
Contents
What is lynch syndrome
Lynch syndrome, often called hereditary nonpolyposis colorectal cancer, is an inherited disorder that increases the risk of many types of cancer, particularly cancers of the colon (large intestine) and rectum, which are collectively referred to as colorectal cancer. Most of these cancers develop before they are 50. People with Lynch syndrome also have an increased risk of cancers of the stomach, small intestine, liver, gallbladder ducts, upper urinary tract, brain, skin and prostate. Additionally, women with this disorder have a high risk of cancer of the ovaries and lining of the uterus (endometrial cancer). People with Lynch syndrome may occasionally have noncancerous (benign) growths (polyps) in the colon, called colon polyps. In individuals with this disorder, colon polyps occur earlier but not in greater numbers than they do in the general population.
Doctors and genetics professionals can check if Lynch syndrome is likely based on your personal and family cancer history using certain criteria. These, known as the Amsterdam criteria and the revised Bethesda guidelines. Mutations in the genes that cause Lynch syndrome can then be tested for with genetic testing.
For people who have colorectal or endometrial cancer, the tumor tissue can be tested for MMR gene changes, or for other changes that can be caused when one of these genes is faulty, which is known as microsatellite instability (or MSI). Having normal findings (no MMR gene changes or MSI) implies that a person probably does not have Lynch syndrome. But if one of these is present, the person may have Lynch syndrome, and is referred for genetic counseling and possible testing.
Someone who is known to carry a gene mutation linked to Lynch syndrome may start screening for colorectal cancer when they are younger (such as during their early 20s), or take other steps to try to prevent cancer from starting (discussed in more detail in Colorectal Cancer). Women with Lynch syndrome may start screening for endometrial cancer or take other steps to try to prevent this cancer. These are discussed in more detail in Endometrial Cancer.
If someone has Lynch syndrome, it means that their close relatives (parents, siblings, and children) have a 50% chance of having a mutation, too. They may wish to be tested, or even without testing they may wish to start screening early for certain cancers or take other precautions to help lower their risk of cancer.
In the United States, about 140,000 new cases of colorectal cancer are diagnosed each year. Approximately 3 to 5 percent of these cancers are caused by Lynch syndrome. The population prevalence of Lynch syndrome has been estimated at 1:440 1.
Facts about Lynch Syndrome 2
- Approximately 3% of colorectal cancers are due to Lynch Syndrome.
- Lynch Syndrome is caused by autosomal dominantly inherited mutations in the mismatch repair (MMR) genes MLH1, MSH2, MSH6 and PMS2.
- Individuals with Lynch Syndrome have a substantial increased risk of developing colorectal cancer. For males, the lifetime risk is 20%-74%, and for females, the risk is 20%-52%. The risk for a second primary colorectal cancer is 15%-20% at 10 years.
- The mean age of onset 42 to 61 years.
- Females with Lynch Syndrome have a 28%- 60% lifetime risk for endometrial cancer.
- First-degree relatives of individuals identified with a Lynch Syndrome gene mutation have a 50% chance to carry the mutation.
Lynch syndrome cancers
In individuals with Lynch syndrome the following lifetime risks for cancer are seen 3:
- Colorectal cancer: 52%-82% (mean age at diagnosis 44-61 years)
- Endometrial cancer in females: 25%-60% (mean age at diagnosis 48-62 years)
- Gastric cancer: 6% to 13% for gastric cancer (mean age at diagnosis 56 years)
- Ovarian cancer: 4%-12% (mean age at diagnosis 42.5 years; ~30% are diagnosed < age 40 years).
The risk for other Lynch syndrome-related cancers is lower, though substantially increased over general population rates.
- Small bowel cancer: 4%-12% (mean age at diagnosis 49 years; male 6% and female 22%).
- Hepatobiliary tract cancer: 0.2%-4% (mean age at diagnosis 54-57 years; male 6% and female 22%).
- Urinary tract cancer: 0.2%-25% (mean age at diagnosis 52-60 years; male 6% and female 22%).
- Brain cancer (e.g. Glioblastoma multiforme): 1%-4% (mean age at diagnosis 50 years; male 6% and female 22%).
- Sebaceous neoplasms: 1%-9% (mean age at diagnosis not reported).
- Pancreas cancer: 0.4%-4% (mean age at diagnosis not reported).
- Prostate cancer: 9%-30% (mean age at diagnosis 59-60 years).
- Breast cancer: 5%-18% (mean age at diagnosis 52 years).
Colorectal cancer
The risk of colorectal cancer associated with MLH1 and MHS2 pathogenic variants is significantly higher than the risk associated with MSH6 or PMS2 pathogenic variants. The mean ages at onset for colorectal cancer in individuals with MSH6 and PMS2 pathogenic variants are also older than for onset for colorectal cancer associated with MLH1 and MSH2 pathogenic variants, 54-63 years and 47-66 years respectively. However, up to 8% of colorectal cancers in PMS2 heterozygotes occur before age 30. Therefore, individuals with MLH6 or PMS2 pathogenic variants should follow the same colorectal cancer screening recommendations as MLH1 and MSH2 heterozygotes. A 2009 study of a Finnish cohort with high compliance with screening found no increase in mortality for individuals with Lynch syndrome compared to relatives without the Lynch syndrome-related pathogenic variant, indicating that annual colonoscopy is effective for the prevention and detection of colorectal cancer 4.
Data on cancer risks for those with an EPCAM deletion is still limited, but Kempers et al 5 reported on findings from 194 individuals with an EPCAM deletion. From this cohort they estimated a 75% (95% CI 65-86) cumulative incidence of CRC by age 70 years. The risk for colorectal cancer did not differ between those who had only a 3′ deletion of EPCAM and those individuals with a deletion that encompassed EPCAM and MSH2. However, only those with an EPCAM deletion that includes MSH2 are at an increased risk for extracolonic cancers 6.
Tumors that are MSI-H, either due to sporadic causes or an underlying germline MMR pathogenic variant, tend to have a better prognosis than MSS tumors 7.
Endometrial cancer
The risk for women with EPCAM deletions that encompass MSH2 is similar to that for individuals with MSH2 pathogenic variants.
Among females with Lynch syndrome who develop both colorectal cancer and endometrial cancer, approximately 50% present first with endometrial cancer 8. The risk for subsequent endometrial cancer to females with Lynch syndrome presenting first with colorectal cancer has been estimated at 26% within ten years of the initial colorectal cancer diagnosis 9.
Overall, a survival advantage similar to that in Lynch syndrome-related colorectal cancer has been reported in Lynch syndrome-related endometrial cancers 10.
Gastric cancer
Intestinal-type adenocarcinoma, the most commonly reported pathology of Lynch syndrome-related gastric cancers 11, differs histologically from the diffuse gastric cancer that is most commonly seen in hereditary diffuse gastric cancer caused by pathogenic variants in CDH1 12. However, Capelle et al 13 reported that up to 20% of Lynch syndrome-related gastric cancers may be the diffuse type. The risk for gastric cancer is higher in individuals with Lynch syndrome who reside in countries with a high incidence of H pylori infection 14.
Ovarian cancer
Ovarian cancer risk to females with a germline MLH1 or MSH2 pathogenic variant has been found to be 4%-20%. The mean age of diagnosis of Lynch syndrome-associated ovarian cancer has been reported at between age 43 and 45 years, although diagnosis at very young ages has been reported. Approximately 30% of Lynch syndrome-associated ovarian cancers are diagnosed before age 35 years 15.
The distribution of pathology types is similar to that seen in sporadic ovarian cancers. Borderline ovarian tumors do not appear to be associated with Lynch syndrome 16.
Small bowel cancer
The duodenum and jejunum are the most common sites for small bowel cancers, with approximately 50% in reach of upper endoscopy 17. The majority of small bowel cancers are adenocarcinomas 17.
Urinary tract cancers
The urinary tract cancers most commonly associated with Lynch syndrome are transitional carcinomas of the ureter and renal pelvis.
Bladder cancer risk is also likely increased in individuals with Lynch syndrome. A study of Dutch individuals with Lynch syndrome demonstrated a relative risk for bladder cancer of 4.4 for males and 2.2 for females; the majority of tumor tissue available for testing was MSI-H and/or had loss of protein expression by IHC 18.
Individuals with Lynch syndrome and a prior diagnosis of colorectal cancer were also at increased risk for subsequent bladder cancer and other urinary tract cancers (kidney, renal pelvis, and ureter) 19.
Risk estimates for urinary tract cancers vary significantly based on gender and the gene involved.
Brain tumors
The most common type of central nervous system tumor is glioblastoma 20. The brain tumors associated with pathogenic variants in an MMR gene are typically MSI-H 21.
Sebaceous neoplasms
Sebaceous neoplasms described in individuals with Lynch syndrome include: sebaceous adenomas, sebaceous epitheliomas, sebaceous carcinomas, and keratoacanthomas. Sebaceous neoplasms associated with Lynch syndrome are typically MSI-H 22. Data on the frequency of sebaceous neoplasms in individuals with Lynch syndrome are limited. Studies have found that between 1% and 9% of individuals with a germline pathogenic variant in an MMR gene have a sebaceous neoplasm 23.
Other Cancers
Other Lynch syndrome-related cancers that have characteristic features have been reported.
Pancreatic cancer. A study by Kastrinos et al 24 based on reported family history found an 8.6-fold increased risk up to age 70 years for pancreatic cancer 24. A prospective study that followed 446 individuals with an MMR pathogenic variant and 1,029 relatives for a median of five years found an increased risk for pancreatic cancer and no increased risk for individuals without a pathogenic variant 25. However, other studies have not demonstrated an increased risk 26. Lynch syndrome has been found to be a rare cause of familial pancreatic cancer 27.
Prostate cancer. Several studies have demonstrated an association with prostate cancer, with the increase in risk ranging from two- to fivefold 28 found that the risk for prostate cancer was increased for males with an MMR pathogenic variant prior to age 60 28, whereas an analysis by Haraldsdottir et al 29 did not find earlier age of onset or a more aggressive phenotype in the prostate cancers occurring in individuals with an MMR pathogenic variant. Pritchard et al 30 identified a pathogenic variant in an MMR gene in four (0.5%) of 692 men with metastatic prostate cancer.
Breast cancer. The relationship between breast cancer and Lynch syndrome is unresolved. A systematic review evaluated 21 studies; 13 did not demonstrate an increased risk for breast cancer in individuals with Lynch syndrome and eight showed an increased risk 19. To date, breast cancer risk has only been evaluated in one prospective study. Individuals with an MMR pathogenic variant were found to have a standard incidence ratio for breast cancer of 3.95 and the median age of breast cancer diagnosis was 56 years. Tumor tissue IHC in 51% of breast cancers in individuals with an MMR pathogenic variant demonstrated loss of expression for the MMR gene with the germline pathogenic variant. Due to the high frequency of breast cancer in the general population, the presence of sporadic breast cancers complicates analysis of the association with Lynch syndrome.
Additional cancer risks. Several other cancer types have been reported to occur in individuals with Lynch syndrome. In some cases, MSI and/or IHC testing of tumor tissue demonstrated concordance between the extracolonic cancer and the molecular diagnosis of the affected individual. While these types of findings suggest that the underlying presence of a pathogenic variant in an MMR gene contributed to the development of the cancer, data are not sufficient to demonstrate that the risk of developing these types of cancers is increased in individuals with Lynch syndrome.
- Several types of sarcomas have been reported in individuals with an MMR pathogenic variant, including fibrous histiocytomas, rhabdomyosarcomas, leiomyosarcoma, and liposarcoma 31. Nilbert et al 31 determined that six of eight sarcomas in individuals with Lynch syndrome exhibited defective MMR, suggesting that sarcomas may also be part of the spectrum of Lynch syndrome tumors. Due to the rarity of sarcomas it may be difficult to determine the magnitude of risk associated with Lynch syndrome.
- Adrenocortical carcinoma has also been reported in families with Lynch syndrome. The most extensive study of this association, performed through a hereditary cancer clinic at the University of Michigan, found that two (1.7%) of 114 individuals presenting with ACC had a family history consistent with Lynch syndrome and had an MMR pathogenic variant identified. This association was further evaluated by case review of 135 individuals with pathogenic MMR variants, which identified two (1.4%) individuals who also had ACC 32.
Lynch syndrome inheritance pattern
Lynch syndrome cancer risk is inherited in an autosomal dominant pattern, which means one inherited copy of the altered gene in each cell is sufficient to increase cancer risk. It is important to note that people inherit an increased risk of cancer, not the disease itself. Not all people who inherit mutations in these genes will develop cancer.
Treatment of colon cancer is surgical removal of the affected part of the colon (colectomy). People with Lynch syndrome should have routine colonoscopies 3.
Other names for Lynch syndrome
- cancer family syndrome
- familial nonpolyposis colon cancer
- hereditary nonpolyposis colorectal cancer
- hereditary nonpolyposis colorectal neoplasms
- HNPCC
Figure 1. Lynch syndrome autosomal dominant inheritance pattern
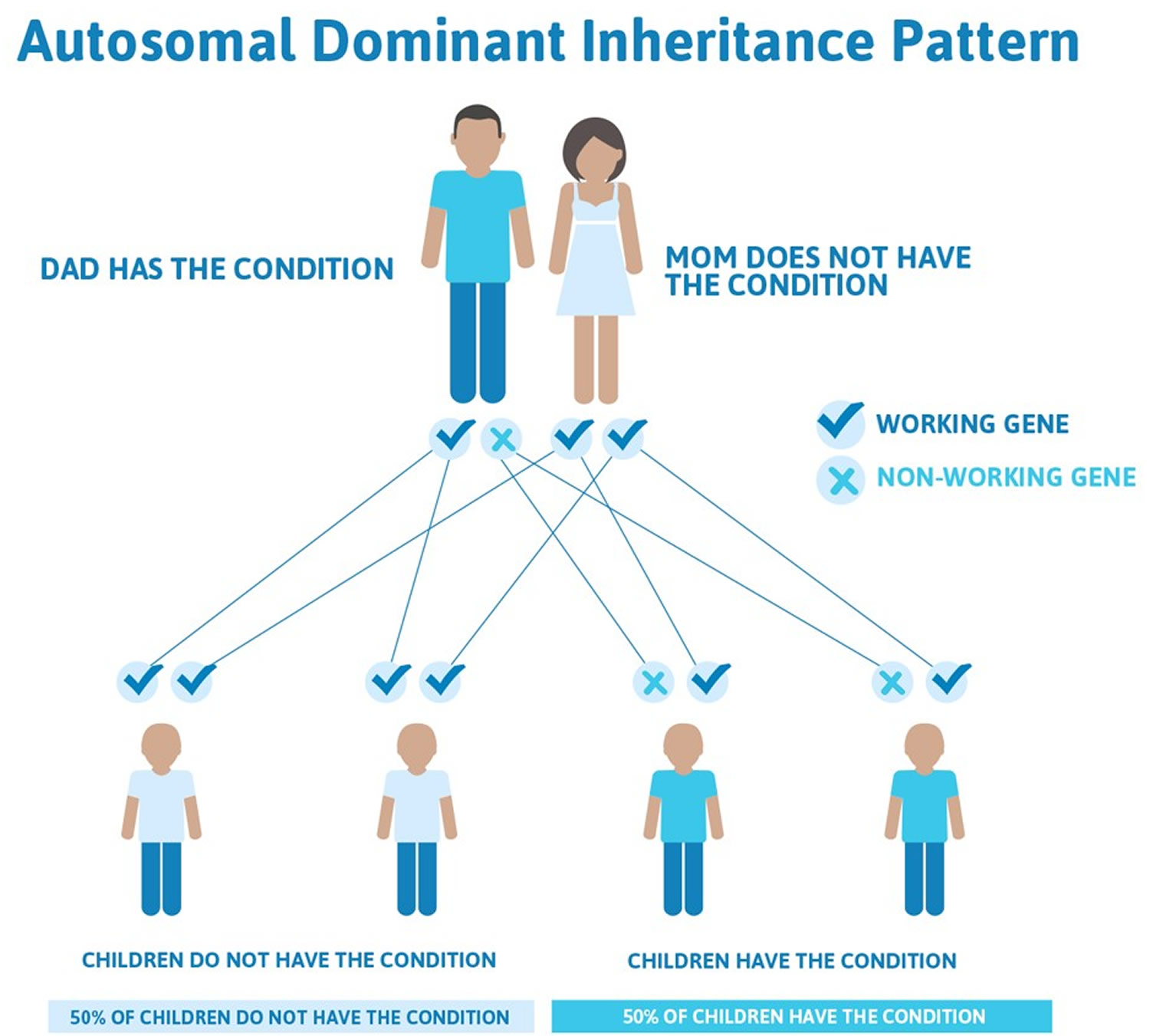
Lynch syndrome testing
People with Lynch syndrome are also at increased risk for some other cancers, such as cancers of the uterus (endometrium), ovaries, stomach, small bowel, pancreas, kidneys, brain, ureters (tubes that carry urine from the kidneys to the bladder), and bile duct.
Amsterdam criteria
Doctors have found that many families with Lynch syndrome tend to have certain characteristics, which are known as the Amsterdam criteria:
- At least 3 relatives have colorectal cancer (or another cancer linked with Lynch syndrome).
- One is a first-degree relative (parent, brother or sister, or child) of the other 2 relatives.
- At least 2 successive generations are involved.
- At least 1 relative had their cancer when they were younger than age 50.
If all of these apply to your family, then you might want to seek genetic counseling. But even if your family history satisfies the Amsterdam criteria, it doesn’t always mean you have Lynch syndrome. Only about half of families who meet the Amsterdam criteria have Lynch syndrome. The other half do not, and although their colorectal cancer risk is about twice as high as normal, it’s not as high as that of people with Lynch syndrome. On the other hand, many families with Lynch syndrome do not meet the Amsterdam criteria.
Revised Bethesda guidelines
A second set of criteria, called the revised Bethesda guidelines, can be used to determine whether a person with colorectal cancer should have their cancer tested for genetic changes that are seen with Lynch syndrome. (These changes are called microsatellite instability or MSI.) These criteria include at least one of the following:
- The person is younger than 50 years.
- The person has or had a second colorectal cancer or another cancer (endometrial, stomach, pancreas, small intestine, ovary, kidney, brain, ureters, or bile duct) linked to Lynch syndrome.
- The person is younger than 60 years, and the cancer has certain characteristics seen with Lynch syndrome when it’s viewed under a microscope.
- The person has a first-degree relative (parent, sibling, or child) younger than 50 who was diagnosed with colorectal cancer or another cancer linked to Lynch syndrome (endometrial, stomach, pancreas, small intestine, ovary, kidney, brain, ureter, or bile duct).
- The person has 2 or more first- or second-degree relatives (aunts, uncles, nieces, nephews, or grandparents) who had colorectal cancer or another Lynch syndrome-related cancer at any age.
If a person with colorectal cancer has any of the Bethesda criteria, testing for MSI may be advised. If MSI is found, the doctor typically will recommend that the patient be tested for Lynch syndrome-associated gene mutations.
It’s important to know that most people who meet the Bethesda criteria do not have Lynch syndrome, and that you can have Lynch syndrome and not meet any of the criteria listed. Not all doctors use the Bethesda guidelines to decide who should have MSI testing. In fact, some experts recommend that all colorectal cancers be tested for MSI. Most doctors recommend genetic testing for Lynch syndrome for anyone whose cancer tests positive for MSI.
Even if you don’t have cancer, your doctor may suspect that Lynch syndrome runs in your family based on cases of colorectal cancer and other cancers associated with this syndrome in your relatives. In that case, your doctor might recommend genetic counseling to evaluate your risk.
In families known to carry a Lynch syndrome gene mutation, doctors recommend that family members who have tested positive for the mutation and those who have not been tested should start colonoscopy screening during their early 20s, or 2 to 5 years younger than the youngest person in the family with a diagnosis (whichever is earlier) to remove any polyps and find any cancers at the earliest possible stage. People known to carry one of the gene mutations may also be given the choice of removal of most of the colon.
Genetic Changes in Lynch syndrome
Variations in the MLH1, MSH2, MSH6, PMS2, or EPCAM gene increase the risk of developing Lynch syndrome.
The MLH1, MSH2, MSH6, and PMS2 genes are involved in the repair of errors that occur when DNA is copied in preparation for cell division (a process called DNA replication). Mutations in any of these genes prevent the proper repair of DNA replication errors. As the abnormal cells continue to divide, the accumulated errors can lead to uncontrolled cell growth and possibly cancer.
Mutations in the EPCAM gene also lead to impaired DNA repair, although the gene is not itself involved in this process. The EPCAM gene lies next to the MSH2 gene on chromosome 2; certain EPCAM gene mutations cause the MSH2 gene to be turned off (inactivated), interrupting DNA repair and leading to accumulated DNA errors.
Although mutations in these genes predispose individuals to cancer, not all people who carry these mutations develop cancerous tumors.
The diagnosis of Lynch syndrome is established in a proband (index case) by identification of a germline heterozygous pathogenic variant in MLH1, MSH2, MSH6, or PMS2 or an EPCAM deletion on molecular genetic testing.
Lynch syndrome is caused by pathogenic variants in genes involved with the mismatch repair (MMR) pathway. This pathway functions to identify and remove single-nucleotide mismatches or insertions and deletion loops. Pathogenic variants in four of the mismatch repair (MMR) genes can cause Lynch syndrome 33. The functions of the mismatch repair (MMR) genes can be disrupted by missense variants, truncating variants, splice site variants, large deletions, or genomic rearrangements. In addition, germline deletions within EPCAM, which is not an MMR gene, can disrupt the MMR pathway by inactivating the adjacent MMR gene MSH2, even though MSH2 itself has not been mutated.
Lynch syndrome testing
The diagnosis of Lynch syndrome can be made on the basis of the Amsterdam Clinical Criteria or on the basis of molecular genetic testing for germline mutations in one of several mismatch repair (MMR) genes 3.
A diagnosis of Lynch syndrome should be suspected in a proband with:
- A diagnosis of colorectal cancer or endometrial cancer and one or more of the following*:
- Colorectal or endometrial cancer diagnosed before age 50 years
- Synchronous or metachronous Lynch syndrome-related cancers (e.g., colorectal, endometrial, stomach, small intestinal, hepatobiliary, renal pelvic, ureteral)
- Colorectal tumor tissue with MSI-high histology (e.g., poor differentiation, tumor-infiltrating lymphocytes, Crohn’s-like lymphocytic reaction, mucinous/signet-ring differentiation, medullary growth pattern)
- Microsatellite instability (MSI) testing showing that tumor tissue (e.g., colon, endometrial) is MSI-high. Microsatellites are stretches of DNA with a repetitive sequence of nucleotides (e.g., AAAAA or CGCGCGCG) that are particularly susceptible to acquiring errors when the MMR gene function is impaired. Cancers arising in cells with defective MMR gene function exhibit an inconsistent number of microsatellite nucleotide repeats when compared to normal tissue, a finding referred to as “microsatellite instability” (MSI).
- Tumor tissue (e.g., colon, endometrial) immunohistochemistry (IHC) demonstrates loss of expression of one or more of the mismatch repair (MMR) gene products: MSH2, MLH1, MSH6, and PMS2. (For information on advantages and disadvantages of IHC testing, click here.)
- At least one first-degree relative with any Lynch syndrome-related cancer diagnosed before age 50 years
- At least two first-degree relatives with any Lynch syndrome-related cancers regardless of age of cancer diagnosis
- A family member with colorectal or endometrial cancer who meets one of the above criteria
Note: Molecular genetic testing ideally begins with a person who has had a Lynch syndrome-related cancer. However, in some families there may be no affected individual who is alive or willing to be tested.
- A family member with a confirmed diagnosis of Lynch syndrome
- A greater-than-5% probability of having a pathogenic variant in one of the genes listed in Table 1 based on risk assessment models
Note: Several risk assessment models including PREMM1,2,6 34 and MMRPro 1 predict the likelihood of identifying a germline pathogenic variant in one of the genes listed in Table 1. Both models have good predictive value in a clinical and population-based setting when using a 5% threshold for testing 35.
*Adapted from revised Bethesda Guidelines and National Comprehensive Cancer Network 36 Guidelines.
Establishing the Diagnosis of Lynch syndrome
The diagnosis of Lynch syndrome is established in a proband by identification of a heterozygous germline pathogenic variant in one of the genes listed in Table 1.
Molecular genetic testing approaches can include a multigene panel, serial single-gene testing, and more comprehensive genomic testing.
A multigene panel that includes MLH1, MSH2, MSH6, and PMS2 as well as EPCAM deletion analysis (see Table 1) and other genes of interest may be considered. Note: (1) The genes included and the sensitivity of multigene panels vary by laboratory and over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
There are two types of multigene panels:
- Off the shelf. These are designed by a laboratory to include genes commonly associated with a broad phenotype (e.g., cardiomyopathy, ataxia, intellectual disability) or a recognizable syndrome with genetic heterogeneity (e.g., Noonan syndrome).
- Custom designed. These include genes selected by a clinician for analysis by clinical sequencing. Results for each gene on the custom multigene panel are reported to the ordering clinician, whereas the results from the remaining genes sequenced (but not requested by the clinician) are not analyzed or included in the final laboratory report. Custom multigene panels offered by some reference laboratories are marketed under names such as XomeDxSlice® and ExomeNext-Select®.
Table 1. Molecular Genetic Testing Used in Lynch Syndrome
| Gene | Proportion of Lynch Syndrome Attributed to Pathogenic Variants in This Gene | Proportion of Pathogenic Variants Detectable by This Method | |
|---|---|---|---|
| Sequence analysis | Gene-targeted deletion/duplication analysis | ||
| MLH1 | 50% | 90%-95% | 5%-10% |
| MSH2 | 40% | <80% | >20% |
| MSH6 | 7%-10% | >95% | <5% |
| PMS2 | <5% | See footnote 1 | See footnote 1 |
| EPCAM | ~1%-3% | See footnote 2 | 100% |
| Unknown | NA | ||
Footnote:
1. Methods to sequence and identify large rearrangements in PMS2 have been developed and improved over time, making it difficult to determine the proportion of pathogenic variant detected by each method in an affected population. Nearly 200 sequence variants and more than 100 large rearrangements in PMS2 have been reported [Human Gene Mutation Database]. Variants detectable by sequence analysis appear to be more common; however, large rearrangements may comprise more than 40%-50% of pathogenic variants in this gene 37.
2. Germline deletions of EPCAM result in silencing of the adjacent MSH2 allele by hypermethylation. The adjacent MSH2 allele itself is not mutated. Sequence analysis of EPCAM is not appropriate for diagnosis of Lynch syndrome.
Recommended screening for Lynch syndrome
Screening for Lynch Syndrome on all newly diagnosed colorectal cancers is recommended by several organizations and it is a Healthy People 2020 Objective. It is already performed in more than a hundred hospitals nationwide.
Screening performed on pathology specimens utilizing immunohistochemistry (IHC) for the four MMR proteins, and/or molecular microsatellite instability (MSI) testing can identify 95% of Lynch Syndrome-associated colorectal cancers 38. While 15%-20% of colorectal cancers will demonstrate abnormal IHC and/or MSI results, additional reflex testing can differentiate somatic vs. germline events.
Screening Methods
Three algorithms are available for Lynch Syndrome screening. All involve the testing of tumor tissue.
MSI (Microsatellite Instability): Approximately 90% of colon tumors from individuals with Lynch Syndrome demonstrate microsatellite instability (MSI), whereas only approximately 15% of sporadic colon tumors do. Thus, MSI testing is useful in determining the likelihood of Lynch Syndrome.
IHC (Immunohistochemistry): Tumors from individuals with Lynch Syndrome are likely to demonstrate loss of mismatch repair protein expression. The pattern of loss observed can provide information about which gene is not functioning properly. As a result, immunohistochemistry (IHC) testing can be helpful both in providing information about the likelihood of Lynch Syndrome and in directing testing for a germline mutation to a specific gene.
MSI and IHC: When used alone, MSI and IHC may miss some cases of Lynch Syndrome. For example, MSI alone may not detect cases of Lynch Syndrome because of MSH6 mutations since MSH6-related tumors are sometimes MSS. On the other hand, IHC alone may not detect cases of Lynch Syndrome because of mutations that result in full length, but dysfunctional, protein.
There is no consensus on whether MSI, IHC, or both is the ideal screening tool for Lynch Syndrome. Some centers start with MSI testing and then perform IHC on tumors that demonstrate microsatellite instability to help direct genetic testing. Others use IHC as the sole screening tool. Some centers perform both tests simultaneously to provide as much information as possible.
Targeted molecular genetic testing on tumor tissue should be considered in individuals with MLH1/PMS2 loss of expression on IHC. Targeted testing includes the following:
- Targeted analysis of BRAF pathogenic variant p.Val600Glu
Note: (1) BRAF p.Val600Glu is not present in Lynch syndrome-associated colorectal tumors. (2) BRAF pathogenic variants are not common in sporadic endometrial cancers; thus, BRAF testing is not helpful in distinguishing endometrial cancers that are sporadic from those that are Lynch syndrome related. - MLH1 promoter methylation analysis on tumor tissue
Note: Lynch syndrome-related cancers do not have hypermethylation of the MLH1 promotor.
Screening approaches include:
- Screen all colorectal cancer with MSI or IHC testing. This was shown to be a cost-effective approach for identifying individuals who should be offered germline molecular genetic testing for Lynch syndrome 39.
- Screen all colorectal cancer and endometrial cancers with MSI or IHC testing 40.
- Use age of onset and pathologic features to predict which individuals are more likely to have a germline MMR pathogenic variant 41.
Impact of screening
- Referral of patients with abnormal screen results for genetic counseling and molecular testing for germline MMR mutations allows for diagnostic confirmation for the patient and accurate testing for family members.
- Identification of a colorectal cancer patient with Lynch Syndrome affects future screening for synchronous colorectal cancer and other Lynch Syndrome-associated malignancies.
- Evidence suggests a diagnosis of Lynch Syndrome may affect surgical and chemotherapeutic management decisions.
- First-degree relatives who test negative for the identified mutation are no longer at increased risk for colorectal cancer or other Lynch Syndrome-related malignancies, nor are their children at risk for Lynch Syndrome.
- Relatives who test positive for the familial mutation require colonoscopy every one to two years beginning at age 25, in addition to screening for non-colonic cancers.
Lynch syndrome symptoms
Most people with Lynch syndrome do not know they have it. Lynch Syndrome is caused by a genetic mutation that increases a person’s risk for certain cancers. One in every 440 Americans has Lynch Syndrome, but the majority of these people have not been diagnosed.
Most people with Lynch syndrome may experience:
- Colon cancer that occurs at a younger age, especially before age 50
- A family history of colon cancer that occurs at a young age
- A family history of cancer that affects the uterus (endometrial cancer)
- A family history of other related cancers, including ovarian cancer, kidney cancer, stomach cancer, small intestine cancer, liver cancer, sweat gland cancer (sebaceous carcinoma) and other cancers
Your doctor can use your family history to estimate your risk.
Your doctor can use a risk prediction model to estimate the chance that you have Lynch syndrome. To estimate your risk, your doctor will need to know the following things:
- Whether you have ever had cancer, and what type(s).
- The approximate age at which each family member was first diagnosed with cancer.
A blood test can confirm the diagnosis. To prevent colorectal cancer, people with Lynch Syndrome should undergo a colonoscopy every 1-2 years, starting in their twenties. They may also consider taking aspirin every day.
People with Lynch syndrome have a mutation of the MMR gene, which means their bodies are less able to fix errors in the DNA. This means that a person with Lynch syndrome is more likely to get certain types of cancer. Lynch syndrome increases the risk of getting colorectal cancer by 80 percent and endometrial cancer by 60 percent. Lynch syndrome can also lead to other cancers, including:
- Cancers of the stomach and small intestine.
- Cancer of the pancreas.
- Cancer of the urinary tract, including kidneys.
- Cancer of the bile ducts.
- Sebaceous (oil) gland tumors.
- Ovarian cancer.
- Skin cancer.
- Glioblastoma multiforme (a type of brain cancer).
Based on this information, your doctor may decide to refer you to a genetic counselor or to order a genetic test. This test uses a small amount of your blood to look for the mutations that cause Lynch syndrome.
You may already know that a family member has Lynch syndrome. If so, tell your doctor. If your relative has already had a genetic test, bringing those results to your doctor can make it easier to find out whether you, too, have Lynch syndrome.
How to Prevent Colorectal Cancer if You Have Lynch Syndrome
Get a colonoscopy every 1 to 2 years, starting in your twenties.
If you have Lynch syndrome, you should get a colonoscopy every one to two years to screen for colorectal cancer. Doing this will reduce your risk of colorectal cancer by 77 percent.
Exactly when should you get your first screening?
Find out the youngest age at which a family member first got colorectal cancer. Then, follow these guidelines:
- If that person was younger than 25 when they were first diagnosed: subtract five from that age. For instance: if they were 24 at the time of diagnosis, get your first colonoscopy at 19.
- If that person was 25 or older when they were first diagnosed: start between the ages of 20 and 25.
Remember, most people with Lynch syndrome do not know they have it. If you have Lynch syndrome, the most important thing to do is start getting screened as soon as possible to keep yourself healthy.
Taking care of yourself through diet, exercise and other lifestyle changes can help improve your overall health.
Take control of your health by trying to:
- Eat a healthy diet full of fruits and vegetables. Choose a variety of fruits and vegetables for your diet. Also, select whole-grain products when possible.
- Exercise regularly. Aim for at least 30 minutes of exercise most days of the week. If you haven’t been active, talk to your doctor before you begin an exercise program. Try gentle exercises like walking or biking to get started.
- Maintain a healthy weight. A healthy diet and regular exercise can help you maintain a healthy weight. If you need to lose weight, talk with your doctor about your options. Eating fewer calories and increasing the amount of exercise you do can help you lose weight. Aim to lose 1 or 2 pounds a week.
- Stop smoking. Smoking increases your risk of several types of cancer and other health conditions. Some evidence indicates smoking may increase the risk of colon cancer in people with Lynch syndrome. If you smoke, stop. Your doctor can recommend strategies to help you quit. You have many options, such as nicotine replacement products, medications and support groups. If you don’t smoke, don’t start.
Talk to your doctor about taking aspirin every day.
- Taking 600 milligrams of aspirin every day may decrease your odds of getting colorectal cancer by 44 percent. It has been shown that daily use of 600mg of aspirin for at least 2 years lowers the risk for colon cancer by almost 60% inindividuals. The cancer risk reduction begins around 45 years after the individuals started taking aspirin. It is still not known what the best amount of aspirin is inorder to decrease the colon cancer risk or the total amount of time that individuals need to take the aspirin.
The ideal dosage of aspirin is not known, and there are risks from taking aspirin at such a high dose. Some people, such as those with stomach bleeding or blood clotting disorders, should not take aspirin regularly. Talk to your doctor to decide whether and how much aspirin to take. Taking aspirin should not replace getting a colonoscopy every one to two years.
Lynch syndrome diagnosis
If it’s suspected that you have Lynch syndrome, your doctor may ask you questions about your family history of colon cancer and other cancers. This may lead to other tests and procedures to diagnose Lynch syndrome.
Family history
A family history of colon cancer and other cancers, particularly when they occur at a younger age, may alert your doctor to the possibility that you or members of your family may have Lynch syndrome.
Your doctor may refer you for further Lynch syndrome evaluation if you have:
- Multiple relatives with any Lynch-associated tumors, including colorectal cancer. Examples of other Lynch-associated tumors include those affecting the endometrium, ovaries, stomach, small intestine, kidney, brain or liver.
- Family members diagnosed with cancer at ages that are younger than the average for their type of cancer.
- More than one generation of family affected by a type of cancer.
Tumor testing
If you or someone in your family has been diagnosed with cancer, special testing may reveal whether the tumor has specific characteristics of Lynch syndrome cancers. Samples of cells from a colon cancer and sometimes from other tumors can be used for tumor testing.
If you or someone in your family has been diagnosed with cancer in the last several years, the hospital that provided care may be able to supply a tissue sample. These tissue samples are often stored for many years.
Tumor testing can reveal whether your cancer was caused by the genes related to Lynch syndrome. Tumor tests include:
- Immunohistochemistry (IHC) testing. IHC testing uses special dyes to stain tissue samples. The presence or absence of staining indicates whether certain proteins are present in the tissue. Missing proteins may tell doctors which mutated gene caused the cancer.
- Microsatellite instability (MSI) testing. Microsatellites are sequences of cellular DNA. In people with Lynch syndrome, there may be errors or instability in these sequences in the tumor.
Positive IHC or MSI test results indicate that you have malfunctions in the genes that are connected to Lynch syndrome. But results can’t tell you whether you have Lynch syndrome because some people develop these gene mutations only in their cancer cells.
People with Lynch syndrome have these gene mutations in all of their cells. Genetic testing can determine whether you have these mutations.
Increasingly, IHC or MSI testing is being offered to anyone diagnosed with colon cancer to look for signs that may indicate Lynch syndrome. Doctors hope this will help identify families with Lynch syndrome that don’t meet the usual criteria for genetic testing.
Genetic testing
Genetic testing looks for changes in your genes that indicate that you have Lynch syndrome. You may be asked to give a sample of your blood for genetic testing. Using special laboratory analysis, doctors look at the specific genes that can have mutations that cause Lynch syndrome.
Results of genetic testing may show:
- A positive genetic test. A positive result, meaning that a gene mutation was discovered, doesn’t mean that you’re certain to get cancer. But it does mean your lifetime risk of developing colon cancer is increased. How much your risk is increased depends on which gene is mutated in your family and whether you undergo cancer screening to reduce your risk of cancer. Your genetic counselor can explain your individual risk to you based on your results.
- A negative genetic test. A negative result, meaning a gene mutation wasn’t found, is more complicated. If other members of your family have Lynch syndrome with a known genetic mutation, but you didn’t have the mutation, your risk of cancer is the same as the general population. If you’re the first in your family to be tested for Lynch syndrome, a negative result may be misleading, since not everyone with Lynch syndrome has a genetic mutation that can be detected with current tests. You could still have a high risk of colon cancer — especially if you have a strong family history of colon cancer or your tumor testing revealed a high likelihood of Lynch syndrome.
- A gene variation of unknown significance. Genetic tests don’t always give you a yes or no answer about your cancer risk. Sometimes your genetic testing reveals a gene variation with an unknown significance. Your genetic counselor can explain the implications of this result to you.
Sometimes genetic testing for Lynch syndrome is done as part of a test for multiple cancer-related genetic mutations. Your genetic counselor can discuss the benefits and risks of genetic testing with you. He or she can explain what genetic testing can tell you and what it can’t.
Lynch syndrome management and treatment
Colon cancer associated with Lynch syndrome is treated similarly to other types of colon cancer. However, surgery for Lynch syndrome colon cancer is more likely to involve the removal of more of the colon, since people with Lynch syndrome have a high risk of developing additional colon cancer in the future. If colon cancer is detected, full colectomy with ileorectal anastomosis is recommended rather than a segmental/partial colonic resection because of the high risk for metachronous cancers. A meta-analysis of six studies including a total of 871 individuals found that based on an average of 91 months’ follow up, the rate of metachronous cancers was 23% among those individuals who had a segmental colectomy, compared to 6% among individuals who had a colectomy (colectomy defined as subtotal or colectomy with ileosigmoid anastomosis) 42.
Your treatment options will depend on the stage and location of your cancer, as well as your own health, age and personal preferences. Treatments for colon cancer may include surgery, chemotherapy and radiation therapy.
Population-based approaches for screening newly diagnosed individuals with colorectal cancer for Lynch syndrome generally rely on analysis of tumor tissue from the surgical resection specimen, but this will not provide information about the diagnosis of Lynch syndrome in time to make decisions about segmental/partial colectomy versus colectomy. For individuals presenting with colorectal cancer before age 50 and/or who have a strong family history, proceeding directly to genetic testing could be considered in order to have information about possible hereditary diagnoses in time for surgical planning.
The other tumors seen in Lynch syndrome are managed as in the general population.
Prophylactic hysterectomy and bilateral salpingo-oophorectomy can be considered after childbearing is completed.
Because screening colonoscopy with polypectomy is an effective preventive measure for colorectal cancer, prophylactic colectomy (removal of the colon prior to the development of cancer) is generally not recommended for individuals with Lynch syndrome.
Aspirin therapy has been shown to decrease the risk for colorectal cancer in individuals with Lynch syndrome.
Cancer screening for people with Lynch syndrome
If you have Lynch syndrome, but haven’t been diagnosed with an associated cancer — sometimes referred to as being a “previvor” — your doctor can develop a cancer-screening plan for you.
Stick to your doctor’s recommended plan. Screening for cancer may help your doctor find tumors at their earliest stages — when they’re more likely to be cured.
Research hasn’t established which cancer screening tests are best for people with Lynch syndrome. As a result, medical groups vary on which tests they recommend. Which tests are best for you may depend on your family history and which gene is causing your Lynch syndrome.
As part of your cancer-screening plan, your doctor may recommend you have:
- Colon cancer screening. A colonoscopy exam allows your doctor to see inside your entire colon and look for areas of abnormal growth that may indicate cancer. Colon cancer screening reduces the risk of dying of colon cancer by removing precancerous growths called polyps. People with Lynch syndrome typically begin colonoscopy screening every year or two starting in their 20s. People with Lynch syndrome tend to develop colon polyps that are more difficult to detect. For this reason, newer colonoscopy techniques may be recommended. High-definition colonoscopy creates more-detailed images and narrow band colonoscopy uses special light to create clearer images of the colon. Chromoendoscopy uses dyes to color colon tissue, which may make it more likely that the flat polyps that tend to occur more often in people with Lynch syndrome are detected.
- Endometrial cancer screening. Women with Lynch syndrome may have an annual endometrial biopsy or ultrasound to screen for cancer beginning in their 30s.
- Ovarian cancer screening. An ultrasound can be used to assess your ovaries and this may be recommended beginning in your 30s. By comparing annual ultrasound images, your doctor may be able to see changes to your ovaries that may indicate cancer. Your doctor may also recommend annual blood tests.
- Urinary system cancer screening. Your doctor may recommend periodic screening for urinary tract cancers. Analysis of a urine sample may reveal blood or cancerous cells.
- Gastrointestinal cancer screening. Your doctor may recommend endoscopy screening for stomach cancer and small intestine cancer. An endoscopy procedure allows your doctor to see your stomach and other parts of your gastrointestinal system.
While research proves the effectiveness of colon cancer screening for reducing the risk of dying of the disease, similar research hasn’t proved the effectiveness of screening for the other types of cancer. Still, experts recommend considering screening for these other types of cancer despite the lack of evidence.
Your doctor may recommend other cancer-screening tests if your family has a history of other cancers. Ask your doctor about what screening tests are best for you.
Aspirin for cancer prevention
Recent studies suggest taking a daily aspirin may reduce the risk of several cancers related to Lynch syndrome. More studies are needed to confirm this. Discuss the potential benefits and risks of aspirin therapy to determine whether this might be an option for you.
Surgery to prevent cancers caused by Lynch syndrome
In certain situations, people with Lynch syndrome may consider surgery to reduce their risk of cancer. Discuss the benefits and risks of preventive surgery with your doctor.
Surgical options for preventing cancer may include:
- Surgery to remove your colon (colectomy). Surgery to remove most or all of your colon will reduce or eliminate the chance that you’ll develop colon cancer. This procedure can be done in a way that allows you to expel waste normally without the need to wear a bag outside of your body to collect waste. Little evidence exists to show that removing your colon has any advantage over frequent cancer screening, in terms of helping you live longer. Yet, some people prefer the peace of mind or may prefer avoiding frequent colonoscopy exams.
- Surgery to remove your ovaries and uterus (oophorectomy and hysterectomy). Preventive surgery to remove your uterus eliminates the possibility that you’ll develop endometrial cancer in the future. Removing your ovaries can reduce your risk of ovarian cancer. Unlike with colon cancer, screening for ovarian cancer and endometrial cancer isn’t proved to reduce the risk of dying of cancer. For this reason, doctors usually recommend preventive surgery for women who have completed childbearing.
Coping and support
Knowing that you or your family members have an increased risk of cancer can be stressful. Helpful ways to cope might include:
- Find out all you can about Lynch syndrome. Write down your questions about Lynch syndrome and ask them at your next appointment with your doctor or genetic counselor. Ask your health care team for further sources of information. Learning about Lynch syndrome can help you feel more confident when making decisions about your health.
- Take care of yourself. Knowing that you have an increased risk of cancer can make you feel as if you can’t control your health. But control what you can. For instance, choose a healthy diet, exercise regularly and get enough sleep so that you wake feeling rested. Go to all of your scheduled medical appointments, including your cancer-screening exams.
- Connect with others. Find friends and family with whom you can discuss your fears. Talking with others can help you cope. Find other trusted people you can talk with, such as clergy members. Ask your doctor for a referral to a therapist who can help you understand your feelings.
- Chen S, Wang W, Lee S, Nafa K, Lee J, Romans K, Watson P, Gruber SB, Euhus D, Kinzler KW, Jass J, Gallinger S, Lindor NM, Casey G, Ellis N, Giardiello FM, Offit K, Parmigiani G. Colon Cancer Family Registry. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA. 2006;296:1479–87. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2538673/[↩][↩]
- Facts about Lynch Syndrome. https://www.lynchscreening.net/development/[↩]
- Kohlmann W, Gruber SB. Lynch Syndrome. Updated 2018 Feb 1. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1211/[↩][↩][↩]
- Järvinen HJ, Renkonen-Sinisalo L, Aktán-Collán K, Peltomäki P, Aaltonen LA, Mecklin JP. Ten years after mutation testing for Lynch syndrome: cancer incidence and outcome in mutation-positive and mutation negative family members. J Clin Oncol. 2009;27:4793–7. https://www.ncbi.nlm.nih.gov/pubmed/19720893[↩]
- Kempers MJ, Kuiper RP, Ockeloen CW, Chappuis PO, Hutter P, Rahner N, Schackert HK, Steinke V, Holinski-Feder E, Morak M, Kloor M, Büttner R, Verwiel ET, van Krieken JH, Nagtegaal ID, Goossens M, van der Post RS, Niessen RC, Sijmons RH, Kluijt I, Hogervorst FB, Leter EM, Gille JJ, Aalfs CM, Redeker EJ, Hes FJ, Tops CM, van Nesselrooij BP, van Gijn ME, Gómez García EB, Eccles DM, Bunyan DJ, Syngal S, Stoffel EM, Culver JO, Palomares MR, Graham T, Velsher L, Papp J, Oláh E, Chan TL, Leung SY, van Kessel AG, Kiemeney LA, Hoogerbrugge N, Ligtenberg MJ. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol. 2011;12:49–55. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3670774/[↩]
- Tutlewska K, Lubinski J, Kurzawski G. Germlie deletions in the EPCAM gene as a cause of Lynch syndrome. Hered Cancer Clin Pract. 2013;11:9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3765447/[↩]
- Kawakami H, Zaanan A, Sinicrope FA. Microsatellite instability testing and its role in the management of colorectal cancer. Curr Treat Options Oncol. 2015;16:30. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4594190/[↩]
- Lu KH, Dinh M, Kohlmann W, Watson P, Green J, Syngal S, Bandipalliam P, Chen LM, Allen B, Conrad P, Terdiman J, Sun C, Daniels M, Burke T, Gershenson DM, Lynch H, Lynch P, Broaddus RR. Gynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet Gynecol. 2005;105:569–74. https://www.ncbi.nlm.nih.gov/pubmed/15738026[↩]
- Obermair A, Youlden DR, Young JP, Lindor NM, Baron JA, Newcomb P, Parry S, Hopper JL, Haile R, Jenkins MA. Risk of endometrial cancer for women diagnosed with HNPCC-related colorectal carcinoma. Int J Cancer. 2010;127:2678–84. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2947566/[↩]
- Maxwell GL, Risinger JI, Alvarez AA, Barrett JC, Berchuck A. Favorable survival associated with microsatellite instability in endometrioid endometrial cancers. Obstet Gynecol. 2001;97:417–22. https://www.ncbi.nlm.nih.gov/pubmed/11239648[↩]
- Aarnio M, Salovaara R, Aaltonen LA, Mecklin JP, Jarvinen HJ. Features of gastric cancer in hereditary non-polyposis colorectal cancer syndrome. Int J Cancer. 1997;74:551–5. https://www.ncbi.nlm.nih.gov/pubmed/9355980[↩]
- Guilford PJ, Hopkins JB, Grady WM, Markowitz SD, Willis J, Lynch H, Rajput A, Wiesner GL, Lindor NM, Burgart LJ, Toro TT, Lee D, Limacher JM, Shaw DW, Findlay MP, Reeve AE. E-cadherin germline mutations define an inherited cancer syndrome dominated by diffuse gastric cancer. Hum Mutat. 1999;14:249–55. https://www.ncbi.nlm.nih.gov/pubmed/10477433[↩]
- Capelle LG, Van Grieken NC, Lingsma HF, Steyerberg EW, Klokman WJ, Bruno MJ, Vasen HF, Kuipers EJ. Risk and epidemiological time trends of gastric cancer in Lynch syndrome carriers in the Netherlands. Gastroenterology. 2010;138:487–92. https://www.ncbi.nlm.nih.gov/pubmed/19900449[↩]
- Park YJ, Shin KH, Park JG. Risk of gastric cancer in hereditary nonpolyposis colorectal cancer in Korea. Clin Cancer Res. 2000;6:2994–8. http://clincancerres.aacrjournals.org/content/6/8/2994.long[↩]
- Watson P, Vasen HF, Mecklin JP, Bernstein I, Aarnio M, Järvinen HJ, Myrhøj T, Sunde L, Wijnen JT, Lynch HT. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123:444–9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2627772/[↩]
- Watson P, Butzow R, Lynch HT, Mecklin JP, Jarvinen HJ, Vasen HF, Madlensky L, Fidalgo P, Bernstein I. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecol Oncol. 2001;82:223–8. https://www.ncbi.nlm.nih.gov/pubmed/11531271[↩]
- Schulmann K, Brasch FE, Kunstmann E, Engel C, Pagenstecher C, Vogelsang H, Kruger S, Vogel T, Knaebel HP, Ruschoff J, Hahn SA, Knebel-Doeberitz MV, Moeslein G, Meltzer SJ, Schackert HK, Tympner C, Mangold E, Schmiegel W. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology. 2005;128:590–9. https://www.ncbi.nlm.nih.gov/pubmed/15765394[↩][↩]
- van der Post RS, Kiemeny LA, Ligtenberg MJ, Witjes JA, Hulsbergen-van de Kaa CA, Bodmer D, Schaap L, Kets CM, van Krieken JH, Hoogerbrugge N. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet. 2010;47:464–70. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2991077/[↩]
- Win AK, Hopper JL, Buchanan DD, Young JP, Tenesa A, Dowty JG, Giles GG, Goldblatt J, Winship I, Boussioutas A, Young GP, Parry S, Baron JA, Duggan D, Gallinger S, Newcomb PA, Haile RW, Le Marchand L, Lindor NM, Jenkins MA. Are the common genetic variants associated with colorectal cancer risk for DNA mismatch repair gene mutation carriers? Eur J Cancer. 2013a;49:1578–87. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3625445/[↩][↩]
- Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet. 2008;124:105–22. https://www.ncbi.nlm.nih.gov/pubmed/18709565[↩]
- Suzui M, Yoshimi N, Hara A, Morishita Y, Tanaka T, Mori H. Genetic alterations in a patient with Turcot’s syndrome. Pathol Int. 1998;48:126–33. https://www.ncbi.nlm.nih.gov/pubmed/9589476[↩]
- Machin P, Catasus L, Pons C, Munoz J, Conde-Zurita JM, Balmana J, Barnadas M, Marti RM, Prat J, Matias-Guiu X. Microsatellite instability and immunostaining for MSH-2 and MLH-1 in cutaneous and internal tumors from patients with the Muir-Torre syndrome. J Cutan Pathol. 2002;29:415–20. https://www.ncbi.nlm.nih.gov/pubmed/12139636[↩]
- South CD, Hampel H, Comeras I, Westman JA, Frankel WL, de la Chapelle A. The frequency of Muir-Torre syndrome among Lynch syndrome families. J Natl Cancer Inst. 2008;100:277–81. https://www.ncbi.nlm.nih.gov/pubmed/18270343[↩]
- Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, Bandipalliam P, Stoffel EM, Gruber SB, Syngal S. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302:1790–5. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4091624/[↩][↩]
- Win AK, Young JP, Lindor NM, Tucker KM, Ahnen DJ, Young GP, Buchanan DD, Clendenning M, Giles GG, Winship I, Macrae FA, Goldblatt J, Southey MC, Arnold J, Thibodeau SN, Gunawardena SR, Bapat B, Baron JA, Casey G, Gallinger S, Le Marchand L, Newcomb PA, Haile RW, Hopper JL, Jenkins MA. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30:958–64. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3341109/[↩]
- Barrow E, Robinson L, Alduaij W, Shenton A, Clancy T, Lalloo F, Hill J, Evans DG. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet. 2009;75:141–9. https://www.ncbi.nlm.nih.gov/pubmed/19215248[↩]
- Gargiulo S, Torrini M, Ollila S, Nasti S, Pastorino L, Cusano R, Bonelli L, Battistuzzi L, Mastracci L, Bruno W, Savarino V, Sciallero S, Borgonovo G, Nyström M, Bianchi-Scarrà G, Mareni C, Ghiorzo P. Germline MLH1 and MSH2 mutations in Italian pancreatic cancer patients with suspected Lynch syndrome. Fam Cancer. 2009;8:547–53. https://www.ncbi.nlm.nih.gov/pubmed/19728162[↩]
- Raymond VM, Mukherjee B, Wang F, Huang SC, Stoffel EM, Kastrinos F, Syngal S, Cooney KA, Gruber SB. Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol. 2013b;31:1713–8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3641694/[↩][↩]
- Haraldsdottir S, Hampel H, Wei L, Wu C, Frankel W, Bekaii-Saab T, de la Chapelle A, Goldberg RM. Prostate cancer incidence in males with Lynch syndrome. Genet Med. 2014;16:553–7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4289599/[↩]
- Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R, Elemento O, Rubin MA, Robinson D, Lonigro R, Hussain M, Chinnaiyan A, Vinson J, Filipenko J, Garraway L, Taplin ME, AlDubayan S, Han GC, Beightol M, Morrissey C, Nghiem B, Cheng HH, Montgomery B, Walsh T, Casadei S, Berger M, Zhang L, Zehir A, Vijai J, Scher HI, Sawyers C, Schultz N, Kantoff PW, Solit D, Robson M, Van Allen EM, Offit K, de Bono J, Nelson PS. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–53. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4986616/[↩]
- Nilbert M, Therkildsen C, Nissen A, Akerman M, Bernstein I. Sarcomas associated with hereditary nonpolyposis colorectal cancer: broad anatomical and morphological spectrum. Fam Cancer. 2009;8:209–13. https://www.ncbi.nlm.nih.gov/pubmed/19130300[↩][↩]
- Raymond VM, Everett JN, Furtado LV, Gustafson SL, Jungbluth CR, Gruber SB, Hammer GD, Stoffel EM, Greenson JK, Giordano TJ, Else T. Adrenocortical carcinoma is a lynch syndrome-associated cancer. J Clin Oncol. 2013a;31:3012–8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3739861/[↩]
- Peltomäki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21:1174–9. https://www.ncbi.nlm.nih.gov/pubmed/12637487[↩]
- Kastrinos F, Steyerberg EW, Mercado R, Balmana J, Holter S, Gallinger S, Siegnund KD, Church JM, Jenkins MA, Lindor NM, Thibodeau SN, Burbidge LA, Wenstrup RJ, Syngal S. The PREMM(1,2,6) Model predicts risk of MLH1, MSH2, and MSH6 germline mutations. Gastroenterology. 2011;140:73–81. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3125673/[↩]
- Kastrinos F, Ojha RP, Leenen C, Alvero C, Mercado RC, Balmaña J, Valenzuela I, Balaguer F, Green R, Lindor NM, Thibodeau SN, Newcomb P, Win AK, Jenkins M, Buchanan DD, Bertario L, Sala P, Hampel H, Syngal S, Steyerberg EW. Lynch syndrome prediction model validation study group. Comparison of prediction models for Lynch syndrome among individuals with colorectal cancer. J Natl Cancer Inst. 2015;108(2). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4862416/[↩]
- Bethesda Guidelines and National Comprehensive Cancer Network. https://www.nccn.org/professionals/physician_gls/default.aspx#colorectal_screening[↩]
- Smith MJ, Urquhart JE, Harkness EF, Miles EK, Bowers NL, Byers HJ, Bulman M, Gokhale C, Wallace AJ, Newman WG, Evans DG. The Contribution of Whole Gene Deletions and Large Rearrangements to the Mutation Spectrum in Inherited Tumor Predisposing Syndromes. Hum Mutat. 2016;37:250–6. https://www.ncbi.nlm.nih.gov/pubmed/26615784[↩]
- https://www.lynchscreening.net/development/[↩]
- Ladabaum U, Wang G, Terdiman J, Blanco A, Kuppermann M, Boland CR, Ford J, Elkin E, Phillips KA. Strategies to identify Lynch syndrome among patients with colorectal cancer: a cost-effectiveness analysis. Ann Intern Med. 2011;155:69–79. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3793257/[↩]
- Mange S, Bellcross C, Cragun D, Duquette D, Gorman L, Hampel H, Jasperson J. Creation of a network to promote universal screening for Lynch syndrome: the Lynch syndrome screening network. J Genet Couns. 2015;24:421–7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4547789/[↩]
- Rabban JT, Calkins SM, Karnezis AN, Grenert JP, Blanco A, Crawford B, Chen LM. Association of Tumor Morphology With Mismatch-repair Protein Status in Older Endometrial Cancer Patients: Implications for Universal Versus Selective Screening Strategies for Lynch Syndrome. Am J Surg Pathol. 2014;38:793–800. https://www.ncbi.nlm.nih.gov/pubmed/24503759[↩]
- Anele CC, Adegbola SO, Askari A, Rajendran A, Clark SK, Latchford A, Faiz OD. Risk of metachronous colorectal cancer following colectomy in Lynch syndrome: a systematic review and meta-analysis. Colorectal Dis. 2017;19:528–36. https://www.ncbi.nlm.nih.gov/pubmed/28407411[↩]





{kind=link}