
Contents
- Hemophilia B
- Hemophilia B cause
- Hemophilia B genetics
- Hemophilia B signs and symptoms
- Hemophilia B complications
- Hemophilia B diagnosis
- Hemophilia B differential diagnosis
- Hemophilia B treatment
- Table 5. Products available to treat people with hemophilia B
- Hemophilia B prophylaxis
- Hemophilia B acute bleeding management
- Hemophilia B inhibitors
- Infants and children with hemophilia B
- Prevention of secondary complications
- Surveillance
- Agents/circumstances to avoid
- Evaluation of relatives at risk
- Hemophilia B pregnancy management
- Hemophilia B prognosis
Hemophilia B
Hemophilia B also called Haemophilia B, Christmas disease or Royal disease is an inherited bleeding disorder that is inherited in X-linked recessive pattern caused by a deficiency in blood clotting factor IX (factor 9) that results in the inability of the blood to clot properly with prolonged bleeding from injuries, tooth extractions, or surgery, and internal bleeding into joints and muscles, which can cause pain and swelling and delayed or recurrent bleeding prior to complete wound healing 1, 2. Hemophilia B primarily follows an X-linked recessive pattern of inheritance. However, researchers have also reported some acquired forms resulting from the development of autoantibodies against factor IX (factor 9) 2. Individuals with hemophilia B have insufficient levels of a blood protein called factor IX (factor 9) or Christmas factor. Factor IX (factor 9) is a clotting factor. Clotting factors are specialized proteins needed for blood clotting, the process by which blood seals a wound to stop bleeding and promote healing. Individuals with hemophilia B do not bleed faster than unaffected individuals, they bleed longer. This is because they are missing or have a decreased amount of factor IX (factor 9) protein involved in blood clotting and are unable to effectively stop the flow of blood from a wound, injury or bleeding site. This is sometimes referred to as prolonged bleeding or a bleeding episode.
Hemophilia B is classified as mild, moderate or severe based upon the activity level of factor IX. In mild hemophilia B cases, bleeding symptoms may occur only after surgery, injury or a dental procedure. In some moderate and most severe hemophilia B cases, bleeding symptoms may occur after a minor injury or spontaneously, meaning without an identifiable cause.
The main signs and symptoms of hemophilia B are 3, 4, 5, 6, 7:
- Easy or excessive bruising from an early age. Bruises may form easily from minor bumps.
- Internal bleeding for no obvious reason, especially in your joints and muscles. That can result in chronic pain, swelling, joint damage, disability and joint deformity at an early age 8, 9, 10
- Greater than normal bleeding after injuries or surgery. Bleeding from cuts, surgery, or dental procedures may take a long time to stop.
- Unexplained nosebleeds.
- Bleeding into the urine or stool can also occur. Gastrointestinal tract and urinary tract bleeding.
- In more severe cases, bleeding can occur without a known cause.
Hemophilia B is caused by a defect in the F9 gene located on the X chromosome and thus it is inherited in an X-linked recessive pattern 11, 12, 13. Because males only have one X-chromosome (males have XY sex chromosome), hemophilia B most commonly affects males, who inherit the X chromosome from their mother. However, in about 30% of cases of hemophilia B, the altered F9 gene occurs spontaneously also called de novo mutation without a previous family history 2. The F9 gene provides instructions for making a protein called coagulation factor IX (factor 9) 11, 12, 13. Coagulation factors are a group of related proteins that are essential for the formation of blood clots (see Figure 2). After an injury, blood clots protect your body by sealing off damaged blood vessels and preventing further blood loss. Coagulation factor IX (factor 9) is made in your liver. The factor IX (factor 9) protein circulates in your bloodstream in an inactive form until an injury that damages blood vessels occurs. In response to injury, coagulation factor IX (factor 9) is activated by another coagulation factor called factor XIa (factor 9 activated). The active protein, sometimes written as coagulation factor IXa (activated factor IX), interacts with coagulation factor VIII (factor 8) and other molecules. These interactions set off a chain of additional chemical reactions that form a blood clot.
Females have 2 X chromosomes (XX chromosome), an X chromosome from the mother and an X chromosome from the father. Since a female has two X chromosomes (XX sex chromosome), in order to have hemophilia B, a female would have to inherit the F9 gene on the X chromosome from each parent. For this reason, it’s very rare for females to get hemophilia B. Usually, a female has only one mutated copy of the F9 gene in her X chromosome (one F9 gene with a mutation and one normal F9 gene), making her a carrier of hemophilia B. A female with one mutated F9 gene and one normal F9 gene is called “heterozygous” or a “carrier”. This means the mutated F9 gene in her X chromosome can be passed down to her children. Boys born to a woman who is a carrier of hemophilia B (one F9 gene with a mutation and one normal F9 gene) have a 50% chance of having hemophilia B. Girls born to a woman who is a carrier of hemophilia B have a 50% chance of being a carrier of hemophilia B. Female hemophilia B F9 gene carriers (one F9 gene with a mutation and one normal F9 gene) do not have symptoms of hemophilia B but may have lower than usual quantities of Factor IX (factor 9) in her blood. However, some females with hemophilia B can have heavy or long periods, which can cause iron deficiency or anemia and heavy bleeding after giving birth. While bleeding symptoms in females are usually milder than those in males with hemophilia B, in rare cases, a female with one hemophilia gene can have bleeding symptoms that are as serious as those of a male with hemophilia.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional. For people with a family history of hemophilia, genetic testing might be used to identify carriers to make informed decisions about becoming pregnant. It’s also possible to determine during pregnancy if your unborn child is affected by hemophilia. However, the testing poses some risks to your unborn child. Discuss the benefits and risks of testing with your doctor.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Newly recommended terminology for female carriers (one F9 gene with a mutation and one normal F9 gene) designates 5 clinical- and laboratory-based categories 14. For females with decreased (≤40%) factor IX clotting activity, the terminology is the same as that used for males with hemophilia B:
- Mild hemophilia B (>5% to <40% factor IX clotting activity)
- Moderate hemophilia A (1%-5% factor IX clotting activity)
- Severe hemophilia A (<1% factor IX clotting activity).
For female carriers (one F9 gene with a mutation and one normal F9 gene) with normal factor IX clotting activity 14:
- Individuals with a bleeding phenotype: “symptomatic hemophilia carriers”
- Individuals who do not have a bleeding phenotype: “asymptomatic hemophilia carriers”
All males with a mutated F9 gene are affected and will have hemophilia B of approximately the same severity as all other affected males in the family; however, other genetic and environmental effects may modify the clinical severity to some extent.
Approximately 30% of female carriers (one F9 gene with a mutation and one normal F9 gene) have factor IX clotting activity below 40% and are at risk for bleeding; mild bleeding can occur in carriers with low-normal factor IX activity 15.
The birth prevalence of hemophilia B has been calculated to be 5 in 100,000 live male births worldwide and 1.5 in 100,000 for severe hemophilia B 16. The birth prevalence is thought to be approximately the same in all countries and all ethnicities, presumably because of the high spontaneous mutation rate of F9 gene and its presence on the X chromosome.
Hemophilia B is about one fifth as prevalent as hemophilia A.
In individuals with severe hemophilia B, spontaneous joint or deep-muscle bleeding is the most frequent sign. Individuals with severe hemophilia B are usually diagnosed during the first two years of life; without prophylactic treatment, they may average up to two to five spontaneous bleeding episodes each month.
Individuals with moderate hemophilia B seldom have spontaneous bleeding; however, they do have prolonged or delayed oozing after relatively minor trauma and are usually diagnosed before age five to six years; the frequency of bleeding episodes varies from once a month to once a year.
Individuals with mild hemophilia B do not have spontaneous bleeding episodes; however, without pre- and postoperative treatment, abnormal bleeding occurs with surgery or tooth extractions; the frequency of bleeding may vary from once a year to once every ten years. Individuals with mild hemophilia B are often not diagnosed until later in life.
In any individual with hemophilia B, bleeding episodes may be more frequent in childhood and adolescence than in adulthood. Approximately 30% of heterozygous females have factor IX clotting activity lower than 40% and are at risk for bleeding (even if the affected family member has mild hemophilia B), although symptoms are usually mild. After major trauma or invasive procedures, prolonged or excessive bleeding usually occurs, regardless of severity.
The diagnosis of hemophilia B is established in individuals with low factor IX clotting activity. Identification of a hemizygous clotting factor IX gene (F9) pathogenic variant on molecular genetic testing in a male proband confirms the diagnosis. Identification of a heterozygous F9 pathogenic variant on molecular genetic testing in a symptomatic female confirms the diagnosis.
If you are the first person in your family to have a suspected bleeding disorder, your doctor will order a series of tests called a coagulation study.
Tests to diagnose hemophilia B include:
- Prothrombin time (PT). A prothrombin time (PT) test evaluates the coagulation factors VII, X, V, II, and I (fibrinogen).
- Partial thromboplastin time (PTT) also known as activated partial thromboplastin time (aPTT). The partial thromboplastin time (PTT) is used to evaluate the coagulation factors XII, XI, IX, VIII, X, V, II (prothrombin), and I (fibrinogen) as well as prekallikrein (PK) and high molecular weight kininogen (HK).
- Serum factor IX activity
Hemophilia B lab results:
- Normal platelet count
- Prolonged activated partial thromboplastin time (aPTT) in severe and moderate hemophilia B. Normal or mildly prolonged aPTT in mild hemophilia B.
- Normal prothrombin time (PT).
People with hemophilia B will exhibit a prolonged activated partial thromboplastin time (aPTT), a normal prothrombin time (PT), and normal platelet levels, indicating an intrinsic pathway disruption. As pregnancy and stress can falsely increase factor IX activity levels, it is essential to recheck the activity levels, if necessary, once these situations have been resolved. Patients with mild factor IX deficiencies may exhibit a normal activated partial thromboplastin time (aPTT). Hematologists may perform mixing studies on blood from people with hemophilia B with an isolated prolonged aPTT to distinguish between a factor deficiency and an inhibitor.
Doctors will also test for factor IX activity on males with a family history of hemophilia, patients without a known family history but with a clinical history consistent with hemophilia, and females who are known or may be genetic carriers. A factor IX activity level below 40% confirms the diagnosis of hemophilia B. Genetic testing for all patients with hemophilia B are done to assist in predicting which patients are likely to develop inhibitors and identifying carrier females in the family. An inhibitor is an antibody that develops against an infused factor and hinders its proper functioning. For individuals with a confirmed diagnosis of hemophilia B, periodic laboratory evaluation involves screening for the presence of factor IX antibodies and testing for transfusion-related infections such as hepatitis and HIV.
The main treatment for severe hemophilia B involves replacing the missing clotting factor IX (factor 9) you need intravenously.
Intravenous infusion of plasma-derived or recombinant factor IX for bleeding episodes should be initiated within an hour of noticing symptoms:
- Dosing is weight based and target levels and duration of treatment vary by the severity of bleeding and/or the risk associated with the surgery or procedure.
- Identify staff members who are expert in performing venipunctures in infants and toddlers because frequent venipunctures may be necessary.
- Parents of children age two to five years with severe hemophilia B should be trained to administer the infusions as soon as is feasible. Home treatment allows for prompt treatment and facilitates prophylactic therapy.
The following formula calculates the appropriate dose for factor IX (factor 9) replacement 17, 2:
- The initial dose for factor IX = [patient’s body weight (in kg)] x [desired factor IX increase (expressed as % in whole number)] x [factor accounting for the volume of redistribution (IU/kg; usually around 1 for factor IX)]
For example, a 45-year-old female with a body weight of 50 kg needs to increase her factor IX level by 100%, which is calculated as 50 x 100 x 1 = 5000 IU of factor IX.
Several strategies and treatment options can be utilized when managing bleeding episodes and dental extractions in patients with hemophilia 2:
- Repeating the dose based on the half-life of the infused product.
- Considering prothrombin complex concentrate if factor IX is unavailable.
- Utilizing antifibrinolytic agents, including tranexamic acid and epsilon-aminocaproic acid, and monoclonal antibodies, such as rituximab, for consideration in cases of mucosal bleeds and dental extractions in patients with hemophilia.
Special considerations for care of infants and children with hemophilia B include the following 18, 19:
- Infant males with a family history of hemophilia B should not be circumcised unless hemophilia B is either excluded or, if present, treated with factor IX concentrate directly before and after the procedure.
- Immunizations should be administered subcutaneously if known to be an effective route; intramuscular injections may be managed with pressure and ice and, if possible, under factor IX coverage.
- Effective dosing of factor IX requires an understanding of different pharmacokinetics in young children.
Young children with severe or moderate hemophilia B should be evaluated at a Hemophilia Treatment Center (accompanied by their parents/guardians) every six to 12 months and as needed to review their history of bleeding episodes and adjust treatment plans. Early signs and symptoms of possible bleeding episodes are reviewed. The assessment should also include a joint and muscle evaluation, an inhibitor screen, viral testing if indicated, a discussion of any other issues related to the individual’s hemophilia B, and family and community support.
In 2022, the U.S. Food and Drug Administration (FDA) approved a gene therapy for patients with hemophilia B with Etranacogene dezaparvovec-drlb (Hemgenix) an adeno-associated virus serotype 5 (AAV5) vector containing a codon-optimized Padua variant of the human F9 gene with a liver-specific promoter 20, 21, 22. This variant of the F9 gene contains a missense mutation that significantly increases F9 activity by 4- to 40-fold. Etranacogene dezaparvovec-drlb (Hemgenix) offers a one-time treatment option for adults with hemophilia B who use factor IX prophylaxis but still experience severe bleeding.
Prophylactic treatment may also be achieved with recent FDA-approved “rebalancing agents” such as marstacimab, concizumab, and fitusiran 23, 24, 25. Non-factor therapies (marstacimab, concizumab, and fitusiran) inhibit inhibitors of coagulation and “rebalance” hemostasis to allow a more normal hemostatic response. They are administered subcutaneously and are used as prophylactic therapy.
- Note: Factor IX concentrate, or a bypassing agent for those with inhibitors, is still needed for breakthrough bleeding and with most surgical procedures.
- Marstacimab, an inhibitor of tissue factor pathway inhibitor, was approved by the FDA in October 2024 for individuals age 12 years and older without inhibitors.
- Concizumab, an inhibitor of tissue factor pathway inhibitor, was approved by the FDA in December 2024 for individuals age 12 years and older with inhibitors.
- Fitusiran, an inhibitor of antithrombin, was approved by the FDA in March 2025 for individuals age 12 years and older with and without inhibitors.
When traveling, people with hemophilia B should wear a medical alert bracelet, carry a supply of factor replacement, and be aware of the location of the nearest hemophilia treatment center if available.
Table 1. Coagulation factors
| Coagulation factor | Other common name |
|---|---|
| I | Fibrinogen |
| II | Prothrombin |
| V | Proaccelerin or labile factor |
| VII | Proconvertin |
| VIII | Antihemophilic factor A |
| IX | Antihemophilic factor B or Christmas factor |
| X | Stuart-Prower factor |
| XI | Plasma thromboplastin antecendent |
| XIII | Fibrin stabilizing factor |
Figure 1. Overview of blood coagulation
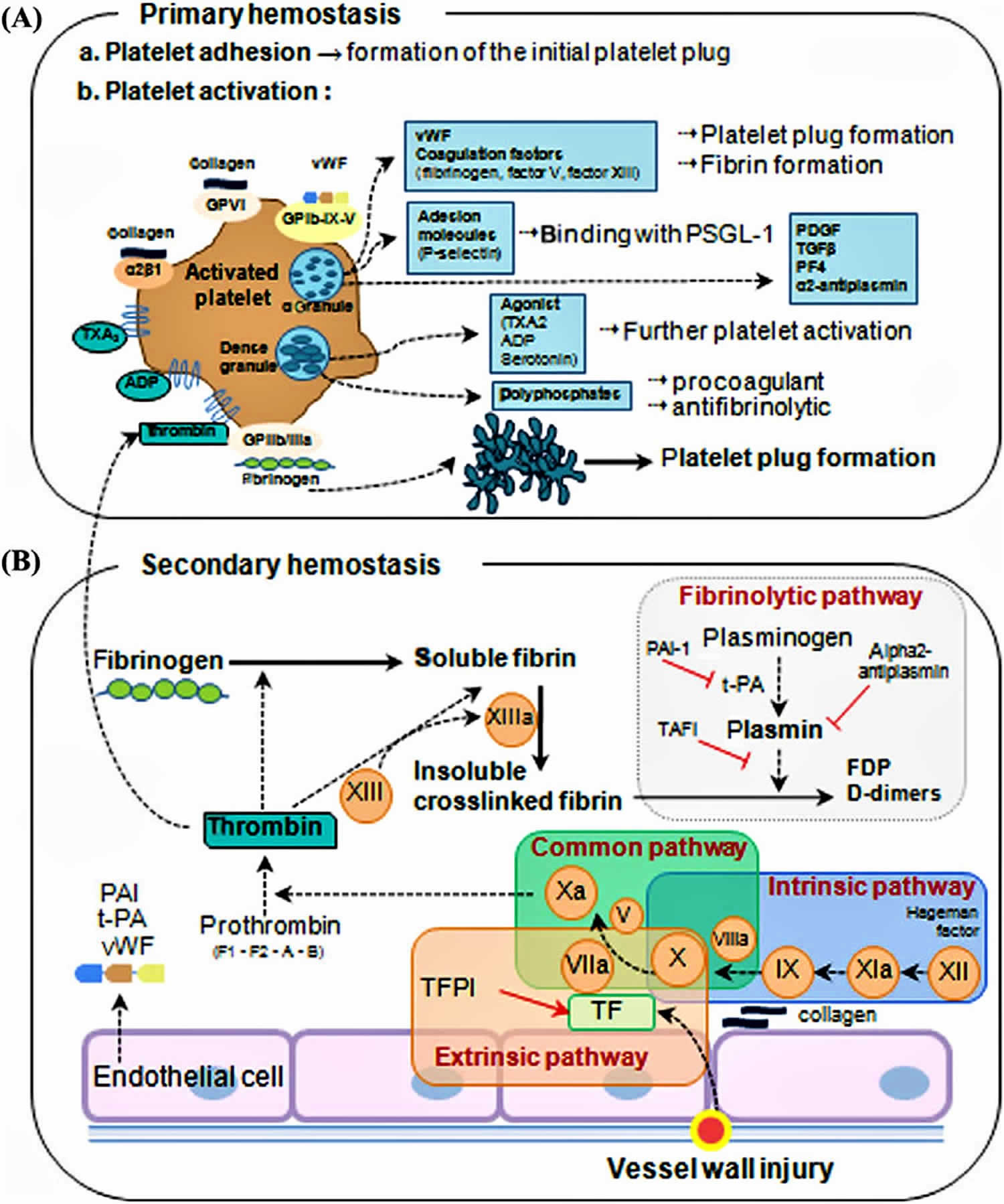
Figure 2. Coagulation cascade
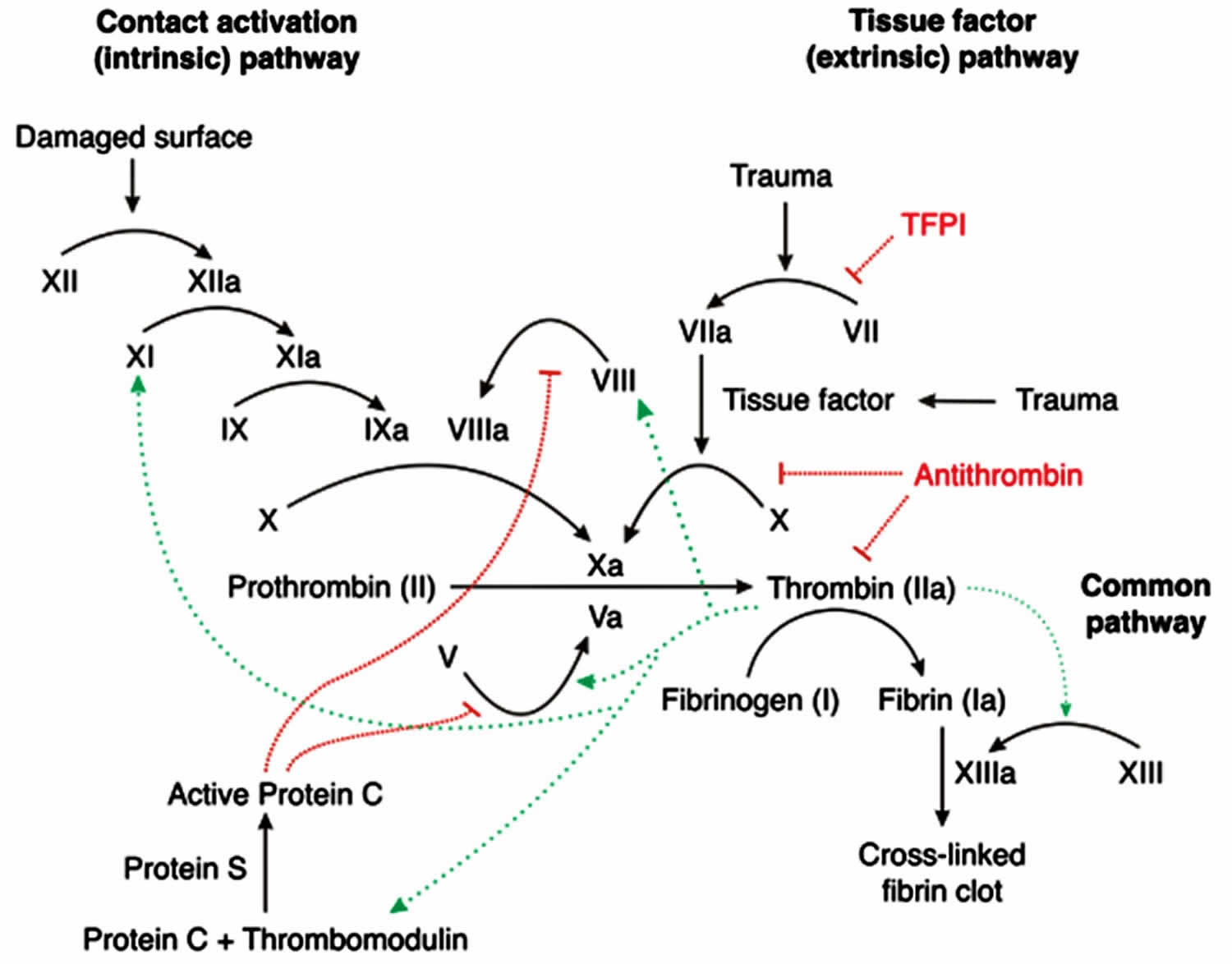
Figure 3. Hemophilia B – X-linked inheritance pattern
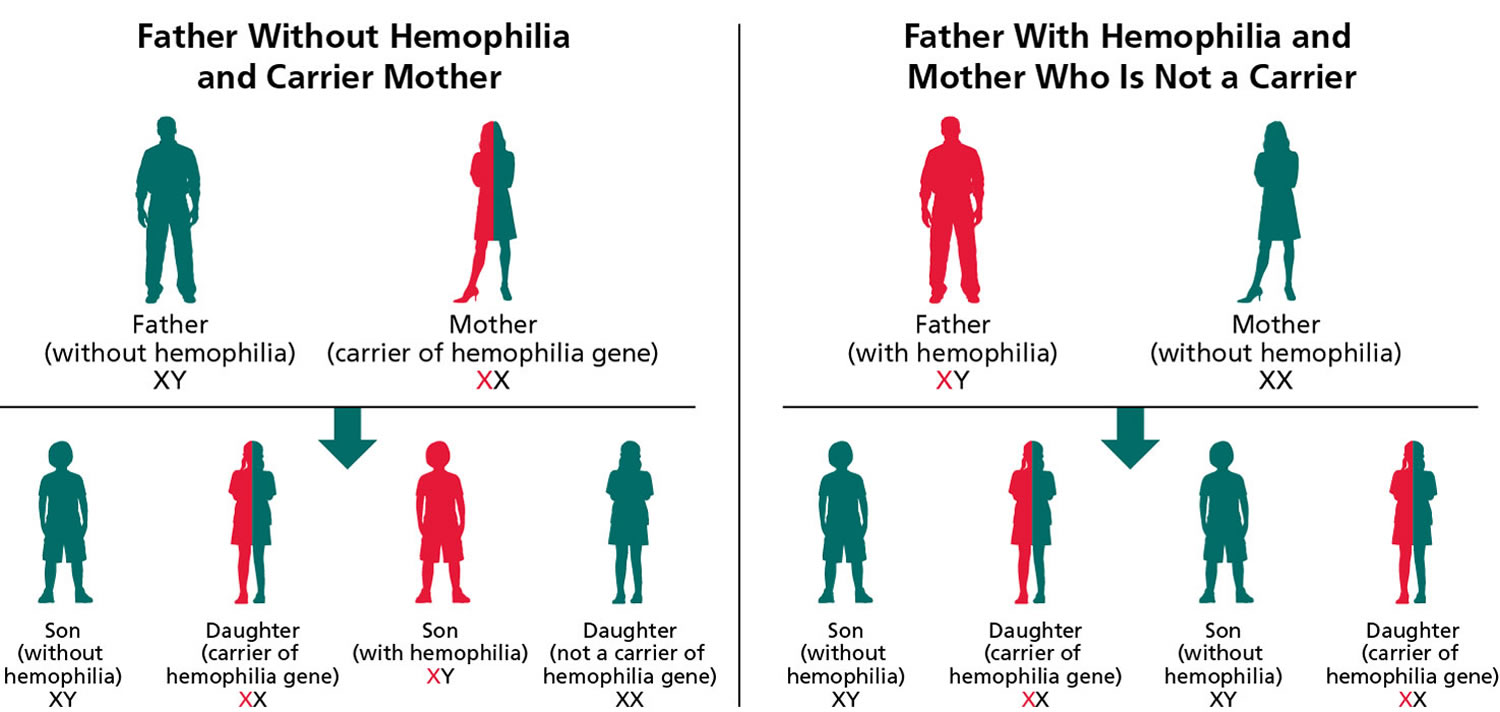
Note: Hemophilia B is inherited in an X-linked manner. Referring to a gene on the X chromosome or to the mode of inheritance in which the causative pathogenic variant is on the X chromosome; hemizygous males will be affected; heterozygous females may or may not be affected depending on the disorder and factors influencing X-chromosome inactivation.
Can people with hemophilia play sport?
Yes. People with hemophilia can safely participate in a wide range of sports 19, 26. Regular exercise and physical activity can strengthen muscles and protect joints, preventing injuries and bleeding episodes 27. As for everyone, physical activity can help people with hemophilia feel better generally, be with friends and have fun. Likewise, it is important that everyone uses the protective equipment that is appropriate to the sport. Generally, it is recommended that people with hemophilia do not do high contact sports like boxing. They can discuss their sporting goals with their Hemophilia Treatment Center and work together on their chosen sport. Ideally, individuals with hemophilia or their family caregivers should consult a physical therapist before engaging in new sports and physical activities to discuss their appropriateness, required protective gear, preventive measures, and required physical skills prior to beginning the activity. This is particularly important if the individual has any joint with recurrent bleeding (i.e., target joint) 28. Target joints can be protected with braces or splints during physical activity, especially in the absence of clotting factor coverage 29, 30.
Non-contact sports such as swimming, walking, jogging, golf, badminton, archery, cycling, rowing, sailing, and table tennis should be encouraged 19, 26. High-contact and collision sports such as soccer, hockey, rugby, boxing, and wrestling, and high-velocity activities such as motocross racing and skiing are not advised due to the potential for life-threatening injuries, unless the individual is on adequate prophylaxis to cover such activities and is well educated on the potential risks 19, 26. Custom-made dental mouthguards should be used by individuals with hemophilia for all contact sports to prevent trauma and injury to teeth and oral soft tissues 31.
For those with significant musculoskeletal dysfunction, weight-bearing activities that promote development and maintenance of good bone density should be encouraged to the extent their joint health permits 32. The choice of activities should reflect the individual’s preferences/interests, physical condition and ability, local contexts, and available resources. Organized sports programs should be encouraged over unstructured sports activities where protective equipment and supervision may be lacking.
Should a child with hemophilia wear protective gear?
Current treatments mean that protective gear for everyday living is not necessary. Wearing appropriate protective gear with some activities is recommended for everyone whether they have hemophilia or not. Examples of standard protective gear are helmets for cycling and motor bike riding, shin pads for soccer, helmets and pads for cricket and mouthgards for water polo and basketball.
Hemophilia B Leyden
There is an unusual form of factor IX (factor 9) deficiency called hemophilia B Leyden. Hemophilia B Leyden is named after the place in the Netherlands where it was first described. Depending upon the particular hemophilia B Leyden mutation present, there are undetectable levels of factor IX (factor 9) present early in life that increase over time. By midlife, these patients have factor IX levels at the low end of the normal range and thus may no longer require treatment for bleeding episodes. Hemophilia B Leyden represents approximately 3% of all hemophilia B cases.
Hemophilia B cause
Hemophilia B is caused by a change (variant or mutation) in the F9 gene located on the X chromosome and it’s passed down from parent in an X-linked recessive pattern. Although most cases of hemophilia B are inherited (passed down) from a parent to a child, about 30% of cases have no previous family history, the altered gene occurs spontaneously without a previous family history called de novo mutation. The F9 gene contains instructions for creating the factor IX (factor 9) protein. Mutations in the F9 gene can lead to deficient levels of functional factor IX (factor 9) protein. The bleeding symptoms associated with hemophilia B occur due to this deficiency.
Hemophilia B is inherited in an X-linked recessive manner. The F9 genes for factor IX (factor 9) are located on the X chromosome. The X chromosome is one of two sex chromosomes, and it contains hundreds of genes that are essential for both male and female development. Females typically have two X chromosomes (XX sex chromosome), while males have one X and one Y chromosome (XY sex chromosome). There are no genes for clotting factors on the Y chromosome.
Males only have 1 X chromosome (XY sex chromosome). Males inherit an X chromosome from the mother and a Y chromosome from the father. Because males have only one X chromosome (males have XY chromosome). A single recessive F9 gene on that X chromosome will cause hemophilia B. This means that hemophilia B almost always occurs in males and is passed from mother to son through one of the mother’s genes. For this reason, most people with hemophilia B are males. Male hemophilia B patients do not transmit hemophilia B to their sons because males only pass their Y chromosome on to their sons. All female children of men with of hemophilia B carry the mutated F9 gene whereas male children do not because female (XX sex chromosome) children of men with of hemophilia B inherited one normal F9 gene from her mother X chromosome and one mutated F9 gene from her father X chromosome.
Females have 2 X chromosomes (XX chromosome), an X chromosome from the mother and an X chromosome from the father. Since a female has two X chromosomes (XX sex chromosome), in order to have hemophilia B, a female would have to inherit the F9 gene on the X chromosome from each parent. For this reason, it’s very rare for females to get hemophilia B. Usually, a female has only one mutated copy of the F9 gene in her X chromosome (one F9 gene with a mutation and one normal F9 gene), making her a carrier of hemophilia B. A female with one mutated F9 gene and one normal F9 gene is called “heterozygous” or a “carrier”. This means the mutated F9 gene in her X chromosome can be passed down to her children. Boys born to a woman who is a carrier of hemophilia B (one F9 gene with a mutation and one normal F9 gene) have a 50% chance of having hemophilia B. Girls born to a woman who is a carrier of hemophilia B have a 50% chance of being a carrier of hemophilia B. Female hemophilia B F9 gene carriers (one F9 gene with a mutation and one normal F9 gene) do not have symptoms of hemophilia B but may have lower than usual quantities of Factor IX (factor 9) in her blood. However, some females with hemophilia B can have heavy or long periods, which can cause iron deficiency or anemia and heavy bleeding after giving birth. While bleeding symptoms in females are usually milder than those in males with hemophilia B, in rare cases, a female with one hemophilia gene can have bleeding symptoms that are as serious as those of a male with hemophilia.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Hemophilia B genetics
Hemophilia B is caused by a change (variant or mutation) in the F9 gene located on the X chromosome and it’s passed down from parent in an X-linked recessive pattern. Although most cases of hemophilia B are inherited (passed down) from a parent to a child, about 30% of cases have no previous family history, the altered gene occurs spontaneously without a previous family history called de novo mutation. The F9 gene contains instructions for creating the factor IX (factor 9) protein. Mutations in the F9 gene can lead to deficient levels of functional factor IX (factor 9) protein. The bleeding symptoms associated with hemophilia B occur due to this deficiency.
Hemophilia B is inherited in an X-linked recessive manner. The F9 genes for factor IX (factor 9) are located on the X chromosome. The X chromosome is one of two sex chromosomes, and it contains hundreds of genes that are essential for both male and female development. Females typically have two X chromosomes (XX sex chromosome), while males have one X and one Y chromosome (XY sex chromosome). There are no genes for clotting factors on the Y chromosome.
Males only have 1 X chromosome (XY sex chromosome). Males inherit an X chromosome from the mother and a Y chromosome from the father. Because males have only one X chromosome (males have XY chromosome). A single recessive F9 gene on that X chromosome will cause hemophilia B. This means that hemophilia B almost always occurs in males and is passed from mother to son through one of the mother’s genes. For this reason, most people with hemophilia B are males. Male hemophilia B patients do not transmit hemophilia B to their sons because males only pass their Y chromosome on to their sons. All female children of men with of hemophilia B carry the mutated F9 gene whereas male children do not because female (XX sex chromosome) children of men with of hemophilia B inherited one normal F9 gene from her mother X chromosome and one mutated F9 gene from her father X chromosome.
Females have 2 X chromosomes (XX chromosome), an X chromosome from the mother and an X chromosome from the father. Since a female has two X chromosomes (XX sex chromosome), in order to have hemophilia B, a female would have to inherit the F9 gene on the X chromosome from each parent. For this reason, it’s very rare for females to get hemophilia B. Usually, a female has only one mutated copy of the F9 gene in her X chromosome (one F9 gene with a mutation and one normal F9 gene), making her a carrier of hemophilia B. A female with one mutated F9 gene and one normal F9 gene is called “heterozygous” or a “carrier”. This means the mutated F9 gene in her X chromosome can be passed down to her children. Boys born to a woman who is a carrier of hemophilia B (one F9 gene with a mutation and one normal F9 gene) have a 50% chance of having hemophilia B. Girls born to a woman who is a carrier of hemophilia B have a 50% chance of being a carrier of hemophilia B. Female hemophilia B F9 gene carriers (one F9 gene with a mutation and one normal F9 gene) do not have symptoms of hemophilia B but may have lower than usual quantities of Factor IX (factor 9) in her blood. However, some females with hemophilia B can have heavy or long periods, which can cause iron deficiency or anemia and heavy bleeding after giving birth. While bleeding symptoms in females are usually milder than those in males with hemophilia B, in rare cases, a female with one hemophilia gene can have bleeding symptoms that are as serious as those of a male with hemophilia.
Male proband (index case). The diagnosis of hemophilia B is established in a male proband by identification of decreased factor IX clotting activity.
- Severe hemophilia B. <1% factor IX
- Moderate hemophilia B. 1%-5% factor IX
- Mild hemophilia B. >5%-40% factor IX
Note:
- The normal range for factor IX clotting activity is approximately 50%-150% 33. Individuals with factor IX clotting activity higher than 40% usually have normal coagulation in vivo. However, some increased bleeding can occur with low to low-normal factor IX clotting activity in hemophilia B carrier females 34.
- Somatic mosaicism in males with hemophilia B has been described 35.
Identification of a hemizygous pathogenic variant in clotting factor IX gene (F9) by molecular genetic testing can help predict the clinical phenotype and allow family studies (see Table 4).
Heterozygous females. The diagnosis of hemophilia B is established by determination of low factor IX clotting activity. Approximately 30% of heterozygous females have a factor IX clotting activity below 40%, regardless of the severity of hemophilia B in their family. Bleeding symptoms may be present in those with factor IX activity in the low-normal range 34.
Carrier status is determined by identification of a heterozygous pathogenic variant in clotting factor IX gene (F9) by molecular genetic testing (see Table 4). Factor IX clotting activity is unreliable in the detection of heterozygous females; the majority of obligate carriers, even of severe hemophilia B, have normal factor IX clotting activities.
Penetrance
All males with a clotting factor IX (F9) gene mutation are affected and will have hemophilia B of approximately the same severity as all other affected males in the family; however, other genetic and environmental effects may modify the clinical severity to some extent.
Approximately 30% of females with one clotting factor IX (F9) gene mutation and one normal F9 gene have a factor IX clotting activity lower than 40% and a bleeding disorder; mild bleeding can occur in female carriers with low-normal factor IX activities 34.
Genotype-Phenotype Correlations
Disease severity
- Large deletions, nonsense mutations, and most frameshift mutations cause severe disease.
- Missense mutations can cause severe, moderate, or mild disease depending on their location and the specific substitutions involved.
Unlike hemophilia A, severe hemophilia B is often caused by a missense mutation and several of these are associated with normal cross-reacting material (factor IX antigen) levels.
Certain missense mutations within the propeptide portion of factor IX enhance sensitivity to warfarin by altering the binding of a gamma-carboxylase responsible for post-translational Gla residue formation 36. Uncommon variants within the carboxylase-binding domain of the propeptide cause increased sensitivity to warfarin anticoagulation in individuals without any baseline bleeding tendency 36.
The variant p.Arg384Leu, a missense gain-of-function change associated with markedly elevated circulating levels of factor IX and venous thrombosis at a young age, has been described in one family 37. This amino acid change has been incorporated into factor IX constructs currently being used in gene therapy clinical trials 38.
In hemophilia B Leyden, more than 20 different causative variants in the proximal clotting factor IX gene (F9) promoter region have been described 39; the severity of disease decreases after puberty; mild disease disappears and severe disease becomes mild, depending on the specific pathogenic variant.
Hemophilia B signs and symptoms
Hemophilia B in the untreated individual is characterized by immediate or delayed bleeding or prolonged oozing after injuries, tooth extractions, or surgery or renewed bleeding after initial bleeding has stopped 40. Muscle hematomas or intracranial bleeding can occur immediately or up to four to five days after the original injury. Intermittent oozing may last for days or weeks after tooth extraction. Prolonged or delayed bleeding or wound hematoma formation after surgery is common. After circumcision, males with hemophilia B of any severity may have prolonged oozing, or they may heal normally. In severe hemophilia B, spontaneous joint bleeding is the most frequent sign.
The age of diagnosis and frequency of bleeding episodes are generally related to the factor IX clotting activity (see Table 3). In any affected individual, bleeding episodes may be more frequent in childhood and adolescence than in adulthood. To some extent, this greater frequency is a function of both physical activity levels and vulnerability during more rapid growth.
Hemophilia B should be suspected in an individual with any of the following clinical and/or laboratory features.
Individuals with severe hemophilia B are usually diagnosed as newborns due to birth- or neonatal-related procedures or during the first year of life 41. In untreated toddlers, bleeding from minor mouth injuries and large “goose eggs” from minor head bumps are common; these are the most frequent presenting symptoms of severe hemophilia B. Intracranial bleeding may also result from head injuries. The untreated child almost always has subcutaneous hematomas; some have been referred for evaluation of possible non-accidental trauma.
As the child grows and becomes more active, spontaneous joint bleeds occur with increasing frequency unless the child is on a prophylactic treatment program. Spontaneous joint bleeds or deep-muscle hematomas initially cause pain or limping before swelling appears. Children and young adults with severe hemophilia B who are not treated have an average of two to five spontaneous bleeding episodes each month. Joints are the most common sites of spontaneous bleeding; other sites include the muscles, kidneys, gastrointestinal tract, brain, and nose. Without prophylactic treatment, individuals with hemophilia B have prolonged bleeding or excessive pain and swelling from minor injuries, surgery, and tooth extractions.
Individuals with moderate hemophilia B seldom have spontaneous bleeding but bleeding episodes may be precipitated by relatively minor trauma. Without pretreatment (as for elective invasive procedures) they do have prolonged or delayed oozing after relatively minor trauma and are usually diagnosed before age five to six years. The frequency of bleeding episodes requiring treatment with factor IX concentrates varies from once a month to once a year. Signs and symptoms of bleeding are otherwise similar to those found in severe hemophilia B.
Individuals with mild hemophilia B do not have spontaneous bleeding. However, without treatment, abnormal bleeding occurs with surgery, tooth extractions, and major injuries. The frequency of bleeding may vary from once a year to once every ten years. Individuals with mild hemophilia B are often not diagnosed until later in life when they undergo surgery or tooth extraction or experience major trauma.
Heterozygous females with a factor IX clotting activity level lower than 40% are at risk for bleeding that is usually comparable to that seen in males with mild hemophilia. However, more subtle abnormal bleeding may occur with baseline factor IX clotting activity levels between 30% and 60% 34, 14.
Table 2. Symptoms Related to Severity of Untreated Hemophilia B
| Clinical Severity | Factor IX Clotting Activity 1 | Symptoms | Usual Age of Diagnosis |
|---|---|---|---|
| Severe | <1% |
| Age ≤2 years |
| Moderate | 1% to 5% |
| Age ≤6 yrs |
| Mild | >5% to 40% |
| Often later in life, depending on hemostatic challenges |
Footnote: 1 Clinical severity does not always correlate with the in vitro assay result.
[Source 1 ]Hemophilia B complications
Complications of untreated bleeding. The leading cause of death related to bleeding is intracranial hemorrhage. The major cause of disability from bleeding is chronic joint disease 42.
- Joint damage can result in chronic pain, swelling, stiffness, and reduced range of motion and disability and joint deformity at an early age. Individuals with Hemophilia A are more likely to suffer from arthritis and more likely to require knee/hip replacement compared with the general population 8, 43.
- Poor mobility, self‐care issues, and inability to perform usual daily activities 44, 45.
- Inability to participate in social or sporting activities 46.
- Higher pain levels and functional impairment associated with anxiety, depression and unemployment 47, 48. Pain/discomfort is an area where most individuals report experiencing ‘extreme’ issues 9. Individuals may experience anger and frustration due to the pain, inconvenience and erratic nature of bleeds 49.
- Anxiety/depression are the areas where most individuals report experiencing ‘extreme’ issues 9.
- Adverse impact on educational achievement and work productivity due to absence and difficulties due to functional impairments and pain 44, 50, 51.
Currently available treatment with clotting factor concentrates is normalizing life expectancy and reducing chronic joint disease for children and adults with hemophilia B. Prior to the availability of such treatment, the median life expectancy for individuals with severe hemophilia B was 11 years (the current life expectancy for affected individuals in several developing countries). Excluding death from HIV, life expectancy for those severely affected individuals receiving adequate treatment was 63 years in 2000 52, having been greatly improved with factor replacement therapy 53.
Since the late 1960s, the mainstay of treatment of bleeding episodes has been factor IX concentrates that initially were derived solely from donor plasma. By the late 1970s, more purified preparations became available, reducing the risk for thrombogenicity. Viral inactivation methods and donor screening of plasmas were introduced by 1990 and a recombinant factor IX concentrate became available shortly thereafter 54. A second recombinant factor IX concentrate was FDA licensed in 2013. Two long-acting modified recombinant factor IX concentrates are now FDA approved, extending the factor IX half-life three- to fivefold compared to unmodified products 55 . HIV transmission from concentrates occurred between 1979 and 1985. Approximately half of these individuals died of AIDS prior to the advent of effective HIV therapy.
Hepatitis B transmission from earlier plasma-derived concentrates was eliminated with donor screening and then vaccination introduced in the 1970s. Most individuals exposed to plasma-derived concentrates prior to the late 1980s became chronic carriers of the hepatitis C virus. Viral inactivation methods implemented in concentrate preparation and donor screening assays developed by 1990 have essentially eliminated hepatitis C transmission from plasma-derived concentrates.
Approximately 2% of individuals with severe hemophilia B develop alloimmune inhibitors to factor IX 56. These individuals usually have partial- or whole-gene deletions or certain nonsense variants. At times, the onset of an alloimmune response has been associated with anaphylaxis to transfused factor IX or development of nephrotic syndrome 57.
Hemophilia B diagnosis
The diagnosis of hemophilia B is established in individuals with low factor IX clotting activity. Identification of a hemizygous clotting factor IX gene (F9) pathogenic variant on molecular genetic testing in a male proband confirms the diagnosis. Identification of a heterozygous F9 pathogenic variant on molecular genetic testing in a symptomatic female confirms the diagnosis.
Note:
- Hemizygous refers to a gene normally present in only a single copy; usually an X-linked gene in a male.
- Heterozygous refers to a variant (distinct from the reference sequence) that comprises one of two alleles of a given gene. An individual with two different alleles at a particular locus (one on each chromosome of a pair), one of which is usually pathogenic.
- X-linked inheritance refers to a gene on the X chromosome or to the mode of inheritance in which the causative pathogenic variant is on the X chromosome; hemizygous males will be affected; heterozygous females may or may not be affected depending on the disorder and factors influencing X-chromosome inactivation.
If you are the first person in your family to have a suspected bleeding disorder, your doctor will order a series of tests called a coagulation study.
Tests to diagnose hemophilia B include:
- Prothrombin time (PT). A prothrombin time (PT) test evaluates the coagulation factors VII, X, V, II, and I (fibrinogen).
- Partial thromboplastin time (PTT) also known as activated partial thromboplastin time (aPTT). The partial thromboplastin time (PTT) is used to evaluate the coagulation factors XII, XI, IX, VIII, X, V, II (prothrombin), and I (fibrinogen) as well as prekallikrein (PK) and high molecular weight kininogen (HK).
- Serum factor IX activity
Hemophilia B lab results:
- Normal platelet count
- Prolonged activated partial thromboplastin time (aPTT) in severe and moderate hemophilia B. Normal or mildly prolonged aPTT in mild hemophilia B.
- Normal prothrombin time (PT).
People with hemophilia B will exhibit a prolonged activated partial thromboplastin time (aPTT), a normal prothrombin time (PT), and normal platelet levels, indicating an intrinsic pathway disruption. As pregnancy and stress can falsely increase factor IX activity levels, it is essential to recheck the activity levels, if necessary, once these situations have been resolved. Patients with mild factor IX deficiencies may exhibit a normal activated partial thromboplastin time (aPTT). Hematologists may perform mixing studies on blood from people with hemophilia B with an isolated prolonged aPTT to distinguish between a factor deficiency and an inhibitor.
Doctors will also test for factor IX activity on males with a family history of hemophilia, patients without a known family history but with a clinical history consistent with hemophilia, and females who are known or may be genetic carriers. A factor IX activity level below 40% confirms the diagnosis of hemophilia B. Genetic testing for all patients with hemophilia B are done to assist in predicting which patients are likely to develop inhibitors and identifying carrier females in the family. An inhibitor is an antibody that develops against an infused factor and hinders its proper functioning. For individuals with a confirmed diagnosis of hemophilia B, periodic laboratory evaluation involves screening for the presence of factor IX antibodies and testing for transfusion-related infections such as hepatitis and HIV.
Hemophilia B Molecular Genetic Testing
Molecular genetic testing approaches can include single-gene testing, use of a multi-gene panel, and more comprehensive genomic testing:
- Single-gene testing. Sequence analysis of clotting factor IX gene (F9) is performed first and followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found.
- A multi-gene panel that includes clotting factor IX gene (F9) and other genes of interest may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and over time. (2) Some multi-gene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multi-gene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of pathogenic variants in genes that do not account for the underlying phenotype. (3) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing based tests.
- More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
Table 3. Molecular Genetic Testing Used in Hemophilia B
| Gene | Test Method | Proportion of Probands with a Pathogenic Variant Detectable by This Method |
|---|---|---|
| F9 | Sequence analysis 3, 4 | 97%-100% 5 |
| Gene-targeted deletion/duplication analysis 6 | 2%-3% 5 |
Footnotes:
3 Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include missense, nonsense, and splice site variants and small intragenic deletions/insertions; typically, exon or whole-gene deletions/duplications are not detected.
4 Routine sequence analysis should detect pathogenic variants in the F9 proximal promoter located immediately upstream of the start codon (e.g., c.-49T>A, one variant associated with hemophilia B Leyden). Detection of disease-associated variants located farther upstream may require a targeted assay 58
5 Mitchell et al 59, Goodeve 60, Johnsen et al 61
5 Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
[Source 1 ]Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with hemophilia B, the following evaluations are recommended if they have not already been completed:
- A personal and family history of bleeding to help predict disease severity
- A joint and muscle evaluation, particularly if the individual describes a history of hemarthrosis or deep-muscle hematomas
- Screening for hepatitis A, B, and C as well as HIV if blood products or plasma-derived clotting factor concentrates were administered prior to 1990
- Baseline complete blood count (CBC) including platelet count and ferritin, especially if there is a history of nose bleeds, gastrointestinal bleeding, mouth bleeding, or in females, heavy menstrual bleeding or postpartum hemorrhage
- Referral to a hemophilia treatment center. For locations:
- Worldwide, see World Federation of Hemophilia 62 at https://wfh.org/find-local-support/#HTCs
- US only, see National Bleeding Disorders Foundation 63 at https://www.bleeding.org/
- Identification of the specific clotting factor IX gene (F9) pathogenic variant in an individual to aid in determining disease severity, the likelihood of inhibitor development, and the risk of anaphylaxis if an inhibitor does develop
- Consultation with a clinical geneticist and/or genetic counselor, particularly if a new diagnosis in the family and for females of childbearing years.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Hemophilia B differential diagnosis
Hemophilia B differential diagnosis include 2:
- Coagulation factor deficiencies, such as hemophilia A (factor VIII) and hemophilia C (factor XI), exhibit similar presentations. Differentiation among the 3 is achieved through coagulation factor assay studies and genetic testing. Hemophilia A and B are X-linked recessive, whereas hemophilia C is autosomal recessive.
- Von Willebrand factor (vWF) deficiency is the most common internal bleeding deficiency, with a defect in platelet plug formation. People with von Willebrand disease are either missing or low in the clotting protein von Willebrand factor (vWF) or it doesn’t work as it’s supposed to 64, 65, 66. For a person to make a successful clot, von Willebrand factor (vWF) binds to factor VIII (factor 8), another clotting protein, and platelets in blood vessel walls. This process will help form a platelet plug during the clotting process. Individuals with von Willebrand disease are not able to form this platelet plug, or it will take longer to form. There are three main types of inherited von Willebrand disease. A fourth type, acquired von Willebrand disease, is not hereditary. A combination of blood tests are used to diagnose von Willebrand disease, including a von Willebrand factor (vWF) antigen test, which measures the amount of von Willebrand factor (vWF) in the blood, tests that measure clotting time and ability to form a clot, and tests measuring platelet function. People with von Willebrand disease have an increased bleeding time, normal or increased partial thromboplastin time (PTT or aPTT [activated partial thromboplastin time]), and normal platelets. Some of these tests may have to be repeated, because the levels of von Willebrand factor (vWF) can change due to stress, exercise, the use of birth control pills, pregnancy, and hyperthyroidism. People with von Willebrand disease usually have less than 50% of normal von Willebrand factor (vWF) in their blood. After a diagnosis of von Willebrand disease is discovered, a molecular genetic testing is done to determine the type.
- Quantitative or qualitative platelet dysfunctions generally manifest as bleeding into mucous membranes and skin, unlike hemophilia. The diagnosis is aided by platelet aggregation studies or electron microscopy. Typical findings include an increased bleeding time and a decrease in platelet count. Platelet dysfunction disorders include immune thrombocytopenia, thrombotic thrombocytopenia, and hemolytic uremic syndrome.
- Disseminated intravascular coagulation (DIC) results in blood clot (thrombosis) and bleeding. Signs and symptoms of disseminated intravascular coagulation (DIC) include a decreased platelet count, increased prothrombin time (PT) and aPTT (activated partial thromboplastin time), elevated fibrin degradation (D-dimer), and reduced fibrinogen levels 67 Usually, a precipitating event such as sepsis, trauma, obstetric complications, acute pancreatitis, acute promylogenous leukemia, or a transfusion triggers disseminated intravascular coagulation (DIC).
- Newborns and patients with prolonged antibiotic use can experience vitamin K deficiency. Vitamin K deficiency manifests as increased prothrombin time (PT) and aPTT (activated partial thromboplastin time), decreased factors II, VII, IX, and X, and proteins C and S, along with normal platelet counts 68.
- Scurvy, a vitamin C deficiency, presents with swollen gums, perifollicular and subperiosteal bleeding, joint bleeding, and poor wound healing 69.
- Ehlers-Danlos syndrome results from a defect in collagen synthesis and mainly presents with mucosal bleeding, hyperextensible skin, and hypermobile joints 70.
- Child abuse can be misidentified and confused with hemophilia. Additional signs of child abuse include different stages of wound healing, malnourishment, subdural hematoma, retinal hemorrhage, and signs of sexual abuse such as sexually transmitted infections and urinary tract infections 71.
Bleeding disorders with Low Factor IX clotting activity
- Combined vitamin K-dependent factor deficiency (VKCFD) (OMIM PS277450) is a rare autosomal recessive bleeding disorder where there is a deficiency in multiple clotting factors, including factors II, VII, IX, and X, as well as the natural anticoagulants protein C, protein S, and protein Z 72, 73, 74, 75. This deficiency leads to an increased tendency for bleeding, usually presenting in childhood with severe bleeding. The primary treatment involves vitamin K administration and may require fresh frozen plasma transfusions for severe bleeding. Coagulation laboratory analysis shows a markedly prolonged prothrombin time (PT) and activated partial thromboplastin time (aPTT). The prolonged prothrombin time (PT), multiple coagulation factor deficiencies, and autosomal recessive inheritance would differentiate this from hemophilia B. Combined vitamin K-dependent factor deficiency (VKCFD) is caused by mutations in the genes encoding either gamma-glutamyl carboxylase (GGCX enzyme; VKCFD type 1 with 2p12) or the vitamin K 2,3-epoxide reductase complex subunit 1 (VKORC1 enzyme; VKCFD type 2 with 16p11.2) in the vitamin K cycle. These two proteins are necessary for gamma-carboxylation, a postsynthetic modification that allows coagulation proteins to display their proper function. The developmental and skeletal anomalies seen in combined vitamin K-dependent factor deficiency (VKCFD) are the result of defective gamma-carboxylation of a number of nonhemostatic proteins.
- Common acquired deficiencies of vitamin K-dependent factors occur in individuals receiving warfarin treatment or those with liver disease. Vitamin K deficiency usually presents in the setting of other illnesses, although it may be solely nutritional. Warfarin therapy is by history. Clinical manifestations of liver disease are usually present when coagulation factors are decreased. These diagnoses can be distinguished from hemophilia B by a prothrombin time (PT) that is prolonged greater than the prolongation of the activated partial thromboplastin time (aPTT) versus an isolated prolonged activated partial thromboplastin time (aPTT) in hemophilia B and multiple coagulation factor deficiencies.
Table 4. Inherited bleeding disorders with Normal Factor IX clotting activity
| Disorder | Gene(s) | Mode of inheritance | Clinical Features | Laboratory Findings / Comment |
|---|---|---|---|---|
| Factor XI deficiency (OMIM 612416) | F11 | Autosomal recessive & autosomal dominant | Both compound heterozygotes & homozygotes may exhibit bleeding similar to that seen in mild or moderate hemophilia B. | Heterozygotes have factor XI coagulant activity 25%-75% of normal; homozygotes have activity <1%-15%. 1 A specific factor XI clotting assay establishes diagnosis. |
| Factor XII (OMIM 234000), prekallikrein (OMIM 612423), or high-molecular-weight kininogen (OMIM 228960) deficiencies | F12 KLKB1 KNG1 | Autosomal recessive | Not assoc with linical bleeding | Can cause prolonged activated partial thromboplastin time (aPTT) |
| Factor XIII deficiency (OMIM 613225, 613235) | F13A1 F13B | Autosomal recessive | Umbilical stump bleeding in >80% of persons. Intracranial bleeding that occurs spontaneously or following minor trauma in 30% of persons. Subcutaneous hematomas, muscle hematomas, defective wound healing, & recurrent spontaneous abortion are also seen. Joint bleeding is rare. | All coagulation screening tests are normal; a screening test for clot solubility or specific assay for factor XIII activity can confirm diagnosis. Bleeding symptoms are reported in persons w/levels <13% by quantitative assay. 2 |
| Prothrombin (factor II) (OMIM 613679), factor V (OMIM 227400), factor X (OMIM 227600), & factor VII (OMIM 227500) deficiencies | F2 F5 F7 F10 | Autosomal recessive | Rare bleeding disorders. Persons may have easy bruising & hematoma formation, epistaxis, heavy menstrual bleeding, & bleeding after trauma & surgery. Hemarthroses are less common. Spontaneous intracranial bleeding can occur. | Factor VII deficiency should be suspected if prothrombin time (PT) is prolonged & activated partial thromboplastin time (aPTT) is normal. Persons with deficiency of factors II, V, or X usually have prolonged prothrombin time (PT) & activated partial thromboplastin time (aPTT), but specific coagulation factor assays establish diagnosis. |
| Hemophilia A | F8 | X-linked | Clinically indistinguishable from hemophilia B | Diagnosis is based on factor VIII clotting activity level <40% in presence of normal von Willebrand factor (vWF) level. |
| Afibrinogenemia (OMIM 202400), hypofibrinogenemia (OMIM 616004), dysfibrinogenemia (OMIM 616004) | FGA FGB FGG | Autosomal recessive & autosomal dominant 3 | Afibrinogenemia is associated signs and symptoms similar to hemophilia B except that bleeding from minor cuts is prolonged due to lack of fibrinogen to support platelet aggregation. Hypofibrinogenemia & dysfibrinogenemia can be associated with mild-to-moderate bleeding symptoms. Rarely, persons with dysfibrinogenemia are at risk for thrombosis. | In dysfibrinogenemia there is discordance between functional & antigenic levels, with the latter usually in normal range. For all fibrinogen disorders, thrombin & reptilase times are almost always prolonged & functional measurements of fibrinogen are ↓. |
| Platelet function disorders incl Bernard-Soulier syndrome (OMIM 231200) & Glanzmann thrombasthenia (OMIM 273800) | GP1BA GP1BB GP9 ITGA2B | Autosomal recessive | In Bernard-Soulier syndrome, Glanzmann thrombasthenia, & storage pool & nonspecific secretory defects: skin & mucous membrane bleeding, recurring epistaxis, gastrointestinal bleeding, heavy menstrual bleeding, & excessive bleeding during or immediately after trauma & surgery. Joint, muscle, & intracranial bleeding is rare. | Diagnosis is established using platelet aggregation assays, flow cytometry, & platelet electron microscopy. |
| Type 1 von Willebrand disease | von Willebrand factor (vWF) | Autosomal dominant | Mucous membrane bleeding incl epistaxis, bleeding w/dental extractions, heavy menstrual & postpartum bleeding, & spontaneous bruises. Also may have trauma- & procedure-related bleeding. | Partial quantitative deficiency of von Willebrand factor (vWF) (low von Willebrand factor (vWF) antigen, low factor VIII clotting activity, & low von Willebrand factor (vWF) activity) (Persons w/hemophilia B have normal von Willebrand factor (vWF) level & normal factor VIII activity.) |
| Type 2A & 2B von Willebrand disease | Autosomal dominant | In type 2A, bleeding as in type 1 von Willebrand disease or may be more severe. In type 2B, bleeding as in type 1 von Willebrand disease or may be more severe. Also may have thrombocytopenia. | Qualitative deficiency of von Willebrand factor (vWF) w/↓ of high-molecular-weight multimers (more loss in type 2A). Measures of von Willebrand factor (vWF) platelet or collagen binding activity are ↓, while von Willebrand factor (vWF) antigen & factor VIII clotting activity may be low-normal to mildly ↓. | |
| Type 2M von Willebrand disease | Autosomal dominant | Bleeding as in type 2A von Willebrand disease | Qualitative deficiency of von Willebrand factor (vWF) w/similar ↓ in function as seen in type 2A, but assoc w/normal multimer pattern | |
| Type 2N von Willebrand disease | Autosomal recessive | Clinically indistinguishable from hemophilia B | von Willebrand factor (vWF) platelet binding is completely normal. Biochemically, type 2N von Willebrand disease is indistinguishable from hemophilia B; however, hemophilia B can be distinguished from type 2N von Willebrand disease by molecular genetic testing. | |
| Type 3 von Willebrand disease | Autosomal recessive | Frequent episodes of mucous membrane bleeding. Joint & muscle bleeding similar to that seen in hemophilia B. | Complete or near-complete quantitative deficiency of von Willebrand factor (vWF). von Willebrand factor (vWF) level is often <1% & factor VIII clotting activity is most commonly 2%-8%. |
Footnotes:
1 Duga et al 76
2 Menegatti et al 77
3 Afibrinogenemia is inherited in an autosomal recessive manner. Hypofibrinogenemia can be inherited in either an autosomal dominant or an autosomal recessive manner. Dysfibrinogenemia is inherited in an autosomal dominant manner.
[Source 1 ]Hemophilia B treatment
The World Federation of Hemophilia has published treatment guidelines for the management of individuals with hemophilia 19, 26. Treatment should be coordinated through a hemophilia treatment center. Individuals in the USA see National Bleeding Disorders Foundation 63; individuals worldwide see World Federation of Hemophilia for locations 78
Treatment of hemophilia B manifestations: Referral to a hemophilia treatment center for assessment, education, genetic counseling, and treatment. Intravenous infusion of plasma-derived or recombinant factor IX for bleeding episodes within an hour of noticing symptoms. Training and home infusions for those with severe hemophilia B.
Intravenous infusion of plasma-derived or recombinant factor IX for bleeding episodes should be initiated within an hour of noticing symptoms:
- Dosing is weight based and target levels and duration of treatment vary by the severity of bleeding and/or the risk associated with the surgery or procedure.
- Identify staff members who are expert in performing venipunctures in infants and toddlers because frequent venipunctures may be necessary.
- Parents of children age two to five years with severe hemophilia B should be trained to administer the infusions as soon as is feasible. Home treatment allows for prompt treatment and facilitates prophylactic therapy.
Pediatric issues. Special considerations for care of infants and children with hemophilia B include the following 79:
- Infant males with a family history of hemophilia B should not be circumcised unless hemophilia B is either excluded or, if present, treated with factor IX concentrate directly before and after the procedure.
- Immunizations should be administered subcutaneously; intramuscular injections should be avoided unless under factor coverage.
- Effective dosing of factor IX requires an understanding of different pharmacokinetics in young children.
Prevention of primary manifestations: For those with severe disease, prophylactic infusions of factor IX concentrate twice weekly to maintain factor IX clotting activity higher than 1% nearly eliminates spontaneous bleeding and prevents chronic joint disease. Some individuals require higher trough levels for this effect. Longer-acting products that allow weekly or biweekly dosing are now available. Choice of product should be individualized based on clinical factors and activity levels. Initiation of prophylactic infusions of factor IX concentrate in young boys before or just after their first few joint bleeds has been shown to nearly eliminate spontaneous bleeding and prevent chronic joint disease 80. Prophylaxis in adults is standard of care in many countries and has been shown to decrease bleeding and improve joint function and quality of life 81.
Table 5. Products available to treat people with hemophilia B
| Product | Proper name | Manufacturer | Indication | Route of administration | Strengths (IU) | Storage |
|---|---|---|---|---|---|---|
| Human plasma-derived coagulation factor IX concentrates to treat hemophilia B | ||||||
| AlphaNine SD | Coagulation factor IX (human) | Grifols Biologicals LLC | Prevention and control of bleeding in patients with factor IX deficiency due to hemophilia B. | Intravenous, 10 mL of sterile water for injection | 500, 1000, or 1500 IU | 2 and 8 °C (36 and 46 °F) for 3 y up to the expiry date |
| Mononine | Coagulation factor IX (human) monoclonal antibody purified | CSL Behring, LLC | Prevention and control of bleeding in factor IX deficiency, also known as hemophilia B or Christmas disease. | Intravenous, 5 or 10 mL of sterile water for injection | 500 or 1000 IU | 2 to 8 °C (36-46 °F) |
| Standard half-life products to treat hemophilia B | ||||||
| Benefix | Coagulation factor IX (recombinant) | Pfizer | Treatment of adults and children with hemophilia B for on-demand treatment and control of bleeding episodes or perioperative management of bleeding. Patients 16 y of age and older with hemophilia B for routine prophylaxis to reduce the frequency of bleeding episodes. | Intravenous | 250, 500, 1000, 2000, or 3000 IU per vial | Room temperature or under refrigeration at a temperature of 2 to 30 °C (36-86 °F) |
| Ixinity | Coagulation factor IX (recombinant) | Medexus Pharma, Inc | On-demand treatment and control of bleeding episodes, perioperative management, and routine prophylaxis to reduce the frequency of bleeding episodes. | Intravenous, 5 or 2 mL of sterile water for injection | 250, 500, 1000, 1500, 2000, or 3000 | 250 IU strength only; store at 2 to 8 °C, other strengths store at 2 to 25 °C |
| Rixubis | Coagulation factor IX (recombinant), nonacog gamma | Takeda Pharmaceuticals U.S.A., Inc | For adults and children with hemophilia B, for (1) control and prevention of bleeding episodes, (2) perioperative management, and (3) routine prophylaxis. | Intravenous, 5 mL of sterile water for injection | 250, 500, 1000, 2000, or 3000 IU per vial | 2 to 8 °C (36-46 °F) for up to 24 mo and room temperature not to exceed 30 °C (86 °F) for up to 12 mo within the 24-mo time period. |
| Extended half-life recombinant products to treat hemophilia B | ||||||
| Alprolix | Coagulation factor IX (recombinant), Fc fusion protein | Sanofi | Adults and children with hemophilia B for on-demand treatment and control of bleeding episodes, perioperative management of bleeding, and routine prophylaxis to reduce the frequency of bleeding episodes. | Intravenous, 5 mL of sterile water for injection | 250, 500, 1000, 2000, 3000, or 4000 IU per vial | 2 to 8 °C (36-46 °F) or at room temperature, not to exceed 30 °C (86 °F) for a single period of up to 6 mo within the expiration date |
| IDelvion | Coagulation factor IX (recombinant), albumin fusion protein | CSL Behring, LLC | Children and adults with hemophilia B for on-demand treatment and control of bleeding episodes, perioperative management of bleeding, and routine prophylaxis to reduce the frequency of bleeding episodes. | Intravenous, 2.5 or 5 mL of sterile water for injection | 250, 500, 1000, 2000, or 3500 IU per vial | In refrigerator or at room temperature 2 to 25 °C (36-77 °F) |
| Rebinyn | Coagulation factor IX (recombinant), GlycoPEGylated | NovoNordisk, Inc | Adults and children with hemophilia B for on-demand treatment and control of bleeding episodes and perioperative management of bleeding. | Intravenous, 4 mL of histidine diluent for injection | 500, 1000, or 2000, IU per vial | 36 to 46 °F (2-8 °C) for up to 24 mo from the date of manufacture until the expiration date stated on the label and may be stored at room temperature not to exceed 86 °F (30 °C) for up to 6 mo within the 24-mo time period. |
| Gene therapy products to treat hemophilia B | ||||||
| Hemgenix | etranacogene (dezaparvovec-drlb) suspension | CSL Behring, LLC | Adults with hemophilia B who currently use factor IX prophylaxis therapy, have a current or historical life-threatening hemorrhage, or have repeated serious spontaneous bleeding episodes. | Intravenous, 1 × 1013 genome copies/mL | 2 × 1013 genome copies/kg of body weight | 2 to 8 °C (36-46 °F). |
| BEQVEZ | fidanacogene (elaparvovec-dzkt) injection | Pfizer, Inc | Adults with moderate to severe hemophilia B (congenital factor IX deficiency) who currently use factor IX prophylaxis therapy, have a current or historical life-threatening hemorrhage, or have repeated serious spontaneous bleeding episodes and do not have neutralizing antibodies to adeno-associated virus serotype Rh74var (AAVRh74var) capsid as detected by an US Food and Drug Administration (FDA)-approved test. | Intravenous, 1 × 1013 vector genomes/mL, and each vial contains an extractable volume of 1 mL | 5 × 1011 vector genomes/kg of body weight | Frozen (−100 °C to −60 °C [−148 °F to −76 °F]) |
Hemophilia B prophylaxis
Prophylactic treatment is recommended by the National Bleeding Disorders Foundation and the World Federation of Hemophilia for individuals with severe hemophilia B and those with moderate or mild hemophilia B with spontaneous bleeding 19, 26, 63. Prophylactic regimens are instituted based on disease severity and may be informed by bleeding symptoms or instituted prior to joint bleeding. Today there are several hemophilia B prophylactic product options and the choice of agent should be individualized based on access and lifestyle 83, 84. The hemophilia B prophylactic product options include plasma-derived factor IX concentrate, recombinant factor IX, and longer-lasting recombinant factor IX 82. The process involves inserting a genetically engineered human factor IX gene into a Chinese hamster ovary cell line to create recombinant factor IX. Using this engineered product eliminates the issue of infectious complications. Fusing factor IX with a monomeric human immunoglobulin Fc domain (rFIXFc), polyethylene glycol, or the gene for albumin extends the half-life of factor IX. Trials have shown that the recombinant factor IX-Fc fusion protein reduces pain in hemophilia B, raises physical activity levels, and improves the quality of life 85.
- Intravenous infusion of factor IX concentrates (recombinant or plasma-derived). Initiation of prophylactic infusions of factor IX concentrate in young children before or just after their first few joint bleeds has been shown to nearly eliminate spontaneous bleeding and prevent chronic joint disease 86. Previously it was recommended that factor IX clotting activity be maintained above 1%, but it is now clear that this goal will not prevent bleeding in many individuals and a personalized approach is recommended 87. Modified recombinant factor IX concentrates extend the half-life 3 to 5 fold, allowing significantly fewer infusions compared to use of standard half-life products 88. Choice of product should be individualized based on clinical factors and activity levels. Parents of children age two to five years with severe hemophilia B should be trained to administer the factor IX concentrates (recombinant or plasma-derived) infusions. Older children should be trained in self-infusion. Home treatment allows for prompt treatment and facilitates prophylactic therapy.
- Experience, primarily in hemophilia A, has shown that lower-dose prophylaxis used in countries with fewer resources can decrease bleeding and improve outcomes 87. In addition, factor IX concentrates are used to treat acute bleeding and prevent bleeding and allow healing in individuals with hemophilia B undergoing procedures. For acute bleeding, treatment should be given as soon as possible after symptoms occur. For those trained in home infusion this can be done promptly in the home. Dosing is weight based; target levels and duration of treatment vary by the severity of bleeding and/or the risk associated with the surgery or procedure.
In 2022, the U.S. Food and Drug Administration (FDA) approved a gene therapy for patients with hemophilia B with Etranacogene dezaparvovec-drlb (Hemgenix) an adeno-associated virus serotype 5 (AAV5) vector containing a codon-optimized Padua variant of the human F9 gene with a liver-specific promoter 20, 21, 22. This variant of the F9 gene contains a missense mutation that significantly increases F9 activity by 4- to 40-fold. Etranacogene dezaparvovec-drlb (Hemgenix) offers a one-time treatment option for adults with hemophilia B who use factor IX prophylaxis but still experience severe bleeding.
Prophylactic treatment may also be achieved with recent FDA-approved “rebalancing agents” such as marstacimab, concizumab, and fitusiran 23, 24, 25. Non-factor therapies (marstacimab, concizumab, and fitusiran) inhibit inhibitors of coagulation and “rebalance” hemostasis to allow a more normal hemostatic response. They are administered subcutaneously and are used as prophylactic therapy.
- Note: Factor IX concentrate, or a bypassing agent for those with inhibitors, is still needed for breakthrough bleeding and with most surgical procedures.
- Marstacimab, an inhibitor of tissue factor pathway inhibitor, was approved by the FDA in October 2024 for individuals age 12 years and older without inhibitors.
- Concizumab, an inhibitor of tissue factor pathway inhibitor, was approved by the FDA in December 2024 for individuals age 12 years and older with inhibitors.
- Fitusiran, an inhibitor of antithrombin, was approved by the FDA in March 2025 for individuals age 12 years and older with and without inhibitors.
When traveling, people with hemophilia B should wear a medical alert bracelet, carry a supply of factor replacement, and be aware of the location of the nearest hemophilia treatment center if available.
Hemophilia B acute bleeding management
Replacing factor IX (factor 9) continues to be the primary treatment for hemophilia B, with the dose determined by the severity of bleeding. The goal is to achieve 30% factor IX (factor 9) activity in patients with mild bleeding, 50% in patients with severe bleeding after trauma or those who need prophylaxis before major dental surgery or other surgery, and 80% to 100% factor IX (factor 9) activity in patients with life-threatening conditions.
The following formula calculates the appropriate dose for factor IX (factor 9) replacement 17, 2:
- The initial dose for factor IX = [patient’s body weight (in kg)] x [desired factor IX increase (expressed as % in whole number)] x [factor accounting for the volume of redistribution (IU/kg; usually around 1 for factor IX)]
For example, a 45-year-old female with a body weight of 50 kg needs to increase her factor IX level by 100%, which is calculated as 50 x 100 x 1 = 5000 IU of factor IX.
Several strategies and treatment options can be utilized when managing bleeding episodes and dental extractions in patients with hemophilia 2:
- Repeating the dose based on the half-life of the infused product.
- Considering prothrombin complex concentrate if factor IX is unavailable.
- Utilizing antifibrinolytic agents, including tranexamic acid and epsilon-aminocaproic acid, and monoclonal antibodies, such as rituximab, for consideration in cases of mucosal bleeds and dental extractions in patients with hemophilia.
Hemophilia B inhibitors
A significant complication in people with hemophilia B receiving factor IX replacement is the development of IgG antibodies that block the activity of the replaced factor IX (factor 9). These inhibitory antibodies develop in response to exogenous factors and affect approximately 3% to 5% of patients with severe hemophilia B 89, 2. Inhibitors occur much less frequently in patients with mild-to-moderate hemophilia B because the body does not recognize the infused factor as a foreign protein. Inhibitors complicate bleeding episodes by reducing responsiveness to replacement factor IX infusions 90. Hemophilia B alloimmune inhibitors occur with the greatest frequency (40%-60%) in individuals with large partial (>50-bp) deletions, whole-gene deletions or early termination (<100 predicted amino acids) variants 91. Missense mutations are rarely associated with inhibitors.
Alloimmune inhibitors to factor IX should be suspected if bleeding does not cease after clotting factor replacement in a previously responsive patient. Anaphylactic reactions to factor IX infusion and and nephrotic syndrome may occur with factor IX inhibitors 92, 57. Because of the risk of severe allergic reactions with development of alloantibodies, it is recommended that the first 20 factor IX replacement treatments be given in a medical setting where resuscitation medications and equipment are available. Risk can be stratified if the genetic variant is known. Those with large partial deletions, complete gene deletions, and early termination variants (<100 predicted amino acids) are at highest risk 88. Screening for alloimmune inhibitors is performed after treatment with factor IX concentrates has been initiated for either bleeding or prophylaxis. Testing for inhibitors should also be performed in any individual with hemophilia B whenever a suboptimal clinical response to treatment is suspected.
Immune tolerance can be challenging to treat, although it can be effectively managed and long-term bypassing therapy, primarily recombinant activated factor VII (rFVIIa), fitusiran, or an investigational drug, may be needed for treatment 93.
Doctors may consider alternatives such as plasmapheresis, bypassing products, or high-dose factor infusions for patients with hemophilia B who develop inhibitors. Recombinant activated factor VIIa contains an activated form of a downstream clotting factor in the coagulation cascade. Activated factor VIIa can activate factor X independently of factor VIII (factor 8) or factor IX (factor 9), but it can increase the risk of thrombosis, which varies from 1% to 10% 94. Plasmapheresis is primarily used to treat individuals with life- or limb-threatening bleeding and may benefit patients with a high titer of an inhibitor. Plasmapheresis can acutely reduce the inhibitor titer, enabling transient use of replacement factor. Another option for individuals with bleeding who have developed an inhibitor is high-dose factor infusions.
Bypassing agents
Bypassing agents are used for the treatment and prevention of bleeding complications in patients with hemophilia A or B who develop factor VIII or factor IX alloantibodies or inhibitors that typically neutralize the function of infused clotting factor concentrates 95. These agents are based on different mechanisms of action to achieve hemostasis, thereby bypassing the need
for factor VIII or factor IX replacement to treat and prevent bleeds 96.
- Recombinant activated factor VIIa (rFVIIa). Recombinant activated factor VIIa (rFVIIa) is a bypassing agent that promotes coagulation through tissue factor-dependent and independent pathways 96. Recombinant activated factor VIIa (rFVIIa) binds to tissue factor to activate factor X and factor IX and allows the coagulation cascade to resume 97, 98.
For people with hemophilia B and an inhibitor with a history of anaphylaxis to factor IX-containing clotting factor concentrates, recombinant activated factor VIIa (rFVIIa) must be administered as activated prothrombin complex concentrate (aPCC) cannot be used. Activated prothrombin complex concentrate (aPCC) is used to treat patients with hemophilia A with inhibitors. Activated prothrombin complex concentrate (aPCC) contains mainly non-activated factor II (prothrombin), factor IX, factor X, and mainly activated factor VII 99, 100, 101.
The World Federation of Hemophilia recommends that patients with hemophilia with an inhibitor should be considered for regular prophylaxis to prevent bleeding events. In addition to bypassing agents, non-factor replacement therapies (e.g., emicizumab) are available that offer new treatment paradigms including for the treatment of inhibitors 26.
Infants and children with hemophilia B
Special considerations for care of infants and children with hemophilia B include the following 18, 19:
- Infant males with a family history of hemophilia B should not be circumcised unless hemophilia B is either excluded or, if present, treated with factor IX concentrate directly before and after the procedure.
- Immunizations should be administered subcutaneously if known to be an effective route; intramuscular injections may be managed with pressure and ice and, if possible, under factor IX coverage.
- Effective dosing of factor IX requires an understanding of different pharmacokinetics in young children.
Young children with severe or moderate hemophilia B should be evaluated at a Hemophilia Treatment Center (accompanied by their parents/guardians) every six to 12 months and as needed to review their history of bleeding episodes and adjust treatment plans. Early signs and symptoms of possible bleeding episodes are reviewed. The assessment should also include a joint and muscle evaluation, an inhibitor screen, viral testing if indicated, a discussion of any other issues related to the individual’s hemophilia B, and family and community support.
Prevention of secondary complications
Recombinant factor IX produced without human- or animal-derived proteins and virucidal treatment of plasma-derived concentrates has eliminated the risk of HIV and hepatitis B and C viruses.
Surveillance
For individuals with severe or moderate hemophilia B, assessments every six to 12 months at a hemophilia treatment center; for individuals with mild hemophilia B, assessments at least every two to three years.
- Older children and adults with severe or moderate hemophilia B benefit from at least annual assessment at a hemophilia treatment center to review bleeding episodes and treatment plans, evaluate joints and muscles, screen for inhibitors, perform viral testing if indicated, provide education, and discuss other issues relevant to the individual’s hemophilia B.
- Individuals with mild hemophilia B can benefit from an assessment at a hemophilia treatment center every one to two years. Affected individuals with comorbidities and other complications or treatment challenges may require more frequent visits.
Screening for alloimmune inhibitors is usually done in those with severe hemophilia B after treatment with factor IX concentrates has been initiated for either bleeding or prophylaxis. Affected individuals at increased risk for inhibitor formation should be closely monitored during initial infusions and additional screening is usually performed up to a few years of age when the genotype is a large partial deletion, complete F9 deletion, or early termination variant (<100 predicted amino acids). Testing for inhibitors should also be performed in any individual with hemophilia B whenever a suboptimal clinical response to treatment is suspected, regardless of disease severity; with hemophilia B, the onset may be heralded by an allergic reaction to infused factor IX concentrate.
Older children and adults with severe or moderate hemophilia B benefit from at least yearly assessments at an hemophilia treatment center and periodic assessments to review bleeding episodes and treatment plans, evaluate joints and muscles, screen for an inhibitor, perform viral testing if indicated, provide education, and discuss other issues relevant to the individual’s hemophilia.
Agents/circumstances to avoid
Circumcision of at-risk males until hemophilia B is either excluded or treated with factor IX concentrate regardless of severity; intramuscular injections; activities with a high risk of trauma, particularly head injury. Individuals with hemophilia B should refrain from using aspirin, and all aspirin-containing products, nonsteroidal anti-inflammatory drugs (NSAIDs), and anticoagulants. Cautious use of other medications and herbal remedies that affect platelet function.
The following should be avoided:
- Circumcision of infant males with a family history of hemophilia B unless hemophilia B is excluded; OR if circumcision is performed on an infant with hemophilia B, the infant should be treated with factor IX concentrate directly before and after the procedure.
- Intramuscular injections
- Activities that involve a high risk of trauma, particularly of head injury
- Medications and herbal remedies that affect platelet function, including aspirin unless there is strong medical indication (e.g., in individuals with atherosclerotic cardiovascular disease). Individuals with severe hemophilia usually require clotting factor prophylaxis to allow aspirin and other platelet inhibitory drugs to be used safely 102.
Older, intermediate purity plasma-derived “prothrombin complex” concentrates should be used cautiously (if at all) in hemophilia B because of their thrombogenic potential.
Evaluation of relatives at risk
Identification of at-risk relatives. To clarify genetic status of females at risk before pregnancy or early in pregnancy in order to facilitate management. A thorough family history may identify other male relatives who are at risk but have not been tested (particularly in families with mild hemophilia B).
At-risk males
Early determination of the genetic status of males at risk. Assay of factor IX clotting activity from a cord blood sample obtained by venipuncture of the umbilical vein (to avoid contamination by amniotic fluid or placenta tissue), assessment of factor IX clotting activity in the neonatal period, or molecular genetic testing for the family-specific F9 gene mutation can establish or exclude the diagnosis of hemophilia B in newborn males at risk. Infant males with a family history of hemophilia B should not be circumcised unless hemophilia B is either excluded or, if present, factor IX concentrate is administered immediately before and after the procedure to prevent delayed bleeding and poor wound healing. The benefit versus the risk of exposure to factor IX concentrate in early childhood should be considered.
- Note:
- The cord blood for factor IX clotting activity assay should be drawn into a syringe containing one-tenth volume of sodium citrate to avoid clotting and to provide an optimal mixing of the sample with the anticoagulant.
- Factor IX clotting activity in cord blood in a normal-term newborn is lower than in adults (mean: ~30%; range: 15% to 50%); therefore, the diagnosis of hemophilia B can be established in an infant with activity lower than 1%, but is ambiguous in an infant with moderately low (15% to 20%) activity.
At-risk females
Determination of genetic status of females at risk. Approximately 30% of “heterozygous” females or female “carriers” with one F9 gene with a mutation and one normal F9 gene have factor IX clotting activity lower than 40% and may have abnormal bleeding. In a Dutch survey of heterozygous females (one F9 gene with a mutation and one normal F9 gene), bleeding symptoms correlated with baseline factor clotting activity; there was suggestion of a very mild increase in bleeding even in those with 40% to 60% factor IX clotting activity 15. Joint range of motion in female carriers with factor VIII or factor IX activity lower than 40% was found to be significantly different from that measured in normal controls and inversely related to factor level 103.
All daughters and mothers of an affected male and other at-risk females should have a baseline factor IX clotting activity assay to determine if they are at increased risk for bleeding (unless they are known to be non-carriers based on molecular genetic testing). Very occasionally, a female will have particularly low factor IX clotting activity that may result from heterozygosity for a F9 gene mutation associated with skewed X-chromosome inactivation or, on rare occasion, compound heterozygosity for two F9 gene mutations.
It is recommended that the genetic status of at-risk females be established prior to pregnancy or as early in a pregnancy as possible.
Hemophilia B pregnancy management
Hemophilia B pregnancy management requires a multidisciplinary team approach, including preconception counseling, careful planning for delivery at a specialized center, and postpartum care to address the risks of bleeding for both the mother and the baby 104, 105, 106, 107, 108, 109. Unlike in hemophilia A where factor VIII as levels can rise, maternal factor IX levels do not generally increase during pregnancy, and heterozygous females are more likely to need factor IX infusion support for delivery and/or to treat or prevent postpartum hemorrhage. In females with hemophilia B, postpartum hemorrhage has been a prominent feature, even in women without heavy menstrual bleeding 110. To prevent postpartum hemorrhage, 1 gm of tranexamic acid given intravenously immediately following cord clamping and then orally for seven to 14 days post partum or as needed with or without factor IX concentrate as indicated by factor IX clotting activity can be used 1.
If the female has a baseline factor IX clotting activity below approximately 40%, she by definition has hemophilia B and is at risk for excessive bleeding, particularly post partum, and may require therapy with factor IX concentrate 110.
Preconception and prenatal planning
- Genetic counseling: Discuss family planning with a hemophilia specialist and a genetic counselor to understand potential risks for the fetus. It is recommended that the carrier status of a female at risk be established prior to pregnancy or as early in a pregnancy as possible. If the female is symptomatic (i.e., has baseline factor IX clotting activity <40%), she will be somewhat protected by the natural rise of factor IX clotting activity during pregnancy, which may even double by the end of the third trimester. The factor IX level should be measured in the third trimester to confirm that the level is in the normal range, and if it is not, a plan for factor replacement therapy should be developed. Postpartum factor IX clotting activity can return to baseline within 48 hours, and postpartum hemorrhage may ensue 111.
- Multidisciplinary team: Assemble a team including a hematologist, an obstetrician, a pediatrician, and an anesthesiologist early in the pregnancy.
- Fetal diagnosis: If the fetus is male, prenatal diagnostic tests like chorionic villus sampling or amniocentesis may be offered to confirm hemophilia, but only after specific mutations have been identified in the family.
- Factor monitoring: Check factor IX levels early in pregnancy and again between 28 and 34 weeks to plan for delivery.
- Delivery plan: Create a detailed, written plan for delivery that outlines obstetric care, the planned mode of delivery, and immediate newborn care. This plan should be shared with the entire care team.
During labor and delivery
- Delivery mode: A spontaneous vaginal delivery is usually preferred unless there are obstetric complications. Controversy remains as to indications for cesarean section versus vaginal delivery 104, 112. In retrospective data analysis of 580 males age birth to two years with hemophilia A and hemophilia B, 17 suffered intracranial hemorrhages with delivery, and all but one were delivered vaginally 113. This finding supports the recommendation of cesarean section for infants with hemophilia. However, 12 of the 17 were born to women not known to be carriers of hemophilia B, suggesting that a planned delivery may mitigate risks. A more recent large study showed a similar risk of intracranial hemorrhage after planned vaginal delivery as reported in the general population 114. The relative risks of cesarean section versus vaginal delivery should be considered and discussed with the family and obstetrician so that a coordinated plan can be developed. Regardless of delivery mode, instrumentation with vacuum assistance or forceps must be avoided.
- Anesthesia: Epidural or spinal anesthesia should only be considered in close consultation with the hematology and anesthesiology teams.
- Factor replacement: The mother may need factor IX infusion just before delivery to ensure adequate levels, depending on the plan established by the multidisciplinary team.
- Avoidance: Avoid invasive procedures like fetal scalp electrodes or operative vaginal delivery if the fetus is potentially severely affected.
Postpartum care
- Maternal care: Continue factor replacement therapy to maintain normal levels for several days after delivery (up to 7 days for a C-section).
- Monitoring: Closely monitor factor levels for up to 21 days postpartum, as they can drop significantly as they return to baseline.
- Postpartum hemorrhage: Active management of the third stage of labor is recommended to prevent postpartum hemorrhage.
- Fetal care: The newborn requires specific care based on fetal status, including a thorough evaluation by a pediatric hematologist.
Hemophilia B prognosis
With treatment, most people with hemophilia B are able to lead a fairly normal life with a normal lifespan. Treatments and prophylaxis currently available allow people with hemophilia born today to live a normal lifespan. If you have hemophilia B, you should have regular checkups with a hematologist. Individuals with hemophilia followed at a Hemophilia Treatment Center have lower mortality than those who are not 115, 116. Key factors for a positive prognosis for people with hemophilia B include regular checkups, access to treatment centers, proper management, and a healthy lifestyle. Receiving regular, adequate treatment is essential for a good prognosis. Without it, life expectancy can be significantly reduced.
In developing countries, inadequate health systems and limited health resources contribute to a mortality rate for patients with hemophilia that remains twice that of the average healthy male 117. Overall, there are still significant disparities in care. In upper-middle-income countries, those born with hemophilia have a 64% lower chance of living a life of average duration and quality, which increases to 77% in middle-income countries and up to 93% in low-income countries 2.
The most common complications of hemophilia B are recurrent bleeding into a joint (hemarthrosis), intracerebral hemorrhage (bleeding into the brain tissue), HIV and hepatitis C infection, and the development of factor inhibitors 2. Recurrent recurrent bleeding into a joint (hemarthrosis) causes synovial membrane inflammation and hypertrophy, ultimately resulting in destructive joint disease. Intracerebral hemorrhages (bleeding into the brain tissue) can lead to chronic neurological disability. HIV and hepatitis C infection can potentially result in death and liver cancer called hepatocellular carcinoma. Other concerns include hypovolemic shock (a medical emergency in which you’ve lost so much blood or fluid and your body can’t send enough of it to all of your organs) caused by iliopsoas muscle bleeding and airway compromise resulting from retropharyngeal bleeding. In addition, individuals with hemophilia experience higher rates of psychosocial and functional impairments and depression than normal healthy individuals. Maturational delays are also reported in children 118.
A positive hemophilia B prognosis also depends on a proactive approach to health, including:
- Regular checkups with a hematologist.
- Participating in activities and exercise that are safe for people with bleeding disorders.
- Managing pain and maintaining a healthy weight to reduce joint strain.
- Having vaccinations such as for hepatitis B, particularly if receiving donated clotting factors.
Potential complications of hemophilia B:
- Inhibitors: Some individuals may develop inhibitors, which are antibodies that make clotting factor treatments less effective. These can often be managed with specific treatments, but may lead to ongoing problems.
- Joint damage: Repeated bleeds can lead to joint damage over time.
- Serious bleeds: Despite treatment, serious bleeding into the brain can still occur in some severe cases, and it is important to be aware of the symptoms and seek immediate medical attention if they appear.
- Konkle BA, Nakaya Fletcher S. Hemophilia B. 2000 Oct 2 [Updated 2025 Aug 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1495[↩][↩][↩][↩][↩]
- Alshaikhli A, Killeen RB, Rokkam VR. Hemophilia B. [Updated 2023 Oct 29]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560792[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- López Fernández MF. Limitations of prophylactic treatment in patients with hemophilia. Blood Coagul Fibrinolysis. 2019 Sep;30(1S Suppl 1):S22-S24. doi: 10.1097/MBC.0000000000000825[↩]
- Mahlangu J. Emicizumab for the prevention of bleeds in hemophilia A. Expert Opin Biol Ther. 2019 Aug;19(8):753-761. doi: 10.1080/14712598.2019.1626370[↩]
- Oldenburg J, Hay CRM, Jiménez-Yuste V, Peyvandi F, Schved JF, Szamosi J, Winding B, Lethagen S. Design of a prospective observational study on the effectiveness and real-world usage of recombinant factor VIII Fc (rFVIIIFc) compared with conventional products in haemophilia A: the A-SURE study. BMJ Open. 2019 May 30;9(5):e028012. doi: 10.1136/bmjopen-2018-028012[↩]
- Salen P, Babiker HM. Hemophilia A. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470265[↩]
- Konkle BA, Nakaya Fletcher S. Hemophilia A. 2000 Sep 21 [Updated 2025 Aug 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1404[↩]
- O’Hara J, Hughes D, Camp C, Burke T, Carroll L, Diego DG. The cost of severe haemophilia in Europe: the CHESS study. Orphanet J Rare Dis. 2017 May 31;12(1):106. doi: 10.1186/s13023-017-0660-y[↩][↩]
- O’Hara J, Walsh S, Camp C, Mazza G, Carroll L, Hoxer C, Wilkinson L. The relationship between target joints and direct resource use in severe haemophilia. Health Econ Rev. 2018 Jan 16;8(1):1. doi: 10.1186/s13561-018-0185-7[↩][↩][↩]
- Manco-Johnson MJ, Abshire TC, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007 Aug 9;357(6):535-44. doi: 10.1056/NEJMoa067659[↩]
- F9 gene. https://medlineplus.gov/genetics/gene/f9/[↩][↩]
- Hemophilia B. https://medlineplus.gov/ency/article/000539.htm[↩][↩]
- Hemophilia B. https://rarediseases.org/rare-diseases/hemophilia-b/[↩][↩]
- van Galen KPM, d’Oiron R, James P, Abdul-Kadir R, Kouides PA, Kulkarni R, Mahlangu JN, Othman M, Peyvandi F, Rotellini D, Winikoff R, Sidonio RF. A new hemophilia carrier nomenclature to define hemophilia in women and girls: Communication from the SSC of the ISTH. J Thromb Haemost. 2021 Aug;19(8):1883-1887. doi: 10.1111/jth.15397[↩][↩][↩]
- Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, van Amstel HK, van der Bom JG, van Diemen-Homan JE, Willemse J, Rosendaal FR. Bleeding in carriers of hemophilia. Blood. 2006 Jul 1;108(1):52-6. doi: 10.1182/blood-2005-09-3879[↩][↩]
- Iorio A, Stonebraker JS, Chambost H, Makris M, Coffin D, Herr C, Germini F; Data and Demographics Committee of the World Federation of Hemophilia. Establishing the Prevalence and Prevalence at Birth of Hemophilia in Males: A Meta-analytic Approach Using National Registries. Ann Intern Med. 2019 Oct 15;171(8):540-546. doi: 10.7326/M19-1208[↩]
- Razmpoosh E, Olasupo OO, Bhatt M, Matino D, Iorio A. Clotting factor concentrates for preventing bleeding and bleeding‐related complications in previously untreated or minimally treated children with hemophilia A or B. Cochrane Database of Systematic Reviews 2025, Issue 8. Art. No.: CD003429. DOI: 10.1002/14651858.CD003429.pub5[↩][↩]
- Chalmers E, Williams M, Brennand J, Liesner R, Collins P, Richards M; Paediatric Working Party of United Kingdom Haemophilia Doctors’ Organization. Guideline on the management of haemophilia in the fetus and neonate. Br J Haematol. 2011 Jul;154(2):208-15. doi: 10.1111/j.1365-2141.2010.08545.x[↩][↩]
- Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020: 26(Suppl 6): 1-158. https://doi.org/10.1111/hae.14046[↩][↩][↩][↩][↩][↩][↩]
- Sekayan T, Simmons DH, von Drygalski A. Etranacogene dezaparvovec-drlb gene therapy for patients with hemophilia B (congenital factor IX deficiency). Expert Opin Biol Ther. 2023 Jul-Dec;23(12):1173-1184. doi: 10.1080/14712598.2023.2282138[↩][↩]
- Pipe SW, Leebeek FWG, Recht M, et al. Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. N Engl J Med. 2023 Feb 23;388(8):706-718. doi: 10.1056/NEJMoa2211644[↩][↩]
- Nathwani AC. Gene therapy for hemophilia. Hematology Am Soc Hematol Educ Program. 2022 Dec 9;2022(1):569-578. doi: 10.1182/hematology.2022000388[↩][↩]
- Matino D, Acharya S, Palladino A, Hwang E, McDonald R, Taylor CT, Teeter J. Efficacy and safety of the anti-tissue factor pathway inhibitor marstacimab in participants with severe hemophilia without inhibitors: results from the phase 3 Basis trial. Blood 2023;142(Supp 1): DOI:.10.1182/blood-2023-181263[↩][↩]
- Matsushita T, Shapiro A, Abraham A, et al. Phase 3 Trial of Concizumab in Hemophilia with Inhibitors. N Engl J Med. 2023 Aug 31;389(9):783-794. doi: 10.1056/NEJMoa2216455[↩][↩]
- Young G, Kavakli K, Klamroth R, Matsushita T, Peyvandi F, Pipe SW, Rangarajan S, Shen MC, Srivastava A, Sun J, Tran H, You CW, Zülfikar B, Menapace LA, Zhang C, Shen Y, Puurunen M, Demissie M, Kenet G. Safety and efficacy of a fitusiran antithrombin-based dose regimen in people with hemophilia A or B: the ATLAS-OLE study. Blood. 2025 Jun 19;145(25):2966-2977. doi: 10.1182/blood.2024027008[↩][↩]
- World Federation of Hemophilia: GUIDELINES FOR THE MANAGEMENT OF HEMOPHILIA 3rd Edition. https://www1.wfh.org/publications/files/pdf-1863.pdf[↩][↩][↩][↩][↩][↩]
- Gomis M, Querol F, Gallach JE, González LM, Aznar JA. Exercise and sport in the treatment of haemophilic patients: a systematic review. Haemophilia. 2009 Jan;15(1):43-54. doi: 10.1111/j.1365-2516.2008.01867.x[↩]
- Seuser A, Boehm P, Kurme A, Schumpe G, Kurnik K. Orthopaedic issues in sports for persons with haemophilia. Haemophilia. 2007 Sep;13 Suppl 2:47-52. doi: 10.1111/j.1365-2516.2007.01507.x[↩]
- Philpott J, Houghton K, Luke A. Physical activity recommendations for children with specific chronic health conditions: Juvenile idiopathic arthritis, hemophilia, asthma and cystic fibrosis. Paediatr Child Health. 2010 Apr;15(4):213-25. doi: 10.1093/pch/15.4.213[↩]
- Querol F, Aznar JA, Haya S, Cid A. Orthoses in haemophilia. Haemophilia. 2002 May;8(3):407-12. doi: 10.1046/j.1365-2516.2002.00637.x[↩]
- ADA Council on Access, Prevention and Interprofessional Relations; ADA Council on Scientific Affairs. Using mouthguards to reduce the incidence and severity of sports-related oral injuries. J Am Dent Assoc. 2006 Dec;137(12):1712-20; quiz 1731. doi: 10.14219/jada.archive.2006.0118[↩]
- Iorio A, Fabbriciani G, Marcucci M, Brozzetti M, Filipponi P. Bone mineral density in haemophilia patients. A meta-analysis. Thromb Haemost. 2010 Mar;103(3):596-603. doi: 10.1160/TH09-09-0629[↩]
- Khachidze M, Buil A, Viel KR, Porter S, Warren D, Machiah DK, Soria JM, Souto JC, Ameri A, Lathrop M, Blangero J, Fontcuberta J, Warren ST, Almasy L, Howard TE. Genetic determinants of normal variation in coagulation factor (F) IX levels: genome-wide scan and examination of the FIX structural gene. J Thromb Haemost. 2006;4:1537–45. http://onlinelibrary.wiley.com/doi/10.1111/j.1538-7836.2006.02024.x/full[↩]
- Plug I, Mauser-Bunschoten EP, Brocker-Vriends AH, van Amstel HK, van der Bom JG, van Diemen-Homan JE, Willemse J, Rosendaal FR. Bleeding in carriers of hemophilia. Blood. 2006;108:52–6. http://www.bloodjournal.org/content/108/1/52.long[↩][↩][↩][↩]
- Ketterling RP, Vielhaber E, Li X, Drost J, Schaid DJ, Kasper CK, Phillips JA 3rd, Koerper MA, Kim H, Sexauer C, Gruppo R, Ambriz R, Paredes R, Sommer SS. Germline origins in the human F9 gene: frequent G:C–>A:T mosaicism and increased mutations with advanced maternal age. Hum Genet. 1999;105:629–40. https://www.ncbi.nlm.nih.gov/pubmed/10647899[↩]
- Oldenburg J, Kriz K, Wuillemin WA, Maly FE, von Welten A, Siegemun A, Keeling DM, Baker P, Chu K, Konkle BA, Lammie B, Albert T, et al. Genetic predisposition to bleeding during oral anticoagulant therapy: evidence for common founder mutations (FIXVal-10 and FIXThr-10) and an independent CpG hotspot mutation. Thromb Haemost. 2001;85:454–7. https://www.ncbi.nlm.nih.gov/pubmed/11307814[↩][↩]
- Simioni P, Tormene D, Tognin G, Gavasso S, Bulato C, Iacobelli NP, Finn JD, Spiezia L, Radu C, Arruda VR. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med. 2009;361:1671. http://www.nejm.org/doi/full/10.1056/NEJMoa0904377[↩]
- Lheriteau E, Davidoff AM, Nathwani AC. Haemophilia gene therapy: progress and challenges. Blood Rev. 2015;29:321–8. https://www.ncbi.nlm.nih.gov/pubmed/26049173[↩]
- Funnell APW, Crossley M. Hemophilia B Leyden and once mysterious cis-regulatory mutations. Trends Genet. 2014;30:18–23. https://www.ncbi.nlm.nih.gov/pubmed/24138812[↩]
- Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388:187–97. http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(15)01123-X/fulltext[↩]
- Kulkarni R, Soucie JM, Lusher J, Presley R, Shapiro A, Gill J, Manco-Johnson M, Koerper M, Mathew P, Abshire T, Dimichele D, Hoots K, Janco R, Nugent D, Geraghty S, Evatt B., Haemophilia Treatment Center Network Investigators. Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Haemophilia. 2009;15:1281–90. https://www.ncbi.nlm.nih.gov/pubmed/19637999[↩]
- Luck JV Jr, Silva M, Rodriguez-Merchan EC, Ghalambor N, Zahiri CA, Finn RS. Hemophilic arthropathy. J Am Acad Orthop Surg. 2004;12:234–45. https://www.ncbi.nlm.nih.gov/pubmed/15473675[↩]
- Chu WM, Ho HE, Wang JD, Chan WC, Liou YS, Ho WC, Hu SY, Tsan YT. Risk of major comorbidities among workers with hemophilia: A 14-year population-based study. Medicine (Baltimore). 2018 Feb;97(6):e9803. doi: 10.1097/MD.0000000000009803[↩]
- Kodra Y, Cavazza M, Schieppati A, De Santis M, Armeni P, Arcieri R, Calizzani G, Fattore G, Manzoli L, Mantovani L, Taruscio D. The social burden and quality of life of patients with haemophilia in Italy. Blood Transfus. 2014 Apr;12 Suppl 3(Suppl 3):s567-75. doi: 10.2450/2014.0042-14s[↩][↩]
- Skinner MW, Chai-Adisaksopha C, Curtis R, Frick N, Nichol M, Noone D, O’Mahony B, Page D, Stonebraker JS, Iorio A. The Patient Reported Outcomes, Burdens and Experiences (PROBE) Project: development and evaluation of a questionnaire assessing patient reported outcomes in people with haemophilia. Pilot Feasibility Stud. 2018 Feb 27;4:58. doi: 10.1186/s40814-018-0253-0[↩]
- Baumann K, Hernandez G, Witkop M, Peltier S, Dunn S, Cutter S, Frick N, Haugstad K, Guelcher C, Frey MJ, Rotellini D, Clark DB, Iyer NN, Cooper DL. Impact of mild to severe hemophilia on engagement in recreational activities by US men, women, and children with hemophilia B: The Bridging Hemophilia B Experiences, Results and Opportunities into Solutions (B-HERO-S) study. Eur J Haematol. 2017 Apr;98 Suppl 86:25-34. doi: 10.1111/ejh.12852[↩]
- Batt K, Boggio L, Neff A, Buckner TW, Wang M, Quon D, Witkop M, Recht M, Kessler C, Iyer NN, Cooper DL. Patient-reported outcomes and joint status across subgroups of US adults with hemophilia with varying characteristics: Results from the Pain, Functional Impairment, and Quality of Life (P-FiQ) study. Eur J Haematol. 2018 Apr;100 Suppl 1:14-24. doi: 10.1111/ejh.13028[↩]
- Kempton CL, Buckner TW, Fridman M, Iyer NN, Cooper DL. Factors associated with pain severity, pain interference, and perception of functional abilities independent of joint status in US adults with hemophilia: Multivariable analysis of the Pain, Functional Impairment, and Quality of Life (P-FiQ) study. Eur J Haematol. 2018 Apr;100 Suppl 1:25-33. doi: 10.1111/ejh.13025[↩]
- Flood E, Pocoski J, Michaels LA, McCoy A, Beusterien K, Sasanè R. Patient-reported experience of bleeding events in haemophilia. Eur J Haematol. 2014 Jun;93 Suppl 75:19-28. doi: 10.1111/ejh.12329[↩]
- Curtis R, Baker J, Riske B, Ullman M, Niu X, Norton K, Lou M, Nichol MB. Young adults with hemophilia in the U.S.: demographics, comorbidities, and health status. Am J Hematol. 2015 Dec;90 Suppl 2:S11-6. doi: 10.1002/ajh.24218[↩]
- Cutter S, Molter D, Dunn S, Hunter S, Peltier S, Haugstad K, Frick N, Holot N, Cooper DL. Impact of mild to severe hemophilia on education and work by US men, women, and caregivers of children with hemophilia B: The Bridging Hemophilia B Experiences, Results and Opportunities into Solutions (B-HERO-S) study. Eur J Haematol. 2017 Apr;98 Suppl 86:18-24. doi: 10.1111/ejh.12851[↩]
- Darby SC, Kan SW, Spooner RJ, Giangrande PL, Hill FG, Hay CR, Lee CA, Ludlam CA, Williams M. Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007;110:815–25. http://www.bloodjournal.org/content/110/3/815.long[↩]
- Tagliaferri A, Rivolta GF, Iorio A, Oliovecchio E, Mancuso ME, Morfini M, Rocino A, Mazzucconi MG, Franchini M, Ciavarella N, Scaraggi A, Valdrè L, Tagariello G, Radossi P, Muleo G, Iannaccaro PG, Biasoli C, Vincenzi D, Serino ML, Linari S, Molinari C, Boeri E, La Pecorella M, Carloni MT, Santagostino E, Di Minno G, Coppola A, Rocino A, Zanon E, Spiezia L, Di Perna C, Marchesini M, Marcucci M, Dragani A, Macchi S, Albertini P, D’Incà M, Santoro C, Biondo F, Piseddu G, Rossetti G, Barillari G, Gandini G, Giuffrida AC, Castaman G, et al. Mortality and causes of death in Italian persons with haemophilia, 1990-2007. Haemophilia. 2010;16:437–46. https://www.ncbi.nlm.nih.gov/pubmed/20148978[↩]
- Monahan PE, Di Paola J. Recombinant factor IX for clinical and research use. Semin Thromb Hemost. 2010;36:498–509. https://www.ncbi.nlm.nih.gov/pubmed/20632248[↩]
- Santagostino E, Martinowitz U, Lissitchkov T, Pan-Petesch B, Hanabusa H, Oldenburg J, Boggio L, Negrier C, Pabinger I, von Depka Prondzinski M, Altisent C, Castaman G, Yamamoto K, Alvarez-Roman MT, Voigt C, Blackman N, Jacobs I., PROLONG-9FP Investigators Study Group. Long-acting recombinant coagulation factor IX albumin fusion protein (rIX-FP) in hemophilia B: results of a phase 3 trial. Blood. 2016;127:1761–9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4825413/[↩]
- Puetz J, Soucie JM, Kempton CL, Monahan PE., Hemophilia Treatment Center Network (HTCN) Investigators. Prevalent inhibitors in haemophilia B subjects enrolled in the Universal Data Collection database. Haemophilia. 2014;20:25–31. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4520536/[↩]
- Chitlur M, Warrier I, Rajpurkar M, Lusher JM. Inhibitors in factor IX deficiency a report of O the ISTH-SSC international FIX inhibitor registry (1997-2006). Haemophilia. 2009;15:1027–31. https://www.ncbi.nlm.nih.gov/pubmed/19515028[↩][↩]
- Funnell AP, Crossley M. Hemophilia B Leyden and once mysterious cis-regulatory mutations. Trends Genet. 2014 Jan;30(1):18-23. doi: 10.1016/j.tig.2013.09.007[↩]
- Mitchell M, Keeney S, Goodeve A. Practical guidelines for the molecular diagnosis of haemophilia B. UK Haemophilia Centre Doctors’ Organisation, the Haemophilia Genetics Laboratory Network and the Clinical Molecular Genetics Society. Available online. 2010. Accessed 1-30-23.[↩]
- Goodeve AC. Hemophilia B: molecular pathogenesis and mutation analysis. J Thromb Haemost. 2015 Jul;13(7):1184-95. doi: 10.1111/jth.12958[↩]
- Johnsen JM, Fletcher SN, Dove A, McCracken H, Martin BK, Kircher M, Josephson NC, Shendure J, Ruuska SE, Valentino LA, Pierce GF, Watson C, Cheng D, Recht M, Konkle BA. Results of genetic analysis of 11 341 participants enrolled in the My Life, Our Future hemophilia genotyping initiative in the United States. J Thromb Haemost. 2022 Sep;20(9):2022-2034. doi: 10.1111/jth.15805[↩]
- Search the Global Treatment Centre Directory. World Federation of Hemophilia https://wfh.org/find-local-support/#HTCs[↩]
- National Bleeding Disorders Foundation https://www.bleeding.org/[↩][↩][↩]
- Holmberg L, Nilsson IM. Von Willebrand’s disease. Annu Rev Med. 1975;26:33-44. doi: 10.1146/annurev.me.26.020175.000341[↩]
- Sabih A, Babiker HM. Von Willebrand Disease. [Updated 2023 Aug 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459222[↩]
- Johnsen J. Von Willebrand Disease. 2009 Jun 4 [Updated 2024 Nov 14]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK7014[↩]
- Saba HI, Morelli GA. The pathogenesis and management of disseminated intravascular coagulation. Clin Adv Hematol Oncol. 2006 Dec;4(12):919-26.[↩]
- Marchili MR, Santoro E, Marchesi A, Bianchi S, Rotondi Aufiero L, Villani A. Vitamin K deficiency: a case report and review of current guidelines. Ital J Pediatr. 2018 Mar 14;44(1):36. doi: 10.1186/s13052-018-0474-0[↩]
- Léger D. Scurvy: reemergence of nutritional deficiencies. Can Fam Physician. 2008 Oct;54(10):1403-6. https://pmc.ncbi.nlm.nih.gov/articles/PMC2567249[↩]
- Malfait F, Wenstrup RJ, De Paepe A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genet Med. 2010 Oct;12(10):597-605. doi: 10.1097/GIM.0b013e3181eed412[↩]
- Jackson J, Carpenter S, Anderst J. Challenges in the evaluation for possible abuse: presentations of congenital bleeding disorders in childhood. Child Abuse Negl. 2012 Feb;36(2):127-34. doi: 10.1016/j.chiabu.2011.09.009[↩]
- Raharimanana A, Cunat S, Falaise C, Oudot C, Fournel A, Dargaud Y. Hereditary Combined Deficiency of the Vitamin K-Dependent Coagulation Factors. Hamostaseologie. 2025 Oct;45(5):396-404. doi: 10.1055/a-2567-3567[↩]
- Hereditary combined deficiency of vitamin K-dependent clotting factors. https://www.orpha.net/en/disease/detail/98434[↩]
- https://www.genome.jp/dbget-bin/www_bget?ds:H00995[↩]
- VITAMIN K-DEPENDENT CLOTTING FACTORS, COMBINED DEFICIENCY OF, 1; VKCFD1. https://www.omim.org/entry/277450[↩]
- Duga S, Salomon O. Congenital factor XI deficiency: an update. Semin Thromb Hemost. 2013 Sep;39(6):621-31. doi: 10.1055/s-0033-1353420[↩]
- Menegatti M, Palla R, Boscarino M, Bucciarelli P, Muszbek L, Katona E, Makris M, Peyvandi F; PRO-RBDD study group. Minimal factor XIII activity level to prevent major spontaneous bleeds. J Thromb Haemost. 2017 Sep;15(9):1728-1736. doi: 10.1111/jth.13772[↩]
- Search the Global Treatment Centre Directory. World Federation of Haemophilia https://wfh.org/find-local-support/#HTCs[↩]
- Chalmers E, Williams M, Brennand J, Liesner R, Collins P, Richards M., Paediatric Working Party of the United Kingdom Haemophilia Doctors’ Organization. Guideline on the management of haemophilia in the fetus and neonate. Br J Haematol. 2011;154:208–15. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2010.08545.x/full[↩]
- Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ML, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, Matsunaga A, Medeiros D, Nugent D, Thomas GA, Thompson AA, McRedmond K, Soucie JM, Austin H, Evatt BL. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–44. http://www.nejm.org/doi/full/10.1056/NEJMoa067659[↩]
- Josephson N.The hemophilias and their clinical management. Hematology Am Soc Hematol Educ Program. 2013;2013:261-7 http://asheducationbook.hematologylibrary.org/content/2013/1/261.long[↩]
- Valentino LA, Santaella ME, Carlson SA, Recht M. Contemporary approaches to treat people with hemophilia: what’s new and what’s not? Res Pract Thromb Haemost. 2025 Jan 31;9(1):102696. doi: 10.1016/j.rpth.2025.102696[↩][↩]
- Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, Ludlam CA, Mahlangu JN, Mulder K, Poon MC, Street A; Treatment Guidelines Working Group on Behalf of The World Federation Of Hemophilia. Guidelines for the management of hemophilia. Haemophilia. 2013 Jan;19(1):e1-47. doi: 10.1111/j.1365-2516.2012.02909.x[↩]
- Djambas Khayat C. Once-weekly prophylactic dosing of recombinant factor IX improves adherence in hemophilia B. J Blood Med. 2016 Nov 30;7:275-282. doi: 10.2147/JBM.S84597[↩]
- Wang X, Yang M, Xu J, Kuai Y, Sun B. Risk analysis of 30-day rebleeding in acute non-variceal upper gastrointestinal bleeding. Arab J Gastroenterol. 2023 May;24(2):136-141. doi: 10.1016/j.ajg.2023.05.001[↩]
- Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007 Aug 9;357(6):535-44. doi: 10.1056/NEJMoa067659[↩]
- Srivastava A, Santagostino E, Dougall A, Kitchen S, Sutherland M, Pipe SW, Carcao M, Mahlangu J, Ragni MV, Windyga J, Llinás A, Goddard NJ, Mohan R, Poonnoose PM, Feldman BM, Lewis SZ, van den Berg HM, Pierce GF; WFH Guidelines for the Management of Hemophilia panelists and co-authors. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020 Aug;26 Suppl 6:1-158. doi: 10.1111/hae.14046. Epub 2020 Aug 3. Erratum in: Haemophilia. 2021 Jul;27(4):699. doi: 10.1111/hae.14308[↩][↩]
- Hart DP, Matino D, Astermark J, Dolan G, d’Oiron R, Hermans C, Jiménez-Yuste V, Linares A, Matsushita T, McRae S, Ozelo MC, Platton S, Stafford D, Sidonio RF Jr, Tiede A. International consensus recommendations on the management of people with haemophilia B. Ther Adv Hematol. 2022 Apr 2;13:20406207221085202. doi: 10.1177/20406207221085202[↩][↩]
- Hay CR, Brown S, Collins PW, Keeling DM, Liesner R. The diagnosis and management of factor VIII and IX inhibitors: a guideline from the United Kingdom Haemophilia Centre Doctors Organisation. Br J Haematol. 2006;133:591–605. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2006.06087.x/full[↩]
- Saba HI, Tran DQ Jr. Challenges and successes in the treatment of hemophilia: the story of a patient with severe hemophilia A and high-titer inhibitors. J Blood Med. 2012;3:17-23. doi: 10.2147/JBM.S30479[↩]
- Goodeve AC. Hemophilia B: molecular pathogenesis and mutation analysis. J Thromb Haemost. 2015;13:1184–95. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4496316/[↩]
- DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br J Haematol. 2007 Aug;138(3):305-15. doi: 10.1111/j.1365-2141.2007.06657.x[↩]
- Astermark J, Holstein K, Abajas YL, Kearney S, Croteau SE, Liesner R, Funding E, Kempton CL, Acharya S, Lethagen S, LeBeau P, Bowen J, Berntorp E, Shapiro AD. The B-Natural study-The outcome of immune tolerance induction therapy in patients with severe haemophilia B. Haemophilia. 2021 Sep;27(5):802-813. doi: 10.1111/hae.14357[↩]
- Shima M. Current status and future prospects of activated recombinant coagulation factor VIIa, NovoSeven®, in the treatment of haemophilia and rare bleeding disorders. Ann Hematol. 2024 Aug;103(8):2647-2658. doi: 10.1007/s00277-023-05287-2[↩]
- Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A; Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014 Nov;12(11):1935-9. doi: 10.1111/jth.12672[↩]
- Negrier C, Dargaud Y, Bordet JC. Basic aspects of bypassing agents. Haemophilia. 2006 Dec;12 Suppl 6:48-52; discussion 52-3. doi: 10.1111/j.1365-2516.2006.01366.x[↩][↩]
- Giansily-Blaizot M, Schved JF. Recombinant human factor VIIa (rFVIIa) in hemophilia: mode of action and evidence to date. Ther Adv Hematol. 2017 Dec;8(12):345-352. doi: 10.1177/2040620717737701[↩]
- NovoSeven® RT (coagulation factor VIIa, recombinant) lyophilized powder for solution, for intravenous use [U.S. prescribing information].
Plainsboro, NJ: Novo Nordisk. https://www.fda.gov/media/70442/download[↩] - Negrier C, Voisin S, Baghaei F, Numerof R, Novack A, Doralt JE, Romanov V, Gringeri A; FEIBA PASS Study group. Global Post-Authorization Safety Surveillance Study: real-world data on prophylaxis and on-demand treatment using FEIBA (an activated prothrombin complex concentrate). Blood Coagul Fibrinolysis. 2016 Jul;27(5):551-6. doi: 10.1097/MBC.0000000000000525[↩]
- FEIBA (anti-inhibitor coagulant complex) for intravenous use, lyophilized powder for solution [U.S. prescribing information]. Lexington, MA: Baxalta US. https://www.fda.gov/media/78852/download[↩]
- Varadi K, Tangada S, Loeschberger M, Montsch P, Schrenk G, Ewenstein B, Turecek PL. Pro- and anticoagulant factors facilitate thrombin generation and balance the haemostatic response to FEIBA(®) in prophylactic therapy. Haemophilia. 2016 Jul;22(4):615-24. doi: 10.1111/hae.12873[↩]
- Angelini D, Konkle BA, Sood SL. Aging among persons with hemophilia: contemporary concerns. Semin Hematol. 2016;53:35–9. https://www.ncbi.nlm.nih.gov/pubmed/26805905[↩]
- Sidonio RF, Mili FD, Li T, Miller CH, Hooper WC, DeBaun MR, Soucie M; Hemophilia Treatment Centers Network. Females with FVIII and FIX deficiency have reduced joint range of motion. Am J Hematol. 2014 Aug;89(8):831-6. doi: 10.1002/ajh.23754[↩]
- Leebeek FWG, Duvekot J, Kruip MJHA. How I manage pregnancy in carriers of hemophilia and patients with von Willebrand disease. Blood. 2020 Nov 5;136(19):2143-2150. doi: 10.1182/blood.2019000964[↩][↩]
- Dunkley SM, Russell SJ, Rowell JA, Barnes CD, Baker RI, Sarson MI, Street AM; Australian Haemophilia Centre Directors’ Organisation. A consensus statement on the management of pregnancy and delivery in women who are carriers of or have bleeding disorders. Med J Aust. 2009 Oct 19;191(8):460-3. doi: 10.5694/j.1326-5377.2009.tb02887.x[↩]
- Moorehead PC, Chan AKC, Lemyre B, Winikoff R, Scott H, Hawes SA, Shroff M, Thomas A, Price VE. A Practical Guide to the Management of the Fetus and Newborn With Hemophilia. Clin Appl Thromb Hemost. 2018 Dec;24(9_suppl):29S-41S. doi: 10.1177/1076029618807583[↩]
- How do I prepare for pregnancy? https://www.haemophilia.org.au/bleeding-disorders/women-with-bleeding-disorders/carrying-the-haemophilia-gene/pregnancy/[↩]
- James AH, Pacheco LD, Konkle BA. Management of pregnant women who have bleeding disorders. Hematology Am Soc Hematol Educ Program. 2023 Dec 8;2023(1):229-236. doi: 10.1182/hematology.2023000475[↩]
- Rodrigues-Martins D, Buchner G, Braga JS. Symptomatic haemophilia A: management during pregnancy and the postpartum period. BMJ Case Rep. 2022 Jan 7;15(1):e245474. doi: 10.1136/bcr-2021-245474[↩]
- Yang MY, Ragni MV. Clinical manifestations and management of labor and delivery in women with factor IX deficiency. Haemophilia. 2004 Sep;10(5):483-90. doi: 10.1111/j.1365-2516.2004.00946.x[↩][↩]
- Lee CA, Chi C, Pavord SR, Bolton-Maggs PH, Pollard D, Hinchcliffe-Wood A, Kadir RA. The obstetric and gynaecological management of women with inherited bleeding disorders–review with guidelines produced by a taskforce of UK Haemophilia Centre Doctors’ Organization. Haemophilia. 2006;12:301–36. http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2516.2006.01314.x/full[↩]
- James AH, Hoots WK. The optimal mode of delivery for the haemophilia carrier expecting an affected infant is caesarean delivery. Haemophilia. 2010;16:420–4. https://www.ncbi.nlm.nih.gov/pubmed/20028425[↩]
- Kulkarni R, Soucie JM, Lusher J, Presley R, Shapiro A, Gill J, Manco-Johnson M, Koerper M, Mathew P, Abshire T, Dimichele D, Hoots K, Janco R, Nugent D, Geraghty S, Evatt B., Haemophilia Treatment Center Network Investigators. Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from The Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Haemophilia. 2009;15:1281–90. https://www.ncbi.nlm.nih.gov/pubmed/19637999[↩]
- Andersson NG, Chalmers EA, Kenet G, Ljung R, Mäkipernaa A, Chambost H; PedNet Haemophilia Research Foundation. Mode of delivery in hemophilia: vaginal delivery and Cesarean section carry similar risks for intracranial hemorrhages and other major bleeds. Haematologica. 2019 Oct;104(10):2100-2106. doi: 10.3324/haematol.2018.209619[↩]
- Soucie JM, Nuss R, Evatt B, Abdelhak A, Cowan L, Hill H, Kolakoski M, Wilber N. Mortality among males with hemophilia: relations with source of medical care. The Hemophilia Surveillance System Project Investigators. Blood. 2000 Jul 15;96(2):437-42. https://doi.org/10.1182/blood.V96.2.437[↩]
- Pai M, Key NS, Skinner M, Curtis R, Feinstein M, Kessler C, Lane SJ, Makris M, Riker E, Santesso N, Soucie JM, Yeung CH, Iorio A, Schünemann HJ. NHF-McMaster Guideline on Care Models for Haemophilia Management. Haemophilia. 2016 Jul;22 Suppl 3:6-16. doi: 10.1111/hae.13008[↩]
- Witkop M, Guelcher C, Forsyth A, Hawk S, Curtis R, Kelley L, Frick N, Rice M, Rosu G, Cooper DL. Treatment outcomes, quality of life, and impact of hemophilia on young adults (aged 18-30 years) with hemophilia. Am J Hematol. 2015 Dec;90 Suppl 2:S3-10. doi: 10.1002/ajh.24220[↩]
- Rodriguez-Merchan EC. Musculoskeletal complications of hemophilia. HSS J. 2010 Feb;6(1):37-42. doi: 10.1007/s11420-009-9140-9[↩]

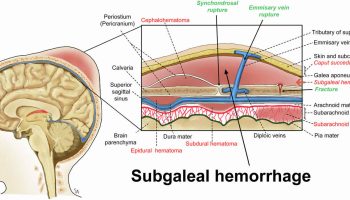
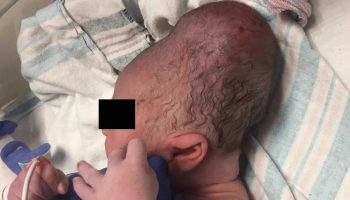
{kind=link}