Contents
Hypogonadism
Hypogonadism is a condition in which the male testes or the female ovaries produce little or no sex hormones 1. Hypogonadism can occur due to a problem in the gonads (testes or ovaries) or primary gonadal failure also known as Primary hypogonadism or HYPERgonadotropic hypogonadism or due to a problem with the hypothalamic-pituitary-gonadal axis known as Secondary hypogonadism or HYPOgonadotropic hypogonadism. These two entities can be distinguished by measuring serum LH and FSH concentrations 2. Primary hypogonadism or hypergonadotropic hypogonadism is characterized by a low serum testosterone level and low sperm count (oligospermia) or complete absence of sperm (azoospermia) in the presence of elevated serum luteinizing hormone (LH) and follicle-stimulating hormone (FSH) concentrations 2. In contrast, secondary hypogonadism or hypogonadotropic hypogonadism can result from failure of the hypothalamic luteinizing-hormone releasing hormone (LHRH) pulse generator or from the inability of the pituitary gland to respond with secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) and is diagnosed in the setting of a low testosterone level and sperm count (oligospermia) in association with low or inappropriately normal serum LH and FSH concentrations 2.
Men with secondary hypogonadism or hypogonadotropic hypogonadism have low levels of androgens (testosterone) in the plasma as well as a lack or delay of sexual maturity, which can cause symptoms such as a lack of libido, depression, increase in adipose tissue, and diminished erectile function or impotence (not being able to obtain or keep an erection that is sufficient for sexual intercourse) 3.
Patients with secondary hypogonadism or hypogonadotropic hypogonadism usually have an issue with gonadotropin-releasing hormone (GnRH) signaling, which then causes a decrease in follicle-stimulating hormone (FSH) and luteinizing hormone (LH) secretion. This decreased FSH and LH contributes to decreased androgen levels and reduced spermatogenesis. Studies have shown that giving these patients pulsatile GnRH or LH (or hCG) and FSH can help increase spermatogenesis and thus increase the sperm concentration in the ejaculate. Even then, most couples will need assisted reproductive technology (ART) to achieve pregnancy 4.
Testosterone therapy, whether by exogenous testosterone replacement or induction of endogenous testosterone production by human chorionic gonadotropin (hCG) is needed in all hypogonadotropic hypogonadism patients 2. Testosterone plays a number of important physiologic roles in the human and are required not only for virilization and normal sexual function, but also for maintenance of both muscle and bone mass, as well as normal mood and cognition.
While testosterone is the primary treatment modality used to induce and maintain secondary sexual characteristics and sexual function in men with hypogonadism, treatment with testosterone does not restore fertility. Therefore, in patients in whom fertility is the treatment goal, induction of gonadotropin secretion by pulsatile GnRH or exogenous gonadotropins is necessary. While hormone therapy is the mainstay of treatment for congenital hypogonadotropic hypogonadism; undescended testicle (cryptorchidism), if present, should be treated surgically with orchidopexy, ideally before the age of 2 years to improve fertility outcomes and reduce the risk of future testicular malignancy 5.
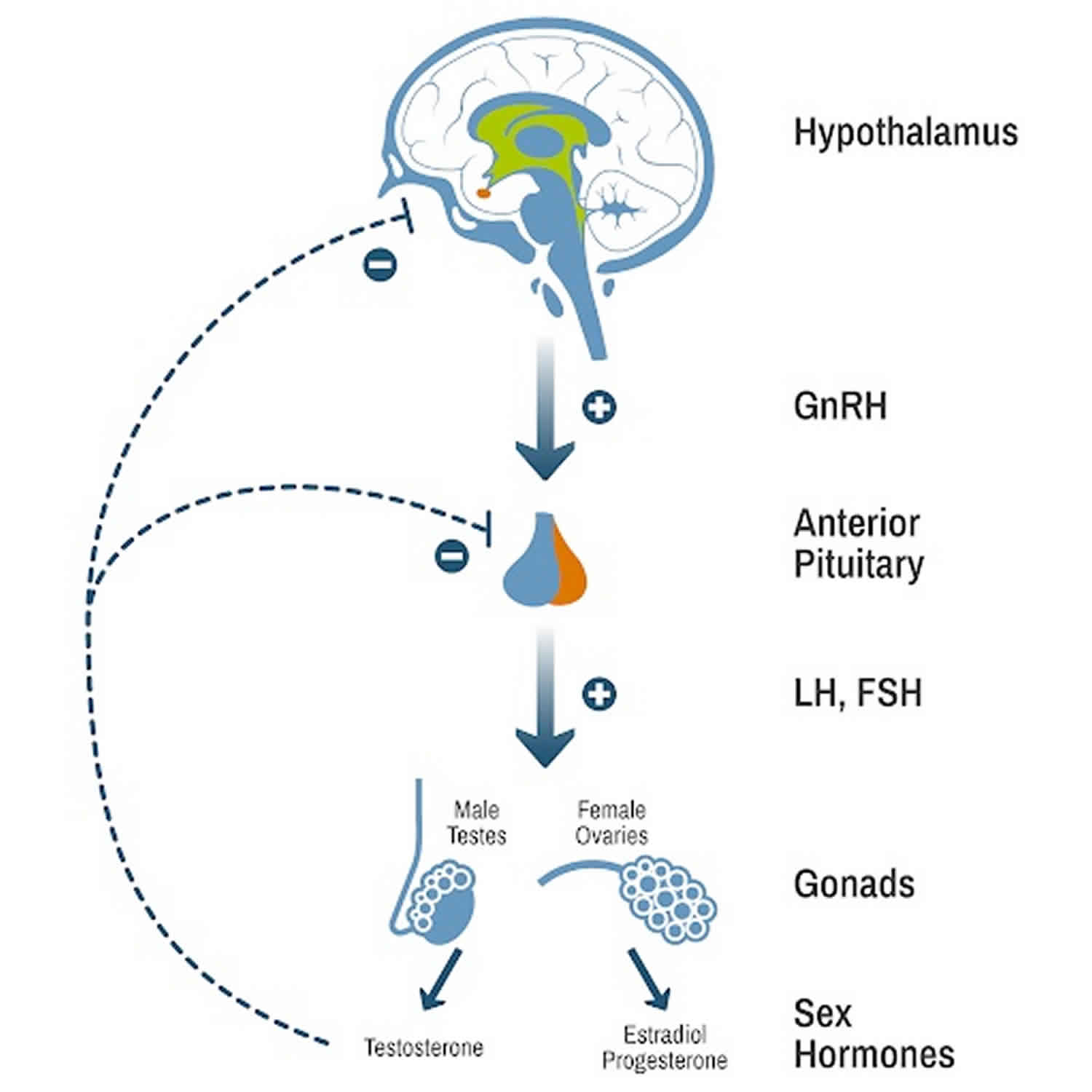
Figure 1. Hypothalamic–pituitary–gonadal axis (HPG axis)
Footnotes: Hypothalamic–pituitary–gonadal (HPG) axis also known as the hypothalamic–pituitary–ovarian/testicular axis refers to the hypothalamus, pituitary gland, and gonadal glands as if these individual endocrine glands were a single entity. The hypothalamic control of reproduction is coordinated through the release of gonadotropin-releasing hormone (GnRH). Pulsatile secretion of gonadotropin-releasing hormone (GnRH) from the hypothalamus is required for both the initiation and maintenance of the hypothalamic–pituitary–ovarian/testicular axis in human. Pulsatile GnRH stimulates secretion of the gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), from the anterior pituitary gonadotropes. Gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH) in turn function at the gonads to stimulate the production of gametes and promote gonadal release of sex steroids [i.e., testosterone (T), estradiol (primarily 17β-estradiol, E2), and progesterone (P4)]. In females, ovarian follicles are stimulated by follicle-stimulating hormone (FSH) to grow and mature; luteinizing hormone (LH) stimulates ovulation and corpus luteum formation. In males, LH stimulates testicular Leydig cells to synthesize and secrete testosterone (T), which in turn maintains spermatogenesis in Sertoli cells through its paracrine action and exerts sexual and anabolic actions. While FSH acts on Sertoli cells to produce androgen-binding protein, which is critical for spermatogenesis initiation and ultimately augments sperm production. Along with guiding reproductive function in the peripheral tissues, these gonadal steroids [i.e., testosterone (T), estradiol (primarily 17β-estradiol, E2) and progesterone (P4)] secreted by ovaries and testis can inhibit gonadotropin-releasing hormone (GnRH)’s hypothalamic synthesis via a feedback loop, hence playing a vital role in regulating reproductive function. In both men and women, gonadal failure results in increased LH, because of loss of the negative feedback of estrogen at the hypothalamus and pituitary in women and from decreases in both androgen and estrogen feedback in men. In response to decreased levels of sex steroids as well as the loss of inhibin, FSH levels are also elevated following gonadal damage. Serum gonadotropin [luteinizing hormone (LH) and follicle-stimulating hormone (FSH)] and sex steroid values will differentiate between reproductive failure at the gonadal level or at the hypothalamic/pituitary level. Sex steroid levels will be low in both, but serum gonadotropin levels will be high in primary gonadal failure and low in those with hypothalamic or pituitary disease. The hallmark of primary gonadal failure from any cause is elevation of gonadotropin [luteinizing hormone (LH) and follicle-stimulating hormone (FSH)] levels, and this is the usual state in postpubertal patients receiving substantial doses of chemotherapy agents. Cranial radiation, on the other hand, may result in significant hypothalamic-pituitary dysfunction and secondary gonadal failure with low serum levels of gonadotropins.
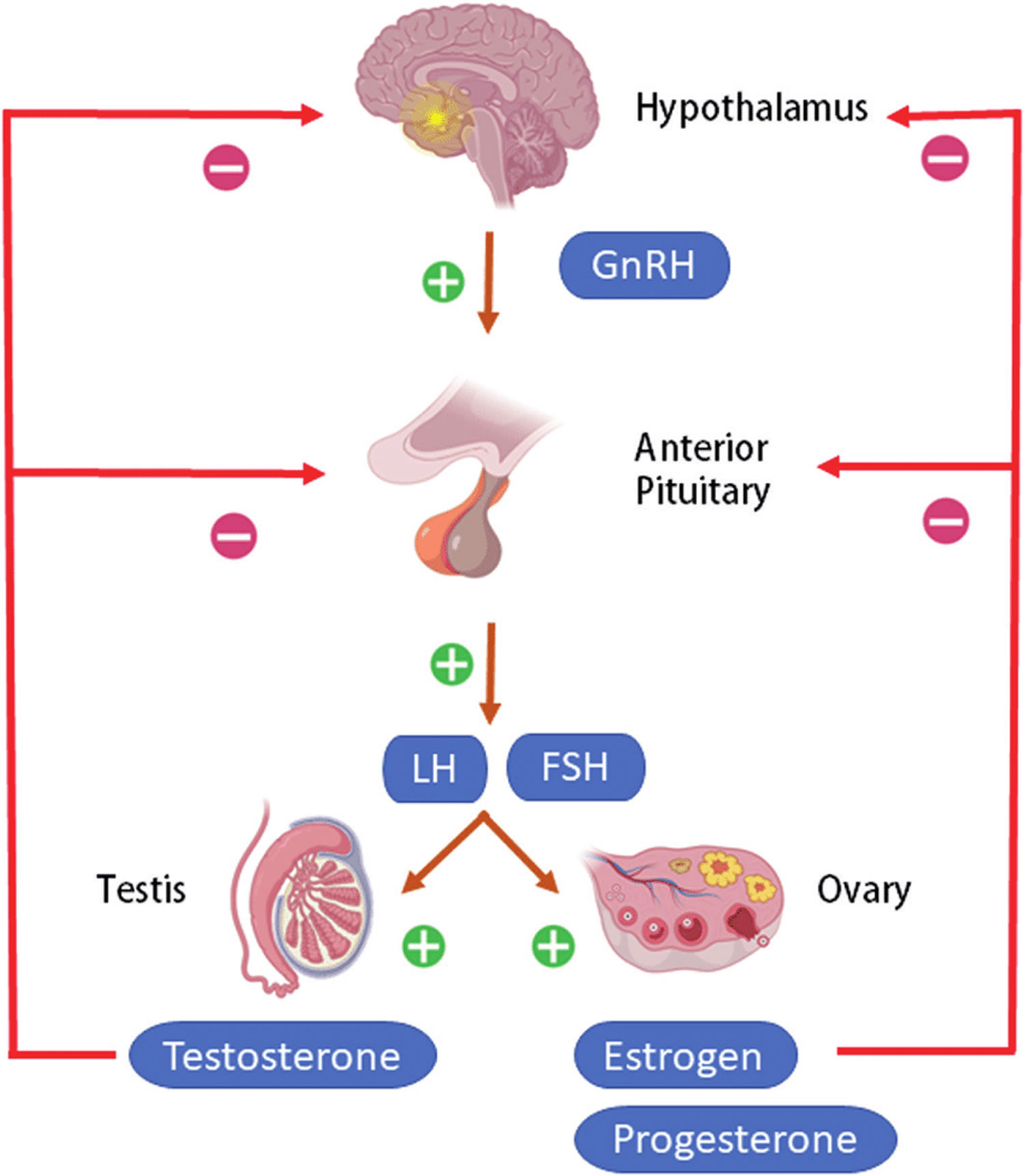
[Source 6 ]Figure 2. The hypothalamus and pituitary gland (anterior and posterior) endocrine pathways and target organs
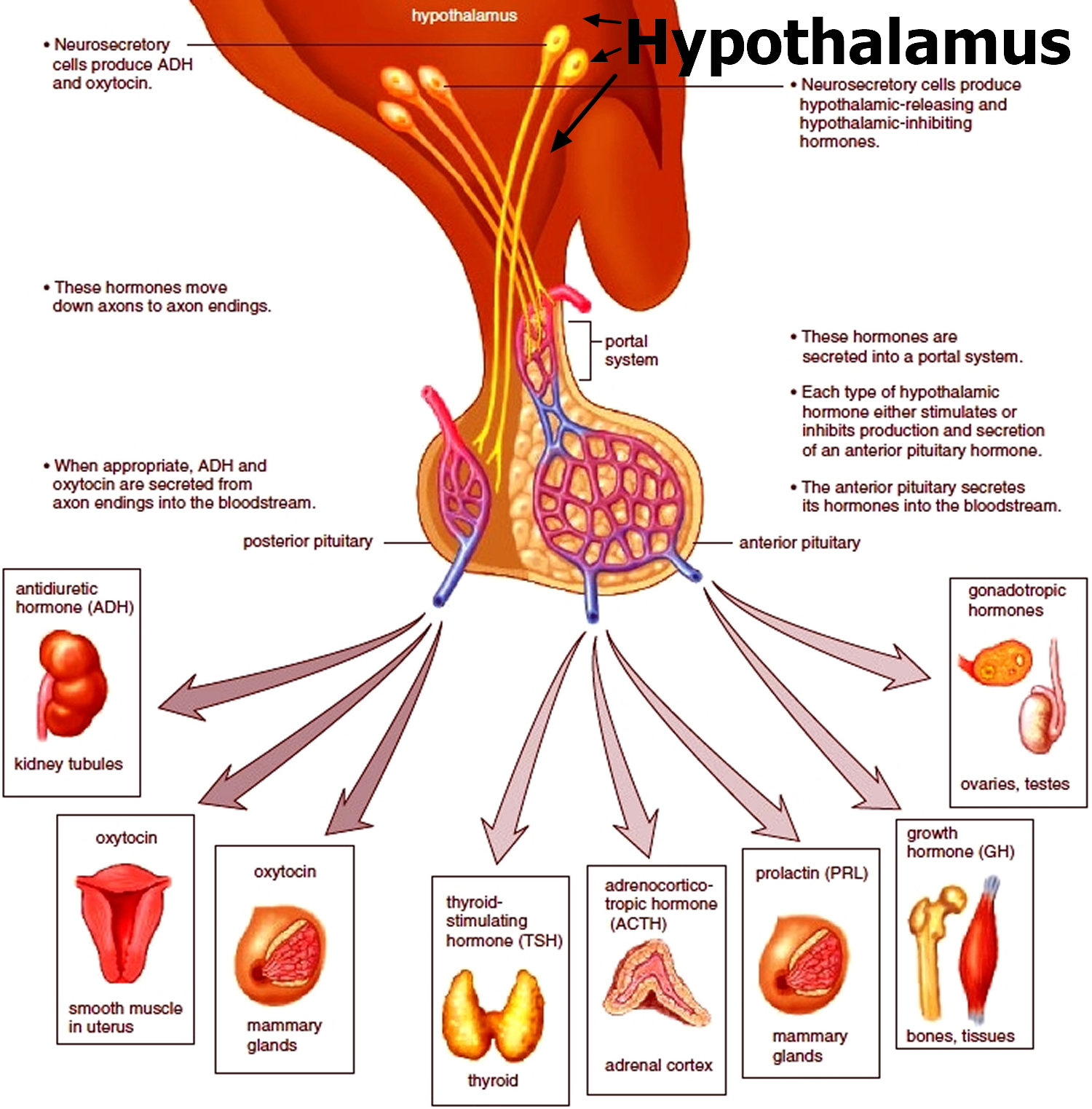
Figure 3. Testosterone production in men
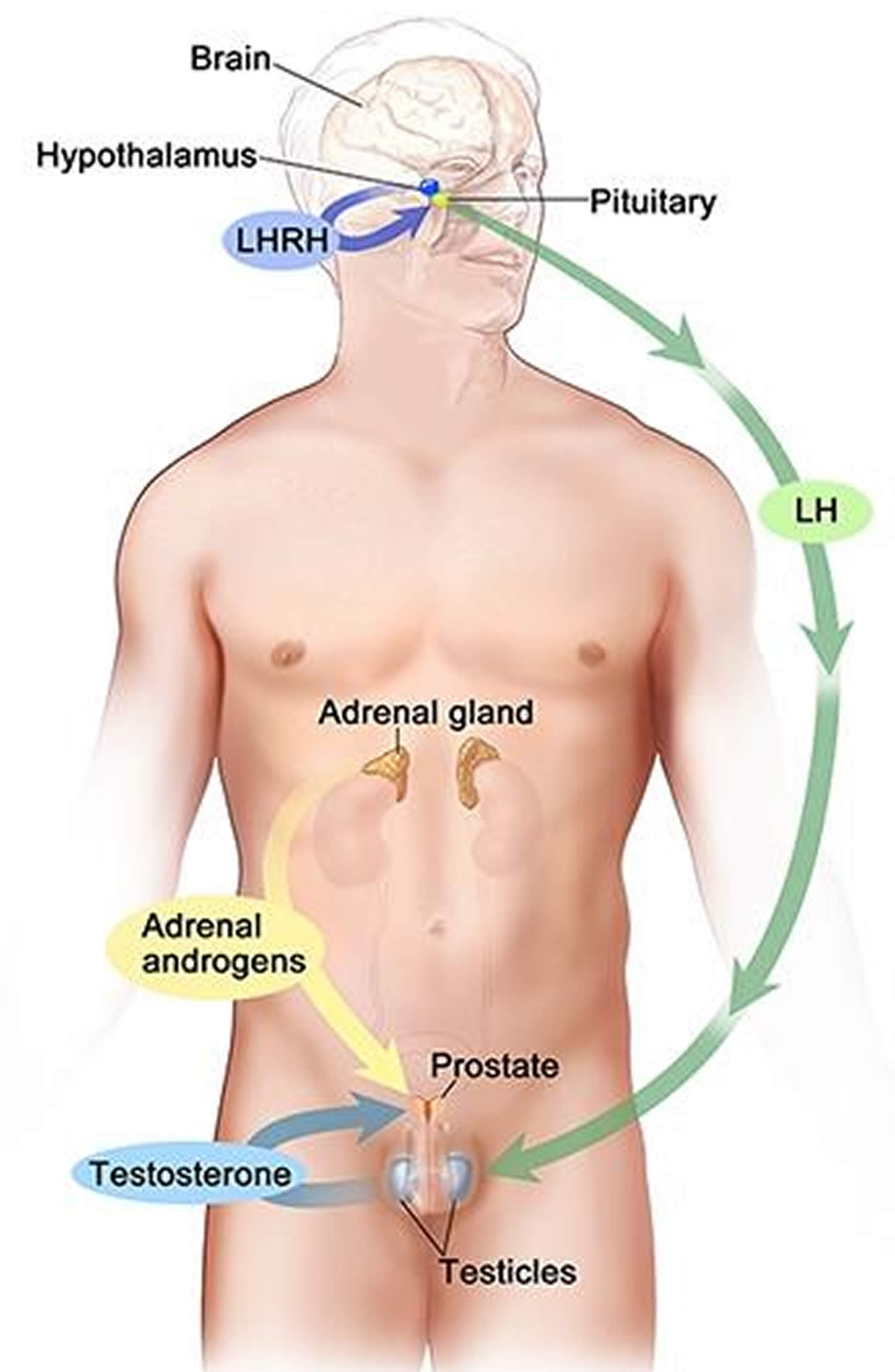
Footnotes: Drawing shows that testosterone (androgen) production is regulated by luteinizing hormone (LH) and luteinizing hormone-releasing hormone (LHRH). The hypothalamus releases luteinizing hormone-releasing hormone (LHRH), which stimulates the release of LH (luteinizing hormone) from the pituitary gland. Luteinizing hormone (LH) acts on Leydig cells in the testes to produce the majority of testosterone in the body. Most of the remaining testosterone are produced by the adrenal glands. Testosterone are taken up by prostate cells, where they either bind to the androgen receptor (AR) directly or are converted to dihydrotestosterone (DHT), which has a greater binding affinity for the androgen receptor than testosterone.
Figure 4. Estrogen and progesterone production in premenopausal women
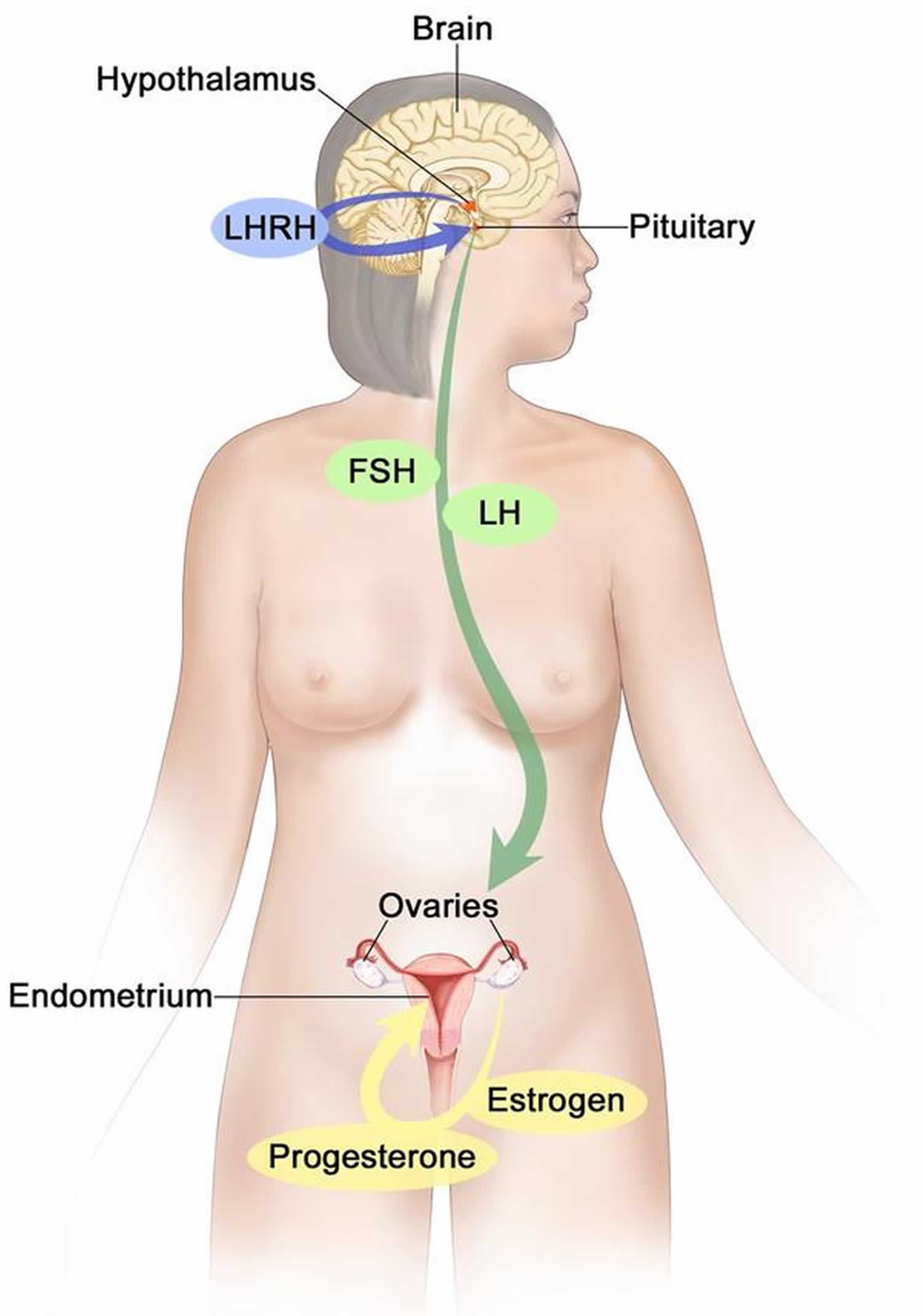
Footnotes: Drawing shows that in premenopausal women, estrogen and progesterone production by the ovaries is regulated by luteinizing hormone (LH) and luteinizing hormone-releasing hormone (LHRH). The hypothalamus releases LHRH, which then causes the pituitary gland to make and secrete LH and follicle-stimulating hormone (FSH). LH and FSH cause the ovaries to make estrogen and progesterone, which act on the endometrium (inner lining of the uterus). When estrogen and progesterone production reaches a certain level during the menstrual cycle, these hormones act on the hypothalamus and pituitary to turn off production of LHRH, LH, and FSH. Estrogen is a steroid hormone that is responsible for the growth and regulation of the female reproductive system and secondary sex characteristics. Estrogen is produced by the granulosa cells of the developing follicle and exerts negative feedback on LH production in the early part of the menstrual cycle. However, once estrogen levels reach a critical level as oocytes mature within the ovary in preparation for ovulation, estrogen begins to exert positive feedback on LH production, leading to the LH surge through its effects on GnRH pulse frequency. Estrogen also has many other effects that are important for bone health and cardiovascular health in premenopausal patients 7.
Hypogonadism causes
There are two basic types of hypogonadism:
- Primary hypogonadism also known as primary gonadal failure or HYPERgonadotropic hypogonadism originates from a problem in the testicles or ovaries.
- Secondary hypogonadism also known as HYPOgonadotropic hypogonadism indicates a problem in the hypothalamus or the pituitary gland — parts of the brain that signal the testicles to produce testosterone. The hypothalamus produces gonadotropin-releasing hormone (GnRH), which signals the anterior pituitary gland to make follicle-stimulating hormone (FSH) and luteinizing hormone (LH). Luteinizing hormone (LH) then signals the testes to produce testosterone.
Either type of hypogonadism can be caused by an inherited (congenital) trait or something that happens later in life (acquired), such as an injury or an infection. At times, primary and secondary hypogonadism can occur together.
Primary hypogonadism (Hypergonadotropic hypogonadism)
Primary hypogonadism common causes include 1:
- Klinefelter syndrome. Klinefelter syndrome results from a congenital abnormality of the sex chromosomes, X and Y. A male normally has one X and one Y chromosome. In Klinefelter syndrome, two or more X chromosomes are present in addition to one Y chromosome. The Y chromosome contains the genetic material that determines the sex of a child and related development. The extra X chromosome that occurs in Klinefelter syndrome causes abnormal development of the testicles, which in turn results in underproduction of testosterone.
- Undescended testicles (cryptorchidism). Before birth, the testicles develop inside the abdomen and normally move down into their permanent place in the scrotum. Sometimes one or both of the testicles aren’t descended at birth. This condition often corrects itself within the first few years of life without treatment. If not corrected in early childhood, it can lead to malfunction of the testicles and reduced production of testosterone.
- Mumps orchitis. A mumps infection involving the testicles that occurs during adolescence or adulthood can damage the testicles, affecting the function of the testicles and testosterone production.
- Hemochromatosis. Too much iron in the blood can cause testicular failure or pituitary gland dysfunction, affecting testosterone production.
- Injury to the testicles. Because the testicles are outside the abdomen, the testicles are prone to injury. Damage to both testicles can cause hypogonadism. Damage to one testicle might not impair total testosterone production.
- Cancer treatment. Chemotherapy or radiation therapy for the treatment of cancer can interfere with testosterone and sperm production. The effects of both treatments often are temporary, but permanent infertility may occur. Although many men regain their fertility within a few months after treatment, preserving sperm before starting cancer therapy is an option for men.
- Gonadectomy (surgical removal of either the testes in males or the ovaries in females)
- Anorchism (congenital absence of both testes)
- Testicular biosynthetic defects – 17β-hydroxylase dehydrogenase deficiency, 5α-reductase deficiency, 17-hydroxylase deficiency
- Gonadal dysgenesis is a genetic condition due to errors in cell division and or alterations in genetic material, leading to complete or partial loss of gonadal development 8. Gonadal dysgenesis can cause impaired development of the gonads, i.e., the testes or ovaries 9. The most notable of gonadal dysgenesis is Turner syndrome, a genetic disorder affecting some females that is caused by a partial or complete loss of an X chromosome (45,X syndrome). Normal females have 46, XX karyotype. Turner syndrome affects 1 in every 2500 live female births, with an array of associated symptoms and complications 10.
- Other rare disorders of sex development (DSDs) – Ovotesticular DSD, XX males
- Sertoli-cell-only syndrome
- LH resistance
- Inactivating mutations
- XX and XY gonadal dysgenesis
- Premature menopause
- Primary ovarian insufficiency
- Autoimmune oophoritis
- Resistant ovary
- Galactosemia
- Glycoprotein syndrome type 1
- FSH-receptor gene mutations
- LH/human chorionic gonadotropin (hCG) resistance
- Polycystic ovarian disease
- Noonan syndrome
Secondary hypogonadism (Hypogonadotropic hypogonadism)
In secondary hypogonadism or hypogonadotropic hypogonadism, the testicles are normal but don’t function properly due to a problem with your pituitary gland or the hypothalamus. Hypogonadotropic hypogonadism (secondary hypogonadism) is caused by a lack of gonadotropin-releasing hormone (GnRH), follicle stimulating hormone (FSH) and luteinizing hormone (LH) that normally stimulate the ovaries or testes. A number of conditions can cause hypogonadotropic hypogonadism 11:
- Kallmann’s syndrome. Kallmann’s syndrome is an abnormal development of the area of the brain called the hypothalamus that controls the secretion of pituitary hormones. This abnormality can also affect the ability to smell (anosmia) and cause red-green color blindness.
- Pituitary disorders. An abnormality in the pituitary gland can impair the release of hormones from the pituitary gland to the testicles, affecting normal testosterone production. A pituitary tumor or other type of brain tumor located near the pituitary gland may cause testosterone or other hormone deficiencies. Also, treatment for a brain tumor, such as surgery or radiation therapy, can affect the pituitary gland and cause hypogonadism.
- Congenital disorders such as Prader-Willi syndrome, Laurence-Moon syndrome, Bardet-Biedl syndrome 12 and Gaucher disease
- High prolactin level (a different hormone released by the pituitary)
- Inflammatory disease. Certain inflammatory diseases, such as sarcoidosis, histiocytosis and tuberculosis, involve the hypothalamus and pituitary gland and can affect testosterone production.
- HIV/AIDS. HIV/AIDS can cause low levels of testosterone by affecting the hypothalamus, the pituitary and the testes.
- Medications. The use of certain drugs, such as opioid pain medications and some steroid (glucocorticoid) medicines, can affect testosterone production.
- Drug use, such as heroin
- Obesity. Being significantly overweight at any age might be linked to hypogonadism.
- Aging. As men age, there’s a slow, progressive decrease in testosterone production. The rate varies greatly.
- Severe stress
- Nutritional problems (both rapid weight gain or weight loss).
Hypogonadism risk factors
Risk factors for hypogonadism include:
- HIV/AIDS
- Damage to the pituitary gland or hypothalamus from surgery, injury, tumor, infection, or radiation
- Previous chemotherapy or radiation therapy
- Aging
- Obesity
- Malnutrition
Hypogonadism can be inherited. If any of these risk factors are in your family health history, tell your doctor.
Hypogonadism signs and symptoms
Hypogonadism can begin during fetal development, before puberty or during adulthood. Signs and symptoms depend on when the condition develops.
For both males and females with hypogonadism, determining whether evidence of a genital abnormality is present at birth or determining the timing and extent of puberty is important. In addition, because Kallmann syndrome (hypogonadotropic hypogonadism and anosmia [lack of a sense of smell]) is a common cause of hypogonadotropic hypogonadism, inquiring about the sense of smell is important.
For females with hypogonadism, determine the age of the first menstrual period (menarche). Menstrual history is important in postpubertal females.
For postpubertal females, inquire about the presence of galactorrhea or milk production from the breast unrelated to pregnancy or breastfeeding. For postpubertal females symptoms suggestive of androgen (testosterone) excess, such as acne or excessive male-pattern hair growth in women after puberty (hirsutism) such as mustache and beard, pubic hair, buttocks, and thighs.
For prepubertal males or females with delayed puberty such as the lack of sexual characteristics by age 13 years in females or ages 13-14 years in males; also, the absence of menstruation at age 16 years, inquire about a family history of constitutional delay of growth and development. Constitutional delay in pubertal development is the most frequent clinical scenario.
Inquire about chronic illness including frequent headaches, intentional or unintentional weight loss, and strenuous exercise.
Hypogonadism signs and symptoms during Fetal development
If the body doesn’t produce enough testosterone during fetal development, the result may be impaired growth of the external sex organs. Depending on when hypogonadism develops and how much testosterone is present, a child who is genetically male may be born with:
- Female genitals
- Genitals that are neither clearly male nor clearly female (ambiguous genitals)
- Underdeveloped male genitals
Hypogonadism signs and symptoms during Puberty
Male hypogonadism can delay puberty or cause incomplete or lack of normal development. It can hamper:
- Development of muscle mass
- Voice deepening
- Growth of body and facial hair
- Growth of the penis and testicles
And it can cause:
- Excessive growth of the arms and legs in relation to the trunk of the body
- Development of breast tissue (gynecomastia)
Hypogonadism signs and symptoms in Adult Males
In adult males, hypogonadism can alter certain masculine physical characteristics and impair normal reproductive function. Early signs and symptoms might include:
- Decreased sex drive (low libido)
- Decreased energy
- Depression
Over time, men with hypogonadism can develop:
- Erectile dysfunction (not being able to obtain or keep an erection that is sufficient for sexual intercourse)
- Infertility
- Decrease in hair growth on the face and body
- Decrease in muscle mass
- Development of breast tissue (gynecomastia)
- Loss of bone mass (osteoporosis)
Severe hypogonadism can also cause mental and emotional changes. As testosterone decreases, some men have symptoms similar to those of menopause in women. These can include:
- Difficulty concentrating
- Hot flashes
Hypogonadism signs and symptoms in Adult Females
Hypogonadism in females can affect the ovaries and their secretion of hormones. Hypogonadism in females can cause:
- Delayed puberty
- Lack of menstruation (amenorrhea)
- Absence of breast development
- Absence of pubic hair
- Vaginal agenesis
- Hot flashes
- Breast discharge
- Short stature
- Infertility
Turner syndrome
The symptoms and severity of Turner syndrome can be quite variable from one person to another. Many features of the disorder are nonspecific, and others may develop slowly over time or can be subtle. It is important to note that affected individuals may not have all the symptoms outlined below. Affected individuals should talk to their physician and medical team about their specific case, associated symptoms and overall prognosis.
Almost all individuals with Turner syndrome exhibit growth failure and attain a final height that is shorter than average (short stature). Children may initially display normal growth, usually for the first few years of life. However, in most cases, the growth rate eventually becomes slower than normal and affected children do not experience normal growth spurts (e.g., no growth spurt during puberty). If untreated, the final height in Turner syndrome is usually less than 5 feet.
Another common feature of Turner syndrome is the failure of the ovaries to develop properly (gonadal dysgenesis). Gonadal dysgenesis can cause the loss of ovarian function early during childhood (premature ovarian failure). Normally, the ovaries produce sex hormones (e.g., estrogen and progesterone) at puberty. These hormones are necessary for the onset of puberty and the proper development of secondary sexual characteristics. Most affected females will require hormone replacement therapy to develop breasts and normal female body contours, undergo proper bone growth and to begin menstruation. Some affected individuals may begin to undergo breast development and may begin menstruating without therapy (spontaneous pubertal development), but most will stop developing sexually and stop menstruating at some later point during their teens or early adulthood.
Intelligence is usually normal in females with Turner syndrome. However, affected females may develop learning disabilities, especially difficulties with visual-spatial relationships. An example would be right-left disorientation. Affected individuals may have difficulties with directional sense, learning math, nonverbal memory and attention. Affected females may also have trouble in certain social situations.
Females with Turner syndrome may develop a variety of distinctive physical features including a short neck with a webbed appearance, a low hairline at the back of the head, low-set ears and narrow fingernails and toenails that are turned upward. A broad chest with widely spaced nipples may occur, which is sometimes referred to as “shield chest.” Some individuals may have swollen, puffy hands and feet. These symptoms may occur due to lymphedema, a condition affecting the lymphatic system. The lymphatic system is a circulatory network of vessels, ducts and nodes that filter and distribute certain protein-rich fluid (lymph) and blood cells throughout the body. Lymphedema is characterized by swelling due to fluid accumulation (edema) in the affected parts of the body.
Additional physical findings may include a receding jaw (retrognathia), crossed eyes (strabismus), lazy eyes (amblyopia), drooping eyelids (ptosis) and a narrow, high-arched roof of the mouth (palate). Some individuals may have skeletal malformations including short bones of the hands, specifically the fourth metacarpals, arms that are turned out at the elbows and flat feet (pes planus). In approximately 10% of people with Turner syndrome, abnormal sideways curvature of the spine (scoliosis) may also occur.
Congenital heart defects may be associated with Turner syndrome, especially in individuals with lymphedema. Such defects may include bicuspid aortic valve, in which the aortic valve has two flaps (leaflets) instead of three. The aorta is the main artery of the heart. The aortic valve regulates blood flow from the heart into the aorta. The flaps open and close to allow the passage of blood. Since there are only two flaps instead of three, the aortic valve does not function properly. A bicuspid aortic valve may or may not cause clinically apparent symptoms. Approximately 5-10% of individuals may have a congenital heart defect known as coarctation of the aorta, a condition characterized by narrowing of the aorta, which causes the heart to pump harder to force blood through constricted area. The condition can be mild and go undiagnosed until adulthood or be more serious, which can be associated with a variety of symptoms including pale skin, irritability, heavy sweating and difficulty breathing. If untreated, severe cases can result in insufficient blood flow to the organs of the body or eventually progress to congestive heart failure.
The heart defects associated with some cases of Turner syndrome can increase the risk of severe, life-threatening complications including high blood pressure of the arteries of the lungs (pulmonary hypertension) or aortic dissection, a condition in which there is a tear in the inner wall of the aorta. Blood rushes into the middle layer of the aorta causing the middle and inner layers to separate (dissect). Aortic dissection can potentially cause the outer wall of the aorta to rupture.
Kidney (renal) abnormalities may occur in some people with Turner syndrome including horseshoe kidneys or absence (agenesis) of a kidney. Kidney abnormalities increase the risk of urinary tract infections and high blood pressure (hypertension).
Liver abnormalities may include a fatty liver.
Some affected individuals may have thyroid disease, which can cause decreased activity of the thyroid (hypothyroidism). Thyroid disease usually occurs because the immune system mistakenly attacks thyroid tissue, a condition known as Hashimoto’s syndrome (autoimmune thyroiditis). Symptoms can vary from one person to another, but can include fatigue, sluggishness, muscle aches, constipation, a hoarse voice and pale, dry skin.
Some individuals with Turner syndrome may have multiple tiny colored spots (pigmented nevi) on their skin. This has not been correlated with an increase in skin cancer.
Affected females may also be prone to infections of the middle ear (otitis media), especially during infancy and early childhood. Chronic otitis media may be associated with hearing loss due to blockage of sound waves (conductive hearing loss). This hearing loss usually resolves as a child ages and ear infections become less frequent. Hearing abnormalities in young children may affect or delay speech development. In adults, hearing loss due to an impaired ability of the auditory nerves to transmit sensory input to the brain (sensorineural hearing loss) may occur and may worsen with age.
Certain individuals with Turner syndrome appear to be at greater risk than the general population for developing certain disorders including diabetes, celiac disease and osteoporosis. Osteoporosis is characterized by a general loss of bone density that can lead to an increased risk of fractures. Gastrointestinal problems including feeding difficulties and gastroesophageal reflux (GERD) may also occur.
Hypogonadism complications
The complications of untreated hypogonadism differ depending on when it develops — during fetal development, puberty or adulthood.
Hypogonadism complications may include:
- Abnormal genitalia
- Enlarged male breasts (gynecomastia)
- Infertility
- Erectile dysfunction (not being able to obtain or keep an erection that is sufficient for sexual intercourse)
- Osteoporosis
- Poor self-image
Early hypogonadism detection in boys can help prevent problems from delayed puberty. Early diagnosis and treatment in men offer better protection against osteoporosis and other related conditions.
Hypogonadism diagnosis
Your doctor will conduct a physical exam and note whether your sexual development, such as your pubic hair, muscle mass and size of your testes, is consistent with your age.
Your doctor will test your blood level of testosterone if you have signs or symptoms of hypogonadism. Because testosterone levels vary and are generally highest in the morning, blood testing is usually done early in the day, before 10 a.m., possibly on more than one day.
If tests confirm that you have low testosterone, further testing can determine if a testicular disorder or a pituitary abnormality is the cause. These studies might include:
- Hormone testing
- Semen analysis
- Pituitary imaging
- Genetic studies
- Testicular biopsy
If hypogonadism is suspected, measurement of serum LH and FSH concentrations can be used to distinguish between hypogonadotropic hypogonadism (problem in the hypothalamus or the pituitary gland) and hypergonadotropic hypogonadism (primary gonadal failure) and guide further evaluation and management. Serum LH and FSH levels are elevated in cases of hypergonadotropic hypogonadism (primary gonadal failure).
Further hormonal testing (ie, measurement of serum prolactin or thyroid function tests) can be ordered based on presentation and history. Obtaining a karyotype can be considered in certain cases.
In the absence of specific symptoms or signs to direct the workup, evaluation for chronic illness includes a complete white blood count, a sedimentation rate, a comprehensive metabolic panel, a celiac screening, thyroid function tests, and a urinalysis.
Males physical exam
Examination of the testes is the most important feature of the physical examination. Determine whether both testes are palpable, their position in the scrotum, and their consistency. Testes size can be quantitated by comparison with testicular models (orchidometer), or their length and width may be measured. Before puberty, testes usually are 1-3 cm³ in volume (approximately 2 cm in length). During puberty, testes grow up to 25 cm³ in size.
Examining the genitalia for hypospadias (a condition in which the opening of the penis is on the underside rather than the tip) is the next important step. Check the scrotum to see if it is completely fused. Hypospadias is usually not related to an endocrine abnormality, but it may be seen in disorders associated with a testosterone biosynthesis defect, partial androgen insensitivity syndrome, or a defect in testicular determination. Finally, evaluate the extent of the biological development of adult male characteristics in young males (virilization).
The presence of micropenis (an atypically small penis that’s diagnosed at birth) suggests Kallmann syndrome or panhypopituitarism.
Puberty should be staged using the Tanner criteria for genitalia, pubic hair, and axillary hair.
Look for signs of Klinefelter syndrome, such as tall stature (especially if the legs are disproportionately long), gynecomastia, small or soft testes, and a eunuchoid body habitus.
Males Lab Tests
For males after puberty, the guidelines of the Endocrine Society require that the diagnosis of hypogonadism be based on symptoms and signs of hypogonadism plus the presence of a low testosterone level measured on at least two occasions 13. Because of the circadian variation in testosterone levels, a morning sample is recommended.
For postpubertal males with total testosterone concentrations near the lower limit of the normal range, measurement of free or bioavailable testosterone using a reliable assay is suggested if there is suspicion of sex hormone binding globulin (SHBG) changes 13.
Obesity decreases serum sex hormone binding globulin (SHBG) concentrations to a degree that is proportionate to the severity of the obesity, thereby decreasing total testosterone values without usually affecting free testosterone. However, morbid obesity (BMI >40 kg/m²) may be associated with a low free testosterone concentration because of hypothalamic hypogonadism. Decreased sex hormone binding globulin (SHBG) concentrations are also seen in insulin resistance, type 2 diabetes, hypothyroidism, glucocorticoid use, and nephrotic syndrome. Increased sex hormone binding globulin (SHBG) concentrations can occur with aging, hyperthyroidism, liver disease, and anticonvulsant use.
Examination of seminal fluid analysis, karyotyping (a type of genetic testing that looks at the size, shape, and number of chromosomes in a sample of cells from your body), and testicular biopsy may also be helpful.
Females physical exam
Examination of the genitalia is important. Determine the extent of the development of secondary male sexual characteristics, which may be adrenal or ovarian in origin and is demonstrated in pubic and armpit hair.
Determine the extent of estrogenization, as evidenced by breast development and maturation of the vaginal mucosa.
Look for signs of Turner syndrome, such as short stature, webbing of the neck (eg, pterygium colli), a highly arched palate, short fourth metacarpals, widely spaced nipples, or multiple pigmented nevi.
Females Lab Tests
Karyotyping (a type of genetic testing that looks at the size, shape, and number of chromosomes in a sample of cells from your body) may be helpful. In females with normal karyotype and elevated gonadotropin levels, measure antiovarian antibody levels to rule out autoimmune disease.
In postpubertal females with signs of acne, hirsutism, and/or amenorrhea or irregular menses, measurement of total and free testosterone and 17-hydroxyprogesterone concentrations is indicated.
Imaging Studies
Magnetic resonance imaging (MRI) of the brain should be considered in cases of loss of smell (anosmia) and suspected hypogonadotropic hypogonadism (problem in the hypothalamus or the pituitary gland). Absence of the olfactory bulbs is associated with Kallmann syndrome.
Furthermore, brain MRI should be ordered in cases of hypogonadotropic hypogonadism that are either isolated or occurring in combination with pituitary defects.
MRI of the pelvis is usually performed in disorders of sex development (DSD) cases, such as androgen insensitivity or ovotesticular DSD, to help delineate the anatomy of internal genitalia. Uterine aplasia in a pubertal girl with normal gonadal function and amenorrhea can be seen in Mayer-Rokitansky-Küster-Hauser syndrome.
Pelvic ultrasonography may be helpful in females.
Bone age may be helpful in the evaluation of adolescents with delayed puberty and provides insight into their growth potential.
Other Tests
Adrenocorticotropic hormone (ACTH) stimulation testing: In patients in whom a form of congenital adrenal hyperplasia is suspected, adrenal steroid synthesis is best evaluated by performing a cosyntropin (ACTH 1-24) stimulation test. Baseline serum adrenocortical hormone levels are measured, then 0.25 mg of cosyntropin is intravenously injected, and serum hormone levels are remeasured after 60 minutes. Precursor product ratios are compared with those in age-matched control subjects to determine whether a steroidogenic defect is involved in sex hormone synthesis.
Luteinizing-hormone releasing hormone (LHRH) stimulation testing: To distinguish between true hypogonadotropic hypogonadism and constitutional delay in growth and maturation, performing a stimulation test with LHRH may be helpful.
- LHRH is intravenously injected, and LH and FSH levels are determined at 15-minute intervals following LHRH administration.
- A shortened version of the study has been used, in which LHRH is subcutaneously injected, and the specimen for LH and FSH levels is taken at 30-40 minutes.
- Obtaining LHRH for testing over the past several years has been difficult. Some centers have substituted testing LH response to aqueous leuprolide.
Testicular tissue testing: If testes are not palpable and whether any testicular tissue is present is unclear, administering human chorionic gonadotropin (hCG) and measuring testosterone response may be helpful.
Procedures
In prepubertal males with delayed puberty, priming with testosterone (usually testosterone enanthate 50 mg IM monthly for a total of 3 months) may lead to puberty initiation and help in the differential diagnosis of hypogonadotropic hypogonadism.
In postpubertal females with amenorrhea, withdrawal bleeding after a 5-10 day course of progestin (such as medroxyprogesterone 10mg hs) indicates adequate estrogen secretion and implies intact gonadal function.
Occasionally, testicular biopsy findings are helpful, particularly if azoospermia or oligospermia is present.
Hypogonadism treatments
Patients with hypogonadism are typically treated with sex steroid replacement. The goals of treatment are:
- To promote the development of and maintain secondary sexual characteristics and normal sexual function
- To build and sustain normal bone and muscle mass
- To assist in the proper psychosocial adjustment of adolescents with hypogonadism
Fertility options can be explored in consultation with a reproductive endocrinologist or urologist. Pulsatile luteinizing-hormone releasing hormone (LHRH) or gonadotropin therapy can induce fertility in individuals with hypogonadotropic hypogonadism.
Patients with hypergonadotropic hypogonadism are traditionally considered infertile. However, men with Klinefelter syndrome may benefit from a consultation with a reproductive urologist and testicular sperm extraction followed by in vitro fertilization (IVF). This technique has allowed men with Klinefelter syndrome to father children. For boys with Klinefelter syndrome who have reached puberty, cryopreservation of semen samples containing very low numbers of spermatozoa is possible and should be offered before testosterone supplementation, since supplementation may suppress spermatogenesis.
Male hypogonadism treatment
Male hypogonadism usually is treated with testosterone replacement to return testosterone levels to normal. Testosterone can help counter the signs and symptoms of male hypogonadism, such as decreased sexual desire, decreased energy, decreased facial and body hair, and loss of muscle mass and bone density.
For older men who have low testosterone and signs and symptoms of hypogonadism due to aging, the benefits of testosterone replacement are less clear.
While you’re taking testosterone, the Endocrine Society recommends that your doctor monitor you for treatment effectiveness and side effects several times during your first year of treatment and yearly after that.
Types of testosterone replacement therapy
Several testosterone formulations are available. Direct comparisons among different testosterone products are still lacking. Patients who are considering testosterone therapy should be adequately informed about the possible risks and benefits of all available testosterone preparations. The final choice should be based on the clinical situation, testosterone formulation availability, and patient needs and expectations 14, 15.
One Food and Drug Administration-approved oral testosterone replacement preparation, testosterone undecanoate (Jatenzo), is absorbed by the lymph system. It might avoid the liver problems seen with other oral forms of testosterone.
Other preparations you might choose, depending on convenience, cost and your insurance coverage, include:
- Testosterone Gel. There are several gels and solutions available, with different ways of applying them. Depending on the brand, you rub the testosterone into your skin on your upper arm or shoulder (AndroGel, Testim, Vogelxo) or apply it to the front and inner thigh (Fortesta). Your body absorbs testosterone through your skin. Don’t shower or bathe for several hours after a gel application, to be sure it gets absorbed. Side effects include skin irritation and the possibility of transferring the medication to another person. Avoid skin-to-skin contact until the gel is completely dry, or cover the area after an application.
- Testosterone Injection. Testosterone cypionate (Depo-Testosterone) and testosterone enanthate are given in a muscle or under the skin. Your symptoms might waver between doses depending on the frequency of injections. You or a family member can learn to give testosterone injections at home. If you’re uncomfortable giving yourself injections, member of your care team can give the injections.
- Testosterone undecanoate (Aveed) is given by deep intramuscular injection, typically every 10 weeks. It must be given at your provider’s office and can have serious side effects.
- Testosterone Patch. A patch containing testosterone (Androderm) is applied each night to your thighs or torso. A possible side effect is severe skin reaction.
- Testosterone Pellets (Testopel): Testosterone-containing pellets are surgically implanted under the skin every three to six months for consistent and long-term dosages. Side effects: pellet coming out through skin, site infection/ bleeding (rare), dose decreasing over time and hypogonadism symptoms possibly returning towards the end of dose period.
- Testosterone Gum and Cheek (buccal cavity). A small putty-like substance, gum-and-cheek testosterone replacement delivers testosterone through the natural depression above your top teeth where your gum meets your upper lip (buccal cavity). This product, taken three times a day, sticks to your gumline and allows testosterone to be absorbed into your bloodstream. It can cause gum irritation.
- Testosterone Nasal. This testosterone gel (Natesto) can be pumped into the nostrils. This option reduces the risk that medication will be transferred to another person through skin contact. Nasal-delivered testosterone must be applied twice in each nostril, three times daily, which might be more inconvenient than other delivery methods.
Testosterone therapy carries various risks, including:
- Increased production of red blood cells (polycythemia or erythrocytosis)
- Acne
- Enlarged breasts
- Sleep disturbances
- Prostate enlargement
- Limited sperm production
Treatment of infertility due to hypogonadism
If a pituitary problem is the cause of your hypogonadism, pituitary hormones can be given to stimulate sperm production and restore fertility. A pituitary tumor may require surgical removal, medication, radiation or the replacement of other hormones.
There’s often no effective treatment to restore fertility in men with primary hypogonadism (primary testicular failure), but assisted reproductive technology (ART) may be helpful. This technology covers a variety of techniques designed to help couples who have been unable to conceive.
Treatment for boys
Treatment of delayed puberty in boys depends on the underlying cause. Three to six months of testosterone supplementation given as an injection can stimulate puberty and the development of secondary sex characteristics, such as increased muscle mass, beard and pubic hair growth, and growth of the penis.
Hypogonadotropic hypogonadism treatment
Hypogonadotropic hypogonadism treatment depends on the source of the problem, but may involve:
- Injections of testosterone (in males)
- Slow-release testosterone skin patch (in males)
- Testosterone gels (in males)
- Estrogen and progesterone pills or skin patches (in females)
- Gonadotropin-releasing hormone (GnRH) injections
- Human chorionic gonadotropin (hCG) injections
Consultation with a reproductive endocrinologist or urologist is required for patients with hypogonadotropic hypogonadism who would like to become fertile. Administration of pulsatile LHRH or gonadotropins in females results in ovulation in 95% of the cases. In males, pulsatile LHRH therapy or hCG alone or in combination with gonadotropins can induce spermatogenesis and results in normal adult male testosterone levels.
Surgical care
Because of the significant risk of gonadoblastoma (a rare benign gonadal tumor) and cancer, gonadal tissue should be removed in females with karyotypes containing a Y chromosome. This situation is observed in females with XY gonadal dysgenesis or in patients with Turner syndrome who have a karyotype that contains a Y chromosome (usually in 1 of 2 or more mosaic karyotypes). Males with nonfunctioning testicular tissue should undergo orchiectomy (a surgical procedure to remove one or both testicles) and replacement with prostheses.
Long-Term Monitoring
Patients with hypogonadism require lifelong treatment, with the exception of persons with congenital hypogonadotropic hypogonadism (spontaneous recovery having been described in 10-20% of these individuals) 16. Patients with hypogonadism receiving hormone replacement therapy are typically evaluated every 6-12 months. Monitoring may include measurement of testosterone concentrations in males, evaluation of bone mass by dual radiographic absorptiometry, and assessment of cardiovascular risk factors.
An increase in red blood cell count (polycythemia) can be a complication of testosterone replacement. For older adult men with testosterone deficiency, the Endocrine Society clinical guidelines recommend monitoring hematocrit values to avoid polycythemia. Also in these individuals, prostate examination and prostate-specific antigen (PSA) measurements should be performed before testosterone therapy and periodically after treatment is instituted 13. Referral to a urologist can be considered based on individual assessment for prostate cancer.
Hypogonadism prognosis
In men, complications of untreated hypogonadism include loss of libido, failure to achieve physical strength, the social implications of failing to go through puberty with peers (if hypogonadism occurs before puberty), and osteoporosis. In addition, if hypogonadism occurs before epiphyseal closure, the result is usually tall stature with a eunuchoid body habitus. Males with hypergonadotropic hypogonadism are typically infertile, although procedures such as testicular sperm extraction have resulted in fertility in Klinefelter syndrome. Men who have hypogonadotropic hypogonadism due to hypothalamic or pituitary dysfunction can potentially become fertile with administration of gonadotropins.
A retrospective study by Baillargeon et al 17 indicated that males with untreated hypogonadism are at increased risk for the development of any rheumatic autoimmune disease, as well as for rheumatoid arthritis and lupus.
In women with hypogonadism, complications include the social implication of failing to go through puberty with peers (if hypogonadism occurs before puberty). An additional concern for untreated women is osteoporosis, which can be avoided with estrogen replacement. Women who have hypogonadotropic hypogonadism because of hypothalamic or pituitary dysfunction can potentially become fertile with administration of gonadotropins. Women with primary hypogonadism (hypergonadotropic hypogonadism) are infertile; however, with in vitro fertilization (IVF) using a donor ovum, these women can carry an infant to term.
Osteoporosis has an earlier onset in individuals with hypogonadism; hence, bone mineral density should be compared with age-matched normative standards, and followed longitudinally. Prescribe treatment using appropriate therapeutic interventions.
- Hypogonadism. https://emedicine.medscape.com/article/922038-overview[↩][↩]
- Hayes F, Dwyer A, Pitteloud N. Hypogonadotropic Hypogonadism (HH) and Gonadotropin Therapy. [Updated 2013 Nov 25]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279078[↩][↩][↩][↩]
- Fraietta R, Zylberstejn DS, Esteves SC. Hypogonadotropic hypogonadism revisited. Clinics (Sao Paulo). 2013;68 Suppl 1(Suppl 1):81-8. doi: 10.6061/clinics/2013(sup01)09[↩]
- Clavijo RI, Hsiao W. Update on male reproductive endocrinology. Transl Androl Urol. 2018 Jul;7(Suppl 3):S367-S372. doi: 10.21037/tau.2018.03.25[↩]
- Canavese F, Mussa A, Manenti M, Cortese MG, Ferrero L, Tuli G, Macchieraldo R, Lala R. Sperm count of young men surgically treated for cryptorchidism in the first and second year of life: fertility is better in children treated at a younger age. Eur J Pediatr Surg. 2009 Dec;19(6):388-91. doi: 10.1055/s-0029-1241171[↩]
- Gupta, Priya & Mahapatra, Archisman & Suman, Anjali & Singh, Rahul. (2021). Effect of Endocrine Disrupting Chemicals on HPG Axis: A Reproductive Endocrine Homeostasis. https://www.researchgate.net/publication/350037153_Effect_of_Endocrine_Disrupting_Chemicals_on_HPG_Axis_A_Reproductive_Endocrine_Homeostasis[↩]
- Tsutsumi R, Webster NJ. GnRH pulsatility, the pituitary response and reproductive dysfunction. Endocr J. 2009;56(6):729-37. doi: 10.1507/endocrj.k09e-185[↩]
- Breehl L, Caban O. Genetics, Gonadal Dysgenesis. [Updated 2023 Aug 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539886[↩]
- Rocha VB, Guerra-Júnior G, Marques-de-Faria AP, de Mello MP, Maciel-Guerra AT. Complete gonadal dysgenesis in clinical practice: the 46,XY karyotype accounts for more than one third of cases. Fertil Steril. 2011 Dec;96(6):1431-4. doi: 10.1016/j.fertnstert.2011.09.009[↩]
- Chacko E, Graber E, Regelmann MO, Wallach E, Costin G, Rapaport R. Update on Turner and Noonan syndromes. Endocrinol Metab Clin North Am. 2012 Dec;41(4):713-34. doi: 10.1016/j.ecl.2012.08.007[↩]
- Hypogonadotropic hypogonadism. https://medlineplus.gov/ency/article/000390.htm[↩]
- Abdulla AB, Niloy AA, Shah TA, Biswas SK, Imran AK, Murshed KM, Ahmed M. Laurence Moon Bardet Biedl Syndrome. Mymensingh Med J. 2009 Jan;18(1 Suppl):S124-128.[↩]
- Bhasin S, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, Montori VM; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010 Jun;95(6):2536-59. doi: 10.1210/jc.2009-2354. Erratum in: J Clin Endocrinol Metab. 2021 Jun 16;106(7):e2848. doi: 10.1210/clinem/dgab311[↩][↩][↩]
- Rastrelli G, Vignozzi L, Corona G, Maggi M. Pharmacotherapy of male hypogonadism. Curr Opin Pharmacol. 2023 Feb;68:102323. doi: 10.1016/j.coph.2022.102323[↩]
- Rastrelli G, Maggi M, Corona G. Pharmacological management of late-onset hypogonadism. Expert Rev Clin Pharmacol. 2018 Apr;11(4):439-458. doi: 10.1080/17512433.2018[↩]
- Hypogonadism Treatment & Management. https://emedicine.medscape.com/article/922038-treatment#d6[↩]
- Baillargeon J, Al Snih S, Raji MA, Urban RJ, Sharma G, Sheffield-Moore M, Lopez DS, Baillargeon G, Kuo YF. Hypogonadism and the risk of rheumatic autoimmune disease. Clin Rheumatol. 2016 Dec;35(12):2983-2987. doi: 10.1007/s10067-016-3330-x[↩]




{kind=link}