
Contents
What is Krabbe disease
Krabbe disease (also called Globoid cell leukodystrophy) is rare inherited metabolic disorder where there is a lack of an enzyme called galactosylceramide beta-galactosidase (galactocerebrosidase); essential enzyme for myelin metabolism. Your body needs this galactocerebroside beta-galactosidase (galactosylceramidase) enzyme to make myelin. Galactosylceramide beta-galactosidase (galactosylceramidase) also degrades galactosylceramide, a major component of myelin, and other terminal beta-galactose–containing sphingolipids, including psychosine (galactosylsphingosine). Increased psychosine levels are believed to lead to widespread destruction of oligodendroglia in the central nervous system and to subsequent demyelination 1.
Myelin surrounds and protects nerve fibers. Without the galactocerebroside beta-galactosidase (GALC) enzyme, myelin breaks down, brain cells die, and nerves in the brain and other body areas do not work properly. Furthermore, without enough galactosylceramide beta-galactosidase (galactocerebrosidase), harmful amounts of lipids (psychosine) build up in various cells and tissues in the body and destroys brain cells. The build-up of these substances damages the nerve cells in the central nervous system, destroying many of them and preventing the repair of others. This can result in progressive damage to the central and peripheral nervous systems resulting in weakness, abnormal muscle tone, immobility, blindness, deafness,and seizures.
Krabbe disease is part of a group of disorders known as leukodystrophies, which result from the loss of myelin (demyelination) in the nervous system. Myelin is the protective covering around nerve cells that ensures the rapid transmission of nerve signals. Krabbe disease is also characterized by abnormal cells in the brain called globoid cells (cells that have more than one nucleus), which are large cells that usually have more than one nucleus that break down the nerve’s protective myelin coating.
Krabbe disease is inherited, which means it is passed down through families. If both parents carry the nonworking copy of the gene related to this condition, each of their children has a 25% (1 in 4) chance of developing the disease. It is an autosomal recessive disorder.
Krabbe disease (Globoid cell leukodystrophy) is thought to affect 1 person in every 100,000 people in the United States. However, there is an unusually high incidence, 6 cases per 1000 live births in the Druze community in Israel.
Krabbe disease most often affects infants, with onset before age 6 months, but can occur in adolescence or adulthood. Symptoms include severe deterioration of mental and motor skills, muscle weakness, hypertonia (inability of a muscle to stretch), myoclonic seizures (sudden, shock-like contractions of the limbs), and spasticity (involuntary and awkward movement). Other symptoms may include irritability, unexplained fever, blindness, difficulty with swallowing, and deafness.
There are four clinical forms of Krabbe’s disease, based on when symptoms of the disease occur.
Type 1: Infantile: begins at age 3 – 6 months
Type 2: Late infantile: begins at age 6 months – 3 years
Type 3: Juvenile: begins at age 3 – 8 years
Type 4: Adult onset: begins any time after 8 years of age
The most common form of Krabbe disease, called the infantile form, usually begins before the age of 1. The infantile form is the most common form and accounts for 85-90% of cases in the Northern European population. The infantile form has an onset at 2-6 months of age and is divided into 3 stages. In the first stage, symptoms include irritability, stiff posture, poor head control, muscle weakness, feeding difficulties, intermittent thumb clasp, episodes of fever without any sign of infection, and developmental delay – delayed mental and physical development.
As Krabbe disease, progresses, muscles continue to weaken, affecting the infant’s ability to move, chew, swallow, and breathe. Affected infants also experience vision loss, respiratory infections and seizures. Because of the severity of the condition, individuals with the infantile form of Krabbe disease rarely survive beyond the age of 2-3 years, generally due to respiratory infections.
Less commonly, Krabbe disease begins in childhood (1-8 years), adolescence (>8 years) or adulthood (late-onset forms). Patients with late infantile /juvenile onset most resemble infantile patients, while the first signs in adult forms are often weakness, gait disturbances (spastic paraparesis or ataxia), burning paresthesias, hemiplegia, and/or vision loss, with or without peripheral neuropathy. Cognitive regression is variable and often absent in adult forms. Vision problems and walking difficulties are the most common initial symptoms in these forms of the disorder, however, signs and symptoms vary considerably among affected individuals. Individuals with late-onset Krabbe disease may survive many years after the condition begins.
Complications of this include infection and respiratory failure which are responsible for most deaths.
Other names for Krabbe disease
- diffuse globoid body sclerosis
- galactosylceramidase deficiency disease
- galactosylceramide lipidosis
- galactosylcerebrosidase deficiency
- galactosylsphingosine lipidosis
- GALC deficiency
- GCL
- GLD
- psychosine lipidosis
Type 1 Krabbe Disease
The infantile or classic form accounts for the vast majority of recognized cases (85-90%) and is considered the prototype of Krabbe disease. The clinical course in patients with the infantile form has 3 stages 2.
Stage 1 includes irritability, hypertonia, hyperesthesia, peripheral neuropathy and arrest of psychomotor development occur following normal early development. Onset usually occurs at age 3-6 months. Feeding difficulties, such as vomiting and reflux, may cause failure to thrive.
In stage 2, rapid psychomotor deterioration, increasing hypertonia, opisthotonus, hyperreflexia, and optic atrophy ensue. Seizures may occur.
In stage 3, severe neurologic impairment often ensues within weeks to months with loss of voluntary movements and persistent decerebrate posturing. Patients become blind, deaf, and unaware of external stimuli. This final stage sometimes is termed the burnt-out stage.
Type 2 Krabbe Disease
Late infantile Krabbe disease follows a similar but less rapid course. After a variable period of normal early development (6 months to 3 years), the patient develops irritability, hypertonia, ataxia, and psychomotor arrest followed by progressive deterioration and vision loss, eventually followed by death.
Type 3 Krabbe Disease
Juvenile Krabbe disease is characterized by later age of onset (3-8 years) and greater variability in the tempo of disease progression. Early normal development is followed by a period of rapid psychomotor regression, although the disease then tends to subside into a slower, but progressive, degeneration.
Type 4 Krabbe Disease
Age of onset of adult Krabbe disease varies widely (8 year through adulthood). This type has a more varied clinical symptomatology and course of progression. Patients may present with signs of peripheral neuropathy, cerebellar dysfunction, spasticity, and impaired higher cortical functioning. Patients with type 4 disease may experience a rapid degenerative course or endure an indolent progression.
Krabbe disease life expectancy
Prognosis of Krabbe Disease (Globoid cell leukodystrophy) is generally poor, but varies depending on the subtype of disease. Most patients with the infantile and late infantile forms of Krabbe’s disease will die within 2-3 years of disease onset. In the juvenile and adult forms disease may not be as severe, and life expectancy may be improved. In late infantile/juvenile patients, the disease is generally fatal 2-7 years after the symptoms begin. People who develop the disease at a later age have survived into adulthood with nervous system disease.
Krabbe disease complications
This disease damages the central nervous system. It can cause:
- Blindness
- Deafness
- Severe problems with muscle tone
The disease is usually life-threatening.
A number of complications — including infections and respiratory difficulties — can develop in children with advanced Krabbe disease. In the later stages of the disease, children become incapacitated, are confined to their beds and eventually lapse into a vegetative state.
Most children who develop Krabbe disease in infancy die before the age of 2, most often from respiratory failure or complications of immobility and markedly decreased muscle tone. Children who develop the disease later in childhood may have a somewhat longer life expectancy, usually between two and seven years after diagnosis.
Krabbe disease cause
Mutations in the GALC gene (14q31) cause Krabbe disease. This gene provides instructions for making an enzyme called galactocerebroside beta-galactosidase (galactosylceramidase), which breaks down certain fats called galactolipids. One galactolipid broken down by galactosylceramidase, called galactosylceramide, is an important component of myelin. Breakdown of galactosylceramide is part of the normal turnover of myelin that occurs throughout life. Another galactolipid, called psychosine, which is formed during the production of myelin, is toxic if not broken down by galactosylceramidase.
GALC gene mutations severely reduce the activity of the galactosylceramidase enzyme. As a result, galactosylceramide and psychosine cannot be broken down. Excess galactosylceramide accumulates in certain cells, forming globoid cells. The accumulation of these galactolipids causes damage to myelin-forming cells, which impairs the formation of myelin and leads to demyelination in the nervous system. Without myelin, nerves in the brain and other parts of the body cannot transmit signals properly, leading to the signs and symptoms of Krabbe disease.
Galactolipids normally exist in cells that produce and maintain the protective coating of nerve cells (myelin). However, an abundance of galactolipids has a toxic effect. Some galactolipids trigger myelin-forming cells to self-destruct.
Other galactolipids are taken up by specialized debris-eating cells in the nervous system called microglia. The process of cleaning up excessive galactolipids transforms these normally helpful cells into abnormal, toxic cells called globoid cells, which promote myelin-damaging inflammation.
The subsequent loss of myelin (demyelination) prevents nerve cells from sending and receiving messages.
Krabbe’s disease is inherited as an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Figure 1. Krabbe disease autosomal recessive inheritance pattern
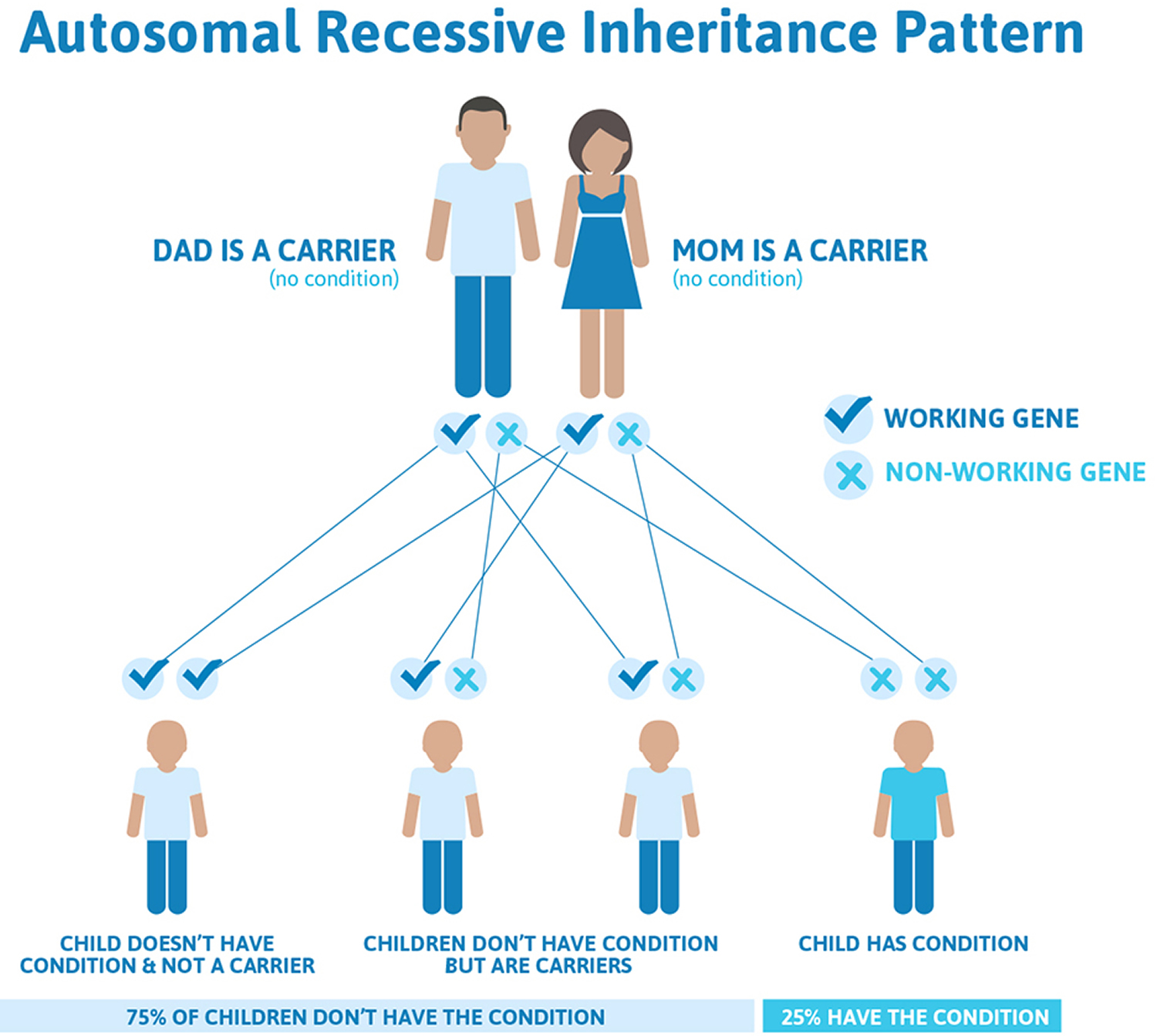
Risk Factors for Krabbe Disease
The gene mutation associated with Krabbe disease only causes the disease if two mutated copies of the gene are inherited. A disease resulting from two mutated copies is called an autosomal recessive disorder.
If each parent has one mutated copy of the gene, the risk for a child would be as follows:
- A 25 percent chance of inheriting two mutated copies, which would result in the disease
- A 50 percent chance of inheriting only one mutated copy, which would result in the child being a carrier of the mutation but would not result in the disease itself
- A 25 percent chance of inheriting two normal copies of the gene.
Krabbe disease prevention
Genetic counseling is recommended for people with a family history of Krabbe disease who are considering having children.
A blood test can be done to see if you carry the gene for Krabbe disease.
Prenatal tests (amniocentesis or chorionic villus sampling) can be done to test a developing baby for this condition.
Genetic testing to understand the risk of having a child with Krabbe disease may be considered in certain situations:
- If one or both parents are likely carriers of a GALC gene mutation because of a known family history of Krabbe disease, a couple may want to have tests to understand the risks in their own family.
- If one child is diagnosed with Krabbe disease, a family may consider genetic tests to identify other children who could develop the disease later in life.
- If the parents are known carriers, they may request a prenatal genetic test to determine if their child is likely to develop the disease.
- Known carriers, who are using in vitro fertilization, may request a genetic test with fertilized eggs before implantation.
Genetic testing should be carefully considered. Ask your doctor about genetic counseling services that can help you understand the benefits, limits and implications of genetic testing.
Krabbe disease symptoms
Signs and symptoms of early onset and late-onset Krabbe disease are described below.
Infantile Krabbe disease
Stage 1 includes the following 3:
- Irritability
- Hypertonia
- Hyperesthesia – Auditory, tactile, and visual
- Peripheral neuropathy
- Hyperpyrexia
- Psychomotor arrest
- Failure to thrive
- Vomiting
- Gastroesophageal reflux
Stage 2 includes the following:
- Hyperreflexia
- Hyporeflexia
- Opisthotonus
- Seizures
- Psychomotor deterioration
- Optic atrophy
- Visual loss
- Sluggish pupillary light response
- Rapid and severe psychomotor deterioration
Stage 3 includes the following:
- Decerebrate posturing
- Blindness
- Deafness
- No voluntary movement
- No interaction with the environment
Common signs and symptoms early in the course of the disease include the following:
- Feeding difficulties
- Unexplained crying
- Extreme irritability
- Fever with no sign of infection
- Declines in alertness
- Delays in typical developmental milestones
- Muscle spasms
- Loss of head control
- Frequent vomiting
As the disease progresses, signs and symptoms become more severe. They may include:
- Seizures
- Loss of developmental abilities
- Progressive loss of hearing and sight
- Rigid, constricted muscles
- Stiff, fixed posture
- Progressive loss of ability to swallow and breathe
Late-onset Krabbe disease
Symptoms include the following 4:
- Paresthesias
- Decreased muscle strength
- Spasticity
- Ataxia
- Paresis
- Psychomotor arrest
- Psychomotor deterioration
- Seizures
- Optic atrophy
- Visual loss
- Blindness
- Unpredictable rate of regression
- Macular cherry red spots were reported in 1 patient. Head circumference may be diminished, although macrocephaly also has been reported 5.
With late onset Krabbe disease, vision problems may appear first, followed by walking difficulties and rigid muscles. Symptoms vary from person to person. Other symptoms may also occur.
Older children and adults
When Krabbe disease develops later in childhood or during adulthood, signs and symptoms can vary widely. They may include:
- Progressive loss of vision
- Difficulty walking (ataxia)
- Decline in thinking skills
- Loss of manual dexterity
- Muscles weakness
As a general rule, the younger the age that Krabbe disease occurs, the faster the disease progresses and the more likely it is to result in death.
Some people diagnosed during adolescence or adulthood may have less severe symptoms, with muscle weakness as a primary condition. They may have no impairment of their thinking skills.
Krabbe disease diagnosis
The health care provider will perform a physical exam and ask about the symptoms.
Tests that may be done include:
- Blood test to look for galactosylceramidase levels. Galactocerebroside beta-galactosidase levels (levels can be measured from the serum, white blood cells, chorionic villi, and fibroblasts).
- CSF (cerebrospinal fluid) total protein – tests the amount of protein in cerebrospinal fluid (CSF)
- Genetic testing may be available for the glycosylceramidase gene (GALC)
- MRI of the head is the best test to reveal abnormal white matter of the brain
- A nerve conduction study assesses the rate at which nerves conduct a signal — essentially how quickly can they send a message. A special device measures the time it takes an electrical impulse to travel from one point on the body to another. When myelin is impaired, nerve conduction is slower.
- Testing for the GALC gene defect on chromosome band 14q31.3 6.
- CT of the head
- Presence of abnormal Globoid cells in biopsy tissue of the nervous system
Figure 2. Krabbe disease MRI (brain MRI)
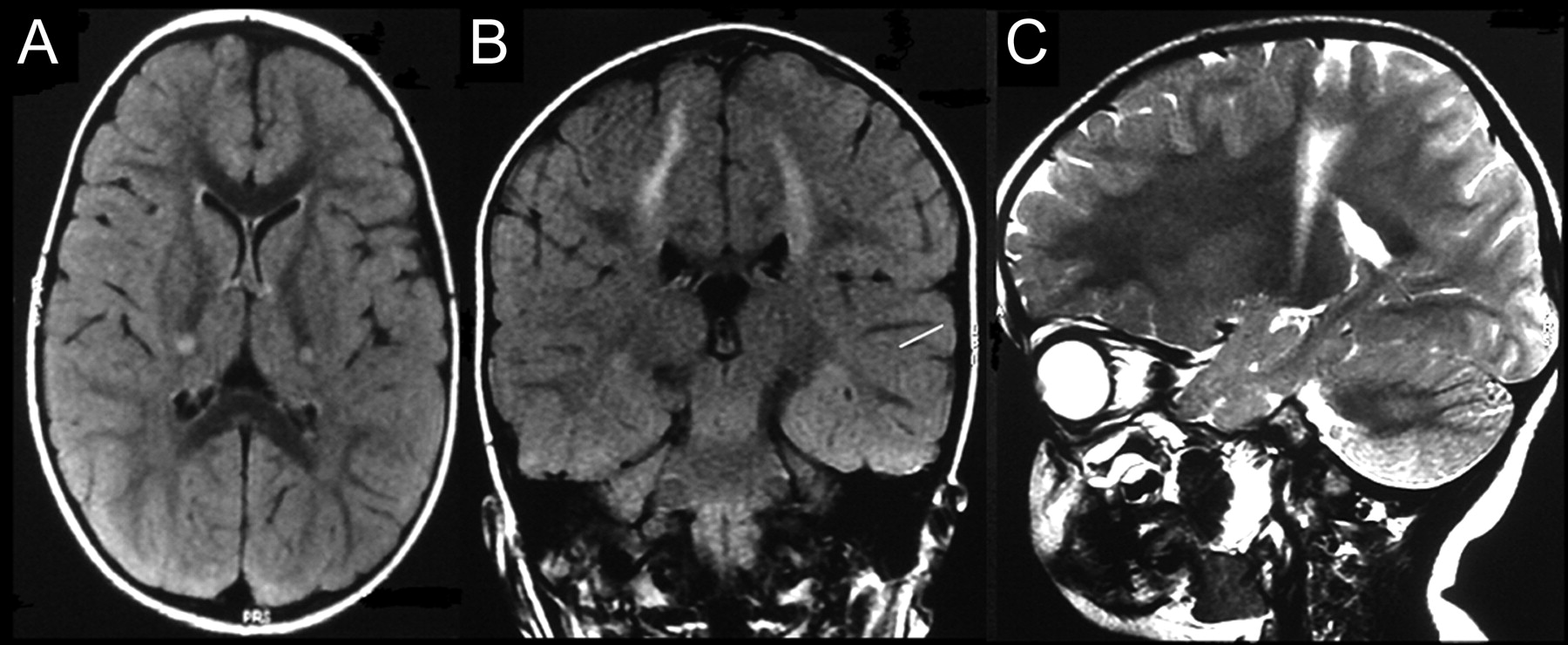
Note: Axial (A) and coronal (B) fluid-attenuated inversion recovery MRI scans show bilateral symmetric hyperintense signal changes involving the corticospinal tracts. (C) Sagittal T2-weighted MRI shows the hyperintense signal along the corticospinal tract from the cortical area extending into the internal capsule.
Figure 3. Krabbe disease MRI (brain MRI)
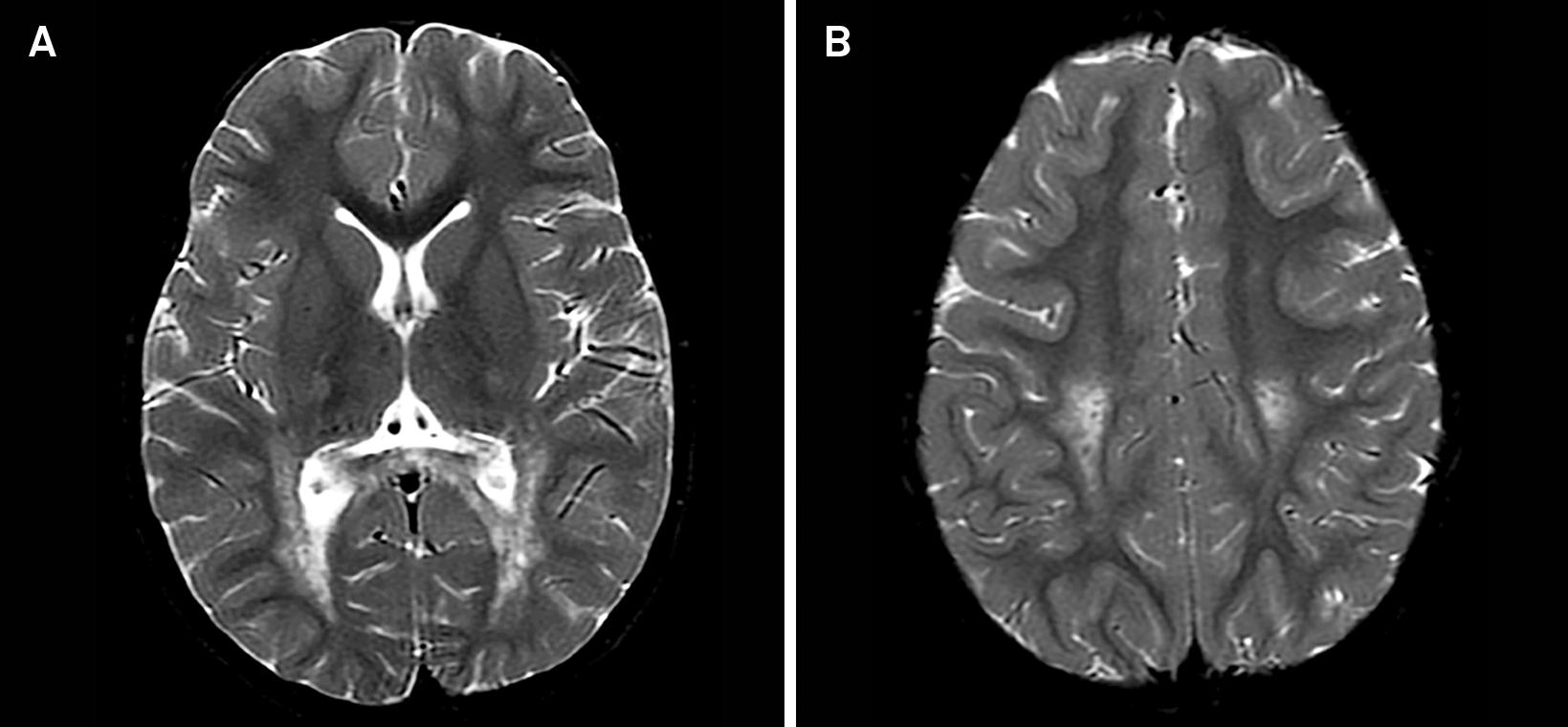
Note: Late-onset Krabbe disease. Axial T2-weighted images (A and B) disclose bilateral and symmetric hyperintensity involving the corticospinal tracts from the centrum semiovale to the posterior limb of the internal capsule. There are similar changes appreciated in the splenium of corpus callosum.
Measurement of GALC activity shows 0%-5% of normal activity in leukocytes or cultured skin fibroblasts. All individuals with deficient GALC enzyme activity have symptoms, which are confirmed with either physical examination or imaging findings that are consistent with the diagnosis of leukodystrophy.
Almost 70 mutations have been identified in the gene responsible for GALC production. Polymorphisms have been identified that may play a considerable role in the resultant phenotypes 7. There are two common mutations. A 30kb deletion has been reported in 40%-45% of individuals with infantile forms of Krabbe disease in northern Europe and in 35% of infantile Krabbe cases among Mexican patients 8. This deletion results in the infantile form of Krabbe, whether in the homozygous or the heterozygous state. However, when it is coupled with the c.857G>A mutation, it always results in the late-onset form of Krabbe disease 9. This c.857G>A variant is common in those with late-onset Krabbe.
Genotype-phenotype correlations are being delineated to provide a molecular explanation for the clinical variability seen in patients with Krabbe disease 10. However, there is no known correlation between enzyme activity and age of onset.
Newborn screening
In some states, a screening test for Krabbe disease is part of a standard set of assessments for newborns. The initial screening test measures GALC enzyme activity. If the enzyme activity is found to be low, follow-up GALC tests and genetic tests are conducted.
The use of newborn screening tests is relatively new. Researchers are still working to understand how best to use these tests, how well the tests lead to an accurate diagnosis and how well they predict the course of the disease.
Krabbe disease treatment
There is no specific treatment for Krabbe disease.
Treatment, therefore, focuses on managing symptoms and providing supportive care. Interventions may include the following:
- Anticonvulsant medications to manage seizures
- Drugs to ease muscle spasticity and irritability
- Physical therapy to minimize deterioration of muscle tone
- Nutritional support, such as the use of a tube to deliver fluids and nutrients directly into the stomach (gastric tube).
Interventions for older children or adults with less severe forms of the disease may include:
- Physical therapy to minimize deterioration of muscle tone
- Occupational therapy to achieve as much independence as possible with daily activities
Stem cell transplantation
Hematopoietic stem cells are specialized cells that can develop into all of the different types of blood cells in the body. These stem cells are also the source of microglia, specialized debris-eating cells that take up residence in the nervous system. In Krabbe disease, microglia are transformed into toxic globoid cells.
In stem cell transplantation, donor stem cells are delivered into the recipient’s bloodstream through a tube called a central venous catheter. The donor stem cells help the body produce healthy microglia that can populate the nervous system and deliver functioning GALC enzymes. This treatment may help restore some degree of normal myelin production and maintenance.
This therapy may improve outcomes in infants if treatment begins before the onset of symptoms — that is, when a diagnosis results from a newborn screening test.
Presymptomatic infants receiving a stem cell transplant have had slower disease progression, but these children still experience significant difficulties with speech, walking and other motor skills.
Older children and adults with mild symptoms also may benefit from this treatment.
Sources for hematopoietic stem cells include:
- Umbilical cord blood
- Donor bone marrow
- Donor circulating (peripheral) blood stem cells
It is too early to know if the new bone marrow can fully restore the brain to health in the small number of patients who have had this treatment.
In the future there may be ‘enzyme replacement therapy’, but it is in the early stages of development as of 2003. Prevention by prenatal or genetic testing is available.
Genetic counseling is recommended for prospective parents with a family history of Krabbe disease. Whether you are a carrier for the disease can be determined by testing your white blood cells or skin cells for decreased galactocerebroside beta-galactosidase levels.
Prenatal diagnosis is possible by measuring galactocerebroside beta-galactosidase levels in cultured amniotic fluid cells or from cultured chorionic villi cells.
Support Groups
Organizations that offer support, educational resources, networking opportunities and services to families dealing with Krabbe disease include the following:
- National Organization for Rare Disorders — rarediseases.org/rare-diseases/leukodystrophy-krabbes
- NIH Genetics Home Reference — ghr.nlm.nih.gov/condition/krabbe-disease
- United Leukodystrophy Foundation — www.ulf.org
- Graziano AC, Cardile V. History, genetic, and recent advances on Krabbe disease. Gene. 2015 Jan 15. 555(1):2-13.[↩]
- Wenger DA, Suzuki K, Suzuki Y, Suzuki K. 147. Galactosylceramide lipidosis: globoid cell leukodystrophy (Krabbe disease). In: Scriver CR, Beaudet AL, Sly WS, Valle D, Vogelstein B (eds) The Metabolic and Molecular Bases of Inherited Disease (OMMBID). McGraw-Hill; New York, NY: McGraw-Hill. 2005.[↩]
- Korn-Lubetzki I, Dor-Wollman T, Soffer D, et al. Early peripheral nervous system manifestations of infantile Krabbe disease. Pediatr Neurol. 2003 Feb. 28(2):115-8.[↩]
- Duffner PK, Barczykowski A, Kay DM, Jalal K, Yan L, Abdelhalim A, et al. Later onset phenotypes of Krabbe disease: results of the world-wide registry. Pediatr Neurol. 2012 May. 46(5):298-306.[↩]
- Nyhan WL, Ozand PT. Krabbe disease/galactosylceramide lipidosis/globoid cell leukodystrophy. Atlas of Metabolic Disease. New York, NY: Chapman & Hall Medical; 1998. 581-5.[↩]
- Oehlmann R, Zlotogora J, Wenger DA, Knowlton RG. Localization of the Krabbe disease gene (GALC) on chromosome 14 by multipoint linkage analysis. Am J Hum Genet. 1993 Dec. 53(6):1250-5.[↩]
- Luzi P, Rafi MA, Wenger DA. Structure and organization of the human galactocerebrosidase (GALC) gene. Genomics. 1995 Mar 20. 26(2):407-9.[↩]
- Rafi MA, Luzi P, Chen YQ, Wenger DA. A large deletion together with a point mutation in the GALC gene is a common mutant allele in patients with infantile Krabbe disease. Hum Mol Genet. 1995;4(8):1285.[↩]
- Wenger DA, Rafi MA, Luzi P. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum Mutat. 1997. 10(4):268-79.[↩]
- Wenger DA, Sattler M, Clark C, McKelvey H. An improved method for the identification of patients and carriers of Krabbe’s disease. Clin Chim Acta. 1974 Oct 30. 56(2):199-206.[↩]





{kind=link}