Contents
- What is progressive supranuclear palsy
What is progressive supranuclear palsy
Progressive supranuclear palsy is a rare degenerative brain disorder that affects movement, vision, speech, and thinking ability (cognition). Deterioration of cells in the brainstem, cerebral cortex, cerebellum and basal ganglia — a cluster of cells deep within your brain — is what causes the coordination and movement issues of progressive supranuclear palsy. The signs and symptoms of progressive supranuclear palsy usually become apparent in mid- to late adulthood, most often in a person’s 60s. Most people with progressive supranuclear palsy survive 5 to 9 years after the disease first appears, although a few affected individuals have lived for more than a decade. The exact prevalence of progressive supranuclear palsy is unknown. Progressive supranuclear palsy was first described as a distinct disorder in 1964, when three scientists published a paper that distinguished the condition from Parkinson’s disease. It is sometimes referred to as Steele-Richardson-Olszewski syndrome, reflecting the combined names of the scientists who defined the disorder.
Progressive supranuclear palsy is under-diagnosed, so it is difficult to know how many people are affected. This disorder is believed to affect approximately 20,000 people in the United States and it may affect about 6 in 100,000 people worldwide. However, far fewer cases have been diagnosed. According to some reports, progressive supranuclear palsy is estimated to affect as many as 5 in 100,000 people. The onset of this disorder occurs between 45 and 75 years of age, with the average age of onset at about 63 years. Males are affected more often than females. The only proven risk factor for progressive supranuclear palsy is age. The condition typically affects people around the age of 60, and is virtually unknown in people under the age of 40.
Loss of balance and frequent falls are the most common early signs of progressive supranuclear palsy. Affected individuals have problems with walking, including poor coordination and an unsteady, lurching gait. Other movement abnormalities develop as the disease progresses, including unusually slow movements (bradykinesia), clumsiness, and stiffness of the trunk muscles. These problems worsen with time, and most affected people ultimately require wheelchair assistance.
Progressive supranuclear palsy is also characterized by abnormal eye movements, which typically develop 3 to 5 years after the walking problem and other movement problems first appear. Restricted up-and-down eye movement (vertical gaze palsy) is a hallmark of this disease. This can interfere with eating or with descending a flight of stairs, among other things. Other eye movement problems include difficulty opening the eyelids (apraxia of eyelid opening) and forceful involuntary closing of the eyelids (blepharospasm), infrequent blinking, and pulling back (retraction) of the eyelids. These abnormalities can lead to blurred vision, an increased sensitivity to light (photophobia), and a staring gaze. Because the main difficulty with the eyes is in aiming them properly, reading often becomes difficult. The patient finds it hard to shift down to the beginning of the next line automatically after reaching the end of the first line. This is very different from just needing reading glasses. An eye doctor unfamiliar with progressive supranuclear palsy may be baffled by the patient’s complaint of being unable to read a newspaper despite normal ability to read the individual letters on an eye chart. Some patients have their mild cataracts extracted in a vain effort to relieve such a visual problem.
Another common visual problem is an inability to maintain eye contact during conversation. This can give the mistaken impression that the patient is senile, hostile, or uninterested. The same eye movement problem can create the symptom of “tunnel vision” and can interfere with driving a car.
Additional features of progressive supranuclear palsy include slow and slurred speech (dysarthria) and trouble swallowing (dysphagia), after 3 or 4 years, on average, although it is the first symptom in a few patients. Swallowing tough foods or thin liquids can become difficult because of throat muscle weakness or incoordination. This tends to occur later than the walking, visual and speech problems, but can become very troublesome if the patient tends to choke on food. Unlike the other difficulties in progressive supranuclear palsy, this one can sometimes pose a danger for the patient – the danger of food going down the wrong pipe into the breathing passages, termed aspiration. Usually, difficulty managing thin liquids precedes difficulty with solid food. This is because in progressive supranuclear palsy, the swallowing muscles have difficulty creating a watertight seal separating the path to the stomach from the path to the lungs. The same is true for the swallowing difficulty of many neurological diseases. For non-neurologic conditions such as stricture of the esophagus, however, the difficulties start with solid foods.
Repeated, minor, often unnoticed episodes of small amounts of food and drink dripping into the lungs can cause pneumonia. Often, it is not apparent to the physician or family that the progressive supranuclear palsy patient’s pneumonia is in fact the result of subtle aspiration. But aspiration pneumonia is the most common cause of death in progressive supranuclear palsy.
Most affected individuals also experience changes in personality and behavior, such as a general loss of interest and enthusiasm (apathy). They develop problems with cognition, including difficulties with attention, planning, and problem solving. As the cognitive and behavioral problems worsen, affected individuals increasingly require help with personal care and other activities of daily living.
Progressive supranuclear palsy is often misdiagnosed as Parkinson disease, Alzheimer disease, corticobasal degeneration and other neurodegenerative disorders.
Figure 1. Human brain
Figure 2. Medial aspect of the human brain
Figure 3. Functional areas of the cerebral cortex
Figure 4. Basal ganglia of human brain
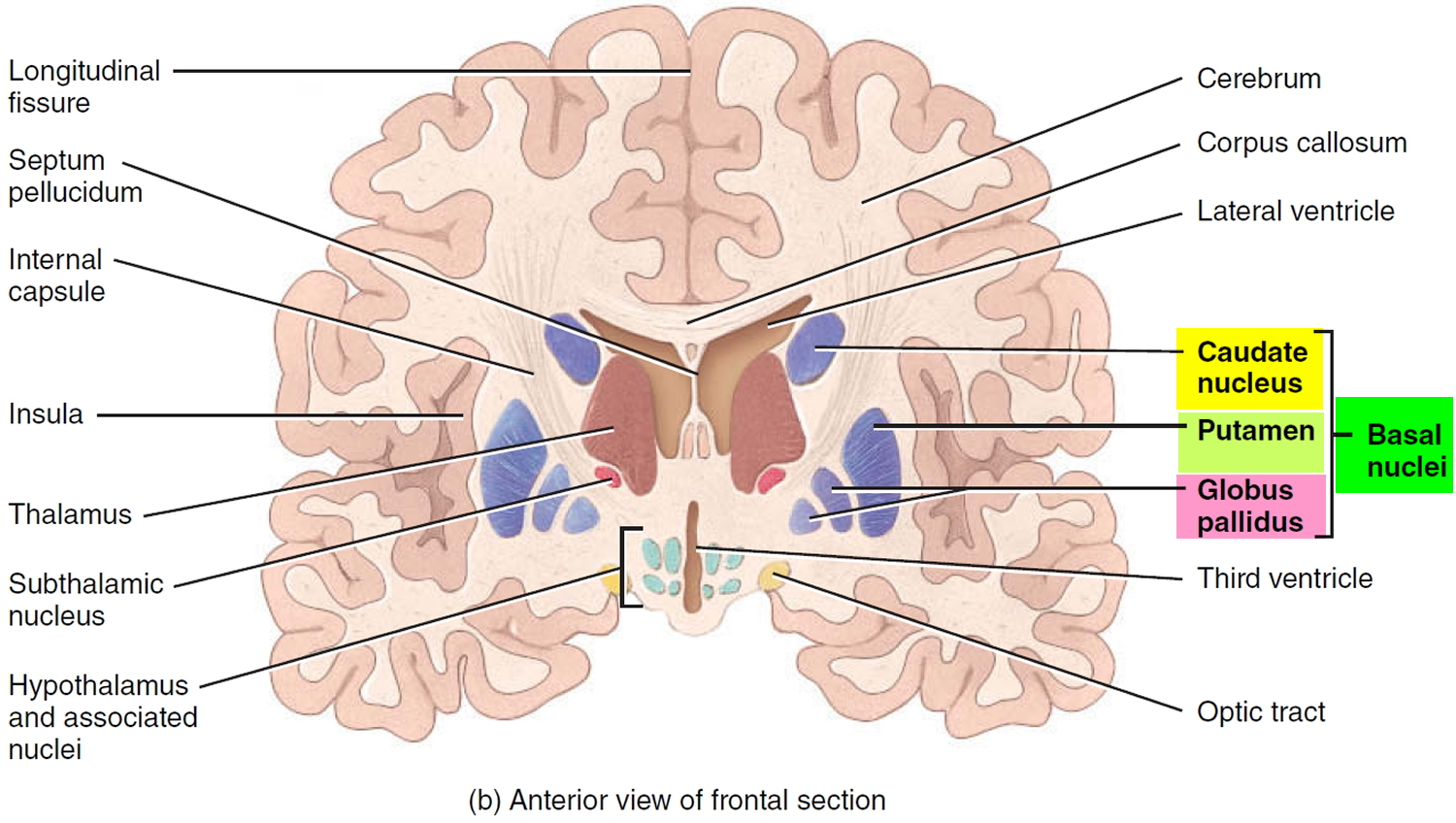
In general, a “palsy” is a weakness or paralysis of a part of the body. The term “supranuclear” refers to the nature of the eye problem in progressive supranuclear palsy. Although some patients with progressive supranuclear palsy describe their symptom as “blurring,” the actual problem is an inability to aim the eyes properly because of weakness or paralysis (palsy) of the muscles that move the eyeballs. These muscles are controlled by nerve cells residing in clusters or “nuclei” near the base of the brain in the brainstem. Most other brain problems that affect eye movements originate in those nuclei, but in progressive supranuclear palsy, the problem lies in parts of the brain that control those eye-movement nuclei themselves. These “higher” control areas are what the prefix “supra” in “supranuclear” refers to.
The symptoms of PSP usually get gradually worse over time. At first, the symptoms can be similar to some other conditions, which makes it difficult to diagnose early on.
Some of the main symptoms of progressive supranuclear palsy include:
- problems with balance and mobility, including frequent falls
- changes in behaviour, such as irritability or apathy (lack of interest)
- muscle stiffness
- an inability to control eye and eyelid movement, including focusing on specific objects or looking up or down at something
- slow, quiet or slurred speech
- difficulty swallowing (dysphagia)
- slowness of thought and some memory problems
The rate at which the symptoms progress can vary widely from person to person.
The exact cause of progressive supranuclear palsy is unknown. Patients with a family history of progressive supranuclear palsy are very rare, and the underlying genetic abnormality is generally unknown in these families. Less than 1% of those with progressive supranuclear palsy have a family member with the same condition. Unlike some other neurological conditions, there does not seem to be a single genetic cause of progressive supranuclear palsy. However the role of genetics in progressive supranuclear palsy is currently under investigation but the likelihood of the condition being passed on through genetic mutations is very small. A variant in the gene for tau protein called the H1 haplotype, located on chromosome 1, has been linked to progressive supranuclear palsy, but this genetic variation is very common and is not enough to cause progressive supranuclear palsy on its own.
Some cases of progressive supranuclear palsy are linked to a mutation or genetic variation in the gene MAPT, which helps to produce (codes for) the tau protein. This gene resides on chromosome 17 (17q21.1). Variants of least three other genes (STX6, EIF2AK3, and MOBP) are associated with an increased the risk of developing progressive supranuclear palsy. The study of genetic mechanisms should eventually lead to effective medical therapies.
Other names for progressive supranuclear palsy
- progressive supranuclear ophthalmoplegia
- PSP
- Richardson’s syndrome
- Steele-Richardson-Olszewski syndrome. Progressive supranuclear palsy was initially described by Steele, Richardson and Olsewski in 1964 and originally bore their names.
- supranuclear palsy, progressive
Does progressive supranuclear palsy lead to dementia like that in Alzheimer’s disease?
Although mental confusion in patients with progressive supranuclear palsy is more apparent than real, most patients do eventually develop some degree of mental impairment. Some are mislabeled as having Alzheimer’s disease. This is not very different from the situation in Parkinson’s disease.
In progressive supranuclear palsy, the dementia, if it does occur, does not feature the memory problem that is so apparent in Alzheimer’s disease. Rather, the dementia of progressive supranuclear palsy is characterized by slowed thought and difficulty synthesizing several different ideas into a new idea or plan. These mental functions are performed mostly by the front part of the brain (the frontal lobes). In Alzheimer’s, on the other hand, the problem is mostly in the part of the brain just above the ears (the temporal lobes), where memory functions are concentrated.
Alzheimer’s disease also includes either difficulty with language (such as trouble recalling correct names of common objects) or difficulty finding one’s way around a previously familiar environment. Fortunately, these symptoms almost never occur in progressive supranuclear palsy. Nevertheless, the “frontal” problems of progressive supranuclear palsy can interfere to a major degree with the ability to function independently, and the patient’s irritability in some cases can make it difficult for caregivers to help.
Slowing of thought can cause major problems for people with progressive supranuclear palsy by making it difficult to partake in conversation. A question may be answered with great accuracy and detail, but with a delay of several minutes. Probably the most important aspect of the dementia of progressive supranuclear palsy is apathy. People with progressive supranuclear palsy seem to lose interest in their surroundings, again creating the impression of loss of thinking ability and interfering with family interactions.
Can I drive?
Depending on your symptoms, you may be able to continue driving for a while with progressive supranuclear palsy. However, you are legally required to disclose your diagnosis to the Department of Motor Vehicles (DMV) and your insurer. You may need to be assessed at a driving centre if you wish to be continue to drive. The Department of Motor Vehicles is likely to seek a medical report from your doctor or specialist. Even with permission to continue driving, you may decide to stop if you feel uncomfortable or no longer in full control of the vehicle. This can be a difficult decision, as it affects your independence and may feel very challenging.
Progressive supranuclear palsy life expectancy
The number of years you can live with progressive supranuclear palsy varies a lot from person to person. It is common to read that life expectancy is six to seven years from onset of progressive supranuclear palsy but this figure does not take into account how very variable the condition is. Progressive supranuclear palsy may appear to move quickly if there are complications such as infection. Although there is no cure for progressive supranuclear palsy yet, there is much that can be done to improve quality of life for the person with the condition. With good attention to medical and nutritional needs, however, most progressive supranuclear palsy patients live well into their 70s and beyond.
Progressive supranuclear palsy gets progressively worse but is not itself directly life-threatening. It does, however, predispose patients to serious complications such as pneumonia secondary to difficulty in swallowing (dysphagia). The most common complications are choking and pneumonia, head injury, and fractures caused by falls. The most common cause of death is pneumonia.
Progressive supranuclear palsy complications
Complications of progressive supranuclear palsy result primarily from hindered muscle movements. These complications may include:
- Falling, which could lead to head injuries, fractures and other injuries.
- Difficulty focusing your eyes, which also can lead to injuries.
- Difficulty sleeping.
- Difficulty looking at bright lights.
- Problems swallowing, which can lead to choking or inhaling food or liquid into your airway (aspiration). Aspiration can develop into pneumonia, the most common cause of death in people with progressive supranuclear palsy.
- Impulsive behaviors — for example, standing up without waiting for assistance — which can lead to falls.
To avoid the hazards of choking, your doctor may recommend a feeding tube. To avoid injuries due to falling, a walker or a wheelchair may be used.
The most common complications are choking and pneumonia, head injury and fractures caused by falls. The most common cause of death is pneumonia.
Progressive supranuclear palsy causes
The cause of progressive supranuclear palsy is not known, but it is a form of tauopathy, in which abnormal phosphorylation of the “tau” protein leads to destruction of vital protein filaments in nerve cells, causing their death. Tau occurs naturally in the brain and is usually broken down before it reaches high levels. The normal function of tau is to help support the internal “skeleton” of the brain cells whose long extensions make contact with other brain cells. In people with progressive supranuclear palsy, the “tau” protein isn’t broken down properly and forms harmful clumps called “neurofibrillary tangles” in brain cells. The amount of abnormal tau in the brain can vary among people with progressive supranuclear palsy, as can the location of these clumps. This means the condition can have a wide range of symptoms.
The condition has been linked to changes in certain genes, but these genetic faults are not inherited and the risk to other family members, including children or siblings of someone with progressive supranuclear palsy, is very low.
Recent work suggests that the disease is at least partly genetic. Many researchers now believe that various genetic and environmental factors contribute to the development of this disorder.
In the medical literature, the word “tauopathies” is used to refer to several neurodegenerative disorders including progressive supranuclear palsy, in which tau is mishandled. Other neurodegenerative disorders classified as tauopathies include corticobasal degeneration and Pick disease.
In most cases, the genetic cause of progressive supranuclear palsy is unknown. Rarely, the disease results from mutations in the MAPT gene. Certain normal variations (polymorphisms) in the MAPT gene have also been associated with an increased risk of developing progressive supranuclear palsy.
The MAPT gene provides instructions for making a protein called tau. This protein is found throughout the nervous system, including in nerve cells (neurons) in the brain. It is involved in assembling and stabilizing microtubules, which are rigid, hollow fibers that make up the cell’s structural framework (the cytoskeleton). Microtubules help cells maintain their shape, assist in the process of cell division, and are essential for the transport of materials within cells.
The signs and symptoms of progressive supranuclear palsy appear to be related to abnormalities in the tau protein. In people with MAPT gene mutations, genetic changes disrupt the protein’s normal structure and function. However, abnormal tau is also found in affected individuals without MAPT gene mutations. The defective tau protein assembles into abnormal clumps within neurons and other brain cells, although it is unclear what effect these clumps have on cell function and survival. Progressive supranuclear palsy is characterized by the gradual death of brain cells, particularly in structures deep within the brain that are essential for coordinating movement. This loss of brain cells underlies the movement abnormalities and other features of progressive supranuclear palsy.
This condition is one of several related diseases known as tauopathies, which are characterized by an abnormal buildup of tau in the brain.
Researchers suspect that other genetic and environmental factors also contribute to progressive supranuclear palsy. For example, the disease has been linked to genetic changes on chromosome 1 and chromosome 11. However, the specific genes involved have not been identified.
Progressive supranuclear palsy hereditary
Most cases of progressive supranuclear palsy are sporadic, which means they occur in people with no history of the disorder in their family. However, some people with this disorder have had family members with related conditions, such as parkinsonism and a loss of intellectual functions (dementia).
When progressive supranuclear palsy runs in families, it can have an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means one copy of an altered gene in each cell is sufficient to cause the disorder.
Fewer than one in 100 people with progressive supranuclear palsy knows of even one other family member with progressive supranuclear palsy. However a variant in the gene on chromosome 17 that encodes the tau protein is more common in progressive supranuclear palsy than in the rest of the population. The variant is called the “H1 haplotype.” About 95% of people with progressive supranuclear palsy have this variant on both of their copies of chromosome 17, while this is true for only about 60% of the general population. So clearly, the H1 haplotype is (nearly) necessary but far from sufficient to cause the disease. There is evidence that what this variant is doing wrong is directing the brain cells to produce too much tau protein. The theory is that the excess tau forms neurofibrillary tangles and that these, or an early, embryonic form of them, damages the cells.
One very large family with progressive supranuclear palsy in multiple members has a variant in a gene other than the tau gene. The specific gene has not yet been identified. Similarly, two other genetic variant that are ordinarily associated with hereditary Parkinson’s disease – the parkin gene (PARK2) and the dardarin gene (PARK8) can in some cases cause changes in the brain very similar to what happens in progressive supranuclear palsy. This means that there may be many different genetic contributors to progressive supranuclear palsy with no single one laying claim to the title of “the progressive supranuclear palsy gene.” It also means that progressive supranuclear palsy, Parkinson’s and perhaps other neurodegenerative disorders may share come causative factors.
Related Disorders
Symptoms of the following disorders can be similar to those of progressive supranuclear palsy. Comparisons may be useful for a differential diagnosis.
Corticobasal degeneration is a rare progressive neurological disorder characterized by cell loss and shrinkage (atrophy) in certain areas of the brain (cerebral cortex and basal ganglia). The symptoms and signs of this disease resemble some patients with progressive supranuclear palsy, and some experts believe that corticobasal degeneration and progressive supranuclear palsy are variations of the same disease. Both are tauopathies.
Multiple system atrophy is a rare progressive neurological disorder characterized by a varying combination of parkinsonism and cerebellar ataxia (poorly coordinated limb movement, unsteady gait and dysarthria). Many patients with multiple system atrophy also develop impaired function of the autonomic nervous system, which controls blood pressure, heart rate, sweating, the bowels and the urinary bladder. The exact cause of multiple system atrophy is unknown.
Shy-Drager syndrome is simply multiple system atrophy with autonomic failure. Most experts no longer use this term.
Parkinson disease is a slowly progressive neurologic condition characterized by involuntary trembling (rest tremor), muscular stiffness or inflexibility (rigidity), slowness of movement (bradykinesia) and difficulty carrying out voluntary movements (akinesia). Degenerative changes occur in areas deep within the brain (substantia nigra and other pigmented regions of the brain), causing a decrease in dopamine levels in the brain. Dopamine is a neurotransmitter, which is a chemical that sends a signal from one nerve cell to another in the brain. Parkinson disease progresses much more slowly than progressive supranuclear palsy and usually is not incapacitating for a decade or more.
Progressive supranuclear palsy stages
Early stage progressive supranuclear palsy
The initial symptoms of progressive supranuclear palsy can include:
- sudden loss of balance when walking, that usually results in repeated falls, often backwards
- muscle stiffness, particularly in the neck
- extreme tiredness
- changes in personality, such as irritability, apathy (lack of interest) and mood swings
- changes in behaviour, such as recklessness and poor judgement
- a dislike of bright lights (photophobia)
- difficulty controlling the eye muscles, particularly problems with looking up and down
- blurred or double vision
Some people have early symptoms that are very similar to those of Parkinson’s disease, such as tremors (involuntary shaking of particular parts of the body) and slow movement.
Mid-stage progressive supranuclear palsy
Over time, the initial symptoms of progressive supranuclear palsy will become more severe.
Worsening balance and mobility problems may mean that walking becomes impossible and a wheelchair is needed. Controlling the eye muscles will become more difficult, increasing the risk of falls and making everyday tasks, such as reading and eating, more problematic.
New symptoms can also develop at this stage, such as:
- slow, quiet or slurred speech
- problems swallowing (dysphagia)
- reduced blinking reflex, which can cause the eyes to dry out and become irritated
- involuntary closing of the eyes (blepharospasm), which can last from several seconds to hours
- disturbed sleep
- slowness of thought and some memory problems
- neck or back pain, joint pain and headaches
Advanced stage progressive supranuclear palsy
As PSP progresses to an advanced stage, people with the condition normally begin to experience increasing difficulties controlling the muscles of their mouth, throat and tongue.
Speech may become increasingly slow and slurred, making it harder to understand. There may also be some problems with thinking, concentration and memory (dementia), although these are generally mild and the person will normally retain an awareness of themselves.
The loss of control of the throat muscles can lead to severe swallowing problems, which may mean a feeding tube is required at some point to prevent choking or chest infections caused by fluid or small food particles passing into the lungs.
Many people with PSP also develop problems with their bowels and bladder functions. Constipation and difficulty passing urine are common, as is the need to pass urine several times during the night. Some people may lose control over their bladder or bowel movements (incontinence).
Progressive supranuclear palsy signs and symptoms
People with progressive supranuclear palsy (PSP) develop a range of difficulties with balance, movement, vision, speech and swallowing.
The condition tends to develop gradually, which means it can be mistaken for another, more common, condition at first.
The symptoms typically become more severe over several years, although the speed at which they worsen varies.
Some of the main symptoms of progressive supranuclear palsy are outlined below. Most people with the condition won’t experience all of these.
Signs and symptoms of progressive supranuclear palsy
The signs and symptoms of progressive supranuclear palsy vary from person to person, but patients generally fall into one of four clinical syndromes (phenotypes):
- Richardson syndrome,
- Atypical Parkinsonism,
- Corticobasal syndrome, and
- Pure akinesia and gait freezing.
- Less commonly, patients present with cognitive loss and no motor signs.
The most common presentation is the Richardson syndrome, consisting of gait and balance impairment, a wide-eyed staring facial expression, abnormal speech, memory and cognitive impairment and a slowing or loss of voluntary eye movement, particularly in the downward direction (supranuclear ophthalmoplegia). Cognitive symptoms include forgetfulness and personality changes, such as loss of interest in formerly pleasurable activities (apathy), impaired attention and concentration, depression, and increased irritability.
Fewer than half of all progressive supranuclear palsy patients are initially diagnosed correctly because many patients do not present with the classic Richardson syndrome. Many of these patients are initially slow and have muscle rigidity and occasionally tremor, resembling Parkinson disease, and they may initially respond somewhat to levodopa. Other patients present with bizarre stiffening (rigidity and dystonia) and loss of voluntary function in one upper limb, as is seen in corticobasal degeneration. Rarely, patients exhibit the syndrome of primary akinesia and gait freezing. These patients exhibit hesitant initiation of gait and a tendency to freeze or stop when turning and when crossing thresholds (doorways). Their eye movements and cognition are normal. Small handwriting and low-volume rapid, mumbling speech (tachyphemia or cluttered speech) are typical and are similar to that which occur in Parkinson disease, but in contrast to Parkinson disease, there is no slowness (bradykinesia) or muscle stiffness (rigidity). Finally, some patients with progressive supranuclear palsy present with cognitive impairment, resembling Alzheimer disease or frontotemporal dementia. Most patients with atypical presentations ultimately develop abnormalities of eye movement, speech, swallowing and gait (Richardson syndrome) in a few years. Thus, the diagnosis of progressive supranuclear palsy typically becomes more certain as the disease progresses.
Impaired eye movements eventually make reading, driving, and interpersonal eye contact difficult or impossible. Abnormal eyelid control causes the eyes to close involuntarily (blepharospasm) for seconds or more, and some affected individuals may not be able to open their eyes (eye opening apraxia), even when a spasm stops. Other patients have trouble closing their eyes or may blink less than normal, causing the eyes to become dry and red.
Muscles of the body may contract involuntarily, causing the affect body part (e.g., the upper or lower limbs) to assume bizarre postures. This is called dystonia. Blepharospasm is a form of dystonia affecting the muscles around the eyes.
A mild or moderate degree of mental impairment eventually occurs in most patients, and this may be misdiagnosed as Alzheimer disease (AD) when it occurs early in the illness, before significant difficulties with speech, balance and eye movements appear.
Some patients experience sleep disturbances such as frequent awakenings and changes in sleeping patterns. Sleep disturbances may be a sign of depression or may be a side effect of a medication. REM (rapid eye movement) sleep behavior disorder is not a characteristic of progressive supranuclear palsy but is a characteristic of dementia with Lewy bodies, Parkinson disease and multiple system atrophy. In REM sleep behavior disorder, patients talk and move during dream sleep, and the movement can result in personal injury or injury to a bed partner.
Progressive supranuclear palsy diagnosis
It can be difficult to diagnose progressive supranuclear palsy, as there’s no single test for it, and the condition can have similar symptoms to a number of others.
There are also many possible symptoms of progressive supranuclear palsy and several different sub-types that vary slightly, making it hard to make a definitive diagnosis in the early stages of the condition.
The diagnosis must be made or confirmed by a consultant with expertise in progressive supranuclear palsy (PSP). This will usually be a neurologist (a specialist in conditions affecting the brain and nerves).
The diagnosis of progressive supranuclear palsy may be suspected based upon a thorough clinical evaluation, a detailed patient history, identification of characteristic physical findings and will be based on the pattern of your symptoms and by ruling out conditions that can cause similar symptoms, such as Parkinson’s disease or a stroke.
Your doctor will need to carry out other tests and scans.
Brain scans
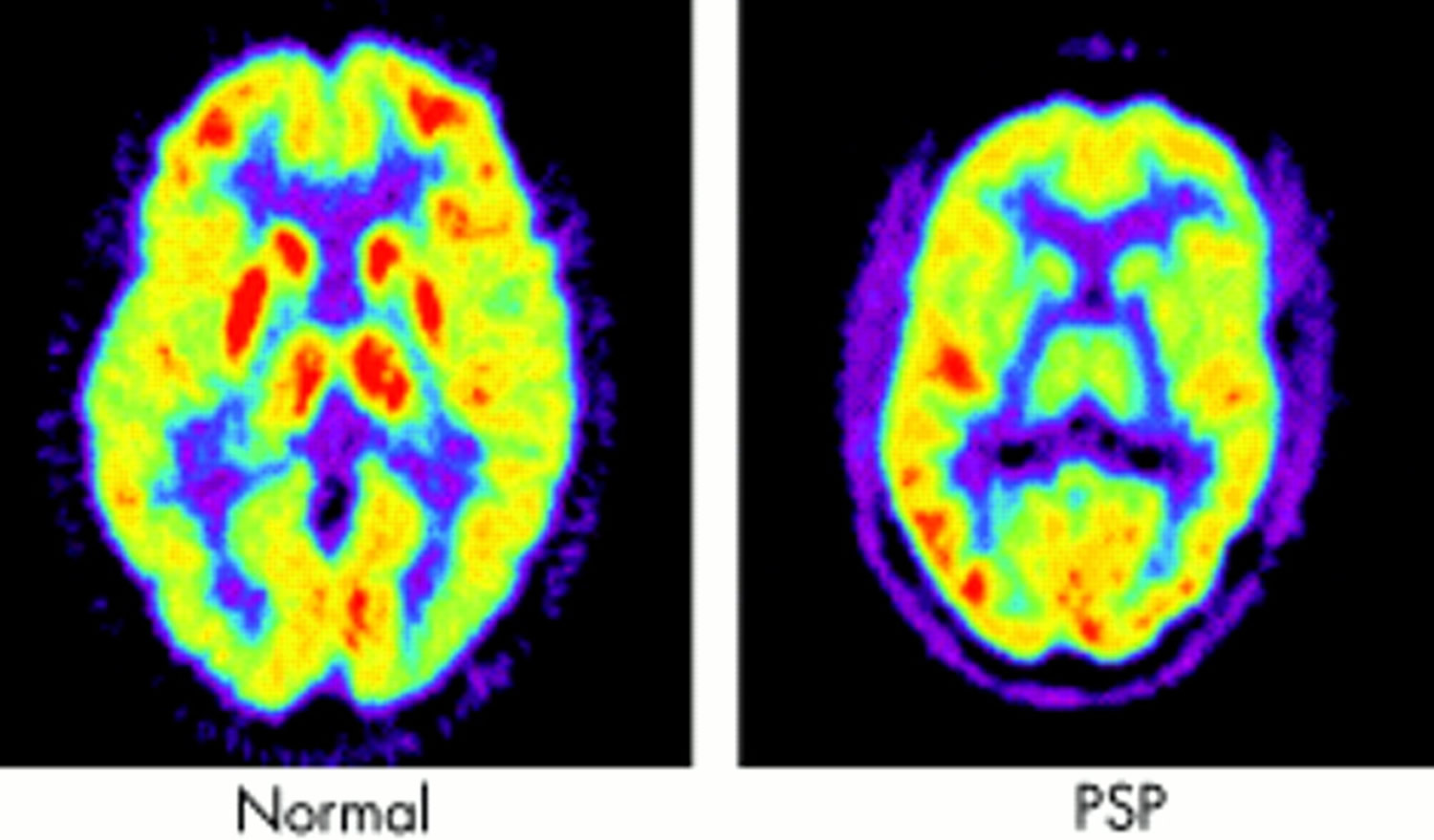
If you have symptoms of progressive supranuclear palsy that suggest there’s something wrong with your brain, it’s likely you’ll be referred for a brain scan.
Types of scan that you may have include:
- magnetic resonance imaging (MRI) scan – where a strong magnetic field and radio waves are used to produce detailed images of the inside of the brain
- positive emission tomography (PET) scan – a scan that detects the radiation given off by a substance injected beforehand
- a DaTscan – where you’re given an injection containing a small amount of a radioactive material and pictures of your brain are taken with a gamma camera
These scans can be useful in ruling out other possible conditions, such as brain tumours or strokes.
MRI scans can also detect abnormal changes to the brain that are consistent with a diagnosis of progressive supranuclear palsy, such as shrinkage of certain areas.
Scans that show the build-up of the tau protein in the brain that’s associated with progressive supranuclear palsy are currently under development.
Ruling out Parkinson’s disease
You may be prescribed a short course of a medication called levodopa to determine whether your symptoms are caused by progressive supranuclear palsy or Parkinson’s disease.
People with Parkinson’s disease usually experience a significant improvement in their symptoms after taking levodopa, whereas the medication only has a limited beneficial effect for some people with progressive supranuclear palsy.
Neuropsychological testing
It’s also likely you’ll be referred to a neurologist and possibly also a psychologist for neuropsychological testing.
This involves having a series of tests that are designed to evaluate the full extent of your symptoms and their impact on your mental abilities.
The tests will look at abilities such as:
- memory
- concentration
- understanding language
- the processing of visual information, such as words and pictures
Most people with progressive supranuclear palsy have a distinct pattern in terms of their mental abilities, including poor concentration, a low attention span and problems with spoken language and processing visual information. Their memory of previously learned facts isn’t usually significantly affected.
Progressive supranuclear palsy treatment
There’s currently no cure for progressive supranuclear palsy (progressive supranuclear palsy) and no treatment to slow it down, but there are lots of things that can be done to help manage the symptoms.
Treatment of progressive supranuclear palsy is symptomatic and supportive.
As progressive supranuclear palsy can affect many different areas of your health, you’ll be cared for by a team of health and social care professionals working together. This is known as a multidisciplinary team.
Members of your multidisciplinary team may include:
- a neurologist – a specialist in conditions that affect the brain and nerves
- a physiotherapist – who can help with movement and balance difficulties
- a speech and language therapist – who can help with speech or swallowing problems
- an occupational therapist – who can help you improve the skills you need for daily activities, such as washing or dressing
- a social worker – who can advise you about the support available from social services
- an ophthalmologist or orthoptist – specialists in treating eye conditions
- a specialist neurology nurse – who may act as your point of contact with the rest of the team
Some of the main treatments that may be recommended are outlined below.
Medication
There are currently no medications that treat progressive supranuclear palsy specifically, but some people in the early stages of the condition may benefit from taking levodopa, amantadine or other medications used to treat Parkinson’s disease.
These medications can improve balance and stiffness for some people with progressive supranuclear palsy, although the effect is often limited and only lasts for up to a few years.
Antidepressants can help with the depression that’s often associated with progressive supranuclear palsy, and some may also help with balance, stiffness, pain and sleep problems.
It’s important to tell your doctor about the symptoms you’re experiencing, so they can consider which of these treatments is best for you.
Physiotherapy
A physiotherapist can give you advice about making the most of your remaining mobility using exercise, while making sure you don’t overexert yourself. Regular exercise may help strengthen your muscles, improve your posture and prevent stiffening of your joints.
Your physiotherapist can advise about equipment that could benefit you, such as a walking frame or specially designed shoes to reduce the risk of slipping and falling.
They can teach you breathing exercises to use when you eat, to reduce your risk of developing aspiration pneumonia (a chest infection caused by food particles falling into your lungs).
Occupational therapy
An occupational therapist (OT) can advise you about how you can increase your safety, and prevent trips and falls during your day-to-day activities.
For example, many people with PSP benefit from having bars placed along the sides of their bath to make it easier for them to get in and out.
The occupational therapist (OT) will also be able to spot potential hazards in your home that could lead to a fall, such as poor lighting, badly secured rugs and crowded walkways and corridors.
Walking aids such as a walker weighted in front and wearing shoes with built-up heels may help in preventing affected individuals from falling backwards.
Speech and language therapy
A speech and language therapist can help you improve your speech and swallowing problems (dysphagia).
They can teach you a number of techniques to help make your voice as clear as possible and can advise you about suitable communication aids or devices that you may need as progressive supranuclear palsy progresses.
Your therapist can also advise you about different swallowing techniques and, working together with a dietitian (see below), they may suggest altering the consistency of your food to make swallowing easier.
As your swallowing problems become more severe, you’ll need additional treatment to compensate for your swallowing difficulties.
When a patient can no longer swallow, a surgical procedure known as percutaneous gastrostomy can be performed, depending upon the patient’s wishes and quality of life. In this procedure, a tube is placed through the skin of the abdomen into the stomach to allow sufficient feeding.
Diet and severe swallowing problems
You may be referred to a dietitian, who will advise you about making changes to your diet, such as including food and liquids that are easier to swallow, while ensuring that you have a healthy, balanced diet.
For example, mashed potatoes are a good source of carbohydrates, while scrambled eggs and cheese are high in protein and calcium.
Feeding tubes may be recommended for severe swallowing problems, where the risk of malnutrition and dehydration is increased. You should discuss the pros and cons of feeding tubes with your family and care team, preferably when your symptoms of dysphagia are at an early stage.
The main type of feeding tube used is called a percutaneous endoscopic gastrostomy (PEG) tube. This tube is placed into your stomach through your abdomen (tummy) during an operation.
Treating eye problems
If you’re having problems controlling your eyelids, injections of botulinum toxin can be used to help relax the muscles of your eyelids. It works by blocking the signals from the brain to the affected muscles. The effects of the injections usually last for up to three months.
If you’re experiencing dry eyes because of reduced blinking, eyedrops and artificial tears can be used to lubricate them and reduce irritation.
Bifocals or special glasses with prisms may be prescribed for some individuals with progressive supranuclear palsy to treat certain difficulties in eyesight (i.e., difficulty looking down). Wearing wraparound, dark glasses can help those who are sensitive to bright light (photophobia).
Palliative care
Palliative care can be offered at any stage of progressive supranuclear palsy, alongside other treatments. It aims to relieve pain and other distressing symptoms while providing psychological, social and spiritual support.
Palliative care can be received:
- in a hospice
- at home or in a residential home
- on a day patient basis in a hospice
- in a hospital
Advanced care planning
Many people with progressive supranuclear palsy consider making plans for the future that outline their wishes (both medical and other decisions) and make them known to both their family and the health professionals involved in their care.
This can be useful in case you’re unable to communicate your decisions later on because you’re too ill, although it’s voluntary and you don’t have to do it if you don’t want to.
Issues that you may want to cover might include:
- if you want to be treated at home, in a hospice or in a hospital when you reach the final stages of progressive supranuclear palsy
- the type of painkillers you would be willing to take
- if you would be willing to use a feeding tube if you were no longer able to swallow food and liquid
- if you’re willing to donate any of your organs after you die
- if you’d be willing to be resuscitated by artificial means if you experienced respiratory failure (loss of lung function)
If you decide to discuss these issues, they can be written down in a number of ways:
- Advance decision to refuse treatment
- Advance statement
- Emergency healthcare plan
- Lasting Power of Attorney
Your care team can provide you with more information and advice about these decisions and how best to record them.
Coping and support
Being told that you have progressive supranuclear palsy can be devastating and difficult to take in.
You may feel numb, overwhelmed, angry, distressed, scared or in denial. Some people are relieved that a cause for their symptoms has finally been found. There’s no right or wrong way to feel – everybody is different and copes in their own way.
Support from your family and care team can help you come to terms with the diagnosis.
Other good sources of information and support for people with PSP:
- CurePSP: https://www.psp.org/
- Progressive Supranuclear Palsy Association UK: https://www.pspassociation.org.uk/
The progressive supranuclear palsy association can give you information and practical advice about living with progressive supranuclear palsy, as well as providing support to help you cope with the emotional impact of the condition.





{kind=link}