Contents
What is oxidative stress
There appears to be a continued public misconception (encouraged by the supplement industry) that free radicals are bad, and that antioxidants are good. Of course, like most phenomena affecting our health, it’s not that simple.
Free radicals are molecules or atoms containing an unpaired electron 1. Unpaired electrons are attention seekers. They really don’t like being alone, so are always searching out other electrons. This makes them highly reactive. Free radicals are like the unstable friend who shows up drunk to the party and starts breaking things. Antioxidants are molecules that are able to donate an electron to the free radical, thus stabilizing it. They’re like the patient friend who is able to convince the free radical not to drive home, takes him to the back room to calm him down, and brings him water and a bucket once he starts throwing up.
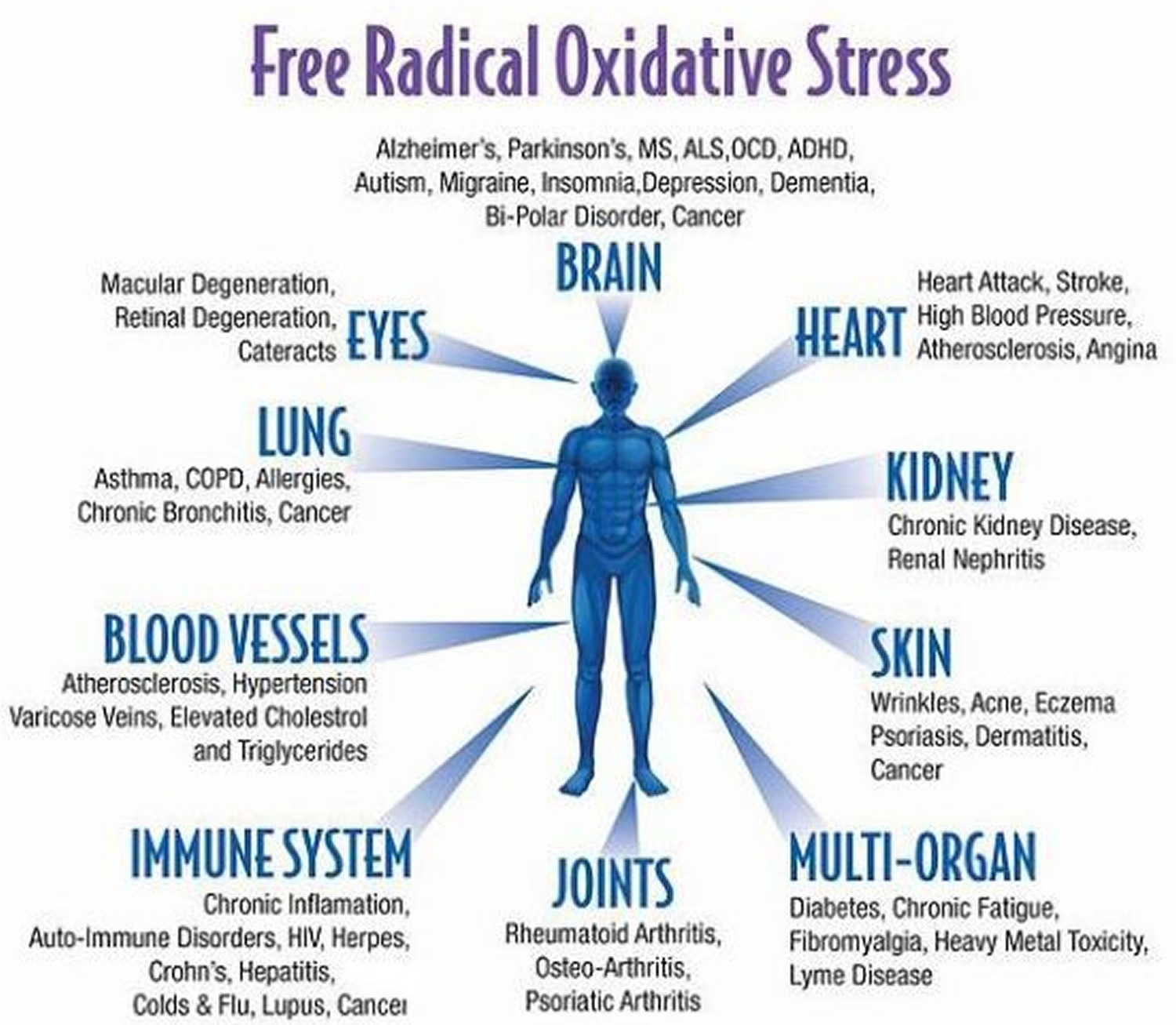
Needless to say, without the presence of antioxidants, free radicals can really ruin the party. The cellular damage that results is called oxidative stress, and is associated with ageing, heart failure, cancer, Alzheimer’s, and many other health problems.
Until recently, the thinking had been that the more antioxidants, the less oxidative stress, because all of those lonely electrons would quickly get paired up before they had the chance to start mucking things up in our cells. But that thinking has changed.
The concept of oxidative stress was first introduced for research in reduction-oxidation (redox) biology and medicine in 1985, in an introductory chapter 1 in a book entitled ‘Oxidative Stress’ 2. Oxidative stress in “an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signalling and control and/or molecular damage” 3. The association of oxidative stress with cardiovascular disease, cancer, metabolic and neurological pathologies and inflammation is well known, but not well understood.
Despite the large body of evidence linking oxidative stress with many common diseases, which is supported by the significant correlation of redox biomarkers with cardiovascular and all-cause mortality 4, direct clinical proof is still lacking that oral therapy with antioxidants, such as vitamin C and E, helps to prevent the development or progression of these diseases. Most large-scale antioxidant clinical trials yielded disappointing results regarding all-cause mortality and in some cases oral antioxidants had detrimental effects 5 and to this date no “antioxidant” is admitted as a drug for clinical use 6. More recent reports in publication channels dedicated to the non-expert or public readership highlighted the potential risks associated with excessive oral consumption of antioxidants [see online publications by Mustain 7 and Riley 8]. Antioxidant therapy was also mentioned among “the science myths that will not die” 9.
The fact that the emergence of aerobic organisms was a consequence of oxygen becoming a significant component of the earth’s atmosphere. The evolution of aerobic organisms was probably the result of their gradual adaptation to oxygen. Antioxidant enzymes probably facilitated part of such adaptation, since strict anaerobes have neither superoxide dismutase (SOD) nor catalase, whereas aerotolerant anaerobes have small levels of those enzymes 10. Aerobic organisms use oxygen to produce the chemical energy they need to survive. Metabolic pathways of aerobic organisms lead to the obligate production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) 11, which are not necessarily bad. Indeed, reactive species are actually needed as signal transduction elements in processes that are essential for life, such as cell growth, differentiation, preconditioning and cell death 12. They also participate in functions such as phagocytosis and stress response 13. This echoes Barry Halliwell’s 14 “All aspects of aerobic life involve free radicals and antioxidants—you cannot escape them nor should you wish to”.
Free radicals are molecules or atoms characterized by possessing an unpaired electron that is always seeking a counterpart to reside inside the parent home or in the home of the other electron. Free radicals can be produced during cellular processes or as a result of the interaction of free radicals, reactive species, or free radical-generating systems with other molecules 15. Many of the ROS and RNS are free radicals. There are also macromolecular radicals that are formed by the effect of oxygen, drug-induced free radicals, irradiation or free radicals on such macromolecules as protein and lipids. For example, it has been demonstrated that γ-irradiated proteins produce electron paramagnetic resonance (EPR)—detectable superoxide and huydroxyl radicals, in the presence of ferrous iron, via their intermediate conversion to hydroperoxydes 16. Such macromolecular radicals lead to cellular dysfunction and decreased cell viability 17. In the presence of hydrogen peroxide (H2O2), aminoglutethimide (xenobiotic) modified myeloperoxidase, transforming this enzyme in a macromolecular free radical; a reaction that has been suggested to have a role in the development of drug-induced agranulocytosis and has been inhibited by polyunsaturated fatty acids 15. Generation of lipid-derived free radicals was demonstrated, by in vivo EPR, in the livers of rats treated with the carcinogen diethylnitrosamine 18. Lipid-derived free radicals have also been demonstrated, through EPR, in culture cells incubated with ferrous iron 19. Free-radical modified lipids 20 and lipid-derived free radicals may play an important role in the development of atherosclerosis.
According to Halliwell and Gutteridge 21 an antioxidant is “any substance that, when present at low concentrations compared to those of an oxidizable substrate, significantly delays or prevents oxidation of that substance”. Just as reactive species are not necessarily bad, antioxidants are not necessarily good. Antioxidants are a kind of matchmaker midwife trying to give or receive an electron to complete the other’s “pair”, but by making a proper pair, antioxidants can themselves become incomplete, and behave as free radicals; i.e., reactive and looking to pair with an unpaired electron. It is known that those “reactive” antioxidants can be stabilized (recycled) by other antioxidants, which in turn become “reactive” and can be recycled in a cascade of “reactivity mitigation” (Figure 1). It is all a matter of equilibrium. It well is accepted that oxidative stress is produced when the equilibrium between reactive species and antioxidants is tilted in favor of the former. However, the equilibrium can be broken also if antioxidant levels exceed those of the reactive species. Dündar 22 proposed the term “antioxidative stress” for such disequilibrium.
Figure 1. Free radical
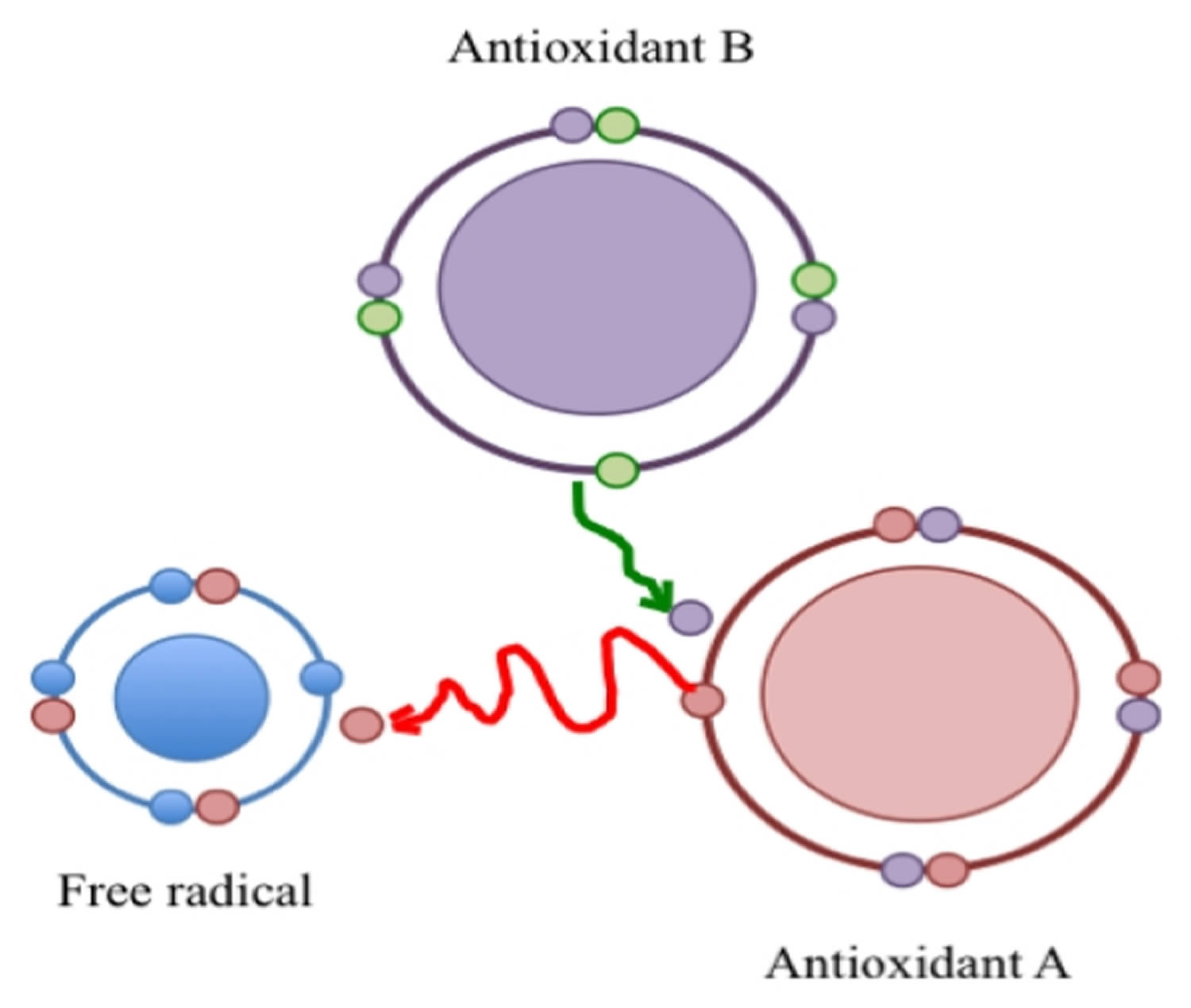
Note: A free radical is a reactive species because it has an unpaired electron. Its reactivity is mitigated by an antioxidant (A) that donates an electron and in turn is converted to a reactive species that is recycled by a second antioxidant (B).
[Source 1]Deregulation of reactive oxygen, nitrogen, and sulfur species (RONSS) affects cellular and organismal well being 23. Nature is certainly wise. Aerobic organisms mitigate reactive species not only with antioxidant molecules that donate or receive electrons but also through reactions catalyzed by antioxidant enzymes. Figure 2, below, illustrates the production of superoxide by pro-oxidant enzymes and the intervention of antioxidant enzymes to stabilize such free radical. Reactive oxygen species (ROS), which are short-lived and very reactive, so they can react with all biological molecules, changing their structure and function. Under physiological conditions, ROS are constantly produced and removed within the cells. Although generally considered as harmful by-products of oxygen metabolism, ROS also have important physiological functions in signal transduction and immunological response 24. A complex interplay between reactive oxygen species (ROS) generation, reactive oxygen species (ROS) signalling and reactive oxygen species (ROS)-induced damage are often species- and tissue-dependent being kept under control of elaborate enzymatic and non-enzymatic antioxidant defence systems 25. However, overproduction of ROS or deregulation of antioxidant systems can lead to localized or systemic oxidative stress 26. In its acute form moderate oxidative stress can boost cellular antioxidant capacities with positive, hormetic effects 27. However, more severe, and especially chronic oxidative stress can lead to many deleterious effects on molecular level generating oxidized forms of proteins, lipids and DNA 28.
Figure 2. Antioxidant reactions
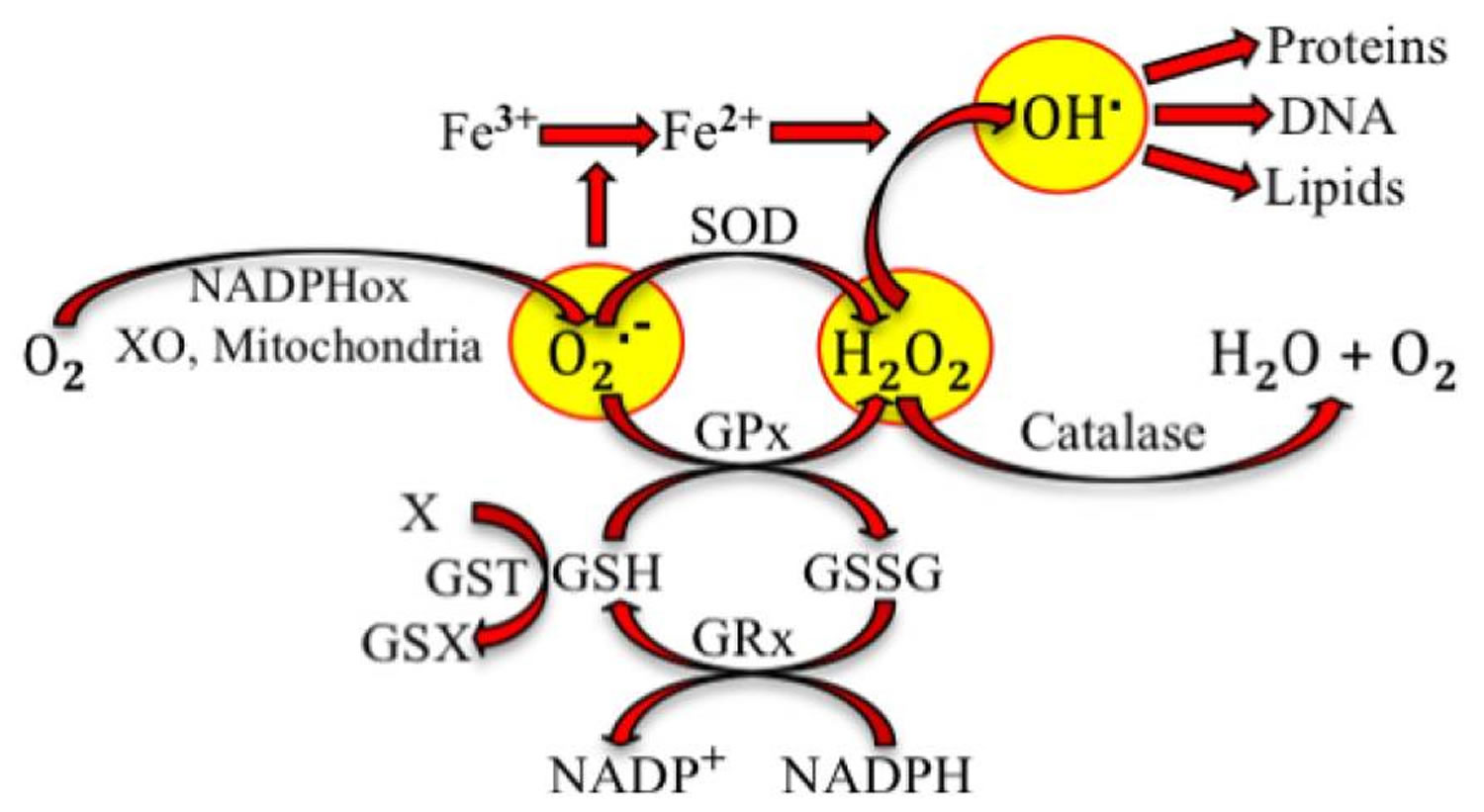
Note: Superoxide radical (O2.−) is produced from oxygen (O2) at complex I and complex III of mitochondria during respiration. It can also be produced through reactions catalyzed by NADPH oxidase (NADPHox) or xanthine oxidase (XO). Superoxide dismutase (SOD) and glutathione peroxide (GPx) catalyze the conversion by superoxide into hydrogen peroxide (H2O2, a reactive oxygen species). GPx requires the oxidation of glutathione (GSH) to oxidized glutathione (GSSG), which is reduced to recycle GSH through the enzyme glutathione reductase (GRx). GSH could be also used in detoxification (metabolism of the toxic X by conjugation with GSH) catalyzed by glutathione S transferase (GST). Superoxide participates in the reduction of ferric iron. Hydroxyl radical (OH.) is formed by the interaction of H2O2 and ferrous ion. OH. oxidizes proteins, DNA and lipids.
[Source 1]Figure 2 is only an example of the different reactions that produce reactive species, which potentially lead to oxidative stress. Villanueva and Kross 1 have taken this very simple sequence of reactions to illustrate how changes can lead to oxidative stress and make a later parallel with the antioxidant-induced stress. Superoxide radical production can be increased if the activity of NADPHox or XO increases. Indeed, it has been postulated and partially shown, that oxidative stress in hypertension is due, at least in part, to the increase of NADPHox’s activity in vascular smooth muscle cells, fibroblasts and endothelial cells 29. Polymorphisms of the genes codifying SOD and GPx have been related to oxidative stress due to decreased activity of the enzymes and high incidence of coronary artery disease 30, ischemic stroke 31 and atherosclerosis 32. Polymorphisms of GST have been associated with cardiovascular events produced in smokers 33.
Estimates of how much oxygen reacts directly to generate free radicals vary; however, typically cited values are around 1.5–5% of the total consumed oxygen 34. These estimates have been questioned by Hansford et al. 35 and Staniek and Nohl 36, who suggested that H2O2 production rates were less than 1% of consumed O2. The rate of generation of H2O2 is dependent on the state of mitochondria as determined by the concentration of ADP, substrates, and oxygen 37. It was observed that maximum lifespan correlates negatively with antioxidant enzyme levels and free-radical production, and positively with the rate of DNA repair 38. While data from studies in invertebrates (e.g., C. elegans and Drosophila) and rodents show a correlation between increased lifespan and resistance to oxidative stress (and in some cases reduced oxidative damage to macromolecules), direct evidence showing that alterations in oxidative damage/stress play a role in aging are limited to a few studies with transgenic Drosophila that overexpress antioxidant enzymes 39. Invertebrate and vertebrate modes should be separated, as the data and results are quite disparate when estimating the role of antioxidants on longevity. According to de Grey 40, “a series of studies over many years has conclusively disproved the hypothesis that longevity in warm-blooded animals (homeotherms) correlates with high levels of antioxidants: in fact, these variables generally exhibit a strong negative cross-species correlation.” It is possible that the antioxidants do not reach the sites of free-radical generation, especially when mitochondria are the primary source of ROS.
Harmful effects of chronic oxidative stress include faster aging, DNA mutations, epigenetic changes that in worst cases cumulate with development and progression of many diseases including malignant alterations and cellular death 41. Many processes at the cellular level depend on the potency to regenerate and maintain the optimal reducing power of the cell, which is mirrored trough cellular redox state/balance 42. When cells have wakened their redox buffering potential or when it is being compromised by overproduction of ROS, cellular redox state shifts more towards oxidation and the cell is likely to be susceptible to oxidative damage. Non-enzymatic antioxidants are usually consumed and enzymatic induced under such circumstances, including utilization of major cellular antioxidant, glutathione (GSH) and nicotinamide adenine dinucleotide phosphate (NADPH) 43.
Working in tandem with endogenous antioxidants, cellular redox balance is also dependent on essential nutrients that are taken by food sources such as vitamins C and E, tannins, carotenes, dietary polyphenols and bioflavonoids, largely originating from vegetable food sources 44. Therefore, success of any organism to defend against harmful oxidative stress largely depends on combination of inherited genetic as well as on the environmental factors. Type of food we eat, smoking habits and alcohol consumption as well as physical exercise, chemical agents and radiation give bases towards occurrence of oxidative stress and the readiness of an organism to defends against it 45.
People who eat fruits and vegetables, which happen to be good sources of antioxidants and other phytochemicals, have a lower risk of heart disease and some neurological diseases 46 and there is evidence that some types of vegetables and fruits in general, protect against a number of cancers 47 as epidemiological studies revealed, without providing the answer whether any specific bioactive molecules within fruit and vegetable have a special contribution on lower incidence. However, 80% of American children and adolescents and 68% of adults do not eat five portions a day 48. Inadequate dietary intakes of vitamins and minerals are widespread, most likely due to excessive consumption of energy-rich, micronutrient-poor, and refined food 49. Even in countries that in the past had high consumption of fruits and vegetables, their consumption has been lowering. The impoverishment of the soil (resulting from the abnormal exploitation of the soil itself, acid rain, increasing desertification, pollution, etc.), the often uncontrolled use of pesticides, the processes of refinement of vegetables, and the processes of transformation, storage and even cooking of foods, can affect the antioxidant content of fruits and vegetables. Today, fruits and vegetables are depleted of some essential micronutrients and antioxidants because of the intensified type of production or because of postharvest processes, transport and storage 50.
Consumption of vegetables and plant-derived foods and beverages has positive effect on the prevention of age associated diseases like coronary heart disease and atherosclerosis as well as for longevity 51. Based on the analysis of available epidemiological data, a daily intake of at least 400 g of fruits and vegetables has been recommended by the World Health Organisation 52. Avoiding calorie-dense refined sugars, saturated fats, and processed foods and replacing them with nutrient-dense but calorie-poor vegetables, fruits, and legumes will result in a vastly increased intake of health-enhancing phytonutrients, including key vitamins and minerals, antioxidants, flavonoids, and other still undiscovered compounds important for our cells to work properly. While there is no formal recommendation for the amount of antioxidants we need 53, the best way to obtain antioxidants is from a varied diet. Foods rich in antioxidants include all fresh and seasonal fruit and vegetables (peppers, apples, onions, pineapple, dark leafy vegetables, flaxseeds, walnuts, pumpkin seeds, and olives), olive oil, and fish. Also omega-3 fatty acids EPA and DHA appear to stabilize mitochondrial membranes 54 as well as magnesium which is required for the oxidation-reduction steps in mitochondria since magnesium deficiency enhances free radical formation 55. Membrane lipids increase their sensitivity to oxidative damage as a function of their unsaturation. Pamplona et al. 56 analyzed fatty acids of liver mitochondria from eight mammals and showed that the total number of double bonds and the peroxidizability index is negatively correlated with maximum life span.
The key to the future success of decreasing oxidative-stress-induced damage should be the suppression of oxidative damage without disruption of the well-integrated antioxidant defense network. The approach to neutralize free radicals with antioxidants should be changed into prevention of free radical formation in the first place. Reducing the free radical formation should be more efficient than trying to neutralize free radicals after they have been produced. Recent evidence emerging from different studies discussed below shows that such possibilities exist.
Oxidative stress causes
High levels of reactive oxygen species (ROS), compared to antioxidant defenses, are considered to play a major role in diverse chronic age-related diseases and aging. It is believed that the oxidative damage is the major senescence process, and the mitochondria are the major source of endogenous reactive oxygen species formation while UV (ultraviolet) radiation, air pollution, and cigarette smoking are the main exogenous sources. The cells of all present aerobic organisms produce the majority of chemical energy by consuming oxygen in their mitochondria. Mitochondria are thus the main site of intracellular oxygen consumption and the main source of ROS formation 57. Mitochondrial ROS sources are represented by the electron transport chain and the nitric oxide synthase reaction. Nonmitochondrial sources of ROS include environmental pollutants, pollutants in food, radiation, or they are the by-products of other metabolic processes in organisms. Majority of free radicals are generated inside the cell rather than coming from the environment 58. Sources of O2−, the one electron reduction product of molecular oxygen, include NAD(P)H oxidases (NOX), xanthine oxidase (XO), mitochondria, and uncoupled nitric oxide synthase (NOS). Additionally, peroxynitrite and nitroxidative stress have also been implicated in various aspects of nitrooxidative cellular damage since peroxynitrite also yields secondary one-electron oxidants 59. Cellular and tissue defenses against ROS include the enzymes superoxide dismutase (Mn-SOD, Cu/Zn-SOD, and extracellular (EC)-SOD), catalase, glutathione peroxidase, peroxiredoxins and the nonenzymatic antioxidants, glutathione (GSH), thioredoxin, ascorbate, α-tocopherol, and uric acid 60. In reality, the oxidative damage potential is greater than antioxidant defence and thus there is a constant small amount of toxic-free radical formation, which escapes the defense of the cell.
Nutritional (or dietary) oxidative stress describes an imbalance between the prooxidant load and the antioxidant defense as a consequence of excess oxidative load or of an inadequate supply of the organism with nutrients 61. In Western societies, a significant part of the day is spent in the postprandial state. Postprandial (postprandial means after eating a meal, while preprandial is before a meal) oxidative stress is characterized by an increased susceptibility of the organism toward oxidative damage after consumption of a meal rich in lipids and/or carbohydrates 62. The generation of excess superoxide due to abundant energy substrate after the meal may be a predominate factor resulting in oxidative stress and a decrease in nitric oxide, which is important to endothelial function. The “three meals a day plus snacks” diet is typical of many modern industrialized countries. In contrast to the continuous availability of food that we currently enjoy, our ancestors were often forced to endure extended periods from many hours to days without food 63. When overeating, high amounts of energy become available and mitochondria do not operate very efficiently and generate more superoxide. The electron transport always proceeds as fast as energy is removed from the gradient. Electron transport is driven by the free energy that is available from the energy carriers, in turn obtained from substrates such as glutamate or Krebs intermediates. It is restricted by the chemiosmotic gradient. Anything that increases the turnover of energy from the gradient increases the rate of electron transport proportionally. ROS production thus plummets when ATP demand is high. Decreased proton concentration or membrane potential brings about a nonlinear and a striking decrease in the level of inadvertent electron leaks, and this likely accounts for ROS decrease 64.
The postprandial increase in plasma lipid hydroperoxide content and oxidative stress is restricted when a meal containing oxidized and oxidizable lipids is consumed together with red wine or with procyanidins 65. Procyanidins can be found in many plants; important sources are red wine, in particular; grape seeds and grape skin, cocoa, pine bark, and green tea.
A mixture of antioxidant compounds are required to provide protection from the oxidative effects of postprandial fats and sugars. No specific antioxidant can be claimed to be the most important because human food consumption varies enormously. However, a variety of polyphenolic compounds derived from plants appear to be effective dietary antioxidants, especially when consumed with high-fat meals 61.
Besides, glucose in the blood increases after consuming a meal, and for this reason, work instead of resting should be performed in order to maintain an appropriate electron flow and avoid electron leaks. The regulation of energy by the body’s circadian rhythms may play a significant role in controlling oxidative stress. In modern society, we are faced with excessive psychological stress, as well as an epidemic of overeating, and the two together appear to have synergistic effects according to Epel 66.
Psychological stress is becoming common in modern society and is known to predispose individuals to several diseases, since stress can diminish the effectiveness of the immune system 67 and possibly also the effectiveness of the antioxidant system and repair processes. It is known that adrenalin is released into the blood when a person faces a stressful situation. If adrenalin is not used for either fight or flight (as it was in the past), it oxidizes in the presence of O2 or promotes redox cycling to form superoxide 67 and increases oxidative stress. Emotional stress in humans associates with higher biomarkers for oxidative stress. Emotional stress increases catecholamine metabolism, which increases oxidative stress by increasing the production of free radicals. Similarly, academic stress (stress due to academic commitments, financial pressures, and lack of time management skills among students) 68 or sleep deprivation 69 associate with lower protective antioxidant levels in blood. Eskiocak et al. 68 demonstrated that glutathione and free sulphydryl levels in seminal plasma decreased in subjects undergoing examination stress. Everson et al. 69 found out that recovery sleep normalized antioxidant content in liver and enhanced enzymatic antioxidant activities in both the liver and the heart. Results of their study link uncompensated oxidative stress to health effects induced by sleep deprivation and provide evidence that restoration of antioxidant balance is a property of recovery sleep. Excessive psychophysical stress should be avoided, and we should take time to relax and enjoy the things we like doing.
In all organisms, melatonin is produced primarily during the daily period of darkness, with only small amounts being synthesized during the day. For this reason, people should sleep long enough and in complete darkness in order to produce necessary amounts of the endogenous powerful antioxidant melatonin 70 and benefit from its particular role in the protection of nuclear and mitochondrial DNA 71. Melatonin is an antioxidant that can easily cross cell membranes and the blood-brain barrier 70. Melatonin is a direct scavenger of OH, O2 −, and NO 72. Unlike other antioxidants, melatonin does not undergo redox cycling, the ability of a molecule to undergo reduction and oxidation repeatedly.
Increased oxidative stress with age may be in part due to a decline in the levels of the endogenous cellular antioxidants, among them also coenzyme Q10 declines significantly with age 73. Coenzyme Q10 is a naturally occurring antioxidant and a prominent component of mitochondrial electron transport chain. The study of Prahl et al. 74 shows significant age-dependent differences in mitochondrial function of keratinocytes isolated from skin biopsies of young and old donors. From the data, researchers postulate that energy metabolism shifts to a predominantly nonmitochondrial pathway and is therefore functionally anaerobic with advancing age. Coenzyme Q is recognized as an obligatory co-factor for the function of uncoupling proteins and a modulator of the transition pore. Furthermore, data reveal that CoQ10 affects expression of genes involved in human cell signaling, metabolism, and transport and some of the effects of exogenously administered CoQ10 may be due to this property 75. It was observed also the influence of CoQ10 on endogenous antioxidant defense by increasing SOD2 and GPx 76.
Oxidative stress symptoms
In order to prevent oxidative damage, all aerobic organisms have developed highly efficient antioxidant strategies during evolution. However, these highly reactive species may be generated accidentally as a result of altered metabolism (e.g. during mitochondrial respiration) or can be formed deliberately (e.g. by professional ROS-producing enzymes at sites of inflammation). Hence, it is not surprising that most metabolic diseases, as well as those pathologies associated with low-grade inflammation, display increased patterns of biomarkers of oxidative stress 77. It is well established that cardiovascular 78, neurodegenerative 79, metabolic 80 and inflammatory diseases 81 are associated with increased oxidative stress [some rare immune diseases are linked to insufficient ROS formation 82].
Also the currently available and consumed antioxidants (e.g., vitamin C, E, and beta-carotene) show inefficacy in ameliorating oxidative stress-mediated diseases. The use of multivitamins/minerals has grown rapidly over the past several decades, and dietary supplements are now used by more than half of the adult population in the United States. On the other hand, some recent studies showed that antioxidant therapy has no effect and can even increase mortality 83, 84, 85, 85. Many clinical trials in which individuals received one or more synthetic antioxidants failed to obtain beneficial results. Results of clinical trials on exogenous antioxidants intake are thus conflicting and contradictory. There are evidently homeostatic mechanisms in cells that govern the amount of allowable antioxidant activity. This indicates that other substances in fruits and vegetables, or a complex mix of substances (e.g., inhibitors of P450 (grapefruit, garlic), inhibitors of cell proliferation (resveratrol, green tea polyphenols, and curcumin), antagonists of estrogen (flavonoids), inhibitors of metastases (flavonoids), and inhibitors of angiogenesis (genistein, epigallocatechin galate), may contribute to the better cardiovascular health and decreased cancer incidence observed in individuals who consume more of fruits and vegetables 86. There is also the problem regarding the dosing of synthetic antioxidants; for example there are claims that RDA levels of vitamins C and E are too low to prevent oxidative stress. Besides, a typical oxidative stress status of an individual is not established yet and is difficult to measure 53. The intake of only one antioxidant could alter the complex system of endogenous antioxidative defense of cells, or alter the necrosis or apoptosis pathways. Changing the level of one antioxidant causes a compensatory change in others, while the overall antioxidant capacity remains unaffected. Dosing cells with exogenous antioxidants might decrease the rate of synthesis or the uptake of endogenous antioxidants, so that the total “cell antioxidant potential” remains unaltered. Cutler 87 described “the oxidative stress compensation model” which explains why dietary supplements of antioxidants have minimum effect on longevity. Cutler explains that most humans are able to maintain their set point of oxidative stress and so no matter how much additional antioxidant supplements they consumed in their diet, further decrease in oxidative stress does not occur. Antioxidant supplements thus do not appear in significantly decrease oxidative stress or/and increase life expectancy in humans 88. For example, several scavengers and neutralizers of peroxynitrite exist, such as peroxiredoxins, metalloporphyrins, simple thiol-based antioxidants (such as mercaptoalkylguanidines, N-acetylcysteine, and dihydrolipoic acid), ebselen, certain selenium-containing proteins (selenoprotein P, glutathione peroxidase), tempol, cabergoline, acetaminophen as well as drugs with demonstrated in vitro peroxynitrite-scavenging effects (hydralazine, pindolol, zileuton, penicillamine, simvastatin, edaravone, propofol, deprenyl, rasagiline, and 1,4-dihydropyridine calcium antagonists), and numerous molecules that are present in natural or dietary products (carotenoids, polyphenol oligomers, and epicatechin), but their in vivo and so their therapeutic potential as peroxynitrite neutralizing agent is low 89.
In contrast, a large number of small to intermediate size cohort studies with short-term and parenteral vitamin C therapy showed highly beneficial effects in various cardiovascular disease settings, and, for vitamin D, positive reports on oral therapy exist [9], [19], [28]. The most likely explanation for the antioxidant /oxidative stress paradox may be that reactive oxygen and nitrogen species (RONS) are not only injurious (oxidative stress) but also modulate important biological functions (redox signaling). Accordingly, chronic, systemic oral therapy with antioxidants will likely interfere with important ROS-mediated cellular processes, such as stress adaptation by ischemic preconditioning [19], [21], [28]. Additional reasons for the failure of large trials on oral antioxidant therapy might comprise the pro-oxidant effects of vitamin C and E radicals, reaction kinetics that are too slow, or lack of effective concentrations at sites of RONS formation. It is also possible that the patients included in these trials had already been exposed to drugs with pleiotropic antioxidant effects (e.g. ACE inhibitors or statins) (all reviewed in [19], [21], [28]). In addition, as shown by the EPIC Norfolk study, large antioxidant clinical trials suffer from suboptimal control of their potential efficacy, such as not measuring the patients’ plasma levels of antioxidants to verify compliance [29]. This study also showed that vitamin C concentrations in the blood inversely correlate with all-cause mortality in healthy volunteers. The inherent problems of vitamin C and E, that were mostly used in clinical trials, were also highlighted by Darley-Usmar and colleagues by a previous review [30]. Moreover, Forman, Davies and Ursini postulated that the nucleophilic tone and para-hormesis (paradoxic oxidative activation of intrinsic defence mechanisms such as the NRF2 pathway) is more important than free radical scavenging properties of antioxidants for their beneficial effects in vivo [31].
Oxidative stress and inflammation
There are complex relations between oxidative stress and many pathophysiological conditions that might have beneficial consequences, as is inflammatory response to pathogens, but could also be harmful, initiating or enhancing degenerative, metabolic and auto-immune diseases or cancer development 90. Hence, pathophysiology of oxidative stress should not be viewed as a hit or miss kind of event, but more as a cumulatively progressive process that gives fertile ground for development and progression of different, mostly stress and age associated diseases 91. In a causally-consequential scheme of pathophysiology oxidative stress can be viewed as a cause and as a consequence of various diseases, giving significance to the idea of personalized and integrative medicine 92. That is in a large part dependent on the assumption that harmful long-term effects of oxidative stress can still be prevented, treated and even reversed if monitored well and in time 93.
One of the goals of the preventive medicine approach should be a definition of the health state of an organism trough a personalized medicine approach and systematic monitoring of oxidative stress occurrence from young age throughout one’s lifespan. The challenges of such an oxidative stress research lies in finding reliable and specific biomarkers of oxidative stress in respect to specific pathophysiological conditions and diseases 94. Direct biomarkers of oxidative stress, ROS, especially free radicals are short-lived, labile and, accordingly, hard to measure. Therefore, more stable, secondary products/metabolites of oxidative stress are mainly utilized in scientific research. They include oxidized forms of proteins, lipids and DNA as well as their degradation products 95. A complementary way of assessing onset of oxidative stress is by determination of antioxidants and their related metabolites 96. So far, biomarkers of oxidative stress were considered to be too variable and highly unspecific for utilization in medical diagnostics of a specific disease. In scientific and clinical research they were mostly utilized to assess involvement of oxidative stress in a certain type of disease, but were usually considered too much unspecific to give specific clues regarding diagnosis or therapy 97.
Antioxidants Do not Change the Evolution of Diseases Related to Oxidative Stress
20,536 patients with hypertension at high cardiovascular risk were followed for five years. Treatment with ascorbic acid, Vitamin E and β-carotene neither reduced blood pressure nor changed mortality or morbidity 98.
Rotterdam study. This involved 4,802 Dutch men and women, ages 55–95, with no history of myocardial infarction (heart attack). Follow up: After 4 years, no association was observed between Vitamin E intake and the risk of heart attack 99.
Scottish Heart Health Study. No protection was associated with Vitamin E. Vitamins C, E and β-carotene had no effect on all-cause mortality 99.
Heart Protection Study. This included 20,536 men and women with coronary heart disease or diabetes, ages 40–80. Follow up: After 5 years, the intake of Vitamin E (600 mg/day), Vitamin C (250 mg/day) and β-carotene (20 mg/day) had no effect 99.
Primary Prevention Project. This program involved 4,495 men and women, with an average age of 64 and at least one risk factor for coronary artery disease, who took a low dose of aspirin (100 mg/day) and Vitamin E (300 mg/day). The study was stopped early because other trials had demonstrated protection with aspirin. Vitamin E had no effect 99.
Heart Outcomes Prevention Evaluation Study (HOPES). This included 2545 women and 6999 men, ≥55, with coronary artery disease or diabetes and one risk factor for atherosclerosis. They received Vitamin E (400 IU/day) or a placebo, angiotensin-converting enzyme inhibitor or a placebo. A follow up after 4.5 years indicated that Vitamin E had no effect 100.
Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI). This included 11,324 Italian men and women who had survived a myocardial infarction (heart attack) within the 3 previous months. They received Omega-3 oils (1 g/day) and/or Vitamin E or no treatment. A follow up after 3–5 years showed that Vitamin E had no effect 101.
Rytter et al. 102 studied 40 patients with type 2 diabetes, who were randomly assigned to one of two groups, one treated with antioxidants extracted from fruits, berries and vegetables (tocopherols, carotenoids and ascorbate), and the other treated with placebo. The authors found that 12 weeks of treatment did not change the metabolic profile, neither oxidative nor inflammatory biomarkers, even though antioxidant systemic levels had increased. Recently, Suksomboon et al. 103 analyzed 9 trials including 418 type 2 diabetic patients who were treated with Vitamin E for 8 weeks. They found that Vitamin E did not improve glycemic control unless the patients began their treatment under bad metabolic control and with a Vitamin E deficiency.
Arain et al. 104 analyzed four clinical trials to evaluate the effects of Vitamin E on colorectal cancer. The trials included 94,069 patients (47,029 received Vitamin E) and had a follow up period of 4 years. The authors concluded that Vitamin E had no effect on colorectal cancer.
Harmful Effects of Antioxidants
We summarize here some of the published studies that show the harmful effects of antioxidant supplements in humans.
In a study made in Australia, 69 hypertensive patients with an ambulatory systolic pressure of >125 mmHg received treatment with Vitamin C (500 mg/day) and grape seed polyphenols (1000 mg/day) for 6 weeks. At the end of the treatment their systolic and diastolic pressures increased and there were no changes in their endothelium dependent vasodilation or oxidative stress biomarkers 105. HDL Atherosclerosis Treatment Study (HATS). 160 men (<70) and women (<63), with low HDL and triglycerides <400 mg/dL. Patients were assigned to one of 4 groups: Simvastatin + niacin with or without Vitamin E (800 IU/day), Vitamin C (1000 mg/day), β carotene (25 mg/day) and selenium (100 μg/day); Antioxidants or placebo. A follow up after 3 years showed that antioxidant treatment blunted HDL (good cholesterol) elevation produced by simvastatin + niacin 106.
The Prostate, Lung, Colorectal and Ovarian cancer Screening Trial (PLCO) was a prospective study of 25,400 postmenopausal U.S. women, ages 55–74, who were followed for 10 years. The results showed that the risk of breast cancer increased significantly (by 20%) in women who had a folic acid supplement (≥400 μg/day) whereas food folate intake was not associated with an increased risk 107. Beta carotene and Retinal Efficacy Trial (CARET). This involved 18,314 U.S. women and men on β carotene (30 mg/day) and Vitamin A (25,000 IU/day) or a placebo. The study had to be stopped two years prematurely because Vitamin-treatment was associated with a 28% greater incidence of lung cancer and 17% more deaths than placebo treatment 108. Alpha Tocopherol Beta Carotene Cancer Prevention Study (ATBC). This covered 29,133 male smokers, ages 50–69, taking α-Tocopherol (50 mg/day) and/or β-carotene (30 mg/day). There was an 8% greater mortality in those men taking the supplements than the men treated with the placebo 109. Bjelakovic et al. 110 published a meta-analysis of antioxidant supplements for the prevention of gastrointestinal cancer. They selected 7 high quality randomized trials (of the 14 trials examined), including 131,727 patients. They found that antioxidants significantly increased overall mortality and did not prevent gastrointestinal cancer. Myung et al. 111 analyzed 31 of 3,327 articles searched, including 22 trials with 161,045 patients (88,610 treated with antioxidants). The authors concluded that antioxidants had no preventive effects on cancer. However, when they evaluated a subgroup of four controlled trials they found that patients receiving antioxidants had a significant increase in bladder cancer.
Antioxidants Vitamin C (12.5 mg/Kg) and N-acetylcysteine (10 mg/Kg) significantly increased the oxidative stress produced by acute exercise in healthy subjects. The effect was attributed to the conversion of ascorbic acid into the ascorbyl radical by reactive species generated during exercise 112. Ristow et al. studied insulin sensitivity, transcriptional regulators of insulin sensitivity and gene expression of SODs, GPx and catalase in 19 trained and 20 untrained healthy young men who were treated with Vitamin E (400 IU/day) + Vitamin C (1000 mg/day) or placebo, and were trained for four weeks. They found that exercise increased insulin sensitivity as well as the gene expression of insulin sensitivity transcriptional regulators and antioxidant enzymes. The effects of exercise were blocked by antioxidant supplements 113. Recently, Peternelj and Combes 114 evaluated 23 studies that reported negative effects of antioxidant supplements on the beneficial effects produced by chronic exercise. They found that antioxidants interfere with vasodilation and increased insulin signaling produced by exercise.
Recently, the Iowa Women’s Health Study results were published 115. The study included 38,772 women, who were older than 60 in 1986. Vitamin and mineral supplement intake was self-reported at different periods. The authors concluded that dietary Vitamin and mineral supplements may be associated with increased mortality, with the effect being worse when the supplements were accompanied by an iron supplement.
Table 1 summarizes the results of the clinical trials described above. Some data command attention. The total number of study patients reporting beneficial antioxidant effects is 194,734 whereas those reporting no, or harmful effects is 554,083 (149,097 and 404,986, respectively). After comparing the trials, it can be concluded that the results probably depended on the population studied (age, gender, health conditions, ethnicity), antioxidants (type, combinations, supplements or dietary), follow-up period, and specific outcome. For instance, comparing the NHS study (beneficial effects) 116, with the Iowa Woman’s Health Study (IWHS, harmful effects) 115, it is seen that, even though the number of participants (all women) is higher in the NHS study (more than double the IWHS), there are significant differences: participants in the NHS study were younger, the follow up period was shorter in the NHS study, the focus of the NHS study was vitamin E whereas the IWHS examined the effect of supplement and dietary vitamin/mineral, outcome in the NHS study was only cardiac heart disease, whereas outcome in the IWHS was mortality.
Table 1. Clinical trials with antioxidants
| Studies reporting beneficial effects of antioxidants | ||||||
|---|---|---|---|---|---|---|
| Study | N | Age (years) | Follow up period | Antioxidant | Main Outcome | References |
| Nurses’ Health Study (NHS) | 87,245 | 34–59 | 8 years | Vitamin E | Significant reduction in CHD risk | 117 |
| Health Professional Follow up Study (HPFS) | 39,910 | 40–75 | 4 years | Vitamin E | Significant reduction in CHD risk | 118 |
| Established Populations for Epidemiological Studies of the Elderly | 11,178 | 67–105 | 6 years | Vitamin E with or without other Vitamins | Vitamin E associated with a significant reduction CHD risk | 119 |
| First National Health and Nutrition Examination Survey (NHANES I) | 11,348 | 25–74 | 10 years | Vitamin C | Inverse correlation between Vitamin C and all-cause CVD death in men; not women | 99 |
| Scottish Heart Health Study | 7869 | 40–59 | 10 years | Vitamins C, E and β-carotene | Significant reduction of CHD; only in men | 120 |
| Meta-analysis | 10,073 | 18–90 | NR | Vitamins C, E and B12, and β-carotene | Preventive effects on cervical neoplasms | 121 |
| Alpha-Tocopherol, Beta carotene Cancer Prevention Study (ATBC) | 27,111 | 50–69 | 16–19.4 years | Alphatocopherol, β-carotene and flavonoids | Alpha-tocopherol was associated with reduced risk of pancreatic and prostate cancer. Flavonoids were associated with decreased risk of pancreatic cancer | 122 |
| Heart Protection Study | 20,536 | 40–80 | 5 years | Vitamins C and E and β-carotene | No reductions in blood pressure, morbidity or mortality | 123 |
| Rotterdam Study | 4802 | 55–95 | 4 years | Vitamins C and E and β carotene | No effects of Vitamin E on the risk of myocardial infarction | 124 |
| Scottish Heart Health Study | 7869 | 40–59 | 10 years | Vitamins C and E and β-carotene | No effects on all-cause mortality | 99 |
| Primary Prevention Project | 4495 | 64 (average) | 3.6 years | Vitamin E and low-dose aspirin | Vitamin E had no beneficial effect. Trial terminated because other studies demonstrated the beneficial effect of aspirin on cardiovascular mortality | 99 |
| Heart Outcomes Prevention Evaluation Study (HOPE) | 9544 | >55 | 4.5 years | Vitamin E | No effect of Vitamin E | 100 |
| Gruppo Italiano per lo Studio della Supravvivenza nell’ Infarto Miocardico (GISSI) | 11,324 | 59.3 (average) | 3.5 years | Vitamin E and omega-3 oils | No effect of Vitamin E | 125 |
| Study on well controlled diabetic patients | 40 | 61.9 | 12 weeks | Extracts of fruits and vegetables | No effect of the extracts | 102 |
| Meta-analysis including studies performed in Type 2 diabetic patients | 418 | 20–80 | 8 weeks | Vitamin E | No effects on metabolic control | 103 |
| Meta-analysis | 94,069 | >49 | 7–10 years | Vitamin E | No effect on colorectal cancer | 104 |
| Randomized, double blind, placebo controlled study in hypertensive patients | 69 | 62 (average) | 6 weeks | Vitamin C and grape seed polyphenols | Increase of blood pressure and no effect on either endothelium dependent vasodilation or oxidative stress | 105 |
| Randomized study in patients with CAD | 169 | 52 (average) | 3 years | One statin, Vitamin C, vitamin E, β-carotene and selenium | Antioxidants blunted the effect of statins on HDL | 106 |
| Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial (PLCO) | 25,400 | 55–74 | 10 years | Folic acid | Folic acid supplementation significantly increased breast cancer | 107 |
| Beta Carotene and Retinal Efficacy Trial (CARET) | 18,314 | 58 (average) | Stopped after 2 years | Vitamin A and β-carotene | Antioxidant treatment was associated with an increased incidence of lung cancer and mortality | 108 |
| Alpha- Tocopherol Beta-Carotene Cancer Prevention Study (ATBC) | 29,133 | 50–69 | 8 years | α-tocopherol, β-carotene | Antioxidants increased the incidence and mortality of lung cancer | 109 |
| Meta-analysis | 131,727 | 55 (average) | 1–12 years | β-carotene, Vitamin A, Vitamin E, selenium | The antioxidant treatment did not prevent gastrointestinal cancer but significantly increased mortality | 110 |
| Meta-analysis | 161,045 | 58.4 (average) | 5.3–5.8 years | β-carotene, Vitamin C, Vitamin E, selenium | Increased risk of bladder cancer in 4 of 22 of the trials included in the analysis | 111 |
| Study in healthy men | 14 | 24.4 (average) | 7 days | Vitamin C and N-Acetylcysteine | Increase of oxidative stress produced by exercise | 112 |
| Study in healthy men | 39 | 25–35 | 4 weeks | Vitamin C and Vitamin E | Antioxidants blocked the increase of insulin sensitivity and expression produced by exercise | 113 |
| Meta-analysis | 338 * | NR | 1 day– 6 weeks | Allopurinol, Coenzyme Q, Vitamins (C, E, B6, B12), α-lipoic acid, β-carotene, lutein, Nacetylcysteine, Selenium and/or Zinc | Any of these effects (compared to groups treated with placebo): ↓ training induced improvement in physical performance, ↓ exercise-induced oxidative stress preconditioning, ↑ CK, ↑ inflammatory biomarkers, prevented beneficial effects on insulin sensitivity and expression | 114 |
| Iowa Woman’s Health Study | 38,772 | >60 | 14 years | Dietary vitamins and mineral supplements | May be associated with increased total mortality risk | 115 |
CAD = Coronary artery disease, CHD = Coronary heart disease, CK = creatine kinase, CVD = Cardiovascular disease, HDL = high density lipoproteins, NR = not reported.
Summary
Altogether, in almost all living organisms ROS cannot simply be considered as detrimental since they contribute to signaling, as well as to maintenance of integrity and homeostasis in different organisms ranging from bacteria to mammals. In the latter, disturbances of ROS homeostasis can contribute to the pathogenesis of several chronic diseases such as atherosclerosis, type II diabetes, and cancer via modulation of diverse processes such as inflammation, and the immune response. The common associated increase in ROS in these diseases points to the therapeutic use of small molecules with antioxidant function; however, so far little or no benefit could be observed from the large scale studies with antioxidant supplementation.
Moreover, a number of clinical trials have shown harmful effects. The most extreme results evidenced increased mortality in individuals taking antioxidant supplements. Although the mechanisms are not explained in those trials, the results suggest that disrupting the delicate balance between antioxidants and reactive species produces antioxidant-induced stress, if the antioxidants overwhelm the physiological production of reactive species.
Though these conclusions remain speculative, the best advice would be to ingest antioxidants from food sources – fruits and vegetables – rather than from self-prescribed supplements. The most probable way of increasing the endogenous antioxidant defense would be by practicing moderate aerobic exercise, as an every day healthy routine. Gene polymorphisms, which condition endogenous antioxidant deficiencies, would be the exception to such advice.
- Villanueva C, Kross RD. Antioxidant-Induced Stress. International Journal of Molecular Sciences. 2012;13(2):2091-2109. doi:10.3390/ijms13022091. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3292009/[↩][↩][↩][↩]
- H. Sies (Ed.), Oxidative Stress, Academic Press, London, 1985, pp. 1–507.[↩]
- Sies H., Jones D. Oxidative stress. In: Fink G., editor. 2nd ed. Vol. 3. Elsevier; Amsterdam: 2007. pp. 45–48. (Encyclopedia of Stress).[↩]
- Wenzel P., Kossmann S., Munzel T., Daiber A. Redox regulation of cardiovascular inflammation – Immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2017.[↩]
- Schmidt H.H., Stocker R., Vollbracht C., Paulsen G., Riley D., Daiber A., Cuadrado A. Antioxidants in translational medicine. Antioxid. Redox Signal. 2015;23:1130–1143.[↩]
- Ghezzi P., Jaquet V., Marcucci F., Schmidt H.H. The oxidative stress theory of disease: levels of evidence and epistemological aspects. Br. J. Pharmacol. 2016. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5446567/[↩]
- P. Mustain Antioxidant Supplements: Too Much of a Kinda Good Thing. https://blogs.scientificamerican.com/food-matters/antioxidant-supplements-too-much-of-a-kinda-good-thing/[↩]
- A. Riley Why vitamin pills don’t work, and may be bad for you. http://www.bbc.com/future/story/20161208-why-vitamin-supplements-could-kill-you[↩]
- Scudellari M. The science myths that will not die. Nature. 2015;528:322–325. https://www.nature.com/news/the-science-myths-that-will-not-die-1.19022[↩]
- An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. McCord JM, Keele BB Jr, Fridovich I. Proc Natl Acad Sci U S A. 1971 May; 68(5):1024-7.[↩]
- Augusto O., Miyamoto S. Oxygen radicals and related species. In: Pantopoulos K., Schipper H.M., editors. Principles of Free Radical Biomedicine. Nova Science Publishers, Inc; 2011.[↩]
- Laranjinha J. Oxidative Stress: From 1980’s to Recent Update. In: Soares R., Costa C., editors. Oxidative Stress, Inflammation and Angiogenesis in the Metabolic Syndrome. Springer, Science + business Media; New York, NY, USA: 2009. pp. 21–32.[↩]
- Some current insights into oxidative stress. Duracková Z. Physiol Res. 2010; 59(4):459-69.[↩]
- The wanderings of a free radical. Halliwell B. Free Radic Biol Med. 2009 Mar 1; 46(5):531-42. https://www.ncbi.nlm.nih.gov/pubmed/19111608/[↩]
- Narwaley M., Michail K., Arvadia P., Siraki A.G. Drug-induced protein free radical formation is attenuated by unsaturated fatty acids by scavenging drug-derived phenyl radical metabolites. Chem. Res. Toxicol. 2011;24:1031–1039.[↩][↩]
- Davies M.J., Fu S., Dean R.T. Protein hydroperoxides can give rise to reactive free radicals. Biochem. J. 1995;305:643–649.[↩]
- Rahmanto A.S., Morgan P.E., Hawkins C.L., Davies M.J. Cellular effects of peptide and protein hydroperoxides. Free Radic. Biol. Med. 2010;48:1071–1078.[↩]
- Yamada K., Yamamiya I., Utsumi H. In vivo detection of free radicals induced by diethylnitrosamine in rat liver tissue. Free Radic. Biol. Med. 2006;40:2040–2046.[↩]
- North J.A., Spector A.A., Buettner G.R. Detection of lipid radicals by electron paramagnetic resonance spin trapping using intact cells enriched with polyunsaturated fatty acid. J. Biol. Chem. 1992;267:5743–5746.[↩]
- Leonarduzzi G., Gamba P., Gargiulo S., Biasi F., Poli G. Inflammation-related gene expression by lipid oxidation-derived products in the progression of atherosclerosis. Free Radic. Biol. Med. 2011;52:19–34.[↩]
- Halliwell B., Gutteridge J.M. The definition and measurement of antioxidants in biological systems. Free Radic. Biol. Med. 1995;18:125–126. https://www.ncbi.nlm.nih.gov/pubmed/7896166[↩]
- Dündar Y., Aslan R. Antioxidative stress. Eastern J. Med. 2000;5:45–47.[↩]
- Egea J, Fabregat I, Frapart YM, et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biology. 2017;13:94-162. doi:10.1016/j.redox.2017.05.007. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5458069[↩]
- Schieber M., Chandel N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014;24:R453–R462.[↩]
- Birben E., Sahiner U.M., Sackesen C., Erzurum S., Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012;5:9–19.[↩]
- Halliwell B. Biochemistry of oxidative stress: figure 1. Biochem. Soc. Trans. 2007;35:1147–1150.[↩]
- Antoncic-Svetina M., Sentija D., Cipak A., Milicic D., Meinitzer A., Tatzber F. Ergometry induces systemic oxidative stress in healthy human subjects. Tohoku J. Exp. Med. 2010;221:43–48.[↩]
- Reuter S., Gupta S.C., Chaturvedi M.M., Aggarwal B.B. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic. Biol. Med. 2010;49:1603–1616.[↩]
- Pechanova O., Simko F. Chronic antioxidant therapy fails to ameliorate hypertension: Potential mechanisms behind. J. Hypertens Suppl. 2009;27:S32–S36.[↩]
- Nemoto M., Nishimura R., Sasaki T., Hiki Y., Miyashita Y., Nishioka M., Fujimoto K., Sakuma T., Ohashi T., Fukuda K., et al. Genetic association of glutathione peroxidase-1 with coronary artery calcification in type 2 diabetes: A case control study with multi-slice computed tomography. Cardiovasc. Diabetol. 2007;6:23:1–23:7.[↩]
- Naganuma T., Nakayama T., Sato N., Fu Z., Soma M., Aoi N., Hinohara S., Doba N., Usami R. Association of extracellular superoxide dismutase gene with cerebral infarction in women: A haplotype-based case-control study. Hereditas. 2008;145:283–292.[↩]
- Samoila O.C., Carter A.M., Futers S.T., Otiman G., Anghel A., Tamas L., Seclaman E. Polymorphic variants of extracellular superoxide dismutase gene in a romanian population with atheroma. Biochem. Genet. 2008;46:634–643.[↩]
- Manfredi S., Federici C., Picano E., Botto N., Rizza A., Andreassi M.G. Gstm1, gstt1 and cyp1a1 detoxification gene polymorphisms and susceptibility to smoking-related coronary artery disease: A case-only study. Mutat. Res. 2007;621:106–112.[↩]
- Casteilla L, Rigoulet M, Pénicaud L. Mitochondrial ROS metabolism: modulation by uncoupling proteins. IUBMB Life. 2001;52(3–5):181–188.[↩]
- Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. Journal of Bioenergetics and Biomembranes. 1997;29(1):89–95.[↩]
- Staniek K, Nohl H. H2O2 detection from intact mitochondria as a measure for one-electron reduction of dioxygen requires a non-invasive assay system. Biochimica et Biophysica Acta. 1999;1413(2):70–80.[↩]
- Kaul N, Forman HJ. Reactive oxygen species in physiology and toxicology: from lipid peroxidation to transcriptional activation. In: Rhodes CR, editor. Toxicology of the Human Environment. New York, NY, USA: Taylor and Francis; 2000. pp. 310–335.[↩]
- Perez-Campo R, López-Torres M, Cadenas S, Rojas C, Barja G. The rate of free radical production as a determinant of the rate of aging: evidence from the comparative approach. Journal of Comparative Physiology. 1998;168(3):149–158.[↩]
- Pérez VI, Bokov A, Remmen HV, et al. Is the oxidative stress theory of aging dead? Biochimica et Biophysica Acta. 2009;1790(10):1005–1014.[↩]
- De Grey ADNJ. Noncorrelation between maximum life span and antioxidant enzyme levels among homeotherms: implications for retarding human aging. Journal of Anti-Aging Medicine. 2000;3(1):25–36.[↩]
- Di Meo S., Reed T.T., Venditti P., Victor V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxid. Med. Cell. Longev. 2016;2016:1245049.[↩]
- Trachootham D., Lu W., Ogasawara M. a., Nilsa R.-D.V., Huang P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008;10:1343–1374.[↩]
- Willcox J.K., Ash S.L., Catignani G.L. Antioxidants and prevention of chronic disease. Crit. Rev. Food Sci. Nutr. 2004;44:275–295.[↩]
- Lobo V., Patil A., Phatak A., Chandra N. Free radicals, antioxidants and functional foods: impact on human health. Pharmacogn. Rev. 2010;4:118–126.[↩]
- Kovacic P., Jacintho J.D. Mechanisms of carcinogenesis: focus on oxidative stress and electron transfer. Curr. Med. Chem. 2001;8:773–796.[↩]
- Stanner SA, Hughes J, Kelly CNM, Buttriss J. A review of the epidemiological evidence for the ’antioxidant hypothesis’ Public Health Nutrition. 2004;7(3):407–422.[↩]
- World Cancer Research Found. http://www.wcrf.org/ [↩]
- Ames BN. DNA damage from micronutrient deficiencies is likely to be a major cause of cancer. Mutation Research. 2001;475(1-2):7–20.[↩]
- Ames BN. Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrient by triage. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(47):17589–17594.[↩]
- López-Torres M, Barja G. Lowered methionine ingestion as responsible for the decrease in rodent mitochondrial oxidative stress in protein and dietary restriction. Possible implications for humans. Biochimica et Biophysica Acta. 2008;1780(11):1337–1347.[↩]
- Sahyoun NR, Jacques PF, Russell RM. Carotenoids, vitamins C and E, and mortality in an elderly population. American Journal of Epidemiology. 1996;144(5):501–511.[↩]
- World Health Organization. http://www.who.int/en/[↩]
- Argüelles S, Gómez A, Machado A, Ayala A. A preliminary analysis of within-subject variation in human serum oxidative stress parameters as a function of time. Rejuvenation Research. 2007;10(4):621–636.[↩][↩]
- Pehowich DJ. Thyroid hormone status and membrane n-3 fatty acid content influence mitochondrial proton leak. Biochimica et Biophysica Acta. 1999;1411(1):192–200.[↩]
- Bada V, Kucharská J, Gvozdjáková A, Herichová I, Gvozdják J. The cytoprotective effect of magnesium in global myocardial ischemia. Bratislavské Lekárske Listy. 1996;97(10):587–595.[↩]
- Pamplona R, Portero-Otín M, Riba D, et al. Low fatty acid unsaturation: a mechanism for lowered lipoperoxidative modification of tissue proteins in mammalian species with long life spans. Journals of Gerontology. 2000;55(6):B286–B291.[↩]
- Ames BN. Delaying the mitochondrial decay of aging. Annals of the New York Academy of Sciences. 2004;1019:406–411.[↩]
- Sastre J, Pallardó FV, García De La Asunción J, Viña J. Mitochondria, oxidative stress and aging. Free Radical Research. 2000;32(3):189–198.[↩]
- Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A. Unraveling peroxynitrite formation in biological systems. Free Radical Biology and Medicine. 2001;30(5):463–488.[↩]
- Pryor WA. Vitamin E and heart disease: basic science to clinical intervention trials. Free Radical Biology and Medicine. 2000;28(1):141–164.[↩]
- Sies H, Stahl W, Sevanian A. Nutritional, dietary and postprandial oxidative stress. Journal of Nutrition. 2005;135(5):969–972.[↩][↩]
- Ursini F, Sevanian A. Postprandial oxidative stress. Biological Chemistry. 2002;383(3-4):599–605.[↩]
- Cutler RG, Mattson MP. Measuring oxidative stress and interpreting its relevance in humans. In: Cutler RG, Rodriguez, editors. Oxidative Stress and Aging. World Scientific; 2003.[↩]
- Arking R. The Biology of Aging, Observations and Principles. 3rd edition. New York, NY, USA: Oxford University Press; 2006.[↩]
- Natella F, Belelli F, Gentili V, Ursini F, Scaccini C. Grape seed proanthocyanidins prevent plasma postprandial oxidative stress in humans. Journal of Agricultural and Food Chemistry. 2002;50(26):7720–7725.[↩]
- Epel ES. Psychological and metabolic stress: a recipe for accelerated cellular aging? Hormones. 2009;8(1):7–22.[↩]
- Halliwell B, Gutteridge J. Free Radicals in Biology and Medicine. 3rd edition. Oxford, UK: Clarendon Press; 1999.[↩][↩]
- Eskiocak S, Gozen AS, Yapar SB, Tavas F, Kilic AS, Eskiocak M. Glutathione and free sulphydryl content of seminal plasma in healthy medical students during and after exam stress. Human Reproduction. 2005;20(9):2595–2600.[↩][↩]
- Everson CA, Laatsch CD, Hogg N. Antioxidant defense responses to sleep loss and sleep recovery. American Journal of Physiology. 2005;288(2):R374–R383.[↩][↩]
- Hardeland R. Antioxidative protection by melatonin: multiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine. 2005;27(2):119–130.[↩][↩]
- Reiter RJ, Tan DX, Manchester LC, Qi W. Biochemical reactivity of melatonin with reactive oxygen and nitrogen species: a review of the evidence. Cell Biochemistry and Biophysics. 2001;34(2):237–256.[↩]
- Poeggeler B, Saarela S, Reiter RJ, et al. Melatonin—a highly potent endogenous radical scavenger and electron donor: new aspects of the oxidation chemistry of this indole accessed in vitro. Annals of the New York Academy of Sciences. 1994;738:419–420.[↩]
- Kalen A, Appelkvist EL, Dallner G. Age-related changes in the lipid compositions of rat and human tissues. Lipids. 1989;24(7):579–584.[↩]
- Prahl S, Kueper T, Biernoth T, et al. Aging skin is functionally anaerobic: importance of coenzyme Q10 for anti aging skin care. BioFactors. 2008;32(1–4):245–255.[↩]
- Ernster L, Forsmark-Andree P. Ubiquinol: an endogenous antioxidant in aerobic organisms. Clinical Investigator. 1993;71(supplement 8):S60–S65.[↩]
- Kim DW, Hwang IK, Kim DW, et al. Coenzyme Q10 effects on manganese superoxide dismutase and glutathione peroxidase in the hairless mouse skin induced by ultraviolet B irradiation. BioFactors. 2007;30(3):139–147.[↩]
- Frijhoff J., Winyard P.G., Zarkovic N., Davies S.S., Stocker R., Cheng D., Knight A.R., Taylor E.L., Oettrich J., Ruskovska T., Gasparovic A.C., Cuadrado A., Weber D., Poulsen H.E., Grune T., Schmidt H.H., Ghezzi P. Clinical relevance of biomarkers of oxidative stress. Antioxid. Redox Signal. 2015;23:1144–1170.[↩]
- Daiber A., Steven S., Weber A., Shuvaev V.V., Muzykantov V.R., Laher I., Li H., Lamas S., Munzel T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2016.[↩]
- Ischiropoulos H., Beckman J.S. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J. Clin. Investig. 2003;111:163–169.[↩]
- Ceriello A. Oxidative stress and diabetes-associated complications. Endocr. Pract. 2006;12(Suppl 1):S60–S62.[↩]
- Karbach S., Wenzel P., Waisman A., Munzel T., Daiber A. eNOS uncoupling in cardiovascular diseases–the role of oxidative stress and inflammation. Curr. Pharm. Des. 2014;20:3579–3594.[↩]
- Aviello G., Knaus U.G. ROS in gastrointestinal inflammation: rescue or Sabotage? Br. J. Pharmacol. 2016.[↩]
- Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. New England Journal of Medicine. 1996;334(18):1150–1155.[↩]
- Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Annals of Internal Medicine. 2005;142(1):37–46.[↩]
- Mursu J, Robien K, Harnack LJ, Park K, Jacobs DR., Jr. Dietary supplements and mortality rate in older women: the iowa women’s health study. Archives of Internal Medicine. 2011;171(18):1625–1633.[↩][↩]
- Lotito SB, Frei B. Consumption of flavonoid-rich foods and increased plasma antioxidant capacity in humans: cause, consequence, or epiphenomenon? Free Radical Biology and Medicine. 2006;41(12):1727–1746.[↩]
- Cutler RG. Genetic stability, dysdifferentiation, and longevity determinant genes. In: Cutler RG, Rodriguez H, editors. Critical Reviews of Oxidative Stress and Damage. River Edge, NJ, USA: World Scientific; 2003.[↩]
- Green LJ. The Dermatologist’s Guide to Looking Younger. Freedom, Calif, USA: Crossing Press; 1999.[↩]
- Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nature Reviews Drug Discovery. 2007;6(8):662–680.[↩]
- Sohal R.S., Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63.[↩]
- A.D. Romano, G. Serviddio, A. de Matthaeis, F. Bellanti, G. Vendemiale, Oxidative stress and aging., J. Nephrol. 23 Suppl 15 (n.d.)S29–S36.[↩]
- Jaganjac M., Čačev T., Čipak A., Kapitanović S., Gall Trošelj K., Zarković N. Even stressed cells are individuals: second messengers of free radicals in pathophysiology of cancer. Croat. Med. J. 2012;53:304–309.[↩]
- Cipak Gasparovic A., Zarkovic N., Zarkovic K., Semen K., Kaminskyy D., Yelisyeyeva O. Biomarkers of oxidative and nitro-oxidative stress: conventional and novel approaches. Br. J. Pharmacol. 2017;174:1771–1783.[↩]
- Egea J., Fabregat I., Frapart Y.M., Ghezzi P., Görlach A., Kietzmann T. European contribution to the study of ROS: a summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS) Redox Biol. 2017;13:94–162.[↩]
- Toyokuni S. The origin and future of oxidative stress pathology: from the recognition of carcinogenesis as an iron addiction with ferroptosis-resistance to non-thermal plasma therapy. Pathol. Int. 2016;66:245–259.[↩]
- Somogyi A., Rosta K., Pusztai P., Tulassay Z., Nagy G. Antioxidant measurements. Physiol. Meas. 2007;28:R41–R55.[↩]
- Frijhoff J., Winyard P.G., Zarkovic N., Davies S.S., Stocker R., Cheng D. Clinical relevance of biomarkers of oxidative stress. 2015;23.[↩]
- Pechanova O., Simko F. Chronic antioxidant therapy fails to ameliorate hypertension: Potential mechanisms behind. J. Hypertens Suppl. 2009;27:S32–S36. https://www.ncbi.nlm.nih.gov/pubmed/19633449[↩]
- Hasnain B.I., Mooradian A.D. Recent trials of antioxidant therapy: What should we be telling our patients? Cleve. Clin. J. Med. 2004;71:327–334. https://www.ncbi.nlm.nih.gov/pubmed/15117174[↩][↩][↩][↩][↩][↩][↩]
- Yusuf S., Dagenais G., Pogue J., Bosch J., Sleight P. Vitamin e supplementation and cardiovascular events in high-risk patients. The heart outcomes prevention evaluation study investigators. N. Engl. J. Med. 2000;342:154–160. http://www.nejm.org/doi/full/10.1056/NEJM200001203420302[↩][↩]
- GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin e after myocardial infarction: Results of the gissi-prevenzione trial. Gruppo italiano per lo studio della sopravvivenza nell’infarto miocardico. Lancet. 1999;354:447–455. https://www.ncbi.nlm.nih.gov/pubmed/10465168[↩]
- Rytter E., Vessby B., Asgard R., Ersson C., Moussavian S., Sjodin A., Abramsson-Zetterberg L., Moller L., Basu S. Supplementation with a combination of antioxidants does not affect glycaemic control, oxidative stress or inflammation in type 2 diabetes subjects. Free Radic. Res. 2010;44:1445–1453. https://www.ncbi.nlm.nih.gov/pubmed/20942575[↩][↩]
- Suksomboon N., Poolsup N., Sinprasert S. Effects of vitamin e supplementation on glycaemic control in type 2 diabetes: Systematic review of randomized controlled trials. J. Clin. Pharm. Ther. 2011;36:53–63. https://www.ncbi.nlm.nih.gov/pubmed/21198720[↩][↩]
- Arain M.A., Abdul Qadeer A. Systematic review on “Vitamin e and prevention of colorectal cancer” Pak. J. Pharm. Sci. 2010;23:125–130. https://www.ncbi.nlm.nih.gov/pubmed/20363687[↩][↩]
- Ward N.C., Hodgson J.M., Croft K.D., Burke V., Beilin L.J., Puddey I.B. The combination of vitamin c and grape-seed polyphenols increases blood pressure: A randomized, double-blind, placebo-controlled trial. J. Hypertens. 2005;23:427–434. https://www.ncbi.nlm.nih.gov/pubmed/15662232[↩][↩]
- Cheung M.C., Zhao X.Q., Chait A., Albers J.J., Brown B.G. Antioxidant supplements block the response of hdl to simvastatin-niacin therapy in patients with coronary artery disease and low hdl. Arterioscler. Thromb. Vasc. Biol. 2001;21:1320–1326. http://atvb.ahajournals.org/content/21/8/1320.long[↩][↩]
- Kim Y.I. Does a high folate intake increase the risk of breast cancer? Nutr. Rev. 2006;64:468–475. https://www.ncbi.nlm.nih.gov/pubmed/17063929[↩][↩]
- Omenn G.S., Goodman G.E., Thornquist M.D., Balmes J., Cullen M.R., Glass A., Keogh J.P., Meyskens F.L., Jr, Valanis B., Williams J.H., Jr, et al. Risk factors for lung cancer and for intervention effects in caret, the beta-carotene and retinol efficacy trial. J. Natl. Cancer Inst. 1996;88:1550–1559. https://www.ncbi.nlm.nih.gov/pubmed/8901853[↩][↩]
- Albanes D., Heinonen O.P., Taylor P.R., Virtamo J., Edwards B.K., Rautalahti M., Hartman A.M., Palmgren J., Freedman L.S., Haapakoski J., et al. Alpha-tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: Effects of base-line characteristics and study compliance. J. Natl. Cancer Inst. 1996;88:1560–1570. https://www.ncbi.nlm.nih.gov/pubmed/8901854[↩][↩]
- Bjelakovic G., Nikolova D., Simonetti R.G., Gluud C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet. 2004;364:1219–1228. https://www.ncbi.nlm.nih.gov/pubmed/15464182[↩][↩]
- Myung S.K., Kim Y., Ju W., Choi H.J., Bae W.K. Effects of antioxidant supplements on cancer prevention: Meta-analysis of randomized controlled trials. Ann. Oncol. 2010;21:166–179. https://www.ncbi.nlm.nih.gov/pubmed/19622597[↩][↩]
- Childs A., Jacobs C., Kaminski T., Halliwell B., Leeuwenburgh C. Supplementation with vitamin c and n-acetyl-cysteine increases oxidative stress in humans after an acute muscle injury induced by eccentric exercise. Free Radic. Biol. Med. 2001;31:745–753. https://www.ncbi.nlm.nih.gov/pubmed/11557312[↩][↩]
- Ristow M., Zarse K., Oberbach A., Kloting N., Birringer M., Kiehntopf M., Stumvoll M., Kahn C.R., Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA. 2009;106:8665–8670. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2680430/[↩][↩]
- Peternelj T.T., Coombes J.S. Antioxidant supplementation during exercise training: Beneficial or detrimental? Sports Med. 2011;41:1043–1069. https://www.ncbi.nlm.nih.gov/pubmed/22060178[↩][↩]
- Mursu J., Robien K., Harnack L.J., Park K., Jacobs D.R., Jr Dietary supplements and mortality rate in older women: The iowa women’s health study. Arch. Intern. Med. 2011;171:1625–1633. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4114071/[↩][↩][↩]
- Stampfer M.J., Hennekens C.H., Manson J.E., Colditz G.A., Rosner B., Willett W.C. Vitamin e consumption and the risk of coronary disease in women. N. Engl. J. Med. 1993;328:1444–1449. http://www.nejm.org/doi/full/10.1056/NEJM199305203282003[↩]
- Ristow M., Schmeisser S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011;51:327–336. https://www.ncbi.nlm.nih.gov/pubmed/21619928[↩]
- Bowman T.S., Bassuk S.S., Gaziano M. Interventional Trials of Antioxidants. In: Hotzman J.L., editor. Atherosclerosis and Oxidant Stress: A New Perspective. Springer; New York, NY, USA: 2007. pp. 25–50.[↩]
- Losonczy K.G., Harris T.B., Havlik R.J. Vitamin e and vitamin c supplement use and risk of all-cause and coronary heart disease mortality in older persons: The established populations for epidemiologic studies of the elderly. Am. J. Clin. Nutr. 1996;64:190–196. https://www.ncbi.nlm.nih.gov/pubmed/8694019[↩]
- Todd S., Woodward M., Tunstall-Pedoe H., Bolton-Smith C. Dietary antioxidant vitamins and fiber in the etiology of cardiovascular disease and all-causes mortality: Results from the scottish heart health study. Am. J. Epidemiol. 1999;150:1073–1080. https://www.ncbi.nlm.nih.gov/pubmed/10568622[↩]
- Myung S.K., Ju W., Kim S.C., Kim H. Vitamin or antioxidant intake (or serum level) and risk of cervical neoplasm: A meta-analysis. BJOG: Int. J. Obstet. Gynaecol. 2011;118:1285–1291. https://www.ncbi.nlm.nih.gov/pubmed/21749626[↩]
- Bobe G., Weinstein S.J., Albanes D., Hirvonen T., Ashby J., Taylor P.R., Virtamo J., Stolzenberg-Solomon R.Z. Flavonoid intake and risk of pancreatic cancer in male smokers (finland) Cancer Epidemiol. Biomark. Prev. 2008;17:553–562. http://cebp.aacrjournals.org/content/17/3/553.long[↩]
- Heart Protection Study Collaborative Group. Mrc/bhf heart protection study of antioxidant vitamin supplementation in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet. 2002;360:23–33. https://www.ncbi.nlm.nih.gov/pubmed/12114037[↩]
- Klipstein-Grobusch K., Geleijnse J.M., den Breeijen J.H., Boeing H., Hofman A., Grobbee D.E., Witteman J.C. Dietary antioxidants and risk of myocardial infarction in the elderly: The rotterdam study. Am. J. Clin. Nutr. 1999;69:261–266. http://ajcn.nutrition.org/content/69/2/261.long[↩]
- GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin e after myocardial infarction: Results of the gissi-prevenzione trial. Gruppo italiano per lo studio della sopravvivenza nell’infarto miocardico. Lancet. 1999;354:447–455. http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(99)07072-5/fulltext[↩]
- Villanueva C, Kross RD. Antioxidant-Induced Stress. International Journal of Molecular Sciences. 2012;13(2):2091-2109. doi:10.3390/ijms13022091. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3292009[↩]





{kind=link}