Contents
What is GABA
GABA is short for Gamma-Aminobutyric Acid, which is a naturally occurring inhibitory neurotransmitter with central nervous system (CNS) inhibitory activity. GABA acts by binding to specific transmembrane receptors in the plasma membrane of both presynaptic and postsynaptic neurons in your brain. This binding causes the opening of ion channels to allow either the flow of negatively-charged chloride ions into the cell or positively-charged potassium ions out of the cell. This will typically result in a negative change in the transmembrane potential, usually causing hyperpolarization. Three general classes of GABA receptor are known 1). These include GABA-A and GABA-C ionotropic receptors, which are ion channels themselves, and GABA-B metabotropic receptors, which are G protein-coupled receptors that open ion channels via intermediaries known as G proteins 2). Activation of the GABA-B receptor by GABA causes neuronal membrane hyperpolarization and a resultant inhibition of neurotransmitter release. In addition to binding sites for GABA, the GABA-A receptor has binding sites for benzodiazepines, barbiturates, and neurosteroids. GABA-A receptors are coupled to chloride ion channels. Therefore, activation of the GABA-A receptor induces increased inward chloride ion flux, resulting in membrane hyperpolarization and neuronal inhibition 3). After release into the synapse, free GABA that does not bind to either the GABA-A or GABA-B receptor complexes can be taken up by neurons and glial cells. Four different GABA membrane transporter proteins (GAT-1, GAT-2, GAT-3, and BGT-1), which differ in their distribution in the central nervous system (CNS), are believed to mediate the uptake of synaptic GABA into neurons and glial cells. The GABA-A receptor subtype regulates neuronal excitability and rapid changes in fear arousal, such as anxiety, panic, and the acute stress response 4). Drugs that stimulate GABA-A receptors, such as the benzodiazepines and barbiturates, have anxiolytic and anti-seizure effects via GABA-A-mediated reduction of neuronal excitability, which effectively raises the seizure threshold. GABA-A antagonists (blockers) produce convulsions in animals and there is decreased GABA-A receptor binding in a positron emission tomography (PET) study of patients with panic disorder. Neurons that produce GABA as their output are called GABAergic neurons and have chiefly inhibitory action at receptors in the vertebrate. Medium spiny neurons are a typical example of inhibitory CNS GABAergic cells. GABA is known for its analgesic effects, anti-anxiety, and hypotensive activity. GABA has also been shown to have excitatory roles in the vertebrate, most notably in the developing cortex.
Although GABA usually induces hyperpolarization in adult neurons, GABA has also been shown to exert depolarizing responses in the immature CNS structures and CNS tumors 5). In particular, GABA increased the proliferation of immature cerebellar granule cells through the activation of GABA-A receptors and voltage-dependent calcium channels 6). Takehara et al. 7) reported that GABA stimulated pancreatic cancer growth through GABRP (GABA-A receptor pi subunit) by increasing intracellular Ca2+ levels and activating the mitogen-activated protein kinase/extracellular signal-regulated kinase cascade. Also, Minuk et al. 8) reported that human hepatocellular carcinoma tissues were depolarized compared with adjacent non-tumor tissues. From the results above, it was deduced that GABA may promote the malignant liver cell lines HepG2 cell proliferation through gamma-aminobutyric acid A receptor θ subunit (GABRQ) by voltage-dependent calcium channels. Interestingly, GABA inhibited the growth of the gamma-aminobutyric acid A receptor θ subunit (GABRQ)-knockdown malignant liver cell lines HepG2 cells. This indicates that GABA activates some other receptors to inhibit the proliferation without gamma-aminobutyric acid A receptor θ subunit (GABRQ), which is identical to some previous reports 9). Hepatocellular carcinoma tissues have increased gamma-aminobutyric acid A receptor θ subunit (GABRQ) receptor expression 10). Knockdown of gamma-aminobutyric acid A receptor θ subunit (GABRQ) expression in receptor-expressing malignant hepatocytes results in attenuated in vitro and in vivo tumor growth 11). Moreover, GABA promotes hepatocyte proliferation through GABRQ. These findings highlight the importance of elucidating the role of GABAergic activity in the pathogenesis of hepatocellular carcinoma. They also raise the potential for new therapeutic and diagnostic approaches to human hepatocellular carcinoma.
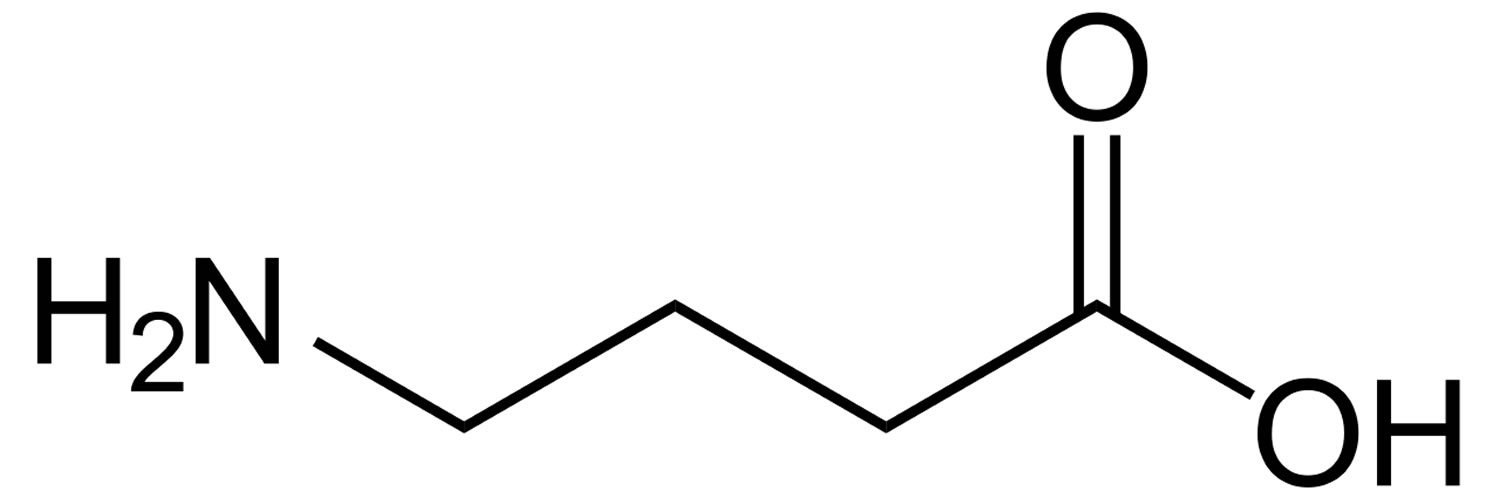
Figure 1. GABA (Gamma-Aminobutyric Acid)
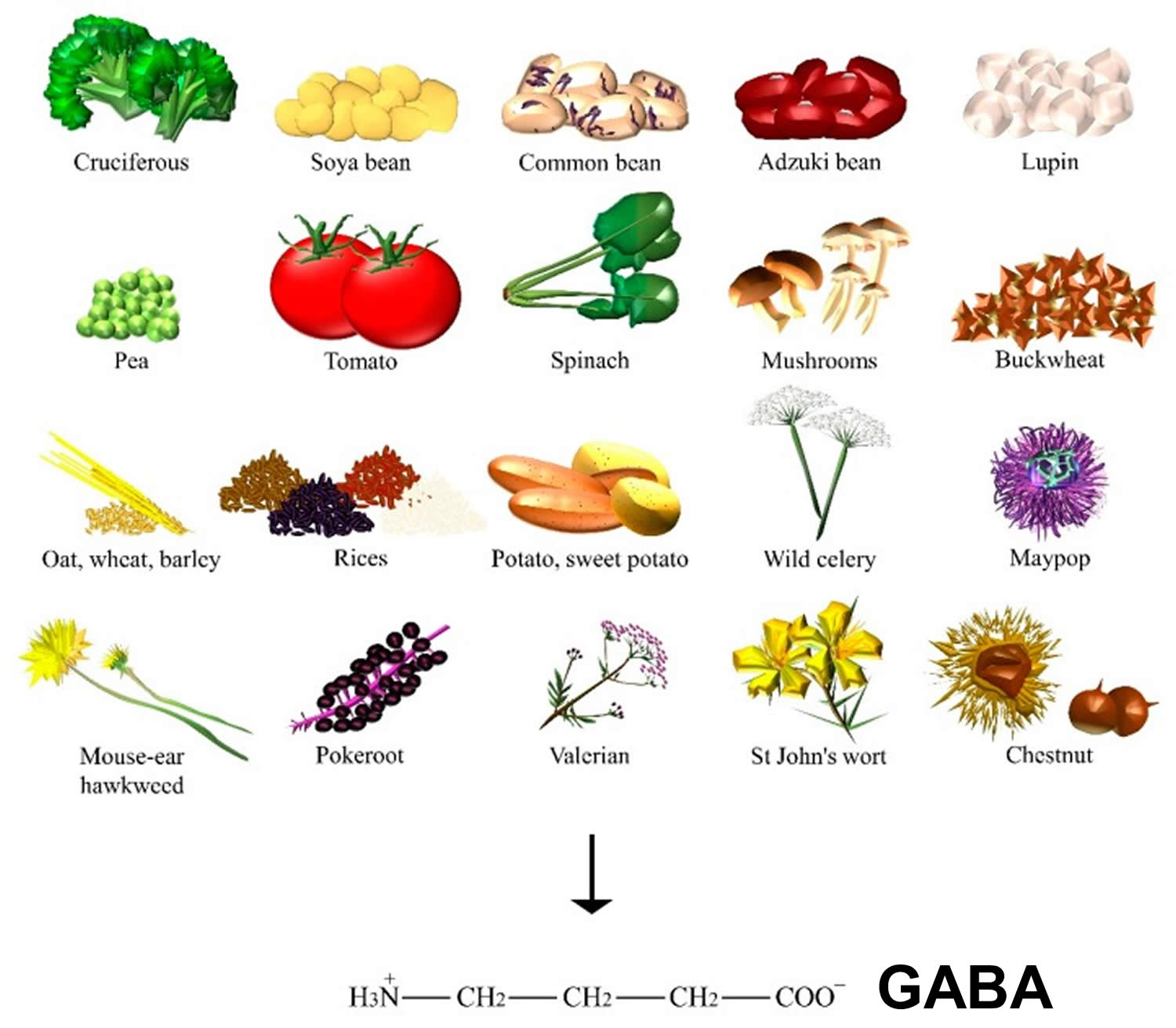
 GABA dietary sources
GABA dietary sources
GABA is found ubiquitously among plants (Figure 2), where GABA can be primarily synthesized from glutamic acid via glutamate decarboxylase enzyme. Levels of GABA were demonstrated to increase in response to biotic and abiotic stresses, such as drought, the presence of salt, wounds, hypoxia, infection, soaking, and germination 12). In particular, sprouts of Lupinus angustifolius L. (that is, lupin) 13), Vigna angularis W. (that is, adzuki bean) 14) and other germinating edible beans, such as Glycine max L. (that is, soya bean) 15), common bean, and pea 16), were reported to increase GABA content when compared to their raw beans. Furthermore, grains of the Gramineae family, such as Avena nuda L. (that is, oat) 17), Triticum aestivum L. (that is, wheat) 18), Hordeum vulgare L. (that is, barley) 19), and many species of the Oryza genus (for example, white, black, brown, and red rice) 20) can also significantly accumulate GABA. Sprouts of Fagopyrum esculentum M. (that is, buckwheat) 21) and the fruits of tomato also contain a substantial amount of this amino acid during the mature green stage 22).
Food technologies and molecular engineering are employed to synthesize GABA through enzymatic or whole-cell biocatalysis, microbial fermentation (for example, GABA soya yogurt 23), black raspberry juice 24), and chemical synthesis 25). Some authors found one of the highest contents on GABA to be 414 nmol/g of dry weight in raw spinach, followed by Solanum tuberosum L. (that is, potato), Ipomoea batatas L. (that is, sweet potato), and Brassica oleracea L. (that is, cruciferous such as kale and broccoli). Mushrooms, such as Lentinula edodes B. (that is, shiitake), and nuts of Castanea genus (that is, chestnut) also showed a significant amount of GABA 26). Among the many types of Chinese teas, the highest content was found in white tea 27). GABA content was found in mistletoe 28), but also in Phytolacca americana L. (that is, pokeroot) 29), Valeriana officinalis L. (that is, valerian), Angelica archangelica L.(that is, wild celery), Hypericum perforatum L. (that is, St John’s wort), Hieracium pilosella L. (that is, mouse-ear hawkweed), and Passiflora incarnata L. (that is, maypop) 30), the latter being used for the relief of mild symptoms of mental stress and as a sleep aid.
Moreover, the benefit from the consumption of GABA-containing vegetables showed the importance of dietary GABA on the sympathetic nerve activity 31). Conversely, there is still discordance over the alleged GABA capacity to cross the blood-brain barrier 32).
Figure 2. GABA dietary sources
GABA Synthesis
GABA synthesis occurs from glutamate or L-glutamate (the principal excitatory neurotransmitter in the brain) via the decarboxylation of L-glutamate by the enzyme L-glutamic acid decarboxylase (GAD)—a single-step, irreversible reaction dependent on the availability of the cofactor pyridoxal-50-phosphate (a vitamer of vitamin B6) 34). It is worth noting that this involves converting the principal excitatory neurotransmitter glutamate into GABA the principal inhibitory one. Gamma-aminobutyric acid (GABA) plays a role in regulating neuronal excitability by binding to its receptors, GABA-A and GABA-B, and thereby causing ion channel opening, hyperpolarization and eventually inhibition of neurotransmission.
Two major isoforms of the enzyme L-glutamic acid decarboxylase (GAD) exist—a 65 kDa isoform (GAD65) and a 67 kDa isoform (GAD67); GAD65 is the major isoform of GAD expressed in the mammalian brain 35), localized primarily to the axon terminals of synaptosomes and interacting readily with the plasma membrane, whereas GAD67 is more widely distributed in the cytosol of cells 36). The differential distribution of the two GAD isoforms corresponds well with the presence of two pools of intracellular GABA—one of which is found in vesicles and the other in the cytosol, and which are released by different mechanisms 37). It has thus been suggested that GAD65, being localized in the synaptic bouton, plays a role in the synthesis of GABA released via a vesicular mechanism 38), whereas GAD67 likely mediates the synthesis of the cytoplasmic pool of GABA 39).
What does GABA do?
GABA (gamma-aminobutyric acid) is the major inhibitory neurotransmitter in the brain 40) and is produced from glutamate by l-glutamic acid decarboxylase (GAD) within GABAergic neurons 41). GABA is then metabolized to succinic acid semialdehyde by GABA transaminase and then to succinate, mainly within astrocytic mitochondria 42). GABA is needed for normal brain function, synaptic plasticity, cortical adaptation and reorganization 43).
GABA is crucially important in the regulation of responsiveness and excitability in human cortical networks 44) and in the synchronization of cortical neuronal signaling activity by networks of cortical interneurons 45). The ubiquity of GABAergic regulation in the CNS gives this neurotransmitter a central role in a very wide range of physiological and biochemical processes—GABAergic control is involved in the regulation of cognition 46), memory and learning 47), motor function 48), circadian rhythms 49), neural development 50), adult neurogenesis 51), and sexual maturation52). Dysfunction in GABAergic signaling is known to be a central factor in the pathogenesis of several neurological disorders, with the role of GABA in epilepsy and the maintenance of the inhibitory-excitatory (E/I) balance in the human cortex being the subject of significant topic in research 53). The contribution of GABAergic dysfunction to
disorders such as epilepsy [genetic epilepsies] 54), Alzheimer’s disease 55), major depressive disorder 56), anxiety 57), autism 58), schizophrenia 59) and bipolar disorder 60) is also known or suspected, with many lines of evidence pointing to the underlying contribution of defects in the signaling system 61). GABAergic inhibition may be one of the mechanisms involved in use-dependent plasticity in the intact human motor cortex 62). Changes in GABA concentration in the sensorimotor cortex during motor learning have been demonstrated 63). One pilot study in relapsing-remitting multiple sclerosis (MS) 64) found that reduced motor performance correlated with increased GABA levels in the sensorimotor cortex. The increased GABA concentration was also associated with increased motor activation on functional MRI. Despite limitations, these in vivo results suggest that cortical reorganization occurring in the sensorimotor cortex in patients with relapsing-remitting multiple sclerosis, as reflected by increased functional MRI response, is linked with increased GABA levels, and is a possible compensatory mechanism that maintains motor function 65). The observed reduced GABA levels in the hippocampus and sensorimotor cortex in patients with secondary progressive multiple sclerosis when compared with healthy controls raises the possibility that GABA may be a marker of neurodegeneration in the brain. The reduction in GABA levels may reflect a combination of reduced GABA receptor levels and decreased density of inhibitory interneuron processes in the motor cortex in patients with progressive multiple sclerosis, which have been described by a previous histological study 66). Lower GABA levels in the sensorimotor cortex of multiple sclerosis patients are associated with reduced motor performance. These findings raise the possibility that altered GABA neurotransmission may be a marker of neurodegeneration, but it may also suggest that GABA is a mechanism of neurodegeneration in progressive multiple sclerosis patients. Thus, the GABAergic system has long been a major target in the development of treatment strategies for these conditions. Following is a brief description of the major components of the GABAergic system (summarized in Figure 3).
Figure 3. GABA signaling system
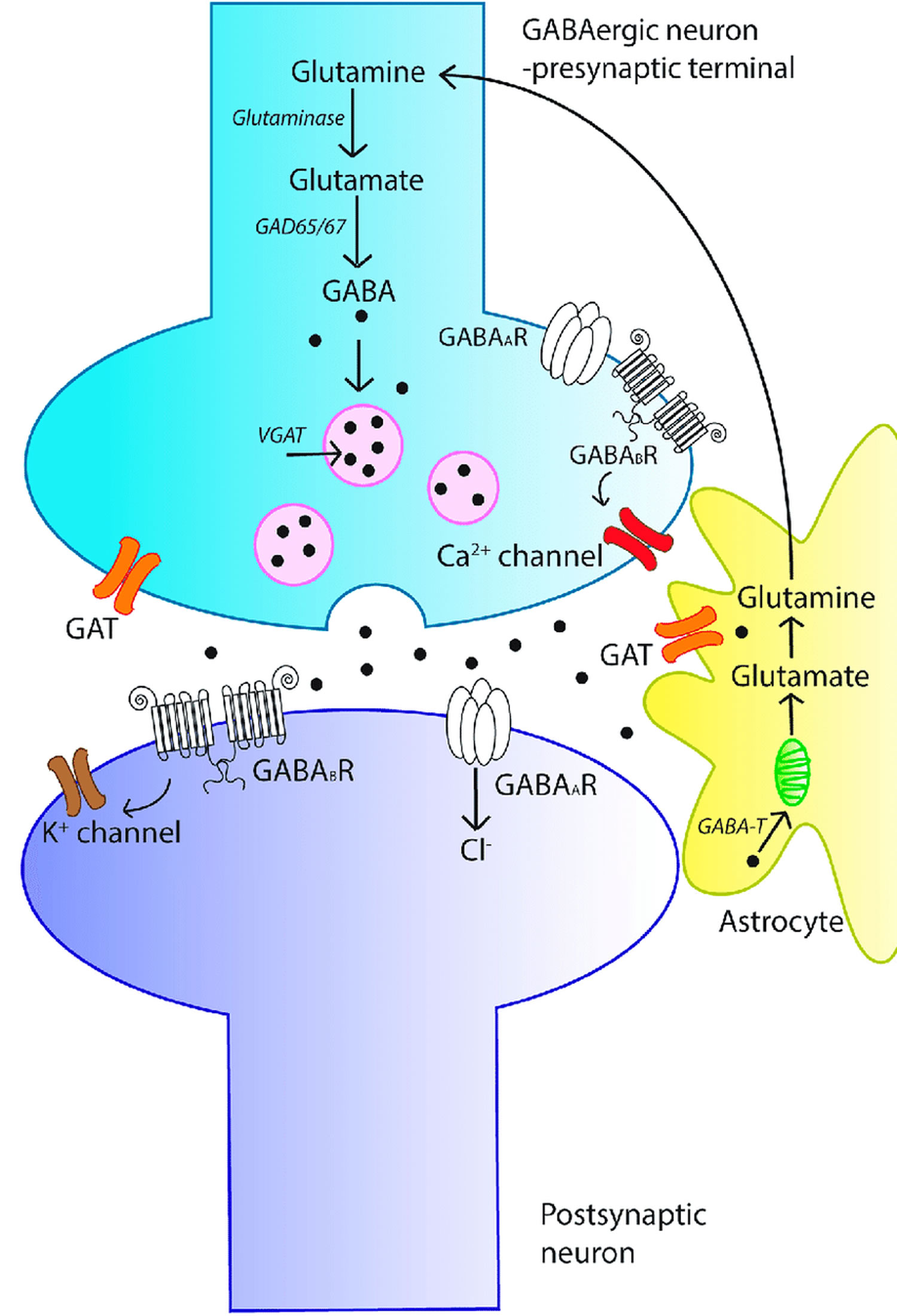
Footnotes: An overview of the γ-aminobutyric acid (GABA) signaling system. The schematic diagram represents a GABAergic synapse and depicts the key aspects of GABAergic signal transduction. GABA is synthesized in the pre-synaptic terminal from glutamate by glutamic acid decarboxylase (GAD). GABA is then recruited into synaptic vesicles via the action of vesicular GABA transporter (vGAT). Following membrane depolarization, GABA is released into the synapse and can bind to either ionotropic GABAA receptors (GABAAR) or metabotropic GABAB receptors (GABABR) on the postsynaptic membrane, resulting in inhibition of the post-synaptic neuron. Released GABA is cleared from the synapse by membrane-bound GABA transporters (GATs), localized to neurons and astrocytes. In astrocytes, GABA is recycled into synaptic vesicles or taken up by mitochondria, where it is metabolized by GABA transaminase (GABA-T) to glutamine for neuronal uptake.
Drugs that act as agonists of GABA receptors (known as GABA analogs or GABAergic drugs), or increase the available amount of GABA typically have relaxing, anti-anxiety, and anti-convulsive effects. GABA is found to be deficient in cerebrospinal fluid (CSF) and the brain in many studies of experimental and human epilepsy. Benzodiazepines (such as Valium) are useful in status epilepticus because they act on GABA receptors. GABA increases in the brain after administration of many seizure medications. Hence, GABA is clearly an antiepileptic nutrient.
Central nervous system (CNS) depressants, a category that includes tranquilizers, sedatives, and hypnotics, are substances that can slow brain activity. This property makes them useful for treating anxiety and sleep disorders. Most central nervous system (CNS) depressants act on the brain by increasing activity at receptors for the inhibitory neurotransmitter GABA (gamma-aminobutyric acid). Although the different classes of depressants work in unique ways, it is through their ability to increase GABA signaling—thereby increasing inhibition of brain activity—that they produce a drowsy or calming effect that is medically beneficial to those suffering from anxiety or sleep disorders 67).
The following are among the medications commonly prescribed for these purposes 68):
- Benzodiazepines, such as diazepam (Valium), clonazepam (Klonopin), and alprazolam (Xanax), are sometimes prescribed to treat anxiety, acute stress reactions, and panic attacks. Clonazepam may also be prescribed to treat seizure disorders. The more sedating benzodiazepines, such as triazolam (Halcion) and estazolam (Prosom) are prescribed for short-term treatment of sleep disorders. Usually, benzodiazepines are not prescribed for long-term use because of the high risk for developing tolerance, dependence, or addiction.
- Non-benzodiazepine sleep medications, such as zolpidem (Ambien), eszopiclone (Lunesta), and zaleplon (Sonata), known as z-drugs, have a different chemical structure but act on the same GABA type A receptors in the brain as benzodiazepines. They are thought to have fewer side effects and less risk of dependence than benzodiazepines.
- Barbiturates, such as mephobarbital (Mebaral), phenobarbital (Luminal), and pentobarbital sodium (Nembutal), are used less frequently to reduce anxiety or to help with sleep problems because of their higher risk of overdose compared to benzodiazepines. However, they are still used in surgical procedures and to treat seizure disorders.
Inhibitors of GAM metabolism can also produce convulsions. Spasticity and involuntary movement syndromes, such as Parkinson’s, Friedreich’s ataxia, tardive dyskinesia, and Huntington’s chorea, are all marked by low GABA when amino acid levels are studied. Trials of 2 to 3 g of GABA given orally have been effective in various epilepsy and spasticity syndromes. Agents that elevate GABA are also useful in lowering hypertension. Three grams orally have been effective in controlling blood pressure. GABA is decreased in various encephalopathies. GABA can reduce appetite and is decreased in hypoglycemics. GABA reduces blood sugar in diabetics. Chronic brain syndromes can also be marked by deficiencies of GABA. Vitamin B6, manganese, taurine, and lysine can increase both GABA synthesis and effects, while aspartic acid and glutamic acid probably inhibit GABA effects.
Low plasma GABA has been reported in some depressed patients and may be a useful trait marker for mood disorders. GABA has an important role in embryonic development, especially facial development, as substantiated by the association of a cleft palate in transgenic mice deficient in GAD67 (glutamate decarboxylase). A recent Japanese population study reported linkage in patients with a nonsyndromic cleft lip with or without a cleft palate and specific GAD67 haplotypes 69).
Unusually high levels of GABA (especially in the brain) can be toxic and GABA can function as both a neurotoxin and a metabotoxin. A neurotoxin is a compound that damages the brain and/or nerve tissue. A metabotoxin is an endogenously produced metabolite that causes adverse health effects at chronically high levels. Chronically high levels of GABA are associated with at least five inborn errors of metabolism, including D-2-hydroxyglutaric aciduria, 4-hydroxybutyric aciduria/succinic semialdehyde dehydrogenase deficiency, GABA-transaminase deficiency, homocarnosinosis, and hyper beta-alaninemia 70). Nearly all of these conditions are associated with seizures, hypotonia, intellectual deficits, macrocephaly, encephalopathy, and other serious neurological or neuromuscular problems 71). Increased levels of GABA seem to alter the function of the GABA-B receptor, which may play a role in the tonic-clonic seizures that are often seen in patients with the above disorders.
GABA Metabolism and Homeostasis
GABA metabolism in the brain occurs through a highly compartmentalized set of enzymatic processes. Neurotransmitter levels at GABAergic and glutamatergic synapses are largely maintained by astrocytes, through their mediation of the glutamate/GABA-glutamine cycle (summarized in Figure 2) 72). Cortical synapses are tightly enveloped by highly specialized astroglial processes 73), where GABA is taken up following synaptic release 74) and catabolized to succinate in a two-step reaction catalyzed by the mitochondrial enzymes GABA transaminase (GABA-T) and succinate semialdehyde dehydrogenase (SSADH). Succinate, being an intermediate component of the tricarboxylic acid (TCA) cycle, is subsequently converted to glutamine (Gln) by glutamine synthase, and glutamine (Gln) is then transported into neurons where it undergoes conversion to glutamate (Glu) 75). It has been estimated that GABA metabolism accounts for ~8–10% of the total flow through the neuronal tricarboxylic acid cycle 76). Most of this glutamate then undergoes conversion to glutamine (Gln) through the action of the strictly astrocyte-specific enzyme glutamine synthetase (GS) 77)]. Glutamine is released by astrocytes and taken up by the closely apposed presynaptic neurons, where it can be re-converted to glutamate through the action of the predominantly neuronally localized enzyme phosphate-activated glutaminase (PAG) 78); this process is regulated by the availability of phosphorylated species such as ATP and GTP 79), by tricarboxylic acid cycle intermediates 80), by cyclic nucleotides such as cAMP and cGMP [86], and by the products of the catalytic reaction (glutamate and NH4) 81), allowing for negative control of this process at a number of different levels. Glutamate is thus readily available for GABA synthesis by glutamic acid decarboxylase (GAD).
Figure 4. GABA metabolic pathway in the brain
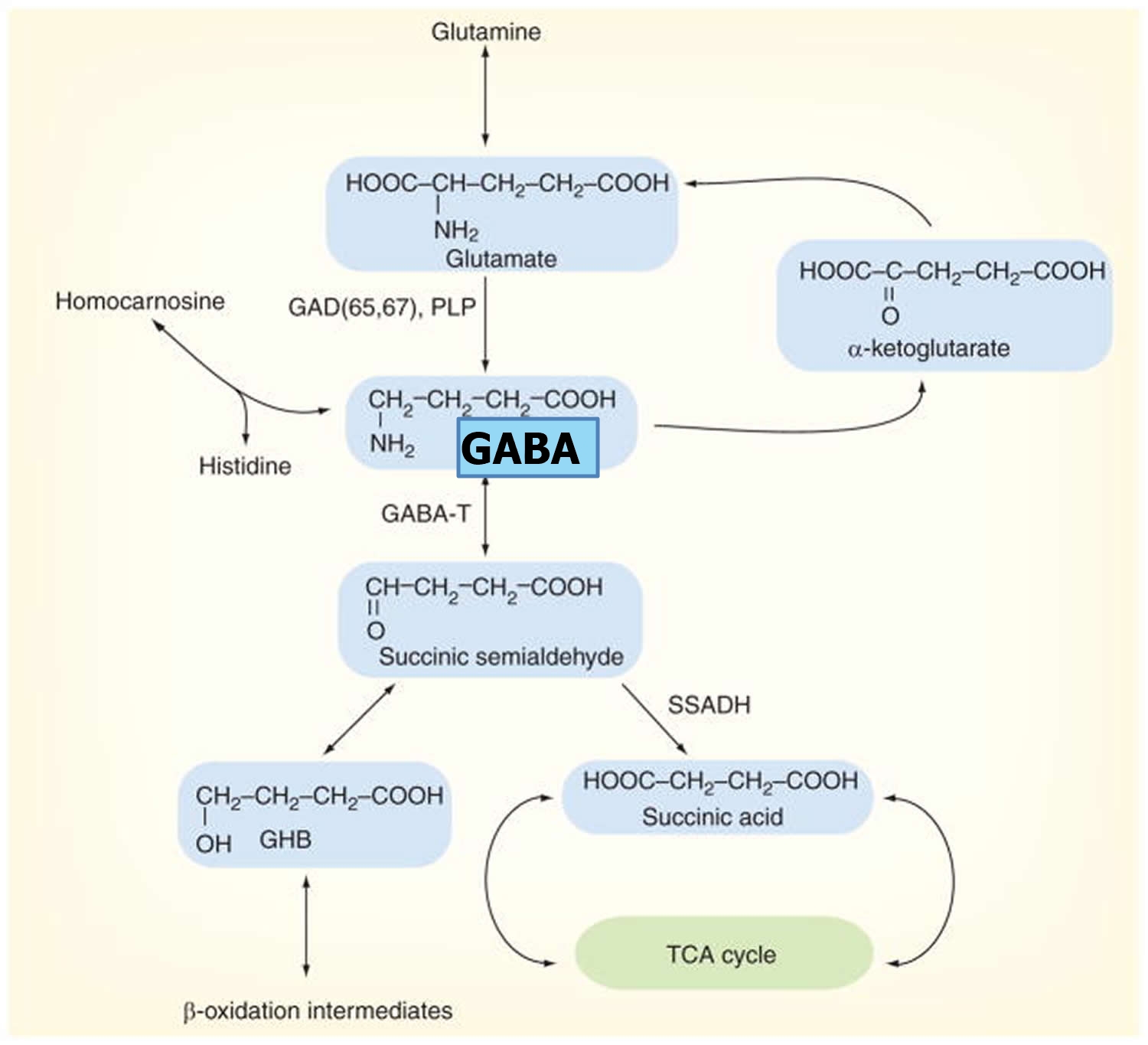
Footnotes: Disorders involving the GABA catabolic pathway are GABA-T deficiency, succinic semi-aldehyde dehydrogenase (SSADH) deficiency and homocarnosinosis; all of these entities invoke neurological dysfunction. Succinic semi-aldehyde dehydrogenase (SSADH) deficiency is the most common, but has a heterogeneous, nonspecific phenotype. Enzymatic deficiency can be documented in SSADH and GABA-T deficiency. Homocarnosine is a dipeptide compound consisting of GABA and histidine.
[Source 82)]Figure 5. GABA synthesis and catabolism in the brain
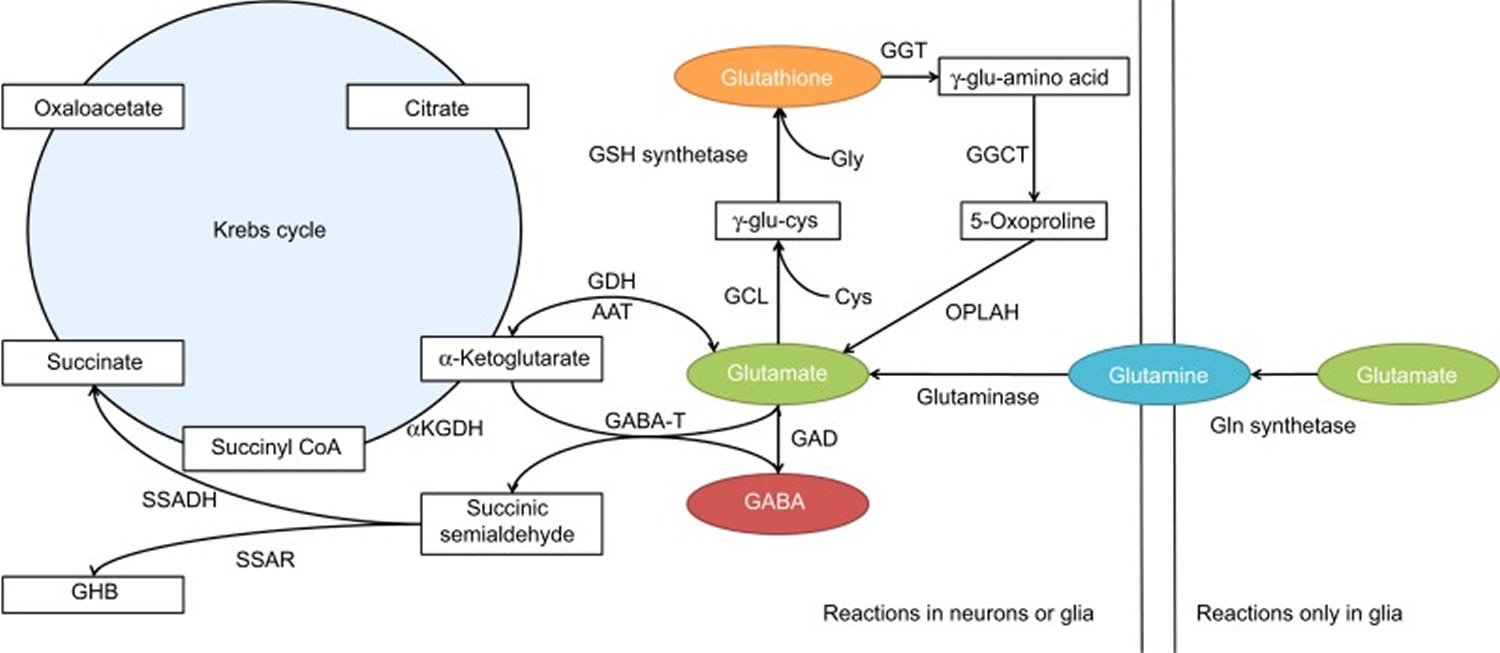
Abbreviations: αKGDH = α-ketoglutarate dehydrogenase; AAT = aspartate aminotransferase; CoA = coenzyme A; Cys = cysteine; GABA = γ-aminobutyric acid; GDH = glutamate dehydrogenase; GABA-T = GABA transaminase; GAD = glutamic acid dehydrogenase; GCL = γ-glutamyl cysteine ligase; Gln = glutamine; Glu = glutamyl; Gly = glycine; GSH = glutathione; GHB = γ-hydroxybutyric acid; GGT = γ-glutamyl transferase; GGCT = γ-glutamyl cyclotransferase; OPLAH = 5-oxoprolinase (adenosine triphosphate-hydrolyzing); SSADH = succinic semialdehyde dehydrogenase; SSAR, succinic semialdehyde reductase.
[Source 83)]Mechanisms of GABA Transport and Synaptic Uptake
The GABAergic system plays an essential role in the fine temporal control of neuronal activity, at the level of individual neurons as well as larger neuronal populations. For this reason, the timing of receptor activation is important, and GABA levels in the extracellular compartment must be carefully regulated 84). The clearance of GABA from the synaptic cleft, and its reuptake into neurons and astrocytes following neurotransmission, occurs through high-affinity GABA uptake systems 85). GABA transport is mediated primarily by four GABA/Na+/Cl-symporters—in humans these are GABA transporter 1 (GAT1), GABA transporter 2 (GAT2), GABA transporter 3 (GAT3) and the betaine-GABA transporter (BGT1). Within neurons, GABA transport into synaptic vesicles is mediated by the vesicular GABA transporter (vGAT) 86). Functionally, the GABA transporters are responsible for the modulation of GABAergic inhibition by terminating the synaptic action of GABA and thus shaping the postsynaptic response to inhibitory presynaptic neurotransmitter release 87). The differing ionic and pharmacological sensitivities of the different GABA transporters, and their differing affinities for GABA transport, make this a very heterogeneous uptake system which can control inhibitory synaptic signaling in a number of ways 88). The GABA transporters are widely distributed throughout the mammalian central nervous system, with individual transporters having unique and sometimes overlapping regional distributions, mainly located on neuronal and glial cell membranes 89).
GABA transporter 1 (GAT1) is the most highly expressed GABA transporter in the mammalian cerebral cortex 90), and is generally considered to be the primary presynaptic neuronal GABA transporter. This transporter is also localized to astrocytic membranes at GABAergic synapses 91). The extensive nature of GABA transporter 1 (GAT1) expression reflects its importance in regulating cortical excitability and information processing at synapses 92). GABA transporter 2 (GAT2) is primarily found in various tissues outside of the CNS (in particular in the proximal tubules of the kidney, in the heart, and in liver hepatocytes), but has a limited distribution in some regions of the brain and in the retina 93). GABA transporter 3 (GAT3) is primarily found in the nervous system, and is localized almost entirely to the processes of astrocytes within the cerebral cortex, indicating that this transporter is responsible for the uptake of GABA into astrocytes rather than neurons 94). Betaine-GABA transporter (BGT1) is primarily a transporter for betaine, and has a lower affinity for GABA than the aforementioned transporters. The distribution of betaine-GABA transporter (BGT1) in the mammalian brain is contentious, but some studies report that it is expressed by astrocytes at extrasynaptic sites, possibly suggesting a role in the regulation of tonic extracellular GABA levels.
GABA, Diseases, and Treatment
Increasing GABA in the brain has for years been the focus of drug development aiming to alleviate the severity of epileptic seizures 95). Initial studies examined the efficacy of administering GABA directly. One study reported a reduction in the amount of seizures in epileptic patients who were administered a very high dose of GABA (0.8 g/kg daily) 96). However, this result was found only in four out of twelve patients. Additionally, the patients in whom the administration of GABA did have an effect were children below the age of 15. This finding is in line with the suggestion that the blood–brain barrier (BBB) permeability to GABA decreases with age 97). Perhaps more importantly, GABA’s half-life is about 17 min in mice 98). If the half-life has a similar short duration in humans, direct administration of GABA is unsuitable as pharmacological treatment of epilepsy.
The GABA analog gabapentin was developed as an anti-epileptic drug. Gabapentin functions by modulating enzymes involved in GABA synthesis. It differs in chemical structure from GABA and its half-life is much longer 99). One proton magnetic resonance spectroscopy (1H-MRS) study in humans has found that the administration of gabapentin increased brain GABA levels by 55.7% 100). Nonetheless, a study exploring the effects of gabapentin in both rat and human neocortical slice preparations suggests that there might be a considerable difference between rodents and humans in the effects on GABA levels: gabapentin was found to increase GABA concentrations by 13% in human neocortical slices, while having no significant effect in rat neocortical slices 101).
Patients with Huntington’s disease also have reduced GABA levels in the brain 102), but administration of GABA to remedy this deficiency has shown mixed results with regards to the reduction of symptoms 103). Of course, that the administration of GABA does not consistently alter the symptoms in complex and multifaceted disorders such as epilepsy and Huntington’s disease, does not necessarily mean that GABA is unable to affect the brain 104).
GABA and Alzheimer’s disease
In the past few decades, many studies have implicated the disruption of cholinergic and glutamatergic neurotransmission in Alzheimer’s disease. Now increasing attention is also being paid to the role of GABAergic dysfunction in Alzheimer’s disease 105). Despite some controversy in the field, there is much evidence to suggest that GABAergic remodeling is a feature of Alzheimer’s disease, being potentially initiated at early stages of disease pathogenesis. There is evidence that alterations in various components of the GABAergic system, including GABA levels, glutamic acid decarboxylase (GAD) activity, GABA currents, and the distribution and subunit composition of GABARs and GABA transporters, might not be occurring simply as a compensatory mechanism in response to glutamate excitotoxicity. Indeed, some alterations might also be caused by the direct effect of Aβ. Thus, the Aβ-induced disruption of GABAergic inhibitory neurotransmission could represent a key mechanism whereby network activity is impaired in Alzheimer’s disease. In this manner, GABAergic remodeling may be involved in E/I balance disruptions that lead to early cognitive deterioration in the Alzheimer’s disease brain. A novel idea emerging from this body of research is the suggestion that the GABAergic system is an important factor in both the early and later stages of disease progression, and is not simply altered as a secondary pathological response. It is thus important to consider both direct and compensatory alterations in GABAergic activity in Alzheimer’s disease. Due to the limitations of previous studies and the inconsistency of previous results, the consequences of these alterations on neural network activity and behavior/cognition are not yet well understood. It is also important to take into account the huge translational gap between animal and in vitro models of Alzheimer’s disease and human clinical trials, and to consider the possibility that currently available Alzheimer’s disease models fail to capture key characteristics of the human disease.
Currently, the only Food and Drug Administration (FDA) approved drugs available to Alzheimer’s disease patients are those targeting either the cholinergic system (e.g. donepezil, tacrine, rivastigmine and galantamine) or the glutamatergic system (e.g. memantine) 106). None of these drugs are curative or disease-modifying, providing only temporary, symptomatic relief, and only to a subset of Alzheimer’s disease patients. As these drugs are only marginally effective, unable to prevent or reverse cognitive decline, and produce many unwanted side effects, there is an ongoing search for novel therapeutic targets 107).
Therefore, there is an urgent need to pursue further research in this area, to enhance our understanding of Alzheimer’s disease-associated alterations in the GABAergic system. Evidence for Alzheimer’s disease-associated GABAergic remodeling along with the failure of anti-glutamatergic and acetylcholinesterase inhibitor therapies to halt the progression of the disease could point to the GABAergic system as a promising therapeutic target for Alzheimer’s disease.
GABA supplement
Gamma-aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the human cortex. In recent years it has become widely available as a food supplement. In Europe and the United States, GABA is considered a “food constituent” and a “dietary supplement,” respectively. As such, manufacturers are not required to provide evidence supporting the efficacy of their products as long as they make no claims with regards to potential benefits in relation to specific diseases or conditions. The food supplement version of GABA is widely available online. Although many consumers claim that they experience benefits from the use of these products, it is unclear whether these supplements confer benefits beyond a placebo effect 108). In recent years researchers have reported a number of placebo-controlled studies in which GABA was administered as a food supplement to healthy participants and participants with a history of acrophobia. One study found an increase in alpha waves in healthy participants and reduced levels of immunoglobulin A (IgA; an indicator of immune system functioning) in participants with a history of acrophobia when they were exposed to heights 109). However, the sample size for the second finding was very small (four participants per group). Another study reported reduced heart rate variability and salivary chromogranin A (CgA) during an arithmetic task compared to a control group after the administration of GABA-enriched chocolate 110). A third study reported less salivary cortisol and CgA than a control group during a psychological stress-inducing arithmetic task. Additionally, participants who received 50 mg of GABA dissolved in a beverage reported less psychological fatigue after completion of the task 111). Finally, in a fourth study, participants were found to show a decrease in alpha waves over time while performing an arithmetic task. This decrease was smaller in the group that orally received GABA (100 mg) compared to a control group 112). By way of comparison, one would have to eat 2.34 kg of uncooked spinach in order to consume a similar amount of GABA, and spinach is relatively rich in GABA compared to other foods 113).
The results of these studies support the claims made by hundreds of consumers of GABA food supplement products and fit with a growing trend in which GABA is administered through everyday (natural) foods 114). However, there are some caveats to consider. First, at least one of the authors in each of these four studies was affiliated with the company that produces the GABA supplement in question. However, a declaration of conflicting interests is lacking in three out of four of these studies. Second, the reported studies used “pharma-GABA,” which is produced for the Asian market through a fermentation process using a strain of lactic acid bacteria, Lactobacillus hilgardii K-3 115). Pharma-GABA has been approved by the FDA as a food ingredient 116). While the manufacturer of pharma-GABA suggests that there are important differences with the synthetic GABA supplement sold online in Western countries, these differences refer to the production process and the occurrence of potentially harmful byproducts in synthetically produced GABA, and not to the chemical structure of the active compound GABA.
Currently, the mechanism of action behind these GABA supplements is unknown 117). It has long been thought that GABA is unable to cross the blood–brain barrier (BBB), but the studies that have assessed this issue are often contradictory and range widely in their employed methods. Accordingly, future research needs to establish the effects of oral GABA administration on GABA levels in the human brain, for example using magnetic resonance spectroscopy. There is some evidence in favor of a calming effect of GABA food supplements, but most of this evidence was reported by researchers with a potential conflict of interest 118).
A recent study by Steenbergen et al. (2015a) with human subjects has shown that the ingestion of synthetic GABA (800 mg) enhanced the ability of prioritized planned actions and inhibitory control 119). However, in view of the lack of evidence with regards to GABA’s blood–brain barrier (BBB) permeability in humans, the mechanism through which GABA might have exerted these effects remains unclear. The same holds for the pharma-GABA studies that were discussed above: none of these effects exclude an indirect of GABA on the brain. The oral intake of these supplements may have exerted these effects through indirect pathways, for example through the enteric nervous system.
Enteric Nervous System Effects of GABA
The bidirectional signaling between the brain and the enteric nervous system is vital in maintaining homeostasis 120). Even though most research thus far has focused on the signaling from the brain to the gut, an increasing number of studies has explored the influence of the gut’s microbiota on the brain. For example, gut microbiota have been shown to improve mood and reduce anxiety in patients with chronic fatigue 121). Similarly, oral intake of probiotics resulted in reduced urinary cortisol and perceived psychological stress 122) and reduced reactivity to sad mood 123) in healthy subjects.
It has been found that certain probiotic strains are able to produce GABA in vivo. Specifically, bacteria from the strains Lactobacillus and Bifidobacterium were effective at increasing GABA concentrations in the enteric nervous system 124). Indeed, both GABA and its receptors are widely distributed through the enteric nervous system 125). Additionally, there is considerable communication between the gut and the brain through the vagal nerve 126). This nerve consists, for the most part, of sensory nerve fibers that relay information about the state of bodily organs to the central nervous system 127).
A study in mice showed that the administration of Lactobacillus rhamnosus (JB-1) consistently modulated the mRNA expression of GABAAα2, GABAAα1, and GABAB1b receptor subunits 128), receptors commonly associated with anxiety-like behavior. Indeed, on a behavioral level the L. rhamnosus (JB-1)-fed mice were less anxious and displayed antidepressant-like behaviors in comparison with controls. Furthermore, the administration of these bacteria reduced the stress-induced elevation of corticosterone compared to the control mice. Importantly, none of these effects were present in mice that underwent vagotomy 129).
In humans, the stimulation of the vagus nerve through transcutaneous vagus nerve stimulation (tVNS) has been used to treat refractory epilepsy 130). This technique has been shown to affect norepinephrine, acetylcholine and GABA concentrations 131). With regards to GABA, VNS seems to increase the level of free GABA in the cerebrospinal fluid 132). Similarly to the administration of synthetic GABA 133), active tVNS was found to enhance the ability of prioritizing and cascading different actions when performing a stop-change paradigm 134).
To summarize, bacteria from the Lactobacillus spp. strain contribute to the formation of GABA in the enteric nervous system. The oral administration of bacteria from this strain can influence GABAergic firing in the mice brain through the vagus nerve. Furthermore, stimulation of the vagal nerve through transcutaneous vagus nerve stimulation (tVNS) has been shown to affect processes thought to be GABAergic in humans. Finally, a similar behavioral effect has been found both for the administration of synthetic GABA and tVNS with regards to action cascading. Even if GABA is unable to cross the blood–brain barrier (BBB) at all in humans, an indirect effect through the enteric nervous system might be a viable route for an effect of GABA food supplements. The link between the oral administration of GABA, the vagal nerve and GABA levels in the brain has not been established yet, but in view of the available evidence it is a promising candidate for future research.
GABA dosage
It is not clear whether GABA taken as a supplement reaches the brain in large enough quantities to have an effect. There isn’t a set dosage for GABA at this time.
In a a double-blind, randomized study involving thirty undergraduate students of the Leiden University (29 females, 1 male, mean age = 19.5 years, range 18–22) participated in the GABA experiment, GABA was proven to have central nervous system action after an oral administration of 800 mg synthetic GABA by modulating fronto-striatal networks 135). Results showed that the administration of GABA, compared to placebo, increased action selection when an interruption (stop) and a change towards an alternative response were required simultaneously, and when such a change had to occur after the completion of the stop process. Therefore, the outcome is consistent with, and further supports, previous findings suggesting that response inhibition processes are modulated by the GABA-ergic system 136). An important limitation of that study is the small sample size, including predominantly female participants. Therefore, further studies are needed in order to verify the reliability and repeatability of their findings in larger samples that are balanced for gender.
GABA supplement side effects
There has not been enough research to uncover the side effects of GABA supplements. Furthermore, there isn’t enough information to be sure about the safety of GABA. For this reason, it’s best to play it safe and not use GABA if you are pregnant or breastfeeding.
References [ + ]

{kind=link}