Contents
What is NADH
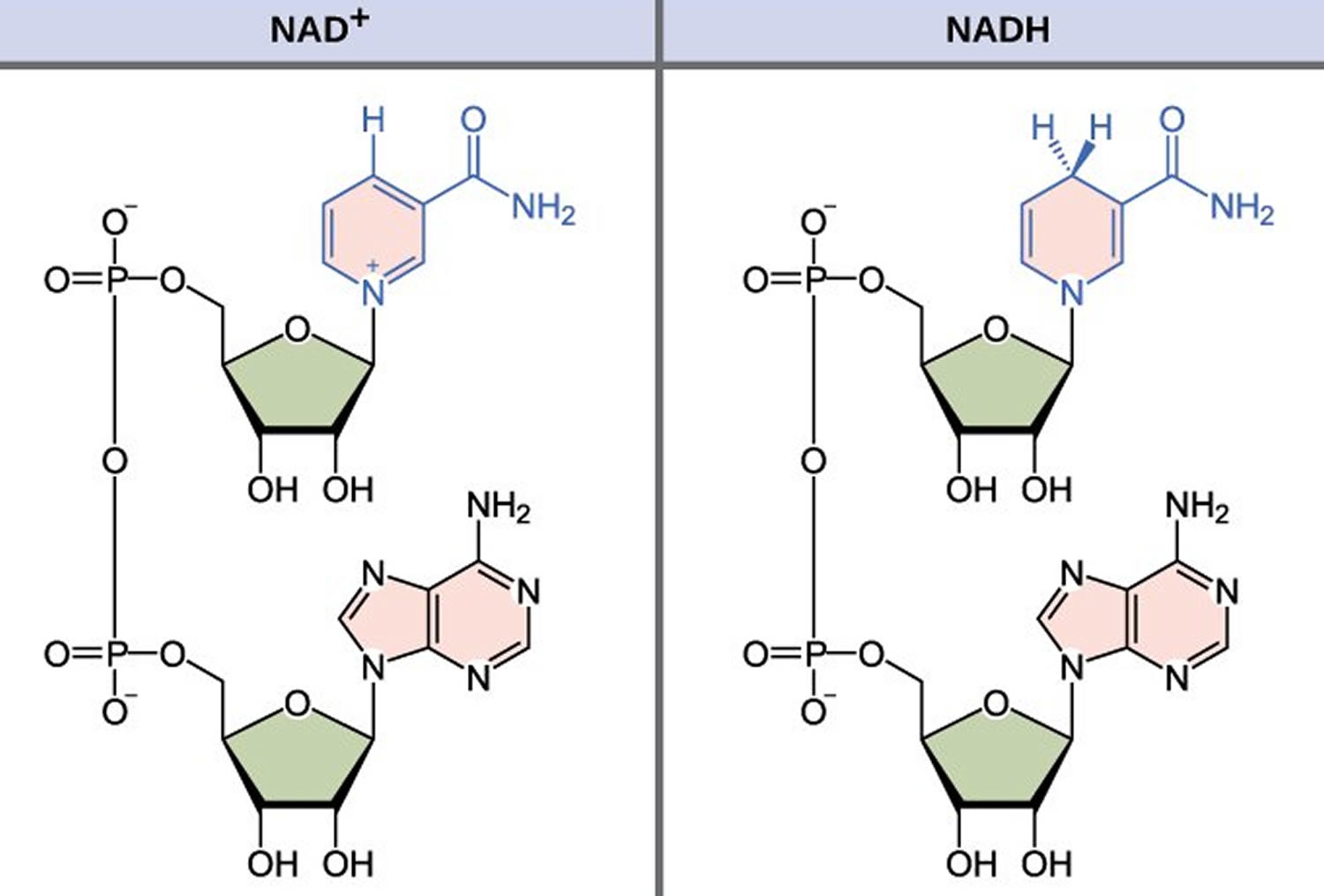
NADH is short for reduced nicotinamide adenine dinucleotide. NADH is a coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). NADH is the reduced form of NAD+ and NAD+ is the oxidized form of NADH 1. NAD+ and NADH participate in reactions such as glycolysis, the tricarboxylic acid cycle (citric acid cycle), and oxidative phosphorylation, participating in multiple redox reactions in cells 2. In addition, it serves as a substrate for several enzymes involved in DNA damage repair, such as the sirtuins (silent information regulator 2 or Sir2) and poly (ADP-ribose) polymerases (PARPs) 3. The human serum NADH concentration was found to be in very small amount (50 nM to 1.2 μM) 4.
Nicotinamide adenine dinucleotide (NAD+) is a coenzyme found in all living cells. NAD+ (nicotinamide adenine dinucleotide) serves both as a critical coenzyme for enzymes that fuel reduction-oxidation reactions, carrying electrons from one reaction to another, and as a cosubstrate for other enzymes such as the sirtuins and poly(adenosine diphosphate–ribose) polymerases. Cellular NAD+ concentrations change during aging, and modulation of NAD+ usage or production can prolong both health span and life span.
The NAD+/NADH ratio also regulates the activity of various metabolic pathway enzymes such as those involved in glycolysis, Kreb’s cycle (also known as tricarboxylic acid cycle or citric acid cycle), and fatty acid oxidation 5. Intracellular NAD+ is synthesized de novo from L-tryptophan, although its main source of synthesis is through salvage pathways from dietary vitamin B3 (Niacin) as precursors. NAD+ is utilized by various proteins including sirtuins (silent information regulator 2), poly ADP-ribose polymerases (PARPs) and cyclic ADP-ribose synthases. The NAD+ pool is thus set by a critical balance between NAD+ biosynthetic and NAD+ consuming pathways. Raising cellular NAD+ content by inducing its biosynthesis or inhibiting the activity of poly ADP-ribose polymerases (PARPs) and cyclic ADP-ribose synthases via genetic or pharmacological means lead to sirtuins activation. Sirtuins (silent information regulator 2) modulate distinct metabolic, energetic and stress response pathways, and through their activation, NAD+ directly links the cellular redox state with signaling and transcriptional events. NAD+ levels decline with mitochondrial dysfunction and reduced NAD+/NADH ratio is implicated in mitochondrial disorders, various age-related pathologies as well as aging.
Sirtuins (silent information regulator 2 or Sir2) proteins are a family of evolutionarily conserved nicotinamide adenine dinucleotide (NAD+)-dependent protein deacylases harboring lysine deacetylase, desuccinylase, demalonylase, demyristoylase and depalmitoylase activity 6 or an ADP-ribosyltransferase activity 7. Mammals contain seven sirtuins (SIRT1–7) that are locacted in different subcellular compartments i.e. nucleus (SIRT1, SIRT6 and SIRT7), cytosol (SIRT2), and mitochondria (SIRT3, SIRT4 and SIRT5) 8 and are implicated in a wide variety of biological functions including control of cellular metabolism and energy homeostasis, aging and longevity, transcriptional silencing, cell survival, proliferation, differentiation, DNA damage response, stress resistance, and apoptosis 9. Since sirtuins are NAD+-dependent enzymes, the availability of NAD+ is one of the key mechanisms that regulate their activity. Sirtuins therefore serve as “metabolic sensors” of the cells as their activity is coupled to changes in the cellular NAD+/NADH redox state, which is largely influenced by the availability and breakdown of nutrients 10. Thus, NAD+ is not only a vital cofactor/coenzyme but also a signaling messenger that can modulate cell metabolic and transcriptional responses. Changes in cellular NAD+ levels can occur due to modulation of pathways involved in NAD+ biosynthesis and consumption. Reduced NAD+ levels have been reported in mitochondrial and age-related disorders, and NAD+ levels also decline with age 11. Boosting cellular NAD+ levels serves as a powerful means to activate sirtuins, and as a potential therapy for mitochondrial as well as age-related disorders.
Figure 1. NADH redox reaction
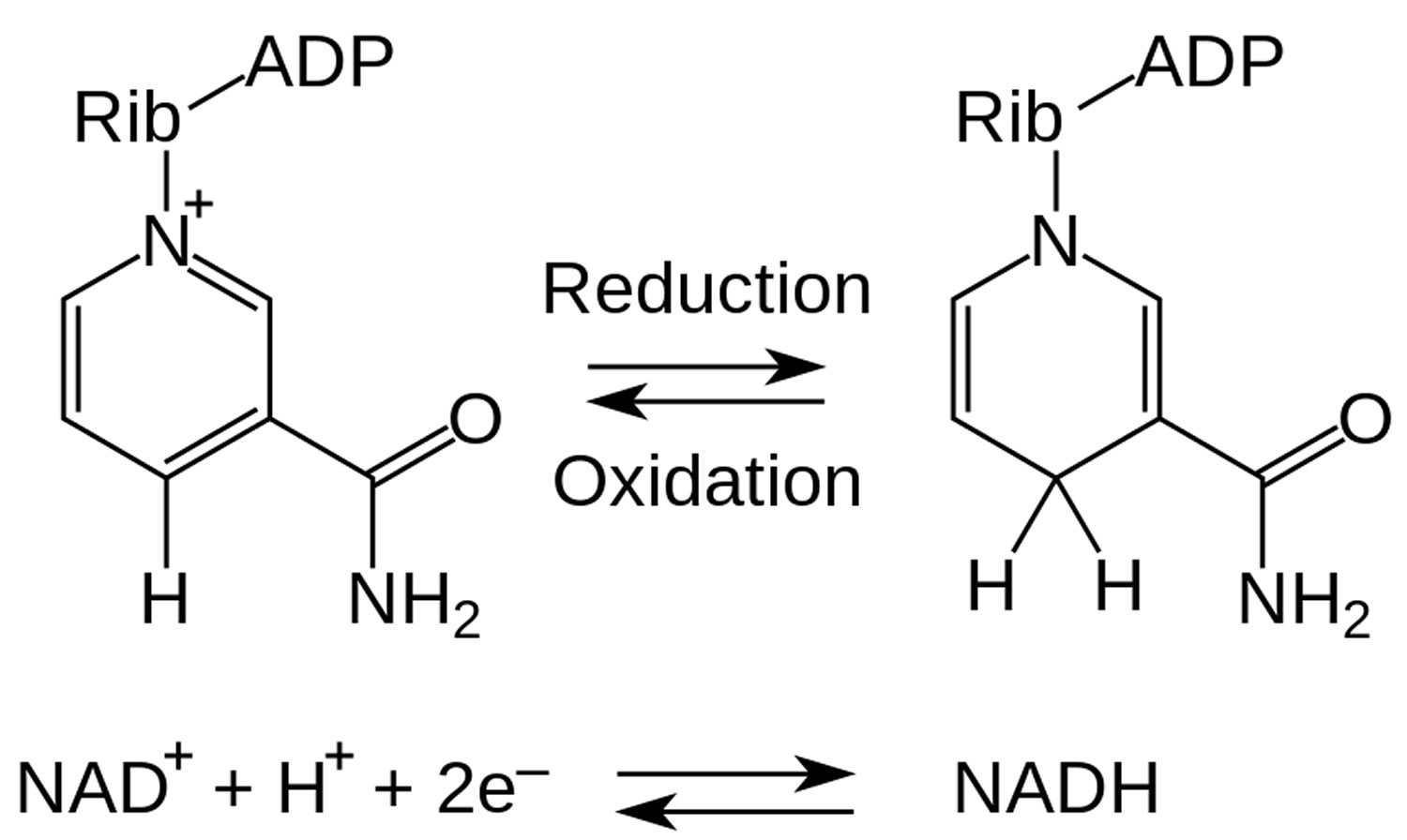
Figure 2. NADH and NAD+ redox metabolism
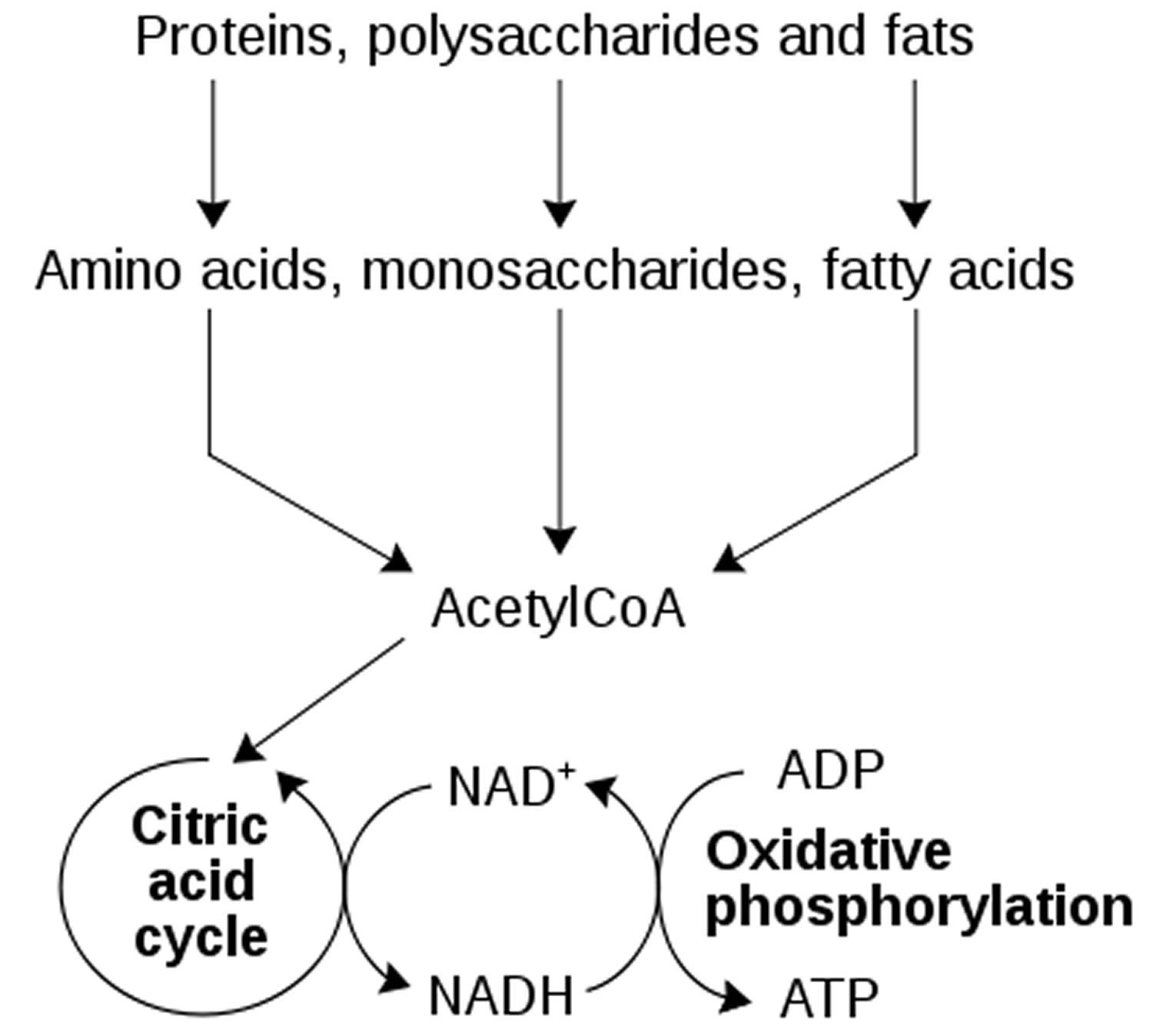
It is known, as aging progresses, nicotinamide adenine dinucleotide (NAD+) levels decrease and are involved in age-related metabolic decline and mitochondrial dysfunction 12. Elevated NADH to NAD+ ratio further suggests that older individuals of both sexes are unable to utilize NADH as effectively as the younger adults. This observation has direct bearing on the mitochondrial oxidation. NADH is perhaps not oxidized efficiently in the older and female adults than the younger individuals, i.e. less of the energy pool (ATP) in the older adults. Recent studies have shown that a reduction in NAD+ is a key factor for the development of age-associated metabolic decline. Increased NAD+ levels in vivo results in activation of pro-longevity and health span-related factors. Also, it improves several physiological and metabolic parameters of aging, including muscle function, exercise capacity, glucose tolerance, and cardiac function in mouse models of natural and accelerated aging.
It has been shown that the cellular NAD+ pool is determined by a balance between the activity of NAD-synthesizing and NAD-consuming enzymes 13. In previous publications, it was demonstrated that expression and activity of the NADase CD38 increases with age and that CD38 is required for the age-related NAD decline and mitochondrial dysfunction via a pathway mediated at least in part by regulation of SIRT3 activity (see Figure 3 below) 14. It was also identified CD38 as the main enzyme involved in the degradation of the NAD precursor nicotinamide mononucleotide (NMN) in vivo. That indicates that CD38 has a key role in the modulation of NAD-replacement therapy for aging and metabolic diseases 14. CD38 was originally identified as a cell-surface enzyme that plays a key role in several physiological processes such as immune response, inflammation, cancer, and metabolic disease 15.
Type 2 diabetes has become an epidemic due to calorie-rich diets overwhelming the adaptive metabolic pathways. One such pathway is mediated by nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in mammalian NAD+ biosynthesis and the NAD+-dependent protein deacetylase SIRT1. The results presented in this study in mice demonstrated that nicotinamide phosphoribosyltransferase (NAMPT)-mediated NAD+ biosynthesis is severely compromised by high fat diet and aging, contributing to the pathogenesis of type 2 diabetes 16. Strikingly, nicotinamide mononucleotide (NMN), a product of the nicotinamide phosphoribosyltransferase (NAMPT) reaction and a key NAD+ intermediate, ameliorates glucose intolerance by restoring NAD+ levels in high fat diet-induced type 2 diabetes mice. Nicotinamide mononucleotide (NMN) also enhances hepatic insulin sensitivity and restores gene expression related to oxidative stress, inflammatory response, and circadian rhythm, partly through SIRT1 activation. Furthermore, NAD+ and NAMPT levels show significant decreases in multiple organs during aging, and nicotinamide mononucleotide (NMN) improves glucose intolerance and lipid profiles in age-induced type 2 diabetes mice 16.
The recent development of potent and specific CD38 inhibitors 17, together with the novel findings highlighting the role of NAD+ replacement therapy and CD38 in age-related diseases such as hearing loss and Alzheimer’s 18, indicate that CD38 inhibition combined with NAD precursors may serve as a potential therapy for metabolic dysfunction and age-related diseases.
Figure 3. NAD decline due to increases in CD38/NADase during aging

NAD+ biosynthesis, consumption and compartmentalization
The mammalian NAD+ biosynthesis occurs via de novo and salvage pathways, and involves four major substrates including the essential amino acid l-tryptophan (Trp), nicotinic acid (NA), nicotinamide (NAM), and nicotinamide riboside (NR) 19. De novo biosynthesis of NAD+ starts from dietary L-tryptophan (Trp) which is catalytically converted to N-formylkynurenine by either indoleamine 2,3-dioxygenase (IDO) or tryptophan 2,3-dioxygenase (TDO) and is the first rate limiting step. N-formylkynurenine is then converted by a series of four enzymatic reactions to α-amino-β-carboxymuconate-ε-semialdehyde (ACMS) which is unstable and hence undergoes either complete enzymatic oxidation or non-enzymatic cyclization to quinolinic acid (see Figure 4). The second rate limiting step involves the catalytic conversion of quinolinic acid to nicotinic acid mononucleotide (NAMN) by quinolinate phosphoribosyl transferase (QPRT). Next, NAMN is converted to nicotinic acid adenine dinucleotide (NAAD) by one of the three isoforms of nicotinamide mononucleotide adenylyltransferase (NMNAT) enzyme. The human NMNAT1 is localized in the nucleus, NMNAT2 is found in the Golgi and cytosol, whereas NMNAT3 is localized in both mitochondria and cytosol 20. The final step of de novo biosynthesis is the amidation of NAAD by NAD synthase (NADS) enzyme (see Figure 4) 19. The de novo pathway contributes only a minor fraction to the total NAD+ pool, however, its importance is stressed by the human disease pellagra which is caused by dietary deficiency of Trp and NAM intermediate, leading to diarrhea, dermatitis, dementia and ultimately death 21. However, pellagra is easily treated by dietary supplementation of L-tryptophan (Trp) or niacin (vitamin B3) (i.e. nicotinic acid, nicotinamide and nicotinamide riboside). The primary source of NAD+ biosynthesis is the salvage or Preiss-Handler pathway which utilizes dietary niacin as precursors (Figure 4). The salvage pathway involves catalytic conversion of nicotinic acid to nicotinic acid mononucleotide by nicotinic acid phosphoribosyltransferase (NAPT), which is subsequently converted to NAD+ by the action of nicotinamide mononucleotide adenylyltransferase (NMNAT) and NAD synthase (NADS) enzymes. The nicotinamide and nicotinamide riboside are converted to nicotinamide mononucleotide (NMN) by the action of nicotinamide phosphoribosyltransferase (NAMPT) and nicotinamide riboside kinase (NRK) enzymes respectively. Finally, nicotinamide mononucleotide (NMN) is enzymatically converted to NAD+ by nicotinamide mononucleotide adenylyltransferase (NMNAT).
The cellular abundance of NAD+ is also regulated by its breakdown since NAD+ serves as a degradation substrate for multiple enzymes including sirtuins, poly ADP-ribose polymerases (PARPs) and cyclic ADP (cADP) ribose synthases which cleave NAD+ to produce nicotinamide and an ADP-ribosyl product 22. For instance, the deacetylase activity of mammalian sirtuins uses NAD+ to cleave the acetyl group from ε–acetyl lysine residues of target proteins to generate nicotinamide and 2′O-acetyl-ADP-ribose. Sirtuins are activated in response to nutrient deprivation or energy deficit which triggers cellular adaptations to improve metabolic efficiency. Poly ADP-ribose polymerases’s are activated in response to DNA damage (e.g. DNA strand breaks) and genotoxic stress, and use NAD+ to catalyze a reaction in which the ADP ribose moiety is transferred to a substrate protein. The cADP-ribose synthases (e.g. CD38 and CD157) use NAD+ to generate cADP-ribose which serves as an intracellular second messenger. The members of poly ADP-ribose polymerases and cADP-ribose synthase family show increased affinity and lower Km for NAD+ compared to sirtuins, indicating that their activation critically impacts intracellular NAD+ levels and determines if it reaches a permissive threshold for sirtuin activation 23. Multiple studies also suggested that PARP activity constitutes the main NAD+ catabolic activity, which drives cells to synthesize NAD+ from de novo or salvage pathways 24.
The intracellular NAD+ levels are typically between 0.2 and 0.5 mM in mammalian cells, and change during a number of physiological processes 23. Since the nucleus, cytosol and mitochondria are equipped with NAD+ salvage enzymes, the compartment-specific NAD+ production activates distinct sirtuins to trigger the appropriate physiological response. The NAD+/NADH levels also vary with the availability of dietary energy and nutrients. For instance, tissue NAD+ levels decrease with energy overload such as high-fat diet 25 and display circadian oscillations with a 24 hour rhythm in the liver, which is regulated by feeding 26. During energetic stress such as exercise, calorie restriction and fasting in mammals, the NAD+ levels increase leading to sirtuin activation, which is associated with metabolic and age-related health benefits (Figure 5) 27. Decreased sirtuins (e.g. SIRT1 and SIRT3) expression is associated with various age-related pathologies 28 and their overexpression has been reported to enhance overall mitochondrial and metabolic health in age-related disorders as well as mitochondrial diseases 29.
Figure 4. NAD+ biosynthesis
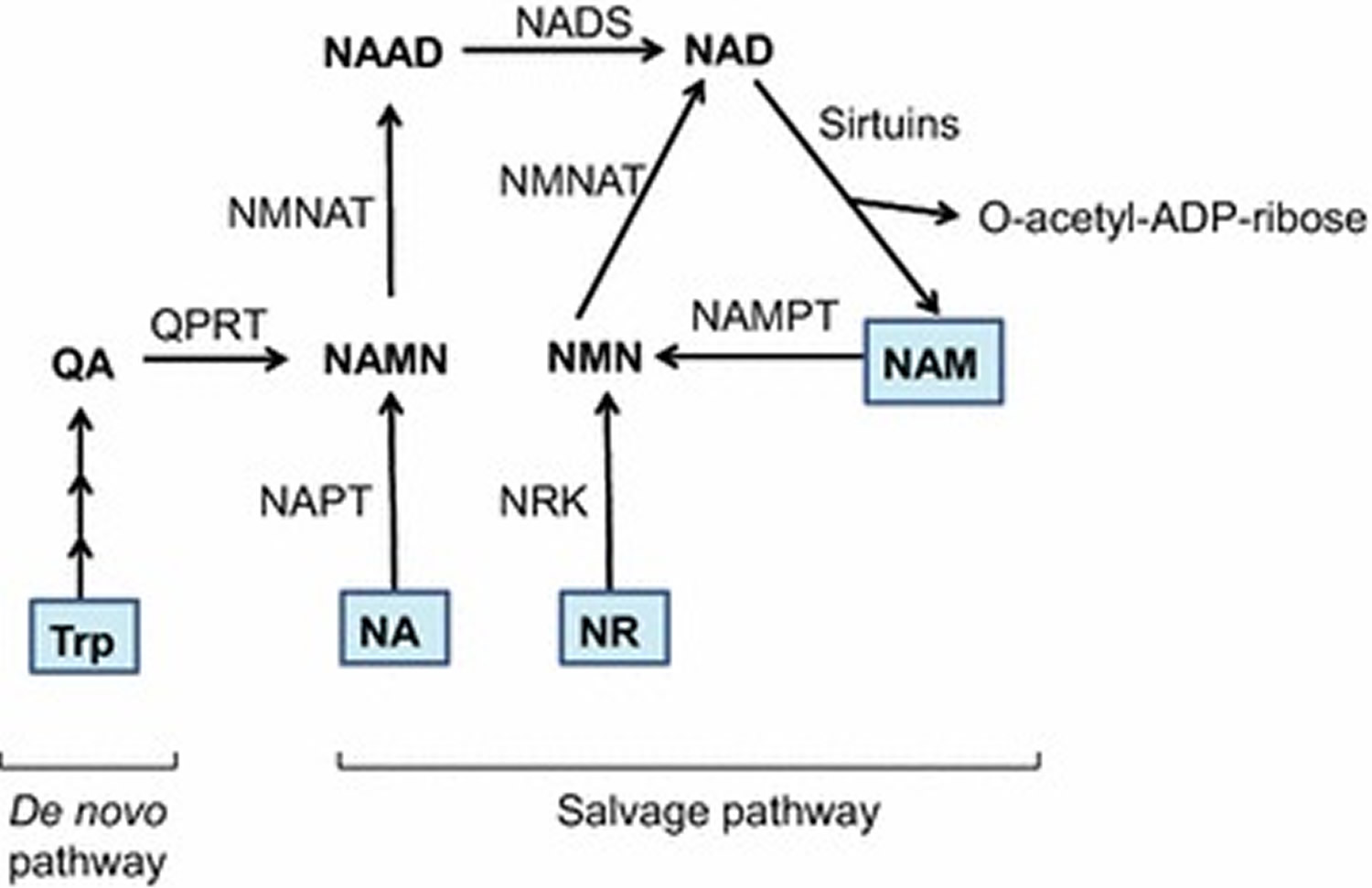
Footnotes: Schematic representation of de novo and salvage pathways for NAD+ biosynthesis. In mammals, the de novo biosynthesis starts from l-tryptophan (Trp) which is enzymatically converted in a series of reactions to quinolinic acid (QA). Through quinolinate phosphoribosyltransferase (QPRT) enzyme activity, QA is converted to nicotinic acid mononucleotide (NAMN), which is then converted to nicotinic acid adenine dinucleotide (NAAD) by nicotinamide mononucleotide adenylyltransferase (NMNAT) enzyme. The final step in de novo biosynthesis is the amidation of NAAD by NAD synthase (NADS) which generates NAD+. The salvage pathway involves NAD+ synthesis from its precursors, i.e. Nicotinic acid (NA), nicotinamide (NAM) or nicotinamide riboside (NR). NA is catalytically converted to NAMN by the action of nicotinic acid phosphoribosyltransferase (NAPT). NAM is converted by nicotinamide phosphoribosyltransferase (NAMPT) to nicotinamide mononucleotide (NMN), which is also the product of phosphorylation of NR by nicotinamide riboside kinase (NRK) enzyme. Finally, NAMN is converted to NAD by the action of NMNAT and NADS enzymes, whereas NMN is converted to NAD by the NMNAT enzyme. Multiple enzymes break-down NAD+ to produce NAM and ADP-ribosyl moiety, however only sirtuins are depicted in this figure
[Source 30]Figure 5. Boosting NAD+ levels is beneficial for health and lifespan
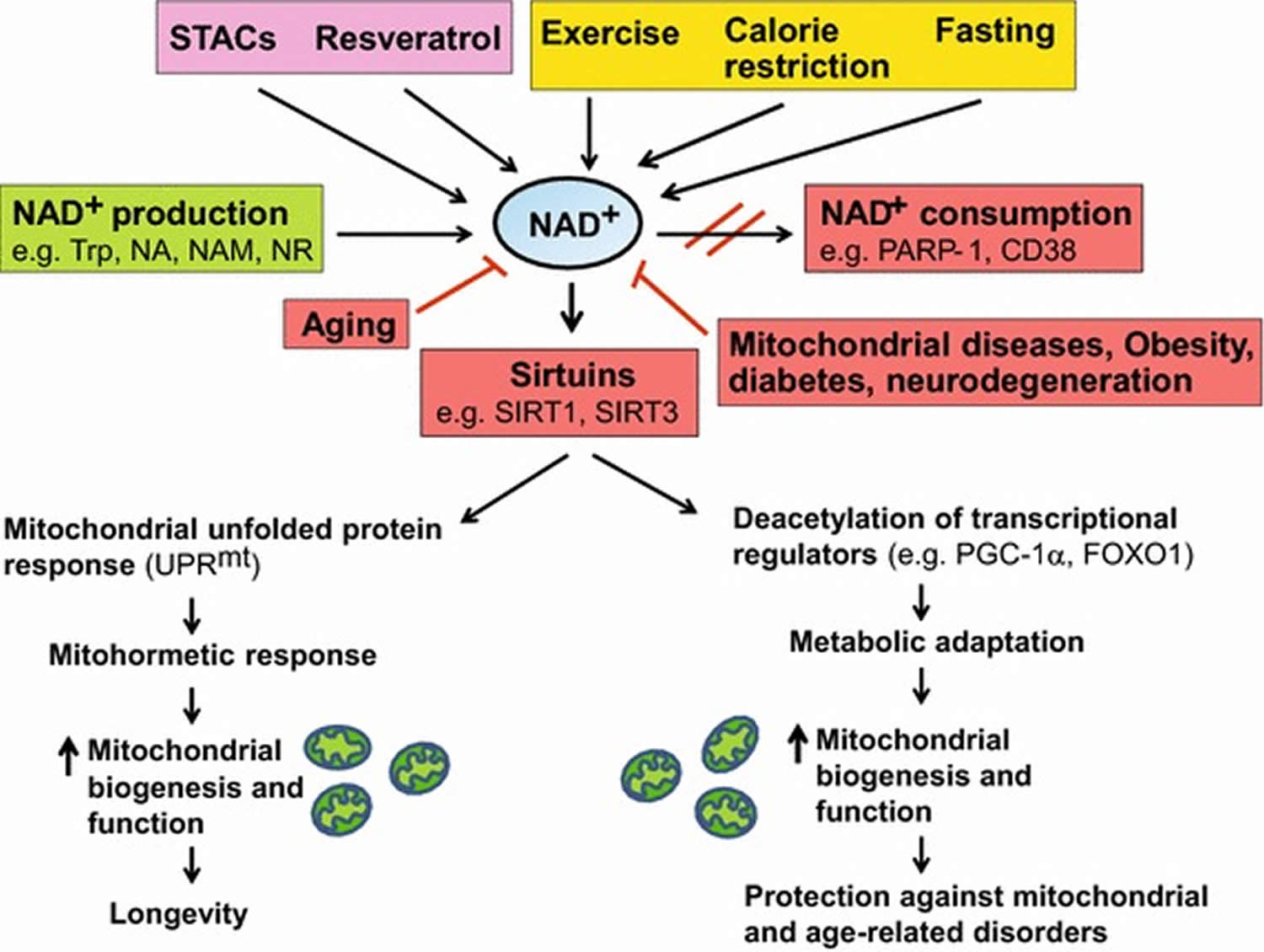
Footnotes: NAD+ is a rate-limiting cofactor for the enzymatic activity of sirtuins. Boosting intracellular NAD+ levels by physiological (e.g. exercise, calorie restriction, fasting) or pharmacological [e.g. resveratrol, sirtuin activating compounds (STACs)] interventions, and inducing NAD+ biosynthesis through supplementation with precursors (e.g. NA, NAM, NR) or inhibition of NAD+ consuming enzymes (e.g. PARP-1, CD38) leads to activation of sirtuins (e.g. SIRT1, SIRT3). SIRT1 deacetylates and activates transcriptional regulators (e.g. PGC-1α, FOXO1), whereas SIRT3 deacetylates and activates multiple metabolic gene targets (e.g. succinate dehydrogenase, superoxide dismutase 2), which in turn regulate mitochondrial biogenesis and function. Supplementation with NR or PARP inhibitors extends lifespan in worms by inducing the UPRmt stress signaling response via Sir-2.1 activation, which then triggers an adaptive mitohormetic response to stimulate mitochondrial function and biogenesis. Improved mitochondrial function associated with mitohormesis or metabolic adaptation can attenuate the impact of mitochondrial diseases, aging as well as age-related metabolic and neurodegenerative disorders. The physiological and pharmacological interventions that boost NAD+ levels are highlighted in yellow and pink respectively whereas the pathways that produce and consume/decrease NAD+ levels are highlighted in green and red respectively
[Source 30]What does NADH do?
NAD+ and its phosphorylated and reduced forms including NADP+, NADH, and NADPH (reduced nicotinamide adenine dinucleotide phosphate) are vital in regulating cellular metabolism and energy production. NAD+ functions as an oxidoreductase cofactor in a wide range of metabolic reactions and modulates the activity of compartment-specific pathways such as glycolysis in the cytosol, and tri-carboxylic acid (TCA) cycle, OXPHOS, fatty acid and amino acid oxidation in the mitochondria. For instance, NAD+ is converted to NADH at the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) step of glycolysis, a pathway that generates pyruvate from glucose 31. In the mitochondrial compartment, NAD+ is converted to NADH at multiple steps in the tricarboxylic acid cycle (citric acid cycle) in which acetyl-coenzyme A is oxidized to carbon dioxide. Mitochondrial NADH is then oxidized by furnishing reducing equivalents to complex I in the ETC through a series of redox reactions that generate ATP from ADP by OXPHOS. The NAD+/NADH ratio thus regulates multiple metabolic pathway enzymes including glyceraldehyde 3-phosphate dehydrogenase (GAPDH), pyruvate dehydrogenase, isocitrate dehydrogenase, α-ketoglutarate dehydrogenase and malate dehydrogenase. In contrast to NAD+/NADH, the NADPH/NADP+ ratios are maintained high in both cytosol and mitochondrial compartments, to maintain a reducing environment 32. NADPH plays a key role in reductive biosynthesis and cellular defense against oxidative damage 33. For instance, NADPH serves as a cofactor for P450 enzymes that detoxify xenobiotics, acts as a terminal reductant for glutathione reductase which maintains reduced glutathione levels during oxidative defense, and also serves as a substrate for NADPH oxidase that generates peroxides for release during oxidative burst processes in the immune system 33.
Mitochondrial disorders represent one of the most common forms of heritable metabolic disease in children 34. Reduced NAD+/NADH ratio is strongly implicated in mitochondrial disorders and, age-related disorders including diabetes, obesity, neurodegeneration and cancer 35. NAD+ levels also decline during aging in multiple models including worms, rodents and human tissue 36. Increasing evidence suggests that boosting NAD+ levels could be clinically beneficial, as it activates the NAD+/sirtuin pathway which yields beneficial effects on multiple metabolic pathways.
Pharmacological activation of NAD+ production has recently been used to treat mouse models of mitochondrial diseases. For instance, treatment of cytochrome c oxidase (COX) deficiency caused by SURF1, SCO2 or COX15 genetic mutations in mice, with AMPK agonist 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), partially rescued mitochondrial dysfunction and improved motor performance 37. These findings could be explained by the fact that AMPK stimulates NAD+ production, consequently activating SIRT1 which promotes energy production and homeostasis 28. Oral administration of NAD+ precursor, NR in mitochondrial myopathy mice harboring a pathogenic mutation in the mtDNA helicase—Twinkle, effectively delayed myopathy progression, by increasing mitochondrial biogenesis, preventing mitochondrial ultrastructural abnormalities, mtDNA deletion formation and activating the mitochondrial unfolded protein (UPRmt) response 38. In addition, NR supplementation and reduction of NAD+ consumption by a specific PARP inhibitor significantly improved mitochondrial respiratory chain defect and exercise intolerance, in a mouse model of COX deficiency caused by SCO2 mutation 29.
Besides improving mitochondrial function, boosting NAD+ levels with resveratrol, nicotinamide riboside or nicotinamide mononucleotide (NMN) also corrects metabolic disturbances in mice caused by high fat diet 28. Nicotinamide mononucleotide (NMN) administration ameliorates glucose intolerance and insulin resistance in diet- and age-induced type 2 diabetic mice 39 and rectifies glucose-stimulated insulin secretion and glucose intolerance in NAMPT-deficient animals, by restoring NAD+ levels 40. Interventions using NAD+ precursors or poly ADP-ribose polymerase inhibitors were also shown to be neuroprotective. For instance treatment with nicotinamide mononucleotide (NMN) or nicotinamide riboside precursors, protected against axonal degeneration and hearing loss in mice 41. Raised NAD+ levels after calorie restriction, nicotinamide or nicotinamide riboside treatment attenuated increase in β-amyloid content and oxidative damage, preventing cognitive decline and neurodegeneration in rodent models of Alzheimer’s disease 42. PARP-1 (poly ADP-ribose polymerase 1) activation also occurs in neurodegenerative DNA repair disorders including xeroderma pigmentosum group A (XPA) and Cockayne syndrome group B, and treatment with specific PARP inhibitors rescues defective phenotypes in XPA mutant worms and Cockayne syndrome group B mutant mice respectively 43. However, PARP-2 (poly ADP-ribose polymerase 2) deleted mice were glucose intolerant and exhibited pancreatic dysfunction, implying that these results may interfere with other beneficial consequences of PARP inhibition, and hence warrant further investigation on the safe clinical use of these inhibitors 44. Because poly ADP-ribose polymerase inhibitors enhance oxidative metabolism and improve metabolic flexibility, these compounds are being tested in phase III trials as anti-cancer agents 45.
Increasing NAD+ levels by treatment with nicotinic acid and nicotinamide precursors has been shown to inhibit metastasis and breast cancer progression in response to mitochondrial complex I defect in mice 46. However, reducing NAD+ bioavailability is reported to have an antineoplastic effect in various tumor cell types, as cancer cells rely on increased central carbon metabolism and biomass production to sustain an unrestricted growth 47. The exact role of sirtuins in cancer remains controversial with dichotomous functions being reported, for example multiple studies have shown that SIRT1, SIRT3 and SIRT5 can act as tumor promoters or tumor suppressors under different cellular conditions, tumor stage and tissue of origin 48. However, SIRT4 is only shown to have a tumor suppressor function 49. Further research is needed to understand why and how certain sirtuins have both oncogenic or tumor-suppressive roles, and how this dual action may be best exploited for cancer management.
Declining NAD+ levels during aging compromise mitochondrial function in multiple model organisms, which can be restored via NAD+ precursor supplementation or poly ADP-ribose polymerase inhibition. For instance, nicotinamide mononucleotide or nicotinamide riboside administration in aged mice or worms respectively, reversed mitochondrial dysfunction by restoring NAD+ levels 50. Moreover, nicotinamide riboside administration or poly ADP-ribose polymerase inhibition in worms extended lifespan by activating the UPRmt response via Sir-2.1 (worm SIRT1 ortholog) and mitonuclear protein imbalance, which in turn induced a mitohormetic response to improve mitochondrial function (Figure 5) 51. Inducing UPRmt genes such as Hsp60 paralogs in Drosophila also prevented mitochondrial and age-dependent muscle dysfunction, thereby promoting longevity 52.
Modulation of NAD+ levels by pharmacological compounds
Besides physiological processes, NAD+ levels can be modulated pharmacologically. Resveratrol—a polyphenolic compound found in red wine has been shown to indirectly stimulate NAD+ production by activating the energy sensor AMP-activated protein kinase (AMPK) 53. Increased NAD+ subsequently stimulates SIRT1 activity, which in turn activates PGC-1α and FOXO family of proteins that govern mitochondrial biogenesis and function (Figure 5) 53. SIRT1 is also amenable to intervention by small molecules such as SIRT1-activating compounds (STACs) that exert beneficial effects on age-related metabolic abnormalities 28. NAD+ levels can be directly raised by supplying NAD+ biosynthetic precursors/intermediates, or by inhibiting NAD+ consuming enzymes with specific inhibitors (Figure 5). For instance, supplementation of nicotinic acid, nicotinamide riboside or nicotinamide mononucleotide compounds increase NAD+ levels in both cultured cells and mouse tissues28. Because nicotinamide riboside can be metabolized both in the nucleus and mitochondria, its supplementation raises the nuclear and mitochondrial NAD+ levels, thereby activating nuclear SIRT1 and mitochondrial SIRT3 respectively 28. Pharmacological activation of NAD+ thus stimulates the activity of multiple sirtuin in a compartment-specific manner to exert its beneficial effects on multiple metabolic pathways which is in contrast to SIRT1 activating compounds’s that specifically stimulate the activity of SIRT1 pathway. Treatment of mice or cultured cells with poly ADP-ribose polymerase and CD38 specific inhibitors has also been shown to induce NAD+ levels that activate sirtuins 45.
Summary
Based on the current evidence, both NAD+ precursors and poly ADP-ribose polymerase inhibitors seem as promising candidates for boosting NAD+ levels in cell culture and animal models. However, there are several key questions that remain unanswered 54.
- First, whether different pharmacological, genetic and physiological manipulations that boosts NAD+ production lead to enhanced activity of all sirtuin enzymes or whether only a few family members are activated, especially considering the fact that some sirtuins may have opposing actions?
- Second, how sirtuins located in different subcellular compartments differ in their enzyme kinetics towards NAD+ availability?
- Third, what may be the optimal dosages, routes of administration, efficacy and bioavailability of compound drugs that raise intracellular NAD+ levels for human application?
Future studies that are directed towards understanding these would be highly relevant in designing therapeutic strategies aimed at selective activation of specific sirtuins, and would also aid in translating the results for human clinical application. It is possible that some of the NAD+ boosting drugs show adverse side effects in humans which could preclude their use and/or may be acceptable for only those inherited conditions that are highly devastating. It is also important to determine if nicotinamide riboside could be valid substitute to avoid undesirable side effects of other NAD+ precursors such as nicotinic acid and nicotinamide, for instance when used as lipid lowering drugs 55.
In addition, future studies are required to examine the UPRmt pathway in vivo in mammalian models to identify key signaling molecules involved in mitochondrial protective mechanisms, which will further advance our understanding of the diseases associated with mitochondrial dysfunction, and will allow discovery of new targets to modulate this pathway. Finally, it remains to be determined whether or not boosting NAD+ levels could extend lifespan in higher organisms. Although much remains to be done, based on the steadily growing evidence, the pharmacological modulation of NAD+ levels via NAD+ precursors and poly ADP-ribose polymerase inhibitors appears to be an attractive and valid strategy to enhance oxidative metabolism and mitochondrial biogenesis, and holds a significant therapeutic potential in the clinical management of mitochondrial and age-related disorders.
- NAD. https://www.ncbi.nlm.nih.gov/mesh/68009243[↩]
- Kanamori KS, de Oliveira GC, Auxiliadora-Martins M, Schoon RA, Reid JM, Chini EN. Two Different Methods of Quantification of Oxidized Nicotinamide Adenine Dinucleotide (NAD+) and Reduced Nicotinamide Adenine Dinucleotide (NADH) Intracellular Levels: Enzymatic Coupled Cycling Assay and Ultra-performance Liquid Chromatography (UPLC)-Mass Spectrometry. Bio-protocol. 2018;8(14):e2937. doi:10.21769/BioProtoc.2937. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6086385/[↩]
- NAD⁺ in aging, metabolism, and neurodegeneration. Verdin E. Science. 2015 Dec 4; 350(6265):1208-13. http://science.sciencemag.org/content/350/6265/1208.long[↩]
- NAD+ and NADH Concentrations in younger and older human adults. FASEB Journal Vol. 20, No. 5, March 2006 https://www.fasebj.org/doi/10.1096/fasebj.20.5.A1357[↩]
- Srivastava S. Emerging therapeutic roles for NAD+ metabolism in mitochondrial and age-related disorders. Clinical and Translational Medicine. 2016;5:25. doi:10.1186/s40169-016-0104-7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4963347[↩]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861[↩]
- Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057[↩]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250[↩]
- Anderson KA, Green MF, Huynh FK, Wagner GR, Hirschey MD. SnapShot: mammalian sirtuins. Cell. 2014;159(956–956):e951[↩]
- Canto C, Auwerx J. NAD+ as a signaling molecule modulating metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:291–298. doi: 10.1101/sqb.2012.76.010439[↩]
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE. 2011;6:e19194. doi: 10.1371/journal.pone.0019194[↩]
- Camacho-Pereira J, Tarragó MG, Chini CCS, et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through a SIRT3-dependent mechanism. Cell metabolism. 2016;23(6):1127-1139. doi:10.1016/j.cmet.2016.05.006. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4911708/[↩][↩]
- Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Yoshino J, Mills KF, Yoon MJ, Imai S. Cell Metab. 2011 Oct 5; 14(4):528-36. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3204926/[↩]
- CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, Chini EN. Cell Metab. 2016 Jun 14; 23(6):1127-1139. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4911708/[↩][↩]
- Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Physiol Rev. 2008 Jul; 88(3):841-86.[↩]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell metabolism. 2011;14(4):528-536. doi:10.1016/j.cmet.2011.08.014. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3204926/[↩][↩]
- Discovery, Synthesis, and Biological Evaluation of Thiazoloquin(az)olin(on)es as Potent CD38 Inhibitors. Haffner CD, Becherer JD, Boros EE, Cadilla R, Carpenter T, Cowan D, Deaton DN, Guo Y, Harrington W, Henke BR, Jeune MR, Kaldor I, Milliken N, Petrov KG, Preugschat F, Schulte C, Shearer BG, Shearer T, Smalley TL Jr, Stewart EL, Stuart JD, Ulrich JC. J Med Chem. 2015 Apr 23; 58(8):3548-71.[↩]
- Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Blacher E, Dadali T, Bespalko A, Haupenthal VJ, Grimm MO, Hartmann T, Lund FE, Stein R, Levy A. Ann Neurol. 2015 Jul; 78(1):88-103. https://www.ncbi.nlm.nih.gov/pubmed/25893674/[↩]
- Canto C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023[↩][↩]
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200[↩]
- Hegyi J, Schwartz RA, Hegyi V. Pellagra: dermatitis, dementia, and diarrhea. Int J Dermatol. 2004;43:1–5. doi: 10.1111/j.1365-4632.2004.01959.x[↩]
- Chini EN. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr Pharm Des. 2009;15:57–63. doi: 10.2174/138161209787185788.[↩]
- Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026[↩][↩]
- Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15. doi: 10.2307/3576299[↩]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022[↩]
- Asher G, Reinke H, Altmeyer M, Gutierrez-Arcelus M, Hottiger MO, Schibler U. Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell. 2010;142:943–953. doi: 10.1016/j.cell.2010.08.016[↩]
- Canto C, Auwerx J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab. 2009;20:325–331. doi: 10.1016/j.tem.2009.03.008[↩]
- Canto C, Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol Rev. 2012;64:166–187. doi: 10.1124/pr.110.003905[↩][↩][↩][↩][↩][↩]
- Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014;19:1042–1049. doi: 10.1016/j.cmet.2014.04.001[↩][↩]
- Srivastava S. Emerging therapeutic roles for NAD+ metabolism in mitochondrial and age-related disorders. Clinical and Translational Medicine. 2016;5:25. doi:10.1186/s40169-016-0104-7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4963347/[↩][↩]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5. New York: W.H. Freeman; 2002.[↩]
- Tischler ME, Friedrichs D, Coll K, Williamson JR. Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch Biochem Biophys. 1977;184:222–236. doi: 10.1016/0003-9861(77)90346-0[↩]
- Pollak N, Dolle C, Ziegler M. The power to reduce: pyridine nucleotides—small molecules with a multitude of functions. Biochem J. 2007;402:205–218. doi: 10.1042/BJ20061638[↩][↩]
- Morava E, van den Heuvel L, Hol F, de Vries MC, Hogeveen M, Rodenburg RJ, Smeitink JA. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67:1823–1826. doi: 10.1212/01.wnl.0000244435.27645.54[↩]
- Mouchiroud L, Houtkooper RH, Auwerx J. NAD(+) metabolism: a therapeutic target for age-related metabolic disease. Crit Rev Biochem Mol Biol. 2013;48:397–408. doi: 10.3109/10409238.2013.789479[↩]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016[↩]
- Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, Schon EA, Lamperti C, Zeviani M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab. 2011;14:80–90. doi: 10.1016/j.cmet.2011.04.011[↩]
- Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsstrom S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med. 2014;6:721–731[↩]
- Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008;7:78–88. doi: 10.1111/j.1474-9726.2007.00355.x[↩]
- Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–375. doi: 10.1016/j.cmet.2007.09.003[↩]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006.[↩]
- Turunc Bayrakdar E, Uyanikgil Y, Kanit L, Koylu E, Yalcin A. Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Abeta(1-42)-induced rat model of Alzheimer’s disease. Free Radic Res. 2014;48:146–158. doi: 10.3109/10715762.2013.857018[↩]
- Scheibye-Knudsen M, Fang EF, Croteau DL, Bohr VA. Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy. 2014;10:1468–1469. doi: 10.4161/auto.29321[↩]
- Bai P, Canto C, Brunyanszki A, Huber A, Szanto M, Cen Y, Yamamoto H, Houten SM, Kiss B, Oudart H, et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011;13:450–460. doi: 10.1016/j.cmet.2011.03.013[↩]
- Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004[↩][↩]
- Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T, Felding-Habermann B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013;123:1068–1081. doi: 10.1172/JCI64264[↩]
- Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome—a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–752. doi: 10.1038/nrc3340[↩]
- Bell EL, Emerling BM, Ricoult SJ, Guarente L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011;30:2986–2996. doi: 10.1038/onc.2011.37[↩]
- Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, Li YH, Wang X, Laurent G, German NJ, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell. 2013;23:450–463. doi: 10.1016/j.ccr.2013.02.024[↩]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037[↩]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188[↩]
- Owusu-Ansah E, Song W, Perrimon N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 2013;155:699–712. doi: 10.1016/j.cell.2013.09.021[↩]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813[↩][↩]
- Srivastava S. Emerging therapeutic roles for NAD+ metabolism in mitochondrial and age-related disorders. Clinical and Translational Medicine. 2016;5:25. doi:10.1186/s40169-016-0104-7 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4963347/[↩]
- Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443[↩]

{kind=link}