Contents
- What are adrenal glands
- Disorders and Diseases of the Adrenal Glands
- What causes adrenal gland disorders ?
- Adrenal gland tumors
- Cushing Syndrome
- What’s the difference between Cushing’s syndrome and Cushing’s disease ?
- What causes Cushing’s syndrome ?
- What are the symptoms of Cushing’s syndrome ?
- How to diagnose Cushing’s syndrome ?
- Is there a cure for Cushing’s syndrome ?
- What are the treatments for Cushing’s syndrome ?
- Cushing’s syndrome can affect fertility in both men and women
- Congenital Adrenal Hyperplasia
- Pituitary Tumors
- Pheochromocytoma and Paraganglioma
- Addison’s disease
- Hyperaldosteronism
- What causes primary hyperaldosteronism ?
- What are the signs and symptoms of primary hyperaldosteronism ?
- How common is primary hyperaldosteronism ?
- Is primary hyperaldosteronism inherited ?
- How is primary hyperaldosteronism diagnosed ?
- How is primary hyperaldosteronism treated ?
- Are there any side-effects to the treatment ?
- What are the longer-term implications of primary hyperaldosteronism ?
- Adrenal Gland Suppression
What are adrenal glands
The adrenal glands are two two triangle-shaped glands that measure about 1.5 inches in height and 3 inches in length. They are located on top of each kidney like a cap and is embedded in the mass of fatty tissue that encloses the kidney (Figure 1 and 2). Their name directly relates to their location (ad—near or at; renes—kidneys) 1.
The adrenal glands, located on the top of each kidney, are responsible for releasing different classes of hormones.
The outer part of the adrenal gland, called the adrenal cortex, produces the hormones cortisol, aldosterone, and hormones that can be changed into testosterone 1. The inner part of the gland, called the adrenal medulla, produces the hormones adrenaline and noradrenaline. These hormones are also called epinephrine and norepinephrine 1.
These hormones—cortisol, aldosterone, adrenaline, and noradrenaline—control many important functions in the body, including:
- Maintaining metabolic processes, such as managing blood sugar levels and regulating inflammation.
- Regulating the balance of salt and water.
- Controlling the “fight or flight” response to stress.
- Maintaining pregnancy.
- Initiating and controlling sexual maturation during childhood and puberty.
The adrenal glands are also an important source of sex steroids, such as estrogen and testosterone.
When the adrenal glands produce more or less hormones than normal, you can become sick. This might happen at birth or later in life 2.
The adrenal glands can be affected by many diseases, such as autoimmune disorders, infections, tumors, and bleeding 2.
Conditions related to adrenal gland problems include:
- Addison disease (also called adrenal insufficiency)
- Congenital adrenal hyperplasia
- Cushing syndrome
- Diabetes – caused by another medical problem
- Glucocorticoid medicines
- Excessive or unwanted hair in women (hirsutism)
- Hump behind shoulders (dorsocervical fat pad)
- Hypoglycemia
- Primary aldosteronism (Conn syndrome)
- Waterhouse-Friderichsen syndrome
Location of adrenal glands
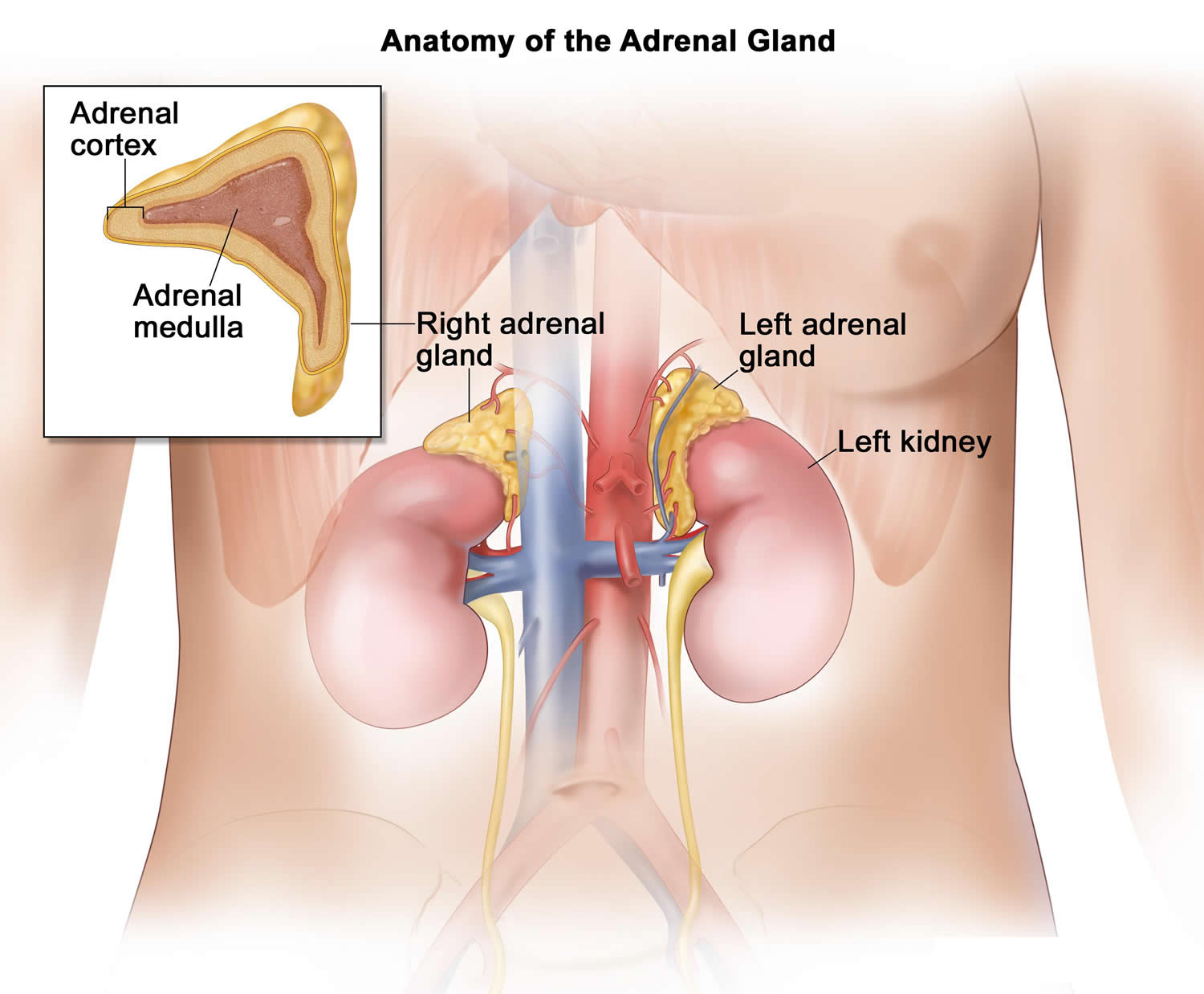
Figure 1. Location of the adrenal glands on top of each kidneys
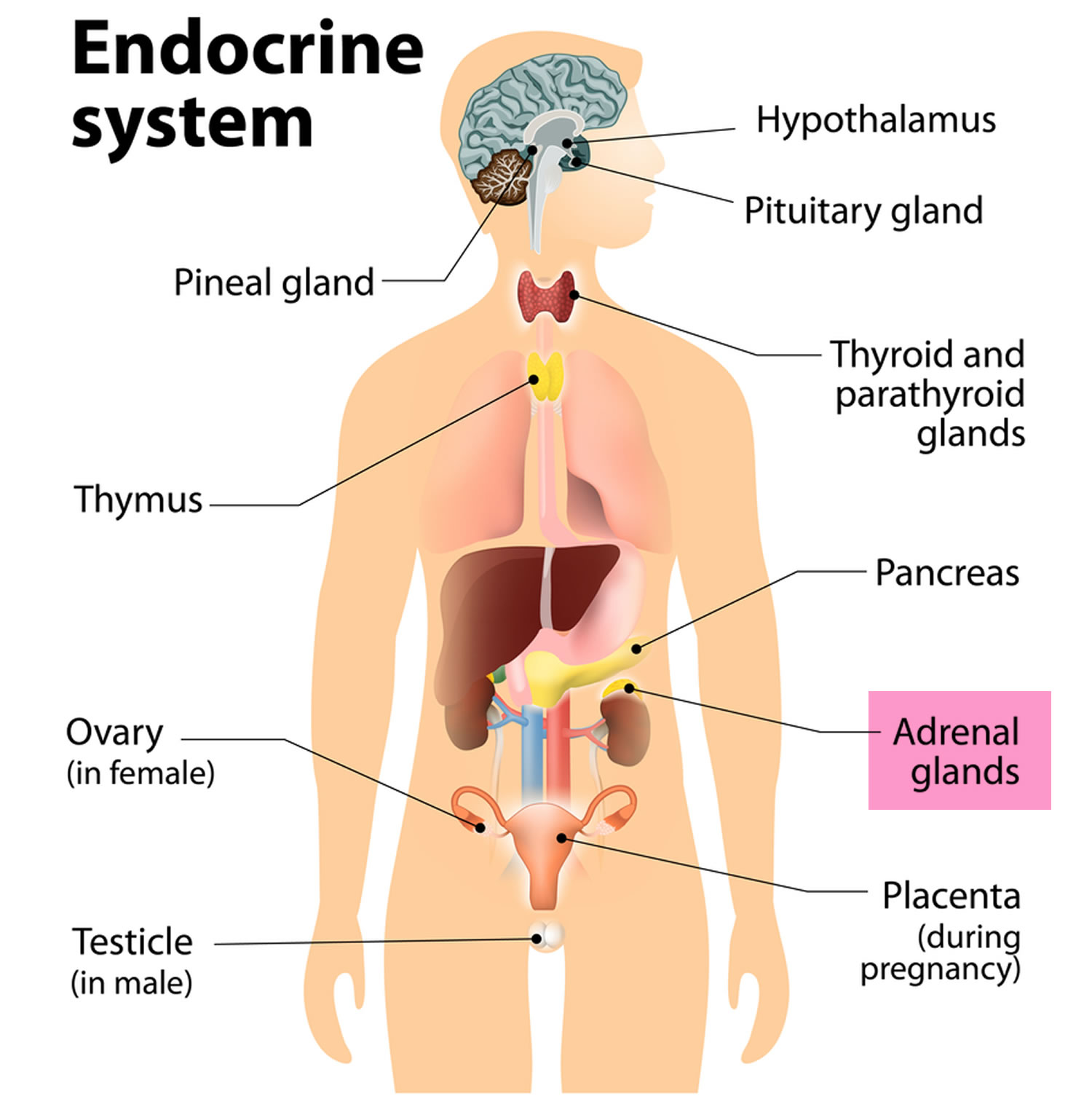
Figure 2. Adrenal gland anatomy
Structure of the adrenal glands
Each adrenal gland is about the size of the top part of the thumb. Each adrenal gland is very vascular and consists of two parts:
- The outer part is the adrenal cortex.
- The central portion is the adrenal medulla.
These regions are not sharply divided, but they are functionally distinct structures that secrete different hormones.
The adrenal cortex and the adrenal medulla have very different functions. One of the main distinctions between them is that the hormones released by the adrenal cortex are necessary for life; those secreted by the adrenal medulla are not.
The adrenal medulla consists of irregularly shaped cells organized in groups around blood vessels. These cells are intimately connected with the sympathetic division of the autonomic nervous system. Adrenal medullary cells are actually modified postganglionic neurons. Preganglionic autonomic nerve fibers control their secretions. The adrenal medulla produces epinephrine and norepinephrine. These hormones are also called adrenaline and noradrenaline.
The adrenal cortex, which makes up the bulk of the adrenal gland, is composed of closely packed masses of epithelial cells, organized in layers. These layers form the outer (glomerulosa), middle (fasciculata), and inner (reticularis) zones of the cortex (Figure 3). As in the adrenal medulla, the cells of the adrenal cortex are well supplied with blood vessels. The adrenal cortex produces steroid hormones such as cortisol, aldosterone, and hormones that can be changed into testosterone.
Figure 3. Adrenal gland microscopic section
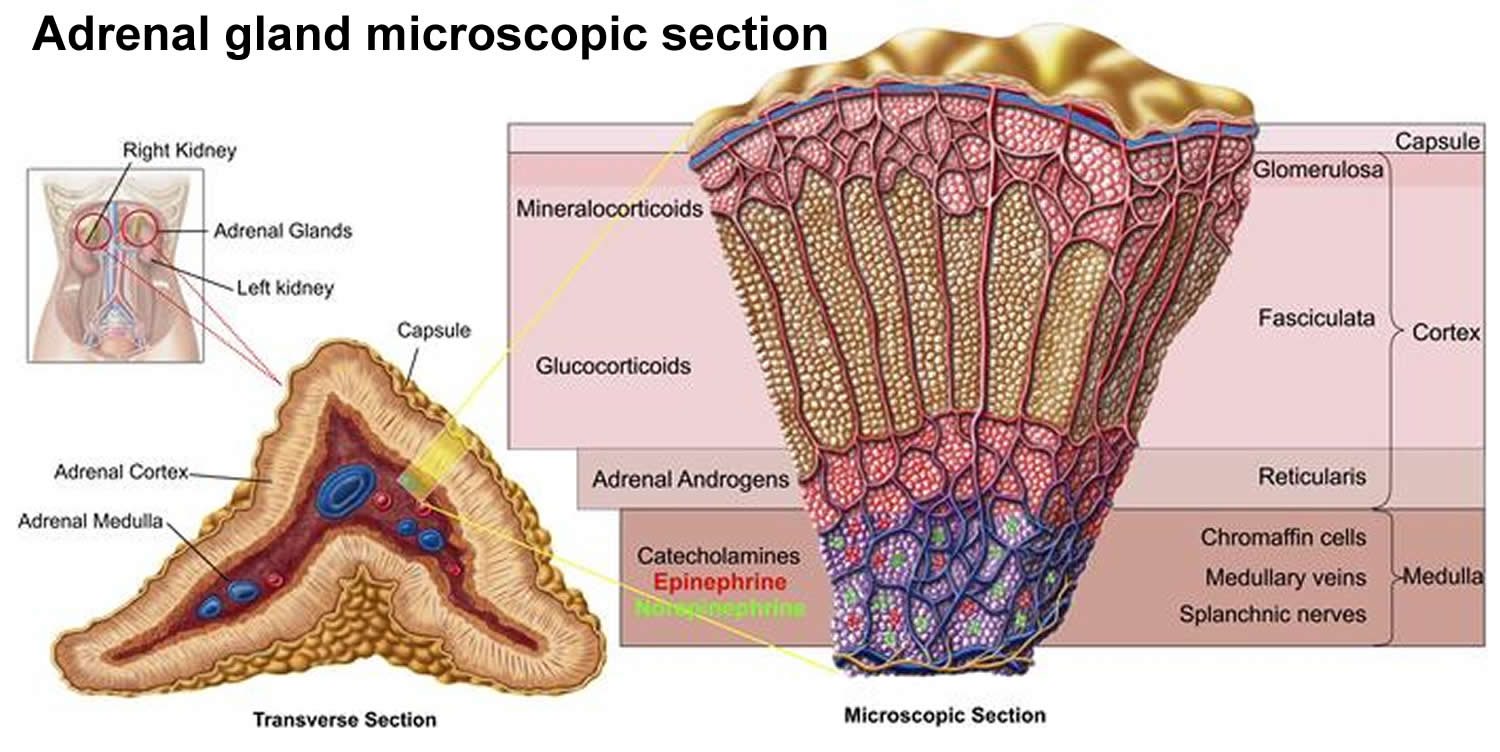
What do adrenal glands do ?
Adrenal Cortex Hormones
The adrenal cortex produces two main groups of corticosteroid hormones—glucocorticoids and mineralcorticoids. The release of glucocorticoids is triggered by the hypothalamus and pituitary gland. Mineralcorticoids are mediated by signals triggered by the kidney.
The principle mineralcorticoid is aldosterone, which maintains the right balance of salt and water while helping control blood pressure. Aldosterone is a hormone produced by the adrenal glands and helps to control the amount of fluid in the body by affecting how much salt and water the kidney retains or excretes. The adrenal glands produce aldosterone in response to another hormone called renin. Renin is produced by specialised cells in the kidney that detect when the body lacks salt; renin released by the kidney signals the adrenal glands to release aldosterone. The kidney detects an increase in aldosterone in the bloodstream and responds by retaining extra salt rather than excreting it in the urine. As the body regains the salt it needs, the level of renin in the bloodstream drops and therefore the amount of aldosterone in the blood also falls, meaning more water is excreted in the urine. This is an example of a feedback system.
When the hypothalamus produces corticotrophin-releasing hormone (CRH), it stimulates the pituitary gland to release adrenal corticotrophic hormone (ACTH). These hormones, in turn, alert the adrenal glands to produce corticosteroid hormones.
Glucocorticoids released by the adrenal cortex include:
- 1) Cortisol: Also known as hydrocortisone, it regulates how the body converts fats, proteins, and carbohydrates to energy. It also helps regulate blood pressure and cardiovascular function.
- 2) Corticosterone: This hormone works with hydrocortisone to regulate immune response and suppress inflammatory reactions.
There is a third class of hormone released by the adrenal cortex, known as sex steroids or adrenal sex hormones. The adrenal cortex releases small amounts of male and female sex hormones. These hormones are male types (adrenal androgens), but some are converted to female hormones (estrogens) in the skin, liver, and adipose tissue. The amounts of adrenal sex hormones are very small compared to the supply of sex hormones from the gonads, but they may contribute to
early development of reproductive organs. However, their impact is usually overshadowed by the greater amounts of hormones (such as estrogen and testosterone) released by the ovaries or testes. Cells in the inner zone of the adrenal cortex produce sex hormones.
The more important actions of cortisol include:
- Inhibition of protein synthesis in tissues, increasing the blood concentration of amino acids.
- Promotion of fatty acid release from adipose tissue, increasing the utilization of fatty acids and decreasing the use of glucose as energy sources.
- Stimulation of liver cells to synthesize glucose from noncarbohydrates, such as circulating amino acids and glycerol, increasing the blood glucose concentration.
These actions of cortisol help keep blood glucose concentration within the normal range between meals. This control is important, because a few hours without food can exhaust the supply of liver glycogen, a major source of glucose.
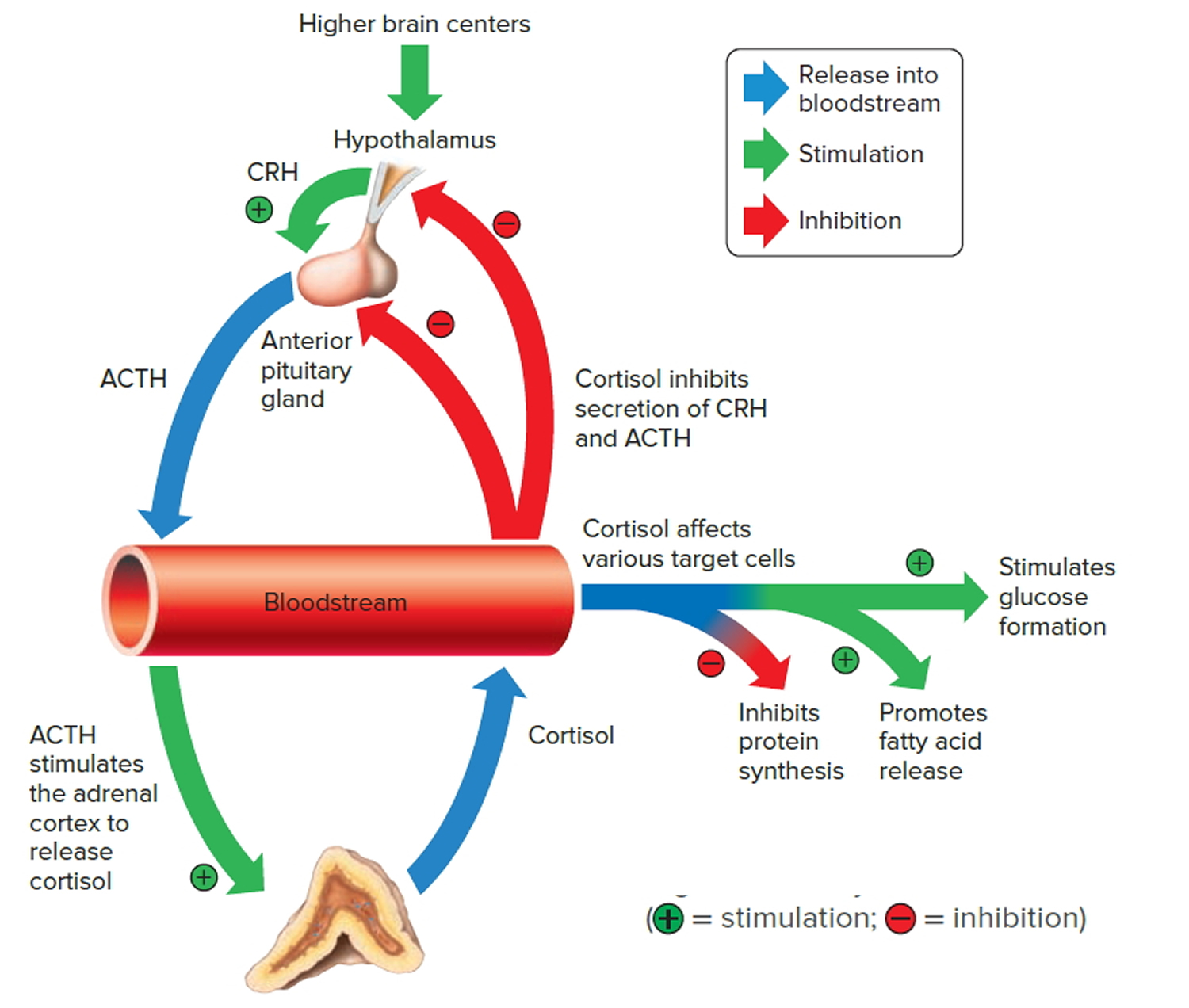
Negative feedback controls cortisol release (see Figure 4). This is much like control of thyroid hormones, involving the hypothalamus, anterior pituitary gland, and adrenal cortex. The hypothalamus secretes corticotropin-releasing hormone (CRH) into the pituitary gland portal veins, which carry CRH to the anterior pituitary, stimulating it to secrete adrenal corticotrophic hormone (ACTH). In turn, ACTH stimulates the adrenal cortex to release cortisol. Cortisol inhibits the release of CRH and ACTH, and as concentrations of these fall, cortisol production drops.
The set point of the feedback mechanism controlling cortisol secretion may change to meet the demands of changing conditions. For example, under stress—as from injury, disease, or emotional upset—information concerning the stressful condition reaches the brain. In response, brain centers signal the hypothalamus to release more corticotropin-releasing hormone (CRH), elevating the blood cortisol concentration until the stress subsides.
Figure 4. Negative feedback regulates cortisol secretion
Adrenal Medulla Hormones
Unlike the adrenal cortex, the adrenal medulla does not perform any vital functions. That is, you don’t need it to live 3. But that hardly means the adrenal medulla is useless. The hormones of the adrenal medulla are released after the sympathetic nervous system is stimulated, which occurs when you’re stressed. As such, the adrenal medulla helps you deal with physical and emotional stress.
You may be familiar with the fight-or-flight response—a process initiated by the sympathetic nervous system when your body encounters a threatening (stressful) situation. The hormones of the adrenal medulla contribute to this response.
Hormones secreted by the adrenal medulla are:
- Epinephrine: Most people know epinephrine by its other name—adrenaline. This hormone rapidly responds to stress by increasing your heart rate and rushing blood to the muscles and brain. It also spikes your blood sugar level by helping convert glycogen to glucose in the liver. (Glycogen is the liver’s storage form of glucose.)
- Norepinephrine: Also known as noradrenaline, this hormone works with epinephrine in responding to stress. However, it can cause vasoconstriction (the narrowing of blood vessels). This results in high blood pressure.
These adrenal medullary hormones have similar molecular structures and physiological functions. In fact, adrenaline (epinephrine), which makes up 80 percent
of the adrenal medullary secretion, is synthesized from noradrenaline (norepinephrine).
The effects of the adrenal medullary hormones resemble those of sympathetic neurons stimulating their effectors. The hormonal effects, however, last up to ten times longer than nervous stimulation because hormones are broken down more slowly than are neurotransmitters. Adrenaline (epinephrine) and noradrenaline (norepinephrine) increase heart rate, the force of cardiac muscle contraction, and blood glucose level. They also dilate airways, which makes breathing easier, elevate blood pressure, and decrease digestive activity.
Impulses arriving on sympathetic preganglionic nerve fibers stimulate the adrenal medulla to release its hormones at the same time that sympathetic impulses are stimulating other effectors. These sympathetic impulses originate in the hypothalamus in response to stress. In this way, adrenal medullary secretions function with the sympathetic division of the autonomic nervous system in preparing the body for energy-expending action, also called “fight-or-flight responses.”
Disorders and Diseases of the Adrenal Glands
Adrenal gland disorders occur when the adrenal glands do not work properly. They can be classified into disorders that occur when too much hormone is produced or when too little hormone is produced 1.
These disorders can occur if there is a problem with the adrenal gland itself, such as a disease, genetic mutation, tumor, or infection. Or, sometimes the disorder results from a problem in another gland, such as the pituitary, which helps to regulate the adrenal gland. In addition, some medications can cause problems with how the adrenal glands function. When the adrenal glands produce too little or too many hormones, or when too many hormones come into the body from an outside source, serious health problems can develop 4.
What causes adrenal gland disorders ?
Adrenal gland disorders are caused by problems with one or both adrenal glands or by problems with other glands, such as the pituitary gland.
Specific disorders can develop when the adrenal glands produce too few or too many hormones, or when too many hormones are introduced from an outside source 5.
There are several types of adrenal gland disorders, each with its own symptoms and treatments.
Adrenal gland tumors
Most adrenal gland tumors—abnormal growths on the adrenal glands—are not cancerous. They often do not cause symptoms or require treatment. However, adrenal gland tumors can produce a variety of different hormones, leading hormone levels to get too high.
Adrenal tumors can cause:
- Cushing’s syndrome, by producing cortisol so that body levels get too high
- Primary hyperaldosteronism, by creating high levels of the hormone aldosterone (controls blood pressure and body salt and potassium levels)
- Pheochromocytoma, by producing too much adrenaline (regulates the “fight-or-flight” response)
Cushing Syndrome
Cushing’s syndrome is a condition that occurs when the body’s tissues are constantly exposed to too much of the hormone cortisol 6. The syndrome is named after a brain surgeon, Harvey Cushing, who identified the condition in 1932 7. Cortisol is produced by the body’s two adrenal glands either in response to stress or when the cortisol levels in the blood are lower than they should be. Cortisol is a type of glucocorticoid or steroid hormone.
In the right amount, cortisol helps the body with several vital tasks:
- Maintaining blood pressure and heart function
- Controlling the immune system
- Converting fat, protein, and carbohydrates into energy
- Raising blood sugar levels as needed
- Controlling bone formation
When the body continually receives or produces too much cortisol, either from medication or as a result of a tumor, Cushing’s syndrome can develop. Many factors influence whether this happens, such as the medication dosage and how long it is taken. Or, in the case of a tumor, how large it grows before it is detected and treated.
The risk of developing Cushing’s syndrome is small; only two to three people per million are identified as having the disease each year 8, 9. Some additional cases might go undiagnosed because the symptoms are mistakenly attributed to other conditions, such as type 2 diabetes or osteoporosis 10. The most common age for Cushing’s syndrome to develop is between the ages of 20 and 50 years. Women are three times more likely than men to get Cushing’s syndrome 8.
Having certain hereditary diseases increases a person’s risk of developing a pituitary tumor 11, which can lead to Cushing’s syndrome. These hereditary diseases include:
- Multiple endocrine neoplasia type 1 (MEN 1) syndrome.
- Carney complex.
- Isolated familial acromegaly.
What’s the difference between Cushing’s syndrome and Cushing’s disease ?
Individuals with Cushing’s disease have Cushing’s syndrome symptoms. However, because their symptoms are caused specifically by a pituitary gland tumor (adenoma) that produces adrenocorticotropic hormone (ACTH), the condition is called Cushing’s disease 12. About 70% of tumor-driven Cushing’s syndrome cases are caused by Cushing’s disease 13.
What causes Cushing’s syndrome ?
Cushing’s syndrome can develop for two reasons:
- Medication containing a cortisol-like synthetic compound called glucocorticoid 14, 15.
- A benign or malignant tumor in the body that makes the adrenal gland produce too much cortisol 14, 15.
Medication
Medication is responsible for most cases of Cushing’s syndrome. It is called an exogenous cause because it originates outside the body. Cortisol-like steroid drugs, or glucocorticoids, are by far the main medications linked to Cushing’s syndrome. These medications are used to treat inflammation caused by one of several sources:
- Allergies
- Asthma
- Autoimmune diseases, where the body’s immune system attacks its own tissues
- Organ transplantation, to help avoid organ rejection
Glucocorticoids, such as prednisone, are effective at reducing inflammatory symptoms. However, taking a high dose for a long time can produce Cushing’s syndrome.
Medroxyprogesterone is another drug that occasionally causes Cushing’s syndrome. This progestin medication is taken to treat abnormal menstruation or irregular vaginal bleeding, or to prevent unusual growth of the uterine (womb) lining.
Tumors
A tumor is an endogenous cause of Cushing’s syndrome, meaning one that originates within the body. Tumors are a much less common cause of Cushing’s syndrome than are medicines.
The tumors that cause Cushing’s symptoms can be either cancerous or noncancerous 15.
Noncancerous (or Benign)
- Pituitary adenoma
- Adrenal adenoma
- Adrenal micronodular hyperplasia
- Adenomas in places other than the pituitary or adrenal glands, mostly in the lungs, pancreas, thyroid, or thymus
Cancerous (or Malignant)
- Adrenal cancer
- Cancer in places other than the pituitary or adrenal glands, mostly in the lungs, pancreas, thyroid, or thymus
How Tumors Can Cause Cushing’s Syndrome
Normally, the pituitary gland in the brain controls how much cortisol the two adrenal glands release into the bloodstream. The pituitary gland signals the adrenal glands by releasing adrenocorticotropic hormone, also known as ACTH or corticotropin. When the adrenal glands sense the ACTH, they produce more cortisol. But a tumor can disrupt that action. Tumors can either produce extra cortisol directly in their own tissue or produce extra ACTH, which triggers production of more cortisol in turn. This process can happen in three different ways:
- A benign pituitary tumor secretes ACTH. This tumor, called a pituitary adenoma, is by far the most common tumor linked to Cushing’s syndrome. As a result, the condition it causes has been given its own name, Cushing’s disease. Pituitary adenomas account for 70% (in adults) to more than 90% (in children) of endogenous (non-medicine-related) Cushing’s syndrome cases 14.
- Adrenal tumors produce cortisol in their tissue, adding to the amount already produced by the adrenal glands. These tumors cause about 15% of endogenous Cushing’s syndrome cases. Children are more likely to have this type of tumor than other tumors, but it is less common in adults. Women are more likely than men to get this type of tumor. These tumors can be adrenal adenomas, micronodular hyperplasia, or cancerous adrenal tumors.
- Some tumors not in the pituitary or adrenal glands can produce ACTH and force more cortisol production. This condition is sometimes called ectopic Cushing’s disease and accounts for about 15% of non-medicine-related Cushing’s syndrome cases 14. The tumors, either benign or cancerous, are mostly found in the lungs, pancreas, thyroid, and thymus.
Familial Cushing’s Syndrome
Individuals with some rare genetic disorders are more vulnerable to tumors in one or more glands that influence cortisol levels. As a result, these people are more likely to develop Cushing’s syndrome 15. Two such conditions are called multiple endocrine neoplasia type 1 and primary pigmented micronodular adrenal disease.
Pseudo-Cushing’s Syndrome
In rare instances, a person may have symptoms and test results that point to Cushing’s, but further testing reveals that he or she does not have the syndrome. This condition is termed “pseudo-Cushing’s syndrome.” Factors that can cause this syndrome are alcohol dependence, depression or other psychiatric disorders, extreme obesity, pregnancy, and poorly controlled diabetes 16, 17.
What are the symptoms of Cushing’s syndrome ?
Most people with Cushing’s syndrome have a range of symptoms, and one person may not have the same symptoms as another individual with Cushing’s 18, 19. The symptoms also might resemble those of other conditions 20.
Physically, someone with Cushing’s might:
- Be heavy or obese above the waist but have thin arms and legs
- Have a round, red face, sometimes referred to as a moon face
- Develop a fat lump between the shoulders, sometimes called a buffalo hump
- Have weak muscles or bones, including osteoporosis, bone pain, and fractures
- Show skin changes, including:
- + Acne or skin infections
- + Reddish-purple stretch marks called striae, which are usually at least ½-inch wide and can appear on the abdomen, buttocks, thighs, arms, and breasts
- + Thin, fragile skin that bruises easily and heals poorly
Additional symptoms can occur in specific groups of people. For example:
- Children grow heavier and grow more slowly than their peers.
- Women may have more hair on their face, neck, chest, abdomen, and thighs. Their menstrual periods may become irregular or stop.
- Men may have lower sex drives, experience impotence, and become less fertile.
The following less common symptoms may also develop:
- Mental changes, such as being depressed, anxious, or moody, or behaving differently
- Severe fatigue
- Headaches
- Thirstiness and increased need to urinate
- High blood pressure
- High blood sugar
- High cholesterol and triglycerides
Overall, the symptoms that most strongly hint at Cushing’s syndrome are fatness around the abdomen, weakness in muscles closest to the torso (such as in the shoulders and hips), wide striae (skin stripes), bruising without being bumped, unexplained osteoporosis, and—in children—slower growth and more weight gain 19, 17.
How to diagnose Cushing’s syndrome ?
Diagnosing Cushing’s syndrome can be complex and difficult. This syndrome is more easily recognized when it is fully developed, but health care providers try to diagnose and treat it well before then. No single laboratory test is perfect in diagnosing the condition, so the health care provider may try different tests.
Diagnosing Cushing’s syndrome often requires several steps. If you are being checked for Cushing’s syndrome, testing may follow this standard order 17, 10:
First Step
You will give your health care provider a complete list of all medications and other treatments you have taken to see if any of them could be responsible for raising your cortisol levels. Make certain your list includes everything containing cortisol-like compounds, such as joint or nerve injections received for pain, “tonics” and herbal medications, high doses of progesterone, and skin creams (including bleaching agents) 17.
Second Step
You will undergo a test that measures cortisol levels, and a laboratory will check to see if the cortisol levels are normal. This test might be one of the following:
- Urine Cortisol Test: Urine samples are collected and tested several times over 24 hours.
- Late-night Salivary Cortisol Test: A special device is used for collecting saliva late at night. The sample can be mailed to a laboratory or delivered to the health care provider.
- A Test Using Dexamethasone: This test might be called a 1 mg overnight suppression test or a 2 mg 48-hour suppression test. Several hours after you take the synthetic glucocorticoid dexamethasone by mouth, your blood level of cortisol is measured.
Third Step
If your results are not normal, your health care provider may do further tests or refer you to a specialist—an endocrinologist —for the tests, which might include:
- Another test from Step 2
- Serum midnight cortisol test, which measures blood levels of cortisol late at night
- Dexamethasone-corticotropin-releasing hormone (Dex-CRH) test, which distinguishes between cells produced by tumors and normal cells
Fourth Step
If your cortisol levels are persistently above normal and you receive a diagnosis of Cushing’s, your health care specialist will check for the cause. Tests might include the following:
- CRH stimulation test
- High-dose dexamethasone suppression test
- Radiologic imaging to see the glands that might be causing symptoms
- Petrosal sinus sampling to visualize the pituitary gland
- Blood sampling to confirm the pituitary gland is making too much ACTH
Different Testing Steps
If you have certain medical conditions, your health care provider may not use some of the tests listed above for reasons of safety or effectiveness. These conditions can include:
- Pregnancy
- Epilepsy
- Renal failure
- Cyclic Cushing’s syndrome, in which cortisol is sometimes normal and sometimes high
- Adrenal incidentaloma
Several kinds of medications unrelated to Cushing’s syndrome may interfere with test results, but your health care provider should be aware of these drugs and know whether you are taking them 17.
Is there a cure for Cushing’s syndrome ?
Untreated Cushing’s syndrome can be life-threatening—fortunately, most people with the syndrome are treated and cured, and have a normal life expectancy 21.
Sometimes, however, treatment for the syndrome does not return people to the same level of health as before 17. For instance, their bones may not regain their previous strength. People who no longer have Cushing’s might be more likely to develop diabetes, high blood pressure, or high cholesterol 22. It also is possible that a person cured of Cushing’s disease might not recover their previous mental strength, including memory, but any functional difference is usually small 23, 24.
People whose Cushing’s syndrome was caused by a tumor need to have regular checkups for the rest of their lives. Surgical removal of a tumor is usually 100% successful, but in rare cases some of the tumor is missed and can grow again 25, 26.
What are the treatments for Cushing’s syndrome ?
Treatment for Cushing’s syndrome depends on the reason for the extra cortisol in the body 26, 27.
Medicine
If Cushing’s syndrome is caused by glucocorticoid medicine taken to treat another disorder, your health care provider will slowly and carefully decrease your dose and give you another medication so that your body can go back to making its own cortisol. However, if you need to continue taking the glucocorticoid, your health care provider will closely monitor you and treat symptoms that might develop, such as high blood sugar, high cholesterol levels, bone thinning, or osteoporosis.
Tumors
If the body is making too much cortisol because of a tumor, treatments may include medication, surgery, radiation, chemotherapy, or a combination of these treatments. Treatments will differ based on where the tumor is located.
Pituitary tumors
The most common treatments for pituitary tumors are:
- Surgery. In most cases, a surgeon removes the tumor through a cut made under the upper lip or at the bottom of the nose, between the nostrils. In other cases, the doctor may cut through the skull to reach the pituitary tumor.
- Radiation therapy. Radiation therapy targets the tumor with high-energy X-rays that kill tumor cells or keep them from growing. It can be used if surgery is not successful at removing all tumor cells.
- Chemotherapy. Chemotherapy uses drugs that kill tumor cells or keep them from growing. Chemotherapy drugs can be taken by mouth or injected.
- Drug therapy. Pituitary tumors can affect hormone levels in two ways. They can produce hormones themselves or crowd out tissue that normally would produce hormones. Drugs can be prescribed to correct these hormone imbalances or reduce too much cortisol.
Adrenal tumor or other tumors
If the tumor is in one of your two adrenal glands, your doctor may attempt to remove it surgically. Often the whole gland is removed. Following surgery, the doctor may prescribe glucocorticoid replacement treatment for 9 to 12 months until the other adrenal gland takes on the functions of the removed gland.
If the tumor cannot be removed, medications can be used to help block the release of cortisol. Radiation therapy usually does not work for cancerous adrenal tumors and is not appropriate for noncancerous tumors.
Very rarely, a surgeon cannot remove all of the cancerous cells causing Cushing’s syndrome. And sometimes a person needs to continue taking glucocorticoid medication to treat an underlying condition, even though the medication is causing Cushing’s syndrome. In both these cases, Cushing’s syndrome symptoms might become worse, and additional symptoms can appear over time, including:
- Persistent fatigue
- Muscle weakness
- Abdominal and facial weight gain
- Depression
- Mood swings
- High blood pressure
- High blood sugar
Health care providers can prescribe medication or other therapy to reduce these symptoms.
Cushing’s syndrome can affect fertility in both men and women
Women
The high levels of cortisol in Cushing’s syndrome disrupt a woman’s ovaries. Her menstrual periods may stop completely or become irregular. As a result, women with Cushing’s syndrome almost always have difficulty becoming pregnant 28, 29, 30. For those who do become pregnant, the risk of miscarriage is high 28, 29, 30.
In rare cases, usually when a woman’s Cushing’s syndrome is caused by a benign adrenal tumor, pregnancy can occur, but it brings high risk for the mother and fetus 28, 29, 30.
Cushing’s syndrome can cause serious and potentially life-threatening effects for the mother and the fetus during pregnancy 31, 32. For example, Cushing’s syndrome raises a woman’s risk of developing pregnancy-related high blood pressure called preeclampsia or eclampsia and/or pregnancy diabetes, which also is called gestational diabetes. Infection and slow healing of any wounds are more likely, as is heart failure. When the syndrome is caused by a tumor, it will be surgically removed as early as possible to reduce any threat 33.
After a woman is treated for Cushing’s syndrome, her ovaries often recover from the effects of too much cortisol. Her regular menstrual cycles will return, and she can become pregnant 34.
In some women, regular periods do not return after they are treated for Cushing’s syndrome. This occurs if surgery removes the part of the pituitary gland involved in reproduction 35. An infertility specialist can prescribe hormone therapy to bring back regular periods, ovulation, and fertility 34.
Men
A man diagnosed with Cushing’s syndrome may have a decline in sperm production and could have reduced fertility 36. He also might experience a lowered sex drive as well as impotence. In addition, some medications used to treat Cushing’s syndrome can reduce fertility.10 However, fertility usually recovers after Cushing’s syndrome is cured and treatment has stopped 37.
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia is a group of inherited disorders of the adrenal glands 38. It affects men and women equally. Both parents must carry the gene in order for a child to be born with congenital adrenal hyperplasia. Researchers have identified the location of the gene that causes the most common forms of congenital adrenal hyperplasia as chromosome 6 39.
Most commonly, congenital adrenal hyperplasia causes male-like characteristics (such as excessive hair growth in girls/women, early hair development in boys) and puberty to occur too early in children. Girls with congenital adrenal hyperplasia may be born with ambiguity of their external genitalia, meaning their genitalia do not look like typical female genitalia.
Congenital adrenal hyperplasia can be categorized as classic or nonclassic. The most common type of congenital adrenal hyperplasia can be life-threatening if it is left undiagnosed and untreated in newborns. Most patients with congenital adrenal hyperplasia must take daily medications to treat the symptoms.
Congenital adrenal hyperplasia is caused by three disturbances:
- Too little cortisol. The adrenal glands of infants born with congenital adrenal hyperplasia cannot make enough of the hormone cortisol. This hormone affects energy levels, blood sugar levels, blood pressure, and the body’s response to stress, illness, and injury.
- Too little aldosterone. In about 75% of cases, infants born with congenital adrenal hyperplasia cannot make enough of the hormone aldosterone, which helps the body maintain the proper level of sodium (salt) and water and helps maintain blood pressure.
- Too much androgens. In certain cases, infants born with congenital adrenal hyperplasia produce too much of male hormones, androgens. Proper levels of these hormones are needed for normal growth and development in both boys and girls.
Congenital adrenal hyperplasia can also cause imbalances in the hormone adrenaline, which affects blood sugar levels, blood pressure, and the body’s response to stress 40, 41.
The hormone imbalances in most cases of congenital adrenal hyperplasia (about 95%) are caused by too little of a substance called 21-hydroxylase. The adrenal glands need 21-hydroxylase to make proper amounts of hormones. This type of congenital adrenal hyperplasia is sometimes referred to as 21-hydroxylase deficiency. In congenital adrenal hyperplasia due to 21-hydroxylase deficiency, the adrenal glands cannot make enough cortisol or aldosterone. In addition, the glands make too much androgen. People with 21-hydroxylase deficiency also may not produce enough adrenaline.
About 5% of cases of congenital adrenal hyperplasia are caused by deficiency in a substance similar to 21-hydroxylase, called 11-hydroxylase. This type of congenital adrenal hyperplasia is sometimes referred to as 11-hydroxylase deficiency. In congenital adrenal hyperplasia due to 11-hydroxylase deficiency, the adrenal glands make too little cortisol and too many androgens. This type of congenital adrenal hyperplasia does not result in aldosterone deficiency.
Other very rare types of congenital adrenal hyperplasia include 3-betahydroxy-steroid dehydrogenase deficiency, lipoid congenital adrenal hyperplasia, and 17-hydroxylase deficiency.
Congenital adrenal hyperplasia can be categorized as classic or nonclassic types based on severity:
- Classic congenital adrenal hyperplasia is more severe than the nonclassic form. It can be life threatening in newborns if it is not diagnosed. Classic congenital adrenal hyperplasia can be caused by either 21-hydroxylase or 11-hydroxylase deficiency. Classic congenital adrenal hyperplasia occurs in about one of every 15,000 births worldwide 42.
- Nonclassic congenital adrenal hyperplasia is sometimes called late-onset congenital adrenal hyperplasia. It is a milder form of the disorder that usually is diagnosed in late childhood or early adolescence. Sometimes, people have nonclassic congenital adrenal hyperplasia and never know it. This form of congenital adrenal hyperplasia is almost always caused by 21-hydroxylase deficiency. Nonclassic congenital adrenal hyperplasia is among the most common genetic disorders. It occurs in about one of every 1,000 people, but could occur in as many as 1 in 20 people in some communities 43. It is more common in Ashkenazi Jews, Hispanics, Italians, and Yugoslavians 40.
What causes congenital adrenal hyperplasia ?
Congenital adrenal hyperplasia is caused by changes (mutations) in one of several genes. These changes lead to deficiencies in 21-hydroxylase or, less commonly, 11-hydroxylase. Both of these are chemicals called enzymes. The adrenal glands need these enzymes to make proper amounts of the hormones: cortisol, aldosterone, androgens, and adrenaline.
How is congenital adrenal hyperplasia inherited ?
The genes for congenital adrenal hyperplasia are passed down from parents to their children. In general, people have two copies of every gene in their bodies. They receive one copy from each parent. For an infant to have congenital adrenal hyperplasia, both copies must have an error that affects an adrenal-gland enzyme.
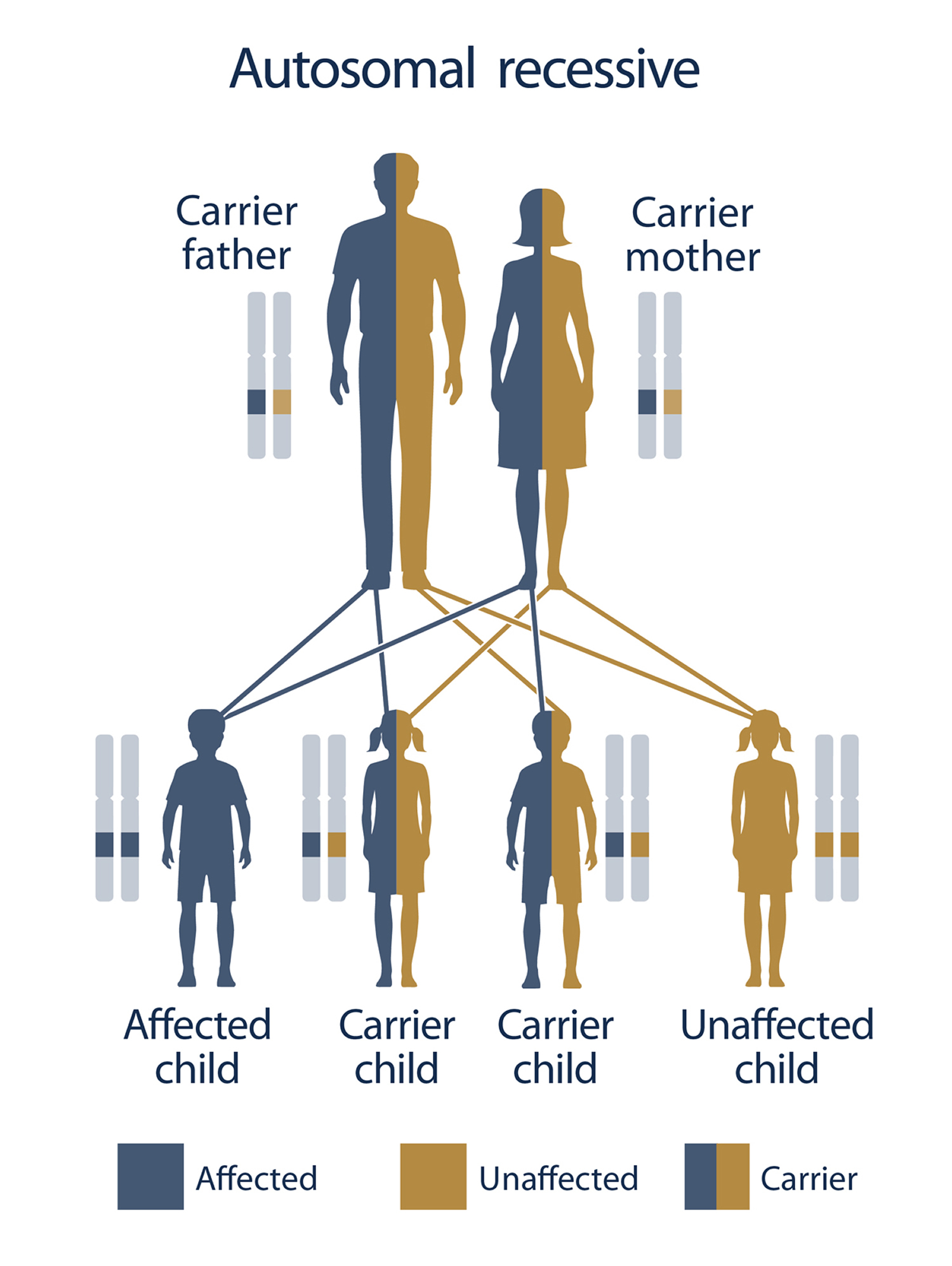
Congenital adrenal hyperplasia is an example of an autosomal recessive disorder:
- Autosomal means the gene is not on the X chromosome or Y chromosome.
- Recessive means that both copies of the gene must have the error for the disease or disorder to occur.
If both parents have congenital adrenal hyperplasia, all of their children will also have it. If each parent carries one affected gene and one normal gene (called a “carrier”), there is a one-in-four chance of their child having congenital adrenal hyperplasia (see Figure 5).
Anyone with congenital adrenal hyperplasia, or from a family in which congenital adrenal hyperplasia has been diagnosed, should consider genetic counseling. Genetic counselors discuss all options for having a child. They explain the risks and benefits of each option.
The genes for congenital adrenal hyperplasia are passed down from parents to their children. In general, people have two copies of every gene in their bodies. They receive one copy from each parent. For an infant to have congenital adrenal hyperplasia, both copies must have an error that affects an adrenal-gland enzyme.
Women with congenital adrenal hyperplasia can get pregnant. In some of the women, high levels of androgens disrupt the regular release of the egg from the ovary, a process known as ovulation. Some women also have irregular menstrual cycles. These problems can make it more difficult to get pregnant. These women often can be helped with medicines. Women with congenital adrenal hyperplasia who want to become pregnant can meet with a reproductive endocrinologist. This is a health care provider who specializes in fertility issues.
Women with congenital adrenal hyperplasia who become pregnant should continue taking their medications.
Men with congenital adrenal hyperplasia can father children. The main challenges for these men are low testosterone (a hormone important for male fertility and sexual function), and growths in the testicles called adrenal rest tissue. These problems can cause reduced sperm production. These issues tend to occur when hormone imbalances are not well controlled with medicines. Men who wish to father children should take all medicines as directed. A health care provider may recommend that males with congenital adrenal hyperplasia who have gone through puberty get an ultrasound of the testicles. The ultrasound provides a picture of the inside of the testicle and can help a health care provider detect abnormal growths. Future ultrasounds can be compared with the original to quickly identify any problems.
Figure 5. Inheritance of an autosomal recessive disorder from carrier parents
What are the symptoms of congenital adrenal hyperplasia ?
Classic congenital adrenal hyperplasia
Symptoms of classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency (95% of classic congenital adrenal hyperplasia cases) can be grouped into two types according to their severity: salt wasting and simple virilizing (also called non-salt wasting).
Symptoms of classic congenital adrenal hyperplasia due to 11-hydroxylase deficiency (5% of classic congenital adrenal hyperplasia cases) are similar to those of simple virilizing congenital adrenal hyperplasia. About two-thirds of people with classic 11-hydroxylase deficiency also have high blood pressure (hypertension) 44.
Salt-wasting congenital adrenal hyperplasia:
Salt-wasting congenital adrenal hyperplasia is the severe form of classic 21-hydroxylase deficiency. In this type of congenital adrenal hyperplasia, the adrenal glands make too little aldosterone, causing the body to be unable to retain enough sodium (salt). Too much sodium is lost in urine (thus the name, “salt-wasting”). If undiagnosed, symptoms of classic salt-wasting congenital adrenal hyperplasia appear within days or weeks of birth and, in some cases, death occurs.
Symptoms may include:
- Dehydration
- Poor feeding
- Diarrhea
- Vomiting
- Heart rhythm problems (arrhythmias)
- Low blood pressure
- Very low blood sodium levels
- Low blood glucose
- Too much acid in the blood, called metabolic acidosis
- Weight loss
- Shock, a condition where not enough blood gets to the brain and other organs. Shock in infants with salt-wasting is called adrenal crisis. Signs include confusion, irritability, rapid heart rate, and/or coma.
Even when carefully treated, children with salt-wasting congenital adrenal hyperplasia are still at risk for adrenal crises when they become ill or are under physical stress. The body needs more than the usual amount of adrenal hormones during illness, injury, or physical stress. This means a child with congenital adrenal hyperplasia must be given more medication during these times to prevent an adrenal crisis.
Salt-wasting congenital adrenal hyperplasia also involves symptoms caused by low cortisol and high androgens. These symptoms may include:
- In female newborns, external genitalia can be ambiguous, i.e., not typical female appearing , with normal internal reproductive organs (ovaries, uterus, and fallopian tubes)
- Enlarged genitalia in male newborns
- Development of certain qualities called virilization in boys or girls before the normal age of puberty, sometimes as early as age 2 or 3. This is a condition characterized by:
- +Rapid growth
- + Appearance of pubic and armpit hair
- + Deep voice
- + Failure to menstruate, or abnormal or irregular menstrual periods (females)
- + Well-developed muscles
- + Enlarged penis (males)
- +Unusually tall height as children, but being shorter than normal as adults
- + Possible difficulties getting pregnant (females)
- + Excess facial hair (females)
- + Early beard (males)
- + Severe acne
- + Benign testicular tumors and infertility (males)
Simple virilizing (non-salt wasting) congenital adrenal hyperplasia:
Simple virilizing congenital adrenal hyperplasia is the moderate form of classic 21-hydroxylase deficiency. This type of congenital adrenal hyperplasia involves less severe aldosterone deficiency. Therefore, there are no severe or life-threatening sodium-deficiency symptoms in newborns. Like salt-wasting congenital adrenal hyperplasia, simple virilizing congenital adrenal hyperplasia involves too little cortisol and too much androgen. Female newborns have ambiguous genitalia and young children display virilization.
Nonclassic congenital adrenal hyperplasia
Almost all cases of nonclassic congenital adrenal hyperplasia are caused by a mild 21-hydroxylase deficiency. Most symptoms of nonclassic congenital adrenal hyperplasia are related to increased androgens. Symptoms can show up in childhood, adolescence, or early adulthood.
Symptoms of nonclassic congenital adrenal hyperplasia can include:
- Rapid growth in childhood and early teens but shorter height than both parents
- Early signs of puberty
- Acne
- Irregular menstrual periods (females)
- Fertility problems (in about 10% to 15% of women)
- Excess facial or body hair in women
- Male-pattern baldness (hair loss near the temples)
- Enlarged penis (males)
- Small testicles (males)
Some people have nonclassic congenital adrenal hyperplasia and never know it because the symptoms are so mild 45, 46.
How to diagnose congenital adrenal hyperplasia ?
During Pregnancy
If a woman already has a child with congenital adrenal hyperplasia and becomes pregnant with the same partner, her fetus has a one in four chance of having congenital adrenal hyperplasia. For this reason, prenatal testing can be done for some forms of congenital adrenal hyperplasia. A health care provider checks for the disorder by using techniques called amniocentesis or chorionic villus sampling 46.
- Amniocentesis. This involves inserting a needle into the womb, through the abdomen, to withdraw a small amount of fluid from the sac that surrounds the fetus. The procedure is usually done between the 15th and 20th week of pregnancy.
- Chorionic villus sampling. This is similar to amniocentesis. A health care provider inserts a needle into the womb, either through the abdomen or the cervix, and extracts a small piece of tissue from the chorionic villi (the tissue that will later become the placenta). This procedure is usually done between the 10th and 12th week of pregnancy.
After a health care provider takes a sample using one of these techniques, he or she will perform a genetic test on the sample. This test will reveal whether the fetus has a gene change that causes congenital adrenal hyperplasia.
Parents may also choose to wait until birth to have the newborn tested. Talking to their health care providers may help parents identify the option that is right for them.
At Birth
All U.S. states have neonatal screening for congenital adrenal hyperplasia. Infants who test positive need to have follow-up testing done to confirm the diagnosis. If, for some reason, the neonatal screening is negative but there is high suspicion for congenital adrenal hyperplasia (such as ambiguous genitalia), further evaluation is also indicated.
Later in Life
Newborns do not show symptoms of nonclassic congenital adrenal hyperplasia, and the test done on newborns does not detect nonclassic congenital adrenal hyperplasia. Nonclassic congenital adrenal hyperplasia is diagnosed in childhood or adulthood, when symptoms appear. To diagnose nonclassic congenital adrenal hyperplasia, a health care provider may:
- Ask whether family members have congenital adrenal hyperplasia.
- Do a physical exam.
- Take blood and urine to measure hormone levels.
- Do a genetic test to determine if the patient has the gene change that causes congenital adrenal hyperplasia.
An X-ray can help to diagnose congenital adrenal hyperplasia in children. Because some children with congenital adrenal hyperplasia grow too quickly, their bones will be more developed than normal for their age 47.
What are the treatments for congenital adrenal hyperplasia ?
Treatments for congenital adrenal hyperplasia include medication and surgery as well as psychological support 48, 40, 49, 46.
Medication
- Classic congenital adrenal hyperplasia
Newborns with classic congenital adrenal hyperplasia should start treatment very soon after birth to reduce the effects of congenital adrenal hyperplasia. Classic congenital adrenal hyperplasia is treated with steroids that replace the low hormones.
- Infants and children usually take a form of cortisol called hydrocortisone.
- Adults take hydrocortisone, prednisone, or dexamethasone, which also replace cortisol.
- Patients with classic congenital adrenal hyperplasia also take another medicine, fludrocortisone, to replace aldosterone.
- Eating salty foods or taking salt pills may also help salt-wasters retain salt 50.
The body needs more cortisol when it is under physical stress. Adults and children with classic congenital adrenal hyperplasia need close medical attention and may need to take more of their medication during these times. They may also need more medication if they:
- Have an illness with a high fever.
- Undergo surgery.
- Sustain a major injury.
People who have classic congenital adrenal hyperplasia need to wear a medical alert identification bracelet or necklace. To alert medical professionals in case of an emergency, the bracelet or necklace should read: “adrenal insufficiency, requires hydrocortisone.” Adults or parents also need to learn how to give an injection of hydrocortisone if there is an emergency.
Patients with classic congenital adrenal hyperplasia need to take medication daily for their entire lives. If a patient stops taking his or her medication, symptoms will return.
The body makes different amounts of cortisol at different times in life, so sometimes a patient’s dose of medication may be too high or too low. Taking too much medication to replace cortisol can cause symptoms of Cushing’s syndrome. These include:
- Weight gain
- Slowed growth
- Stretch marks on the skin
- Rounded face
- High blood pressure
- Bone loss
- High blood sugar
It is important to alert the health care provider if these symptoms appear so that he or she can adjust the medication dose.
- Nonclassic congenital adrenal hyperplasia
People with nonclassic congenital adrenal hyperplasia may not need treatment if they do not have symptoms. Individuals with symptoms are given low doses of the same cortisol replacing medication taken by people with classic congenital adrenal hyperplasia.
Symptoms of nonclassic congenital adrenal hyperplasia that signal that the patient may need treatment are:
- Early puberty
- Excess body hair
- Irregular menstrual periods (females)
- Infertility
It may be possible for patients with nonclassic congenital adrenal hyperplasia to stop medication as adults if their symptoms go away.
Surgery
- Classic congenital adrenal hyperplasia
Girls who are born with ambiguous external genitalia usually have surgery. For example, surgery is necessary if changes to the genitals have affected urine flow. Surgery to make the genitals look more female also can be done.
The Endocrine Society, which supports hormonal research and clinical practice, recommends that this feminizing surgery be considered during infancy. If it is done, the group recommends choosing an experienced surgeon who practices in a center that sees many congenital adrenal hyperplasia cases 51.
The Congenital Adrenal Hyperplasia Research, Education & Support (CARES) Foundation strongly recommends delaying surgery until:
- The child is medically stable,
- Parents are fully informed of the risks and benefits, and
- A surgeon with expertise in this type of procedure is found.
Parents should also find a psychologist, social worker, or other mental health professional to support them in their decision making. It is important to find an experienced mental health provider whose expertise includes working with children who have congenital adrenal hyperplasia and their special needs 52.
- Nonclassic congenital adrenal hyperplasia
Girls with nonclassic congenital adrenal hyperplasia have normal genitals, so they do not need surgery.
Pituitary Tumors
Pituitary tumors are abnormal growths of cells in the tissue of the pituitary gland. The pituitary gland is a bean-shaped organ in the center of the brain, just behind and between the eyes. The pituitary gland causes the release of hormones in the body that control growth, metabolism, response to stress, and sexual and reproductive function 53.
Doctors and scientists classify pituitary tumors according to whether or not they spread beyond the pituitary gland 54:
- Pituitary adenomas are benign, meaning they are noncancerous and do not spread to other parts of the body. Most pituitary tumors fall into this category. Despite being benign, pituitary adenomas can make the pituitary gland produce too much or too little of certain hormones, causing health problems.
- Pituitary carcinomas are malignant. This means they can spread beyond the pituitary gland into the brain or spinal cord, or into other parts of the body. Very few pituitary tumors are carcinomas.
Their size:
- Microadenomas are smaller than 1 centimeter. Most pituitary adenomas are microadenomas.
- Macroadenomas are 1 centimeter or larger.
Whether they secrete hormones 54:
Functioning pituitary tumors (also called secretory tumors) produce levels of hormones that are too high. Most pituitary tumors are functioning tumors. The symptoms they cause are due to the excessive levels of hormones they produce. These hormones play important roles in the healthy functioning of the body:
- Prolactin causes a woman’s breasts to make milk during and after pregnancy.
- Adrenocorticotropic hormone (ACTH) is involved in the body’s response to stress.
- Growth hormone helps control body growth and metabolism.
- Thyroid-stimulating hormone is involved in growth, body temperature, and heart rate.
Nonfunctioning pituitary tumors (also called nonsecretory tumors) do not produce hormones. They can press on or damage the pituitary gland and prevent it from secreting adequate levels of hormones.
For more information on Pituitary tumor go to What does the pituitary gland do ?
Pheochromocytoma and Paraganglioma
What is pheochromocytoma ?
Pheochromocytoma is a rare tumor that develops in the adrenal glands. Pheochromocytoma can occur at any age. However, it is diagnosed most frequently between the ages of 30 and 50 55. Up to 20% of pheochromocytomas are diagnosed in children 56. The number of new cases of pheochromocytoma is estimated to be two to eight cases per million people per year 57, although the actual prevalence may be higher 58. For malignant pheochromocytoma, the incidence was 93 cases per 400 million people in the United States in 2002 59.
Pheochromocytomas may be found in one or both adrenal glands and may spread, or metastasize, beyond the adrenal glands. Pheochromocytomas develop from the center of the adrenal gland, in an area called the adrenal medulla, which secretes catecholamines 60.
Hormones that are normally produced by the adrenal medulla, catecholamines, help to regulate heart rate, blood pressure, and the body’s responses to stress. Pheochromocytomas release additional catecholamines, causing higher than normal amounts in the body 61. Changes in hormone levels produce some of the clinical signs and life-threatening symptoms of pheochromocytoma.
About 70% of pheochromocytomas are benign (non-cancerous or non-metastatic); 30% are malignant (cancerous or metastatic) and spread to other parts of the body 62. Common sites to which malignant pheochromocytomas spread, or metastasize, include the liver, lungs, bone, and distant lymph nodes 61.
Pheochromocytomas are equally common in males and females 55.
Pheochromocytomas are diagnosed in people of all races but are less common in African Americans 63.
What is paraganglioma ?
Paragangliomas are tumors originating from neuronal tissue; they were formerly called extra-adrenal pheochromocytomas. There are developmentally two subgroups of these tumors: parasympathetic paragangliomas and sympathetic paragangliomas. These subgroups differ in the type of tissue from which they form (parasympathetic versus sympathetic) and also in their location and hormonal production. Parasympathetic tissue is important for certain body processes, including salivation, urination, and digestion. Sympathetic tissue forms the tissue important for “fight-or-flight” responses.
The group of paragangliomas that develop from parasympathetic-associated tissue in the head and neck are usually referred to as “head and neck paragangliomas.” These tumors can be locally invasive but usually do not metastasize or produce catecholamines (stress hormones). Signs and symptoms of head and neck paragangliomas are usually due to the tumor mass itself instead of the secreted catecholamines.
Paragangliomas that develop from sympathetic neuronal tissue are usually localized in the chest, abdomen, or pelvis. These tumors often have excessive hormone secretion, which makes them very similar to pheochromocytomas. As a result of excessive hormone secretion, paragangliomas often cause the signs and symptoms described below (e.g., heart palpitations, irregular heartbeat, hypertension, and sweating). Sympathetic (or extra-adrenal) paragangliomas generally tend to be more malignant than pheochromocytomas (localized to the adrenal gland) 64.
What are common symptoms of pheochromocytoma ?
Pheochromocytoma causes a variety of signs and symptoms, including (in alphabetical order):
- Abdominal pain
- Constipation
- Chest pain
- Dizziness
- Elevated blood sugar
- Facial flushing (redness)
- High blood pressure
- Increased respiratory rate
- Nausea
- Nervousness, anxiety, and irritability
- Pale skin tone
- Rapid heart rate and heart palpitations
- Severe headaches
- Sweating
- Visual disturbances
- Weight loss
What causes pheochromocytoma?
Approximately one-third of pheochromocytoma cases occur when patients inherit a mutated gene from their parents 65. Studies have linked several genes to the disease, but researchers are not sure how these genes contribute to the formation of this tumor 66.
The remaining two-thirds of cases are spontaneous and are not associated with a family history. However, genetic inheritance may play a role in the development of the disease through unknown genes. For example, in one study, a significant percentage of patients (7.5% to 27%) with sporadic pheochromocytoma had genetic mutations that have been linked to other family-inherited syndromes 67.
Children are at risk of developing pheochromocytoma/paraganglioma if they inherit one of the known disease-causing mutated genes from their parents. The penetrance of the disease, or the percentage of individuals with a known genetic mutation who develop the disease, varies with different mutations.
Pregnancy may be complicated by the clinical signs of pheochromocytoma. In pregnant women, high blood pressure, cardiovascular problems, and seizures, which occur in patients with pheochromocytoma, could affect the health of the unborn child 55.
Genetic testing is available for pheochromocytoma. Genetic testing is especially recommended for some groups of patients who 68:
- Have a family history of pheochromocytoma
- Have clinical features of syndromes associated with pheochromocytoma
- Have multiple tumors or an extra-adrenal tumor (tumor is at a location other than the adrenal glands) (paraganglioma)
- Are diagnosed before age 50
- Have specific biochemical results
Genetic testing may also be recommended for other patients depending upon family history, the patient’s age, and the location of the patient’s tumor.
How to diagnose pheochromocytoma
A health care provider uses blood and urine tests that measure catecholamines and/or their metabolites to diagnose pheochromocytoma. Metabolites are biochemical substances that form when another is broken down in the body. Higher than normal amounts of these biochemical substances in the blood and/or urine can be an indication of the presence of a pheochromocytoma/paraganglioma.
Pheochromocytomas can secrete all, none, or any combination of catecholamines (epinephrine, norepinephrine, dopamine) and their metabolites, which are called metanephrines (metanephrine, normetanephrine, methoxytyramine) 69.
Tumors can also be found accidentally during non-related imaging studies. The location of a pheochromocytoma can be determined by using several imaging methods, including computed tomography (CT) and magnetic resonance imaging (MRI). CT scans use X-rays to produce detailed images of the inside of the body, while MRI uses magnetic waves to produce these pictures. A third imaging method that can be used to detect pheochromocytomas is MIBG (metaiodobenzylguanidine) scintigraphy. During this procedure, MIBG, a compound containing a small amount of radioactivity, is injected into a vein and is picked up by pheochromocytoma cells, but not normal cells 69. The body is scanned with a scanner that detects the MIBG. Any MIBG that is seen with the scanner can indicate the presence of pheochromocytoma cells.
What are the treatments for pheochromocytoma ?
Standard treatments for pheochromocytoma include 70, 71:
- Surgical removal of the tumor
- Medications (chemotherapy) designed to kill tumor cells
- Radiotherapy: utilizing radio waves to destroy the tumors
- Medications to control the signs and symptoms of the disease.
The prognosis for patients with a completely removed tumor is excellent 72. The 5-year survival rate for patients with benign pheochromocytomas is between 86% and 92%.
In patients with malignant pheochromocytomas, the 5-year survival rate may be less than 50%, although prospective studies are missing 73. Life expectancy varies for patients with malignant pheochromocytoma depending on where the tumor has spread. Survival is generally less than 5 years when tumors have spread to the liver and lung and somewhat longer when tumors have spread to the bones 74.
Types of Therapies for Pheochromocytoma:
Ninety percent of patients are cured by surgery to remove benign pheochromocytoma tumors 70. Surgery for tumor removal is typically done by laparoscopy, during which a small incision is made in the abdomen. During surgery to remove the tumor, the physician will usually examine nearby organs to determine whether the pheochromocytoma has spread to other parts of the body.
Medications are prescribed to treat the clinical signs and symptoms of pheochromocytoma.
Commonly prescribed medications include the following 70:
- Alpha-adrenergic blockers to lower blood pressure
- Beta blockers for controlling rapid, irregular pulse
For tumors that are successfully removed, blood pressure and hormone levels usually return to normal over the weeks immediately following surgery.
Is there a cure for pheochromocytoma ?
The success of pheochromocytoma treatment depends upon several factors; the most important include: presence of metastasis, genetics, location, and overall extent of the disease. Malignant pheochromocytoma can only be determined by the presence of metastasis or tumor spreading (tumors in locations such as the bone, liver, lungs, or lymph nodes).
The only curative treatment for pheochromocytoma is complete surgical removal of the tumor. The long-term prognosis of patients after resection of a single sporadic pheochromocytoma is excellent. However, it is common for a patient to continue to have high blood pressure after surgery (nearly 50% of patients).
Recurrence (or return) of the disease occurs in 17% of patients, with half of these patients showing signs of the malignant form of the disease. Recurrence happens more often in patients with paragangliomas (33% have recurrence) than in those with pheochromocytomas (14%). Additionally, patients with family members who have the disease have recurrence more often (33%) than patients who do not (13%) 72.
There is currently no cure for malignant pheochromocytoma. Radiation therapy, or the use of radio waves to destroy tumors, can assist in shrinking some malignant tumors 70. Tumor shrinkage can lessen the clinical signs and symptoms of pheochromocytoma by reducing the production of hormones and in some cases may allow for surgery.
Although not curative, medications are used to control the clinical signs and symptoms of both benign and malignant pheochromocytoma.
Addison’s disease
This rare disorder develops when the adrenal glands do not make enough cortisol 75. In most cases of Addison’s disease, the body also doesn’t make enough of the hormone aldosterone 75.
Addison’s is an autoimmune disease—a condition in which the immune system, which is supposed to protect the body, mistakenly attacks the body’s own tissues and cells. In the case of Addison’s disease, this reaction results in damage to the adrenal glands 76. In the long term, this damage can get worse until eventually the adrenal glands aren’t working at all.
One in 15,000 people have Addison’s disease, so it is a rare condition. Around two to three times more women than men get this disease, because autoimmunity is more common in women. The peak age of the start of Addison’s disease is between 30 and 50, but it can happen at any age. Those people with other autoimmune diseases, such as Graves’ disease, hypothyroidism, type 1 diabetes, pernicious anaemia and vitiligo, have a greater risk of also developing autoimmune Addison’s disease.
Very rare genetic disorders may cause different effects such as abnormal development of the adrenal glands, problems with production of adrenal hormone or failure of the adrenal glands to respond to the pituitary adrenocorticotropic hormone.
What causes Addison’s disease ?
By far the most common cause of Addison’s disease is autoimmunity. This is known as autoimmune Addison’s disease. Normally the body’s immune system attacks invading viruses and bacteria to defend the body. In cases of autoimmunity, the immune system makes a mistake, attacking and destroying the adrenal cortex as if it was an infection.
Rare causes of Addison’s disease include infections such as tuberculosis, removal of the adrenal glands by surgery, bleeding into the adrenal glands (for instance after abdominal injuries), cancer of the adrenal glands and genetic defects such as adrenoleukodystrophy.
The pituitary gland produces hormones that affect the adrenal gland. If the pituitary gland stops working properly, this can cause secondary adrenal insufficiency.
What are the signs and symptoms of Addison’s disease ?
Symptoms of Addison’s disease can vary, depending on what causes the disease.
The signs and symptoms of Addison’s disease usually appear very gradually, as it takes months or years for the adrenal cortex to be destroyed significantly enough to cause symptoms.
Symptoms typically include 77, 78:
- Weight loss
- Weakness
- Extreme fatigue
- Nausea and/or vomiting
- Low blood pressure
- Patches of darker skin just like a suntan
- Craving for salt
- Dizziness upon standing
- Depression
- New scars may heal with too much colour around them and this can also be noticed in skin creases on the palms of the hands and inside the mouth.
- In women, loss of body hair may occur and periods may stop.
How to diagnose Addison’s disease
There may be some clues from simple blood tests, such as a low sodium level or a high potassium level in the blood. Diagnosis depends on showing that the secretion of cortisol is low and that the adrenal glands themselves are damaged. The most commonly used test is called the short synacthen test, where a hormone similar to pituitary adrenocorticotropic hormone is injected into the bloodstream to stimulate the adrenal glands. The cortisol level in the blood is measured immediately before the injection is given and again after 30–60 minutes. For more information on diagnosis, see our information leaflet on adrenal insufficiency in the topical issues section of this website.
If the adrenal glands are healthy, cortisol production in the second sample will exceed a certain level, commonly 500–550 nmol/l. By contrast, failing adrenal glands will not be able to produce this amount of cortisol. It is important that this test is carried out under the supervision of an endocrinologist.
Drawing only baseline blood samples for cortisol without injecting adrenocorticotropic hormone to stimulate cortisol production, is only of very limited value in the diagnosis of Addison’s disease as this does not reflect the ability of the adrenal glands to respond to stress with increased production of cortisol. Stress such as surgery or injury modifies cortisol production. This means that a certain cortisol concentration may be appropriate in a relaxed patient, but much too low for a severely distressed patient. Only a dynamic function test, commonly the short synacthen test described above, can give a conclusive answer, supported by the clinical judgement of an experienced endocrinologist.
Measuring adrenocorticotropic hormone levels in the blood can show whether the adrenal glands themselves are damaged, as damage causes the hormone level to rise. Antibodies which act against the adrenal glands can be measured in the blood to show that autoimmunity is the cause of the adrenal damage. If other causes besides autoimmunity are suspected, further tests are needed, such as a scan of the adrenal glands. Testing is usually done as an outpatient unless the case is critical because of an adrenal crisis.
How is Addison’s disease treated ?
Treatment of Addison’s disease is usually managed as an outpatient. It consists of cortisol replacement using hydrocortisone tablets (usually taken two to three times a day) and aldosterone replacement using fludrocortisone tablets (usually taken once a day). The exact medication regime will vary depending on the individual and should be reviewed at regular intervals.
Adrenal sex hormone replacement can sometimes be offered to women because there is some evidence that replacing adrenal sex hormones with a drug called dehydroepiandrosterone might improve sex drive and general wellbeing. However, side-effects such as acne and facial hair growth can occur, and more research is needed in this area. It is not required for men as they also produce the sex hormones from their testes.
When a patient has an adrenal crisis, they require immediate admission to hospital as an emergency. Treatment consists of immediate hydrocortisone injections, fluid and sugar replacement and careful monitoring.
Are there any side-effects to the treatment ?
When properly monitored and controlled, there should be no side-effects to treatment. In Addison’s disease the body cannot cope adequately with stress, so people with Addison’s disease will not naturally produce enough cortisol if they become ill. Normally this is helped by doubling their dose of hydrocortisone for three days after any significant illness, stress or surgery.
A Medic-Alert necklace or bracelet to alert doctors should be worn by people with Addison’s disease in case they are brought into hospital unconscious. If someone with Addison’s disease cannot take tablets because of vomiting, they need to take the emergency hydrocortisone injection supplied and they must contact a doctor straightaway so that hydrocortisone can be continued to be given by injection. When travelling to places where hospital care may be difficult to find in an emergency, it is essential to carry hydrocortisone for injection and the patient and any companions must know how to give this injection.
What are the longer-term implications of Addison’s disease ?
Although Addison’s disease is a lifelong condition, it can be very successfully treated with daily medication and patients can lead full and active lives.
There are no long-term implications provided that tablets are taken, the advice given above is followed, and treatment is regularly monitored (at least once a year). At these visits, checks should be made on the hydrocortisone emergency kit to ensure it is within date of expiry and that patients know how to use this. People with Addison’s disease are at increased risk of developing other autoimmune diseases (such as thyroid disease) and these should be checked for at annual hospital appointments.
Hyperaldosteronism
Aldosterone is a hormone produced by the adrenal glands and helps to control the amount of fluid in the body by affecting how much salt and water the kidney retains or excretes. The adrenal glands produce aldosterone in response to another hormone called renin. Renin is produced by specialised cells in the kidney that detect when the body lacks salt; renin released by the kidney signals the adrenal glands to release aldosterone. The kidney detects an increase in aldosterone in the bloodstream and responds by retaining extra salt rather than excreting it in the urine. As the body regains the salt it needs, the level of renin in the bloodstream drops and therefore the amount of aldosterone in the blood also falls, meaning more water is excreted in the urine. This is an example of a feedback system.
Hyperaldosteronism refers to any state where there are excessive or inappropriate levels of aldosterone in the bloodstream.
- In primary hyperaldosteronism (Conn syndrome), aldosterone secretion is inappropriately high for the level of body salt and blood volume regardless of the renin level in the blood (which is usually suppressed).
- Secondary hyperaldosteronism occurs when the kidney produces too much renin. This is often seen in patients with chronic low blood volume such as in cardiac, liver or renal disease; the kidney mistakes the low blood supply for dehydration and produces excess renin.
This disorder occurs when the body produces too much aldosterone, a hormone that controls blood pressure and regulates the body’s salt and potassium levels. There are two causes of hyperaldosteronism. The extra aldosterone is produced either by a non-cancerous tumor, which typically affects one adrenal gland, or by abnormal growth of both glands, a condition called “hyperplasia.” 79
There are no known gene mutations associated with this disorder at this time. However, rarely, hyperaldosteronism can run in families.
What causes primary hyperaldosteronism ?
Hyperaldosteronism may be due either to diffuse swelling/overgrowth (hyperplasia) affecting either, but much more commonly both, of the adrenal glands; or, a small tumour within the gland (called an adrenal adenoma). In both cases, there is excessive secretion of aldosterone. These tumours are invariably small, benign, and do not spread or invade other areas in the way malignant tumours can.
Rarely, the condition is hereditary – so called glucocorticoid or dexamethasone-suppressible hyperaldosteronism. Rarer still, large adrenal carcinomas may secrete aldosterone. In all conditions, the feedback system fails and aldosterone secretion continues despite a low blood renin level.
What are the signs and symptoms of primary hyperaldosteronism ?
The diagnosis is made as a secondary cause of high blood pressure; specific symptoms are often few.
Patients with primary hyperaldosteronism are often diagnosed very late as the symptoms can be subtle. Most commonly, high blood pressure as a result of water and salt retention is found and a doctor will often suspect primary hyperaldosteronism if high blood pressure is not responding to multiple medications. About one-third of patients with primary hyperaldosteronism will also have a low blood level of potassium, which the kidney excretes in exchange for salt. Occasionally, this may cause symptoms such as cramps, weakness and excessive thirst. A common scenario is the development of a low potassium level when drugs such as thiazide diuretics are used to treat what was initially thought to be essential hypertension.
How common is primary hyperaldosteronism ?
The prevalence of hyperaldosteronism is debated. Initial studies reported that it probably affects 0.1–1% (1 in 1,000 to 1 in 100) of all patients with high blood pressure. However, with the wider use of the plasma aldosterone/plasma renin ratio as a screening test in patients with hypertension, estimates of the prevalence have risen. Some studies suggest 5–10% of patients with high blood pressure have primary aldosteronism; and in those patients whose blood pressure is resistant to drug therapy, it may be as high as 25%.
Adenomas are more frequent in women, and the diagnosis is most commonly made between the ages of 30 and 40.
Is primary hyperaldosteronism inherited ?
The most common causes of hyperaldosteronism (adrenal hyperplasia and benign adrenal tumours) are not inherited. The rare form – glucocorticoid-suppressible hyperaldosteronism – is inherited as an autosomal dominant condition.
How is primary hyperaldosteronism diagnosed ?
Primary hyperaldosteronism is suspected if a patient has high blood pressure that does not respond to medication (often three or four drugs used together). Your doctor may be more suspicious if the potassium level in the blood is low. A strong history of high blood pressure and/or stroke in the family may also raise suspicion. Your doctor would normally refer you to an endocrinologist at this point, who will organise a blood test to check the levels of renin and aldosterone. To make this more accurate, your doctor may have to change the medications you already take as they can affect the results of this test. The blood test is most often performed at 9 a.m. in the sitting or lying position. In primary hyperaldosteronism, the aldosterone level is significantly raised, whilst the renin level is normally low. If the renin level is normal or high, it is likely that the patient has secondary hyperaldosteronism.
To confirm the diagnosis of hyperaldosteronism, a ‘salt challenge’ test may be carried out. In this test, aldosterone levels are checked after giving a salt solution drip directly into the bloodstream, or by increasing the amount of salt in the diet for a few days before rechecking aldosterone levels. In both cases, a normally healthy person’s aldosterone level should fall. If this does not happen, primary hyperaldosteronism is confirmed.
To find a cause, your doctor will organise a scan of the adrenal glands to look for a benign tumour or gland swelling. If there is doubt, a day-case radiology procedure can be performed under local anaesthetic using a fine cannula (narrow tube), to measure the amount of aldosterone in each of the veins coming from the two adrenal glands. A much higher level from one gland than the other suggests a tumour on one side.
If glucocorticoid-suppressible hyperaldosteronism is suspected, the doctor may give you some synthetic corticosteroids (‘steroids’) to try and lower aldosterone levels, or may recommend genetic testing.
How is primary hyperaldosteronism treated ?
It is recommended that adrenal tumours should be removed if the patient is considered fit enough for surgery. This is performed via keyhole surgery – a process called laparoscopic adrenalectomy – in many hospitals and is usually very safe with few complications. In virtually all cases, potassium levels are normalised with successful surgery, and over 70% of patients have an improvement in blood pressure, with 50% of patients coming off blood pressure medication completely.
Surgery is usually not recommended if the cause is swelling of both glands, since this will have little impact on future blood pressure. Also, the adrenal glands produce other hormones that are very important to normal body functions. In these situations, medications that block the effect of aldosterone causing salt and water retention in the kidney can be used.
Are there any side-effects to the treatment ?
The medication most commonly used to treat primary hyperaldosteronism (spironolactone) can cause high blood potassium and low blood salt (hyponatraemia) as it blocks the action of aldosterone. It can affect periods in women and in men it can also cause impotence and enlargement of breast tissue, as it blocks the action of testosterone. Eplerenone is an alternative drug that works through the same mechanism as spironolactone but without the sex hormone side-effects; eplerenone is much more expensive.
Pregnant women or those of child bearing age would usually not be offered spironolactone as it can have serious effects on the developing baby.
What are the longer-term implications of primary hyperaldosteronism ?
The main consequence of primary hyperaldosteronism is high blood pressure, which leads to an increased risk of heart disease and strokes, as well as disease of the peripheral blood vessels, the eyes, and the kidneys. However, treatment for hyperaldosteronism can be very effective and is primarily aimed at reducing these long-term complications of high blood pressure. Patients who require long-term medication for this condition may need regular follow-up appointments with their doctor.
Adrenal Gland Suppression
The normal activity of the adrenal glands can be suppressed—or reduced—when people take steroid medications (medicines that act like cortisol in the body) such as prednisone, hydrocortisone, or dexamethasone 80. Steroid medications, most often prednisone, may be prescribed to treat certain types of arthritis, severe allergic reactions, asthma, autoimmune diseases, and other conditions 81.
Ordinarily, someone taking steroids takes gradually lower and lower doses as time goes by until they stop taking the drug completely. This is called “tapering” the dose. When steroid medications are stopped suddenly, especially after being taken for several weeks or more, the adrenal glands may be unable to produce steroid hormones (most importantly, cortisol) in sufficient amounts for several weeks or even months 82. This situation can cause health problems because of the imbalance of hormone levels that continues until the adrenal glands start functioning normally again.
- US Department of Health and Human Services. National Institutes of Health. About Adrenal Gland Disorders. https://www.nichd.nih.gov/health/topics/adrenalgland/conditioninfo/Pages/default.aspx[↩][↩][↩][↩]
- U.S. National Library of Medicine. Medline Plus. Adrenal glands. https://medlineplus.gov/ency/article/002219.htm[↩][↩]
- Vertical Health Endocrine Web. An Overview of the Adrenal Glands. https://www.endocrineweb.com/endocrinology/overview-adrenal-glands[↩]
- U.S. National Library of Medicine. Medline Plus. Adrenal Gland Disorders. https://medlineplus.gov/adrenalglanddisorders.html[↩]
- New York Times Health Guide. (2008). Exogenous adrenal insufficiency. http://www.nytimes.com/health/guides/disease/exogenous-adrenal-insufficiency/overview.html[↩]
- Stewart, P. M., and Krone, H. P. The adrenal cortex. In: Kronenberg H.M., Shlomo, M., Polonsky, K.S, Larsen, P.R., eds. Williams. Textbook of Endocrinology. 12th ed. Philadelphia, Pa: Saunders Elsevier; 2011:chapter 15.[↩]
- Cushing Exhibit Online – Yale School of Medicine. http://library.medicine.yale.edu/[↩]
- Steffensen, C., Bak, A. M., Rubeck, K. Z., & Jørgensen J. O. (2010). Epidemiology of Cushing’s syndrome. Neuroendocrinology, 92 (Suppl 1), 1-5. https://www.ncbi.nlm.nih.gov/pubmed?term=20829610[↩][↩]
- Lindholm, J., Juul, S., Jørgensen, J. O. L, Astrup, J., Bjerre, P., Feldt-Rasmussen, U., et al. (2001). Incidence and late prognosis of Cushing’s syndrome: A population-based study. Journal of Clinical Endocrinology and Metabolism, 86(1), 117-123. https://www.ncbi.nlm.nih.gov/pubmed?term=11231987[↩]
- Boscaro, M., & Arnaldi, G. (2009). Approach to the patient with possible Cushing’s syndrome. Journal of Clinical Endocrinology and Metabolism, 94(9), 3121-3131. https://www.ncbi.nlm.nih.gov/pubmed?term=19734443[↩][↩]
- Melmed, S. (2011). Pathogenesis of pituitary tumors. Nature Reviews Endocrinology, 7, 257-266.[↩]
- National Institute of Health. Cushing Syndrome: Other FAQs. https://www.nichd.nih.gov/health/topics/cushing/conditioninfo/pages/faqs.aspx[↩]
- Nieman, L. K., & Ilias, I. (2005). Evaluation and treatment of Cushing’s syndrome. Journal of American Medicine, 118(12), 1340-1346. https://www.ncbi.nlm.nih.gov/pubmed?term=16378774[↩]
- Nieman L. K., & Ilias, I. (2005). Evaluation and treatment of Cushing’s syndrome. Journal of American Medicine, 118(12), 1340–1346. https://www.ncbi.nlm.nih.gov/pubmed?term=16378774[↩][↩][↩][↩]
- Mayo Foundation for Medical Education and Research. Cushing syndrome. http://www.mayoclinic.org/diseases-conditions/cushing-syndrome/home/ovc-20197169[↩][↩][↩][↩]
- Batista, D. L., Courcoutsakis, N., Riar, J., Keil, M. F., & Stratakis, C. A. (2008). Severe obesity confounds the interpretation of low-dose dexamethasone test combined with the administration of ovine corticotrophin-releasing hormone in childhood Cushing syndrome. Journal of Clinical Endocrinology and Metabolism, 93(11), 4323-4330.[↩]
- Nieman, L. K., Biller, B. M. K., Findling, J. W., Newell-Price, J., Savage, M. O., et al. (2008). The diagnosis of Cushing’s syndrome: An Endocrine Society clinical practice guideline. https://academic.oup.com/jcem/article-lookup/doi/10.1210/jc.2008-0125[↩][↩][↩][↩][↩][↩]
- Batista, D. L., Riar, J., Keil, M., & Stratakis, C. A. (2007). Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Pediatrics, 120(3), e575-e586.[↩]
- Nieman, L. K., & Ilias, I. (2005) Evaluation and treatment of Cushing’s syndrome. Journal of American Medicine, 118(12), 1340–1346. https://www.ncbi.nlm.nih.gov/pubmed?term=16378774[↩][↩]
- National Library of Medicine, MedlinePlus. Cushing’s syndrome. https://medlineplus.gov/cushingssyndrome.html [↩]
- Nieman, L. K., & Ilias, I. (2005) Evaluation and treatment of Cushing’s syndrome. Journal of American Medicine, 118(12), 1340-1346. https://www.ncbi.nlm.nih.gov/pubmed?term=16378774[↩]
- Lindsay, J. R., Nansel, T., Baid, S., Gumowski, J., & Nieman, L. K. (2006). Long-term impaired quality of life in Cushing’s syndrome despite initial improvement after surgical remission. Journal of Clinical Endocrinology and Metabolism, 91, 447-453.[↩]
- Tiemensma, J., Kokshoorn, N. E., Biermasz, N. R., Keijser, B. J., Wassenaar, M. J., Middelkoop, H. A., et al. (2010). Subtle cognitive impairments in patients with long-term cure of Cushing’s disease. Journal of Clinical Endocrinology and Metabolism, 95(6), 2699-2714. https://www.ncbi.nlm.nih.gov/pubmed?term=20371667[↩]
- Keil, M. F., Merke, D. P., Gandhi, R., Wiggs, E. A., Obunse, K., & Stratakis, C.A. (2009). Quality of life in children and adolescents 1-year after cure of Cushing syndrome: A prospective study. Clinical Endocrinology, 71(3), 326-333.[↩]
- Steffensen, C., Bak, A. M., Rubeck, K. Z., & Jørgensen J. O. (2010). Epidemiology of Cushing’s syndrome. Neuroendocrinology, 92(Suppl 1), 1-5. https://www.ncbi.nlm.nih.gov/pubmed?term=20829610[↩]
- Graversen, D., Vestergaard, P., Stochholm, K., Gravholt, C. H., & Jørgensen, J. O. (2012). Mortality in Cushing’s syndrome: A systematic review and meta-analysis. European Journal of Internal Medicine, 23(3), 278-282. https://www.ncbi.nlm.nih.gov/pubmed?term=22385888[↩][↩]
- National Library of Medicine, MedlinePlus. Cushing’s syndrome. https://medlineplus.gov/cushingssyndrome.html[↩]
- Abraham, M. R., & Smith, C. V. (n.d.). Adrenal disease and pregnancy. http://emedicine.medscape.com/article/127772-overview#aw2aab6b6[↩][↩][↩]
- Lindsay, J. R., Jonklaas, J., Oldfield, E. H., & Nieman, L. K. (2005). Cushing’s syndrome during pregnancy: Personal experience and review of the literature. Journal of Clinical Endocrinology and Metabolism, 90(5), 3077. https://www.ncbi.nlm.nih.gov/pubmed?term=15705919[↩][↩][↩]
- Pickard, J., Jochen, A. L., Sadur, C. N., & Hofeldt, F. D. (1990). Cushing’s syndrome in pregnancy. Obstetrical & Gynecological Survey, 45(2), 87-93. https://www.ncbi.nlm.nih.gov/pubmed?term=2405312[↩][↩][↩]
- Abraham, M. R., & Smith, C. V. Adrenal disease and pregnancy. http://emedicine.medscape.com/article/127772-overview#aw2aab6b6[↩]
- Buescher, M. A. (1996). Cushing’s syndrome in pregnancy. Endocrinologist, 6, 357-361.[↩]
- Ezzat, S., Asa, S. L., Couldwell, W. T., Barr, C. E., Dodge, W. E., Vance M. L., et al. (2004). The prevalence of pituitary adenomas: A systematic review. Cancer, 101(3), 613-619. https://www.ncbi.nlm.nih.gov/pubmed/15274075[↩]
- libansky, A. (n.d.). Pregnancy after cure of Cushing’s disease. https://csrf.net/doctors-articles/reproductive-issues/pregnancy-after-cure-of-cushings-disease/[↩][↩]
- Biddie, S. C., Conway-Campbell, B. L, & Lightman, S. L. (2012). Dynamic regulation of glucocorticoid signalling in health and disease. Rheumatology, 51(3), 4034-4112. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3281495/[↩]
- Jequier, A.M. Endocrine infertility. In Male infertility: A clinical guide (2nd ed.). Cambridge University Press, 2011: chap 20, pages 187-188.[↩]
- Stewart, P. M., & Krone, N. P. (2011). The adrenal cortex. In Kronenberg, H. M., Shlomo, M., Polonsky, K. S., Larsen P. R. (Eds.). Williams textbook of endocrinology (12th ed.). (chap. 15). Philadelphia, PA: Saunders Elsevier.[↩]
- National Institutes of Health. Congenital Adrenal Hyperplasia. https://www.nichd.nih.gov/health/topics/cah/Pages/default.aspx[↩]
- CARES Foundation. (2007). Overview: What is congenital adrenal hyperplasia (CAH)? http://www.caresfoundation.org/what-is-cah/[↩]
- NIH Clinical Center. Patient education: Facts about CAH. https://www.cc.nih.gov/ccc/patient_education/pepubs/cah.pdf[↩][↩][↩]
- Texas Department of State Health Services. (2001). Congenital adrenal hyperplasia: A handbook for parents. http://www.dshs.texas.gov/newborn/hand_cah.shtm[↩]
- National Adrenal Diseases Foundation. CAH: Diagnosis. http://www.nadf.us/adrenal-diseases/congenital-adrenal-hyperplasia-cah/[↩]
- National Adrenal Diseases Foundation. CAH: Nonclassical. http://www.nadf.us/adrenal-diseases/congenital-adrenal-hyperplasia-cah/[↩]
- Medscape Reference. (2011). C-11 Hydroxylase deficiency. http://emedicine.medscape.com/article/117012-overview[↩]
- National Adrenal Diseases Foundation. (n.d.). Adrenal diseases—Congenital adrenal hyperplasia (CAH): The facts you need to know. http://www.nadf.us/adrenal-diseases/congenital-adrenal-hyperplasia-cah/[↩]
- Screening, Technology and Research in Genetics (STAR-G) Project. Genetic fact sheet for parents: Congenital adrenal hyperplasia. http://www.newbornscreening.info/Parents/otherdisorders/CAH.html[↩][↩][↩]
- National Library of Medicine, Medline Plus. Congenital adrenal hyperplasia. https://medlineplus.gov/ency/article/000411.htm[↩]
- National Library of Medicine, MedLine Plus. Congenital adrenal hyperplasia. https://medlineplus.gov/ency/article/000411.htm[↩]
- The Hormone Foundation. (2010). Patient guide to congenital adrenal hyperplasia. http://www.hormone.org/diseases-and-conditions/adrenal/congenital-adrenal-hyperplasia[↩]
- National Adrenal Diseases Foundation. CAH: Standard treatment. http://www.nadf.us/adrenal-diseases/congenital-adrenal-hyperplasia-cah/[↩]
- The Endocrine Society. (2010). Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society clinical practice guideline. Journal of Clinical Endocrinology & Metabolism, 95(9), 4133–4160.[↩]
- Congenital Adrenal Hyperplasia Research Education & Support Foundation. (2007). Surgery considerations for girls with classical CAH. http://www.caresfoundation.org/dosing/surgery/[↩]
- National Institute of Neurological Disorders and Stroke. (2010). NINDS pituitary tumors information page. https://www.ninds.nih.gov/Disorders/All-Disorders/Pituitary-Tumors-Information-Page[↩]
- National Cancer Institute. Pituitary tumors treatment. https://www.cancer.gov/types/pituitary/patient/pituitary-treatment-pdq[↩][↩]
- Eunice Kennedy Shriver National Institute of Child Health and Human Development. The proper diagnosis, treatment, genetics, and research of pheochromocytoma and paraganglioma: Overview. https://science.nichd.nih.gov/confluence/display/pheo/Overview[↩][↩][↩]
- King, K. S., Prodanov, T., Kantorovich, V., Fojo, T., Hewitt, J. K., Zacharin, M., et al., (2011). Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. Journal of Clinical Oncology 29: 4137-4142.[↩]
- Timmers, H. J., Gimenez-Roqueplo, A. P., Mannelli, M., & Pacak, K. (2009).Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocrine-Related Cancer 16: 391-400.[↩]
- Pacak, K., Eisenhofer, G., Ahlman, H., Bornstein, S. R., Gimenez-Roqueplo, A. P., Grossman, A. B., et al. (2007). Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nature Clinical Practice Endocrinology & Metabolism 3: 92-102.[↩]
- Grogan, R. H., Mitmaker, E. J., & Duh, Q. Y. (2011). Changing paradigms in the treatment of malignant pheochromocytoma. Cancer Control 18: 104-112.[↩]
- National Cancer Institute, PDQ Cancer Information Summaries. Pheochromocytoma and paraganglioma treatment. https://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0033247/[↩]
- Pacak, K. (2011). Phaeochromocytoma: a catecholamine and oxidative stress disorder. Endocrine Regulation 45: 65-90.[↩][↩]
- Parenti, G., Zampetti, B., Rapizzi, E., Ercolino, T., Giachè, V., & Mannelli, M. (2012). Updated and new perspectives on diagnosis, prognosis, and therapy of malignant pheochromocytoma/paraganglioma. Journal of Oncology, 2012, 872713.[↩]
- Karasek, D., Shah, U., Frysak, Z., Stratakis, C., Pacak, K. (In press). An update on the genetics of pheochromocytoma. Journal of Human Hypertension , doi:10.1038/jhh.2012.20[↩]
- Eisenhofer, G., Lenders J. W., Siegert, G., Bornstein, S. R., Friberg, P., Milosevic, D., et al., (2012). Plasma methoxytyramine: A novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. European Journal of Cancer, 48: 1739–1749.[↩]
- Eunice Kennedy Shriver National Institute of Child Health and Human Development. The proper diagnosis, treatment, genetics, and research of pheochromocytoma and paraganglioma: Genetic screening. https://science.nichd.nih.gov/confluence/display/pheo/Genetic+Screening[↩]
- Karasek, D., Shah, U., Frysak, Z., Stratakis, C., Pacak, K. (In press). , An update on the genetics of pheochromocytoma. Journal of Human Hypertension , doi:10.1038/jhh.2012.20.[↩]
- Pacak, K., Lenders, J. W. M., & Eisenhofer, G. (2007). Pheochromocytoma: diagnosis, localization, and treatment. Malden, MA: Wiley-Blackwell.[↩]
- Karasek, D., Shah, U., Frysak, Z., Stratakis, C., Pacak, K. (In press). An update on the genetics of pheochromocytoma. Journal of Human Hypertension , doi:10.1038/jhh.2012.20.[↩]
- Pacak, K., & Eisenhofer G. (2007). An assessment of biochemical tests for the diagnosis of pheochromocytoma. Nature Clinical Practice Endocrinology & Metabolism. 3: 744-745.[↩][↩]
- National Cancer Institute. Pheochromocytoma and paraganglioma treatment. https://www.cancer.gov/types/pheochromocytoma/patient/pheochromocytoma-treatment-pdq[↩][↩][↩][↩]
- Waguespack, S. G., Rich, T., Grubbs, E., Ying, A. K., Perrier, N. D., Ayala-Ramirez, M., et al. (2010). A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. Journal of Clinical Endocrinology and Metabolism. 95(5), 2023–2037. PMID 20215394[↩]
- Lenders, J. W., Eisenhofer, G., Mannelli, M., & Pacak, K. (2005.) Phaeochromocytoma. Lancet 366: 665-675.[↩][↩]
- Blake, M. A. (2011). Pheochromocytoma. http://emedicine.medscape.com/article/124059-overview[↩]
- Waguespack, S. G., Rich, T., Grubbs, E., Ying, A. K., Perrier, N. D., Ayala-Ramirez, M., et al. (2010) A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. Journal of Clinical Endocrinology and Metabolism 95: 2023-2037.[↩]
- Society for Endocrinology. (2015). Addison’s disease. http://www.yourhormones.info/endocrine-conditions/addisons-disease.aspx[↩][↩]
- National Library of Medicine. Addison’s disease. https://medlineplus.gov/addisondisease.html[↩]
- National Library of Medicine. Medline Plus. Addison disease. https://medlineplus.gov/addisondisease.html[↩]
- Society for Endocrinology. Addison’s disease. http://www.yourhormones.info/endocrine-conditions/addisons-disease.aspx[↩]
- Society for Endocrinology. Hyperaldosteronism. http://www.yourhormones.info/endocrine-conditions/primary-hyperaldosteronism/[↩]
- National Library of Medicine. Medline Plus. Cushing syndrome. https://medlineplus.gov/ency/article/000410.htm[↩]
- National Library of Medicine. Prednisone. https://www.ncbi.nlm.nih.gov/pubmedhealth/PMHT0011828/[↩]
- American Society of Clinical Oncology. Pituitary gland tumor. http://www.cancer.net/cancer-types/pituitary-gland-tumor[↩]
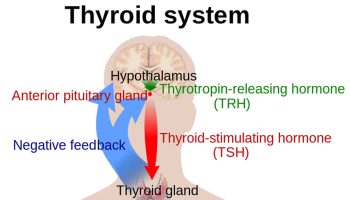

{kind=link}