolin K. A 2011 analysis showed that some of the red yeast rice products on the market contain very little monacolin K. These products may have little or no effect on blood cholesterol levels. Therefore, even though the participants in the clinical trials were able to lower their cholesterol levels by taking red yeast rice, you might not be able to achieve the same results.
In one of the clinical trials, the tested product produced a cholesterol-lowering effect greater than would be expected based on its monacolin K content. Further investigations, supported by the National Center for Complementary and Integrative Health, suggested that other monacolins or other substances present in the product may have contributed to its cholesterol-lowering effect.
This systematic review 13 assessed the efficacy and safety of red yeast rice versus simvastatin in the management of dyslipidemia. The evidence suggests that red yeast rice and simvastatin have similar effects in reducing total cholesterol, LDL “bad” cholesterol and triglyceride and increasing HDL “bad” cholesterol. The same adverse events reported between the red yeast rice and simvastatin groups were upset stomach 14, nausea 15, abdominal distention 15, aspartate aminotransferase (AST) increased 15, alanine aminotransferase (ALT) increased 16 and anorexia 17. One trial 18 reported ‘gastrointestinal symptoms’ as an adverse event in both treatment groups with no further description of symptoms. Dry mouth 16 and creatine kinase increased 19 were reported only in the simvastatin group. However, because the included trials in this review were of low quality, the review authors are unable to conclusively suggest replacing simvastatin with red yeast rice for the management of dyslipidaemia particularly as red yeast rice is not standardized in active drug content 13. Furthermore, all trial subjects were recruited from Chinese populations 13. Hence, the results of this review may not be applicable to other populations. Therefore, the beneficial effects of red yeast rice over simvastatin need to be established using trials with larger sample size, diverse population, improved methodology and longer duration of treatment 13.
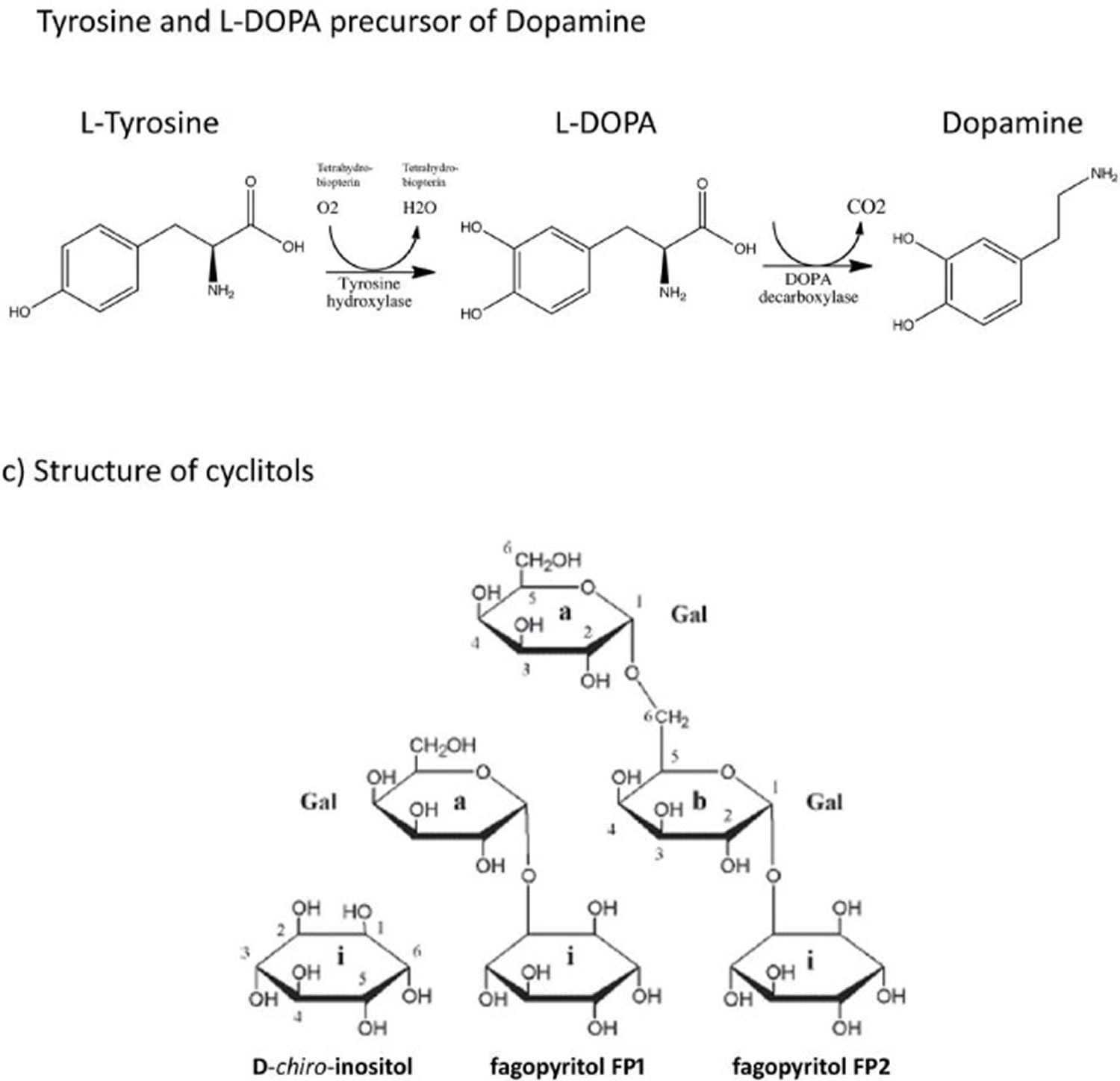
These properties of red yeast rice are due to its monacolins content, a family of naturally occurring substances that inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate‐limiting step in cholesterol synthesis. Monacolins content in red yeast rice is usually around 0.4% w/w; of this about 90% consists of monacolin K (also known as Mevinolin) which is chemically identical to lovastatin. Other chemical components of red yeast rice are fatty acids and the pigments monascidin A, ankaflavin, monascorubrine and monascorubramine. A further pigment citrinin, a nephrotoxic mycotoxin, can be produced by some strains of Monascus 20. A dose of red yeast rice containing about 5–7 mg of monacolin K is considered as effective in lowering cholesterol as 20–40 mg of pure lovastatin, probably owing to the presence of other active monacolins or to an increased bioavailability of lovastatin when given as red yeast rice 21. Clinical studies suggest that red yeast rice has the potential to reduce serum LDL “bad” cholesterol levels by 10% to 33% 22, 23 and total cholesterol, triglyceride and as well as at increasing HDL “good” cholesterol in people with moderate hypercholesterolemia, but those indicators return to the baseline level when the treatment is discontinued 24. This phenomenon is similar to statins, which could be explained by the main components of red yeast rice (monacolins, which are capable of inhibiting the HMG-CoA reductase enzyme). In addition to lovastatin, most red yeast rice preparations contain other active substances such as Coenzyme Q10 and isoflavones 25. Moreover, a recent study showed that the oral bioavailability of lovastatin is significantly improved in red yeast rice products as a result of a higher dissolution rate and reduced crystallinity. This indicates that probably the activity of red yeast rice is much higher than predicted based on the very low doses generally given to patients 26. However, whether red yeast rice preparations should be used as drugs or dietary supplements is still inconclusive. In the US, the FDA recognizes these supplements as drugs when they contain a standardized, specific amount of lovastatin 27. Despite the lipid regulating benefits, some other positive effects of red yeast rice have also been found in studies, such as improving endothelial function and insulin resistance in patients with mild or moderate hypercholesterolemia, reducing high-sensitivity C-reactive Protein (hs-CRP) and markers of vascular remodeling in Italian subjects. Moreover, red yeast rice was proved effective, safe and well tolerated in nephrotic dyslipidemia both in adults and children and in familial hypercholesterolemia children 28.
Tolerability of Red Yeast Rice Products
Two studies supported by National Center for Complementary and Integrative Health have indicated that some people who had been unable to tolerate statin drugs because of side effects (muscle pain or weakness) were able to tolerate red yeast rice. It is uncertain whether the smaller amount of monacolin K in the red yeast rice products, as compared with the amounts of active ingredients in the drugs, accounted for the greater tolerability or whether other factors were responsible.
Red yeast rice dosage
Based on Chinese clinical trials, these are the dosage used: in 19 trials prescribed Xuezhikang 600 mg once daily, one trial used Xuezhikang 600 mg three times daily if the serum total cholesterol or triglyceride is still higher after having been prescribed for 6 weeks (600 mg twice daily in previous 6 weeks) 29, one trial 30 prescribed Xuezhikang 300 mg three times daily, and one trial 31 prescribed Xuezhikang 1200 mg at night. The duration of treatment ranged from 4 weeks to 7 years 32. According to the systematic review of Xuezhikang authors (Shang Q, Liu Z, Chen K, Xu H, Liu J. A Systematic Review of Xuezhikang, an Extract from Red Yeast Rice, for Coronary Heart Disease Complicated by Dyslipidemia. Evidence-based Complementary and Alternative Medicine : eCAM. 2012;2012:636547. doi:10.1155/2012/636547. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3332166/)), “before recommending Xuezhikang as an alternative to statins in coronary heart disease patients, more rigorous trials with high quality are needed to give high level of evidence, especially for comparing the effectiveness and safety between Xuezhikang and statins”.
Xuezhikang is a partially purified extract of fermented red yeast rice (Monascus purpureus). It is composed of 13 kinds of natural statins, unsaturated fatty acids, ergosterol, amino acids, flavonoids, alkaloid, trace element, and so forth 32.
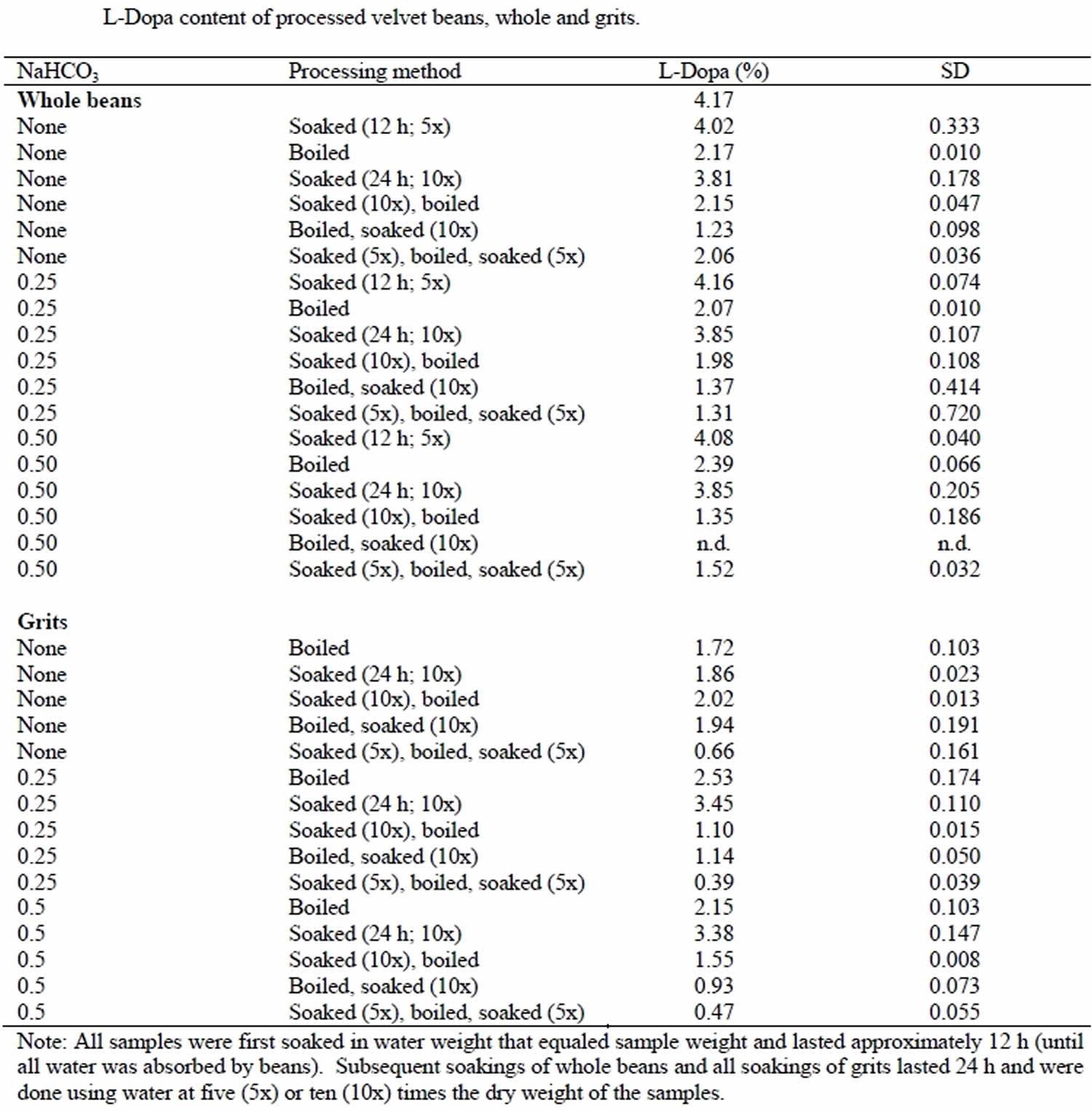
In other clinical trials conducted outside of China, their results are listed in Table 1. The red yeast rice dose per day used range from 500 mg to 3600 mg 33. This meta-analysis included 13 random, placebo-controlled trials containing 804 participants. Red yeast rice showed significant lowering effects on total total cholesterol, triglyceride, and LDL “bad” cholesterol, but did not show a significant increasing effect on HDL “good” cholesterol compared with the placebo group which was different from the result of the last meta-analysis where most of the trials used Xuezhikang as intervention 34. That Chinese meta-analysis suggested that the lipid modification effects of red yeast rice preparations appeared to be similar to simvastatin, atorvastatin, pravastatin, lovastatin or fluvastatin 34.
Table 1. Basic characteristics of included red yeast rice trials
| References | clinical trial sites | Sample size(I/C) | Intervention | Control | Doses of red yeast rice per day | Doses of lovastatin | Other ingredients | Duration of treatment |
| Ogier 2013 35 | France | 19/20 | Compounds | placebo | 500 mg | 2 mg | SCdP, artichoke leaf extract | 16 weeks |
| Barrat 2013 36 | France | 50/50 | Compounds | placebo | 500 mg | 2 mg | policosanol, artichoke leaf extract | 16 weeks |
| Barrat 2012 37 | France | 15/15/15 | Compounds | placebo | I1:500 mg/I2:1000 mg | I1:2 mg/I2:4 mg | policosanol, artichoke leaf extract | 4 weeks |
| Lee 2012 38 | Taiwan | 54/52 | Compounds | placebo | 1110 mg | No mention | bitter gourd, chlorella, soybean, licorice | 12 weeks |
| Karl 2012 39 | USA | 26/25 | Compounds | placebo | 1200 mg | 4.8 mg | niacin, phytosterol esters, L-carnitine, vitamin C,CoQ-10 | 8 weeks |
| Higashikawa 2012 40 | Japan | 28/27 | Compounds | placebo | 900 mg | 2 mg | Garlic | 12 weeks |
| Marazzi 2011 41 | Italy | 40/40 | Compounds | placebo | 200 mg | No mention | berberine, policosanol, folic acid, CoQ-10, astaxanthin | 12 months |
| Bogsrud 2010 42 | Norway | 22/20 | Red yeast rice | placebo | 1200 mg | 7.2 mg | no | 16 weeks |
| Affuso 2010 43 | Italy | 25/25 | Compounds | placebo | 200 mg | 3 mg | berberine, policosanols | 6 weeks |
| Yang 2009 44 | Taiwan | 19/10 | Compounds | placebo | 1200 mg | No mention | nattokinase | 6 months |
| Becker 2009 45 | America | 31/31 | Red yeast rice | placebo | 3600 mg | 6.12 mg | no | 24 weeks |
| Huang 2007 46 | Taiwan | 39/40 | Red yeast rice | placebo | 1200 mg | 11.4 mg | no | 8 weeks |
| Heber 1999 47 | USA | 42/41 | Red yeast rice | placebo | 2400 mg | 7.2 mg | no | 12 weeks |
Abbreviations: RYR = Red yeast rice, I = Intervention group, C = Control group, I1 = high dose group, I2 = low dose group.
[Source 33]Red yeast rice side effects
Statin drugs such as lovastatin are associated with various side effects such as headache, dizziness, rash, upset stomach, and hepatic dysfunction 3. The most common adverse effect is muscle weakness, which can be a sign of more serious myopathy or, in rare cases, rhabdomyolysis. Double-blind, controlled clinical trials have demonstrated that red yeast rice is effective and well tolerated in a wide range of patients 48; however, case reports have linked it to muscular myopathy and rhabdomyolysis.
Red yeast rice safety
- The same types of side effects that can occur in patients taking lovastatin as a drug can also occur in patients who take red yeast rice products that contain monacolin K. Potential side effects include myopathy (muscle symptoms such as pain and weakness), rhabdomyolysis (a condition in which muscle fibers break down, releasing substances into the bloodstream that can harm the kidneys), and liver toxicity. Each of these three side effects has been reported in people who were taking red yeast rice.
- Red yeast rice supplements should not be used while pregnant or breastfeeding.
- Lovastatin can interact with a variety of drugs to increase the risk of rhabdomyolysis; these drugs include other cholesterol-lowering agents, certain antibiotics, the antidepressant nefazodone, drugs used to treat fungal infections, and drugs used to treat HIV infection. Red yeast rice containing monacolin K could interact with drugs in the same way.
- If the process of culturing red yeast rice is not carefully controlled, a substance called citrinin can form. Citrinin has been shown to cause kidney failure in experimental animals and genetic damage in human cells. In a 2011 analysis of red yeast rice products sold as dietary supplements, 4 of 11 products were found to contain this contaminant.
Red yeast rice caused or exacerbated myopathy marked by elevated serum creatine kinase levels. Rhabdomyolysis, the most severe adverse effect associated with statins, occurred in a renal transplant patient who used red yeast rice while concomitantly taking cyclosporine 49. Lovastatin as a prescription drug is contraindicated in pregnancy and is a Category X agent. Category X means studies in animals or humans have demonstrated fetal abnormalities and/or there is positive evidence of human fetal risk based on adverse reaction data from investigational or marketing experience, and the risks involved in use of the drug in pregnant women clearly outweigh potential benefits. This labeling is a main reason for the FDA’s rejection of the application submitted by Merck to sell lovastatin over the counter 50. Red yeast rice, when used by pregnant women, places the fetus at unnecessary risk of central nervous system defects during the first trimester. Although red yeast rice contains a lower dose of lovastatin compared with the FDA-approved product, the risk posed may be similar.
Overall, the findings from Adverse Reactions Surveillance System of Natural Health Products raise the hypothesis that the safety profile of red yeast rice is similar to synthetic statins:
- Myopathies (muscle pain, creatine kinase and transaminases elevation) 51,
- Cutaneous reactions, gastrointestinal and liver reactions emerged as a potential safety signals of red yeast rice and have been reported as not uncommon adverse reactions associated with statins 52.
These data are also in line with cases collected from WHO‐Vigibase and reports received by the ANSES French Agency, where muscle‐ and liver‐related adverse reactions were largely reported with red yeast rice 30. In 14 cases (27%), the reactions were serious and mostly required hospitalization, especially those related to liver injury. Most of the reactions involved women, probably because they use more dietary supplements than men 53. Notably, some patients who showed muscular adverse reactions associated with statin switched to red yeast rice supplements as an alternative and experienced similar adverse effects 54. The time to onset of muscle‐ and liver‐related side effects was relatively rapid: one third of cases occurred during the first month of treatment, and two‐thirds within 60 days.
Therefore, you should know that monacolin K contained in red yeast rice is identical to lovastatin, and you should consider early monitoring of your liver function and become aware of signs of muscle injury. Furthermore, consumers should be discouraged from using red yeast rice preparations as self‐medication, particularly if they have experienced previous adverse reactions to statins 54. When self‐prescribed, without medical advice and monitoring and possibly for the long term, patients should be aware they are consuming an active substance, with both therapeutic and toxic effects. In fact, even serious adverse reactions, such as hepatitis or rhabdomyolysis, can remain asymptomatic for long periods with the risk of organ failure progression. In contrast, when statins are taken under medical control, blood tests to check creatine kinase and organ function are performed periodically so that statin use can be stopped as soon as abnormal results are shown.
The potential safety signals of myopathies and liver injury raise the hypothesis that the safety profile of red yeast rice is similar to that of statins.
Red yeast rice complications
Musculoskeletal and connective tissue disorders
Muscular pain, with or without indication of creatine phosphokinase (creatine kinase) increase, occurred in 19 patients, one case of rhabdomyolysis was also recorded. Muscle pain affected generally the lower extremities and cramps were sometimes present. The increase in creatine kinase values was mild in some cases (about 200 U/L or less), high in others (from 288 to 732 U/L) and very high (up to ten times the normal concentrations) in two patients; in the rhabdomyolysis case, the creatine kinase level reached 12,245 U/L. The latency of the reaction was variable, ranging between 9 days and more than one year; however, about one third of the reactions occurred within 30 days and 63% within two months 54. Several patients were taking medications, most of which are not known to be associated with muscular disorders, with exception of venlafaxine 55; this evidence was taken into account in the causality assessment. Noteworthy, some patients were using red yeast rice preparations as an alternative option, being intolerant to statins, while some patients previously experienced myopathy and creatine kinase increase associated with statin use; patient #15, who showed rhabdomyolysis, presented a previous identical reaction after taking statins; in these cases red yeast rice use was considered a positive rechallenge.
The causality assessment, according to the WHO scale, resulted as possible in 8 cases, probable in 11 cases and certain in case #15 54.
Gastrointestinal disorders
Gastrointestinal reactions occurred in 12 patients and consisted mostly in dyspepsia, nausea, vomiting and abdominal pain, sometimes in diarrhea 54. The reactions occurred within one or two days or after 3–4 weeks of treatment 54. The reactions were in general not serious, and only one case needed hospitalization 54. Dechallenge was always positive 54. Nevertheless, in two patients the reaction, even if improved, was persistent. Five patients had a positive rechallenge. Some cases were judged as possible because similar reactions were reported for concomitant drugs; for example, abdominal pain has been reported in more than 1% of hypertensive patients who received moexipril/hydrochlorotiazide. In causality assessment a conservative approach was used, taking in account that gastrointestinal symptoms such as nausea, vomiting and impaired digestion do not represent specific medical disorders and are common side‐effects of drugs, a positive response to withdrawal and a positive rechallenge.
Hepatobiliary disorders
Hepatic reactions occurred in ten patients and consisted in acute hepatitis (six patients), that required hospitalization, or in increase of hepatic enzymes. These six cases fulfilled criteria for drug‐induced liver injury, according to the international Expert Working Group 56. In one patient increase in aspartate aminotransferase (AST) occurred together with creatine phosphokinase (creatine kinase) increase. Nine cases were compatible with the definition of drug‐induced liver injury, as recommended by the International Drug Safety Department 57.
Time to onset was between two weeks and one year of treatment, 37% of the reactions occurred within one month and 75% within two months 54. Transaminases ranged from about twice to 80 times the upper normal level 54. In all cases, except for two in which it was not reported, dechallenge was positive. Patients were taking other drugs or dietary supplements, but generally these were not known to be associated with liver injury 54. Case 26 regarded a 49‐year‐old woman who had consumed Armolipid plus® for 50 days. She was hospitalized for suspected myocardial infarction (heart attack); the infarction was excluded, while an acute hepatitis was diagnosed with alanine aminotransferase (ALT) and aspartate aminotransferase (AST) values corresponding to 46 and 57 times the upper normal concentrations, respectively. Total bilirubin was normal, viral and autoimmune serology was negative and other drug and non‐drug causes of hepatitis were excluded; rhabdomyolysis was also excluded. The supplement consumption was suspended and the values decreased, returning to normal after 17 days. The causality assessment resulted in a score of 7 (probable).
Case 27 was a 68‐year‐old woman who had a history of hypercholesterolemia, for which she had been taking the supplement Armolipid plus® for 2 years. An increase of liver enzymes (AST = 87; ALT = 168) was discovered during a routine blood test, and an additional laboratory test reported total bilirubin 0.5, gamma-glutamyl transferase (GGT) 53, ALP 109. Creatine kinase was normal, thus suggesting that the increase in transaminases did not have a muscular aetiology. The ultrasound scan did not show clinically important signs of hepato‐biliary damage, except for moderate steatosis and lithiasis (previously documented). Major infective hepatitis was excluded, no recent flu‐like episodes or gastro‐intestinal symptoms were documented, no history of pre‐existing liver or biliary disease, blood transfusion or alcohol abuse was documented. Concomitant drugs included levothyroxine (for 7 years) and chenodeoxycholic acid (for 5 years). The product was suggested by healthcare professionals because of the patient’s complaints about symptomatic myopathy, probably related to the administration of statins. Notably, no elevation of creatine kinase or hepatic transaminases was previously documented when the woman was treated with statins. The supplement was suspended. Laboratory blood test, performed after less than 4 weeks, gave normal values for AST, ALT, gamma-glutamyl transferase (GGT) and ALP. Based on transaminases values, the liver injury was classified as hepatocellular and the application of the CIOMS/RUCAM scale resulted in a score of 5 (possible).
Skin and subcutaneous disorders
Nine cutaneous reactions associated with red yeast rice products were collected. Most of them occurred with a 1–4‐day latency, the others occurred after 10 days, 3 weeks or about 2 months of treatment 54. Four cases were serious and required hospitalization 54. In one case, which involved a 57‐year‐old woman, the reaction consisted in Pemphigus vulgaris, diagnosed by titration of anti‐desmoglein 1 and 3 antibodies and by biopsy. The reaction occurred 2 weeks after discontinuation of the supplement and needed a pharmacological therapy. The patient was not taking other drugs, but as she had experienced a previous episode of Pemphygus eight years before, then the reaction could be considered a relapse and the causality was judged as unlikely. Another patient a 75‐year‐old woman was admitted to hospital with severe urticaria, rash and edema of the lips. The reaction improved after discontinuation of the red yeast rice supplement but recurred after 6 days, without rechallenge. It is to be underlined that this patient, even without predisposing conditions, was taking various medications (cardioaspirin, amiloride plus hydrochlorothiazide, triazolam, metoprolol, lansoprazole, risedronate), but, most importantly, received flu vaccine 5 days before the onset of the reaction, so, also in this case, the causality was judged as unlikely. In other cases, the causality was judged as possible despite a positive outcome and dechallenge, because patients were taking other drugs, some of which (amiloride plus hydrochlorothiazide, lansoprazole) have been reported to induce cutaneous adverse reactions 58. Another patient was hospitalized for DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome, a severe drug‐induced reaction 59. Laboratory tests suggested that the patient had a Herpes simples virus infection, a condition associated with DRESS 60. Cases of DRESS are described also for atorvastatine 61, a lipid‐lowering drug belonging to the statins class (as monacolin contained in red yeast rice), as well as for omeprazole 62, a proton pump inhibitor similar to pantoprazole that the patient was consuming. Based on these considerations, the causality between DRESS and red yeast rice preparation was judged as possible. In the other cutaneous reactions the causality was considered as probable on the basis of the positive dechallenge, outcome and absence of alternative causes.
Others miscellaneous reactions
A case involved a 78‐year‐old woman who was taking warfarin and showed an increase of INR (international normalized ratio) after consumption of the red yeast rice supplement Armolipid plus®. The dose of warfarin was reduced and the INR value returned to normal. In this case a pharmacodynamic interaction between the anticoagulant drug and red yeast rice supplement can be hypothesized, even if the dechallenge was not done 54. Furthermore, three patients showed nausea, vertigo and hazy vision, tingling of extremities and tachycardia; the latter required hospitalization 54. Dechallenge was always positive and other causes were excluded, so the causality was judged as probable in all of them 54. Nevertheless, it has to be pointed out that in one case the supplement consumed (Fisiostatin®) contained, besides red yeast rice, green tea [Camellia sinensis (L.) O. Kuntze], a source of caffeine that could be responsible for the adverse reaction 54.
- Smith DJ, Olive KE. Chinese red rice–induced myopathy. South Med J. 2003;96(12):1265–1267. https://www.ncbi.nlm.nih.gov/pubmed/14696880[↩]
- Lachenmeier DW, Monakhova YB, Kuballa T et al. NMR evaluation of total statin content and HMG‐CoA reductase inhibition in red yeast rice (Monascus spp.) food supplements. Chin Med, 2012;7:8.[↩]
- Klimek M, Wang S, Ogunkanmi A. Safety and Efficacy of Red Yeast Rice (Monascus purpureus) as an Alternative Therapy for Hyperlipidemia. Pharmacy and Therapeutics. 2009;34(6):313-327. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2697909/[↩][↩]
- Wang J, Lu Z, Chi J et al. Multicenter clinical trial of the serum lipid‐lowering effects of a Monascus purpureus (red yeast) rice preparation from traditional Chinese medicine. Curr Ther Res, 1997;58:964–978.[↩][↩]
- Huang CF, Li TC, Lin CC, Liu CS, Shih HC, Lai MM. Efficacy of Monascus purpureus Went rice on lowering lipid ratios in hypercholesterolemic patients. Eur J Cardiovasc Prev Rehabil, 2007;14:438–440.[↩]
- Heber D, Yip I, Ashley JM, Elashoff DA, Elashoff RM, Go VLW. Cholesterol‐lowering effects of a proprietary chinese red‐yeast‐rice dietary supplement. Am J Clin Nutr, 1999;69:231–236.[↩][↩]
- Ma J, Li Y, Ye Q, Li J, Hua Y, Ju D, et al. Constituents of red yeast rice, a traditional Chinese food and medicine. J Agric Food Chem 2000; 48: 5220–5225 https://www.ncbi.nlm.nih.gov/pubmed/11087463[↩]
- Moriarty PM, Roth EM, Karns A, Ye P, Zhao SP, Liao Y, et al. Effects of Xuezhikang in patients with dyslipidemia: a multicenter, randomized, placebo‐controlled study. J Clin Lipidol 2014; 8: 568–575.[↩]
- Gonnelli S, Caffarelli C, Stolakis K, Cuda C, Giordano N, Nuti R. Efficacy and tolerability of a nutraceutical combination (red yeast rice, policosanols, and berberine) in patients with low‐moderate risk hypercholesterolemia: a double‐blind, placebo‐controlled study. Curr Ther Res Clin Exp 2014; 77: 1–6.[↩]
- Marazzi G, Pelliccia F, Campolongo G, Quattrino S, Cacciotti L, Volterrani M, et al. Usefulness of nutraceuticals (Armolipid plus) versus ezetimibe and combination in statin‐intolerant patients with dyslipidemia with coronary heart disease. Am J Cardiol 2015; 116: 1798–1801[↩]
- Ong YC, Aziz Z. Systematic review of red yeast rice compared with simvastatin in dyslipidaemia. J Clin Pharm Ther 2016; 41: 170–179.[↩]
- Childress L, Gay A, Zargar A, Ito MK. Review of red yeast rice content and current Food and Drug Administration oversight. J Clin Lipidol 2013; 7: 117–122.[↩]
- Systematic review of red yeast rice compared with simvastatin in dyslipidaemia. Journal of Clinical Pharmacy and Therapeutics Volume 41, Issue 2, April 2016. https://doi.org/10.1111/jcpt.12374[↩][↩][↩][↩]
- Chen L, Liu J. The effects of xuezhikang on hypercholesterolemia. Herald Med, 2002;21:31–32. Chinese.[↩]
- Hua D. Comparative observation of simvastatin and xuezhikang for patients with hyperlipidemia. Hebei Med, 2008;14:879–881. Chinese.[↩][↩][↩]
- Li KL. Comparison study on the effect of xuezhikang and simvastatin for treatment of primary hypercholesterolemia. China Med Herald, 2006;3:22–23. Chinese.[↩][↩]
- Quan SL, Wang W, Qu XW. The effect of xuezhikang and simvastatin on lipid of hypercholesterolemia. China Clinical Pract Med, 2008;2:91–92. Chinese.[↩]
- Qi MY, Zhang J, Xiao JG. Clinical observation of xuezhikang on treatment of 112 cases of hypercholesterolemia. Pract Clin Med, 2004;5:20–22. Chinese.[↩]
- Li LH, Deng GL, Chen YZ. Clinical comparative study of simvastatin and xuezhikang and pravastatin in common doses on primary hyperlipidemia. J Chongqing Med University, 2005;30:278–284. Chinese.[↩]
- Flajs D, Peraica M. Toxicological properties of citrinin. Arh Hig Rada Toksikol 2009; 60: 457–464.[↩]
- Burke FM. Red yeast rice for the treatment of dyslipidemia. Curr Atheroscler Rep 2015; 17: 495.[↩]
- Ross SM (2012) Red yeast rice: efficacy and tolerability of Monascus purpureus yeast, for treatment of hyperlipidemia in patientswith statin-associated myalgias. Holist Nurs Pract 26: 173–175. https://www.ncbi.nlm.nih.gov/pubmed/22517353[↩]
- Venero CV, Venero JV, Wortham DC, Thompson PD (2010) Lipid-lowering efficacy of red yeast rice in a population intolerant to statins. Am J Cardiol 105: 664–666. https://www.ncbi.nlm.nih.gov/pubmed/20185013[↩]
- Lu JH, Bonovich K, Colfer H, Davidson M, Dujovne CA, et al. (2013) Clinical study on cholesterol-lowering effect of RYR Cholestin among Americans with moderate hy-percholesterolemia. Shanghai Journal of Preventive Medicine 25: 501–506.[↩]
- Yang CW, Mousa SA (2012) The effect of red yeast rice (Monascus purpureus) in dyslipidemia and other disorders. Complement Ther Med 20: 466–474.[↩]
- Chen CH1, Yang JC, Uang TD, Lin CJ (2013) Improved dissolution rate and oral bioavailability of lovastatin in red yeast rice products. Int J Pharm 444: 18–24[↩]
- Gordon RY, Cooperman T, Obermeyer W, Becker DJ (2010) Marked variability of monacolin levels in commercial red yeast rice products: buyer beware! Arch Intern Med. 170: 1722–1727.[↩]
- Guardamagna O, Abello F, Baracco V, Stasiowska B, Martino F (2011) The treatment of hypercholesterolemic children: efficacy and safety of a combination of red yeast rice extract andpolicosanols. Nutr Metab Cardiovasc Dis 21: 424–429 https://www.ncbi.nlm.nih.gov/pubmed/20153154[↩]
- Qi BL, Zhang GJ, Suo XX. Efficiency rate of Xuezhikang for angina pectoris. Chinese Journal of Primary Medicine and Pharmacy. 2001;8(6):p. 547.[↩]
- Zhang X. Clinical effect of atorvastatin calcium tablets for coronary heart disease complicated with hyperlipidemia. Hebei Medical Journal. 2011;33(6):882–883.[↩]
- Shang XB. Clinical observation of Xuezhikang and atorvastatin for patients with coronary heart disease complicated with dyslipidemia on serum lipid and hemorheology. Guangxi Medical Journal. 2007;29(8):1158–1159.[↩]
- Shang Q, Liu Z, Chen K, Xu H, Liu J. A Systematic Review of Xuezhikang, an Extract from Red Yeast Rice, for Coronary Heart Disease Complicated by Dyslipidemia. Evidence-based Complementary and Alternative Medicine : eCAM. 2012;2012:636547. doi:10.1155/2012/636547. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3332166/[↩][↩]
- Li Y, Jiang L, Jia Z, et al. A Meta-Analysis of Red Yeast Rice: An Effective and Relatively Safe Alternative Approach for Dyslipidemia. Calabresi L, ed. PLoS ONE. 2014;9(6):e98611. doi:10.1371/journal.pone.0098611. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4045580[↩][↩]
- Liu J, Zhang J, Shi Y, Grimsgaard S, Alraek T, et al. (2006) Chinese red yeast rice (Monascus purpureus) for primary hyperlipidemia: a meta-analysis of randomizedcontrolled trials. Chin Med 1: 4. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1761143/[↩][↩]
- Ogier N, Amiot MJ, Georgé S, Maillot M, Mallmann C, et al. (2013) LDL-cholesterol-lowering effect of a dietary supplement with plant extracts in subjects with moderatehypercholesterolemia. Eur J Nutr 52: 547–557.[↩]
- Barrat E, Zaïr Y, Ogier N, Housez B, Vergara C, et al. (2013) A combined natural supplement lowers LDL cholesterol in subjects with moderate untreatedhypercholesterolemia: a randomized placebo-controlled trial. Int J Food Sci Nutr 64: 882–889.[↩]
- Barrat E, Zaïr Y, Sirvent P, Chauveau P, Maudet C, et al. (2012) Effect on LDL-cholesterol of a large dose of a dietary supplement with plant extracts in subjects with untreatedmoderate hypercholesterolaemia: a randomised, double-blind, placebo-controlled study. Eur J Nutr 52: 1843–1852.[↩]
- Lee IT, Lee WJ, Tsai CM, Su IJ, Yen HT, et al. (2012) Combined extractives of red yeast rice, bitter gourd, chlorella, soy protein, and licorice improve total cholesterol, low-density lipoprotein cholesterol, and triglyceride in subjects with metabolic syndrome. Nutr Res 32: 85–92.[↩]
- Karl M, Rubenstein M, Rudnick C, Brejda J (2012) A multicenter study of nutraceutical drinks for cholesterol (evaluating effectiveness and tolerability). J Clin Lipidol 6: 150–158.[↩]
- Higashikawa F, Noda M, Awaya T, Ushijima M, Sugiyama M (2012) Reduction of serum lipids by the intake of the extract of garlic fermented with Monascus pilosus: a randomized,double-blind, placebo-controlled clinical trial. Clin Nutr 31: 261–266.[↩]
- Marazzi G, Cacciotti L, Pelliccia F, Iaia L, Volterrani M, et al. (2011) Long-term effects of nutraceuticals (berberine, red yeast rice, policosanol) in elderly hypercholesterolemicpatients. Adv Ther 28: 1105–1113.[↩]
- Bogsrud MP, Ose L, Langslet G, Ottestad I, Strøm EC, et al. (2010) HypoCol (red yeast rice) lowers plasma cholesterol – a randomized placebo controlled study. Scand Cardiovasc J 44: 197–200.[↩]
- Affuso F, Ruvolo A, Micillo F, Saccà L, Fazio S (2010) Effects of a nutraceutical combination (berberine, red yeast rice and policosanols) on lipid levels and endothelialfunction randomized, double-blind, placebo-controlled study. Nutr Metab Cardiovasc Dis 20: 656–661.[↩]
- Yang NC, Chou CW, Chen CY, Hwang KL, Yang YC (2009) Combined nattokinase with red yeast rice but not nattokinase alone has potent effects on blood lipids in humansubjects with hyperlipidemia. Asia Pac J Clin Nutr 18: 310–317[↩]
- Becker DJ, Gordon RY, Halbert SC, French B, Morris PB, et al. . (2009) Red yeast rice for dyslipidemia in statin-intolerant patients: a randomized trial.Ann Intern Med 150: : 830–839, W147–149.[↩]
- Huang CF, Li TC, Lin CC, Liu CS, Shih HC, et al. (2007) Efficacy of Monascus purpureus Went rice on lowering lipid ratios in hypercholesterolemic patients. Eur J Cardiovasc Prev Rehabil 14: 438–440.[↩]
- Heber D, Yip I, Ashley JM, Elashoff DA, Elashoff RM, et al. (1999) Cholesterol-lowering effects of a proprietary Chinese red-yeast-rice dietary supplement. Am J Clin Nutr 69: 231–236.[↩]
- Lu Z, Kou W, Du B, et al. Effect of Xuezhikang, an extract from red yeast Chinese rice, on coronary events in a Chinese population with previous myocardial infarction. Am J Cardiol. 2008;101(12):1689–1693.[↩]
- Prasad GV, Wong T, Meliton G, et al. Rhabdomyolysis due to red yeast rice (Monascus purpureus) in a renal transplant recipient. Transplantation. 2002;74(8):1200–1201. https://www.ncbi.nlm.nih.gov/pubmed/12438974[↩]
- Dyer O. FDA rejects sale of over the counter statins. BMJ. 2005;330(7484):164. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC545018/[↩]
- Perdices EV, Medina‐Caliz I, Hernando S, Ortega A, Martin‐Ocana F, Navarro JM, et al. Hepatotoxicity associated with statin use: analysis of the cases included in the Spanish Hepatotoxicity Registry. Rev Esp Enferm Dig 2014; 106: 246–254.[↩]
- Wilinski J, Dabrowski M. Safety and tolerability of the use of atorvastatin 40 mg in common daily practice in short‐term observation in 3227 patients. Przegl Lek 2013; 70: 373–376.[↩]
- Schwab S, Heier M, Schneider A, Fischer B, Huth C, Peters A, et al. The use of dietary supplements among older persons in southern Germany – results from the KORA‐age study. J Nutr Health Aging 2014; 18: 510–519.[↩]
- Mazzanti G, Moro PA, Raschi E, Da Cas R, Menniti‐Ippolito F. Adverse reactions to dietary supplements containing red yeast rice: assessment of cases from the Italian surveillance system. British Journal of Clinical Pharmacology. 2017;83(4):894-908. doi:10.1111/bcp.13171. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5346868/[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Huang SS, Yang HY, Lin YC, Chan CH. Low‐dose venlafaxine‐induced severe rhabdomyolysis: a case report. Gen Hosp Psychiatry 2012; 34: 436–437.[↩]
- Aithal GP, Watkins PB, Andrade RJ, Larrey D, Molokhia M, Takikawa H, et al. Case definition and phenotype standardization in drug‐induced liver injury. Clin Pharmacol Ther 2011; 89: 806–815.[↩]
- Danan G, Benichou C. Causality assessment of adverse reactions to drugs – I. A novel method based on the conclusions of international consensus meetings: application to drug‐induced liver injuries. J Clin Epidemiol 1993; 46: 1323–1330. https://www.ncbi.nlm.nih.gov/pubmed/8229110[↩]
- Thestrup‐Pedersen K. Adverse reactions in the skin from anti‐hypertensive drugs. Dan Med Bull 1987; 34 (Suppl 1): 3–5.[↩]
- Avancini J, Maragno L, Santi CG, Criado PR. Drug reaction with eosinophilia and systemic symptoms/drug‐induced hypersensitivity syndrome: clinical features of 27 patients. Clin Exp Dermatol 2015; 40: 851–859.[↩]
- Descamps V, Ranger‐Rogez S. DRESS syndrome. Joint Bone Spine 2014; 81: 15–21.[↩]
- Gressier L, Pruvost‐Balland C, Dubertret L, Viguier M. Atorvastatin‐induced drug reaction with eosinophilia and systemic symptoms (DRESS). Ann Dermatol Venereol 2009; 136: 50–53.[↩]
- Bourneau‐Martin D, Leclech C, Jamet A, Drablier G, Trenque T, Juengel K, et al. Omeprazole‐induced drug reaction with eosinophilia and systemic symptoms (DRESS). Eur J Dermatol 2014; 24: 413–415.[↩]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}