Contents
What is ophthalmoplegia
Ophthalmoplegia is defined as weakness or paralysis of the muscles that move the eye. Ophthalmoplegia is a complex disease, due to the influence of congenital causes and acquired factors, which usually lead to organic diseases of the eye movement system and the eye muscles 1). Ophthalmoplegia can cause extraocular muscles partial or complete paralysis 2). Ophthalmoplegia is usually caused by a variety of factors, due to the third, fourth, and sixth cranial nerve paresis, that may be associated with cavernous sinus lesion 3), intracranial aneurysm 4), diabetes 5), Tolosa-Hunt syndrome 6), migraine 7), Kearns-Sayre syndrome 8), myasthenia gravis 9), trauma 10), etc.
Ophthalmoplegia can be categorized as oculomotor cranial nerve palsy, trochlear cranial nerve palsy, or abducens nerve palsy, all conditions in which the third, fourth, and sixth cranial nerves are affected. The overall prevalence of ophthalmoplegia cases was 0.32% among diabetic patients 11), along with 0.5% in the Italian studies in 2011 12), and 0.5% in the Japanese studies. Which are 10 times more frequent than nondiabetic subjects 13).
It should be noted that ophthalmoplegia can also lead to strabismus, diplopia, headache, and ptosis 14), which seriously affect the quality of life and appearance of patients.
Up to now, the treatment for ophthalmoplegia includes medical therapy and surgery, such as tetracycline 15), corticosteroid treatment 16), botulinum toxin 17), vitamin 18), reconstruction of the lateral orbital wall 19), combination of modest blepharoplasty, and frontalis suspension 20). Most of the interventions are symptomatic treatments, and the efficacy concerning recovery of ophthalmoplegia has not been adequately assessed 21).
Internuclear ophthalmoplegia
Internuclear ophthalmoplegia presents as the inability to perform conjugate side gaze and produces an ophthalmoplegia due to damage to the nerve fiber bundle (interneuron) that connects the 6th cranial nerve (CN VI) nucleus on one side of the pons to the medial rectus subnucleus of the 3rd cranial nerve (CN III) in the contralateral midbrain 22). This interneuron nerve fiber bundle is called the medial longitudinal fasciculus (MLF). The medial longitudinal fasciculus can be damaged by any lesion (e.g., demyelinating, ischemic, neoplastic, inflammatory) in the pons or midbrain. The medial longitudinal fasciculus is supplied blood by branches of the basilar artery and ischemia in the vertebrobasilar system can produce an ischemic internuclear ophthalmoplegia 23).
The medial longitudinal fasciculus is a heavily myelinated nerve tract connecting the oculomotor nucleus (cranial nerve 3) of the ipsilateral side (same side) with the paramedian pontine reticular formation (PPRF) and cranial nerve 6 of the contralateral (opposite side) pons. Thus, demyelinating lesions in the midbrain or pons often produce a unilateral or bilateral internuclear ophthalmoplegia in young patients.
The internuclear ophthalmoplegia is characterized clinically by an ispilesional adduction deficit (partial or complete) with a contralateral, dissociated, horizontal abducting saccade on attempted gaze to the contralesional side. Hering’s law of equal innervation has been hypothesized as a possible explanation for the dissociated contralateral horizontal gaze evoked nystagmus in the abducting eye. Increased innervation to the underacting adducting muscle would result in an enhanced stimulus to the contralateral abducting muscle 24).
The internuclear ophthalmoplegia can be unilateral or bilateral and may present with or without (neurologically isolated) other brainstem findings.
Internuclear ophthalmoplegia pathway
Typically, the paramedian pontine reticular formation (PPRF) receives information from the higher cortical centers such as the frontal eye fields, occipital and parietal lobes and the superior colliculus. From the PPRF the signal travels to the ipsilateral nucleus for the abducens nerve (cranial nerve 6). The abducens nucleus would then send an excitatory signal through the medial longitudinal fasciculus (MLF) to the contralateral medial rectus muscle (cranial nerve 3). The activation of the contralateral medial rectus and ipsilateral lateral rectus muscle produces horizontal conjugate eye movement. The side of the internuclear ophthalmoplegia is named by the side of the adduction deficit, which is ipsilateral to the MLF lesion. Because of the close proximity of the the MLF to the midline, bilateral involvement may not be uncommon (bilateral internuclear ophthalmoplegia).
Figure 1. Internuclear ophthalmoplegia pathway
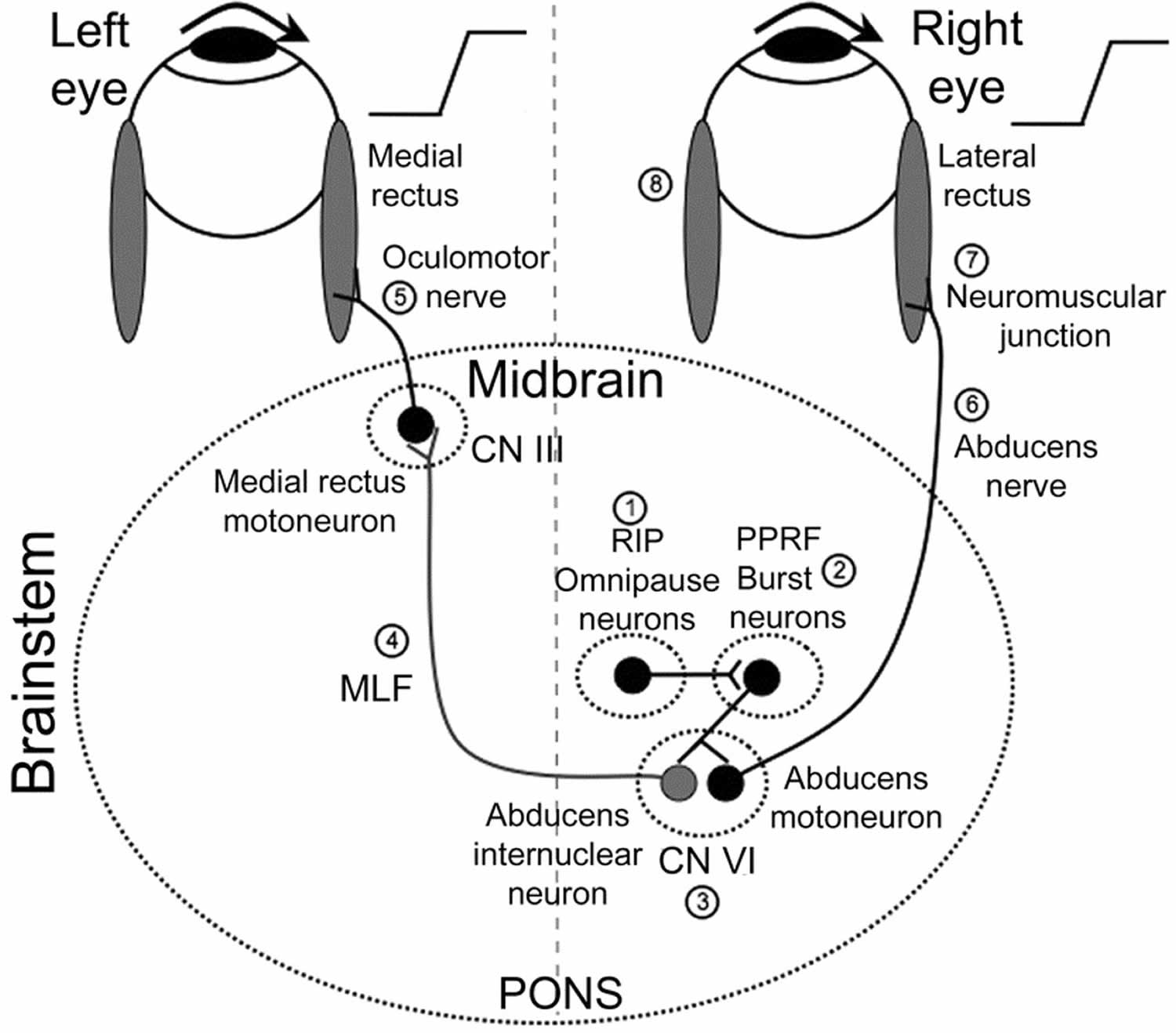
Footnotes: Premotor burst neurons, lying in the paramedian pontine reticular formation (PPRF) (2), are inhibited by omnipause neurons in the nucleus raphe interpositus (RIP) (1). When a saccade needs to be generated, this inhibition ceases. Premotor burst neurons project a pulse of innervation to the abducens nucleus (cranial nerve VI) (3). Abducens motoneurons project the pulse of innervation via the sixth nerve (6) to the right lateral rectus, which contracts rapidly to generate an abducting saccade of the right eye. Abducens internuclear neurons project the pulse of innervation, via the medial longitudinal fasciculus (MLF; internuclear pathway) (4) to medial rectus motoneurons, which, in turn, innervate the left medial rectus via the third nerve (5) to generate a fast adducting saccade of the left eye. Also shown are the neuromuscular junction (7) and the horizontal recti muscles (8) sites.
CN III = cranial nerve 3 (oculomotor nerve); CN VI = cranial nerve 6 (abducens nerve)
[Source 25)]Figure 2. Bilateral internuclear ophthalmoplegia
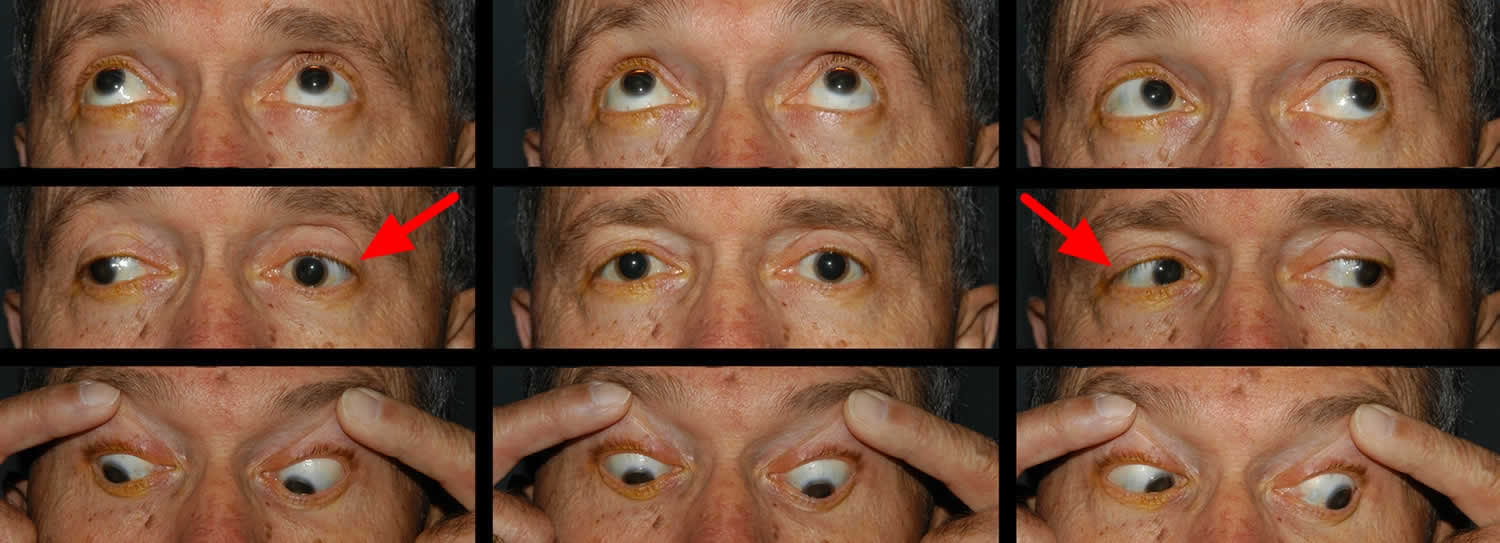
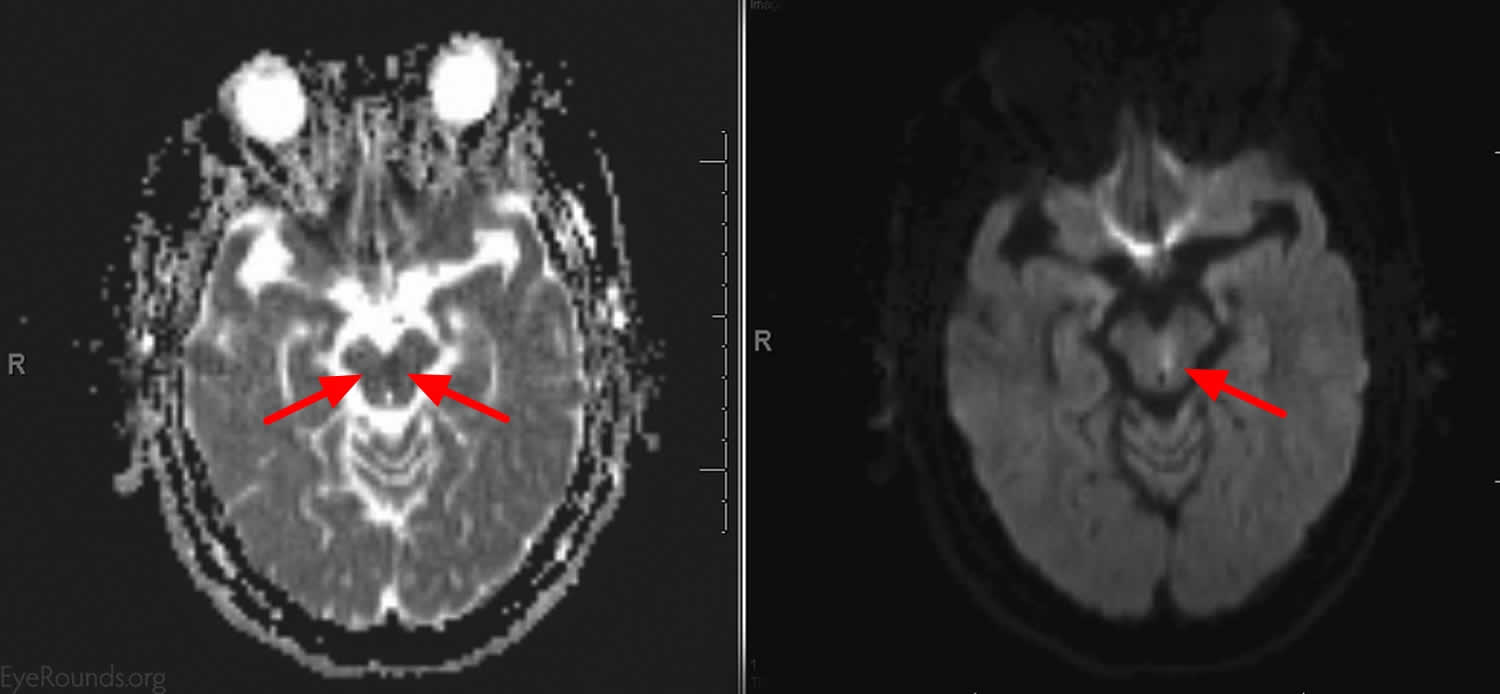
Footnote: This patient presented with an acute onset of binocular horizontal diplopia that was most noticable in right gaze and difficulty tracking objects as they move to the right. He has vascular risk factors. He had an adduction deficit of the left eye that was overcome with convergence. There was abducting nystagmus of the right eye and a slowed adducting saccade on the left. Imaging showed a small focus of diffusion restriction with corresponding low apparent diffusion coefficient (ADC) consistent with an acute infarct in the left inferior medial midbrain just anterior to the cerebral aquaduct that is along the left MLF.
[Source 26)]Figure 3. Bilateral internuclear ophthalmoplegia (brain MRI scan of patient in Figure 2)
Internuclear ophthalmoplegia causes
A lesion in the medial longitudinal fasciculus (MLF) interrupts the neural communication to the cranial nerve 3 (CN III) subnuclei that allows for conjugate horizontal gaze arising from the final common pathway for horizontal gaze (cranial nerve 6 nuclei). An internuclear ophthalmoplegia is a common presentation of multiple sclerosis (MS) in younger patients. In older people, stroke is a more common cause. Less common causes for an internuclear ophthalmoplegia include traumatic, neoplastic, inflammatory (e.g., sarcoid, Behcet’s disease, lupus), or infectious (e,g., cryptococcosis, Borrelia burgdorferi (Lyme disease) 28). Internuclear ophthalmoplegia is rare in children but may result from neoplasms (e.g., medulloblastomas or pontine gliomas) or similar causes to adults 29).
In one review of internuclear ophthalmoplegia, some of the most common causes included:
- Infarction – 38%- Unilateral internuclear ophthalmoplegia in 87% of cases;
- Multiple sclerosis in 34%-Unilateral in 27%; and
- Unusual causes included trauma, tentorial herniation, infection, tumor, iatrogenic injury, hemorrhage, vasculitis 30).
Associated Syndromes
Wall-Eyed Bilateral Internuclear Ophthalmoplegia
Wall-Eyed Bilateral Internuclear Ophthalmoplegia (WEBINO) exists when there is bilateral damage to the MLF. This damage causes a primary position exotropia (eyes are looking at the opposite “wall.”, thus the possibly outdated term “wall eyed”). The most common cause is infarction of the midbrain in older patients and demyelinating disease in young patients 31). The exotropia is likely decompensation of fusional mechanisms and the symptomatic deviation is not present in every case of bilateral internuclear ophthalmoplegia.
Wall-Eyed Monocular Internuclear Ophthalmoplegia
Wall-Eyed Monocular Internuclear Ophthalmoplegia (WEMINO) is a less common variant of internuclear ophthalmoplegia, similar to the Wall-Eyed Bilateral Internuclear Ophthalmoplegia above. As in Wall-Eyed Bilateral Internuclear Ophthalmoplegia (WEBINO), patients with a unilateral MLF lesion (monocular internuclear ophthalmoplegia) have a primary position symptomatic deviation 32).
One and one half syndrome
This syndrome occurs when there is a lesion to the MLF and the PPRF or cranial nerve 6 nucleus on the same side resulting in an internuclear ophthalmoplegia in one eye and an ipsilateral horizontal gaze palsy 33).
Eight and a Half Syndrome
This syndrome is characterized by having one-and-a-half syndrome and a facial fascicular nerve (cranial nerve 7) palsy. The fascicle of cranial nerve 7 wraps around the nucleus of cranial nerve 6 in the dorsal pons. There is conjugate horizontal gaze palsy on looking to one side followed by internuclear ophthalmoplegia on looking to the opposite side, along with unilateral facial weakness. The close proximity of the PPRF, facial nerve nucleus and MLF located in the dorsal pons makes this syndrome much more likely. The lesion is most often vascular or demyelinating in the dorsal tegmentum of the caudal pons 34).
Half and Half Syndrome
A syndrome that consists of an internuclear ophthalmoplegia in one eye combined with an ipsilateral cranial nerve 6 fascicular involvement with sparing of the sixth nerve nucleus. Thus, there is “half” of a horizontal gaze palsy (internuclear ophthalmoplegia) plus an additional “half” (abduction deficit from cranial nerve 6 fascicular palsy).
Posterior internuclear ophthalmoplegia (Lutz)
This syndrome is a rare ophthalmoplegia, either bilateral or unilateral that exhibits contralateral adducting eye (rather than abducting eye) nystagmus with abduction restriction on physical exam. It is the reverse of the typical internuclear ophthalmoplegia, and although the lesion localization is not consistent, it likely is due to cranial nerve 6 pre-nuclear input asymmetry 35).
Internuclear ophthalmoplegia diagnosis
The diagnosis is made clinically with testing of the eye’s ability to perform conjugate movements.
Imaging tests such as a CT scan or MRI are ordered after a diagnosis is made to discover where the damage is located so that the physician can then assess which
route to take based on what is causing the damage. If the etiology is inflammation or infection, high dose corticosteroids can help. If the etiology is MS (multiple sclerosis) and not relapsing-remitting, then treatment becomes harder. In general MRI is superior to CT scan for evaluation of internuclear ophthalmoplegia.
Internuclear ophthalmoplegia prognosis
Dalfampridine, a potassium channel blocker prescribed for gait impairment was used in a case series and the authors reported improvement in saccades and ocular motility in patients with internuclear ophthalmoplegia secondary to demyelination in Multiple sclerosis. Improved neuronal conduction along MLF has been discussed as a possible explanation for this effect seen 36).
The prognosis of most patients with an internuclear ophthalmoplegia is good but the final outcome depends in part on treatment of the underlying cause. Ischemic and demyelinating internuclear ophthalmoplegia typically recover. Patients with Wall-Eyed Bilateral Internuclear Ophthalmoplegia (WEBINO) or Wall-Eyed Monocular Internuclear Ophthalmoplegia (WEMINO) may benefit from patching, prism, or strabismus surgery to correct any residual primary position symptomatic deviation that does not recover 37).
Progressive external ophthalmoplegia
Chronic progressive external ophthalmoplegia is a mitochondrial myopathy characterized by slowly progressive, symmetric ophthalmoplegia and ptosis, which can be inherited, or it can occur sporadically (due to a new mutation in an individual with no history of the condition in the family) 38). Chronic progressive external ophthalmoplegia typically appears in adults between ages 18 and 40 and slowly worsens over time 39). The first sign of progressive external ophthalmoplegia is typically drooping eyelids (ptosis), which can affect one or both eyelids. As ptosis worsens, affected individuals may use the forehead muscles to try to lift the eyelids, or they may lift up their chin in order to see. Another characteristic feature of progressive external ophthalmoplegia is weakness or paralysis of the muscles that move the eye (ophthalmoplegia). Affected individuals have to turn their head to see in different directions, especially as the ophthalmoplegia worsens. People with progressive external ophthalmoplegia may also have general weakness of the muscles used for movement (myopathy), particularly those in the neck, arms, or legs. The weakness may be especially noticeable during exercise (exercise intolerance). Muscle weakness may also cause difficulty swallowing (dysphagia).
Progressive external ophthalmoplegia is a condition caused by defects in mitochondria, which are structures within cells that use oxygen to convert the energy from food into a form cells can use. This process is called oxidative phosphorylation. Although most DNA is packaged in chromosomes within the nucleus (nuclear DNA), mitochondria also have a small amount of their own DNA, called mitochondrial DNA or mtDNA. This DNA contains genes essential for oxidative phosphorylation.
When the muscle cells of affected individuals are stained and viewed under a microscope, these cells usually appear abnormal. These abnormal muscle cells contain an excess of cell structures called mitochondria and are known as ragged-red fibers.
Although muscle weakness is the primary symptom of progressive external ophthalmoplegia, this condition can be accompanied by other signs and symptoms. In these instances, the condition is referred to as progressive external ophthalmoplegia plus (PEO+). Additional signs and symptoms can include hearing loss caused by nerve damage in the inner ear (sensorineural hearing loss), weakness and loss of sensation in the limbs due to nerve damage (neuropathy), impaired muscle coordination (ataxia), a pattern of movement abnormalities known as parkinsonism, and depression.
Progressive external ophthalmoplegia is part of a spectrum of disorders with overlapping signs and symptoms. Similar disorders include ataxia neuropathy spectrum and Kearns-Sayre syndrome. Like progressive external ophthalmoplegia, the other conditions in this spectrum can involve weakness of the eye muscles. However, these conditions have many additional features not shared by most people with progressive external ophthalmoplegia.
People with Kearns-Sayre syndrome have progressive external ophthalmoplegia, which is weakness or paralysis of the eye muscles that impairs eye movement and causes drooping eyelids (ptosis). Affected individuals also have an eye condition called pigmentary retinopathy, which results from breakdown (degeneration) of the light-sensing tissue at the back of the eye (the retina) that gives it a speckled and streaked appearance 40). The retinopathy may cause loss of vision. In addition, people with Kearns-Sayre syndrome have at least one of the following signs or symptoms: abnormalities of the electrical signals that control the heartbeat (cardiac conduction defects), problems with coordination and balance that cause unsteadiness while walking (ataxia), or abnormally high levels of protein in the fluid that surrounds and protects the brain and spinal cord (the cerebrospinal fluid or CSF). The use of Tensilon (edrophonium) is worrisome in these patients as it may precipitate heart block and arrhythmias. It is also worrisome that these patients may suffer sudden cardiac death, even if cardiac evaluation results are normal. For this reason, some electrophysiologists recommend prophylactic pacemaker placement for these patients.
People with Kearns-Sayre syndrome may also experience muscle weakness in their limbs, deafness, kidney problems, or a deterioration of cognitive functions (dementia). Affected individuals often have short stature. In addition, diabetes mellitus is occasionally seen in people with Kearns-Sayre syndrome 41). Patients with chronic progressive external ophthalmoplegia must be evaluated for this potentially fatal syndrome and treated accordingly.
A related condition called ophthalmoplegia-plus may be diagnosed if an individual has many of the signs and symptoms of Kearns-Sayre syndrome but not all the criteria are met.
Treatment of the ptosis in chronic progressive external ophthalmoplegia is primarily surgical. The use of coenzyme Q has been reported but has not been shown to be effective in treating this condition 42).
Chronic progressive external ophthalmoplegia signs and symptoms
Signs of chronic progressive external ophthalmoplegia:
- Ptosis with poor levator function
- Loss of Bell’s reflex (eyes roll upwards when lids are closed)
- Symmetric ophthalmoplegia
- Kearns-Sayre syndrome may include: pigmentary retinopathy, cerebellar ataxia, deafness, and endocrine disorders (e.g., diabetes or gonadal dysfunction)
Chronic progressive external ophthalmoplegia symptoms:
- Chronic progressive external ophthalmoplegia
- Reduced superior visual field
- Exposure keratopathy from poor blink
- NO DIPLOPIA
- Kearns-Sayre syndrome
- Possible ataxia, deafness, endocrine problems and sudden cardiac death
Figure 4. Chronic progressive external ophthalmoplegia and ptosis
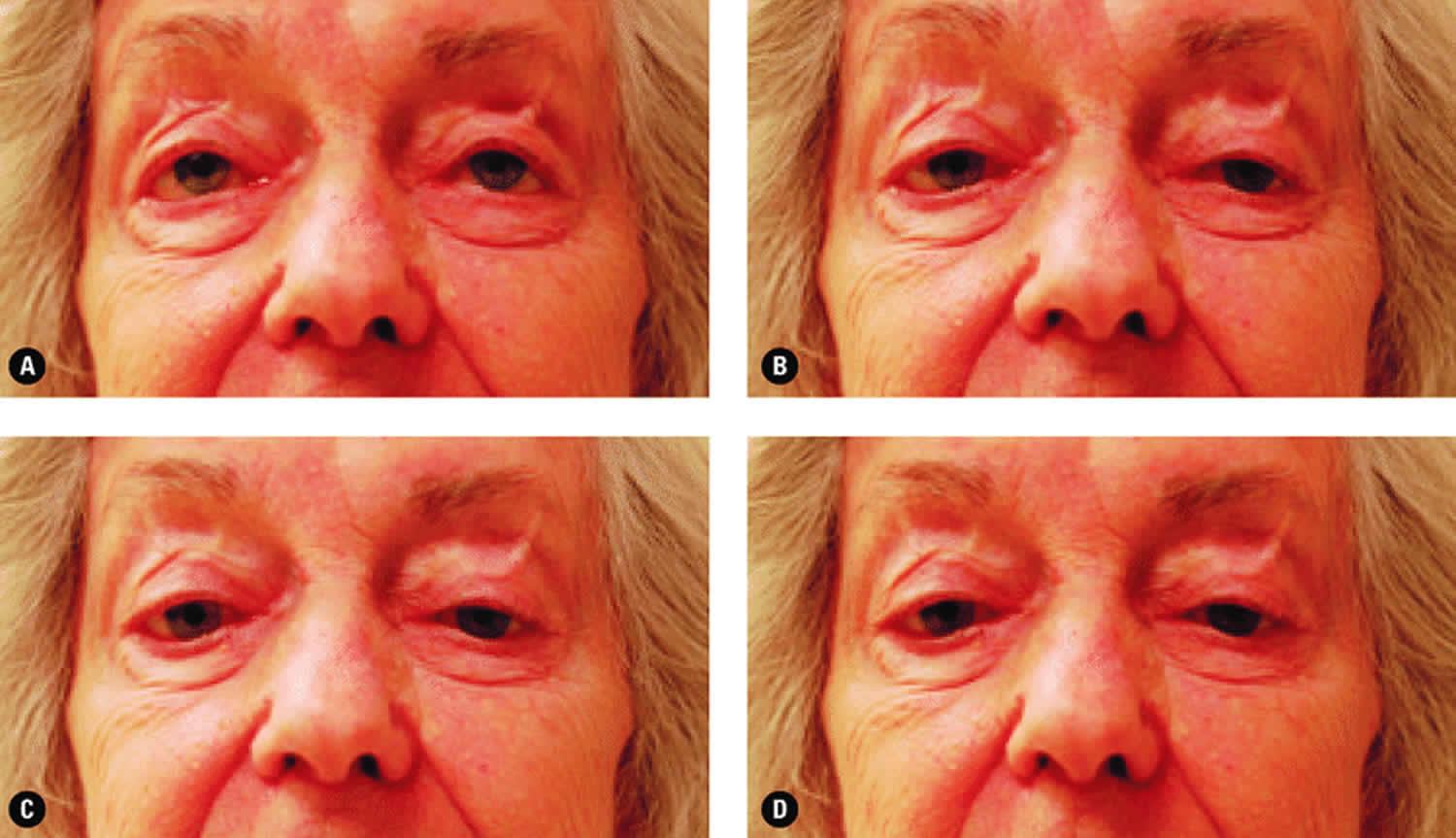
Footnote: The ptosis evident and the lack of eye movement demonstrated when the patient was asked to look up (A), to look left (B), to look right (C), and to look down (D) both indicate the presence of a mitochondrial disease.
[Source 43)]Chronic progressive external ophthalmoplegia causes
Chronic progressive external ophthalmoplegia (chronic progressive external ophthalmoplegia) can be inherited, or it can occur sporadically (due to a new mutation in an individual with no history of the condition in the family).
Chronic progressive external ophthalmoplegia is considered a “mitochondrial disorder.” This is because all the genetic mutations that can cause chronic progressive external ophthalmoplegia ultimately result in dysfunction of the mitochondria, which are structures in our cells that produce energy required for normal cell function. While most of your DNA is located in the cell’s center (nuclear DNA), some of your DNA is located within the mitochondria (mitochondrial DNA) 44). Chronic progressive external ophthalmoplegia can be caused by mutations in any of several genes, which may be located in mitochondrial DNA or nuclear DNA. It has different inheritance patterns depending on the gene involved in the affected individual 45).
Unlike nuclear DNA which is inherited from both the mother and the father, mitochondrial DNA is inherited from only the mother. In chronic progressive external ophthalmoplegia, the affected mitochondria (i.e., the ones carrying the mutations) are found only in the skeletal muscle cells. These mitochondrial DNA mutations are almost always sporadic (occurring by chance for the first time in the affected individual). Nuclear gene mutations that cause chronic progressive external ophthalmoplegia may be inherited in an autosomal recessive or autosomal dominant manner, depending on the gene involved 46). The risk for other family members to be affected depends on the genetic cause and the inheritance pattern in the family.
Progressive external ophthalmoplegia can result from mutations in one of several different genes. In some cases, mutations in nuclear DNA are responsible for the condition, including mutations in the POLG, TWNK, RRM2B, and SLC25A4 genes, among others. These genes are critical for the production and maintenance of mtDNA. Although the mechanism is unclear, mutations in these genes lead to the deletion of large segments of mtDNA in muscle cells. The size of the deleted region can range from 2,000 to 10,000 DNA building blocks (nucleotides).
In other cases, the condition is caused by a single large deletion of mtDNA that is not associated with a mutation in a nuclear DNA gene.
Less commonly, mutations that change single nucleotides in genes found in mtDNA, such as the MT-TL1 gene, cause progressive external ophthalmoplegia. These mutations occur in genes that provide instructions for making molecules called transfer RNAs. Transfer RNAs help assemble protein building blocks (amino acids) into functioning proteins. The transfer RNAs associated with progressive external ophthalmoplegia are present in mitochondria and help assemble the proteins that carry out the steps of oxidative phosphorylation.
Researchers have not determined how deletions of mtDNA or mutations in mtDNA genes lead to the specific signs and symptoms of progressive external ophthalmoplegia, although the features of the condition are probably related to impaired oxidative phosphorylation. It has been suggested that eye muscles are commonly affected by mitochondrial defects because they are especially dependent on oxidative phosphorylation for energy.
Inheritance Pattern
Progressive external ophthalmoplegia can have different inheritance patterns depending on the gene involved.
When the nuclear genes POLG, TWNK, RRM2B, or SLC25A4 are involved, progressive external ophthalmoplegia is usually inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Certain mutations in the POLG or RRM2B gene can also cause a form of the condition that is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
When this condition is caused by mutations in the MT-TL1 gene and other mitochondrial transfer RNA genes, it is inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mtDNA. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can inherit disorders resulting from mtDNA mutations only from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
Single, large deletions of mtDNA are typically not inherited but occur during the formation of a mother’s egg cells or in early development of the embryo. Individuals with these mutations usually have no history of the disorder in their family.
Chronic progressive external ophthalmoplegia treatment
Ptosis caused by chronic progressive external ophthalmoplegia can be corrected by surgery, or by using glasses that have a “ptosis crutch” to lift the upper eyelids 47). Strabismus surgery can be helpful in carefully selected patients if diplopia (double vision) occurs 48).
Some individuals with a deficiency of coenzyme Q10 have chronic progressive external ophthalmoplegia as an associated abnormality. Coenzyme Q10 is important for normal mitochondrial function. In individuals with this deficiency, supplemental coenzyme Q10 has been found to improve general neurologic function and exercise tolerance. However, coenzyme Q10 has not been shown to improve the ophthalmoplegia or ptosis in people who have isolated chronic progressive external ophthalmoplegia 49).
- Cardiac workup (Kearns-Sayre syndrome)
- Electromyography (EMG) + muscle biopsy (Ragged Red Fibers)
- Ptosis repair: Dynarod sling 50)
- Adjustable
- Irradiated fascia lata sling: 20% ptosis recurrence rate over 7 years among 132 lids with myogenic ptosis; only 1 with significant post-op exposure 51)
Chronic progressive external ophthalmoplegia prognosis
Chronic progressive external ophthalmoplegia can be an isolated condition, or it can occur as part of other underlying conditions, such as ataxia neuropathy spectrum and Kearns-Sayre syndrome. These conditions may involve not only chronic progressive external ophthalmoplegia, but various additional features that are not shared by most individuals with chronic progressive external ophthalmoplegia in isolation. Individuals with isolated chronic progressive external ophthalmoplegia generally have a normal life expectancy. While symptoms tend to worsen over time, the specific symptoms and their severity can vary greatly from person to person. Therefore, when symptoms first appear, the course of the condition is very difficult to predict 52).
For individuals with additional symptoms or another underlying condition associated with chronic progressive external ophthalmoplegia, the prognosis depends on the specific signs and symptoms present and/or the outlook associated with the underlying condition in the affected individual. For this reason, obtaining an accurate diagnosis is very important.
References [ + ]


{kind=link}