Contents
What is CVID
CVID (common variable immunodeficiency) is a type of primary immune deficiency disease in which you have low levels of several of the proteins (antibodies) that help you fight infections. CVID is characterized by low levels of serum IgG, IgA, and/or IgM, with a loss of antobody production and an increased risk of infections 1. CVID also is known as hypogammaglobulinemia (low blood immunoglobulins), adult-onset agammaglobulinemia (absent blood immunoglobulins), late-onset hypogammaglobulinemia, and acquired agammaglobulinemia. CVID leaves you open to recurrent infections in your ears, sinuses and respiratory system, and increases your risk of digestive disorders, blood disorders and cancer.
The World Health Organization (WHO) recognizes more than 200 different forms of primary immune deficiency diseases ranging from relatively common to quite rare affecting approximately 500,000 people in the United States. CVID is estimated to affect 1 in 25,000 to 1 in 50,000 people worldwide, although the prevalence can vary across different populations 2.
Signs and symptoms of CVID may appear during childhood or adolescence, though most people with CVID are diagnosed in their twenties or thirties. Because the immune system is slow to mature, the diagnosis of CVID is generally not made until after the age of 4. The condition can be inherited, or it can be acquired during your lifetime. Less than 10% of CVID are inherited 3. In some of these families, possible loci for dominant CVID genes on chromosomes 4q and 16q by genetic linkage studies have been identified. There is also rare evidence of recessive diseases involving B-cell genes (CD19) 4 and T-cell genes (ICOS) 5 in patients with these types of primary antibody failures. In addition, some polymorphisms in genes needed for B-cell survival (TACI and Msh5) are thought to be associated with CVIDs, due to a higher gene prevalence in patients compared with the general population 6.
People with CVID may experience frequent bacterial and viral infections of the upper airway, sinuses, and lungs. Acute lung infections can cause pneumonia, and long-term lung infections may cause a chronic form of bronchitis known as bronchiectasis, which is characterized by thickened airway walls colonized by bacteria.
People with CVID also may have diarrhea, problems absorbing food nutrients, reduced liver function, and impaired blood flow to the liver. Autoimmune problems that cause reduced levels of blood cells or platelets also may occur. People with CVID may develop an enlarged spleen and swollen glands or lymph nodes. Painful swollen joints in the knee, ankle, elbow, or wrist also can develop. In addition, people with CVID may have an increased risk of developing some cancers.
The standard of care in CVID is replacement Ig given at frequent intervals for life 7 and this replacement reduces the number of bacterial infections 8 and probably enhances survival 9. However, immune globulin does not seem to protect against or treat the largely noninfectious complications such as functional and structural lung disease, autoimmunity, granulomatous disease, liver diseases and hepatitis, gastrointestinal inflammatory disease, or the development of cancer or lymphoma that are found in varying percentages of patients in different series 10.
There are many different types of CVID that are distinguished by genetic cause. People with the same type of CVID may have varying signs and symptoms. Patients can quite often be classified into 2 main groups by CVID phenotypes that are apparently stable over time. These 2 groups contain subjects with a history of infections and those who may have infections, but in addition, a variety of inflammatory and/or autoimmune conditions. For the latter set of patients, these medical problems have emerged as the manifestations of CVID that are most difficult to handle because one or more forms of immune suppression may be needed. This categorization becomes apparent when studying the large collections of patients assembled in Europe and in the United States. In the largest European study, 334 subjects from the European Society for Immune Deficiency 11 were followed for an average of 25.5 years; 71% had one or more of the inflammatory/autoimmune complications and the reminder had infections only. In the US group, 476 patients studied from one medical center, 68% had one or more of these manifestations and the rest had infections as the only manifestation 12.
In the US group, the most prevalent of these conditions, even while on Ig therapy, was progressive lung disease in 28.5%, conceivably a residual of longstanding infections. However, in these cases, lymphocytic and/or granulomatous pulmonary infiltrates led to lung failure, suggesting an aggressive inflammatory component. More difficult to understand are the hematologic and organ-specific autoimmune diseases that were diagnosed in 12%-46% of subjects in the European Society for Immune Deficiency report 11, in 28.6% of the US subjects 12 and in up to 20% of a cohort examined in France 13. As in all previous studies, for unclear reasons, the most common autoimmune diseases in CVID were immune thrombocytopenia followed by autoimmune hemolytic anemia. Other autoimmune conditions in the US study included anticardiolipin Ab, antiphospholipid syndrome, diabetes mellitus, inflammatory bowel disease, pernicious anemia, rheumatoid arthritis, juvenile rheumatoid arthritis, uveitis, multiple sclerosis, neutropenia, primary biliary cirrhosis, systemic lupus erythematosus, autoimmune thyroid disease, vasculitis, psoriasis, and vitiligo (Table 1).
Table 1. CVID Phenotypes and conditions
| Associated condition (N = 473) | n | Percentage |
|---|---|---|
| Infections only (no complications) | 151 | 31.9 |
| Chronic lung disease (functional/structural) | 135 | 28.5 |
| Autoimmunity | 134 | 28.6 |
| Gastrointestinal disease | 73 | 15.4 |
| Granulomatous disease | 46 | 9.7 |
| Liver disease/hepatitis | 43 | 9.1 |
| Lymphomas and other lymphoid malignancies | 39 | 8.2 |
| Splenectomy | 39 | 8.2 |
| Other cancers | 33 | 6.9 |
CVID causes
The cause of CVID is unknown in at least 90% of affected individuals, as a genetic cause has been identified in about 10%. Sporadic cases, with no apparent history of the disorder in the family, are the commonest form. It is likely that CVID is caused by both environmental and genetic factors, a complex interaction of environmental and genetic components (multifactorial inheritance), but genes that are involved in the development and function of immune cells are believed to be the primary cause.
While the specific environmental factors are unclear, the genetic influences in CVID are believed to be mutations in genes that are involved in the development and function of immune system cells called B cells. B cells are specialized white blood cells that help protect the body against infection. When B cells mature, they produce special proteins called antibodies (also known as immunoglobulins). These proteins attach to foreign particles, marking them for destruction. Mutations in the genes associated with CVID result in dysfunctional B cells that cannot make sufficient amounts of antibodies.
In about 10 percent of cases, a genetic cause for CVID is known. Mutations in at least 13 genes have been associated with CVID. The most frequent mutations occur in the TNFRSF13B gene. The protein produced from this gene plays a role in the survival and maturation of B cells and in the production of antibodies. TNFRSF13B gene mutations disrupt B cell function and antibody production, leading to immune dysfunction. Other genes associated with CVID are also involved in the function and maturation of immune system cells, particularly of B cells; mutations in these genes account for only a small percentage of cases.
All individuals with CVID have a shortage (deficiency) of two or three specific antibodies. Some have a deficiency of the antibodies called immunoglobulin G (IgG) and immunoglobulin A (IgA), while others, in addition to lacking IgG and IgA, are also deficient in immunoglobulin M (IgM). A shortage of these antibodies makes it difficult for people with this disorder to fight off infections. Abnormal and deficient immune responses over time likely contribute to the increased cancer risk. In addition, vaccines for diseases such as measles and influenza do not provide protection for people with CVID because they cannot produce an antibody response.
Most cases of CVID are sporadic and occur in people with no apparent history of the disorder in their family. These cases probably result from a complex interaction of environmental and genetic factors.
In rare cases, CVID is inherited in an autosomal recessive pattern, which means both copies of a gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
In a few cases, this condition is inherited in an autosomal dominant pattern, which means one copy of an altered gene in each cell is sufficient to cause the disorder.
When CVID is caused by mutations in the TNFRSF13B gene, it is often sporadic and the result of a new mutation in the gene that occurs during the formation of reproductive cells (eggs or sperm) or in early embryonic development. When TNFRSF13B gene mutations are inherited, they can cause either autosomal dominant CVID or autosomal recessive CVID.
Not all individuals who inherit a gene mutation associated with CVID will develop the disease. In many cases, affected children have an unaffected parent who has the same mutation. Additional genetic or environmental factors are likely needed for the disorder to occur.
CVID life expectancy
The life expectancy of individuals with CVID varies depending on the severity and frequency of illnesses they experience. Most people with CVID live into adulthood. Reduced survival was associated with age at diagnosis, lower baseline IgG, higher IgM, and fewer peripheral B cells. The risk of death was 11 times higher for patients with noninfectious complications. Mortality was associated with lymphoma, any form of hepatitis, functional or structural lung impairment, and gastrointestinal disease with or without malabsorption, but not with bronchiectasis, autoimmunity, other cancers, granulomatous disease, or previous splenectomy.
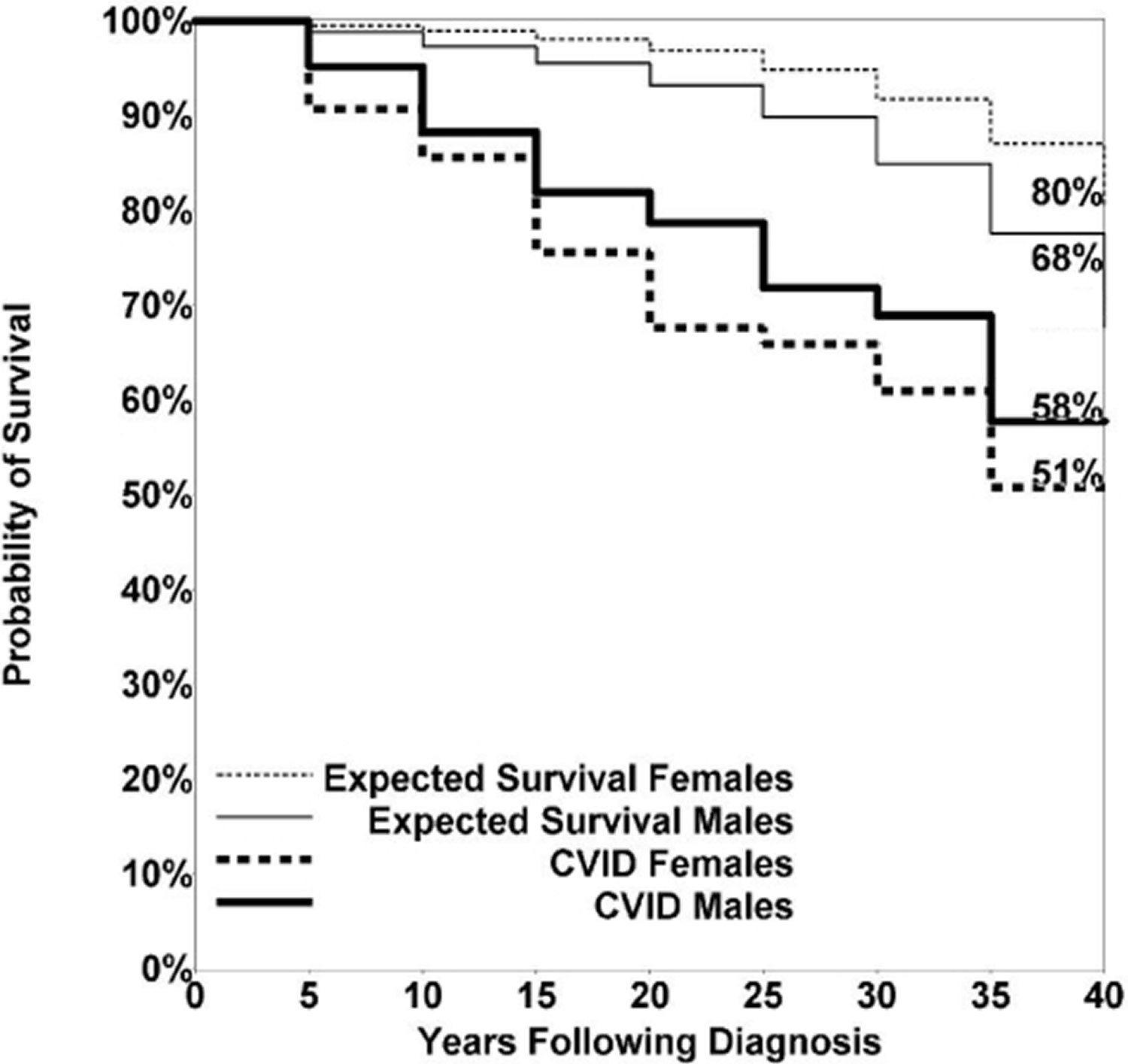
For the US study, 58% of female patients with CVID survived more than 4 decades, compared with 80% of age-matched female subjects; 53% of males survived compared with 68% of age-matched male subjects 14. Due to the heterogeneity of the population followed over this period, the median age at death was 44 years (range, 10-90) for female subjects and 42 years (range, 9-79) for male subjects: not significantly different. The predominant causes of death included respiratory failure from chronic lung disease, lymphoid or other malignancies, or overwhelming infections. However, as also seen in the European Society for Immune Deficiency study 11, in the US group, the risk of death in follow-up was nearly 11 times higher for CVID patients with 1 or more of the noninfectious complications outlined above than for subjects who had infections only 14. In fact, 89 of the 93 subjects who died had one or more of the noninfectious complications and only the 4 others had infections only.
Figure 1. CVID Life expectancy
CVID prognosis
In a study spanning four decade involving 473 subjects in New York 15 411 subjects with known follow-up (87% of the cohort), 93 patients (19.6%) had died. The median age at death was 44 years for females (range, 10-90 years) and 42 years for males (range, 9-79 years), not significantly different 15. The predominant causes of death included respiratory failure from chronic lung disease, lymphoid or other malignancy, or overwhelming infections (Table 2).
Patients with gastrointestinal disease; liver diseases and hepatitis; lymphoma; chronic lung disease, including radiologic or functional lung disease, or both; or malabsorption had reduced survival, compared with CVID patients without these particular complications 15. In contrast, patients with any of the autoimmune conditions, cancers other than lymphoma, history of splenectomy, presence of granulomatous disease, or the development of bronchiectasis alone did not have significantly reduced survival over the 4 decades of study. Kaplan-Meier survival curves also affirmed these observations, showing significantly reduced survival for patients with gastrointestinal disease, malabsorption, chronic lung disease, liver diseases and hepatitis and lymphoma, but not for subjects with a history of autoimmunity, cancer other than lymphomas, previous splenectomy, granuloma, or the radiologic observation of bronchiectasis with lack of other findings 15.
Table 2. CVID Causes of death
| Cause of death | No. (n = 93) | Comments |
|---|---|---|
| Lung failure | 34 | 14 of these from respiratory failure, 3 after lung transplant |
| Lymphoma | 17 | 2 cases with coexistent lung disease and 1 with coexistent liver disease contributing to death |
| Cancers | 10 | 2 lung, 2 stomach, 1 ovarian, 1 esophageal, 1 carcinoid, 1 oral, 1 breast, 1 colon |
| Liver disease | 8 | 1 after liver transplant, 4 infectious hepatitis |
| Other infections | 5 | Pneumocystis pneumonia, measles, nocardia brain abscess, multiple anaerobes, meningitis |
| Heart disease | 4 | 1 with coexisting lung disease |
| Aplastic anemia | 4 | 1 with coexisting neurodegenerative disease |
| Unknown cause of death | 4 | |
| Progressive multifocal leukoencephalopathy | 2 | |
| Vasculitis | 1 | |
| Neurodegenerative disease | 1 | |
| Autonomic amyloidosis | 1 | |
| Accidental death | 1 | |
| Suicide | 1 |
CVID symptoms
People with CVID are highly susceptible to infection from foreign invaders such as bacteria, or more rarely, viruses and often develop recurrent infections, particularly in the lungs, sinuses, and ears. Pneumonia is common in people with CVID. Over time, recurrent infections can lead to chronic lung disease. Affected individuals may also experience infection or inflammation of the gastrointestinal tract, which can cause diarrhea and weight loss. Abnormal accumulation of immune cells causes enlarged lymph nodes (lymphadenopathy) or an enlarged spleen (splenomegaly) in some people with CVID. Immune cells can accumulate in other organs, forming small lumps called granulomas.
Approximately 25 percent of people with CVID have an autoimmune disorder, which occurs when the immune system malfunctions and attacks the body’s tissues and organs. The blood cells are most frequently affected by autoimmune attacks in CVID; the most commonly occurring autoimmune disorders are immune thrombocytopenia purpura, which is an abnormal bleeding disorder caused by a decrease in cell fragments involved in blood clotting called platelets, and autoimmune hemolytic anemia, which results in premature destruction of red blood cells. Other autoimmune disorders such as rheumatoid arthritis can occur. Individuals with CVID also have a greater than normal risk of developing certain types of cancer, including a cancer of immune system cells called non-Hodgkin lymphoma and less frequently, stomach (gastric) cancer.
Some patients with CVID who may not be receiving optimal immunoglobulin replacement therapy may also develop a painful inflammation of one or more joints. This condition is called polyarthritis. In the majority of these cases, the joint fluid does not contain bacteria. To be certain that the arthritis is not caused by a treatable infection; the joint fluid may be removed by needle aspiration and studied for the presence of bacteria. In some instances, a bacterium called Mycoplasma may be the cause and can be difficult to diagnose. The typical arthritis associated with CVID may involve the larger joints such as knees, ankles, elbows and wrists. The smaller joints, like the finger joints, are rarely affected. Symptoms of joint inflammation usually disappear with adequate immunoglobulin therapy and appropriate antibiotics. In some patients, however, arthritis may occur even when the patient is receiving adequate immunoglobulin replacement.
Lymphoma and other malignancies
The overall incidence of malignancies in CVID appears to have increased, but the data for cancers other than lymphoma are difficult to separate out. In one study of 416 CVID subjects in Australia, there was a 5-fold increase in cancer overall, but lymphoma/ leukemia was 31.5% of the total 16. The incidence of stomach cancer may be increased as well, but recent comparative data appear to show a reduced incidence over prior studies 17. In the US report 17, 8.2% of CVID patients had a lymphoid malignancy, all B cell in type. As noted in earlier reports 18, 19 lymphoma was more common in females than males. Of these, non-Hodgkin B-cell lymphomas were the most common, with some of these being further classified into specific B-cell phenotypes, including mucosa-associated lymphoid tissue lymphoma, marginal zone lymphoma, and T cell–rich B-cell EBV-associated lymphoma. Monoclonal B lymphocytosis was noted in one patient. Lymphomas in CVID are usually extranodal, B cell in type, and, unlike lymphomas in other congenital immune defects, are more common in subjects in the fourth to seventh decade of life and are usually EBV negative 18. Cancers of other sorts also developed in 33 patients (7%) in this study 17. Other large series have not noted this high a percentage of lymphoma: 1.6% in Italy 20, 3% in the European Society for Immune Deficiency study 11, 3.8% in a previous United Kingdom study, and 2% in a combined Danish/Swedish cohort 21.
Table 3. CVID selected complications
| Associated condition | No. | % of cohort (n = 473) |
|---|---|---|
| Infections only (no complications) | 151 | 31.9 |
| Chronic lung disease (functional/structural) | 135 | 28.5 |
| Bronchiectasis | 53 | 11.2 |
| Autoimmunity | 134 | 28.6 |
| ITP | 67 | 14.2 |
| AIHA | 33 | 7 |
| Evans syndrome | 20 | 4.2 |
| Rheumatoid arthritis | 15 | 3.2 |
| Anti-IgA antibody | 7 | 1.5 |
| Alopecia | 5 | 1.1 |
| Neutropenia, pernicious anemia, anticardiolipin antibody, antiphospholipid syndrome, diabetes mellitus, juvenile rheumatoid arthritis, uveitis, multiple sclerosis, systemic lupus erythematosis, autoimmune thyroid disease, lichen planus, vasculitis, vitiligo, psoriasis | < 5 | < 1 |
| Gastrointestinal disease | 73 | 15.4 |
| Malabsorption | 28 | 5.9 |
| Inflammatory bowel disease (Crohn disease, ulcerative colitis, ulcerative proctitis) | 20 | 4.2 |
| Chronic diarrhea | 9 | 1.9 |
| Idiopathic mucosal inflammation | 6 | 1.3 |
| Nodular lymphoid hyperplasia | 5 | 1.1 |
| Gastrointestinal bleeding, irritable bowel syndrome, partial gastrectomy, diverticulitis, esophagitis | 1 | < 1 |
| Liver disease/hepatitis | 43 | 9.1 |
| Hepatitis C | 9 | 1.9 |
| Liver granuloma | 8 | 1.7 |
| Idiopathic liver disease | 8 | 1.7 |
| Non-A, non-B hepatitis* | 6 | 1.3 |
| Chronic hepatitis of unknown origin | 5 | 1.1 |
| Primary biliary cirrhosis | 3 | < 1 |
| Nodular regenerative hyperplasia | 2 | < 1 |
| Hepatitis B, cirrhosis of unknown etiology | 1 | < 1 |
Note: *Diagnosed before availability of the hepatitis C PCR.
[Source 15]Table 4. CVID granulomatous disease by location
| Tissue location | No. (n = 46) |
|---|---|
| Lung | 20 |
| Multiple locations (ie, liver, lung, and spleen) | 7 |
| Lymph node | 6 |
| Liver | 4 |
| Skin | 3 |
| Spleen | 2 |
| Bone marrow | 1 |
| Brain | 1 |
| Neck tissue | 1 |
| Operative site | 1 |
Table 5. CVID Lymphoma and selected outcomes
| Lymphoma type | No. (n = 39) | Outcome |
|---|---|---|
| Non-Hodgkin lymphoma, B-cell type, not further classified | 23 | 11 died of lymphoma, 12 alive |
| Diffuse large B-cell lymphoma | 3 | 2 died of lymphoma, 1 also had severe lung disease, 1 alive |
| Hodgkin disease | 4 | 3 developed B-cell lymphoma years after treatment for Hodgkin disease, 2 of these died of lymphoma, 2 alive |
| MALT | 5 | 3 no treatment, 2 chemotherapy, 5 alive |
| Marginal zone lymphoma/monoclonal B lymphocytosis | 1 | No treatment given, 1 alive |
| Monoclonal B lymphocytosis | 1 | No treatment given, 1 alive |
| Diffuse poorly differentiated lymphoma with IgM-κ macroglobulinemia | 1 | 1 died of lymphoma |
| T cell–rich B cell EBV + lymphoma | 1 | 1 died of lymphoma |
Table 6. CVID – other cancers and selected outcomes
| Malignancy type | No. (n = 33) | Outcome |
|---|---|---|
| Breast cancer | 9 | 1 died of breast cancer, 2 died of other causes, 6 alive |
| Gastric cancer | 3 | 2 died of gastric cancer, 1 alive |
| Melanoma | 3 | 1 died of other causes, 2 alive |
| Malignancy of unknown primary | 3 | 3 alive |
| Colon cancer | 2 | 1 died of colon cancer, 1 died of other causes |
| Lung cancer | 2 | 2 died of lung cancer |
| Oral cancer | 2 | 1 died of oral cancer, 1 alive |
| Skin cancer | 2 | 2 alive |
| Hepatic carcinoid tumor | 1 | 1 died of carcinoid tumor |
| Colon, prostate cancer | 1 | 1 alive |
| Prostate, skin cancer | 1 | 1 alive |
| Thyroid cancer | 1 | 1 alive |
| Vaginal cancer | 1 | 1 alive |
| Ovarian cancer | 1 | 1 died of ovarian cancer |
| Esophageal cancer | 1 | 1 died of esophageal cancer |
Table 7. CVID – Splenectomy reasons and selected outcomes
| Reason | No. (n = 39) | Outcome |
|---|---|---|
| ITP | 12 | 11 cases resolved ITP, 1 case complicated by postoperative sepsis but this patient was not yet on immunoglobulin therapy |
| Hypersplenism | 9 | 6 cases resolved with no sequelae, 2 cases resolved hypersplenism but later developed liver failure with portal hypertension of unclear etiology, 1 case later developed bone marrow failure |
| AIHA | 6 | 4 cases resolved AIHA, 2 cases complicated by fistulae, 1 from the pancreas to the back, 1 to the stomach and abdominal wall |
| Hodgkin disease staging | 2 | 2 patients received chemotherapy for Hodgkin disease, both later died of lymphoma |
| Enlarged spleen and hypersplenism | 2 | 1 case resolved, 1 case improved |
| Unknown reason | 2 | 1 patient eventually developed lymphoma |
| Lymphoma | 1 | Patient found to have lymphoma |
| ITP/AIHA | 1 | Patient needed rituxumab for recurrence |
| Enlarged spleen | 1 | Splenomegaly with no other pathology |
| Abscess of spleen | 1 | Patient with splenic abscess with extension to psoas muscle |
| Enlarged spleen, presumed lymphoma | 1 | Patient with granulomata on pathology after splenectomy, no lymphoma found |
| Presumed lymphoma (medical error) | 1 | Spleen lost; pathology unknown, but patient given chemotherapy |
CVID diagnosis
In most patients, CVID is diagnosed based upon a thorough clinical evaluation, identification of characteristic symptoms and physical findings, a detailed patient and family history, and a pattern of immune system defects confirmed by laboratory testing.
Confirmation of certain immunologic abnormalities plays an essential role in establishing the diagnosis of CVID. The diagnosis of CVID is primarily established by testing for low blood (serum) IgG immunoglobulin concentrations ranging from severely reduced (<100 mg/dL) to just below adult normal range (500-1200 mg/dL). In addition, laboratory testing may reveal normal or, in some cases, reduced numbers of circulating B cells. Failure of certain B cells to appropriately mature into antibody-producing plasma cells may also be detected. Specialized laboratory tests may also help to determine the exact nature of the immune defect (e.g., B cell, helper T cell, suppressor T cell, or B and T cell defects). In many cases, x-ray, examination of the small intestine (enteroscopy), or surgical removal (biopsy) of small samples of tissue from lymph nodes may reveal certain abnormalities (e.g., nodular lymphoid hyperplasia). In addition, in some cases, specialized imaging tests followed by biopsy and microscopic examination may confirm the presence of granular, inflammatory nodules (noncaseating granulomas) within tissue of the skin, lungs, spleen, and/or liver.
Another part of the diagnosis of CVID is to determine if there is a lack of functional antibody. This is done by measuring serum levels of antibody, against vaccine antigens such as tetanus or diphtheria, measles, mumps, or rubella, hemophilus or pneumococcal polysaccharide. Patients with CVID have very low or absent antibody levels to most of these vaccines. Immunization with killed vaccines is used to measure antibody function, and this functional testing is crucial prior to beginning treatment. These tests also help the physician decide if the patient will benefit from immunoglobulin replacement therapy and can be key in obtaining insurance authorization for this therapy. The number of B- and T-lymphocytes may also be determined and their function tested in tissue cultures.
Most subjects have normal numbers of peripheral blood B cells (but these appear immature); reduced numbers of memory B cells identified by the surface marker CD27; and, in concert with the low serum IgG and IgA, even more reduced numbers of isotype-switched memory B cells (IgD-IgM-CD27+) 22. One the other hallmarks of CVID is a relative or complete loss of plasma cells in bone marrow and other tissues such as the lamina propria of the gastrointestinal tract 23 in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544–1551 http://www.bloodjournal.org/content/99/5/1544.long)). In terms of genetics, autosomal-recessive genetic mutations in genes important for B-cell function, including inducible costimulatory 24, CD19 25, B-cell activating factor receptor 26, CD20 27, CD21 28 and CD81 29 have been identified in a small number of patients, more commonly in circumstances of consanguinity. CD27 deficiency was also noted in 2 brothers with persistent EBV viremia and hypogammaglobulinemia in one brother, suggestive more of a combined immune defect 30. Although heterozygous and homozygous mutations in the gene for the BCR transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) have been found in 8%-10% of subjects 31 and are significantly associated with CVID 32, some of the same mutations may also be found in healthy controls and non-immunodeficient relatives, suggesting that these are disease-associated polymorphisms 33. For this reason, examining this gene is not considered diagnostic of CVID or predictive of the development of immune deficiency. Recent genome-wide studies have demonstrated the unique genetic characteristics of this immune defect, showing both novel chromosomal associations and highly significant copy number loss and gain 34.
CVID treatment
The treatment of Common Variable Immune Deficiency requires the coordinated efforts of a team of specialists who may need to systematically and comprehensively plan an affected individual’s treatment. Such specialists may include physicians who diagnose and treat disorders of the blood (hematologists), the digestive tract (gastroenterologists), and/or the lungs (pulmonologists); specialists in the treatment of immune system disorders (immunologists); and/or other health care professionals.
CVID is treated with intravenous immunoglobulin infusions or subcutaneous (under the skin) immunoglobulin injection to partially restore immunoglobulin levels. The immunoglobulin given by either method provides antibodies from the blood of healthy donors. Immunoglobulin is extracted from a large pool of human plasma; it consists mostly of IgG and contains all the important antibodies present in the normal population. The frequent bacterial infections experienced by people with CVID are treated with antibiotics. Other problems caused by CVID may require additional, tailored treatments.
Individuals with CVID who experience adverse reactions to intravenous gammaglobulin may benefit from administration of medications that block the effects of the chemical histamine (antihistamines), which is released during allergic reactions, or nonsteroidal anti-inflammatory agents (NSAIDs). Rarely, hydrocortisone, a corticosteroid medication, may be needed prior to gammaglobulin therapy. Because corticosteroids may actually suppress an already weakened immune system, NSAIDs may be helpful in controlling autoimmune-like symptoms while avoiding the use of corticosteroids. However, after being immunoglobulin therapy for several months, most patients no longer require any premedications.
Some researchers have recommended that when a patient is diagnosed with an autoimmune disease, the possibility of an underlying CVID should be evaluated before the administration of immunosuppressive drugs for the autoimmune disease.
Antibiotic medications often prove beneficial for the treatment of various bacterial infections associated with CVID. Patients with irregularities involving the malabsorption of vitamin B12 may also benefit from monthly B12 injections.
Affected individuals with severely low levels of circulating platelets may be cautioned to avoid the use of aspirin, since this medication may interfere with the ability of platelets to assist in the blood-clotting process. In addition, as is the case with individuals affected by many other primary immunodeficiency disorders, individuals with CVID should not receive live virus vaccines since there is the remote possibility that the vaccine strains of virus may cause disease as a result of their defective immune systems.
Surveillance for complications include periodic complete blood count (CBC), and differential white blood counts to detect lymphoma, annual thyroid examination and thyroid function testing, annual lung (pulmonary) function testing beginning about age eight to ten years, biopsy of enlarged lymphoid tissue, and other imaging techniques for assessment of granulomatous disease and gastrointestinal complications.
Genetic counseling is recommended for affected individuals and their family members if a rare autosomal recessive type of CVID is suspected or confirmed. Other treatment is symptomatic and supportive.
Inflammatory bowel disease remains a significant problem in 19%-32% CVID, leading to chronic, even severe diarrhea characterized by weight loss, steatorrhea, and malabsorption 18. On biopsy, the gastrointestinal mucosa contains excess intraepithelial lymphocytes, villous blunting, lymphoid aggregates, granulomas, crypt distortion, and a characteristic lack of plasma cells. Another common feature is villous flattening in the small intestine, suggesting celiac sprue, wheat exclusion is not likely to be helpful. Nodular lymphoid hyperplasia (containing an expanded number of B cells but reduced plasma cells) is common and can impair the absorption of nutrients. Nutrient replacement, broad-spectrum antibiotics, or, in some cases, oral budesonide can be of use in refractory cases.
Monitoring patients over time
One of the questions that have arisen is how to follow CVID subjects over time. Due to the heterogeneity of this disease, there are no set rules aside from regularly scheduled follow-ups, and periodic monitoring of serum IgG levels. Full chemistry panels and complete blood counts are also important to check for small or larger problems that can arise over time. Monitoring subjects for lung disease has been controversial and there is no current consensus, but most agree that chest X-ray at baseline is more than reasonable. However, because radiosensitivity has been demonstrated in CVID 35, MRIs appear to provide a satisfactory means of follow-up 36. For more frequent monitoring of patients with chronic cough and/or known lung damage, doctors do complete lung functions including carbon monoxide diffusion as a means of assessing lung damage at yearly intervals. Gastrointestinal diseases will be evident with symptoms of diarrhea and/or weight loss and this can be evaluated with appropriate measures. According to some researchers, the data do not suggest that routine endoscopy is required, although patients with suggestive gastrointestinal symptoms should have appropriate upper and/or lower endoscopy with examination for Helicobacter pylori or other mucosal changes. Specific monitoring for autoimmunity is not required because routine blood counts and general medical oversight will reveal characteristic symptoms. Lymphadenopathy is very common in CVID and evaluation of these is not simple; when new nodes appear and persist, biopsy may be required. However, lymphomas are more commonly extranodal and appear in unusual locations such as the lungs or mucosal-associated tissues such as the gastrointestinal tract, and are thus not amenable to standard follow-up measures. Bone marrow examinations to look for lymphoma have not been revealing, with the exception of advanced cases in which the diagnosis was already established by other means.
- Cunningham-Rundles C. The many faces of common variable immunodeficiency. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2012;2012:301-305. doi:10.1182/asheducation-2012.1.301. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4066657/[↩]
- Primary immunodeficiencies: 2009 update. International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies., Notarangelo LD, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, Etzioni A, Hammartröm L, Nonoyama S, Ochs HD, Puck J, Roifman C, Seger R, Wedgwood J. J Allergy Clin Immunol. 2009 Dec; 124(6):1161-78. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2797319/[↩]
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol 1999;92:34-48.[↩]
- van Zelm M, Reisli I, van der Burg M, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med 2006;354:1901-1912.[↩]
- Salzer U, Maul-Pavicic A, Cunningham-Rundles C, et al. ICOS deficiency in patients with common variable immunodeficiency. Clin Immunol 2004;113:234-240.[↩]
- Pan-Hammarström Q, Salzer U, Du L, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet 2007;39:429-430.[↩]
- Orange JS, Hossny EM, Weiler CR, et al. Use of intravenous immunoglobulin in human disease: a review of evidence by members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol. 2006;117(4 suppl):S525–553. https://www.ncbi.nlm.nih.gov/pubmed/16580469[↩]
- Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125(6):1354–1360. https://www.ncbi.nlm.nih.gov/pubmed/20471071[↩]
- Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–286 http://www.bloodjournal.org/content/112/2/277.long[↩]
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92(1):34–48. https://www.ncbi.nlm.nih.gov/pubmed/10413651[↩]
- Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, Fieschi C, Thon V, Abedi MR, Hammarstrom L. Blood. 2008 Jul 15; 112(2):277-86. http://www.bloodjournal.org/content/112/2/277.long[↩][↩][↩][↩]
- Morbidity and mortality in common variable immune deficiency over 4 decades. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3286343/Blood. 2012 Feb 16; 119(7):1650-7.[↩][↩][↩]
- Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. J Allergy Clin Immunol. 2004 Aug; 114(2):415-21. https://www.ncbi.nlm.nih.gov/pubmed/15316526/[↩]
- Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119:1650–1657. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3286343/[↩][↩]
- Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650-1657. doi:10.1182/blood-2011-09-377945. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3286343/[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Vajdic CM, Mao L, van Leeuwen MT, Kirkpatrick P, Grulich AE, Riminton S. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood. 2010;116:1228–1234 http://www.bloodjournal.org/content/116/8/1228.long[↩]
- Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119:1650–1657 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3286343/[↩][↩][↩]
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. https://www.ncbi.nlm.nih.gov/pubmed/10413651[↩][↩][↩]
- Cunningham-Rundles C, Lieberman P, Hellman G, Chaganti RS. Non-Hodgkin lymphoma in common variable immunodeficiency. Am J Hematol. 1991;37:69–74. https://www.ncbi.nlm.nih.gov/pubmed/1829873[↩]
- Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27:308–316. https://www.ncbi.nlm.nih.gov/pubmed/17510807[↩]
- Mellemkjaer L, Hammarstrom L, Andersen V, et al. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: a combined Danish and Swedish study. Clin Exp Immunol. 2002;130:495–500. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1906562/[↩]
- Sánchez-Ramón S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B cells in common variable immunodeficiency: Clinical associations and sex differences. Clin Immunol. 2008;128(3):314–321 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2692232/[↩]
- Warnatz K, Denz A, Drager R, et al. Severe deficiency of switched memory B cells (CD27(+)IgM(−)IgD(−[↩]
- Grimbacher B, Hutloff A, Schlesier M, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4:261–268 https://www.ncbi.nlm.nih.gov/pubmed/12577056[↩]
- van Zelm MC, Reisli I, van der Burg M, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–1912 http://www.nejm.org/doi/10.1056/NEJMoa051568[↩]
- Warnatz K, Salzer U, Rizzi M, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. 2009;106:13945–13950. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2722504/[↩]
- Kuijpers TW, Bende RJ, Baars PA, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. 2010;120:214–222 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2798692/[↩]
- Thiel J, Kimmig L, Salzer U, et al. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J Allergy Clin Immunol. 2012;129:801–810. https://www.ncbi.nlm.nih.gov/pubmed/22035880[↩]
- van Zelm MC, Smet J, Adams B, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;120:1265–1274 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2846042/[↩]
- van Montfrans JM, Hoepelman AI, Otto S, et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol. 2012;129:787–793. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3294016[↩]
- Pan-Hammarström Q, Salzer U, Du L, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39:429–430 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2931279/[↩]
- Salzer U, Bacchelli C, Buckridge S, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113:1967–1976. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2651012/[↩]
- Conley ME, Dobbs AK, Farmer DM, et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. 2009;27:199–227. https://www.ncbi.nlm.nih.gov/pubmed/19302039[↩]
- Orange JS, Glessner JT, Resnick E, et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. J Allergy Clin Immunol. 2011;127:1360–1367 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3646656[↩]
- Vorechovský I, Scott D, Haeney MR, Webster DA. Chromosomal radiosensitivity in common variable immune deficiency. Mutat Res. 1993;290:255–264 https://www.ncbi.nlm.nih.gov/pubmed/7694117[↩]
- Serra G, Milito C, Mitrevski M, et al. Lung MRI as a possible alternative to CT scan for patients with primary immune deficiencies and increased radiosensitivity. Chest. 2011;140:1581–1589. https://www.ncbi.nlm.nih.gov/pubmed/21622550[↩]





 immunoglobulin injection to partially restore immunoglobulin levels.){kind=link}