Contents
What is myasthenia gravis
Myasthenia gravis is a rare chronic neuromuscular autoimmune disease caused by a breakdown in communication between your nerves and muscles. The main symptom of myasthenia gravis is muscle weakness that increases during periods of activity and improves after rest. With myasthenia gravis, it is the voluntary or striated muscles that are weakened. Voluntary muscles are the muscles that you can control, where the message is transmitted via your nervous system to contract the muscle. They are muscles that control your eye and eyelid movements, facial expression, chewing, talking, and swallowing are often, but not always, involved. The muscles that control breathing and neck and limb movements may also be affected. Involuntary heart muscle and smooth muscles of the gut, urinary bladder, blood vessels, and uterus are not involved in myasthenia gravis. Nor does myasthenia gravis affect your mental capacity.
The name myasthenia gravis comes from the Greek ‘myasthenia’, meaning muscle weakness, and the Latin ‘gravis’, meaning severe.
Myasthenia gravis is not directly inherited nor is it contagious. Occasionally, myasthenia gravis may occur in more than one member of the same family.
Myasthenia gravis is an autoimmune disease. This means it is caused by your body’s immune system. People with myasthenia gravis are more likely than others to have relatives with other autoimmune diseases, such as Graves’ disease, rheumatoid arthritis and lupus. It is not really understood why some people get autoimmune diseases and not others.
The myasthenia gravis annual incidence is about 0.25-2 patients per 100,000 1 and the prevalence about 40 per million people. Myasthenia gravis occurs in all ethnic groups and both genders but most commonly affects young adult women (under 40) and older men (over 60). Myasthenia gravis can however occur at any age.
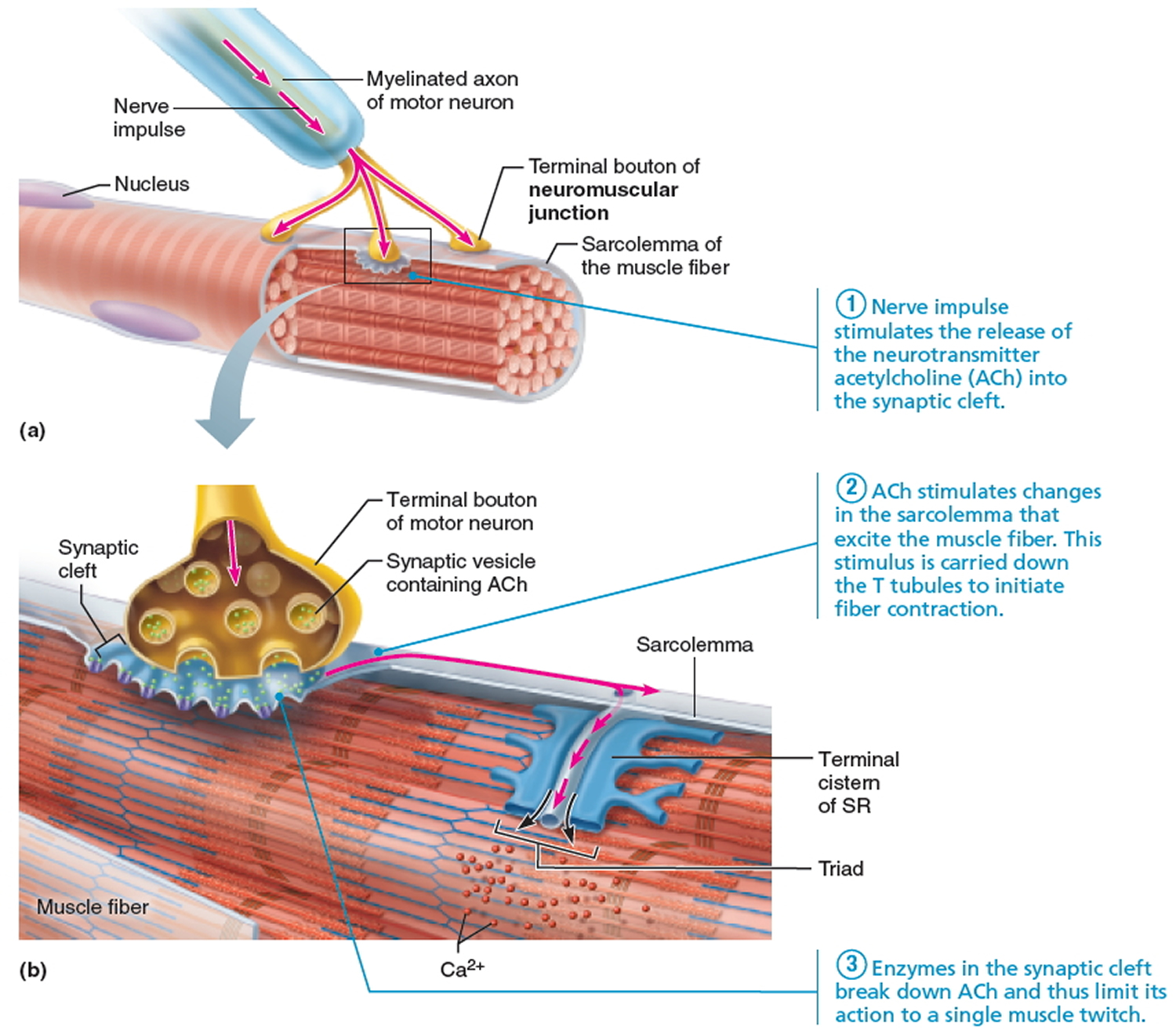
For a muscle to contract, the chemical acetylcholine (ACh) normally transmits nerve impulses to muscle fibers at the place where the nerve and muscle connect (the neuromuscular junction). The physical weakness of the myasthenic’s muscle is caused by a defect at this neuromuscular junction. However, whilst the he myasthenic has a problem with the transmission of nerve impulses to the muscle, the nerves and muscles themselves may remain normal.
Research has shown that most myasthenics form abnormal antibodies against the acetylcholine receptor (sites at which the chemical can be received on the surface of the muscle). The number of acetylcholine receptors in a myasthenic is reduced due to an attack on the receptors by the body’s immune system – if receptors are missing, the response of the muscle to the nerve impulses is poor, and so weakness occurs. Scientists are investigating what triggers the body to develop an autoimmune response. In many myathenics, the thymus gland appears to be involved.
Figure 1. Neuromuscular junction
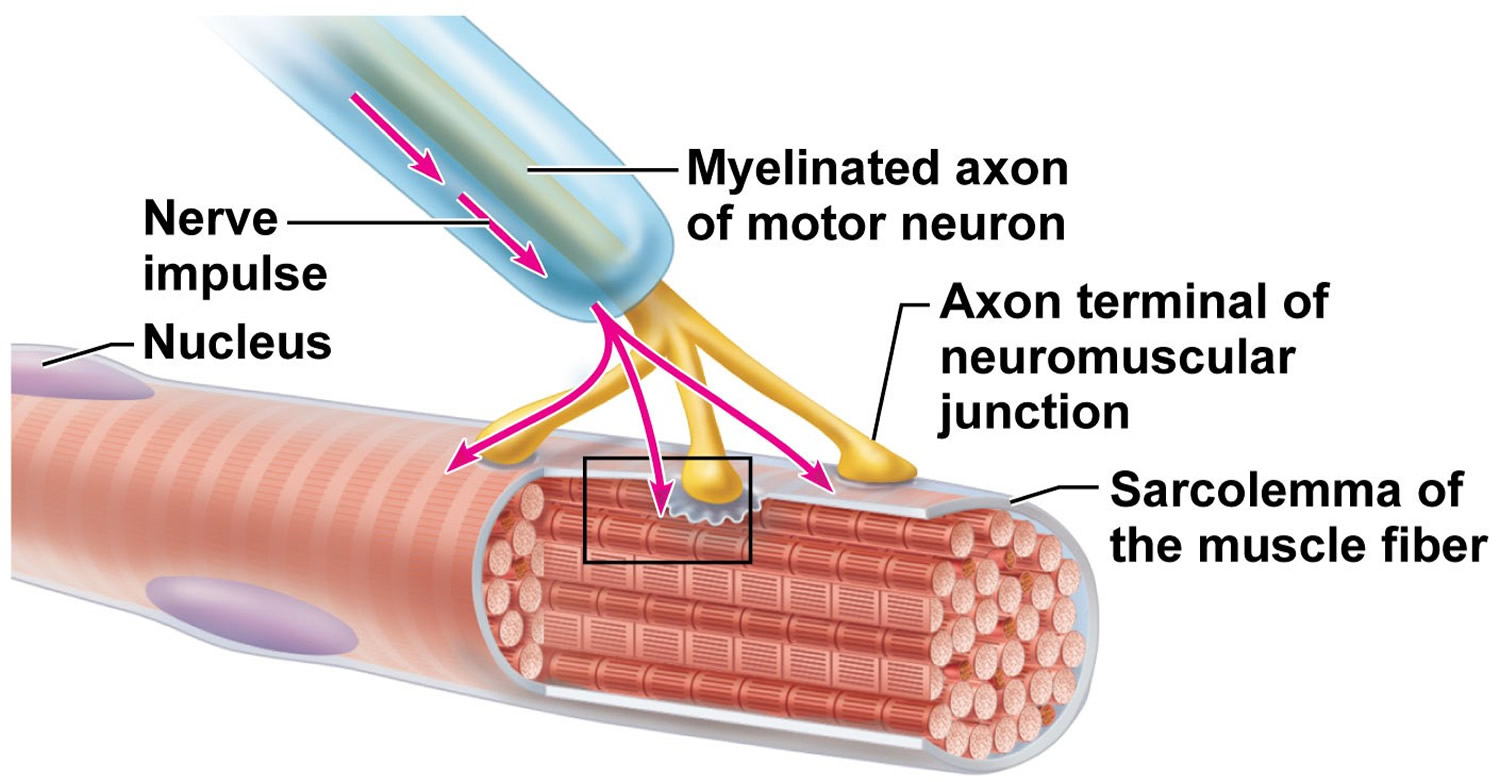
Figure 2. Neuromuscular junction and acetylcholine (ACh)
Babies can also get a form of myasthenia gravis from their mothers. In neonatal myasthenia gravis, the baby may acquire immune proteins (antibodies) from a mother affected with myasthenia gravis. Generally, cases of neonatal myasthenia gravis are temporary and the child’s symptoms usually disappear within few weeks after birth. Around 12% of babies born to myasthenic mothers suffer from neonatal myasthenia where the baby develops a feeble cry, poor sucking, and respiratory distress. These symptoms disappear over the first few months of life. It is rare that infants born to non-myasthenic mothers have myasthenic symptoms – these infants are said to have congenital myasthenia. Myasthenia gravis in juveniles is common. Children who develop myasthenia gravis are indistinguishable from adults myasthenia gravis.
Myasthenia gravis often begins in your eye muscles or elsewhere on your face. Myasthenia gravis can make it hard to control your eye and eyelid movements, and to talk, chew and swallow.
The onset of myasthenia gravis may be sudden, with severe and generalized muscle weakness, but more often the symptoms in the early stages are subtle and variable, making it difficult to diagnose correctly. Some have only ocular myasthenia involving the eye muscles and eyelids; others have mainly swallowing difficulties or slurred speech; others have generalised myasthenia gravis affecting many muscle groups. They might come and go.
Muscle weakness may develop over a few days or weeks, or remain at the same level for long periods of time. The severity varies at different times of the day, and from person to person. The maximum extent of involvement in an individual patient usually manifests itself within the first 5 to 7 years, and thereafter it tends not to be progressive, even though muscle involvement and severity of weakness may still fluctuate from hour-to-hour and day-to-day.
Common symptoms of myasthenia gravis include:
- difficulty walking normally
- your arms, hands, fingers, legs or neck are weak
- the expression on your face changes
- shortness of breath and occasionally serious breathing difficulties
- having trouble swallowing
- droopy eyelids
- double vision
- difficulty making facial expressions
- problems with chewing and difficulty swallowing
- slurred speech
Myasthenia gravis often affects the eyes and face first, but usually spreads to other parts of the body over time.
- Call your local emergency number for an ambulance immediately if you have worsening severe breathing or swallowing difficulties, as you may need emergency treatment in hospital.
Figure 1. Myasthenia gravis (note droopy eyelids)
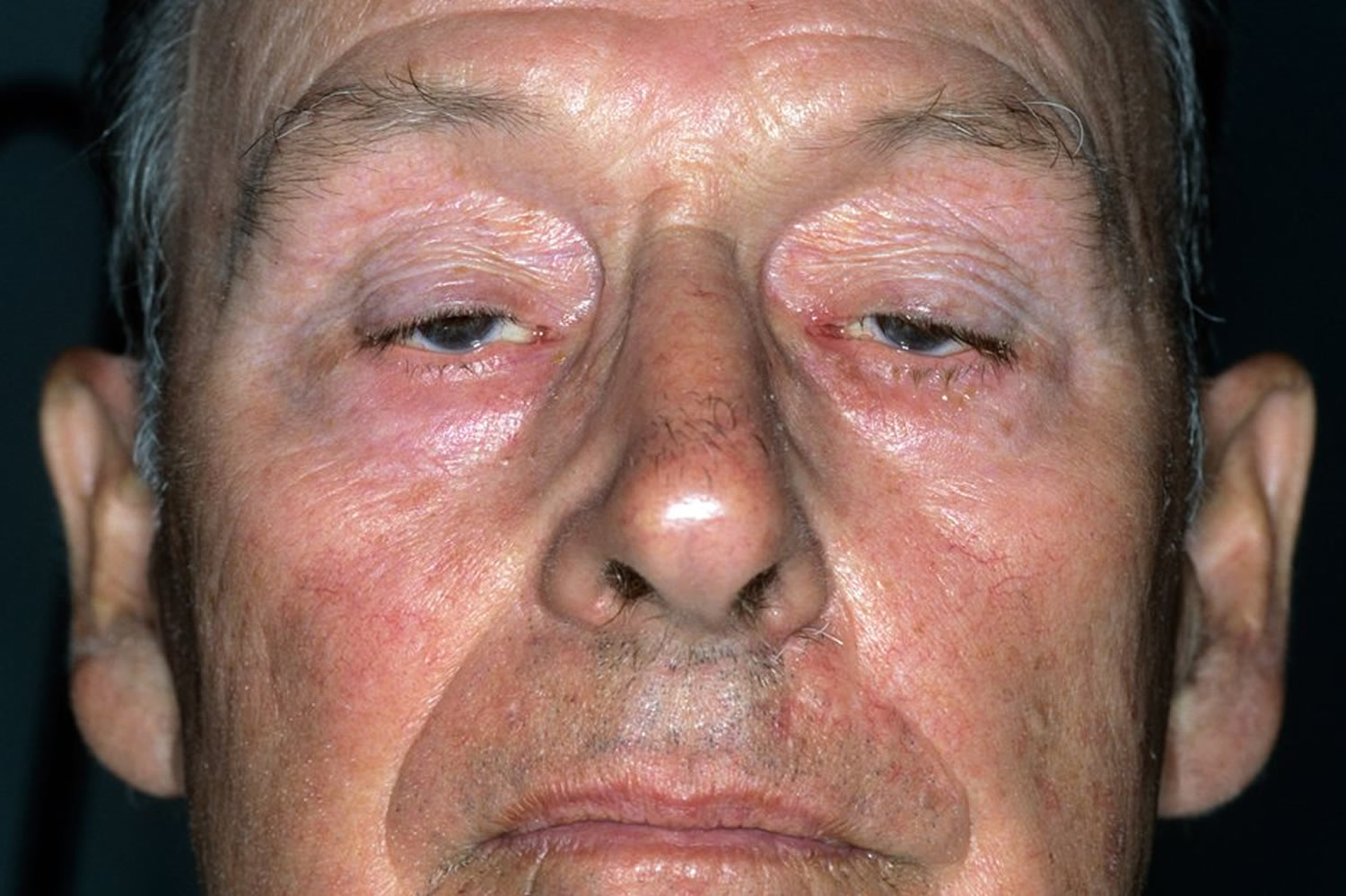
The severity of the weakness varies from person to person. It tends to be worse when you’re tired and gets better after resting.
In some people, the symptoms can also have a number of other triggers, such as stress, infections and certain medicines.
If you notice any of these symptoms, see your doctor.
Unfortunately, a delay in diagnosis of one or two years is not unusual in cases of myasthenia gravis. Because weakness is a common symptom of many other disorders, the diagnosis is often missed in people who experience only mild weakness or in those whose weakness is restricted to only a few muscles. The signs the physician looks for are impairment of eye movements or muscle weakness without any changes in the individual’s ability to feel things. If the doctor suspects myasthenia gravis, several tests are available to confirm the diagnosis.
Most people with myasthenia gravis say that the muscle weakness is often worse when they are active and improves after they rest.
Myasthenia gravis can affect the muscles that control breathing in some people.
Myasthenic crisis is a medical emergency that develops when the breathing control muscles are severely weakened. It is usually triggered by an infection, emotional stress, or a bad reaction to medication.
There is no cure for myasthenia gravis, but treatment can help relieve signs and symptoms, such as weakness of arm or leg muscles, double vision, drooping eyelids, and difficulties with speech, chewing, swallowing and breathing.
Your doctor will determine which treatment may be most appropriate for you based on several factors, including:
- Your age
- Severity of your condition
- Location of muscles affected
- Other existing medical conditions
Myasthenia gravis crisis
Myasthenic crisis is a complication of myasthenia gravis characterized by worsening muscle weakness, resulting in respiratory failure that requires intubation and mechanical ventilation 2. A more comprehensive definition of myasthenic crisis also includes post-surgical patients, in whom exacerbation of muscle weakness from myasthenia gravis causes a delay in extubation 3.
Fifteen to 20% of myasthenic patients are affected by myasthenic crisis at least once in their lives 2. The median time to first myasthenic crisis from onset of myasthenia gravis ranges from 8-12 months 4. However, myasthenic crisis may be the initial presentation of myasthenia gravis in one-fifth of patients 4. Overall, women are twice as likely as men to be affected. A bimodal distribution of myasthenic crisis is seen. An early peak prior to age 55 affects women 4:1, whereas a later peak after age 55 affects women and men equally 5. The average age of admission with myasthenic crisis is almost 59 years. Patients in crisis requiring endotracheal intubation spend a median of 17 days in the hospital. Eighteen percent of patients admitted with myasthenic crisis will require discharge to a rehabilitation center 6.
Advances in mechanical ventilation and critical care have been paramount in improving mortality associated with myasthenic crisis. During the early 1960s, respiratory care of these patients was transitioned from negative external pressure ventilation to positive pressure ventilation in an intensive care unit. The mortality rate from myasthenic crisis declined from 42% in the early 1960s to 6% by the late 1970s, and the median age at death increased 7. Currently, mortality is 4% and is primarily the result of comorbid medical conditions 5.
Respiratory support is imperative in the management of myasthenic crisis. Two-thirds to 90% of patients with myasthenic crisis require intubation and mechanical ventilation 8. Over 20% of patients require intubation during evaluation in the emergency department, and almost 60% are intubated after admission to an intensive care unit 8. Elective intubation of a myasthenic patient with impending respiratory failure is favored over emergent intubation. Once intubated, patients should be placed on an assisted ventilator setting with tidal volumes of 8-10 cc/kg ideal body weight and pressure support of 8-15 cm H2O to prevent atelectasis and to minimize the work of breathing. The degree of support required is ultimately patient dependent 9.
Weaning from the ventilator should be initiated after the patient demonstrates clinical improvement, typically at a vital capacity of more than 15 mL/kg 10. Improvement in the strength of neck flexors and other adjunct muscles usually is associated with improvement in bulbar and respiratory muscle strength and can be a useful tool for assessing clinical improvement 10. Patients should be transitioned to a spontaneous mode of ventilation (eg, pressure support ventilation) in which all breaths are patient initiated. Pressure support can then gradually be decreased to minimal settings. If the patient does not tolerate weaning, an assisted ventilator setting should be reinstituted 9.
It remains unclear when first to attempt extubation after myasthenia gravis crisis. Only half of patients are extubated at 13 days 5. In 1 series, 3 independent risk factors for prolonged intubation (>14 days) were identified: age >50 years, peak vital capacity <25 mL/kg on post-intubation days 1 to 6, and a serum bicarbonate ≥30 mmol/L. All of the patients with no risk factors were intubated for less than 2 weeks, whereas 88% of the patients with all 3 risk factors had prolonged intubation. Patients with a prolonged intubation were hospitalized 3 times longer and were less likely to be functionally independent upon discharge 5. Thymoma is also associated with a worse prognosis in myasthenic crisis 8.
Fluctuating weakness and pulmonary complications often confound the decision to extubate 11. A maximal expiratory pressure has been demonstrated to independently predict extubation success. Extubation failure is most commonly associated with a weak cough and inadequate airway clearance 12. Older age, atelectasis, and pneumonia are also associated with extubation failure 4. Tracheostomy placement ranges from 14%-40% 4.
Reintubation occurs more than one-fourth of the time 11. Acidosis, decreased forced vital capacity (FVC), atelectasis, and need for noninvasive ventilatory support are predictors of reintubation.21 Two retrospective studies found atelectasis in all patients requiring reintubation 11. To prevent atelectasis, early intubation, aggressive chest physiotherapy, and frequent suctioning should be implemented and high positive end-expiratory pressure given while the patient is mechanically ventilated 13. Reintubaton is a significant event because patients requiring reintubation have significantly longer ICU and hospital stays 4.
Fever is the most common complication associated with myasthenic crisis. Infectious complications include pneumonia, bronchitis, urinary tract infections, Clostridium difficile colitis, bacteremia, and sepsis 5. When compared to patients admitted for non-crisis myasthenia gravis, patients admitted with myasthenic crisis are more likely to experience sepsis, deep vein thrombosis, and cardiac complications including congestive heart failure, acute myocardial infarction, arrhythmias, and cardiac arrest. These complications, however, are not independent predictors of mortality 6. In 1 series, atelectasis, C. difficile colitis, transfusion-dependent anemia, and congestive heart failure were independently associated with a longer duration of myasthenic crisis, but not with a longer duration of intubation 5.
Common precipitants of myasthenic crisis are shown in Table 1. The most common precipitant is infection 5. One series documented infection in 38% of patients presenting with myasthenic crisis; most commonly, the infection was bacterial pneumonia followed by a bacterial or viral upper respiratory infection 5. Other precipitants include aspiration pneumonitis, surgery, pregnancy, perimenstrual state, certain medications (see below), and tapering of immune-modulating medications 5. Other antecedent factors include exposure to temperature extremes, pain, sleep deprivation, and physical or emotional stress 14. Approximately one-third to one-half of patients may have no obvious cause for their myasthenic crisis 5.
Table 1. Precipitants of Myasthenia gravis crisis
| Stressors | Medications |
|---|---|
| Physical stressors | α-Interferon |
| Aspiration pneumonitis | Antibiotics |
| Infection | Aminoglycosides |
| Perimenstrual state | Gentamicin |
| Pregnancy | Streptomycin |
| Sleep deprivation | Ampicillin |
| Surgery | Macrolides |
| Environmental Stressors | Erythromycin |
| Emotional stress | Quinolones |
| Pain | Ciprofloxacin |
| Temperature extremes | Polymyxin |
| Tapering of immune-modulating medications | Antiepileptics Gabapentin Phenytoin |
| β-Adrenergic antagonists | |
| Calcium channel antagonists | |
| Contrast media | |
| Magnesium Methimazole | |
| Prednisone | |
| Procainamide | |
| Quinidine |
Myasthenia gravis crisis treatment
The 2 primary pharmacologic therapies available for myasthenic crisis are intravenous immunoglobulin (IVIG) and plasma exchange (Table 2). A typical course of IVIg is 400 mg/kg daily for 5 days 16. Patients should be screened for IgA deficiency to avoid anaphylaxis from IVIG 17. The most common side effect associated with IVIg is headache. Other complications include fever, nausea, IV site discomfort, rash, malaise, aches and pain. More serious adverse events can include aseptic meningitis, hypertension, cardiac arrhythmia, thrombocytopenia, and thrombotic events, including stroke, myocardial infarction, and pulmonary embolism.47,48 For PE, 5 exchanges (1 plasma volume or 3-4 L per exchange) are usually performed every other day over 10 days 16. Replacement fluid is generally a solution of normal saline/5% albumin. Venous access is obtained via large-bore peripheral catheters, or temporary or tunneled double-lumen central venous catheters. Infection and bleeding from obtaining central venous access occurs in less than 2% 18. Common complications from PE include fever, symptoms from hypocalcemia (primarily paresthesias), a transient decrease in blood pressure, and tachycardia 18. Other more serious, but less common, adverse effects include cardiac arrhythmia, myocardial infarction, and hemolysis 19. Response to treatment generally occurs after 2 days with Plasma Exchange and after 4-5 days with IVIG 20. For both treatments, an effect can be seen for several weeks 10. If there is insufficient or no response to treatment, Plasma Exchange can be given after IVIg, and IVIg can be administered after Plasma Exchange 17. Although there is a theoretical concern that sequential use of Plasma Exchange after IVIg might result in removal of the IVIg, removal is usually done when there has been insufficient response to IVIg, and removal of IVIg is therefore of little concern.
Table 2. Comparison of Intravenous Immunoglobulin to Plasma Exchange in treating myasthenia gravis crisis
| Intravenous Immunoglobulin (IVIG) | Plasma Exchange | |
|---|---|---|
| Dose | 400 mg/kg × 5 d | One plasma exchange every other day over 10 d |
| Response | Improvement in 4-5 d; effect for 4-8 wk | Improvement in 2 d; effect for 3-4 wk |
| Advantages | More readily available | Faster treatment response |
| Disadvantages | Slower treatment response | Need for special venous access, equipment, and personnel |
| Contraindications | IgA deficiency | Hemodynamic instability, unstable coronary disease, current internal bleeding |
| Serious complications | Aseptic meningitis, cardiac arrhythmia, thrombocytopenia, thrombotic events | Hemodynamic instability, cardiac arrhythmia, myocardial infarction, hemolysis |
Corticosteroids are used in conjunction with IVIg and Plasma Exchange. High-dose prednisone (60-100 mg daily or 1-1.5 mg/kg/d) may be initiated concurrently with IVIg or Plasma Exchange, since prednisone begins to work after 2 weeks, at which point the effects of Plasma Exchange and IVIg are waning. Enteral administration of corticosteroids is preferred, and initiation of prednisione may be deferred until after the patient is extubated if the patient is rapidly improving with IVIg or Plasma Exchange treatment 21. Initial worsening from high-dose prednisone is treated with continued ventilatory support 14. The mean time to improvement with prednisone in 1 series of patients with myasthenia gravis exacerbation was 13 days. Eighty-five percent of patients showed improvement within 3 weeks. Worsening of symptoms with the initiation of corticosteroids is not predictive of overall response to corticosteroids 22. Once the patient has begun to show improvement, the prednisone dose can be decreased and gradually converted to alternate-day dosing 14. The most common side effects from prednisone are a Cushingoid appearance, cataracts, and weight gain 22. Infection, poorly controlled diabetes, and severe osteoporosis are relative contraindications to instituting corticosteroids.
Cyclosporine may be considered after initiation of IVIg or Plasma Exchange in patients who cannot tolerate or who are refractory to corticosteroids. However, the onset of action of cyclosporine is 1-2 months. Other immunomodulating agents, azathioprine and mycophenolate, are not useful in myasthenic crisis because of their long latency of action 21.
Abnormal laboratory values that could affect muscle strength should also be corrected. Potassium, magnesium, and phosphate depletion can all exacerbate myasthenic crisis and should be repleted. Hematocrit less than 30% might affect weakness by decreasing oxygen-carrying capacity 9. Adequate nutrition is important to avoid a negative energy balance and worsening of muscle strength 9.
Myasthenia gravis prognosis
It is difficult to predict the course of myasthenia gravis. It is generally thought that the course of myasthenia gravis tends to be defined in the first several years of illness. Symptoms of myasthenia gravis usually progress to maximum severity within the first few years. After this periods of time, patients usually stabilize or improve. The maximum extent of involvement in an individual patient usually manifests itself within the first 5 to 7 years, and thereafter it tends not to be progressive, even though muscle involvement and severity of weakness may still fluctuate from hour-to-hour and day-to-day.
Patients over the age of 40, those with a short history of severe disease, and those with thymoma have a worse prognosis. In a few cases, myasthenia gravis may cause severe weakness resulting in acute respiratory failure; however, most patients can expect to lead normal or nearly normal lives.
Spontaneous remissions without treatment are rare as are drug free remissions following immunosuppressive medication treatment alone. Thymectomy is felt to induce some drug free remissions, however, no controlled trials have been done and none are likely to be done.
These days, myasthenia gravis is not considered fatal. It can be life threatening if muscle weakness interferes with breathing. It is treatable, but at this stage, not curable. Treatments provide patients with a marked relief of symptoms and often allow them to lead full, normal lives.
Myasthenia gravis life expectancy
With current treatment, however, most cases of myasthenia gravis are not as ‘grave’ as the name implies. Advances in medical care have greatly reduced the mortality rate from respiratory failure in myasthenia gravis patients. In fact, for the majority of individuals with myasthenia gravis, life expectancy is not decreased by the disorder.
With treatment, the outlook for most patients is bright. They will have significant improvement of their muscle weakness and can expect to lead normal or nearly normal lives. Some cases of myasthenia gravis may go into remission temporarily and muscle weakness may disappear completely so that medications can be discontinued. Stable, long-lasting complete remissions are the goal of surgery.
Myasthenia gravis is normally not a degenerative disease – that is, with continued use, the muscle itself should not deteriorate. A distinctive feature of myasthenia gravis, however, is fluctuating weakness of muscles, made worse by use of those muscles and improved by rest of the same muscles. Consequently, a person with myasthenia gravis experiences phases of muscular weakness that alternate with periods of normal health.
The severity varies at different times of the day, and from person to person.
Myasthenia gravis complications
Complications of myasthenia gravis are treatable, but some can be life-threatening.
Myasthenic crisis
Myasthenic crisis is a life-threatening condition that occurs when the muscles that control breathing become too weak to do their jobs. Emergency treatment is needed to provide mechanical assistance with breathing. Medications and blood-filtering therapies help people to again breathe on their own.
Thymus tumors
About 15 percent of people with myasthenia gravis have a tumor in their thymus, a gland under the breastbone that is involved with the immune system. Most of these tumors, called thymomas, aren’t cancerous (malignant).
Other disorders
People with myasthenia gravis are more likely to have the following conditions:
- Underactive or overactive thyroid. The thyroid gland, which is in the neck, secretes hormones that regulate your metabolism. If your thyroid is underactive, you may have difficulties dealing with cold, weight gain and other issues. An overactive thyroid can cause difficulties dealing with heat, weight loss and other issues.
- Autoimmune conditions. People with myasthenia gravis may be more likely to have autoimmune conditions, such as rheumatoid arthritis or lupus.
Myasthenia gravis causes
Myasthenia gravis is an autoimmune disease – a disease where the immune system, which normally protects the body from foreign organisms, mistakenly attacks itself. Myasthenia gravis is most frequently associated with antibodies against acetylcholine receptors (AChR) in the post-synaptic motor end plate 23. A second form of myasthenia gravis, usually seen in young women, involves antibodies against muscle-specific tyrosine kinase (MuSK) 24. The muscle-specific receptor tyrosine kinase protein (MuSK) is involved in forming the nerve-muscular junction. When antibodies block the function of this protein, it may lead to myasthenia gravis. Research continues to study how the antibodies inhibiting this protein are related to the development of myasthenia gravis.
A third group of patients has antibodies to neither acetylcholine receptors (AChR) nor muscle-specific tyrosine kinase (MuSK), and these patients are considered seronegative or antibody-negative myasthenia gravis. Clinically, these patients are similar to patients with acetylcholine receptors (AChR) antibodies 24. Antibodies against another protein, called lipoprotein-related protein 4, may play a part in the development of myasthenia gravis.
Genetic factors also may be associated with myasthenia gravis.
Rarely, mothers with myasthenia gravis have children who are born with myasthenia gravis (neonatal myasthenia gravis). If treated promptly, children generally recover within two months after birth.
Some children are born with a rare, hereditary form of myasthenia, called congenital myasthenic syndrome.
Factors that can worsen myasthenia gravis
- Fatigue
- Illness
- Stress
- Some medications — such as beta blockers, quinidine gluconate, quinidine sulfate, quinine (Qualaquin), phenytoin (Dilantin), certain anesthetics and some antibiotics.
Risk factors for myasthenia gravis
The weakness may be exacerbated by pregnancy, decreased potassium, change of climate, emotion, exercise, gentamicin and other drugs.
It is associated with thymic tumor (thymoma), hypothyroidism, rheumatoid arthritis and SLE (systemic lupus erythematosus).
Myasthenia gravis symptoms
The onset of myasthenia gravis may be sudden and often not immediately recognized. In most cases, the first noticeable symptom is weakness of the eye muscles. In others, difficulty in swallowing and slurred speech may be the first signs.
Muscle weakness may develop over a few days or weeks, or remain at the same level for long periods of time. The degree of muscle weakness involved in myasthenia gravis varies greatly among patients; it may be limited to eye muscles but may take a more generalized form in which many muscles – sometimes including those that control breathing – are affected.
Initial symptoms of myasthenia gravis may include difficulty speaking (dysarthria), difficulty swallowing (dysphagia), a change in facial expression, drooping of one or both eyelids (ptosis), and blurred or double vision (diplopia) due to weakness of the muscles that control eye movements. Patients often have nasal-sounding speech and weak neck muscles that give the head a tendency to fall forward or backward. These symptoms occur in about 90% of myasthenia gravis cases, and are usually intermittent (i.e. come and go), they may disappear for weeks and then recur.
Generalized weakness often develops in the trunk, arms, and legs within a year of onset. Usually arm muscles are affected most severely. Symptoms may include unstable or waddling gait, weakness in arms, hands, fingers, legs and shortness of breath. Muscle weakness tends to worsen as the day progresses, especially after prolonged activity.
Pregnancy can improve, worsen, or have little effect on myasthenia gravis symptoms. Frequently, symptoms first occur during pregnancy or after delivery.
A myasthenic crisis, usually triggered by infection, fever, emotional stress or adverse reaction to medication, may occur when weakness affects the muscles that control breathing. The patient requires a respirator for assisted ventilation.
- Call your local emergency number for an ambulance immediately if you have worsening severe breathing or swallowing difficulties, as you may need emergency treatment in hospital.
Eyes, eyelids and face
Most people with myasthenia gravis have weakness in the muscles of the eyes, eyelids and face.
This can cause:
- droopy eyelids – affecting one or both eyes
- double vision
- difficulty making facial expressions
In around one in five people, the condition only affects the eye muscles. This is known as “ocular myasthenia”.
But for most people, the weakness spreads to other parts of the body over a few weeks, months or years.
If you’ve only had symptoms affecting your eyes for two years or more, it’s unusual for other parts of your body to be affected later on.
Swallowing, speaking and breathing
If the weakness affects the muscles in the mouth, throat and chest, it can cause:
- difficulty chewing
- slurred speech
- a husky, quiet or nasal-sounding voice
- difficulty swallowing
- choking and accidentally inhaling bits of food, which can lead to repeated chest infections
- shortness of breath, particularly when lying down or after exercise
Some people with myasthenia gravis also experience severe breathing difficulties, known as a “mysathenic crisis”.
Limbs and other parts of the body
The weakness caused by myasthenia gravis can also spread to other parts of the body, including the neck, arms and legs.
This can cause:
- difficulty holding the head up
- difficulty with physical tasks, such as lifting, getting up from sitting to standing, climbing stairs, brushing teeth or washing hair
- a waddling walk
- aching muscles after using them
The weakness tends to be worse in the upper body than in the legs and feet.
Myasthenia gravis diagnosis
To diagnose your condition, your doctor will review your symptoms and your medical history and conduct a physical examination. Your doctor may conduct several tests, including:
Neurological examination
Your doctor may check your neurological health by testing your:
- Reflexes
- Muscle strength
- Muscle tone
- Senses of touch and sight
- Coordination
- Balance
The key sign that points to the possibility of myasthenia gravis is muscle weakness that improves with rest.
Myasthenia gravis test
Tests to help confirm the diagnosis may include:
Edrophonium test
Injection of the chemical edrophonium chloride (Tensilon) may result in a sudden, although temporary, improvement in your muscle strength. This is an indication that you may have myasthenia gravis.
Edrophonium chloride blocks an enzyme that breaks down acetylcholine, the chemical that transmits signals from your nerve endings to your muscle receptor sites.
Ice pack test
If you have a droopy eyelid, your doctor may conduct an ice pack test. In this test, a doctor places a bag filled with ice on your eyelid. After two minutes, your doctor removes the bag and analyzes your droopy eyelid for signs of improvement. Doctors may conduct this test instead of the edrophonium test.
Blood analysis
A blood test may reveal the presence of abnormal antibodies that disrupt the receptor sites where nerve impulses signal your muscles to move.
Repetitive nerve stimulation
In this nerve conduction study, doctors attach electrodes to your skin over the muscles to be tested. Doctors send small pulses of electricity through the electrodes to measure the nerve’s ability to send a signal to your muscle.
To diagnose myasthenia gravis, doctors will test the nerve many times to see if its ability to send signals worsens with fatigue.
Single-fiber electromyography (EMG)
Electromyography (EMG) measures the electrical activity traveling between your brain and your muscle. It involves inserting a fine wire electrode through your skin and into a muscle to test a single muscle fiber.
Imaging scans
Your doctor may order a CT scan or an MRI to check if there’s a tumor or other abnormality in your thymus.
Pulmonary function tests
Your doctor may perform pulmonary function tests to evaluate whether your condition is affecting your breathing.
Myasthenia gravis treatment
Myasthenia gravis is one of the most treatable neuromuscular disorders. The choice of treatment depends on several factors, including age, overall health, severity of disease, and rate of disease progression.
A neurologist will determine the appropriate treatment for each patient depending on the severity of the weakness, which muscles are affected, the individual’s age and other medical problems.
Doctors use a variety of treatments, alone or in combination, to relieve symptoms of myasthenia gravis.
Medications
- Cholinesterase inhibitors. Medications such as pyridostigmine (Mestinon) enhance communication between nerves and muscles. These medications don’t cure the underlying condition, but they may improve muscle contraction and muscle strength. Possible side effects may include gastrointestinal upset, nausea, and excessive salivation and sweating.
- Corticosteroids. Corticosteroids such as prednisone inhibit the immune system, limiting antibody production. Prolonged use of corticosteroids, however, can lead to serious side effects, such as bone thinning, weight gain, diabetes and increased risk of some infections.
- Immunosuppressants. Your doctor may also prescribe other medications that alter your immune system, such as azathioprine (Imuran), mycophenolate mofetil (CellCept), cyclosporine (Sandimmune, Neoral), methotrexate (Trexall) or tacrolimus (Prograf). Side effects of immunosuppressants can be serious and may include nausea, vomiting, gastrointestinal upset, increased risk of infection, liver damage and kidney damage.
Patients taking an excess of acetylcholinesterase inhibitors may precipitate a cholinergic crisis characterized by both muscarinic and nicotinic toxicity 21. Symptoms may include an increase in perspiration, lacrimation, salivation and pulmonary secretions, nausea, vomiting, diarrhea, bradycardia, and fasciculations 21. Although cholinergic crisis is an important consideration in the evaluation of the patient in myasthenia gravis crisis, it is uncommon 21. One retrospective series of patients with myasthenic crisis found none of the patients had cholinergic crisis 5. Regardless of whether myasthenic or cholinergic crisis is suspected, acetylcholinesterase inhibitors should be significantly lowered or discontinued to avoid excessive pulmonary secretions in the setting of respiratory distress 25.
Symptoms of Cholinergic Crisis
- Nicotinic toxicity
- Muscle weakness
- Fasciculations
- Muscarinic toxicity
- Diaphoresis
- Excessive tearing
- Increased oral and pulmonary secretions
- Nausea and vomiting
- Diarrhea
- Bradycardia
Although corticosteroids can be used in the treatment of myasthenia gravis, initial treatment with prednisone led to an exacerbation of myasthenia gravis in almost half of patients in 1 series 22. The incidence of myasthenic crisis resulting from corticosteroids ranges from 9%-18% 26. Thus, commencement of corticosteroids for the treatment of myasthenia gravis should always occur in a hospital setting, where respiratory function can be monitored 22. Predictors of exacerbation from prednisone include older age, lower score on the Myasthenia Severity Scale (a clinical assessment tool), and bulbar symptoms 26.
Intravenous therapy
- Plasmapheresis. This procedure uses a filtering process similar to dialysis. Your blood is routed through a machine that removes the antibodies that block transmission of signals from your nerve endings to your muscles’ receptor sites. However, the beneficial effects usually last only a few weeks. After repeated treatments, it may be difficult for doctors to gain access to your vein. They may need to implant a long, flexible tube (catheter) into your chest to conduct the procedure. Other risks associated with plasmapheresis include a drop in blood pressure, bleeding, heart rhythm problems or muscle cramps. Some people may also develop an allergic reaction to the solutions used to replace the plasma.
- Intravenous immunoglobulin (IVIg). This therapy provides your body with normal antibodies, which alters your immune system response. IVIg has a lower risk of side effects than do plasmapheresis and immune-suppressing therapy. However, it may take about a week to start working, and the benefits usually last no more than three to six weeks. Side effects, which usually are mild, may include chills, dizziness, headaches and fluid retention.
- Monoclonal antibody. Rituximab (Rituxan) is an intravenous medication that is used in some cases of myasthenia gravis. This drug depletes certain white blood cells, altering the immune system and improving myasthenia gravis. Rituximab is usually given in infusions at an infusion center or done in a hospital on an outpatient basis. Repeat infusions are often done over a few weeks. Occasionally the infusions are repeated months later.
Surgery
About 15 percent of the people with myasthenia gravis have a tumor in their thymus gland, a gland under the breastbone that is involved with the immune system. If you have a tumor, called a thymoma, doctors will conduct surgery to remove your thymus gland (thymectomy).
Thymic hyperplasia is common in young myasthenic patients with positive acetylcholine receptors (AChR) antibodies, especially women. Thymic tumors, found in 15% of patients with myasthenia gravis and in 32% of patients with myasthenic crisis, should be treated with thymectomy 23. Patients with non-thymomatous myasthenia gravis can consider thymectomy to improve the likelihood of improvement or remission of the disease. One retrospective study found that myasthenic patients who had undergone thymectomy had fewer incidences of myasthenic crisis and less-severe episodes 27.
If you don’t have a tumor in the thymus gland, surgery to remove the thymus gland may improve your myasthenia gravis symptoms. The surgical removal of the thymus gland (which lies in the upper chest area beneath the breastbone and plays an important part in the early development of the immune system) improves symptoms in more than 50% of patients and may cure some by rebalancing the immune system. It may eliminate your symptoms, and you may be able to stop taking medications for your condition. However, you may not notice the benefits of a thymectomy for several years, if at all.
A thymectomy may be performed as an open surgery or as a minimally invasive surgery.
In an open surgery, your surgeon splits the central breast bone (sternum) to open your chest and remove your thymus gland.
Surgeons may perform minimally invasive surgery to remove the thymus gland, which uses smaller incisions. Minimally invasive thymectomy may include:
- Video-assisted thymectomy. In one form of this surgery, surgeons make a small incision in your neck and use a long thin camera (video endoscope) and small instruments to visualize and remove the thymus gland through your neck. Alternatively, surgeons may make a few small incisions in the side of your chest. Doctors use a video scope and small instruments to conduct the procedure and remove the thymus gland through these incisions.
- Robot-assisted thymectomy. In a robot-assisted thymectomy, surgeons make several small incisions in the side of your chest. Surgeons conduct the procedure to remove the thymus gland using a robotic system, which includes a camera arm and mechanical arms.
Benefits of these procedures may include less blood loss, less pain, lower mortality rates and shorter hospital stays compared with open surgery.
Postoperative myasthenic crisis is common after thymectomy; the incidence ranges from 12% to 34% 28. Postoperative crisis in these patients has been related to a history of myasthenic crisis, preoperative presence of bulbar weakness, preoperative serum AChR antibody levels >100 nmol/L, and intraoperative blood loss of >1 L 29.
Home remedies
Supplementing your medical care with these approaches may help you make the most of your energy and cope with the symptoms of myasthenia gravis:
- Adjust your eating routine. Try to eat when you have good muscle strength. Take your time chewing your food, and take a break between bites of food. Small meals eaten several times a day may be easier to handle. Also, try eating mainly soft foods and avoid foods that require more chewing, such as raw fruits or vegetables.
- Use safety precautions at home. Install grab bars or railings in places where you may need support, such as next to the bathtub or next to steps. Keep your floors clean, and move any loose rugs out of areas where you may walk. Outside your home, keep paths, sidewalks and driveways cleared of leaves, snow and other potential debris that could cause you to stumble.
- Use electric appliances and power tools. You may lose energy quickly when conducting tasks. Try using an electric toothbrush, electric can openers and other electrical tools to perform tasks when possible.
- Wear an eye patch. Consider wearing an eye patch if you have double vision, as this can help relieve the problem. Try wearing the eye patch while you write, read or watch television. Periodically switch the eye patch to the other eye to help reduce eyestrain.
- Plan. If you have chores, shopping or errands to do, plan the activity to coincide with the time at which you have the most energy. Also, try to reduce extra walking in your house when working on projects, as it may reduce your energy.
Coping and support
For people with myasthenia gravis and their family members, coping with the disease may be difficult.
If you have myasthenia gravis, find ways to relax. Stress may worsen your condition.
Also, ask for help with tasks if you need it. Your family and friends may be able to assist you with tasks that are difficult.
If you’re a family member of someone with myasthenia gravis, try to be understanding of your loved one’s emotions as he or she adjusts to the condition. Read about myasthenia gravis and learn about what your family member is experiencing.
You and your family members may benefit from participating in a support group. A support group may offer a place for you to meet people who understand what you and your family members are going through.
- Myasthenia gravis. Vincent A, Palace J, Hilton-Jones D. Lancet. 2001 Jun 30; 357(9274):2122-8. http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(00)05186-2/fulltext[↩]
- Ropper AH, Gress DR, Diringer MN, Green DM, Mayer SA, Bleck TP. Treatment of the Critically Ill Patient With Myasthenia Gravis. Neurological and Neurosurgical Intensive Care. 4th ed. Philadelphia, PA: Lipincott Williams & Wilkins; 2004:299–311[↩][↩]
- Bedlack RS, Sanders DB. On the concept of myasthenic crisis. J Clin Neuromuscul Dis. 2002;4:40–42 https://www.ncbi.nlm.nih.gov/pubmed/19078687[↩]
- Rabinstein AA, Mueller-Kronast N. Risk of extubation failure in patients with myasthenic crisis. Neurocrit Care. 2005;3:213–215 https://www.ncbi.nlm.nih.gov/pubmed/16377831[↩][↩][↩][↩][↩]
- Thomas CE, Mayer SA, Gungor Y, et al. Myasthenic crisis: clinical features, mortality, complications, and risk factors for prolonged intubation. Neurology. 1997;48:1253–1260 https://www.ncbi.nlm.nih.gov/pubmed/9153452[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology. 2009;72:1548–1554 https://www.ncbi.nlm.nih.gov/pubmed/19414721[↩][↩]
- Cohen MS, Younger D. Aspects of the natural history of myasthenia gravis: crisis and death. Ann N Y Acad Sci. 1981;377:670–677 https://www.ncbi.nlm.nih.gov/pubmed/6951491 [↩]
- O’Riordan JI, Miller DH, Mottershead JP, Hirsch NP, Howard RS. The management and outcome of patients with myasthenia gravis treated acutely in a neurological intensive care unit. Eur J Neurol. 1998;5:137–142 https://www.ncbi.nlm.nih.gov/pubmed/10210824[↩][↩][↩]
- Kirmani JF, Yahia AM, Qureshi AI. Myasthenic crisis. Curr Treat Options Neurol. 2004;6:3–15 https://www.ncbi.nlm.nih.gov/pubmed/14664765[↩][↩][↩][↩]
- Meriggioli MN. Myasthenia gravis: immunopathogenesis, diagnosis, and management. Continuum: Lifelong Learning in Neurology. 2009;15:35–62[↩][↩][↩]
- Seneviratne J, Mandrekar J, Wijdicks EF, Rabinstein AA. Predictors of extubation failure in myasthenic crisis. Arch Neurol. 2008;65:929–933 https://www.ncbi.nlm.nih.gov/pubmed/18625860[↩][↩][↩]
- Wu JY, Kuo PH, Fan PC, Wu HD, Shih FY, Yang PC. The role of non-invasive ventilation and factors predicting extubation outcome in myasthenic crisis. Neurocrit Care. 2009;10:35–42 https://www.ncbi.nlm.nih.gov/pubmed/18810663[↩]
- Varelas PN, Chua HC, Natterman J, et al. Ventilatory care in myasthenia gravis crisis: assessing the baseline adverse event rate. Crit Care Med. 2002;30:2663–2668 https://www.ncbi.nlm.nih.gov/pubmed/12483056[↩]
- Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve. 2004;29:484–505 https://www.ncbi.nlm.nih.gov/pubmed/15052614[↩][↩][↩]
- Wendell LC, Levine JM. Myasthenic Crisis. The Neurohospitalist. 2011;1(1):16-22. doi:10.1177/1941875210382918. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3726100/[↩][↩]
- Ahmed S, Kirmani JF, Janjua N, et al. An update on myasthenic crisis. Curr Treat Options Neurol. 2005 Mar;7(2):129–141 https://www.ncbi.nlm.nih.gov/pubmed/15676116[↩][↩]
- Younger DS, Raksadawan N. Therapy in neuromuscular disease. Neurol Clin. 2001;19:205–215, vii https://www.ncbi.nlm.nih.gov/pubmed/11471765[↩][↩]
- Shemin D, Briggs D, Greenan M. Complications of therapeutic plasma exchange: a prospective study of 1,727 procedures. J Clin Apher. 2007;22:270–276 https://www.ncbi.nlm.nih.gov/pubmed/17722046[↩][↩]
- Rodnitzky RL, Goeken JA. Complications of plasma exchange in neurological patients. Arch Neurol. 1982;39:350–354 https://www.ncbi.nlm.nih.gov/pubmed/7092611[↩]
- Gold R, Schneider-Gold C. Current and future standards in treatment of myasthenia gravis. Neurotherapeutics. 2008;5:535–541 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4514693/[↩]
- Lacomis D. Myasthenic crisis. Neurocrit Care. 2005;3:189–194 https://www.ncbi.nlm.nih.gov/pubmed/16377829[↩][↩][↩][↩][↩]
- Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol. 1984;15:291–298 https://www.ncbi.nlm.nih.gov/pubmed/6721451[↩][↩][↩][↩]
- Chaudhuri A, Behan PO. Myasthenic crisis. QJM. 2009;102:97–107 https://www.ncbi.nlm.nih.gov/pubmed/19060020[↩][↩]
- Vincent A, Leite MI. Neuromuscular junction autoimmune disease: muscle specific kinase antibodies and treatments for myasthenia gravis. Curr Opin Neurol. 2005;18:519–525 https://www.ncbi.nlm.nih.gov/pubmed/16155434[↩][↩]
- Juel VC. Myasthenia gravis: management of myasthenic crisis and perioperative care. Semin Neurol. 2004;24:75–81 https://www.ncbi.nlm.nih.gov/pubmed/15229794[↩]
- Bae JS, Go SM, Kim BJ. Clinical predictors of steroid-induced exacerbation in myasthenia gravis. J Clin Neurosci. 2006;13:1006–1010 https://www.ncbi.nlm.nih.gov/pubmed/17074487[↩][↩]
- Soleimani A, Moayyeri A, Akhondzadeh S, Sadatsafavi M, Tavakoli Shalmani H, Soltanzadeh A. Frequency of myasthenic crisis in relation to thymectomy in generalized myasthenia gravis: a 17-year experience. BMC Neurol. 2004;4:12. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC518967/[↩]
- Kas J, Kiss D, Simon V, Svastics E, Major L, Szobor A. Decade-long experience with surgical therapy of myasthenia gravis: early complications of 324 transsternal thymectomies. Ann Thorac Surg. 2001;72:1691–1697 https://www.ncbi.nlm.nih.gov/pubmed/11722066[↩]
- Watanabe A, Watanabe T, Obama T, et al. Prognostic factors for myasthenic crisis after transsternal thymectomy in patients with myasthenia gravis. J Thorac Cardiovasc Surg. 2004;127:868–876 https://www.ncbi.nlm.nih.gov/pubmed/15001919[↩]





{kind=link}