Contents
What is methemoglobinemia
Methemoglobinemia (MetHb) is a potentially life-threatening blood disorder in which an abnormal amount of methemoglobin (MetHb) is produced 1, 2, 3, 4, 5, 6, 7. Hemoglobin (Hb) is the protein in red blood cells that carries and distributes oxygen to your body. Oxygen is essential to life, and your body need a certain amount of oxygen to function properly. Oxygen enters your body through your nose or mouth when you breathe (inhale) and passes through your lungs into your bloodstream. Once in your bloodstream, oxygen is carried by the hemoglobin (Hb) in red blood cells to cells all over your body. All of your cells need oxygen to create energy efficiently, and your body needs energy to fulfill all of its processes, such as digestion and even thinking. Your body tightly regulates the amount of oxygen saturation in your blood, because low blood oxygen levels (hypoxemia) can lead to many serious conditions and damage to individual organ systems, especially your brain and heart. Methemoglobin (MetHb) is a form of hemoglobin (Hb) where some or all of the four iron (Fe) within the hemoglobin molecule has been oxidized from the reduced ferrous (Fe2+) state to the oxidized ferric (Fe3+) state 8. Ferric iron (Fe3+) is unable to bind and transport oxygen. This change in the iron molecule from the reduced ferrous (Fe2+) state to the oxidized ferric (Fe3+) state prevents the hemoglobin from effectively carrying oxygen to your body’s tissues, potentially leading to a condition called methemoglobinemia 9. Normally, red blood cells contain less than 2 percent methemoglobin as a naturally occurring oxidized metabolite of hemoglobin. However, when methemoglobin (MetHb) levels increase with methemoglobinemia, the blood is not able to release oxygen effectively to body tissues resulting in functional anemia. The end result of these changes is decreased oxygen delivery leading to tissue hypoxia. The resulting lack of oxygen throughout the body can cause symptoms such as pale or blue-colored skin or cyanosis.
Normal red blood cells contain molecules of iron-containing hemoglobin, which deliver oxygen to the body’s tissues. The iron in hemoglobin is the reduced ferrous (Fe2+), but it can spontaneously become the oxidized ferric (Fe3+). Hemoglobin that contains the oxidized ferric (Fe3+) iron is called methemoglobin (MetHb), and it cannot deliver oxygen.
Methemoglobinemia condition can be:
- Passed down through families (inherited or congenital) also known as autosomal recessive methemoglobinemia, congenital methemoglobinemia or hereditary methemoglobinemia. Congenital methemoglobinemia or hereditary methemoglobinemia are caused by biallelic mutations in the CYB5R3 gene that codes the NADH cytochrome b5 reductase enzyme also known as NADH diaphorase, which is very rare and the actual incidence is not known 10. To date, more than 80 different disease‐causing gene variants in the CYB5R3 gene have been reported 11, 12, 10. Increased frequency of hereditary methemoglobinemia has been found in Siberian Yakuts, Athabaskans, Eskimos, and Navajo 13. Cytochrome-b5 reductase enzyme utilizes NADH formed during glycolysis to reduce methemoglobin back to functional hemoglobin 14. In addition to the autosomal recessive methemoglobinemia subtypes, a rare methemoglobin group called hemoglobin M disease (M group variants of hemoglobin), results from autosomal dominant mutations (variants) in the genes encoding alpha or beta globin proteins of hemoglobin due to substitution of an amino acid with tyrosine in the alpha‐globin (HBA1, HBA2), beta‐globin (HBB), or gamma‐globin (HBG1, HBG2) 1. Multiple variants of hemoglobin M have been described (Boston, Fort Ripley, Hyde Park, Iwate, Kankakee, Osaka, Saskatoon) 15, 16. In very rare cases, inherited methemoglobinemia is caused by deficiency of the electron acceptor cytochrome b5 17. This causes methemoglobinemia associated with ambiguous genitalia due to a homozygous variant in the CYB5A gene, encoding the microsomal cytochrome b5 18. This condition is due to isolated 17,20‐lyase deficiency since the cofactor cytochrome b5 is required for optimal 17,20‐lyase activity 19.
- Acquired methemoglobinemia caused by exposure to certain drugs, chemicals, or foods (acquired). The most common drugs are benzocaine and lidocaine 20, 21, 22.
Acquired methemoglobinemia is much more common than the inherited methemoglobinemia and, is the result of exposure to substances that cause oxidation of the hemoglobin either directly or indirectly. This exposure results in the production of methemoglobin (hemoglobin with oxidized ferric (Fe3+) iron) that exceeds the body’s capacity to convert the ferric (Fe3+) iron within the hemoglobin back to its ferrous (Fe2+) state. Acquired methemoglobinemia may be due to exposure to direct oxidizing agents (e.g. benzocaine and prilocaine), indirect oxidation (e.g. nitrates), or metabolic activation (e.g. aniline and dapsone) 23. Classic examples include patient exposure to benzocaine in endoscopy suite and infantile exposure to nitrites from groundwater and wells in rural areas 24.
Most cases of acquired methemoglobinemia are due to accidental exposure to a chemical or through the use of topical or local anesthetics 8. A single center review of 28,478 transesophageal echocardiograms found the incidence of methemoglobinemia to be 0.067% 21. A systematic review of cases of local anesthetic-related methemoglobinemia found that benzocaine was an agent in two-thirds of cases 25. This higher association with benzocaine-containing products has led the United States Food and Drug Administration (FDA) to release multiple advisories regarding the use of benzocaine-containing oral products 26.
Under normal physiologic circumstances, NADPH (nicotinamide adenine dinucleotide phosphate)-MetHb reductase contributes very little to the reduction of methemoglobin, but under oxidative stress, the function of this alternative reduction pathway (NADPH-MetHb reductase) can be enhanced by the presence of exogenous electron donors, such as methylene blue. Methemoglobinemia secondary to accidental exposure to a chemical or through the use of topical or local anesthetics occurs when cytochrome-b5 reductase (CYB5R) ability to reduce ferric (Fe3+) hemoglobin, or methemoglobin, is overwhelmed by the induced oxidant stress. The result is increasing concentrations of methemoglobin leading to methemoglobinemia.
Percentage of methemoglobin is calculated by dividing the concentration of methemoglobin by the concentration of total hemoglobin. Percentage of methemoglobin is likely a better indicator of illness severity than overall concentration, as underlying medical conditions play an important role 8. For example, a methemoglobin concentration of 1.5 g/dL may represent a percentage of 10% in an otherwise healthy patient with a baseline hemoglobin of 15 mg/dL, whereas the presence of the same concentration of 1.5 g/dL of methemoglobin in an anemic patient with a baseline hemoglobin of 8 g/dL would represent a percentage of 18.75% 8. The healthy patient will be left with a functional hemoglobin concentration of 13.5 g/dL and potentially remain asymptomatic while the anemic patient with a functional hemoglobin concentration 6.5 g/dL may be severely symptomatic with a methemoglobin of less than 20%. This may be further compounded by the “functional hemoglobin’s” decreased ability to release oxygen in the presence of methemoglobin. Anemia, congestive heart failure, chronic obstructive pulmonary disease (COPD), and essentially any pathology that impairs the ability to deliver oxygen may worsen the symptoms of methemoglobinemia 8.
In the otherwise healthy person, cyanosis may be clinically evident with a methemoglobin as low as 10% 9. The classic appearance of “chocolate brown blood” can be present at as low as 15% methemoglobin. As the percentage of methemoglobinemia approaches 20%, the patient may experience anxiety, light-headedness, and headaches 8. At methemoglobin levels of 30% to 50%, there may be abnormally rapid breathing (tachypnea), confusion, and loss of consciousness 8. Approaching 50% methemoglobinemia, the patient is at risk for seizures, dysrhythmias, metabolic acidosis, and coma 8. Levels above 70% methemoglobinemia are often fatal 27.
Figure 1. Methemoglobin (MetHb)
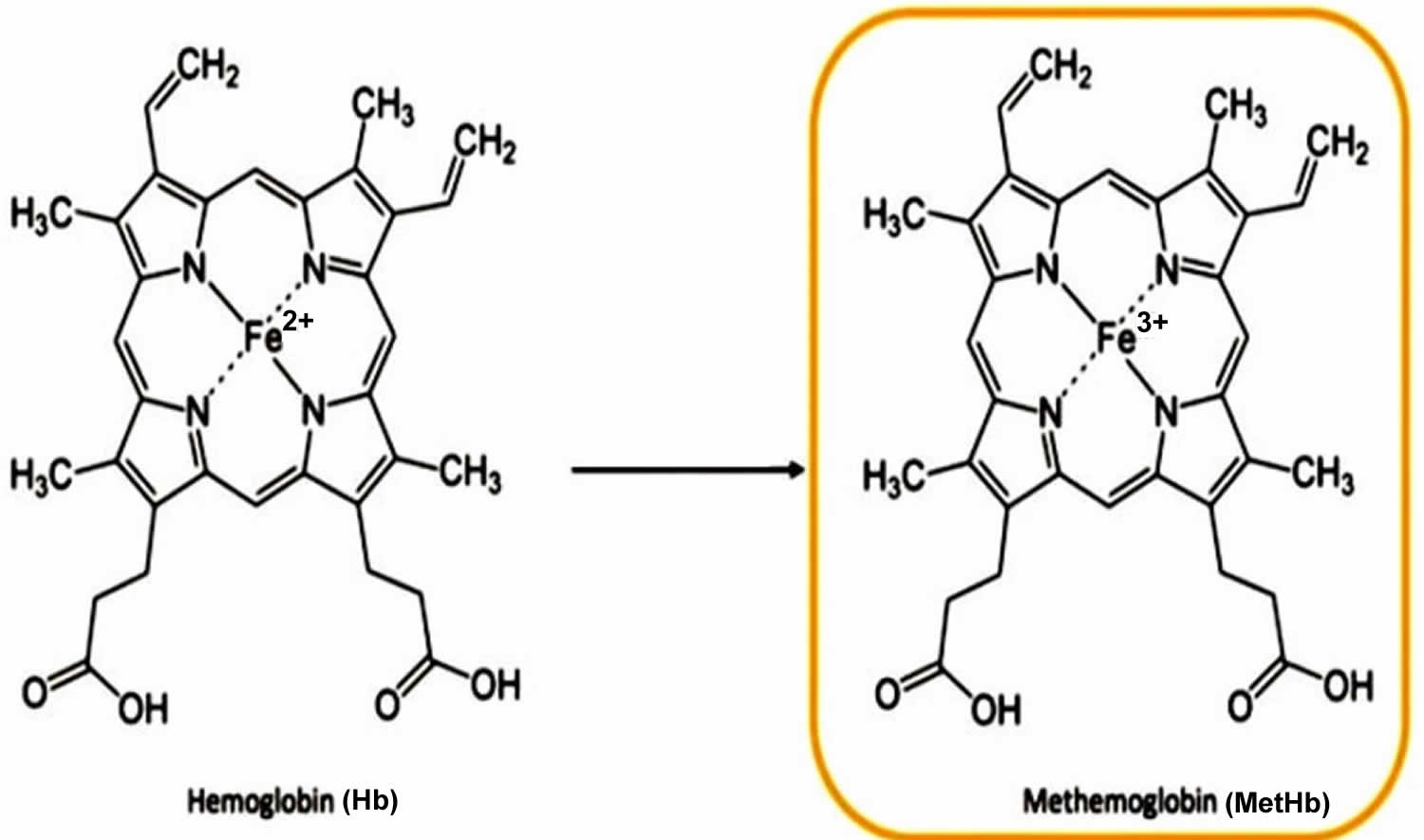
Figure 2. Methemoglobin formation
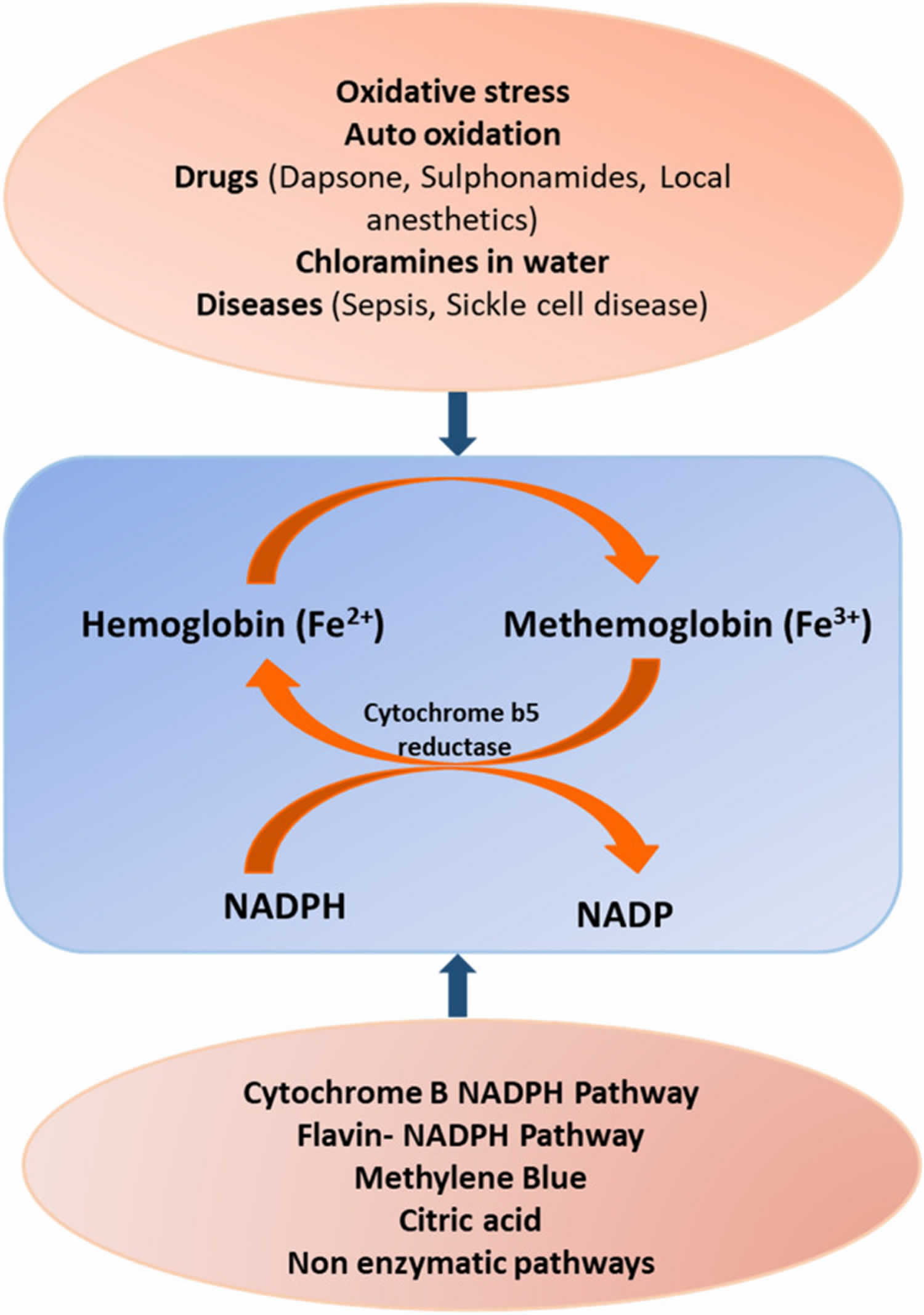
Footnotes: Pathophysiology of methemoglobin (MetHb) formation showing oxidation of hemoglobin to methemoglobin and its reversal to hemoglobin by reductase and the factors involved. Methylene blue works as cofactor in transferring electron to methemoglobin (MetHb) from NADPH 28, 29. Methylene blue is oxidized into leucomethylene blue by accepting an electron from NADPH (NADPH-methemoglobin reductase), which it then delivers to methemoglobin (Fe3+), converting it into hemoglobin (Fe2+) 30. Response to treatment is typically seen in 30 to 60 minutes and can be re-dosed if needed.
[Source 31 ]What is hemoglobin?
Hemoglobin (Hb or Hgb) is the iron-containing protein found in all red blood cells (RBCs) that gives red blood cells their characteristic red color and it carries oxygen (O2) throughout the body. Hemoglobin enables red blood cells to bind to oxygen in the lungs and carry oxygen to tissues and organs throughout your body. Hemoglobin also helps transport a small portion of carbon dioxide (CO2), a product of cell metabolism, from tissues and organs to the lungs, where it is exhaled 32.
Hemoglobin is made up of heme, which is the iron-containing portion, and globin chains, which are proteins. The globin protein consists of chains of amino acids, the “building blocks” of proteins. One portion of hemoglobin called heme is the molecule with iron at the center (see Figure 1). Another portion is made of up four protein chains called globins. Depending on their structure, the globin chains are designated as alpha (α), beta (ß), delta (δ), and gamma (γ). Each of the four globin chains holds a heme group containing one iron atom. .
Not all hemoglobin is the same. Different types of hemoglobin are classified according to the type of globin chains they contain. The type of globin chains present is important in hemoglobin’s ability to transport oxygen.
Normal hemoglobin types include:
- Hemoglobin A (Hb A) – this is the predominant type of hemoglobin (Hb) in adults, makes up about 95%-98% of hemoglobin found in adults; Hb A contains two alpha (α) protein chains and two beta (ß) protein chains.
- Hemoglobin A2 (Hb A2) – makes up about 2-3.5% of hemoglobin (Hb) found in adults; it has two alpha (α) and two delta (δ) protein chains.
- Hemoglobin F (Hb F, fetal hemoglobin) – makes up to 1%-2% of hemoglobin (Hb) found in adults; it has two alpha (α) and two gamma (γ) protein chains. Hemoglobin F (Hb F, fetal hemoglobin) is the primary hemoglobin produced by a developing baby (fetus) during pregnancy. Its production usually falls to a low level within a year after after birth and reaches adult level within 1-2 years.
Under normal circumstances, adult blood contains 4 types of hemoglobin: oxygenated hemoglobin or oxyhemoglobin, hemoglobin that contains no oxygen (reduced hemoglobin) or deoxyhemoglobin, hemoglobin transporting carbon dioxide or carboxyhemoglobin, and oxidized hemoglobin or methemoglobin. The last 2 are usually found in small amounts in blood.
Under normal circumstances, most of the hemoglobin iron exists in the ferrous (Fe+2) state, and a small fraction of the hemoglobin present in erythrocytes undergoes oxidation. As a result of this process, some of the hemoglobin iron is converted to the ferric (Fe+3) state, forming methemoglobin. Oxidized hemoglobin cannot bind or carry oxygen. The physiologic level of methemoglobin is less than or equal to 1% of total hemoglobin concentration, and when greater than 1% of hemoglobin is oxidized to methemoglobin, a hemoglobin deficiency occurs. In normal blood, the methemoglobin reductase system maintains methemoglobin in equilibrium with deoxygenated hemoglobin 33.
Figure 3. Red blood cell hemoglobin
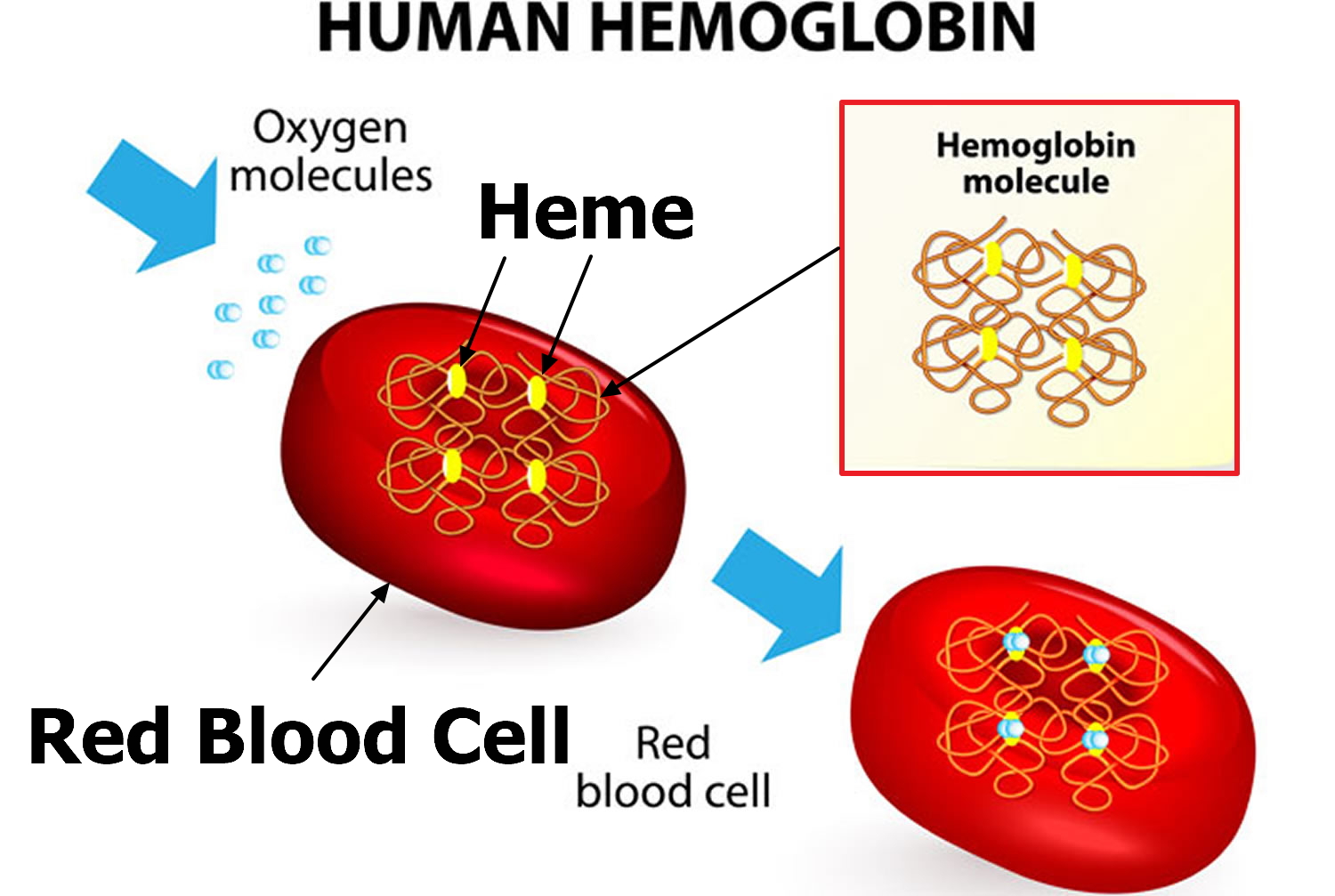
Figure 4. Structure of hemoglobin
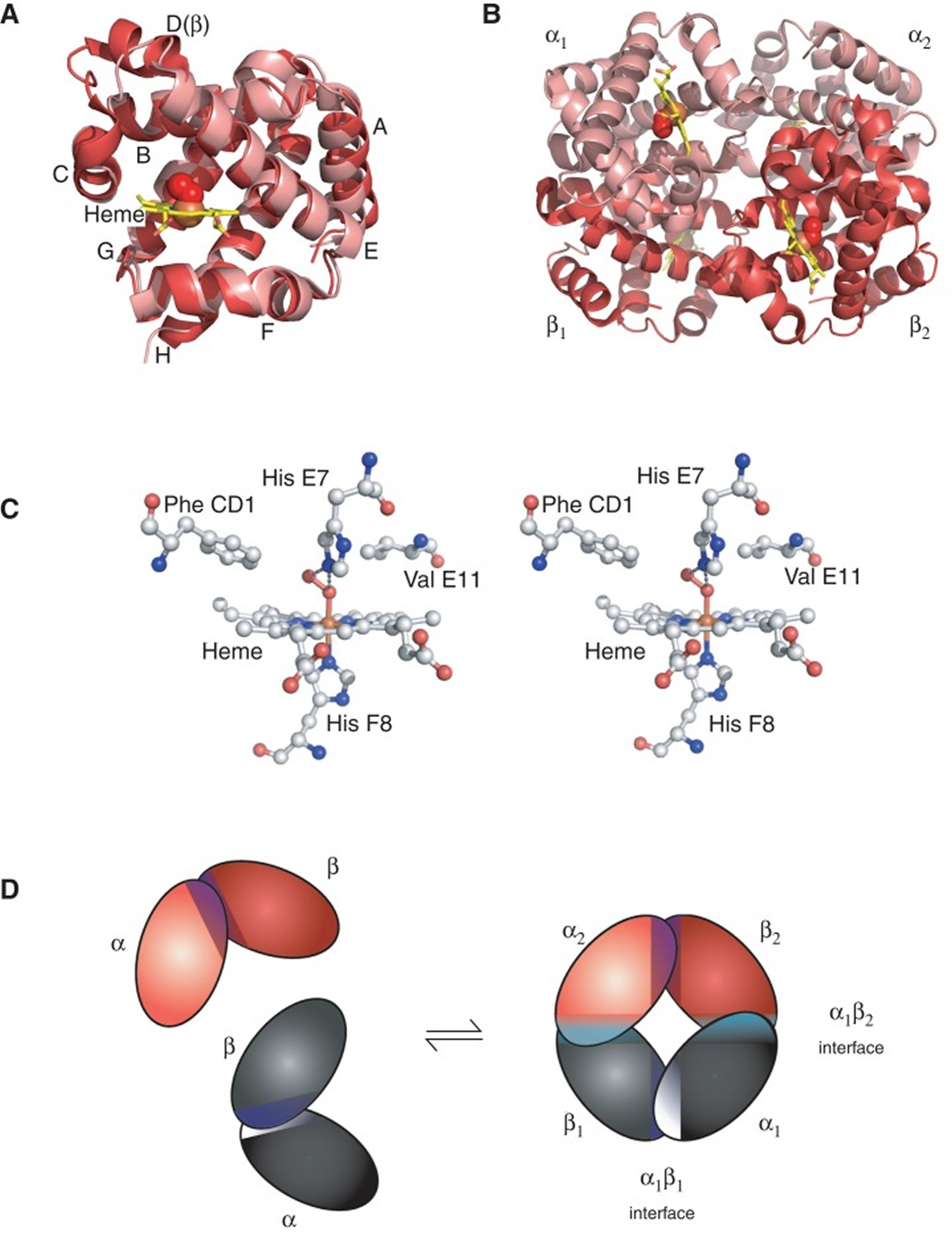 Footnotes: The structure of hemoglobin (Hb). (A) The α (pink) and β (red) hemoglobin (Hb) subunits have conserved α-helical folds. Helices are labeled A–H from the amino terminus. The α subunit lacks helix D. (B) The high O2 affinity R state quaternary structure of hemoglobin (Hb) with O2 (red spheres) bound at all four heme sites (protoporphyrin-IX as yellow sticks, with central iron atom as orange sphere). (C) Stereo (wall-eye) diagram of the heme pocket of β showing the proximal (F8) and distal (E7) histidines and selected residues in the distal heme pocket that influence ligand binding and autoxidation. (D) hemoglobin (Hb) tetramer is assembled from two identical αβ dimers (shown in red and gray for clarity). In the tetramer, each subunit makes contact with the unlike chain through a high affinity dimerization α1β1 interface and a lower affinity α1β2 dimer–tetramer interface (cyan).
Footnotes: The structure of hemoglobin (Hb). (A) The α (pink) and β (red) hemoglobin (Hb) subunits have conserved α-helical folds. Helices are labeled A–H from the amino terminus. The α subunit lacks helix D. (B) The high O2 affinity R state quaternary structure of hemoglobin (Hb) with O2 (red spheres) bound at all four heme sites (protoporphyrin-IX as yellow sticks, with central iron atom as orange sphere). (C) Stereo (wall-eye) diagram of the heme pocket of β showing the proximal (F8) and distal (E7) histidines and selected residues in the distal heme pocket that influence ligand binding and autoxidation. (D) hemoglobin (Hb) tetramer is assembled from two identical αβ dimers (shown in red and gray for clarity). In the tetramer, each subunit makes contact with the unlike chain through a high affinity dimerization α1β1 interface and a lower affinity α1β2 dimer–tetramer interface (cyan).What causes methemoglobinemia
Methemoglobinemia can result from either inherited (passed down through families or congenital) or acquired processes 8.
Acquired methemoglobinemia
Acquired methemoglobinemia is more common than the inherited forms. Acquired methemoglobinemia occurs in some people after they are exposed to certain chemicals, toxins and drugs that cause oxidation of the hemoglobin either directly or indirectly, including 34, 35:
- Anesthetics such as benzocaine, bupivacaine, lidocaine, prilocaine
- Methylene blue
- Nitrobenzene
- Certain antibiotics (including dapsone and chloroquine)
- Nitrites (used as additives to prevent meat from spoiling)
- Adulterants used in cocaine (local anesthetics, phenacetin)
- Amyl nitrite (poppers)
- Isobutyl nitrite
- Sodium nitrite
- Aniline
- Chloramine
- Metoclopramide
- Nitrate
- Nitric oxide
- Nitrous oxide (laughing gas)
- Nitroglycerin
- Nitroprusside
- Nitrofuran
- Paraquat/monolinuron
- Phenazopyridine (Pyridium)
- Quinones (e.g., chloroquine)
- Rifampin
- Sulfonamides (e.g., sulfamethoxazole)
- Certain foods, such as spinach, beets or carrots contain natural nitrates in large amounts. These foods should not be given to children younger than 6 months of age.
Drugs or toxins that can cause methemoglobinemia 36:
- Acetanilid
- Alloxan
- Aniline
- Arsine
- Benzene derivatives
- Benzocaine
- Bivalent copper
- Bismuth subnitrate
- Bupivacaine hydrochloride
- Chlorates
- Chloroquine
- Chromates
- Clofazimine
- Dapsone
- Dimethyl sulfoxide
- Dinitrophenol
- Exhaust fumes
- Ferricyanide
- Flutamide
- Hydroxylamine
- Lidocaine hydrochloride
- Metoclopramide hydrochloride
- Methylene blue
- Naphthalene
- Nitrates
- Nitric oxide
- Nitrites
- Nitrofuran
- Nitroglycerin
- Sodium nitroprusside
- Paraquat
- Phenacetin
- Phenazopyridine hydrochloride
- Phenol
- Phenytoin
- Prilocaine hydrochloride
- Primaquine phosphate
- Rifampin
- Silver nitrate
- Sodium valproate
- Smoke inhalation
- Sulfasalazine
- Sulfonamides
- Trinitrotoluene
Footnote: *Certain drugs are more likely to cause methemoglobinemia than others. These are dapsone, local anesthetics, phenacetin, and antimalarial drugs. Screening everybody for methemoglobinemia before exposing them to these drugs is impractical because of the rarity of the condition and because a growing number of drugs are implicated in its causation.
Table 1. Agents known to induce methemoglobinemia
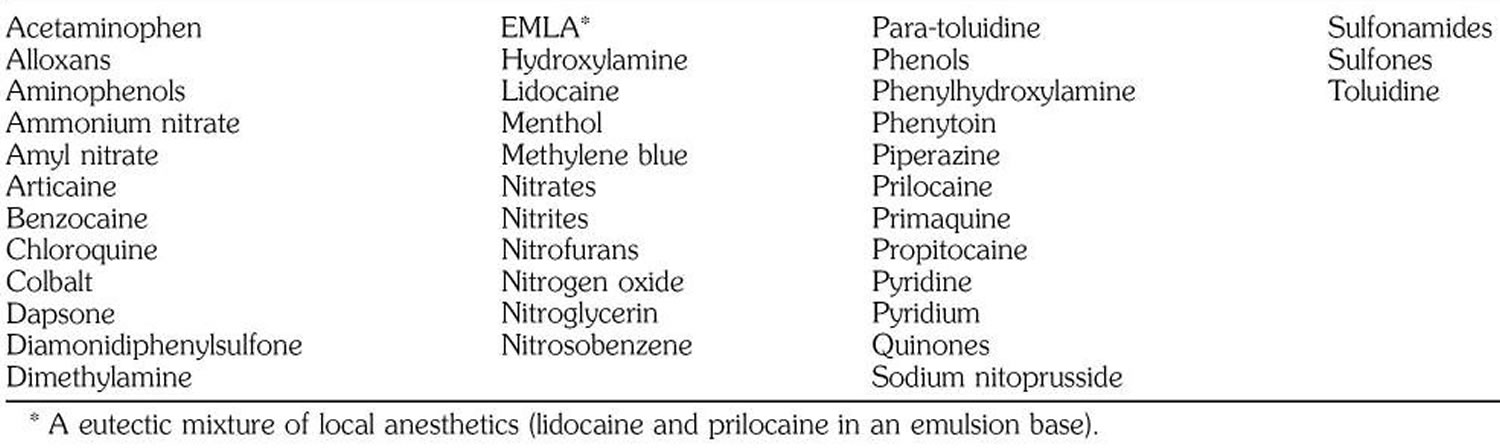
Exposure to certain drugs, toxins and chemical results in the production of methemoglobin that exceeds the body’s capacity to convert the iron within the hemoglobin back to its ferrous (Fe2+) state. Acquired methemoglobinemia may be due to exposure to direct oxidizing agents (e.g. benzocaine and prilocaine), indirect oxidation (e.g. nitrates), or metabolic activation (e.g. aniline and dapsone) 23. Classic examples include patient exposure to benzocaine in endoscopy suite and infantile exposure to nitrites from groundwater and wells in rural areas 24, 38.
Some recreational drugs are also associated with acquired methemoglobinemia, including amyl nitrate (poppers), nitrous oxide (laughing gas) and adulterants used in cocaine (local anesthetics, phenacetin). These can be associated with very high levels of methemoglobinemia (>90%) and fatalities have been reported 39.
Drugs that induce methemoglobinemia often also cause breakdown of red blood cells (hemolysis). Newborn infants are particularly susceptible to the development of methemoglobinemia because the activity of cytochrome b5 reductase is lower compared to adult period 40. Therefore, methemoglobinemia is observed in infants because of toxic materials, such as aniline dyes used on diapers 41 and the ingestion of nitrate‐contaminated water 42, 43 and even of consumption of silver beets and incorrect storage of homemade purées of mixed vegetables in infants 44.
Inhaled nitric oxide (NO) is approved for treatment of infants with pulmonary hypertension because of its vasodilatory effect on pulmonary vessels. During the binding and release of nitric oxide from hemoglobin, methemoglobin is formed at a higher rate. In a study of 163 infants with pulmonary hypertension treated with inhaled nitric oxide, methemoglobin was above 5% in one and between 2.5% and 5% in 16 infants 45.
Large amounts of nitric oxide are released in patients with sepsis. Nitric oxide is converted to methemoglobin and nitrate. It has been reported that methemoglobin levels are significantly higher in patients with sepsis than in nonseptic patients 46.
Methemoglobinemia has been reported in young infants (<6 months) in whom severe metabolic acidosis develops from diarrhea and dehydration 47. Young infants may be particularly susceptible to this complication because of their low stomach acid production, large number of nitrite-reducing bacteria, and the relatively easy oxidation of fetal hemoglobin. Small infants have lower erythrocyte levels of cytochrome b reductase 48. Higher intestinal pH of infants may promote the growth of gram-negative organisms that convert dietary nitrates to nitrites.
Methemoglobinemia has been reported in diarrhea induced by hypersensitivity to cow’s milk proteins 49. It has also been reported in association with renal tubular acidosis 50.
Benzocaine-induced methemoglobinemia
A wide variety of agents are known to induce methemoglobinemia (see Table 1), including lidocaine. Benzocaine has been reported to cause methemoglobinemia when applied to infants as an ointment or a rectal suppository 51 and when used topically to the perineal area 52. It has also been associated with methemoglobinemia after its use as a lubricant on endotracheal, bronchoscopic, and esophageal tubes 53. The particular preparation of benzocaine used in this case contained 20% benzocaine (Hurricane) 54.
Methemoglobinemia has also been observed after the use of other local anesthetics. Prilocaine has been implicated most frequently 55. Cyanosis can develop after administration of a dose as low as 500 mg for major nerve block 56. Methemoglobinemia has been also reported after topical use of a eutectic mixture of the local anesthetics lidocaine and prilocaine in children 57. Other commonly used drugs known to cause methemoglobinemia are phenacetin, acetaminophen, vasodilators, sulfonamides, and dapsone.
The onset of methemoglobinemia may be immediate or delayed.
Inherited methemoglobinemia
Inherited methemoglobinemia are caused biallelic mutations in the CYB5R3 gene that codes the NADH cytochrome b5 reductase enzyme also known as NADH diaphorase 10. To date, more than 80 different disease‐causing gene variants in the CYB5R3 gene have been reported 11, 12, 10. Increased frequency of hereditary methemoglobinemia has been found in Siberian Yakuts, Athabaskans, Eskimos, and Navajo 13. Cytochrome-b5 reductase enzyme utilizes NADH formed during glycolysis to reduce methemoglobin back to functional hemoglobin 14.
There are 2 forms of inherited methemoglobinemia. Inherited forms of methemoglobinemia are due to autosomal recessive inheritence in the CYB5R3 genes that code the cytochrome b5 reductase enzyme (CYB5R) 8. The first form of inherited methemoglobinemia is passed on by both parents is also called autosomal recessive congenital methemoglobinemia. The parents usually do not have the condition themselves (autosomal recessive inheritance pattern). They carry the gene that causes the methemoglobinemia. The CYB5R3 gene provides instruction for making an enzyme called cytochrome b5 reductase 3. This enzyme is involved in transferring negatively charged particles called electrons from one molecule to another. Two versions (isoforms) of cytochrome b5 reductase 3 enzyme are produced from the CYB5R3 gene. The soluble isoform is present only in red blood cells, and the membrane-bound isoform is found in all other cell types. The membrane-bound cytochrome b5 reductase 3 isoform is embedded in the membranes of various cellular compartments and is widely used in the body. This cytochrome b5 reductase 3 isoform is necessary for many chemical reactions, including the breakdown and formation of fatty acids, the formation of cholesterol, and the breakdown of various molecules and drugs 58.
There are two types of inherited methemoglobinemia:
- Congenital methemoglobinemia type 1 also called erythrocyte cytochrome B5 reductase deficiency occurs when only the red blood cells lack the cytochrome B5 reductase enzyme. Congenital methemoglobinemia type 1 is mainly due to missense mutations (variants) in the CYB5R3 gene that cause a production of an unstable cytochrome B5 reductase enzyme purely in the red blood cells, associated with methemoglobin levels above 25%, cyanosis, headache, fatigue, and dyspnea. In these cases, cyanosis may be the only sign since most of congenital methemoglobinemia type 1 patients are asymptomatic.
- Congenital methemoglobinemia type 2 also called generalized cytochrome B5 reductase deficiency occurs when all cells lack the cytochrome B5 reductase enzyme 59. Congenital methemoglobinemia type 2 is caused by CYB5R3 gene mutations that lead to either low expression or low activity of the cytochrome B5 reductase enzyme in all the tissues and associated with alterations in the lipid metabolism and neurological involvement. In congenital methemoglobinemia type 2, 8% to 40% of the hemoglobin is in the form of methemoglobin (MetHb) 10. Congenital methemoglobinemia type 2 is associated with high morbidity and mortality because of severe neurologic involvements 1.
- In addition to the autosomal recessive methemoglobinemia subtypes above, a rare methemoglobin group called M group variants of hemoglobin or hemoglobin M disease, results from autosomal dominant mutations (variants) in the genes encoding alpha or beta globin proteins of hemoglobin due to substitution of an amino acid with tyrosine in the alpha‐globin (HBA1, HBA2), beta‐globin (HBB), or gamma‐globin (HBG1, HBG2) 1. In most forms of the hemoglobin M disease, tyrosine is substituted for either the proximal or the distal histidine. Tyrosine can form an iron‐phenolate complex that resists ferric (Fe3+) reduction to the divalent ferrous (Fe2+) state by the metabolic systems of the red blood cell. Patients with hemoglobin M disease are cyanotic but usually otherwise asymptomatic 1. In hemoglobin M disease, structural abnormalities in the globin portion of the molecule cause heme iron to auto‐oxidize 1, 10. The mutation in the alpha‐globin (HBA1, HBA2), beta‐globin (HBB), or gamma‐globin (HBG1, HBG2) leads to easier oxidation of the iron to the ferric (Fe3+) state and allows for the stabilization of iron in the ferric (Fe3+) state. Patients with hemoglobin M disease usually have methemoglobin levels between 15 to 30% and remain asymptomatic 15. Only one parent needs to pass on the abnormal gene for the child to inherit the disease (autosomal dominant inheritance pattern) 60. At least 13 variants of hemoglobin M have been reported (Boston, Fort Ripley, Hyde Park, Iwate, Kankakee, Osaka, Saskatoon) 61, 15, 62, 16. Four hemoglobin M diseases are a consequence of substitution of tyrosine for histidine in the proximal and distal sites of the alpha and beta chains. These four hemoglobin M diseases have been designated by the geographic names of their discovery, i.e. Boston, Saskatoon, Iwate, and Hyde Park. Analogous His→Tyr substitutions in the γ chain of fetal Hb (HbF) have also been documented and have been designated Hb F‐M‐Osaka 19 and Hb FM-Fort Ripley 63. Patients with hemoglobin M develop cyanosis. For the alpha‐globin variants, the dusky color is evident at birth, while the clinical manifestations of beta‐globin variants become evident only after beta chains have replaced the fetal gamma (γ) chains at 6 to 9 months of age. Hemolytic anemia with jaundice can be present, as observed in hemoglobin M Saskatoon and hemoglobin M Hyde Park 64. Additionally, some patients with unstable hemoglobin also have elevated methemoglobin (MetHb) levels associated with hemolytic anemia 65, 66. This is the case of hemoglobin Chile (β28 Leu → Met), an unstable hemoglobin characterized by chronic methemoglobinemia 67.
- In very rare cases, inherited methemoglobinemia is caused by deficiency of the electron acceptor cytochrome b5 17. This causes methemoglobinemia associated with ambiguous genitalia due to a homozygous variant in the CYB5A gene, encoding the microsomal cytochrome b5 18. This condition is due to isolated 17,20‐lyase deficiency since the cofactor cytochrome b5 is required for optimal 17,20‐lyase activity 19.
Table 2. Inherited methemoglobinemia (congenital methemoglobinemia)
| Disease | Transmission | Cyanosis | Anemia | Other symptoms | Gene(s) | Methemoglobin (MetHb) level % | CYB5R activity | Hb electrophoresis/HPLC |
|---|---|---|---|---|---|---|---|---|
| Methemoglobinemia type 1 | Autosomal recessive | Yes since birth | No | ‐ | CYB5R3 | 20–30 | Decreased | Normal |
| Methemoglobinemia type 2 | Autosomal recessive | Yes since birth | No | Neurological involvement | CYB5R3 | 8–40 | Decreased | Normal |
| Methemoglobinemia type 4 | Autosomal recessive | Yes | No | 46,XY disorder of sexual differentiation Ambiguous genitalia | CYB5A | 12–19 | Normal | Normal |
| Hemoglobin M disease | Autosomal dominant | Yes since birth or after HbF/A switching | Yes | ‐ | HBA1, HBA2, HBB, HBG1, HBG2 | 12–25 | Normal | Abnormal |
| Unstable hemoglobin | Autosomal dominant | Yes | Yes | ‐ | HBA1, HBA2, HBB, HBG1, HBG2 | Variable (Stressor induced) | Normal | Normal or Abnormal |
Methemoglobinemia prevention
Genetic counseling is suggested for couples with a family history of methemoglobinemia and are considering having children.
Babies 6 months or younger are more likely to develop methemoglobinemia. Therefore, homemade baby food purees made from vegetables containing high levels of natural nitrates, such as carrots, beetroots, or spinach should be avoided.
Methemoglobinemia symptoms
Methemoglobinemia should be considered in the setting of shortness of breath (dyspnea), cyanosis and low level of oxygen in the blood (hypoxemia) that is refractory to supplemental oxygen, especially in the setting of exposure to a known oxidative agent 8. However, the presentation may vary in severity from minimally symptomatic to severe. The clinical presentation of methemoglobinemia is based on a spectrum illness that is associated with cyanosis, pallor, fatigue, weakness, headache, central nervous system depression, metabolic acidosis, seizures, dysrhythmias, coma, and death 8. The degree of symptom severity is multifactorial and depends on the patient’s percentage of methemoglobin, the rate at which methemoglobin was accumulated, the individual’s ability to intrinsically clear it, and the underlying health status of the patient. Duration and magnitude of exposure to an oxidizing agent may also play a role 8.
Key symptoms of methemoglobinemia are related to the methemoglobin (MetHb) levels and the resulting hypoxia. For inherited conditions, methemoglobin (MetHb) levels range between 10% and 30% that accounts for the occurrence of cyanosis and dark brown blood as main signs. At these levels of methemoglobin (MetHb), patients are generally asymptomatic or may present with headaches, tachycardia, and mild shortness of breath (dyspnea). A normal methemoglobin fraction is about 1% (range, 0% to 3%). Symptoms associated with higher levels of methemoglobin (MetHb) are as follows 68:
- Less than 10% methemoglobin: None (patients with underlying diseases may have more symptoms at lower level)
- Methemoglobin 10% to 20%: Slight discoloration (eg, pale, gray, blue) of the skin
- Methemoglobin 20% to 30%: Anxiety, headache, tachycardia, lightheadedness
- Methemoglobin 30% to 50%: Dyspnea, weakness, confusion, chest pain
- Methemoglobin 50% to 70%: Arrhythmias; altered mental status, delirium, seizures, coma; profound acidosis
- Methemoglobin greater than 70%: Usually, death 69
The physical examination of patients with suspected methemoglobinemia should include examination of the skin and mucous membranes. Vital signs should be documented, and mental status should be assessed. Careful attention should be paid to the cardiac, respiratory, and circulatory examinations to assess for evidence of an underlying disease (either congenital or acquired).
Methemoglobinemia physical findings may include the following 70:
- Discoloration of the skin and blood (the most striking physical finding)
- Cyanosis – This occurs in the presence of 1.5 g/dL (10%) of methemoglobin (as compared with 5 g/dL of deoxygenated hemoglobin)
- Seizures
- Coma
- Dysrhythmia (eg, bradyarrhythmia or ventricular dysrhythmia)
- Acidosis
- Cardiac or neurologic ischemia
- Pallor of the skin or conjunctiva (suggestive of anemia and possible hemolysis)
- Skeletal abnormalities and intellectual disability
Symptoms of acquired methemoglobinemia include:
- Bluish coloring of the skin
- Headache
- Fatigue
- Shortness of breath
- Lack of energy.
Symptoms of congenital methemoglobinemia type 1 include:
- Bluish coloring of the skin
Symptoms of congenital methemoglobinemia type 2 include:
- Developmental delay
- Failure to thrive
- Intellectual disability
- Microcephaly
- Seizures
Symptoms of hemoglobin M disease include:
- Bluish coloring of the skin (cyanosis)
Congenital methemoglobinemia type 1 (autosomal recessive hereditary methemoglobinemia type 1)
In congenital methemoglobinemia type 1, the methemoglobin (MetHb) levels are typically 20% to 30% 1. Congenital methemoglobinemia type 1 is a generally rare disorder with frequencies of up to 1 per 1000 newborns in some isolated populations with founder mutations. The most striking feature of congenital methemoglobinemia type 1 is the blue discoloration (cyanosis), which typically may be very intense and give a lavender or slate‐gray appearance. The blue discoloration is apparent all over the body, particularly the lips, nose, cheeks, and buccal mucosa and does not improve with supplemental oxygen. This is present from birth and is persistent without much variation unless it has been treated 1. Most often, there are no other clinical features and no impairment of cardiorespiratory function. There are some reports of patients with headaches, tachycardia and mild shortness of breath (dyspnea), attributed to reduced blood oxygenation, and possibly associated mild polycythemia 71. Anecdotally, it is reported that fatigue and breathlessness may become more common with increasing age, possibly related to deteriorating cardiovascular function 1.
Congenital methemoglobinemia type 2 (autosomal recessive hereditary methemoglobinemia type 2)
In congenital methemoglobinemia type 2, the methemoglobin (MetHb) levels levels are typically 20% to 30% 1. Congenital methemoglobinemia type 2 is a very rare disorder, but case reports and small case series suggest that congenital methemoglobinemia type 2 is much more severe with the cyanosis accompanied by neurological dysfunction such as intellectual deficit, baby’s head is significantly smaller than expected (microcephaly), growth retardation, opisthotonus (spasm of the muscles causing backward arching of the head, neck, and spine), crossed eyes (strabismus) and increased muscle tone (hypertonia), which usually becomes evident during the first four months of life 72, 73. Microcephaly is nearly always present. Typical neurological features include abnormally low muscle tone in the trunk or core muscles (axial hypotonia), variable dystonia (involuntary muscle contractions that cause repetitive or twisting movements), movement disorder characterized by a combination of chorea (rapid, jerky, and involuntary movements) and athetosis (slow, writhing, and twisting motions) and opisthotonos (spasm of the muscles causing backward arching of the head, neck, and spine). Typically, psychomotor skills do not progress beyond those expected of a one‐year‐old, and patients do not learn to speak or walk 1. Growth retardation is common, compounded by difficulties feeding and swallowing 1. Crossed eyes (strabismus) occurs in more than 80% of patients, with significant numbers also having seizures. Neurological problems are not typically progressive, but life expectancy is reduced, predominantly due to swallowing difficulties and respiratory complications. Death typically occurs in the first decade of life, although some patients survive into adulthood 73.
Hemoglobin M disease
Hemoglobin M disease patients have no signs and symptoms other than the blue discoloration of their skin and mucous membranes (cyanosis). The cyanosis is similar to that in enzymopenic methemoglobinemia, with lavender blue appearance of skin. The main distinguishing feature is that there can be a family history suggesting autosomal dominant inheritance, as illustrated by the recognition of an autosomal dominant condition called “Kochikuro” (black mouth) in Japan since the 1800s, which was subsequently shown to be caused by Hb M‐Iwate 74. Methemoglobin (MetHb) levels are reported to vary from 12.5% to 25%, with levels possibly higher in beta‐globin variants, such as Hb M‐Saskatoon, than alpha‐globin variants, such as Hb M‐Iwate, although there is considerable overlap 1.
Methemoglobinemia possible complications
Complications of methemoglobinemia include:
- Shock
- Seizures
- Death
Methemoglobinemia diagnosis
Methemoglobinemia is a clinical diagnosis based on history and presenting symptoms, including hypoxemia refractory to supplemental oxygen and the likely presence of chocolate-colored blood 8. Methemoglobinemia diagnosis is confirmed by arterial or venous blood gas with co-oximetry, which will speciate hemoglobin to determine the methemoglobin concentration and percentage 26.
Your blood oxygen level (blood oxygen saturation) is the amount of oxygen you have circulating in your blood. There are two main ways to measure or test blood oxygen levels: through a blood draw test also known as an arterial blood gas (ABG) test and through pulse oximetry using an oximeter. A blood draw test provides much more information about your oxygen levels than an pulse oximeter does.
A baby with methemoglobinemia will have a bluish skin color (cyanosis) at birth or shortly afterward. Your doctor will perform blood tests to diagnose methemoglobinemia. Laboratory studies that may be ordered include the following 75:
- Studies to rule out hemolysis – Complete blood count (CBC), reticulocyte counts, lactate dehydrogenase (LDH), indirect bilirubin, haptoglobin
- Studies to test for organ failure and general end-organ dysfunction – Liver function tests, electrolyte concentrations, blood urea nitrogen (BUN), creatinine
- Urine pregnancy tests
- Heinz body preparation (indicative of oxidative injury to the erythrocyte)
- Hemoglobin electrophoresis to identify hemoglobin M (Hb M); some difficult cases require DNA sequencing of the globin chain gene or mass spectrometry for diagnosis
- Specific enzyme assays for causative deficiencies
- Bedside tests for methemoglobinemia – Examination of blood color on white filter paper after exposure to room air or after aerating a tube of blood with 100% oxygen; if the blood remains dark with these maneuvers, then methemoglobinemia is likely
- Serum levels of nitrites or other offending drugs
Oxygen-carrying capacity of the blood may be determined with the help of the following 75:
- Arterial blood gas (ABG): Blood test to check levels of gases in the blood
- Pulse oximetry (SpO2): Typically less accurate than CO-oximetry in the setting of methemoglobinemia, with the exception of newer multiwavelength pulse oximeters
- CO-oximetry (if available): Measures concentration of oxyhemoglobin, carboxyhemoglobin, methemoglobin, and reduced hemoglobin
A pulse oximeter can also measure blood oxygen saturation levels through a small clip that’s usually placed on your finger or toe. An oximeter reading only indicates what percentage of your blood is saturated with oxygen, known as the SpO2 level, as well as your heart rate. It’s a quick and harmless way to check if someone’s blood oxygen level is too low. However, oxygen saturation (SpO2) measurements cannot be utilized to directly calculate the severity of methemoglobinemia. The traditional dual wave-length pulse oximetry is inaccurate in the setting of methemoglobinemia because these pulse oximeters measure the absorbance of light at two wavelengths at 660 and 940 nm 8. The ratio of this absorbance allows the distinction between oxyhemoglobin and deoxyhemoglobin, with the expressed percentage (SpO2), indicating the measured amount of hemoglobin that is oxygenated 8. Methemoglobin has high absorbance at both of these wavelengths, leading to the interference that causes an inaccurate SpO2 reading 8. When the level of methemoglobin approaches 30% to 35%, the ratio of absorbance of light (A660/A940) becomes 1.0. A ratio of absorbance (A660/A940) of 1.0 reads as a SpO2 of 85% 76. There is a disproportional, inverse relationship between methemoglobin concentration and SpO2, and despite SpO2 being consistently depressed, it is generally a falsely elevated indication of true oxygen saturation that varies depending on a specific device.
Whereas SpO2 measurements are inaccurate and depressed from wavelength interference, often to 75% to 90% even with supplemental oxygen, SaO2 calculations are falsely normal due to the assumption that all hemoglobin is either oxyhemoglobin or deoxyhemoglobin 26 The difference between the depressed SpO2 measurement and the falsely normal SaO2 calculation is known as the “saturation gap” 8. This additional diagnostic clue should hint at the presence of a hemoglobinopathy, but is nonspecific and cannot be used to confirm a diagnosis of methemoglobinemia 8. A saturation gap greater than 5% presents in cases of elevated abnormal forms of hemoglobin such as carboxyhemoglobin, methemoglobin, and sulfhemoglobin 77.
The key diagnostic tests in methemoglobinemia diagnosis are methemoglobin evaluation, measurement of CYB5R activity and DNA sequencing of CYB5R3 gene. Methemoglobin (MetHb) levels are best measured using the change of absorbance of methemoglobin (MetHb) at 630 nm that occurs when cyanide is added, converting the methemoglobin (MetHb) to cyan‐MetHb, as initially described in the spectrophotometric method by Evelyn‐Malloy 78, 79. Human blood contains less than 1% methemoglobin (MetHb). There is a direct correlation between methemoglobin (MetHb) levels and symptoms. CYB5R activity is best measured using ferricyanide as a receptor, measuring the rate of oxidation of NADH 80. In congenital CYB5R3 deficiency, the residual enzyme activity is usually less than 20% of normal. DNA sequencing of CYB5R3 gene allows characterization of the defect and confirmation of the diagnosis.
Electrophoresis at pH 7.1, can be used to identify hemoglobin M disease caused by mutations affecting alpha‐, beta‐, or rarely, gamma‐ globin genes. More than 13 different pathogenic variants have been reported to be associated with hemoglobin M disease in the HbVar database (https://globin.bx.psu.edu/hbvar/). However, when suspecting a hemoglobin M‐variant, targeted sequencing of the alpha‐ and the beta‐globin genes is commonly selected as the technically simpler, more widely available and more specific diagnostic procedure. Direct sequencing of the globin genes is also appropriate, and can be useful in the presence of transfused blood or to exclude electrophoretically silent variants.
Other studies that may be considered are as follows 75:
- Potassium cyanide test to distinguish methemoglobin from sulfhemoglobin
- CT of the head
- Chest radiography to exclude pulmonary or cardiac disease
- Echocardiography to determine the presence of congenital heart disease
Methemoglobinemia treatment
Treatment of methemoglobinemia includes removal of the inciting agent and consideration of treatment with methylene blue (tetramethylthionine chloride). High flow oxygen delivered by non-rebreather mask increases oxygen delivery to tissues and enhances the natural degradation of methemoglobin.
People with hemoglobin M disease don’t have symptoms. So, they may not need treatment.
Methylene Blue (Methylthioninium chloride) is used to treat severe methemoglobinemia. Treatment decision should be made on clinical presentation and not withheld for confirmational laboratory values. The methylene blue dose is 1 to 2 mg/kg (0.1-0.2 mL/kg of 1% solution) intravenously over 5 minutes 14. Methylene blue dose can be repeated in 30 to 60 minutes if significant symptoms or methemoglobin levels remain above the treatment threshold.
In cases of acquired methemoglobinemia, treatment with methylene blue should occur when methemoglobin exceeds 20% to 30%, or at lower levels, if the patient is symptomatic 8.
Methylene blue acts by reacting within red blood cell to form leukomethylene blue, which is a reducing agent of oxidized hemoglobin converting the ferric iron (Fe3+) back to its oxygen carrying ferrous state (Fe2+) 81. Methylene blue usually works rapidly and effectively through its interaction with the secondary pathway of methemoglobin reduction, where NADPH-MetHb reductase reduces methylene blue to leukomethylene blue using NADPH from the G6PD-dependent hexose monophosphate shunt 8. Leukomethylene blue then acts as an electron donor to reduce methemoglobin to hemoglobin.
For methemoglobinemia due to drug exposure, traditional first-line therapy consists of an infusion of methylene blue, whose action depends on the availability of reduced nicotinamide adenine nucleotide phosphate (NADPH) within the red blood cells. Intravenous administration of methylene blue (1–2 mg/kg) as a 1% solution over 5 minutes quickly relieves cyanosis due to methemoglobinemia. Intravenous methylene blue is indicated for methemoglobin fractions over 30% and at lower fractions in patients with anemia or cardiovascular disease. The dose may be repeated if no clinical response is observed within 1 hour. A dose greater than 7 mg/kg of methylene blue by itself can cause methemoglobinemia 82. Supplemental oxygen should also be administered. After an acute exposure to an oxidizing agent, treatment should be considered when the methemoglobin is 30% in an asymptomatic patient and 20% in a symptomatic patient 83. Patients with anemia or cardiorespiratory problems should be treated at lower levels of methemoglobin. Methemoglobinemia due to hemoglobin M does not respond to ascorbic acid or methylene blue.
Nicotinamide adenine dinucleotide phosphate (NADPH) helps in the conversion of methylene blue to leucomethylene blue. This end product helps reduce methemoglobin to normal hemoglobin 84. Dextrose should be given 85 because the major source of NADH in the red blood cells is the catabolism of sugar through glycolysis. Dextrose is also necessary to form NADPH through the hexose monophosphate shunt, which is necessary for methylene blue to be effective.
Methylene blue is an oxidant; its metabolic product leukomethylene blue is the reducing agent. Therefore, large doses of methylene blue may result in higher levels of methylene blue rather than the leukomethylene blue, which will result in hemolysis and, paradoxically, methemoglobinemia in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency 86. Patients with G6PD deficiency also may not produce sufficient NADPH to reduce methylene blue to leukomethylene blue; thus, methylene blue therapy may be ineffective in these patients 86.
Methylene blue may be unsafe in people who have or may be at risk for glucose-6-phosphate dehydrogenase (G6PD) deficiency. They should not take this medicine. If you or your child has G6PD deficiency, always tell your doctor before getting treatment.
Some drugs, such as dapsone, benzocaine, and aniline, produce a rebound methemoglobinemia, in which methemoglobin levels increase 4 to 12 hours after successful methylene blue therapy 87.
Vitamin C also known as ascorbic acid may also be used to reduce the level of methemoglobin 88. Ascorbic acid or Vitamin C is a natural water‐soluble vitamin which reduces excessive oxidative stress. High-dose ascorbic acid (vitamin C), up to 10 g/dose intravenously, can be considered to treat methemoglobin. However, ascorbic acid (vitamin C) is generally ineffective because the reaction rate is too slow for it to be effective when used alone as it generally requires multiple doses and may take 24 hour or longer to lower methemoglobin (MetHb) levels and not considered standard of care 8, 10, 89. Ascorbic acid is the treatment of choice when methylene blue (Methylthioninium chloride) is not available 90 and in cases of methemoglobinemia in a G6PD deficiency person 91. Dosing is not standardized. Doses in adults have ranged from 0.5 g every 12 hour × 16 doses, 1 g every 12 hour × 14 doses, 1.5‐2 g iv × 3–4 infusions, 5 g every 6 hour × 6 doses, or even 10 g × one dose, 63 while doses in children have ranged from 0.5 g every 12 hr × 16 doses and 1 g every 4 hr × 8 doses 88. Note that high dose ascorbic acid (vitamin C) administration is associated with increased urinary excretion of oxalate. In the presence of kidney disease, high dose ascorbic acid may be predisposed to kidney failure due to hyperoxaluria 92.
Alternative treatments for refractory acquired methemoglobinemia include hyperbaric oxygen therapy, red blood cell transfusion, exchange transfusions and hemodialysis 93, 94. Exchange transfusion is reserved for patients in whom methylene blue therapy is ineffective.
N-Acetylcysteine, cimetidine, and ketoconazole are experimental therapies in the treatment of methemoglobinemia that have shown some promising results 95. In vitro studies show N‐acetylcysteine can act as a cofactor to enhance reduction and increase intracellular glutathione. N-Acetylcysteine has been suggested for use in patients with methemoglobinemia and G6PD deficiency and acetaminophen induced methemoglobinemia 91.
In most cases of mild acquired methemoglobinemia, no treatment is needed. But you should avoid the medicine or chemical that caused the problem. Severe cases may need a blood transfusion.
Inherited methemoglobinemia treatment
In hereditary methemoglobinemia due to NADH cytochrome b5 reductase enzyme (NADH diaphorase) deficiency, treatment of cyanosis is primarily for cosmetic reasons. However, many patients or parents strongly desire treatment for themselves or their children, respectively. Treatment options, benefits and risks should be discussed accordingly.
Methylene Blue (Methylthioninium chloride) with or without ascorbic acid (vitamin C) is prescribed in more severe cases of congenital methemoglobinemia 9, 10, 96. In some countries, oral methylene blue (Methylthioninium chloride) 100–300 mg per day is administered with dose adjustment according to MetHb levels. Of note, Hemoglobin M disease are not typically responsive to methylene blue (Methylthioninium chloride). Some Unstable hemoglobin diseases, such hemoglobin-Cheverly, hemoglobin-Evans, may allow water to enter the homophobic heme pocket during stressor events, therefore methemoglobinemia is only intermittent. If significantly elevated, these subgroup of Unstable hemoglobin diseases could respond to methylene blue (Methylthioninium chloride) 97.
Riboflavin also known as vitamin B2 can accelerate reduction of methemoglobin (MetHb) levels via the nicotinamide adenine dinucleotide‐flavin reductase system. There are limited data available on the use of riboflavin in hereditary methemoglobinemia. Described cases administer riboflavin at a dose of 20–30 mg/day 98, or 20 mg three times a day 99.
How is methylene blue given?
Methylene blue is injected into a vein through an IV. A doctor will give you this injection. The IV infusion can take up to 30 minutes to complete.
Administration of methylene blue for both children and adults experiencing methemoglobinemia is done intravenously at a dose of 1 mg/kg of a 1% solution over 5 to 30 minutes 100.
Your breathing, blood pressure, oxygen levels, kidney function, and other vital signs will be watched closely while you are receiving methylene blue. Your blood will also need to be tested to help your doctor determine that the medicine is working.
You may only need to receive one dose of methylene blue. If you do need a second dose, it can be given 1 hour after your first dose.
Methylene blue will most likely cause your urine or stools to appear blue or green in color. This is a normal side effect of the medication and will not cause any harm. However, this effect may cause unusual results with certain urine tests.
As an oxidizing agent, methylene blue can actually precipitate methemoglobinemia or hemolysis in high doses or when ineffectively reduced 8. Methylene blue administration in a patient taking a serotonergic drugs such as Selective Serotonin Reuptake Inhibitors (SSRIs), Serotonin-Norepinephrine Reuptake Inhibitors (SNRIs), Monoamine Oxidase Inhibitors (MAOIs), and Tricyclic Antidepressants (TCAs) due to the Monoamine Oxidase Inhibitor (MAOI) may precipitate serotonin syndrome (serotonin toxicity) 101. Methylene blue carries a Black Box warning for risk of serious or fatal serotonergic syndrome when used in combination with serotonergic drugs (eg, selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs], and monoamine oxidase inhibitors [MAOIs]) or opioids.
Caution should also be practiced with treating neonates as they are also very sensitive to oxidizing agents. Also, methylene blue is a United States Food and Drug Administration (FDA) pregnancy category X drug, indicating that studies have shown concrete evidence of human fetal risk and possible intestinal atresia.
Diagnostic studies utilizing methylene blue in utero via intraamniotic injection and postnatally in doses of 2–4 mg/kg in premature infants has led to hemolysis and methemoglobinemia in non‐G6PD‐deficient infants 102, 103.
Methylene blue should be used cautiously in patients with renal failure and in anesthetized patients where it may inhibit guanylate cyclase, decreasing nitric oxide‐mediated vasodilatation leading to systemic and pulmonary hypertension.
If methylene blue administration is ineffective after the second dose, underlying conditions including, but not limited to, glucose-6-phosphate dehydrogenase (G6PD) deficiency and NADPH-MetHb reductase deficiency should be considered as reasons for refractoriness to treatment. However, methemoglobinemia alone is not an indication to screen for these disease processes.
When treatment with methylene blue is ineffective or not recommended, additional options may include ascorbic acid (Vitamin C), exchange transfusion, hyperbaric oxygen therapy 104, 105.
Methemoglobinemia prognosis
In severe cases, methemoglobinemia prognosis is determined by the degree of anoxic end-organ damage. Death occurs when methemoglobin fractions approach 70% 69. Death can also occur at lower methemoglobin levels in patients with significant comorbidities. Complications of methemoglobinemia may include heart attack (myocardial infarction), seizure, coma, and death.
People with congenital methemoglobinemia type 1 and hemoglobin M disease often do well. Congenital methemoglobinemia type 2 is more serious. It often causes death within the first few years of life.
People with acquired methemoglobinemia often do very well once the drug, food, or chemical that caused the problem is identified and avoided. Most patients with methemoglobinemia respond well to treatment and can be discharged after brief period of observation.
Anyone with persistent symptoms after initial treatment or exacerbated underlying medical conditions should be considered for admission.
- Iolascon A, Bianchi P, Andolfo I, et al. SWG of red cell and iron of EHA and EuroBloodNet. Recommendations for diagnosis and treatment of methemoglobinemia. Am J Hematol. 2021 Dec 1;96(12):1666-1678. doi: 10.1002/ajh.26340[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- McQueen CA, ed. Comprehensive Toxicology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2017. Vol 12.[↩]
- Benz EJ Jr, Ebert BL. Hemoglobin Variants Associated With Hemolytic Anemia, Altered Oxygen Affinity, and Methemoglobinemias. In: Hoffman R, Benz EJ Jr, Slberstein LE, Heslop H, Weitz J, Anastasi J, eds. Hematology: Basic Principles and Practice. 7th ed. Philadelphia, PA: Elsevier/Saunders; 2018. 608-15.[↩]
- Prchal J. Chapter 51: Methemoglobinemia and Other Dyshemoglobinemias. 10th ed. McGraw Hill; 2021.[↩]
- Hall A.H., Kulig K.W., Rumack B.H. Drug- and chemical-induced methaemoglobinaemia. Clinical features and management. Med. Toxicol. 1986;1(4):253–260. doi: 10.1007/BF03259842[↩]
- Rehman H.U. Methemoglobinemia. West. J. Med. 2001;175(3):193–196. doi: 10.1136/ewjm.175.3.193[↩]
- Sahu KK, Mishra A. Methemoglobinemia: Challenges in Diagnosis and Management. J Assoc Physicians India. 2019 Aug;67(8):94.[↩]
- Ludlow JT, Wilkerson RG, Nappe TM. Methemoglobinemia. [Updated 2023 Aug 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537317[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med. 1999 Nov;34(5):646-56. doi: 10.1016/s0196-0644(99)70167-8[↩][↩][↩]
- Percy MJ, Lappin TR. Recessive congenital methaemoglobinaemia: cytochrome b(5) reductase deficiency. Br J Haematol. 2008 May;141(3):298-308. doi: 10.1111/j.1365-2141.2008.07017.x[↩][↩][↩][↩][↩][↩][↩][↩]
- Dekker J, Eppink MH, van Zwieten R, de Rijk T, Remacha AF, Law LK, Li AM, Cheung KL, van Berkel WJ, Roos D. Seven new mutations in the nicotinamide adenine dinucleotide reduced-cytochrome b(5) reductase gene leading to methemoglobinemia type I. Blood. 2001 Feb 15;97(4):1106-14. doi: 10.1182/blood.v97.4.1106[↩][↩]
- Nicolas-Jilwan M. Recessive congenital methemoglobinemia type II: Hypoplastic basal ganglia in two siblings with a novel mutation of the cytochrome b5 reductase gene. Neuroradiol J. 2019 Apr;32(2):143-147. doi: 10.1177/1971400918822153[↩][↩]
- Burtseva TE, Ammosova TN, Protopopova NN, Yakovleva SY, Slobodchikova MP. Enzymopenic Congenital Methemoglobinemia in Children of the Republic of Sakha (Yakutia). J Pediatr Hematol Oncol. 2017 Jan;39(1):42-45. doi: 10.1097/MPH.0000000000000705[↩][↩]
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J. 2011 Nov;104(11):757-61. doi: 10.1097/SMJ.0b013e318232139f[↩][↩][↩]
- Curry S. Methemoglobinemia. Ann Emerg Med. 1982 Apr;11(4):214-21. doi: 10.1016/s0196-0644(82)80502-7[↩][↩][↩]
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem. 2005 Feb;51(2):434-44. doi: 10.1373/clinchem.2004.035154[↩][↩]
- Hegesh E, Hegesh J, Kaftory A. Congenital methemoglobinemia with a deficiency of cytochrome b5. N Engl J Med. 1986 Mar 20;314(12):757-61. doi: 10.1056/NEJM198603203141206[↩][↩]
- Kok RC, Timmerman MA, Wolffenbuttel KP, Drop SL, de Jong FH. Isolated 17,20-lyase deficiency due to the cytochrome b5 mutation W27X. J Clin Endocrinol Metab. 2010 Mar;95(3):994-9. doi: 10.1210/jc.2008-1745[↩][↩]
- Idkowiak J, Randell T, Dhir V, Patel P, Shackleton CH, Taylor NF, Krone N, Arlt W. A missense mutation in the human cytochrome b5 gene causes 46,XY disorder of sex development due to true isolated 17,20 lyase deficiency. J Clin Endocrinol Metab. 2012 Mar;97(3):E465-75. doi: 10.1210/jc.2011-2413[↩][↩]
- McGuigan MA. Benzocaine-induced methemoglobinemia. Can Med Assoc J. 1981 Oct 15;125(8):816. https://pmc.ncbi.nlm.nih.gov/articles/instance/1862717/pdf/canmedaj01353-0018a.pdf[↩]
- Kane GC, Hoehn SM, Behrenbeck TR, Mulvagh SL. Benzocaine-induced methemoglobinemia based on the Mayo Clinic experience from 28 478 transesophageal echocardiograms: incidence, outcomes, and predisposing factors. Arch Intern Med. 2007 Oct 8;167(18):1977-82. doi: 10.1001/archinte.167.18.1977[↩][↩]
- O’Donohue WJ, Moss LM, Angelillo VA. Acute Methemoglobinemia Induced by Topical Benzocaine and Lidocaine. Arch Intern Med. 1980;140(11):1508–1509. doi:10.1001/archinte.1980.00330220067023[↩]
- Bradberry SM. Occupational methaemoglobinaemia. Mechanisms of production, features, diagnosis and management including the use of methylene blue. Toxicol Rev. 2003;22(1):13-27. doi: 10.2165/00139709-200322010-00003[↩][↩]
- Johnson CJ, Kross BC. Continuing importance of nitrate contamination of groundwater and wells in rural areas. Am J Ind Med. 1990;18(4):449-56. doi: 10.1002/ajim.4700180416[↩][↩]
- Guay J. Methemoglobinemia related to local anesthetics: a summary of 242 episodes. Anesth Analg. 2009 Mar;108(3):837-45. doi: 10.1213/ane.0b013e318187c4b1[↩]
- Nappe TM, Pacelli AM, Katz K. An Atypical Case of Methemoglobinemia due to Self-Administered Benzocaine. Case Rep Emerg Med. 2015;2015:670979. doi: 10.1155/2015/670979[↩][↩][↩]
- Wilkerson RG. Getting the blues at a rock concert: a case of severe methaemoglobinaemia. Emerg Med Australas. 2010 Oct;22(5):466-9. doi: 10.1111/j.1742-6723.2010.01336.x[↩]
- Brown C, Bowling M. Methemoglobinemia in bronchoscopy: a case series and a review of the literature. J Bronchology Interv Pulmonol. 2013;20:241–246. doi: 10.1097/LBR.0b013e3182a125de[↩]
- Singh S, Sethi N, Pandith S, Ramesh GS. Dapsone-induced methemoglobinemia: “Saturation gap”-The key to diagnosis. J Anaesthesiol Clin Pharmacol. 2014;30:86–88. doi: 10.4103/0970-9185.125710[↩]
- Ashurst JV, Wasson MN, Hauger W, Fritz WT. Pathophysiologic mechanisms, diagnosis, and management of dapsone-induced methemoglobinemia. J Am Osteopath Assoc. 2010 Jan;110(1):16-20.[↩]
- Mannemuddhu SS, Ali R, Kadhem S, Ruchi R. Unusual cause of persistent dyspnea in a patient with nephrotic syndrome: dapsone-induced methemoglobinemia. CEN Case Rep. 2021 Aug;10(3):336-340. doi: 10.1007/s13730-020-00565-8[↩]
- Thom CS, Dickson CF, Gell DA, Weiss MJ. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harbor Perspectives in Medicine. 2013;3(3):a011858. doi:10.1101/cshperspect.a011858. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3579210/[↩]
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med. 1999;34:646–656.[↩]
- King A., Menke N., Katz K. Toxic hemoglobinopathies in the emergency department. EM Critical Care. 2013;3(6):1–17.[↩]
- Ashurst J, Wasson M. Methemoglobinemia: a systematic review of the pathophysiology, detection, and treatment. Del Med J. 2011 Jul;83(7):203-8.[↩]
- Rehman HU. Methemoglobinemia. West J Med. 2001;175(3):193-6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1071541/[↩]
- Hegedus F, Herb K. Benzocaine-induced methemoglobinemia. Anesth Prog. 2005;52(4):136-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1586795/[↩]
- Askew GL, Finelli L, Genese CA, Sorhage FE, Sosin DM, Spitalny KC. Boilerbaisse: an outbreak of methemoglobinemia in New Jersey in 1992. Pediatrics 1994;94: 381-384.[↩]
- Hunter L, Gordge L, Dargan PI, Wood DM. Methaemoglobinaemia associated with the use of cocaine and volatile nitrites as recreational drugs: a review. Br J Clin Pharmacol. 2011 Jul;72(1):18-26. doi: 10.1111/j.1365-2125.2011.03950.x[↩]
- Lo SC, Agar NS. NADH-methemoglobin reductase activity in the erythrocytes of newborn and adult mammals. Experientia. 1986 Dec 1;42(11-12):1264-5. doi: 10.1007/BF01946415[↩]
- Graubarth J, Bloom CJ, Coleman FC, Solomon HN. Dye poisoning in the nursery: a review of seventeen cases. JAMA. 1945;128(16):1155‐1157.[↩]
- Knobeloch L, Proctor M. Eight blue babies. WMJ. 2001;100(8):43-7.[↩]
- Lukens JN. The Legacy of Well-Water Methemoglobinemia. JAMA. 1987;257(20):2793–2795. doi:10.1001/jama.1987.03390200133028[↩]
- Sanchez-Echaniz J, Benito-Fernández J, Mintegui-Raso S. Methemoglobinemia and consumption of vegetables in infants. Pediatrics. 2001 May;107(5):1024-8. doi: 10.1542/peds.107.5.1024[↩]
- Hamon I, Gauthier-Moulinier H, Grelet-Dessioux E, Storme L, Fresson J, Hascoet JM. Methaemoglobinaemia risk factors with inhaled nitric oxide therapy in newborn infants. Acta Paediatr. 2010 Oct;99(10):1467-73. doi: 10.1111/j.1651-2227.2010.01854.x[↩]
- Ohashi K, Yukioka H, Hayashi M, Asada A. Elevated methemoglobin in patients with sepsis. Acta Anaesthesiol Scand 1998;42: 713-716.[↩]
- Pollack ES, Pollack CV. Incidence of subclinical methemoglobinemia in infants with diarrhea. Ann Emerg Med 1994;24: 652-656.[↩]
- Hjelt K, Lund JT, Scherling B, et al. Methaemoglobinaemia among neonates in a neonatal intensive care unit. Acta Paediatr 1995;84: 365-370.[↩]
- Catalan Munoz M, Carrasco Sanchez P, Gentles MG, et al. Methemoglobinemia, acidemia and diarrhea induced by hypersensitivity to cow’s milk proteins [in Spanish]. An Esp Pediatr 1996;44: 295-296.[↩]
- Sager S, Grayson GH, Feig SA. Methemoglobinemia associated with acidosis of probable renal origin. J Pediatr 1995;126: 59-61.[↩]
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med. 1999;34:646–656[↩]
- Ferraro-Borgida MJ, Mulhern SA, DeMeo MO, Bayer MJ. Methemoglobinemia from perineal application of an anesthetic cream. Ann Emerg Med. 1996;27:785–788.[↩]
- Novaro GM, Aronow HD, Militello MA, Garcia MJ, Sabik EM. Benzocaine-induced methemoglobinemia: experience from a high-volume transesophageal echocardiography laboratory. J Am Soc Echocardiogr. 2003;16:170–175.[↩]
- Spielman FJ, Anderson JA, Terry WC. Benzocaine-induced methemoglobinemia during general anesthesia. J Oral Maxillofac Surg. 1984;42:740–743.[↩]
- Klos CP, Hays GL. Prilocaine-induced methemoglobinemia in a child with Shwachman syndrome. J Oral Maxillofac Surg. 1985;43:621–623[↩]
- Warren RE, Van de Mark TB, Weinberg S. Methemoglobinemia induced by high doses of prilocaine. Oral Surg. 1974;37:866.[↩]
- Sinisterra S, Miravet E, Alfonso I, Soliz A, Papazian O. Methemoglobinemia in an infant receiving nitric oxide after use of eutectic mixture of local anesthetic. J Pediatr. 2002;141:285–286.[↩]
- CYB5R3 gene. https://medlineplus.gov/genetics/gene/cyb5r3[↩]
- Kedar PS, Gupta V, Warang P, Chiddarwar A, Madkaikar M. Novel mutation (R192C) in CYB5R3 gene causing NADH-cytochrome b5 reductase deficiency in eight Indian patients associated with autosomal recessive congenital methemoglobinemia type-I. Hematology. 2018 Sep;23(8):567-573. doi: 10.1080/10245332.2018.1444920[↩]
- Spears F, Banerjee A. Hemoglobin M variant and congenital methemoglobinemia: methylene blue will not be effective in the presence of hemoglobin M. Can J Anaesth. 2008 Jun;55(6):391-2. doi: 10.1007/BF03021499[↩]
- Kulozik AE. Hemoglobin variants and the rarer hemoglobin disorders. Pediatric Hematology; Wiley; 2006:231‐254.[↩]
- van Zwieten R, Verhoeven AJ, Roos D. Inborn defects in the antioxidant systems of human red blood cells. Free Radic Biol Med. 2014 Feb;67:377-86. doi: 10.1016/j.freeradbiomed.2013.11.022[↩]
- Priest JR, Watterson J, Jones RT, Faassen AE, Hedlund BE. Mutant fetal hemoglobin causing cyanosis in a newborn. Pediatrics. 1989 May;83(5):734-6.[↩]
- Wild B, Bain BJ. Investigation of abnormal haemoglobins and thalassaemia. Dacie and Lewis Practical Haematology. 10th ed. Churchill Livingstone; 2006:271‐310.[↩]
- Agarwal AM, Prchal JT. Methemoglobinemia and other dyshemoglobinemias. In: Kaushansky K, Lichtman MA, Prchal JT, et al., eds. Williams Hematology, 9e; McGraw‐Hill; 2015.[↩]
- Stamatoyannopoulos G, Nute PE, Giblett E, Detter J, Chard R. Haemoglobin M Hyde Park occurring as a fresh mutation: diagnostic, structural, and genetic considerations. J Med Genet. 1976 Apr;13(2):142-7. https://pmc.ncbi.nlm.nih.gov/articles/instance/1013374/pdf/jmedgene00309-0062.pdf[↩]
- Hojas-Bernal R, McNab-Martin P, Fairbanks VF, Holmes MW, Hoyer JD, McCormick DJ, Kubik KS. Hb Chile [beta28(B10)Leu–>Met]: an unstable hemoglobin associated with chronic methemoglobinemia and sulfonamide or methylene blue-induced hemolytic anemia. Hemoglobin. 1999 May;23(2):125-34. doi: 10.3109/03630269908996157[↩]
- Methemoglobinemia. https://emedicine.medscape.com/article/204178-overview[↩]
- Mun SH, Park GJ, Lee JH, Kim YM, Chai HS, Kim SC. Two cases of fatal methemoglobinemia caused by self-poisoning with sodium nitrite: A case report. Medicine (Baltimore). 2022 Feb 18. 101 (7):e28810.[↩][↩]
- Methemoglobinemia Clinical Presentation. https://emedicine.medscape.com/article/204178-clinical#b3[↩]
- Jaffé ER. Hereditary methemoglobinemias associated with abnormalities in the metabolism of erythrocytes. Am J Med. 1966 Nov;41(5):786-98. doi: 10.1016/0002-9343(66)90037-4[↩]
- Autosomal recessive methemoglobinemia. https://www.orpha.net/en/disease/detail/621[↩]
- Ewenczyk C, Leroux A, Roubergue A, Laugel V, Afenjar A, Saudubray JM, Beauvais P, Billette de Villemeur T, Vidailhet M, Roze E. Recessive hereditary methaemoglobinaemia, type II: delineation of the clinical spectrum. Brain. 2008 Mar;131(Pt 3):760-1. doi: 10.1093/brain/awm337[↩][↩]
- Shibata S, Miyaji T, Iuchi I, Tamura A. Substitution of tyrosine for histidine (87 in the alpha‐chain of hemoglobin M‐Iwate). Nihon Ketsueki Gakkai Zasshi. 1964;27:13‐18.[↩]
- Methemoglobinemia Workup. https://emedicine.medscape.com/article/204178-workup[↩][↩][↩]
- Chan ED, Chan MM, Chan MM. Pulse oximetry: understanding its basic principles facilitates appreciation of its limitations. Respir Med. 2013 Jun;107(6):789-99. doi: 10.1016/j.rmed.2013.02.004[↩]
- Akhtar J, Johnston BD, Krenzelok EP. Mind the gap. J Emerg Med. 2007 Aug;33(2):131-2. doi: 10.1016/j.jemermed.2006.11.016[↩]
- Evelyn KE, Malloy HT. Microdetermination of oxyhemoglobin, methemoglobin, and sulfhemoglobin in a single sample of blood. J Biol Chem. 1938;126(2):655‐662.[↩]
- Beutler E. Carboxyhemoglobin, methemoglobin, and sulfhemoglobin determinations. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. Williams Hematology. McGraw‐Hill; 1995.[↩]
- Beutler E. Red Cell Metabolism: A Manual of Biochemical Methods. Grune & Stratton; 1984.[↩]
- Methemoglobinemia: a case study. Boylston M, Beer D. Crit Care Nurse. 2002 Aug; 22(4):50-5. http://ccn.aacnjournals.org/content/22/4/50.long[↩]
- Methemoglobinemia and hemolysis after enteral administration of methylene blue in a preterm infant: relevance for pediatric surgeons. Allegaert K, Miserez M, Lerut T, Naulaers G, Vanhole C, Devlieger H. J Pediatr Surg. 2004 Jan; 39(1):E35-7.[↩]
- Price D. Methemoglobinemia. In: Goldfrank LR, Flomenbaum NE, Lewin NA, Weisman RS, Howland MA, Hoffman RS, eds. Goldfrank’s Toxicologic Emergencies. 6th ed. Old Tappan, NJ: Appleton & Lange; 1998: 1507-1523.[↩]
- Adeyinka A, Kondamudi NP. Cyanosis. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482247[↩]
- Regulatory factors in methylene blue catalysis in erythrocytes. Roigas H, Zoellner E, Jacobasch G, Schultze M, Rapoport S. Eur J Biochem. 1970 Jan; 12(1):24-30.[↩]
- Studies of the efficacy and potential hazards of methylene blue therapy in aniline-induced methaemoglobinaemia. Harvey JW, Keitt AS. Br J Haematol. 1983 May; 54(1):29-41.[↩][↩]
- Glucose-6-phosphate dehydrogenase deficiency. Beutler E. N Engl J Med. 1991 Jan 17; 324(3):169-74.[↩]
- Sahu KK, Dhibar DP, Gautam A, Kumar Y, Varma SC. Role of ascorbic acid in the treatment of methemoglobinemia. Turk J Emerg Med. 2016 Aug 5;16(3):119-120. doi: 10.1016/j.tjem.2016.07.003[↩][↩]
- Breakey VK, Gibson QH. Familial idiopathic methaemoglobinaemia. Lancet. 1951;1(6661):935‐938. doi: 10.1016/s0140-6736(51)92452-x[↩]
- Rino PB, Scolnik D, Fustiñana A, Mitelpunkt A, Glatstein M. Ascorbic acid for the treatment of methemoglobinemia: the experience of a large tertiary care pediatric hospital. Am J Ther. 2014 Jul-Aug;21(4):240-3. doi: 10.1097/MJT.0000000000000028[↩]
- Rehman A, Shehadeh M, Khirfan D, Jones A. Severe acute haemolytic anaemia associated with severe methaemoglobinaemia in a G6PD-deficient man. BMJ Case Rep. 2018 Mar 28;2018:bcr2017223369. doi: 10.1136/bcr-2017-223369[↩][↩]
- Lee KW, Park SY. High-dose vitamin C as treatment of methemoglobinemia. Am J Emerg Med. 2014 Aug;32(8):936. doi: 10.1016/j.ajem.2014.05.030[↩]
- Patnaik S, Natarajan MM, James EJ, Ebenezer K. Methylene blue unresponsive methemoglobinemia. Indian J Crit Care Med. 2014 Apr;18(4):253-5. doi: 10.4103/0972-5229.130582[↩]
- Golden PJ, Weinstein R. Treatment of high-risk, refractory acquired methemoglobinemia with automated red blood cell exchange. J Clin Apher. 1998;13(1):28-31. doi: 10.1002/(sici)1098-1101(1998)13:1<28::aid-jca6>3.0.co;2-b[↩]
- Wright RO, Magnani B, Shannon MW, Woolf AD. N-Acetylcysteine reduces methemoglobin in vitro. Ann Emerg Med 1996;28: 499-503.[↩]
- Zorc JJ, Kanic Z. A cyanotic infant: true blue or otherwise? Pediatr Ann. 2001 Oct;30(10):597-601. doi: 10.3928/0090-4481-20011001-08[↩]
- Rangan A, Savedra ME, Dergam-Larson C, Swanson KC, Szuberski J, Go RS, Porter TR, Brunker SE, Shi M, Nguyen PL, Hoyer JD, Oliveira JL. Interpreting sulfhemoglobin and methemoglobin in patients with cyanosis: An overview of patients with M-hemoglobin variants. Int J Lab Hematol. 2021 Aug;43(4):837-844. doi: 10.1111/ijlh.13581[↩]
- Kaplan JC, Chirouze M. Therapy of recessive congenital methaemoglobinaemia by oral riboflavine. Lancet. 1978 Nov 11;2(8098):1043-4. doi: 10.1016/s0140-6736(78)92357-7[↩]
- Hirano M, Matsuki T, Tanishima K, Takeshita M, Shimizu S, Nagamura Y, Yoneyama Y. Congenital methaemoglobinaemia due to NADH methaemoglobin reductase deficiency: successful treatment with oral riboflavin. Br J Haematol. 1981 Mar;47(3):353-9. doi: 10.1111/j.1365-2141.1981.tb02802.x[↩]
- Bistas E, Sanghavi DK. Methylene Blue. [Updated 2023 Jun 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557593[↩]
- Ramsay RR, Dunford C, Gillman PK. Methylene blue and serotonin toxicity: inhibition of monoamine oxidase A (MAO A) confirms a theoretical prediction. Br J Pharmacol. 2007 Nov;152(6):946-51. doi: 10.1038/sj.bjp.0707430[↩]
- McEnerney JK, McEnerney LN. Unfavorable neonatal outcome after intraamniotic injection of methylene blue. Obstet Gynecol. 1983 Mar;61(3 Suppl):35S-37S.[↩]
- Allegaert K, Miserez M, Lerut T, Naulaers G, Vanhole C, Devlieger H. Methemoglobinemia and hemolysis after enteral administration of methylene blue in a preterm infant: relevance for pediatric surgeons. J Pediatr Surg. 2004 Jan;39(1):E35-7. doi: 10.1016/j.jpedsurg.2003.09.045[↩]
- Grauman Neander N, Loner CA, Rotoli JM. The Acute Treatment of Methemoglobinemia in Pregnancy. J Emerg Med. 2018 May;54(5):685-689. doi: 10.1016/j.jemermed.2018.01.038[↩]
- Cho Y, Park SW, Han SK, Kim HB, Yeom SR. A Case of Methemoglobinemia Successfully Treated with Hyperbaric Oxygenation Monotherapy. J Emerg Med. 2017 Nov;53(5):685-687. doi: 10.1016/j.jemermed.2017.04.036[↩]




{kind=link}