Contents
What is neuroblastoma
Neuroblastoma is a form of cancer that is made up of cells that are found in nerve tissues of the body. These cells are called neuroblasts and they are early nerve cells of the sympathetic nervous system, so they can be found anywhere along this system.
A little more than 1 out of 3 neuroblastomas start in the adrenal glands. About 1 out of 4 begin in sympathetic nerve ganglia in the abdomen. Most of the rest start in sympathetic ganglia near the spine in the chest or neck, or in the pelvis.
Rarely, a neuroblastoma has spread so widely by the time it is found that doctors can’t tell exactly where it started.
- Neuroblastoma is by far the most common cancer in infants (less than 1 year old). It accounts for about 6% of all cancers in children. There are about 700 new cases of neuroblastoma each year in the United States. This number has remained about the same for many years.
- The average age of children when they are diagnosed is about 1 to 2 years. In rare cases, neuroblastoma is detected by ultrasound even before birth.
- Neuroblastoma occurs most commonly in infants and children under 5 years of age with nearly 90% of cases are diagnosed by age 5.
- Neuroblastoma is very rare in in children over 10 years of age.
- Very rarely, neuroblastoma occurs in adults.
- In children aged 6 months or younger, the disease sometimes goes away without treatment.
- Neuroblastoma is usually found when the tumor begins to grow and cause signs or symptoms. By the time it is diagnosed, the cancer has usually metastasized (spread to other parts of the body).
- In about 2 of 3 cases, the neuroblastoma has already spread to the lymph nodes or to other parts of the body when it is diagnosed.
There is a wide range in how neuroblastomas behave. Some grow and spread quickly, while others grow slowly. Sometimes, in very young children, the cancer cells die for no reason and the tumor goes away on its own. In other cases, the cells sometimes mature on their own into normal ganglion cells and stop dividing. This makes the tumor a ganglioneuroma. Ganglioneuroma is a benign (non-cancerous) tumor made up of mature ganglion and nerve sheath cells.
Ganglioneuroblastoma is a tumor that has both malignant and benign parts. It contains neuroblasts (immature nerve cells) that can grow and spread abnormally, similar to neuroblastoma, as well as areas of more mature tissue that are similar to ganglioneuroma.
Ganglioneuromas are usually removed by surgery and looked at carefully under a microscope to be sure they don’t have areas of malignant cells (which would make the tumor a ganglioneuroblastoma). If the final diagnosis is ganglioneuroma, no other treatment is needed. If it’s found to be a ganglioneuroblastoma, it’s treated the same as a neuroblastoma.
The sympathetic nervous system
To understand neuroblastoma, it helps to know about the sympathetic nervous system, which is where these tumors start.
About the sympathetic nervous system
The nervous system consists of the brain, spinal cord, and the nerves that reach out from them to all areas of the body. The nervous system is essential for thinking, sensation, and movement, among other things.
Part of the nervous system also controls body functions we are rarely aware of, such as heart rate, breathing, blood pressure, digestion, and other functions. This part of the nervous system is known as the autonomic nervous system.
The sympathetic nervous system is part of the autonomic nervous system. It includes:
- Nerve fibers that run along either side the spinal cord.
- Clusters of nerve cells called ganglia (plural of ganglion) at certain points along the path of the nerve fibers.
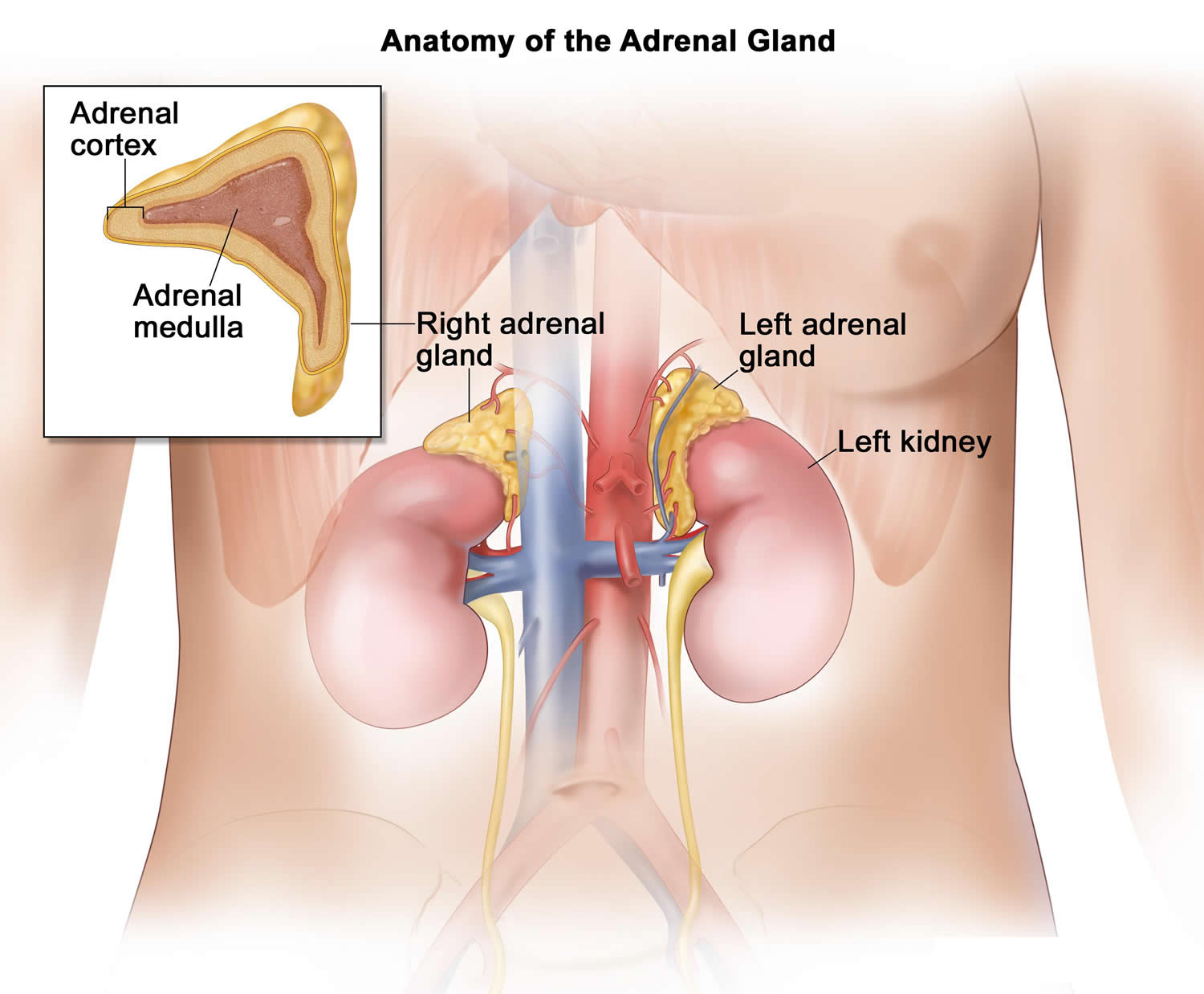
- Nerve-like cells found in the medulla (center) of the adrenal glands. The adrenals are small glands that sit on top of each kidney. These glands make hormones (such as adrenaline [epinephrine]) that help control heart rate, blood pressure, blood sugar, and how the body reacts to stress.
The main cells that make up the nervous system are called nerve cells or neurons. These cells interact with other types of cells in the body by releasing tiny amounts of chemicals (hormones). This is important, because neuroblastoma cells often release certain hormones that can cause symptoms
Figure 1. Sympathetic nervous system
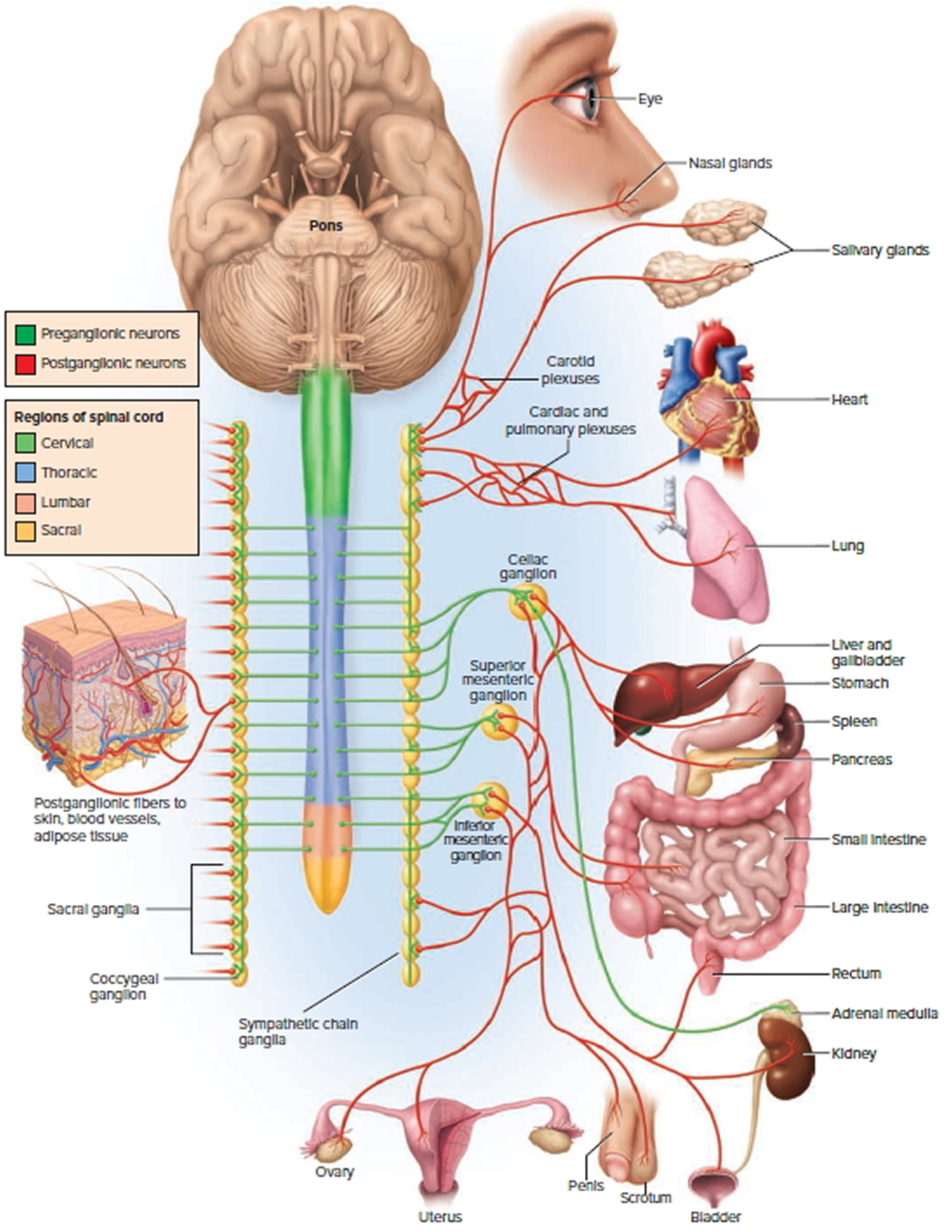
Figure 2. Sympathetic nervous system preganglionic and postganglionic neurons
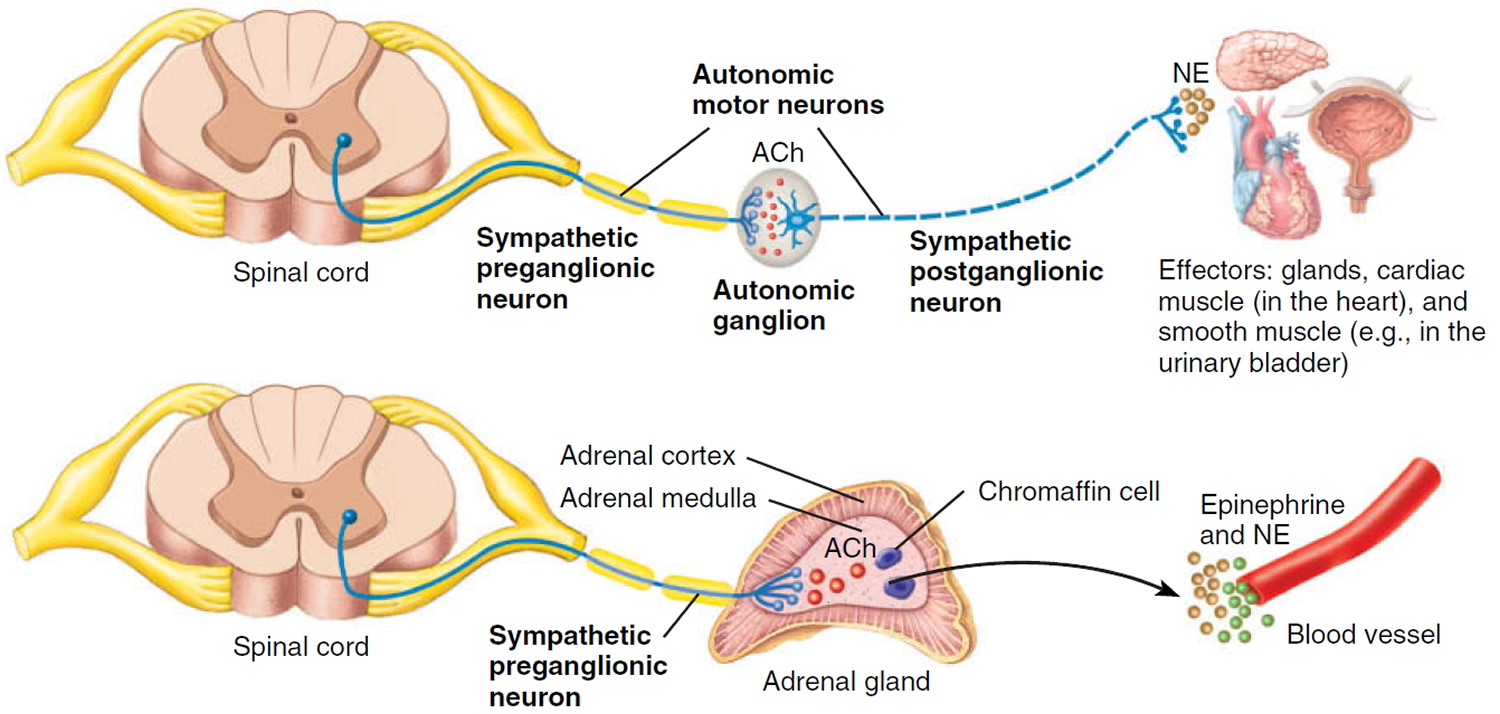
Figure 3. Adrenal gland location and anatomy
Neuroblastoma causes
The causes of most neuroblastomas are not known. But researchers have found important differences between neuroblastoma cells and the normal neuroblasts (early forms of nerve cells) from which they develop. They have also found differences between neuroblastomas that are likely to respond to treatment and those that have a poor prognosis (outlook). These differences (known as prognostic markers) are sometimes helpful in choosing the best treatment.
Both nerve cells and cells of the medulla (center) of the adrenal gland develop from neuroblasts in the fetus. Neuroblastomas develop when normal fetal neuroblasts fail to become mature nerve cells or adrenal medulla cells. Instead, they continue to grow and divide.
Neuroblasts may not have matured completely in babies by the time they are born. In fact, studies have shown that there are small clusters of neuroblasts in the adrenal glands of some infants less than 3 months old. Most of these eventually mature into nerve cells or simply die off and do not form neuroblastomas. Sometimes, neuroblasts remaining in very young infants continue to grow and then form tumors. Some can even spread to other parts of the body. But many of these tumors will still eventually mature into nerve tissue or go away on their own.
However, as children get older, it becomes less likely that these cells will mature and more likely that they will grow into a cancer. By the time neuroblastomas are large enough to be felt or cause symptoms, most can no longer mature on their own and will grow and spread unless treated.
The failure of some neuroblasts to mature and to stop growing is due to abnormal DNA inside the cells. DNA is the chemical in each of your cells that makes up your genes – the instructions for how your cells function. The DNA inside your cells is in long string-like structures called chromosomes.
Some genes contain instructions for controlling when your cells grow, divide into new cells, and die. Certain genes that help cells grow, divide, or stay alive are called oncogenes. Others that slow down cell division or cause cells to die at the right time are called tumor suppressor genes. Cancers can be caused by DNA changes that turn on oncogenes or turn off tumor suppressor genes. These gene changes can be inherited from a parent (as is sometimes the case with childhood cancers), or they may happen during a person’s lifetime as cells in the body divide to make new cells.
In most cases, neuroblastoma cells have chromosome changes (such as having too many or too few chromosomes or missing part of a chromosome) that are likely to affect certain genes. Scientists are still trying to determine which genes are affected by these chromosome changes, as well as how these changes affect the growth of neuroblastoma cells.
In rare cases, neuroblastoma seems to occur because of gene changes inherited from a parent. Inherited changes in the ALK oncogene seem to account for most cases of hereditary neuroblastoma. A small number of inherited neuroblastomas are caused by changes in PHOX2B, a gene that normally helps nerve cells mature.
Still, most neuroblastomas are not caused by inherited DNA changes. They are the result of gene changes that happen early in the child’s development, often before birth. These changes are found only in the child’s cancer cells, so they will not be passed on to his or her children. For example, about 10% to 15% of sporadic (non-inherited) neuroblastomas have changes in the ALK gene. But in many neuroblastomas the exact genes affected are not known.
Some gene changes seem to affect how quickly a neuroblastoma is likely to grow. For example, neuroblastoma cells sometimes have extra copies of an oncogene called MYCN, which is often a sign that the tumor will grow quickly and be harder to treat. On the other hand, the NTRK1 gene (which makes the TrkA protein) is often overactive in the cells of neuroblastomas that have a better outlook. Researchers recently found that neuroblastoma cells in older children are more likely to have changes in the ATRX tumor suppressor gene. Tumors with this gene change tend to grow more slowly, but they are also harder to cure. This may help explain why younger children with neuroblastoma tend to do better long term than children who are older when they are diagnosed.
Researchers have found some of the gene changes that may lead to neuroblastoma, but it’s still not clear what causes these changes. Some gene changes may be inherited. Some might have unknown outside causes, but others may just be random events that sometimes happen inside a cell, without having an outside cause. There are no known lifestyle-related or environmental causes of neuroblastomas at this time, so it’s important to remember that there is nothing these children or their parents could have done to prevent these cancers.
Risk Factors for Neuroblastoma
A risk factor is anything that affects your chance of getting a disease such as cancer. Different cancers have different risk factors.
Lifestyle-related risk factors such as body weight, physical activity, diet, and tobacco use play a major role in many adult cancers. But these factors usually take many years to influence cancer risk, and they are not thought to play much of a role in childhood cancers, including neuroblastomas.
No environmental factors (such as exposures during the mother’s pregnancy or in early childhood) are known to increase the chance of getting neuroblastoma.
Age
Neuroblastoma is most common in very young children, but it is still rare even in this age group. It is very rare in people over the age of 10 years.
Heredity
In about 1% to 2% of all neuroblastomas, children inherit an increased risk of developing neuroblastoma from a parent. But most neuroblastomas do not seem to be inherited.
Children with the familial form of neuroblastoma (those with an inherited tendency to develop this cancer) usually come from families with one or more members who had neuroblastoma as infants. The average age at diagnosis of familial cases is younger than the age for sporadic (not inherited) cases.
Children with familial neuroblastoma sometimes develop 2 or more of these cancers in different organs (for example, in both adrenal glands or in more than one sympathetic ganglion). It’s important to distinguish neuroblastomas that start in more than one organ from neuroblastomas that have started in one organ and then spread to others (metastatic neuroblastomas). When tumors develop in several places at once it suggests a familial form. This might mean that family members should consider genetic counseling and testing. Both familial and sporadic neuroblastoma can spread to other organs.
Neuroblastoma prevention
The risk of many adult cancers can be reduced with certain lifestyle changes (such as staying at a healthy weight or quitting smoking), but at this time there are no known ways to prevent most cancers in children.
The only known risk factors for neuroblastoma (age and heredity) cannot be changed. There are no known lifestyle-related or environmental causes of neuroblastomas at this time.
Some studies suggest that having mothers take prenatal multi-vitamins or folic acid might lower the risk of neuroblastoma, but further research is needed to confirm this.
If there is a history of neuroblastoma in your family, you may want to talk with a genetic counselor about your children’s risks of developing the disease. It is important to remember, though, that familial neuroblastoma is very rare.
Can Neuroblastoma Be Found Early?
Researchers have studied whether screening infants for neuroblastoma might find these tumors earlier and lead to better treatment results. Screening is testing for a disease, such as cancer, in people who don’t have any symptoms. One way to screen for neuroblastoma is to test children’s urine for certain substances made by neuroblastoma tumors.
Studies have not found neuroblastoma screening to be helpful. Testing infants when they were 6 months old did find many tumors that wouldn’t have normally been diagnosed. But most of these tumors were of a type that probably would have gone away or matured into benign (non-cancerous) tumors on their own. These tumors probably would never have caused any problems. The screening didn’t lower the number of cancers found at advanced stages or save lives.
What’s more, finding tumors that would never cause serious problems may needlessly frighten parents and can lead to unnecessary tests and surgery in children whose tumors would have gone away or matured on their own if left alone.
For these reasons, most experts do not recommend screening for neuroblastoma in infants who are not at increased risk of the disease.
In rare instances, neuroblastoma is found before birth during an ultrasound, a test that uses sound waves to create an image of the internal organs of a fetus. Ultrasounds are usually done to estimate the age of a fetus, predict the date of birth, and look for certain common birth defects. Improvements in ultrasound technology or other tests may lead to more accurate prenatal (before birth) testing for this disease.
Neuroblastoma is sometimes found incidentally in young children without any symptoms during tests done to find other childhood diseases. These children will usually have a good outcome, and some may not even need treatment.
But most often, neuroblastoma is first detected because of signs or symptoms the child is having.
Neuroblastoma signs and symptoms
The signs and symptoms of neuroblastoma vary widely, depending on the size of the tumor, where it is, how far it has spread, and if the tumor cells secrete hormones.
Many of the signs and symptoms below are more likely to be caused by something other than neuroblastoma. Still, if your child has any of these symptoms, check with your doctor so the cause can be found and treated, if needed.
Signs or symptoms caused by the main neuroblastoma tumor
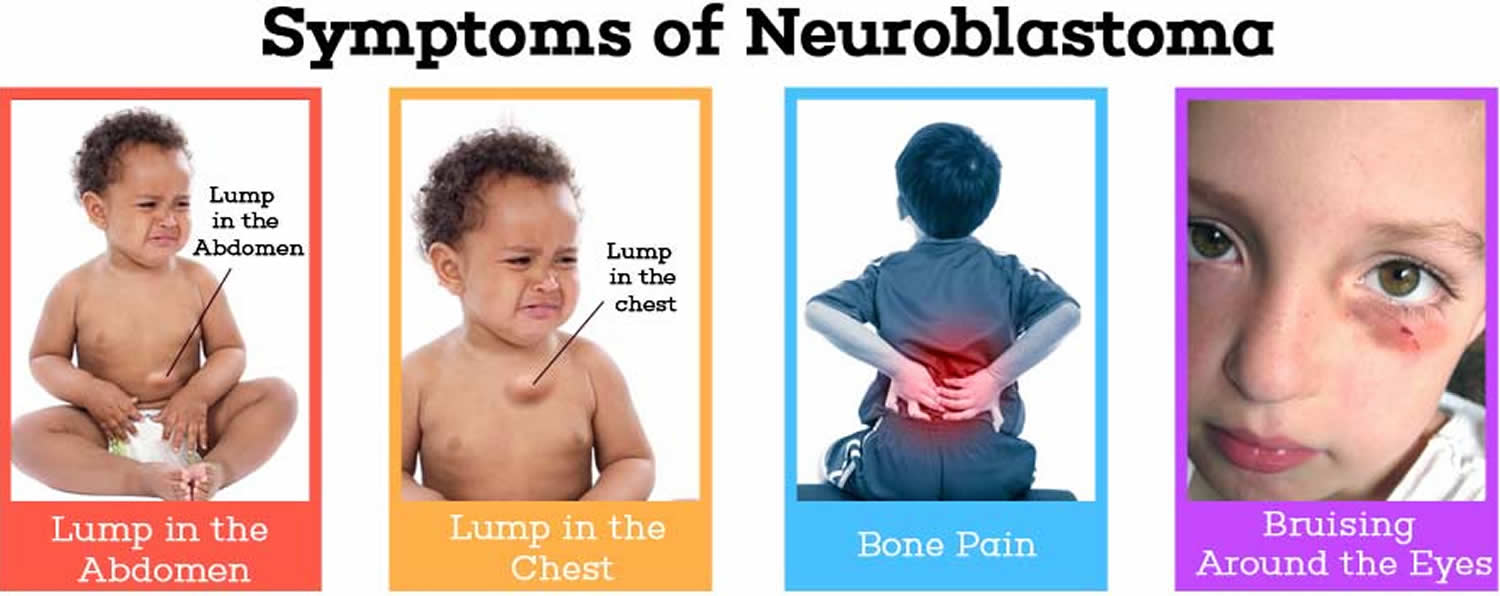
- Tumors in the abdomen (belly) or pelvis: One of the most common signs of a neuroblastoma is a large lump or swelling in the child’s abdomen. The child might not want to eat (which can lead to weight loss). If the child is old enough, he or she may complain of feeling full or having belly pain. But the lump itself is usually not painful to the touch. Sometimes, a tumor in the abdomen or pelvis can affect other parts of the body. For example, tumors that press against or grow into the blood and lymph vessels in the abdomen or pelvis can stop fluids from getting back to the heart. This can sometimes lead to swelling in the legs and, in boys, the scrotum. In some cases the pressure from a growing tumor can affect the child’s bladder or bowel, which can cause problems urinating or having bowel movements.
- Tumors in the chest or neck: Tumors in the neck can often be seen or felt as a hard, painless lump. If the tumor is in the chest, it might press on the superior vena cava (the large vein in the chest that returns blood from the head and neck to the heart). This can cause swelling in the face, neck, arms, and upper chest (sometimes with a bluish-red skin color). It can also cause headaches, dizziness, and a change in consciousness if it affects the brain. The tumor might also press on the throat or windpipe, which can cause coughing and trouble breathing or swallowing. Neuroblastomas that press on certain nerves in the chest or neck can sometimes cause other symptoms, such as a drooping eyelid and a small pupil (the black area in the center of the eye). Pressure on other nerves near the spine might affect the child’s ability to feel or move their arms or legs.
Signs or symptoms caused by cancer spread to other parts of the body
About 2 out of 3 neuroblastomas have already spread to the lymph nodes or other parts of the body by the time they are found.
Lymph nodes are bean-sized collections of immune cells found throughout the body. Cancer that has spread to the lymph nodes can cause them to swell. These nodes can sometimes be felt as lumps under the skin, especially in the neck, above the collarbone, under the arm, or in the groin. Enlarged lymph nodes in children are much more likely to be a sign of infection than cancer, but they should be checked by a doctor.
Neuroblastoma often spreads to bones. A child who can talk may complain of bone pain. The pain may be so bad that the child limps or refuses to walk. If it spreads to the bones in the spine, tumors can press on the spinal cord and cause weakness, numbness, or paralysis in the arms or legs. Spread to the bones around the eyes is common and can lead to bruising around the eyes or cause an eyeball to stick out slightly. The cancer can also spread to other bones in the skull, causing bumps under the scalp.
If the cancer spreads to the bone marrow (the inner part of certain bones that makes blood cells), the child may not have enough red blood cells, white blood cells, or blood platelets. These shortages of blood cells can result in tiredness, irritability, weakness, frequent infections, and excess bruising or bleeding from small cuts or scrapes.
Rarely, large tumors can start to break down, leading to a loss of clotting factors in the blood. This can result in a high risk of serious bleeding, which is known as a consumption coagulopathy and can be life threatening.
A special widespread form of neuroblastoma (known as stage 4S) occurs only during the first few months of life. In this special form, the neuroblastoma has spread to the liver, to the skin, and/or to the bone marrow (in small amounts). Blue or purple bumps that look like small blueberries may be a sign of spread to the skin. The liver can become very large and can be felt as a mass on the right side of the belly. Sometimes it can grow large enough to push up on the lungs, which can make it hard for the child to breathe. Despite the fact that the cancer is already widespread when it is found, stage 4S neuroblastoma is very treatable, and often shrinks or goes away on its own. Almost all children with this form of neuroblastoma can be cured.
Signs or symptoms caused by hormones from the tumor
Neuroblastomas sometimes release hormones that can cause problems with tissues and organs in other parts of the body, even though the cancer has not spread to those tissues or organs. These problems are called paraneoplastic syndromes.
Symptoms of paraneoplastic syndromes can include:
- Constant diarrhea
- Fever
- High blood pressure (causing irritability)
- Rapid heartbeat
- Reddening (flushing) of the skin
- Sweating
An uncommon set of symptoms is called the opsoclonus-myoclonus-ataxia syndrome or “dancing eyes, dancing feet.” The child has irregular, rapid eye movements (opsoclonus), twitch-like muscle spasms (myoclonus), and appears uncoordinated when standing or walking (ataxia). He or she may also have trouble speaking. For reasons that are not clear, neuroblastomas that cause this syndrome tend to be less life-threatening than other forms of the disease.
Neuroblastoma diagnosis
Neuroblastomas are usually found when a child is brought to the doctor because of signs or symptoms he or she is having. If a tumor is suspected, tests will be needed to confirm the diagnosis.
Medical history and physical exam
If your child has signs or symptoms that might be caused by a neuroblastoma (or another tumor), the doctor will want to get a complete medical history to learn more about the symptoms. The doctor might also ask if there’s a family history of any type of cancer.
The doctor will examine your child for possible signs of a neuroblastoma and other health problems. For example, the doctor may be able to see or feel an abnormal mass or swelling in the body or may find a child has lumps or bumps under the skin or high blood pressure. Neuroblastomas can sometimes grow close to the spinal cord, which can affect movement and strength in the child’s arms and legs, so the doctor will pay close attention to these.
Some signs that could be caused by neuroblastoma, such as fever and enlarged lymph nodes, are much more likely to be caused by an infection, so the doctor might look for other signs of infection at first.
If the history and exam suggest a child might have a neuroblastoma (or another type of tumor), other tests will be done. These could include blood and urine tests, imaging tests, and biopsies. These tests are important because many of the symptoms and signs of neuroblastoma can also be caused by other diseases, such as infections, or even other types of cancer.
Blood and urine catecholamine tests
Sympathetic nerve cells normally release hormones called catecholamines, such as epinephrine (adrenaline) and norepinephrine (noradrenaline), which enter the blood. Eventually the body breaks these down into metabolites (smaller pieces), which then pass out of the body in the urine.
Neuroblastoma cells can also make these hormones. In most cases, neuroblastoma cells make enough catecholamines to be detected by blood or urine tests. The 2 catecholamine metabolites most often measured are:
- Homovanillic acid (HVA)
- Vanillylmandelic acid (VMA)
Other lab tests
If neuroblastoma is suspected or has been found, your child’s doctor will probably order blood tests to check blood cell counts, liver and kidney function, and the balance of salts (electrolytes) in the body. A urinalysis (urine test) may also be done to further check kidney function.
Imaging tests
Imaging tests use x-rays, magnetic fields, sound waves, or radioactive substances to create pictures of the inside of the body. Imaging tests can be done for a number of reasons, including:
- To help find out if a suspicious area might be cancerous
- To learn how far cancer has spread
- To help determine if treatment has been effective
Most children who have or might have neuroblastoma will have one or more of these tests.
Children with neuroblastoma are often very young, so it can be hard to do some of these tests.
Ultrasound
Ultrasound is often one of the first tests done in small children if a tumor is suspected, because it is fairly quick and easy, it does not use radiation, and it can often give the doctor a good view inside the body, especially in the abdomen (belly).
This test uses sound waves to create pictures of organs or masses inside the body. For this test, your child lies on a table (or sits on your lap) while a small wand called a transducer is placed on the skin over the belly (which is first lubricated with gel). The wand gives off sound waves and picks up the echoes as they bounce off organs. The echoes are converted by a computer into a black and white image on a screen. The test is not usually painful, but it might cause some discomfort if the transducer is pressed down hard on the belly.
Ultrasound is used most often to look for tumors in the abdomen. (It’s not used to look in the chest because the ribs block the sound waves.) Ultrasound can detect if kidneys have become swollen because the outflow of urine has been blocked by enlarged lymph nodes or a mass. It can also be used to help guide a biopsy needle into a suspected tumor to get a sample for testing. It is particularly useful in checking to see if tumors in the abdomen are shrinking.
The pictures from ultrasound aren’t as detailed as those from some other tests, so even if a tumor is found, CT or MRI scans (described below) might still be needed.
X-rays
The doctor may order an x-ray of the chest or another part of the body as an early test if a child is having symptoms but it’s not clear what might be causing them. But the images might not always be detailed enough to spot tumors.
If neuroblastoma has already been diagnosed, x-rays can be useful to see if cancer has spread to certain bones. An x-ray of the head may be done to see if cancer has spread to the skull bones. An MIBG scan or a bone scan (described below) is usually better for looking at the bones in the rest of the body, but x-rays may be used in infants, where these scans might not be possible.
A standard chest x-ray may be done if doctors suspect that the tumor has invaded the lungs, but a CT or MRI scan of the chest can show the area in more detail.
Computed tomography (CT or CAT) scan
CT scans are often used to look for neuroblastoma in the abdomen, pelvis, and chest.
The CT scan is an x-ray test that produces detailed cross-sectional images of parts of the body. Instead of taking one picture, like a regular x-ray, a CT scanner takes many pictures as it rotates around your child while he or she lies on a table. A computer then combines these pictures into images showing slices of the part of the body being studied. Unlike a regular x-ray, a CT scan creates detailed images of the soft tissues in the body.
Before the test, your child may be asked to drink a contrast solution and/or get an IV (intravenous) injection of a contrast dye. This helps better outline structures in the body. The contrast may cause some flushing (a feeling of warmth, especially in the face). Some people are allergic and get hives. Rarely, more serious reactions like trouble breathing or low blood pressure can occur. Be sure to tell the doctor if your child has any allergies or has ever had a reaction to any contrast material used for x-rays.
CT scans take longer than regular x-rays. A CT scanner has been described as a large donut, with a narrow table in the middle opening. Your child will need to lie still on the table while the scans are being done. During the test, the table slides in and out of the scanner. Younger children may be sedated (given medicine to make them sleepy) before the test to reduce movement and help make sure the pictures come out well.
CT-guided needle biopsy: CT scans can also be used to help guide a biopsy needle into a tumor. For this procedure, the child lies on the CT scanning table while a radiologist advances a biopsy needle through the skin and toward the mass. CT scans are repeated until the needle is within the mass. A biopsy sample is then removed and looked at under a microscope. In children, this procedure is always done under general anesthesia (where the child is asleep).
Magnetic resonance imaging (MRI) scan
MRI scans provide detailed images of soft tissues in the body. These scans are very helpful in looking at the brain and spinal cord. They may be slightly better than CT scans for seeing the extent of a neuroblastoma tumor, especially around the spine, but this test can be harder to do in small children.
MRI scans use radio waves and strong magnets to create the images instead of x-rays, so there is no radiation. A contrast material called gadolinium may be injected into a vein before the scan to better see details, but this is needed less often than with a CT scan. It usually does not cause allergic reactions, but it can cause other problems in children with kidney disease, so doctors are careful when they use it.
MRI scans take longer than CT scans, often up to an hour. For most MRI machines, your child has to lie inside a narrow tube, which is confining and can be distressing. Newer, more open MRI machines may be an option in some cases, but they still require the child to stay still for long periods of time. The MRI machine also makes loud buzzing and clicking noises that may be disturbing. Younger children are often given medicine to help keep them calm or even asleep during the test.
MIBG scan
This scan uses a form of the chemical meta-iodobenzylguanidine (MIBG) that contains a small amount of radioactive iodine. MIBG is similar to norepinephrine (noradrenaline), a hormone made by sympathetic nerve cells. It is injected into a vein and travels through the blood, and in most patients it will attach to neuroblastoma cells anywhere in the body. Several hours or days later, the body is scanned with a special camera to look for areas that picked up the radioactivity. This helps doctors tell where the neuroblastoma is and whether it has spread to the bones and/or other parts of the body.
This test is preferred by many doctors as a standard test in children with neuroblastoma. It can be repeated after treatment to see if it has been effective. It is also good to know if the tumor takes up the MIBG because in some cases, this radioactive molecule can be used at higher doses to treat the neuroblastoma.
Positron emission tomography (PET) scan
For a PET scan, a radioactive substance (usually a type of sugar related to glucose, known as FDG) is injected into the blood. The amount of radioactivity used is very low and will pass out of the body within a day or so. Because cancer cells in the body are growing quickly, they absorb large amounts of the radioactive sugar. After about an hour, your child will be moved onto a table in the PET scanner. He or she will lie on the table for about 30 minutes while a special camera creates a picture of areas of radioactivity in the body. Younger children may be given medicine to help keep them calm or even asleep during the test. The picture from a PET scan is not as detailed as a CT or MRI scan, but it can provide helpful information about the whole body.
Some newer machines can do a PET and CT scan at the same time (PET/CT scan). This lets the doctor compare areas of higher radioactivity on the PET scan with the more detailed appearance of that area on the CT scan.
Bone scan
A bone scan can help show if a cancer has spread to the bones, and can provide a picture of the entire skeleton at once. Neuroblastoma often causes bone damage, which a bone scan can find. This test used to be done routinely, but in some centers it has been replaced by use of MIBG or PET scans.
For this test, a small amount of low-level radioactive material (technetium-99) is injected into a vein. The amount of radioactivity used is very low and will pass out of the body within a day or so. The substance settles in areas of damaged bone throughout the skeleton over the course of a couple of hours. Your child then lies on a table for about 30 minutes while a special camera detects the radioactivity and creates a picture of the skeleton. Younger children may be given medicine to help keep them calm or even asleep during the test.
Areas of active bone changes attract the radioactivity and appear as “hot spots” on the skeleton. These areas may suggest cancer, but other bone diseases can also cause the same pattern. To help tell these apart, other imaging tests such as plain x-rays or MRI scans, or even a bone biopsy might be needed.
Biopsies
Exams and tests might strongly suggest a child has neuroblastoma, but a biopsy (removing some of the tumor for viewing under a microscope and other lab testing) is often done to be sure.
During a biopsy, the doctor removes a sample of the tumor mass. In adults, biopsies are sometimes done using local anesthetic (numbing medicine), but in children they are more often done while the child is under general anesthesia (asleep). There are 2 main types of biopsies:
- Incisional (open or surgical) biopsy: This type of biopsy is done by removing a piece of the tumor through an incision (cut) in the skin. For tumors deep in the body this may be done laparoscopically using long, thin surgical tools inserted through small cuts in the skin.
- Needle (closed) biopsy: For this type of biopsy, a thin, hollow needle is placed through the skin and into the tumor to remove a small sample. If the tumor is deep within the body, CT scans or ultrasound can be used to help guide the needle into the tumor.
The biopsy samples are sent to a lab, where they are viewed under a microscope by a pathologist (a doctor with special training in identifying cancer cells). Some neuroblastomas are easily recognized when looked at by experienced doctors. But some may be hard to tell apart from other types of children’s cancers. In these cases, special lab tests must be done to show the tumor is a neuroblastoma.
Other lab tests may also be done on neuroblastoma samples to help determine how quickly the tumor is likely to grow.
Bone marrow aspiration and biopsy
Neuroblastoma often spreads to the bone marrow (the soft inner parts of certain bones). If blood or urine levels of catecholamines are increased, then finding cancer cells in a bone marrow sample is enough to diagnose neuroblastoma (without getting a biopsy of the main tumor). If neuroblastoma has already been diagnosed by a biopsy done elsewhere in the body, bone marrow tests are done to help determine the extent of the disease.
A bone marrow aspiration and biopsy are usually done at the same time. In most cases the samples are taken from the back of both of the pelvic (hip) bones.
Even when the area is numbed with local anesthetic, these tests can be painful, so in most cases the child is also given other medicines to reduce pain or even be asleep during the procedure.
For a bone marrow aspiration, a thin, hollow needle is inserted into the bone and a syringe is used to suck out a small amount of liquid bone marrow.
A bone marrow biopsy is usually done just after the aspiration. A small piece of bone and marrow is removed with a slightly larger needle that is pushed down into the bone. Once the biopsy is done, pressure is applied to the site to help stop any bleeding.
Samples from the bone marrow are sent to a lab, where they are looked at and tested for the presence of cancer cells.
Neuroblastoma Staging
The stage of a cancer describes how far it has spread. Your child’s treatment and prognosis (outlook) depend, to a large extent, on the cancer’s stage.
The stage of the neuroblastoma is based on results of physical exams, imaging tests, and biopsies of the main tumor and other tissues. The results of surgery are sometimes used in staging as well.
For neuroblastoma, several other factors also affect prognosis, including a child’s age and certain tests of blood and tumor specimens. These prognostic factors are not used to determine the stage of the cancer, but they are used along with the stage to determine which risk group a child falls into, which in turn affects treatment options.
The stages and risk groups for neuroblastoma are complex and can be confusing. If you are unsure about what these mean for your child, ask your child’s doctor to explain them to you in a way you can understand.
International Neuroblastoma Staging System
A staging system is a standard way for the cancer care team to sum up the extent of the cancer. Since the mid-1990s, most cancer centers have used the International Neuroblastoma Staging System (INSS) to stage neuroblastoma. This system takes into account the results of surgery to remove the tumor. In simplified form, the stages are:
- Stage 1: The cancer is still in the area where it started. It is on one side of the body (right or left). All visible tumor has been removed completely by surgery (although looking at the tumor’s edges under the microscope after surgery may show some cancer cells). Lymph nodes outside the tumor are free of cancer (although nodes enclosed within the tumor may contain neuroblastoma cells).
- Stage 2A: The cancer is still in the area where it started and on one side of the body, but not all of the visible tumor could be removed by surgery. Lymph nodes outside the tumor are free of cancer (although nodes enclosed within the tumor may contain neuroblastoma cells).
- Stage 2B: The cancer is on one side of the body, and may or may not have been removed completely by surgery. Nearby lymph nodes outside the tumor contain neuroblastoma cells, but the cancer has not spread to lymph nodes on the other side of the body or elsewhere.
- Stage 3: The cancer has not spread to distant parts of the body, but one of the following is true:
- The cancer cannot be removed completely by surgery and it has crossed the midline (defined as the spine) to the other side of the body. It may or may not have spread to nearby lymph nodes.
- The cancer is still in the area where it started and is on one side of the body. It has spread to lymph nodes that are relatively nearby but on the other side of the body.
- The cancer is in the middle of the body and is growing toward both sides (either directly or by spreading to nearby lymph nodes) and cannot be removed completely by surgery.
- Stage 4: The cancer has spread to distant sites such as distant lymph nodes, bone, liver, skin, bone marrow, or other organs (but the child does not meet the criteria for stage 4S).
- Stage 4S (also called “special” neuroblastoma): The child is younger than 1 year old. The cancer is on one side of the body. It might have spread to lymph nodes on the same side of the body but not to nodes on the other side. The neuroblastoma has spread to the liver, skin, and/or the bone marrow. However, no more than 10% of marrow cells are cancerous, and imaging tests such as an MIBG scan do not show that the cancer has spread to the bones or the bone marrow.
- Recurrent: While not formally part of the staging system, this term is used to describe cancer that has come back (recurred) after it has been treated. The cancer might come back in the area where it first started or in another part of the body.
International Neuroblastoma Risk Group Staging System
A risk-group staging system now coming into use is known as the International Neuroblastoma Risk Group Staging System (INRGSS). It is similar to the INSS, but it does not use the results of surgery to help define the stage. This lets doctors determine a stage before surgery, based on the results of imaging tests (usually a CT or MRI scan, and an MIBG scan), as well as exams and biopsies. The stage can then be used to help predict how resectable the tumor is – that is, how much of it can be removed with surgery.
The International Neuroblastoma Risk Group Staging System uses image-defined risk factors (IDRFs), which are factors seen on imaging tests that might mean the tumor will be harder to remove. This includes things like the tumor growing into a nearby vital organ or growing around important blood vessels.
The International Neuroblastoma Risk Group Staging System divides neuroblastomas into 4 stages:
- L1: A tumor that has not spread from where it started and has not grown into vital structures as defined by the list of image-defined risk factors. It is confined to one body compartment, such as the neck, chest, or abdomen.
- L2: A tumor that has not spread far from where it started (for example, it may have grown from the left side of the abdomen into the left side of the chest), but that has at least one image-defined risk factor.
- M: A tumor that has spread (metastasized) to a distant part of the body (except tumors that are stage MS).
- MS: Metastatic disease in children younger than 18 months with cancer spread only to skin, liver, and/or bone marrow. No more than 10% of marrow cells are cancerous, and an MIBG scan does not show spread to the bones and/or the bone marrow.
Prognostic markers
Prognostic markers are features that help predict whether the child’s outlook for cure is better or worse than would be predicted by the stage alone. The following markers are used to help determine a child’s prognosis.
Age
Younger children (under 12-18 months) are more likely to be cured than older children.
Tumor histology
Tumor histology is based on how the neuroblastoma cells look under the microscope. Tumors that contain more normal-looking cells and tissues tend to have a better prognosis and are said to have a favorable histology. Tumors whose cells and tissues look more abnormal under a microscope tend to have a poorer prognosis and are said to have an unfavorable histology.
DNA ploidy
The amount of DNA in each cell, known as ploidy or the DNA index, can be measured using special lab tests, such as flow cytometry or imaging cytometry. Neuroblastoma cells with about the same amount of DNA as normal cells (a DNA index of 1) are classified as diploid. Cells with increased amounts of DNA (a DNA index higher than 1) are termed hyperdiploid.
In infants, hyperdiploid cells tend to be associated with earlier stages of disease, respond better to chemotherapy, and usually predict a more favorable prognosis (outcome) than diploid cells. Ploidy is not as useful a factor in older children.
MYCN gene amplifications
MYCN is an oncogene, a gene that helps regulate cell growth. Changes in oncogenes can make cells grow and divide too quickly, as with cancer cells.
Neuroblastomas with too many copies (amplification) of the MYCN oncogene tend to grow quickly and are less likely to mature. Children whose neuroblastomas have this feature tend to have a worse prognosis than other children with neuroblastoma.
Other markers
These markers are not used to help determine risk groups at this time, but they are still important and may influence the treatment of a child with neuroblastoma.
- Chromosome changes: Tumor cells that are missing certain parts of chromosomes 1 or 11 (known as 1p deletions or 11q deletions) may predict a less favorable prognosis. It is thought that these chromosome parts, which are missing in many neuroblastomas, may contain important tumor suppressor genes, but more studies are needed to verify this. Having an extra part of chromosome 17 (17q gain) is also linked with a worse prognosis. This probably means that there is an oncogene in this part of chromosome 17. Understanding the importance of chromosome deletions/gains is an active area of neuroblastoma research.
- Neurotrophin (nerve growth factor) receptors: These are substances on the surface of normal nerve cells and on some neuroblastoma cells. They normally allow the cells to recognize neurotrophins – hormone-like chemicals that help the nerve cells mature. Neuroblastomas that have more of certain neurotrophin receptors, especially the nerve growth factor receptor called TrkA, may have a better prognosis.
- Serum markers: Serum (blood) levels of certain substances can be used to help predict prognosis.
Neuroblastoma cells release ferritin, a chemical that is an important part of the body’s normal iron metabolism, into the blood. Patients with high ferritin levels tend to have a worse prognosis.
Neuron-specific enolase (NSE) and lactate dehydrogenase (LDH) are made by some types of normal cells as well as by neuroblastoma cells. Increased levels of NSE and LDH in the blood are often linked with a worse outlook in children with neuroblastoma.
A substance on the surface of many nerve cells known as ganglioside GD2 is often increased in the blood of neuroblastoma patients. Although the usefulness of GD2 in predicting prognosis is unknown, it may turn out to be more important in treating neuroblastoma.
Children’s Oncology Group (COG) risk groups
The Children’s Oncology Group (COG) uses the major prognostic factors discussed in the staging section, combined with the International Neuroblastoma Staging System (INSS) stage of the disease, to place children into 3 different risk groups: low, intermediate, and high. These risk groups are used to help predict how likely it is that a child can be cured. For example, a child in a low-risk group can often be cured with limited treatment, such as surgery alone. With children in higher risk groups, the chance of cure is not as great, so more intensive treatment is often needed.
These risk groups are based on what is now known about neuroblastoma and how it is treated. As new research provides more information, these risk groups may change over time. For example, in recent treatment recommendations the age cut-off for some of these categories has been revised from up to 12 months (365 days) to up to 18 months (547 days).
Low risk
- All children who are Stage 1
- Any child who is Stage 2A or 2B and younger than age 1
- Any child who is Stage 2A or 2B, older than age 1, whose cancer has no extra copies of the MYCN gene
- Any child who is Stage 4S (younger than age 1), whose cancer has favorable histology, is hyperdiploid (excess DNA) and has no extra copies of the MYCN gene
Intermediate risk
- Any child who is Stage 3, younger than age 1, whose cancer has no extra copies of the MYCN gene
- Any child who is Stage 3, older than age 1, whose cancer has no extra copies of the MYCN gene and has favorable histology (appearance under the microscope)
- Any child who is Stage 4, younger than age 1, whose cancer has no extra copies of the MYCN gene
- Any child who is Stage 4S (younger than age 1), whose cancer has no extra copies of the MYCN gene and has normal DNA ploidy (number of chromosomes) and/or has unfavorable histology
High risk
- Any child who is Stage 2A or 2B, older than age 1, whose cancer has extra copies of the MYCN gene
- Any child who is Stage 3, younger than age 1, whose cancer has extra copies of the MYCN gene
- Any child who is Stage 3, older than age 1, whose cancer has extra copies of the MYCN gene
- Any child who is Stage 3, older than 18 months of age, whose cancer has unfavorable histology
- Any child who is Stage 4, whose cancer has extra copies of the MYCN gene regardless of age
- Any child who is Stage 4 and older than 18 months
- Any child who is Stage 4 and between 12 and 18 months old whose cancer has extra copies of the MYCN gene, unfavorable histology, and/or normal DNA ploidy (a DNA index of 1)
- Any child who is Stage 4S (younger than age 1), whose cancer has extra copies of the MYCN gene
International Neuroblastoma Risk Group (INRG) classification
As with the staging system described in the previous section, a newer risk group classification system, the International Neuroblastoma Risk Group (INRG) classification, is now being studied and may soon replace the Children’s Oncology Group (COG) system above. This system is based on the newer International Neuroblastoma Risk Group Staging System (INRGSS), which includes the image-defined risk factors (IDRFs), as well as many of the prognostic factors listed in the staging section, such as:
- The child’s age
- Tumor histology (how the tumor looks under the microscope)
- The presence or absence of MYCN gene amplification
- Certain changes in chromosome 11 (known as an 11q aberration)
- DNA ploidy (the total number of chromosomes in the tumor cells)
The International Neuroblastoma Risk Group (INRG) classification uses these factors to put children into 16 different pre-treatment groups (lettered A through R). Each of these pretreatment groups falls into 1 of 4 overall risk groups:
- Very low risk
- Low risk
- Intermediate risk
- High risk
This system has not yet been widely adopted, but it is being researched in new treatment protocols.
Neuroblastoma survival rate
Survival rates are a way to get an idea of the outlook for children with a certain type of cancer. Some parents may want to know the statistics for children in similar situations, but others may not find the numbers helpful, or may even not want to know them.
The 5-year survival rate refers to the percentage of children who live at least 5 years after their cancer is diagnosed. Of course, many children live much longer than 5 years (and many are cured).
In order to get 5-year survival rates, doctors have to look at children who were treated at least 5 years ago. Improvements in treatment since then may result in a better outlook for children now being diagnosed with neuroblastoma.
Survival rates are based on previous outcomes of large numbers of people who had the disease, but they cannot predict what will happen in any particular child’s case. The risk group of a child’s cancer is important in estimating their outlook. But many other factors can also affect a child’s outlook, such as their age, the location of the tumor, and how well the cancer responds to treatment. Your child’s doctor can tell you how the numbers below might apply to your child, as he or she knows your situation best.
Survival by Children’s Oncology Group (COG) risk group
- Low-risk group: Children in the low-risk group have a 5-year survival rate that is higher than 95%.
- Intermediate-risk group: In children in the intermediate-risk group, the 5-year survival rate is around 90% to 95%.
- High-risk group: The 5-year survival rate in children in the high-risk group is around 40% to 50%.
Neuroblastoma treatment
Treating neuroblastoma is complex and often requires the expertise of many different doctors, nurses, and other health professionals. The doctors on the treatment team often include:
- A pediatric cancer surgeon
- A pediatric oncologist (doctor who uses chemotherapy and other medicines to treat childhood cancers)
- A pediatric radiation oncologist (doctor who uses radiation therapy to treat cancer in children)
Many other specialists may be involved in your child’s care as well, including physician assistants, nurse practitioners, nurses, psychologists, social workers, rehabilitation specialists, and other health professionals.
The types of treatment used for neuroblastoma can include:
- Surgery
- Chemotherapy
- Radiation therapy
- High-dose chemotherapy/radiation therapy and stem cell transplant
- Retinoid therapy
- Immunotherapy
Treatment of neuroblastoma depends on the risk group of the cancer, the child’s age, and other factors, and might include more than one type of treatment.
Your child’s cancer care team will discuss the treatment options with you. It’s important to discuss these options and their possible side effects with your child’s doctors so you can make an informed decision.
Surgery
Surgeons use scalpels and other surgical tools to remove cancer cells. In children with low-risk neuroblastoma, surgery to remove the tumor may be the only treatment needed.
Whether the tumor can be completely removed depends on its location and its size. Tumors that are attached to nearby vital organs — such as the lungs or the spinal cord — may be too risky to remove.
In intermediate-risk and high-risk neuroblastoma, surgeons may try to remove as much of the tumor as possible. Other treatments, such as chemotherapy and radiation, may then be used to kill remaining cancer cells.
Chemotherapy
Chemotherapy uses chemicals to destroy cancer cells. Chemotherapy targets rapidly growing cells in the body, including cancer cells. Unfortunately, chemotherapy also damages healthy cells that grow quickly, such as cells in the hair follicles and in the gastrointestinal system, which can cause side effects.
Children with intermediate-risk neuroblastoma often receive a combination of chemotherapy drugs before surgery to improve the chances that the entire tumor can be removed.
Children with high-risk neuroblastoma usually receive high doses of chemotherapy drugs to shrink the tumor and to kill any cancer cells that have spread elsewhere in the body. Chemotherapy is usually used before surgery and before bone marrow stem cell transplant.
Radiation therapy
Radiation therapy uses high-energy beams, such as X-rays, to destroy cancer cells.
Children with low-risk or intermediate-risk neuroblastoma may receive radiation therapy if surgery and chemotherapy haven’t been helpful. Children with high-risk neuroblastoma may receive radiation therapy after chemotherapy and surgery, to prevent cancer from recurring.
Radiation therapy primarily affects the area where it’s aimed, but some healthy cells may be damaged by the radiation. What side effects your child experiences depends on where the radiation is directed and how much radiation is administered.
Stem cell transplant
Children with high-risk neuroblastoma may receive a transplant using their own blood stem cells (autologous stem cell transplant).
Before the stem cell transplant, your child undergoes a procedure that filters and collects stem cells from his or her blood. The stems cells are stored for later use. Then high doses of chemotherapy are used to kill any remaining cancer cells in your child’s body. Your child’s stem cells are then injected into your child’s body, where they can form new, healthy blood cells.
Immunotherapy
Immunotherapy is the use of medicines to help a patient’s own immune system recognize and destroy cancer cells more effectively.
Children with high-risk neuroblastoma may receive immunotherapy drugs that stimulate the immune system to kill the neuroblastoma cells.
Several types of immunotherapy are now being studied for use against neuroblastoma.
Monoclonal antibodies are man-made versions of immune system proteins that can be made to attack a very specific target. They can be injected into the body to seek out and attach to cancer cells.
A monoclonal antibody called dinutuximab (Unituxin) attaches to GD2, a substance found on the surface of many neuroblastoma cells. This antibody can be given together with cytokines (immune system hormones) such as GM-CSF and interleukin-2 (IL-2) to help the child’s immune system recognize and destroy neuroblastoma cells. This antibody is now part of the routine treatment for many children with high-risk neuroblastoma, often after a stem cell transplant.
Possible side effects
Side effects of dinutuximab treatment can include:
- Nerve pain (which can sometimes be severe)
- Leaking of fluid in the body (which can lead to low blood pressure, fast heart rate, shortness of breath, and swelling)
- Allergic reactions (which can lead to airway swelling, trouble breathing, and low blood pressure)
- Vomiting
- Diarrhea
- Infections
Newer treatments
Doctors are studying a newer form of radiation therapy that may help control high-risk neuroblastoma. The treatment uses a radioactive form of the chemical metaiodobenzylguanidine (MIBG). When injected in to the bloodstream, the MIBG travels to the neuroblastoma cells and releases the radiation.
MIBG therapy is sometimes combined with chemotherapy or stem cell transplant. After receiving an injection of the radioactive MIBG, your child will need to stay in a special hospital room until the radiation leaves his or her body in the urine. MIBG therapy usually takes a few days.
Retinoid Therapy for Neuroblastoma
Retinoids are chemicals that are related to vitamin A. They are known as differentiating agents because they are thought to help some cancer cells mature (differentiate) into normal cells.
In children with high-risk neuroblastoma, treatment with a retinoid called 13-cis-retinoic acid (isotretinoin) reduces the risk of the cancer coming back after high-dose chemotherapy and stem cell transplant. Most doctors now recommend 6 months of 13-cis-retinoic acid after the transplant. This drug is taken as a capsule, twice a day for 2 weeks, followed by 2 weeks off.
Researchers are now trying to develop more effective retinoids and to define the exact role of this approach in treating neuroblastoma.
Possible side effects
The most common side effect of 13-cis-retinoic acid is dry and cracked lips. Dry skin or eyes are also possible, as are nosebleeds, muscle and joint pains, and changes in the nails.





{kind=link}