Contents
What is thrombocytopenia
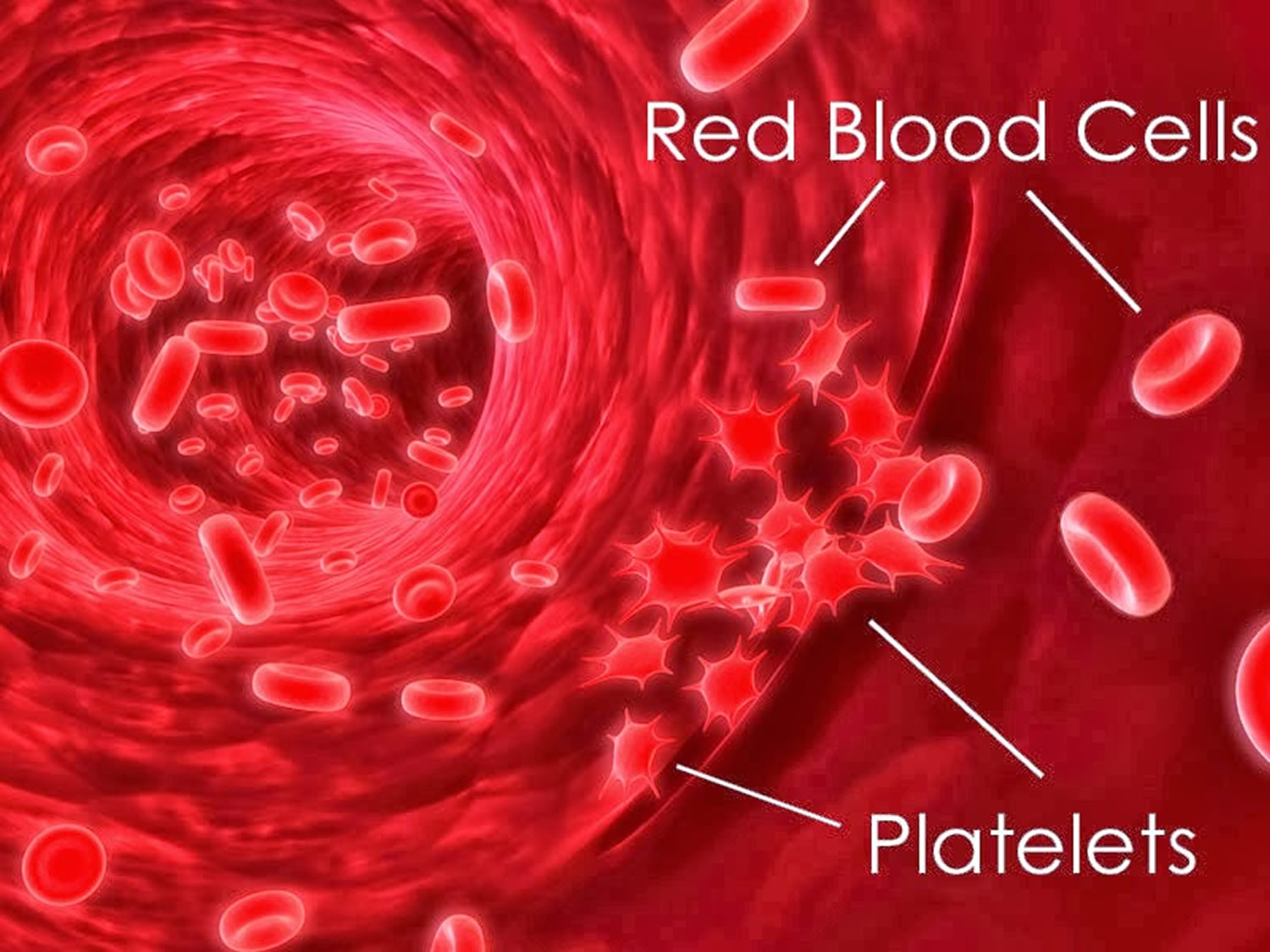
Thrombocytopenia refers to an abnormally low number of platelets (thrombocytes) in the blood. Platelets (thrombocytes) are small anucleated blood cells, derived from precursor megakaryocytes in bone marrow 1, that play an important role in blood clotting by clumping and forming plugs when necessary 2.
Platelets are produced in the bone marrow and circulate in the blood. When there is an injury to a blood vessel and bleeding begins, platelets are the first elements to help to stop bleeding. They do so in three ways. They:
- Adhere to the injury site
- Clump together (aggregate) with other platelets, forming a temporary plug
- Release compounds that stimulate further aggregation and the eventual formation of a blood clot
These reactions result in the formation of a loose platelet plug in a process called primary hemostasis. At the same time, activated platelets support the coagulation cascade, a series of steps that involves the sequential activation of proteins called clotting factors. This is called secondary hemostasis and the two processes result in the formation of a stable clot that remains in place until the injury has healed.
Normally, you have anywhere from 150,000 to 450,000 platelets per microliter of circulating blood. Because each platelet lives only about 10 days, your body continually renews your platelet supply by producing new platelets in your bone marrow. If for any reason your blood platelet count falls below normal, the condition is called thrombocytopenia.
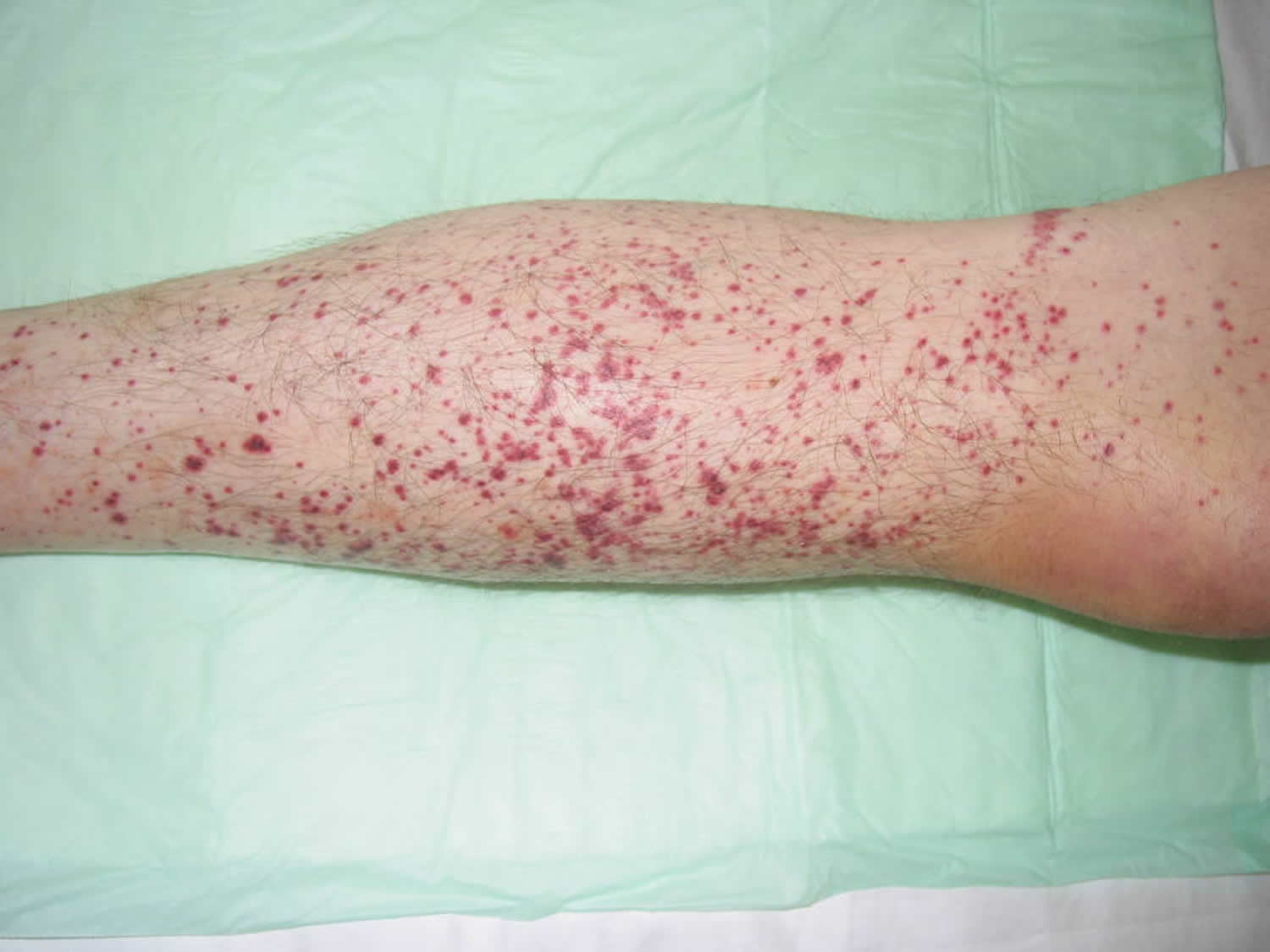
As the major role of platelets is to help the blood to clot, thrombocytopenia may lead to abnormal bleeding due your blood not clotting properly. This can result in bleeding into the skin, spontaneous bruising and the development of petechiae. If the platelet count falls below 20,000 per microliter, spontaneous bleeding may occur and is considered a life-threatening risk. A person with a very low count may be given platelets through a transfusion.
Thrombocytopenia often occurs as a result of a separate disorder, such as leukemia or an immune system problem. Or it can be a side effect of taking certain medications. It affects both children and adults. Mildly decreased platelet counts may be seen in women before menstruation. Up to 5% of pregnant women may have a lower platelet count at term.
Thrombocytopenia may be mild and cause few signs or symptoms. In rare cases, the number of platelets may be so low that dangerous internal bleeding occurs. Bruising for no apparent reason, bleeding from the nose, mouth, or rectum without obvious injury, excessive or prolonged menstrual periods, or the inability to stop a small wound from bleeding within a reasonable period of time may indicate a platelet deficiency. Treatment options are available.
Figure 1. Platelets
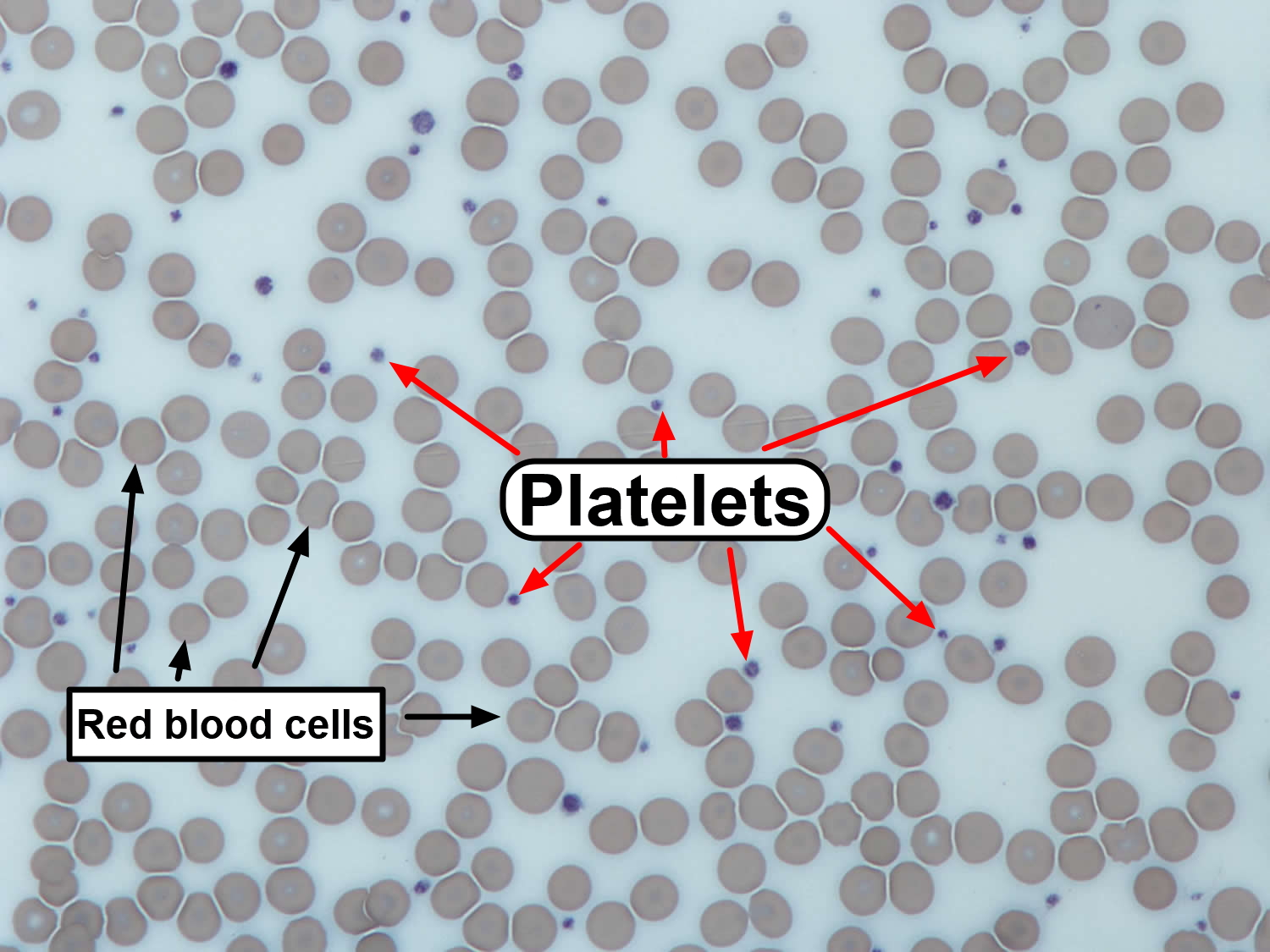
Figure 2. Thrombocytopenia – bleeding into the skin (purpura) – rash of purple spots that do not disappear when pressure is applied to the skin
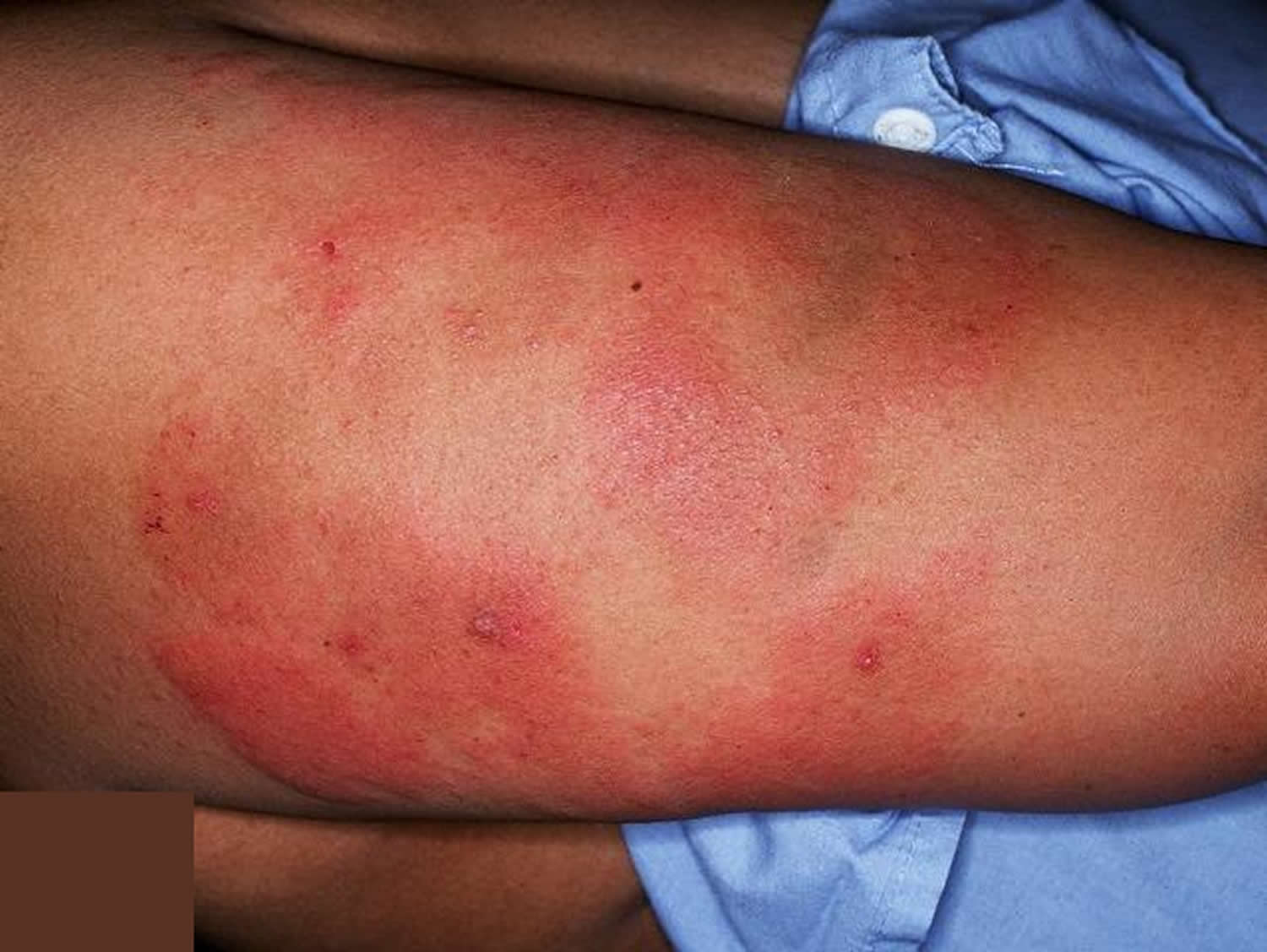
Figure 3. Thrombocytopenia – bleeding into the skin (petechiae) – rash of small red spots that do not disappear when pressure is applied to the skin
Autoimmune thrombocytopenia
Autoimmune thrombocytopenia is caused by an abnormal immune response, targeting one’s own platelets. Autoimmune thrombocytopenia is categorized into primary immune thrombocytopenia (ITP), drug-induced thrombocytopenia and infection associated thrombocytopenia 3.
Idiopathic thrombocytopenia
Idiopathic thrombocytopenic purpura (ITP) is the most common with an incidence of 1–2.5 per 10,000 individuals 4 and the degree of thrombocytopenia can range from mild to severe 5. It has been shown that both platelet destruction and impaired platelet production may contribute to low platelet counts, although the exact contributions by each remain unclear 6. To date, autoantibodies are considered to be the predominant effector in thrombocytopenia. In adult idiopathic thrombocytopenic purpura patients, approximately 70% of detectable platelet autoantibodies are directed against αIIbβ3 integrin, and roughly 20%–40% have specificity for the GPIbα complex, or both.70 Interestingly, there is a subpopulation of idiopathic thrombocytopenic purpura patients who are thrombocytopenic but have no detectable antibodies. One proposed explanation is that in this group of patients, idiopathic thrombocytopenic purpura is mediated by CD8+ T cells (i.e. cytotoxic T-lymphocytes) 7. On the other hand, it may be that the current antibody detection systems are not optimal and may be unable to detect some of the autoantibodies that recognize conformation-dependent epitopes. The conformations of these platelet antigens may be changed during the anticoagulant treatment and sample preparations, subsequently losing their binding sites for some antibodies 8. This may be particularly important for αIIbβ3 integrin, since divalent cations play important roles in maintaining integrin structure and function 9. Divalent cation chelators (e.g. sodium citrate, and EDTA) in the anti-coagulated blood may generate false negative results and decrease the autoantibody detection.
Platelet clearance mediated by anti-platelet autoantibodies typically result in platelet phagocytosis by Fcγ-receptor bearing cells of the reticuloendothelial system (RES) such as macrophages, and the majority of these opsonized platelets are cleared in the spleen. However some idiopathic thrombocytopenic purpura patients, particularly those with anti-GPIbα antibodies who do not respond well to treatments 10, may mediate platelet clearance through an Fc-independent pathway 11, leading to alternative sites of platelet clearance 12. A recent study found that platelet desialylation 13 may occur after antibody binding, particularly those platelets opsonized by anti-GPIbα antibodies 14. This mechanism leads to platelet clearance in the liver via Ashwell-Morell receptors on hepatocytes, which is fundamentally different from the classical Fc-FcγR-dependent macrophage phagocytosis in spleen 14. This discovery may be important not only in basic research, but also for diagnosis and treatment of refractory idiopathic thrombocytopenic purpura 15. It is currently unknown whether this novel Fc-independent platelet clearance pathway also occurs in fetuses and contributes to fetal and neonatal alloimmune thrombocytopenia.
Idiopathic thrombocytopenic purpura (ITP) is a disorder that can lead to easy or excessive bruising and bleeding. The bleeding results from unusually low levels of platelets.
Idiopathic thrombocytopenic purpura, which is also called immune thrombocytopenia, affects children and adults. Children often develop idiopathic thrombocytopenic purpura after a viral infection and usually recover fully without treatment. In adults, the disorder is often long term.
If you don’t have signs of bleeding and your platelet count isn’t too low, you may not need any treatment. In rare cases, the number of platelets may be so low that dangerous internal bleeding occurs. A rare complication of idiopathic thrombocytopenic purpura is bleeding into the brain, which can be fatal. Treatment options are available.
Idiopathic thrombocytopenic purpura symptoms
Idiopathic thrombocytopenic purpura may have no signs and symptoms. When they do occur, they may include:
- Easy or excessive bruising (purpura)
- Superficial bleeding into the skin that appears as a rash of pinpoint-sized reddish-purple spots (petechiae), usually on the lower legs
- Bleeding from the gums or nose
- Blood in urine or stools
- Unusually heavy menstrual flow
Pregnancy
In pregnant women with idiopathic thrombocytopenic purpura, the condition doesn’t usually affect the baby. But the baby’s platelet count should be tested soon after birth.
If you’re pregnant and your platelet count is very low or you have bleeding, you have a greater risk of heavy bleeding during delivery. In such cases, you and your doctor may discuss treatment to maintain a stable platelet count, taking into account the effects on your baby.
Idiopathic thrombocytopenic purpura Causes
In some people thrombocytopenia is caused by the immune system mistakenly attacking and destroying platelets. If the cause of this immune reaction is unknown, the condition is called idiopathic thrombocytopenic purpura. Idiopathic means “of unknown cause.”
In most children with idiopathic thrombocytopenic purpura, the disorder follows a viral illness, such as the mumps or the flu. It may be that the infection triggers the immune system malfunction.
Increased breakdown of platelets
In people with idiopathic thrombocytopenic purpura, antibodies produced by the immune system attach themselves to the platelets, marking the platelets for destruction. The spleen, which helps your body fight infection, recognizes the antibodies and removes the platelets from your system. The result of this case of mistaken identity is a lower number of circulating platelets than is normal.
A normal platelet count is generally between 150,000 and 450,000 platelets per microliter of circulating blood. People with idiopathic thrombocytopenic purpura often have platelet counts below 20,000. Because platelets help the blood clot, as their number decreases, your risk of bleeding increases. The greatest risk is when your platelet count falls very low — below 10,000 platelets per microliter. At this point, internal bleeding may occur even without any injury.
Risk factors for idiopathic thrombocytopenic purpura
Idiopathic thrombocytopenic purpura can occur in anyone at almost any age, but these factors increase the risk:
- Your sex. Women are two to three times more likely to develop idiopathic thrombocytopenic purpura than men are.
- Recent viral infection. Many children with idiopathic thrombocytopenic purpura develop the disorder after a viral illness, such as mumps, measles or a respiratory infection.
Idiopathic thrombocytopenic purpura diagnosis
To diagnose idiopathic thrombocytopenic purpura, your doctor will try to exclude other possible causes of bleeding and a low platelet count, such as an underlying illness or medications you or your child may be taking.
Your doctor will also ask you about your or your child’s medical history, perform a physical exam and run one or more of the following tests:
- Complete blood count (CBC). This common blood test is used to determine the number of blood cells, including platelets, in a sample of blood. With idiopathic thrombocytopenic purpura, white and red blood cell counts are usually normal, but the platelet count is low.
- Blood smear. This test is often used to confirm the number of platelets observed in a complete blood count. A sample of blood is placed on a slide and observed under a microscope.
- Bone marrow exam. This test may be used to help identify the cause of a low platelet count, though the American Society of Hematology doesn’t recommend this test for children with idiopathic thrombocytopenic purpura. Platelets are produced in the bone marrow — soft, spongy tissue in the center of large bones. In some cases, a sample of bone tissue and the enclosed marrow is removed in a procedure called a bone marrow biopsy. Or your doctor may do a bone marrow aspiration, which removes some of the liquid portion of the marrow. In many cases, both procedures are performed at the same time (bone marrow exam). In people with idiopathic thrombocytopenic purpura, the bone marrow will be normal because a low platelet count is caused by the destruction of platelets in the bloodstream and spleen — not by a problem with the bone marrow.
Idiopathic thrombocytopenic purpura treatment
People with mild idiopathic thrombocytopenic purpura may need nothing more than regular monitoring and platelet checks. Children usually improve without treatment. Most adults with idiopathic thrombocytopenic purpura will eventually need treatment, as the condition often becomes severe or long-term (chronic) idiopathic thrombocytopenic purpura.
Treatment may include a number of approaches, such as medications to boost your platelet count or surgery to remove your spleen (splenectomy). Talk with your doctor about the risks and benefits of your treatment options. Some people find that the side effects of treatment are more burdensome than the effects of the disease itself.
Medications
Your doctor will talk with you about medications or supplements you take and whether you need to stop using any that might inhibit platelet function. Examples include aspirin, ibuprofen (Advil, Motrin IB, others), ginkgo biloba and warfarin (Coumadin).
Your doctor may prescribe one or more of the following medications to treat idiopathic thrombocytopenic purpura:
- Drugs that suppress your immune system. Your doctor will likely start you on an oral corticosteroid, such as prednisone. This drug may help raise your platelet count by decreasing the activity of your immune system. Once your platelet count is back to a safe level, you can gradually discontinue taking the drug under the direction of your doctor. In general, this takes about two to six weeks. The problem is that many adults experience a relapse after discontinuing corticosteroids. A new course of corticosteroids may be pursued, but long-term use of these medications isn’t recommended because of the risk of serious side effects. These include cataracts, high blood sugar, increased risk of infections and thinning of bones (osteoporosis).
- Injections to increase your blood count. If corticosteroids don’t help, your doctor may give you an injection of immune globulin (IVIG). This drug may also be used if you have critical bleeding or need to quickly increase your blood count before surgery. The effect usually wears off in a couple of weeks. Possible side effects include headache, vomiting and low blood pressure.
- Drugs that boost platelet production. Thrombopoietin receptor agonists — such as romiplostim (Nplate) and eltrombopag (Promacta) — help your bone marrow produce more platelets. Possible side effects include headache, dizziness, nausea or vomiting, and an increased risk of blood clots.
- Other immune-suppressing drugs. Rituximab (Rituxan) helps reduce the immune system response that’s damaging platelets, thus raising the platelet count. Possible side effects include low blood pressure, fever, sore throat and rash.
Emergency treatment
Although rare, severe bleeding can occur with idiopathic thrombocytopenic purpura. Emergency care usually includes transfusions of platelet concentrates, intravenous corticosteroid (methylprednisolone) and intravenous immune globulin.
Treatments for resistant disease
If your condition persists despite treatment, your doctor may suggest other drugs that suppress the immune system or boost platelet production:
- Removal of your spleen. If your condition is severe or persists despite initial drug treatment, your doctor may suggest surgical removal of your spleen (splenectomy). This quickly eliminates the main source of platelet destruction in your body and improves your platelet count, though it doesn’t work for everyone. Serious post-surgical complications sometimes occur, and not having a spleen permanently increases your susceptibility to infection. Splenectomy is rarely a treatment choice for children with idiopathic thrombocytopenic purpura because they often get better without treatment.
- Other drugs. Azathioprine (Imuran, Azasan) has been used to treat idiopathic thrombocytopenic purpura. But it can cause significant side effects, and its effectiveness has yet to be proved. Possible side effects include fever, headache, nausea and vomiting, and muscle pain.
Neonatal alloimmune thrombocytopenia
Neonatal alloimmune thrombocytopenia is caused by maternal antibodies raised against alloantigens carried on fetal platelets 16. Differences in fetal and maternal human platelet antigens (HPAs) can lead to maternal immunization and destruction of the fetal platelets, a condition named fetal and neonatal alloimmune thrombocytopenia 17. The human platelet antigens (HPAs) are located on platelet membrane glycoprotein receptors. These glycoproteins play fundamental roles in platelet functions, such as adhesion and aggregation. Most human platelet antigens (HPAs) are based on single-nucleotide polymorphisms resulting in amino acid substitutions localized on the main platelet receptors: integrin αIIbβ3 (GPIIb/IIIa, CD41/CD61: the fibrinogen receptor), the GPIb-IX-V complex (CD42, von Willebrand factor receptor), and the GPIa/IIa complex (α2β1, CD29, the collagen receptor) 18. HPA-15 is the only exception; this biallelic system is carried by the platelet membrane protein CD109, which is a part of the transforming growth factor-β receptor system 19.
Figure 4. Neonatal alloimmune thrombocytopenia antigens – Antigens known to trigger maternal sensitization leading to neonatal alloimmune thrombocytopenia are carried on four different platelet membrane glycoproteins (GP) and glycoprotein complexes. Structural domains identified by crystallographic studies are shown schematically.
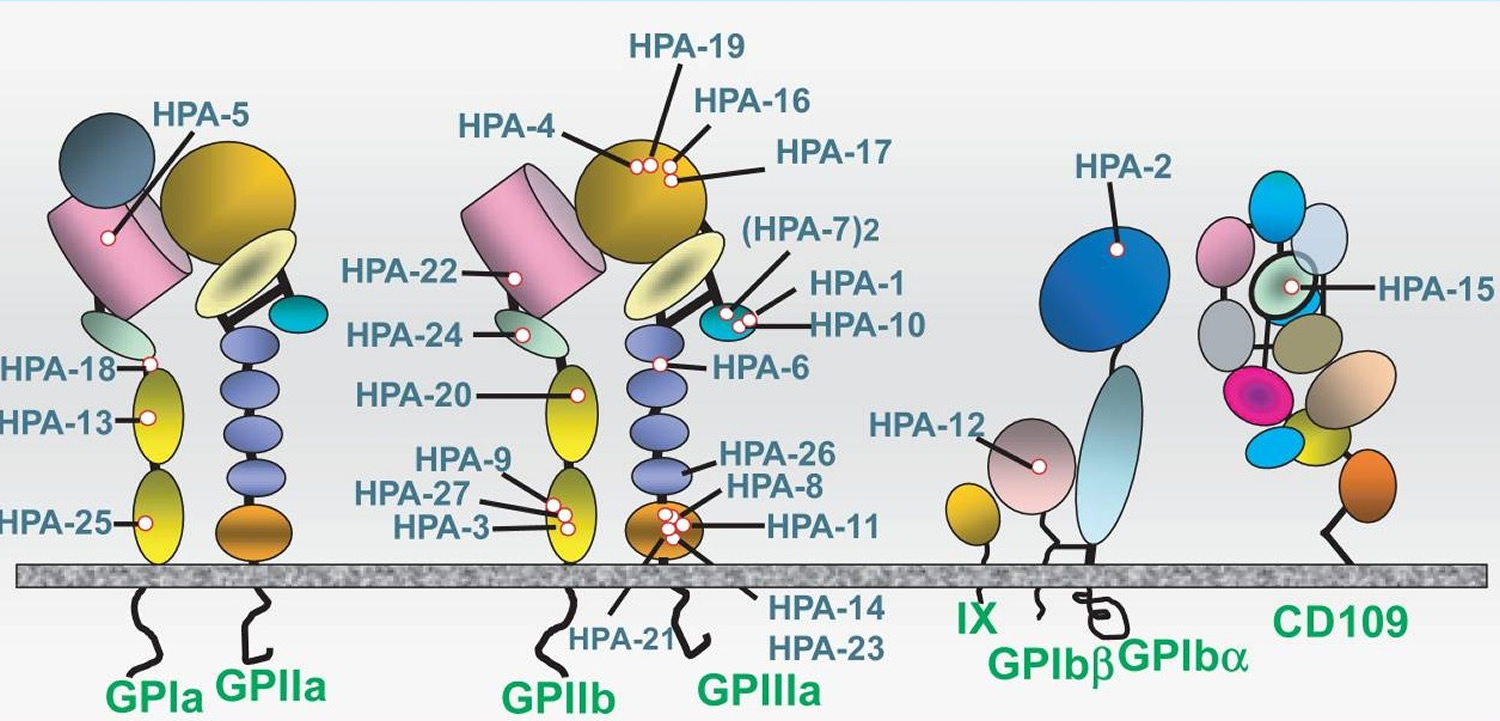
Figure 5. Pathophysiology of maternal human platelet antigens (HPA-1) alloimmunization
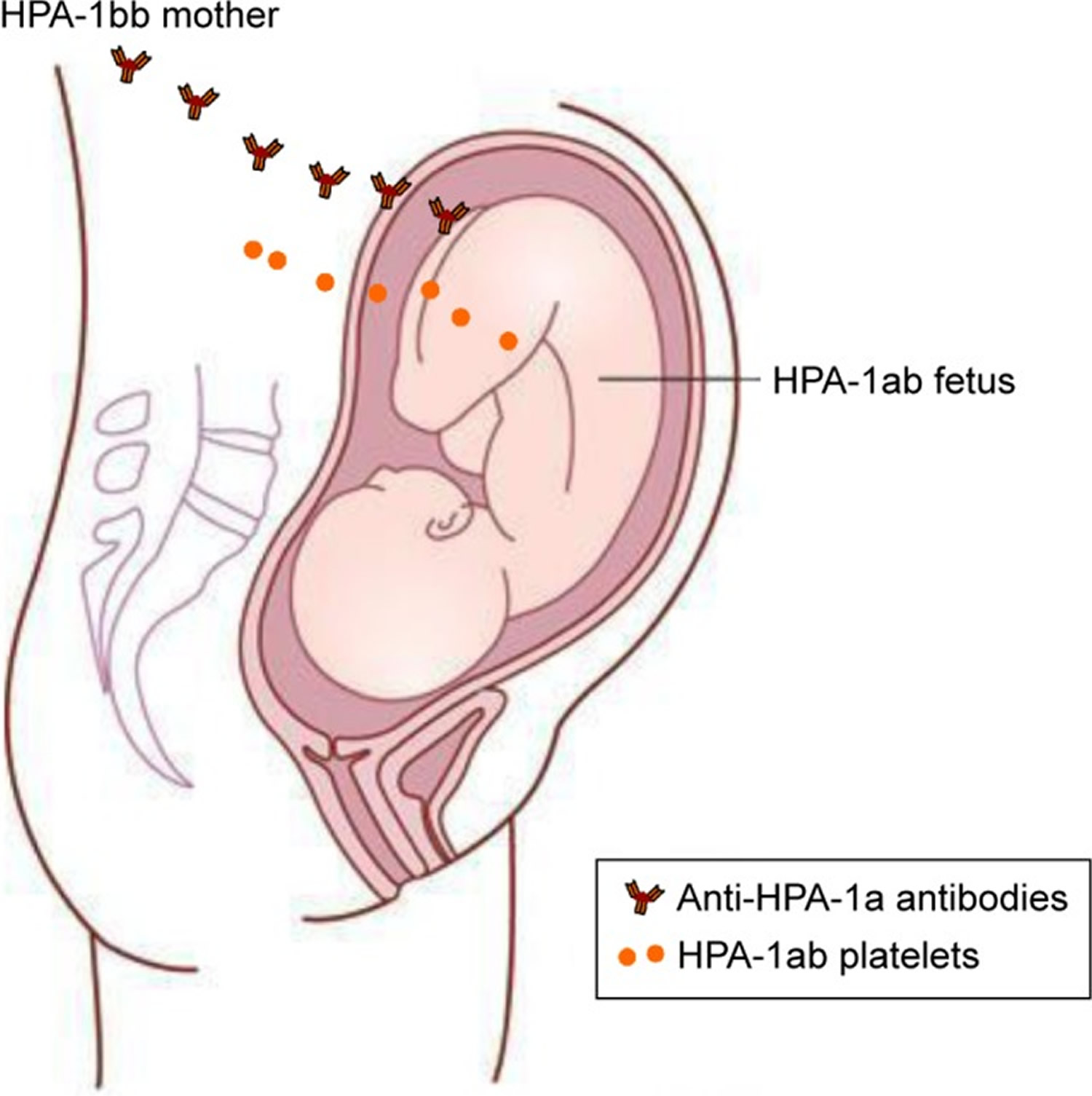
Notes: In an HPA-1bb mother who is pregnant with an HPA-1ab fetus, fetal platelets/fetal platelet antigen may enter the maternal circulation and lead to the production of anti-HPA-1a antibodies in the mother.
Fetal and neonatal alloimmune thrombocytopenia is reported to occur in ~1 per 1,000 live born neonates 17. The major risk is intracranial hemorrhage in the fetus or newborn, which is associated with severe neurological complications or death 20. Since no countries have yet implemented a screening program to detect pregnancies at risk, the diagnosis is typically established after the birth of a child with symptoms.
Fetal and neonatal alloimmune thrombocytopenia is not a common pregnancy complication, but carries a significant risk of severe fetal and/or neonatal complications and has been recognized as the major cause of primary hemorrhagic morbidity and mortality in fetuses and newborns. In neonatal intensive care units, severe thrombocytopenia (platelet count <50,000 platelets per microliter) is reported in 5%–22% of children 21. Most of these cases have underlying causes such as prematurity, congenital infections, maternal immune thrombocytopenic purpura, or chronic fetal hypoxia 21. However, in otherwise healthy term newborns with isolated severe thrombocytopenia, the most frequent cause is fetal and neonatal alloimmune thrombocytopenia 22. The condition occurs in ~1 per 1,000 births in Caucasian populations 23.
Neonatal alloimmune thrombocytopenia diagnosis
The diagnosis of fetal and neonatal alloimmune thrombocytopenia requires that the fetus/neonate carries a platelet alloantigen that the mother lacks, and to which she has made detectable antibodies 24. The current gold standard for the detection of platelet-specific antibodies is the monoclonal antibody-specific immobilization of platelet antigen (MAIPA) assay 25, a sensitive and specific capture immunoassay. Quantitation of anti-HPA-1a antibodies is done using a modified MAIPA assay 26. Other HPA alloantibody specificities are normally not quantified. Other techniques are also possible to use. For instance, different Luminex bead-based assays for the detection of anti-HPA-1a antibodies have been tested and are used by some 27. Furthermore, low-avidity anti-HPA-1a antibodies may be detected using surface plasmon resonance technology 28.
Cordocentesis has been used in order to identify thrombocytopenic fetuses requiring intrauterine platelet transfusions and also to help decide on antenatal maternal treatment. Because of high risk of procedure-related complications 29, avoidance of invasive procedures is currently recommended 30.
Assays for noninvasive prenatal testing to detect fetal HPA-1a DNA in maternal plasma have been developed and are in use in many research laboratories, but are not yet implemented in routine clinical practice in most countries 31. Determination of paternal zygosity may be relevant. If the father is typed and found to be HPA-1aa, the pregnancy will always be HPA-1 incompatible and fetal HPA-1 genotyping is not necessary. Whereas if the father is HPA-1ab, there is a 50% chance of the fetus being HPA-1bb and compatible with the mother, and in this situation knowing the fetal HPA-1 genotype would be clinically helpful.
Neonatal alloimmune thrombocytopenia signs and symptoms
The suspicion of fetal and neonatal alloimmune thrombocytopenia is typically raised when a newborn develops widespread skin petechiae shortly after birth and blood tests show severe thrombocytopenia. Most fetal and neonatal alloimmune thrombocytopenia cases present with platelet counts well below 50,000 platelets per microliter. Clinical symptoms range from no symptoms to limited or widespread skin petechiae or purpura to symptoms of extra- or intracranial hemorrhage (ICH).
Neonatal alloimmune thrombocytopenia treatment
Intravenous immunoglobulin
Previous severe fetal and neonatal alloimmune thrombocytopenia, with or without bleeding complications, is currently used clinically to determine the risk of severe fetal and neonatal alloimmune thrombocytopenia in subsequent pregnancies, and as such serves as the major basis in antenatal management planning. Weekly intravenous immunoglobulin (IVIG) treatment to the mother starting from the second trimester is the first-line therapy of choice in most of the Western countries and is administered when the risk of fetal and neonatal alloimmune thrombocytopenia is considered to be high 30. The treatment is considered effective when the neonatal platelet count is increased or intracranial hemorrhage (ICH) avoided in a subsequent pregnancy compared with the previous fetal and neonatal alloimmune thrombocytopenia pregnancy, with the underlying assumption that fetal and neonatal alloimmune thrombocytopenia gets worse in younger siblings. In a recent Norwegian prospective study 32, the natural course of fetal and neonatal alloimmune thrombocytopenia in several subsequent pregnancies was reported for the first time. Tiller et al 17 data showed that younger siblings of fetal and neonatal alloimmune thrombocytopenia-affected children had unchanged or higher neonatal platelet counts without antenatal treatment in the majority of subsequent pregnancies. Therefore, this study 17 did not support the common opinion that the outcome after HPA-1a alloimmunization is generally worse in the next pregnancy. Thus, increased neonatal platelet count in a subsequent fetal and neonatal alloimmune thrombocytopenia pregnancy may not always reflect an antenatal treatment effect. Delbos et al 33 reported that efficacy of intravenous immunoglobulin (IVIG) treatment may be dependent on maternal HLA-DRB4*0101 haplotype, also suggesting the possible use of several HLA-DR haplotypes as predictive markers of clinical outcome.
Corticosteroids
Some clinicians use systemic corticosteroids alongside intravenous immunoglobulin (IVIG) as a means of supporting the action of intravenous immunoglobulin (IVIG). Dexamethasone is not recommended due to risk of oligohydramnios at higher doses 34 and lack of effect at lower doses 35. Prednisone is therefore the recommended choice; however, the potential benefits versus risks deserves further evaluation 36.
Cordocentesis
Repeat cordocentesis to measure fetal platelet count followed by intrauterine platelet transfusions in case of severe fetal thrombocytopenia was commonly performed previously, but is nowadays abandoned by many countries due to high risk of procedure-related complications 37. Some countries still combine diagnostic fetal blood sampling (FBS) with maternal intravenous immunoglobulins (IVIGs), with or without a second fetal blood sampling to decide on treatment effect and mode of delivery. However, a complete noninvasive management strategy is advocated by most.
Mode of delivery
Whether delivery by caesarean section prevents intracranial hemorrhage (ICH) in fetal and neonatal alloimmune thrombocytopenia affected neonates is not really known 38. Vaginal delivery is advised by some as an option when fetal platelet count is >50,000 platelets per microliter 35. However, this approach requires fetal blood sampling in order to know the fetal platelet count around the time of delivery. A Dutch pilot study of 32 pregnancies where an older sibling had fetal and neonatal alloimmune thrombocytopenia without intracranial hemorrhage (ICH) found that vaginal delivery was not associated with an increased risk of intracranial hemorrhage 39. The current policy in the Netherlands is to induce vaginal delivery at 37–38 weeks without fetal blood sampling first. If the woman has a previous caesarean section, they may sometimes do a fetal blood sampling before inducing delivery. If the previous child had intracranial hemorrhage, delivery is performed by elective caesarean section at 36 weeks. The intervention part of the Norwegian screening study consisted of delivering all HPA-1a alloimmunized women by caesarean section at 36–38 weeks followed by immediate transfusion of HPA-1 compatible platelets if the newborn was severely thrombocytopenic 40. Delivery by caesarean section was one of several interventions in this study, and the isolated effect of delivery mode is therefore difficult to assess. Still, mortality and morbidity were significantly reduced in the screening and intervention population compared with historical controls 41. The current management guideline in Norway recommends delivery by elective caesarean section around 38 weeks if the maternal anti-HPA-1a antibody level is ≥3 IU/mL, irrelevant of previous obstetric history.
Since the vast majority of intracranial hemorrhage (ICH) caused by fetal and neonatal alloimmune thrombocytopenia is found to occur before delivery, and little data support the idea that intracranial hemorrhage due to fetal and neonatal alloimmune thrombocytopenia tend to occur in connection with delivery, it is difficult to argue that the risk of intracranial hemorrhage is affected by mode of delivery. The risk of intracranial hemorrhage when older siblings did not suffer from intracranial hemorrhage is reported to be 7% 24. A larger study sample is therefore needed before we can conclude whether vaginal delivery for this patient group is safe or not. In many places, HPA-1bb platelets are not available in the blood banks on a daily basis. A planned delivery – whether vaginally or by surgery – is therefore important in order to have appropriate platelets available in case of a severely thrombocytopenic newborn.
Postnatal management of the newborn
The majority of fetal and neonatal alloimmune thrombocytopenia-affected newborns will not have suffered intracranial hemorrhage before delivery. Preventing intracranial hemorrhage by increasing platelet count above a certain threshold is therefore considered a neonatal emergency, and prompt correction by platelet transfusion should be done based on clinical suspicion without awaiting laboratory confirmation of the diagnosis. However, it is not clear what threshold should be used to trigger platelet transfusion. Previous reports also suggest that neonates with HPA-5b incompatibility may be at risk of bleeding at higher platelet counts compared with HPA-1 incompatibility. It is recommended to give compatible platelet concentrates instead of random donor platelets, due to both larger platelet increment and longer half-life of the transfused platelets 42. Random donor platelets may be used when compatible platelets are not available 42. The use of intravenous immunoglobulin (IVIG) as treatment to increase neonatal platelet count varies, but is often recommended as supplemental therapy for 1–3 days depending on platelet increment response of transfusions 43. Other management options include corticosteroid therapy, but documentation of effect is poor 44. It is also recommended that all babies with severe fetal and neonatal alloimmune thrombocytopenia should have a cranial ultrasound for the detection of intracranial hemorrhage 45.
In summary, there is consensus on postnatal correction of severe thrombocytopenia, but antenatal treatment management protocols vary. Knowledge gaps on the natural history of fetal and neonatal alloimmune thrombocytopenia, together with lack of randomized controlled trials evaluating the effects of different treatment options, are probably a major reason for the struggle to have common management protocols. Still, nobody questions the severity of this disorder. Therefore, there is no doubt that a preventive approach hindering the mother from becoming HPA-1a alloimmunized in the first place would be welcomed by all.
Thrombocytopenia causes
Thrombocytopenia can be inherited or it may be caused by a number of medications or conditions. Whatever the cause, circulating platelets are reduced by one or more of the following processes: trapping of platelets in the spleen, decreased platelet production or increased destruction of platelets.
Trapped platelets
The spleen is a small organ about the size of your fist located just below your rib cage on the left side of your abdomen. Normally, your spleen works to fight infection and filter unwanted material from your blood. An enlarged spleen — which can be caused by a number of disorders — may harbor too many platelets, causing a decrease in the number of platelets in circulation.
Decreased production of platelets
Platelets are produced in your bone marrow. If production is low, you may develop thrombocytopenia.
Disorders in which the bone marrow cannot produce enough platelets:
- Leukemia, lymphoma, or another cancer that has spread (metastasized) to the bone marrow—people with cancers often experience excessive bleeding due to a significantly decreased number of platelets. As the number of cancer cells increases in the bone marrow, normal bone marrow cells are crowded out, resulting in fewer platelet-producing cells.
- Some types of anemia. Aplastic anemia—a condition in which the production of all blood cells is significantly reduced.
- Viral infections such as mononucleosis, hepatitis C, HIV or measles
- Chemotherapy or radiation therapy, which may affect the bone marrow’s ability to produce platelets
- Heavy alcohol consumption
- Long-term bleeding problems (e.g., chronic bleeding from stomach ulcers)
- Cirrhosis
- Exposure to toxic chemicals, such as pesticides, arsenic, or benzene
Increased breakdown of platelets
Some conditions can cause your body to use up or destroy platelets more rapidly than they’re produced. This leads to a shortage of platelets in your bloodstream. Examples of such conditions include:
- Pregnancy. Gestational thrombocytopenia caused by pregnancy is usually mild and improves soon after childbirth.
- Immune thrombocytopenia. This type is caused by autoimmune diseases, such as lupus and rheumatoid arthritis. The body’s immune system produces antibody against platelets – mistakenly attacks and destroys platelets. If the exact cause of this condition isn’t known, it’s called idiopathic thrombocytopenic purpura or also known as immune thrombocytopenic purpura. This type more often affects children.
- Autoimmune disorders, such as lupus, where the body’s immune system produces antibodies that attack its own organs or tissues, causing increased destruction of platelets
- Bacteria in the blood. Severe bacterial infections involving the blood (bacteremia) may lead to destruction of platelets. Sepsis, especially that caused by a serious bacterial infection with Gram-negative bacteria.
- Thrombotic thrombocytopenic purpura. This is a rare condition that occurs when small blood clots suddenly form throughout your body, using up large numbers of platelets.
- Hemolytic uremic syndrome. This rare disorder causes a sharp drop in platelets, destruction of red blood cells and impairment of kidney function. Sometimes it can occur in association with a bacterial Escherichia coli (E. coli) infection, such as may be acquired from eating raw or undercooked meat.
- Medications. Certain medications can reduce the number of platelets in your blood. Sometimes a drug confuses the immune system and causes it to destroy platelets. Certain drugs, such as aspirin and ibuprofen, quinine, anticonvulsants, heparin, some antibiotics (including sulfa-containing antibiotics), colchicine and indomethacin, H2-blocking agents, hydralazine, isoniazid, quinidine, thiazide diuretics, and tolbutamide, are just a few that have been associated with drug-induced decreased platelet counts. Heparin-induced thrombocytopenia (HIT) results in low platelets when a person who is on or received heparin therapy develops an antibody.
Platelet consumption may be observed in various diseases and conditions. For example, disseminated intravascular coagulation (DIC), thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS) can result in fewer circulating platelets in the blood.
Thrombocytopenia complications
Dangerous internal bleeding can occur when your platelet count falls below 10,000 platelets per microliter. Though rare, severe thrombocytopenia can cause bleeding into the brain, which can be fatal.
Thrombocytopenia symptoms
Thrombocytopenia signs and symptoms may include:
- Easy or excessive bruising (purpura) (see Figure 2 above)
- Superficial bleeding into the skin that appears as a rash of pinpoint-sized reddish-purple spots (petechiae), usually on the lower legs (see Figure 3 above)
- Prolonged bleeding from cuts
- Bleeding from your gums or nose
- Blood in urine or stools
- Unusually heavy menstrual flows
- Fatigue
- Enlarged spleen
- Jaundice
Thrombocytopenia diagnosis
Your doctor may use the following tests and procedures to determine whether you have thrombocytopenia:
- Blood test. A complete blood count determines the number of blood cells, including platelets, in a sample of your blood. In adults, normal platelet count is 150,000 to 450,000 platelets per microliter of blood. If the complete blood count finds you have fewer than 150,000 platelets, you have thrombocytopenia.
- Platelet function tests are a group of assays that use specialized equipment to measure the ability of platelets to aggregate and promote clotting in a sample of blood. There are a variety of tests available but no one test that identifies all problems with platelet function. Also, there is no widespread agreement on which test(s) is best for each circumstance.
- Physical exam, including a complete medical history. Your doctor will look for signs of bleeding under your skin and feel your abdomen to see if your spleen is enlarged. He or she will also ask you about illnesses you’ve had and the types of medications and supplements you’ve recently taken.
Your doctor may suggest that you undergo other tests and procedures to determine the cause of your condition, depending on your signs and symptoms.
Thrombocytopenia treatment
People with mild thrombocytopenia may not need treatment. For example, they may not have symptoms or the condition clears up on its own.
Some people develop severe or long-term (chronic) thrombocytopenia. Depending on what’s causing your low platelet count, treatments may include:
- Treating the underlying cause of thrombocytopenia. If your doctor can identify a condition or a medication that’s causing your thrombocytopenia, addressing that cause may clear up your thrombocytopenia. For example, if you have heparin-induced thrombocytopenia, your doctor will direct you to stop using heparin and prescribe a different blood-thinning drug. Your thrombocytopenia may persist for a week or more despite stopping all heparin therapy.
- Blood or platelet transfusions. If your platelet level becomes too low, your doctor can replace lost blood with transfusions of packed red blood cells or platelets.
- Medications. If your condition is related to an immune system problem, your doctor may prescribe drugs to boost your platelet count. The first-choice drug may be a corticosteroid. If that doesn’t work, he or she may try stronger medications to suppress your immune system.
- Surgery. If other treatment options don’t help, your doctor may recommend surgery to remove your spleen (splenectomy).
- Plasma exchange. Thrombotic thrombocytopenic purpura can result in a medical emergency requiring plasma exchange.
Home remedies
If you have thrombocytopenia, try to:
- Avoid activities that could cause injury. Ask your doctor which activities are safe for you. Contact sports, such as boxing, martial arts and football, carry a high risk of injury.
- Drink alcohol in moderation, if at all. Alcohol slows the production of platelets in your body. Ask your doctor whether it’s okay for you to drink alcohol.
- Use caution with over-the-counter medications. Over-the-counter pain medications, such as aspirin and ibuprofen (Advil, Motrin IB, others) can impair platelet function.
- Dynamic visualization of thrombopoiesis within bone marrow. Junt T, Schulze H, Chen Z, Massberg S, Goerge T, Krueger A, Wagner DD, Graf T, Italiano JE Jr, Shivdasani RA, von Andrian UH. Science. 2007 Sep 21; 317(5845):1767-70. https://www.ncbi.nlm.nih.gov/pubmed/17885137/[↩]
- Platelets in thrombosis and hemostasis: old topic with new mechanisms. Wang Y, Andrews M, Yang Y, Lang S, Jin JW, Cameron-Vendrig A, Zhu G, Reheman A, Ni H. Cardiovasc Hematol Disord Drug Targets. 2012 Dec; 12(2):126-32. https://www.ncbi.nlm.nih.gov/pubmed/23030445[↩]
- Liebman H. Other immune thrombocytopenias. Semin Hematol. 2007;44(4 suppl 5):S24–S34. https://www.ncbi.nlm.nih.gov/pubmed/18096469[↩]
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol. 2009;83:83–89. https://www.ncbi.nlm.nih.gov/pubmed/19245532[↩]
- Rodeghiero F, Ruggeri M. ITP and international guidelines: what do we know, what do we need? Presse Med. 2014;43(4 Pt 2):e61–67. https://www.ncbi.nlm.nih.gov/pubmed/24656296[↩]
- Semple JW, Provan D, Garvey MB, Freedman J. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr Opin Hematol. 2010;17:590–595. https://www.ncbi.nlm.nih.gov/pubmed/20739879[↩]
- Chow L, Aslam R, Speck ER, et al. A murine model of severe immune thrombocytopenia is induced by antibody- and CD8+ T cell-mediated responses that are differentially sensitive to therapy. Blood. 2010;115:1247–1253. http://www.bloodjournal.org/content/115/6/1247.long[↩]
- Allen DL, Metcalfe P, Kaplan C, et al. Sensitivity of assays for the detection of HPA-1a antibodies: results of an international workshop demonstrating the impact of cation chelation from integrin alphaIIbbeta3 on three widely used assays. Vox Sang. 2013;105:167–173. https://www.ncbi.nlm.nih.gov/pubmed/23662600[↩]
- Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Ann Rev Immunol. 2007;25:619–647. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1952532/[↩]
- Peng J, Ma SH, Liu J, et al. Association of autoantibody specificity and response to intravenous immunoglobulin G therapy in immune thrombocytopenia: a multicenter cohort study. J Thromb Haemost. 2014;12:497–504. https://www.ncbi.nlm.nih.gov/pubmed/24517219[↩]
- Webster ML, Zhu G, Li Y, Ni H. Fc-independent phagocytosis: implications for intravenous IgG therapy in immune thrombocytopenia. Cardiovasc Hematol Disord Drug Targets. 2008;8:278–282. https://www.ncbi.nlm.nih.gov/pubmed/19075638[↩]
- Li J, van der Wal DE, Zhu L, et al. Fc-independent phagocytosis: implications for IVIG and other therapies in immunemediated thrombocytopenia. Cardiovasc Hematol Disord Drug Targets. 2013;13:50–58. https://www.ncbi.nlm.nih.gov/pubmed/23082940[↩]
- Jansen AJ, Peng J, Zhao HG, Hou M, Ni H. Sialidase inhibition to increase platelet counts: a new treatment option for thrombocytopenia. Am J Hematol. 2015;90(5):E94–E95. https://www.ncbi.nlm.nih.gov/pubmed/25615710[↩]
- Li J, van der Wal DE, Zhu G, et al. Platelet desialylation: a novel mechanism of Fc-independent platelet clearance and a potential diagnostic biomarker and therapeutic target in immune thrombocytopenia. Paper presented at: 56th ASH Annual Meeting; December 4–6, 2014; San Francisco, CA.[↩][↩]
- Li J, Callum JL, Lin Y, Zhou Y, Zhu G, Ni H. Severe platelet desialylation in a patient with glycoprotein Ib/IX antibodymediated immune thrombocytopenia and fatal pulmonary hemorrhage. Haematologica. 2014;99:e61–63. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3971097/[↩]
- Peterson JA, McFarland JG, Curtis BR, Aster RH. Neonatal alloimmune thrombocytopenia: pathogenesis, diagnosis and management. British journal of haematology. 2013;161(1):3-14. doi:10.1111/bjh.12235. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3895911/[↩]
- Tiller H, Husebekk A, Ahlen MT, Stuge TB, Skogen B. Current perspectives on fetal and neonatal alloimmune thrombocytopenia – increasing clinical concerns and new treatment opportunities. International Journal of Women’s Health. 2017;9:223-234. doi:10.2147/IJWH.S90753. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5402885/[↩][↩][↩][↩]
- Landau M, Rosenberg N. Molecular insight into human platelet antigens: structural and evolutionary conservation analyses offer new perspective to immunogenic disorders. Transfusion. 2010;51(3):558–569. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3084503/[↩]
- Finnson KW, Tam BY, Liu K, et al. Identification of CD109 as part of the TGF-beta receptor system in human keratinocytes. FASEB J. 2006;20(9):1525–1527. https://www.ncbi.nlm.nih.gov/pubmed/16754747[↩]
- Bussel JB. Alloimmune thrombocytopenia in the fetus and newborn. Semin Thromb Hemost. 2001;27(3):245–252. https://www.ncbi.nlm.nih.gov/pubmed/11446658[↩]
- Stanworth SJ, Clarke P, Watts T, et al. Prospective, observational study of outcomes in neonates with severe thrombocytopenia. Pediatrics. 2009;124(5):e826–e834. https://www.ncbi.nlm.nih.gov/pubmed/19841111[↩][↩]
- Sainio S, Jarvenpaa AL, Renlund M, Riikonen S, Teramo K, Kekomaki R. Thrombocytopenia in term infants: a population-based study. Obstet Gynecol. 2000;95(3):441–446. https://www.ncbi.nlm.nih.gov/pubmed/10711560[↩]
- Kamphuis MM, Paridaans NP, Porcelijn L, Lopriore E, Oepkes D. Incidence and consequences of neonatal alloimmune thrombocytopenia: a systematic review. Pediatrics. 2014;133(4):715–721. http://pediatrics.aappublications.org/content/133/4/715.long[↩]
- Berkowitz RL, Bussel JB, McFarland JG. Alloimmune thrombocytopenia: state of the art 2006. Am J Obstet Gynecol. 2006;195:907–913. https://www.ncbi.nlm.nih.gov/pubmed/16875656[↩][↩]
- Kiefel V, Santoso S, Weisheit M, Mueller-Eckhardt C. Monoclonal antibody-specific immobilization of platelet antigens (MAIPA): a new tool for the identification of platelet-reactive antibodies. Blood. 1987;70:1722–1726. http://www.bloodjournal.org/content/70/6/1722.long[↩]
- Bertrand G, Jallu V, Gouet M, et al. Quantification of human platelet antigen-1a antibodies with the monoclonal antibody immobilization of platelet antigens procedure. Transfusion. 2005;45:1319–1323. https://www.ncbi.nlm.nih.gov/pubmed/16078919[↩]
- Porcelijn L, Huiskes E, Comijs-van Osselen I, et al. A new bead-based human platelet antigen antibodies detection assay versus the monoclonal antibody immobilization of platelet antigens assay. Transfusion. 2014;54(6):1486–1492. https://www.ncbi.nlm.nih.gov/pubmed/24299453[↩]
- Socher I, Andrei-Selmer C, Bein G, Kroll H, Santoso S. Low-avidity HPA-1a alloantibodies in severe neonatal alloimmune thrombocytopenia are detectable with surface plasmon resonance technology. Transfusion. 2009;49(5):943–952. https://www.ncbi.nlm.nih.gov/pubmed/19175553[↩]
- Radder CM, Brand A, Kanhai HH. Will it ever be possible to balance the risk of intracranial haemorrhage in fetal or neonatal alloimmune thrombocytopenia against the risk of treatment strategies to prevent it? Vox Sang. 2003;84(4):318–325. https://www.ncbi.nlm.nih.gov/pubmed/12757506[↩]
- Kamphuis MM, Oepkes D. Fetal and neonatal alloimmune thrombocytopenia: prenatal interventions. Prenat Diagn. 2011;31(7):712–719. https://www.ncbi.nlm.nih.gov/pubmed/21618560[↩][↩]
- Wienzek-Lischka S, Krautwurst A, Frohner V, et al. Noninvasive fetal genotyping of human platelet antigen-1a using targeted massively parallel sequencing. Transfusion. 2015;55(6 Pt 2):1538–1544. https://www.ncbi.nlm.nih.gov/pubmed/25873286[↩]
- Tiller H, Husebekk A, Skogen B, Kjeldsen-Kragh J, Kjaer M. True risk of fetal/neonatal alloimmune thrombocytopenia in subsequent pregnancies: a prospective observational follow-up study. BJOG. 2016;123(5):738–744. https://www.ncbi.nlm.nih.gov/pubmed/25752647[↩]
- Delbos F, Bertrand G, Croisille L, Ansart-Pirenne H, Bierling P, Kaplan C. Fetal and neonatal alloimmune thrombocytopenia: predictive factors of intracranial hemorrhage. Transfusion. 2016;56(1):59–66. quiz 58. https://www.ncbi.nlm.nih.gov/pubmed/26469867[↩]
- Bussel JB, Berkowitz RL, McFarland JG, Lynch L, Chitkara U. Antenatal treatment of neonatal alloimmune thrombocytopenia. N Engl J Med. 1988;319(21):1374–1378. http://www.nejm.org/doi/full/10.1056/NEJM198811243192103[↩]
- Bussel JB, Berkowitz RL, Lynch L, et al. Antenatal management of alloimmune thrombocytopenia with intravenous gamma-globulin: a randomized trial of the addition of low-dose steroid to intravenous gamma-globulin. Am J Obstet Gynecol. 1996;174:1414–1423. https://www.ncbi.nlm.nih.gov/pubmed/9065105[↩][↩]
- Rayment R, Brunskill SJ, Soothill PW, Roberts DJ, Bussel JB, Murphy MF. Antenatal interventions for fetomaternal alloimmune thrombocytopenia. Cochrane Database Syst Rev. 2011;5:CD004226. https://www.ncbi.nlm.nih.gov/pubmed/21563140[↩]
- Overton TG, Duncan KR, Jolly M, Letsky E, Fisk NM. Serial aggressive platelet transfusion for fetal alloimmune thrombocytopenia: platelet dynamics and perinatal outcome. Am J Obstet Gynecol. 2002;186(4):826–831. https://www.ncbi.nlm.nih.gov/pubmed/11967515[↩]
- Rayment R, Brunskill SJ, Soothill PW, Roberts DJ, Bussel JB, Murphy MF. Antenatal interventions for fetomaternal alloimmune thrombocytopenia. Cochrane Database of Systematic Reviews 2011, Issue 5. Art. No.: CD004226. DOI: 10.1002/14651858.CD004226.pub3. http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD004226.pub3/full[↩]
- van den AE, Oepkes D, Brand A, Kanhai HH. Vaginal delivery for fetuses at risk of alloimmune thrombocytopenia? BJOG. 2006;113(7):781–783. https://www.ncbi.nlm.nih.gov/pubmed/16827760[↩]
- Kjeldsen-Kragh J, Killie MK, Tomter G, et al. A screening and intervention program aimed to reduce mortality and serious morbidity associated with severe neonatal alloimmune thrombocytopenia. Blood. 2007;110(3):833–839. http://www.bloodjournal.org/content/110/3/833.long[↩]
- Valentin N, Vergracht A, Bignon JD, et al. HLA-DRw52a is involved in alloimmunization against PL-A1 antigen. Hum Immunol. 1990;27(2):73–79. https://www.ncbi.nlm.nih.gov/pubmed/2298610[↩]
- Allen D, Verjee S, Rees S, Murphy MF, Roberts DJ. Platelet transfusion in neonatal alloimmune thrombocytopenia. Blood. 2007;109(1):388–389. http://www.bloodjournal.org/content/109/1/388.long[↩][↩]
- te Pas AB, Lopriore E, van den Akker ES, et al. Postnatal management of fetal and neonatal alloimmune thrombocytopenia: the role of matched platelet transfusion and IVIG. Eur J Pediatr. 2007;166(10):1057–1063. https://www.ncbi.nlm.nih.gov/pubmed/17177068[↩]
- Blanchette VS, Johnson J, Rand M. The management of alloimmune neonatal thrombocytopenia. Baillieres Best Pract Res Clin Haematol. 2000;13:365–390. https://www.ncbi.nlm.nih.gov/pubmed/11030040[↩]
- Roberts I, Stanworth S, Murray NA. Thrombocytopenia in the neonate. Blood Rev. 2008;22(4):173–186. https://www.ncbi.nlm.nih.gov/pubmed/18433954[↩]





{kind=link}