Contents
Bardet-Biedl syndrome
Bardet-Biedl syndrome also called BBS is a rare complex genetic disorder that affects many parts of the body characterized by retinal dystrophy (a progressive eye disorder that leads to blindness, characterized by tunnel vision and night blindness), truncal obesity, polydactyly (extra fingers and/or toes), genitourinary and kidney abnormalities, learning difficulties, developmental delay, speech and language difficulties and hypogonadism 1, 2, 3, 4. Individuals with Bardet-Biedl syndrome (BBS) have a retinal degeneration similar to retinitis pigmentosa (RP). The retina is a light-sensitive layer of tissue at the back of your eyes that converts images into electrical signals for your brain. Rod and cone photoreceptors in the retina convert light into electrical signals that the brain interprets as vision. People with Bardet-Biedl syndrome (BBS) with retinal degeneration that is characterized by rod-cone dystrophy experience a gradual decline in their vision, because the photoreceptors (specialized cells in the retina that convert light into electrical signals that the brain can use for vision) degenerate. People with Bardet-Biedl syndrome (BBS) also have at least three additional non-eye features such as intellectual disability, truncal obesity (a condition where fat is disproportionately distributed onto the abdomen and chest rather than the arms and legs), polydactyly (a condition where a person has more than the normal number of fingers or toes), hypogonadism (a condition where the body’s sex glands [ovaries in women or testicles in men], produce little to no sex hormones), or kidney abnormalities as primary signs and symptoms 1, 2, 5. However, the signs and symptoms of Bardet-Biedl syndrome vary among affected individuals, even among members of the same family 6, 7.
In 1866, Laurence and Moon described four affected siblings with retinal dystrophy, obesity, and mental retardation 8. Three of them (males) also had small external genitalia and an abnormal gait 8. In 1920s, Bardet 9, 10 and Biedl 11, 12 separately reported a similar clinical features in individuals, who also had polydactyly (extra fingers and/or toes) and the condition was coined Laurence-Moon-Bardet-Biedl syndrome. The overlapping features of these cases suggested that the two disorders, Laurence-Moon-Bardet-Biedl syndrome and Bardet-Biedl syndrome (BBS) represented variable expression of a single condition 13, 14, 15 although this is controversial 16, 17. However, it is now generally considered that Bardet-Biedl syndrome (BBS) and Laurence-Moon syndrome (LMS) are distinct conditions 5.
Bardet-Biedl syndrome can result from mutations in at least 26 different BBS genes 1, 3, 18. The proteins produced from BBS genes are are known or suspected to play critical roles in the maintenance and function of cilia. Cilia are microscopic, finger-like projections that stick out from the surface of many types of cells. Cilia are involved in cell movement and many different chemical signaling pathways. Cilia are also necessary for the perception of sensory input such as sight, hearing, and smell.
Mutations in BBS genes lead to problems with the structure and function of cilia. Defects in these cell structures probably disrupt important chemical signaling pathways during development and lead to abnormalities of sensory perception. Researchers believe that defective cilia are responsible for most of the features of Bardet-Biedl syndrome.
About one-quarter of all cases of Bardet-Biedl syndrome result from mutations in the BBS1 gene. Another 20 percent of cases are caused by mutations in the BBS10 gene. The other BBS genes each account for only a small percentage of all cases of Bardet-Biedl syndrome. In about 25 percent of people with Bardet-Biedl syndrome, the cause of the disorder is unknown.
In individuals with Bardet-Biedl syndrome who have mutations in one of the BBS genes, mutations in additional genes may be involved in causing or modifying the course of the disorder. Studies suggest that these modifying genes may be known BBS genes or other genes. The additional genetic changes could help explain the variability in the signs and symptoms of Bardet-Biedl syndrome. However, this phenomenon appears to be uncommon, and it has not been found consistently in scientific studies.
Bardet-Biedl syndrome (BBS) is more common than Laurence-Moon syndrome, with a prevalence of 1 in 125,000 to 1 in 160,000 newborns in North America and Europe 15, 6, 1. Bardet-Biedl syndrome (BBS) is more common on the island of Newfoundland (off the east coast of Canada), where it affects an estimated 1 in 17,000 newborns 1. Bardet-Biedl syndrome (BBS) also occurs more frequently, due to increased marriages among consanguineous, affecting about 1 in 13,500 newborns in the Bedouin population of Kuwait; 1:6900 in Jahra district and 1:3700 in Faroe Islands 19, 20, 21, 22, 1.
Vision loss is one of the major features of Bardet-Biedl syndrome 1, 2. Most patients with Bardet-Biedl syndrome will experience the loss of light-sensing tissue at the back of the eye called the retina 5. The retina is part of the eye involved in detecting and decoding incoming images. Incoming light is focused onto the retina at the back of the eye. The retina is composed of cells called “rods and cones”. They translate incoming light into nerve impulses the brain can use. This gradual loss of the rod and cone cells on the retina is described as “retinal dystrophy”. Symptoms associated with cone-rod dystrophy may not become apparent until 7 or 8 years of age when children begin to complain of problems with night vision with an inability to see in dimly lit environments, such as a sidewalk lit only by streetlights 5. This “night blindness” may progress to variable degrees by blind spots that develop in the side (peripheral) vision. In most people, the vision becomes progressively weaker through the first and second decades of life 5. Over time, these blind spots enlarge and merge to produce tunnel vision. Affected individuals often first lose peripheral vision, and see only what is directly in front of their focus point. They see in what is termed ‘tunnel vision’ 5. Most people with Bardet-Biedl syndrome also eventually lose central vision (poor visual acuity) and become legally blind by mid-teens or early adulthood 5. In some people, the degeneration of the retina may follow a characteristic course, referred to as “retinitis pigmentosa” (RP). Retinitis pigmentosa (RP) begins with night blindness, followed by a loss of the ability to discriminate colors from one another, and finally to a progressive tunnel vision 5. Additional effects on the eye characteristic to individuals with Bardet-Biedl syndrome include: lazy eye (strabismus), clouding of the lens of the eyes (cataracts), and an increased pressure within the eyes that can result in damage to the optic nerve conducting signals to the brain (glaucoma) 5.
Truncal obesity is another characteristic feature of Bardet-Biedl syndrome 1, 2, 5. The term ‘truncal obesity’ refers to a condition where fat is disproportionately distributed onto the abdomen and chest rather than the arms and legs 5. Individuals can be described as having an apple-shape body type. Weight is usually normal at birth but weight gain is quickly evident through the first year of life in as many as 90% of people with Bardet-Biedl syndrome 5. Complications of obesity can include type 2 diabetes, high blood pressure (hypertension), and abnormally high cholesterol levels (hypercholesterolemia). Type 2 diabetes has been estimated to affect up to 45% of patients with Bardet-Biedl syndrome 5. High blood pressure (hypertension), and abnormally high cholesterol levels (hypercholesterolemia) may further complicate problems with the heart and blood vessels seen in patients with Bardet-Biedl syndrome. The heart functions as a pump for the blood, moving the blood through the vessels that bring it throughout the body. The heart relies on valves that keep the flow moving in the forward direction. With age, stiffening of the heart valves is completely normal. The stiffening is due to calcium laying down on the valves, and this process if described by the word “stenosis”. People with Bardet-Biedl syndrome may experience stenosis of their heart valves prematurely 5. People with Bardet-Biedl syndrome may also have defects of the heart’s muscular walls 5. The heart muscle is designed such that every motion is smoothly orchestrated. Defects in the heart muscle predispose people with Bardet-Biedl syndrome to heart beat abnormalities, referred to as “arrhythmias” 5.
Other major signs and symptoms of Bardet-Biedl syndrome include the presence of extra fingers or toes (polydactyly), intellectual disability or learning problems, and abnormalities of the genitalia. People with Bardet-Biedl syndrome may be born with an extra digit near the pinky or an extra toe near the fifth “little” toe (polydactyly). This finding occurs in approximately 70 percent of patients. Specifically, the presence of an extra toe is more common than that of an extra finger 5. In medical terminology, this is described as ‘postaxial polydactyly’. Fingers and toes may also show webbing, called “syndactyly”. Syndactyly is especially common between the second and third toes 5. Fingers and toes may occasionally be abnormally short in length. This characteristic is called “brachydactyly” 5. The feet may overall be short in length, of wide width and carry a flat arch.
Another major feature of Bardet-Biedl syndrome is a small size and poor function of the male gonads, termed “testicular hypogonadism”. This may manifest as a small penis, failure of the testes to descend into the scrotum termed “cryptorchidism” or a delay in the onset of puberty. Undescended testicles are a concern because they are associated with a greater risk for testicular cancer and should be surgically managed. An undescended testicle needs to be treated surgically with a procedure called orchiopexy before a child is 2 years old to increase his chance for fertility later in life. Affected females may also have complex genital and urinary tract abnormalities. Affected females may demonstrate an underdeveloped uterus, fallopian tubes, or ovaries. Menstruation cycles are often delayed from the average first age of onset and may also follow an irregular cycle. Pregnant women with Bardet-Biedl syndrome should be followed closely by obstetricians that are well trained in dealing with high-risk pregnancies.
Problems with fertility arise in both men and women. Most affected males produce reduced amounts of sex hormones (testosterone), and they are usually unable to father biological children (infertile) 1, 2.
Some individuals with Bardet-Biedl syndrome may also have abnormalities of the structure and function of the kidneys, which can be serious or life-threatening 1, 2. Kidney defects are highly variable but generally result in an accumulation of urine in the kidneys that results in inappropriate pressures within the kidneys, leading to stretching of important structures 5. The dilation resulting from this fluid accumulation is called “hydronephrosis” 5. It can be monitored by medical imaging such as ultrasound, abdominal x-ray, etc. One common risk that accompanies hydronephrosis includes bacterial infection of the kidneys. The inflammation associated with infection of the kidneys is called “pyelonephritis” 5. These complications to kidney functions can often predispose individuals with Bardet-Biedl syndrome to end-stage renal disease (ESRD) also known as kidney failure. Other clinical feature of Bardet-Biedl syndrome include the development of kidney cysts and damage to the microscopic filtration unit of the kidney 5. Kidneys are responsible for filtering the blood and damage to the filtration systems will result in urine that is dark red blood, possibly even foamy. In scenarios of kidney failure, patients may require dialysis and kidney transplantation. In the scenario that a patient requires kidney transplantation, the use of kidney-protective immunosuppressive medications have been associated with an extra increase in weight gain. This extra weight gain can further complicate any pre-existing diabetes and heart conditions.
Mild-to-moderate learning difficulties are common in individuals with Bardet-Biedl syndrome 5. Often, learning disabilities are attributed to weakened cognitive capacity. Some individuals affected with Bardet-Biedl syndrome may have true learning disabilities due to dysfunction of brain development 5. However, it is important to be sure that suspected disabilities (eg: delayed speech or reading skills) are not due to underlying vision impairment 5. Neurological impairments may manifest in poor coordination, gross and fine motor skills, and social milestones (eg: ability to play complicated games with other children). Many patients report a significant degree of clumsiness and often walk with legs in a wide-based stance. Walking heel-to-toe may be difficult 5.
Additional features of Bardet-Biedl syndrome can include impaired speech, delayed development of motor skills such as standing and walking, behavioral problems such as emotional immaturity and inappropriate outbursts, and clumsiness or poor coordination 1, 2. Distinctive facial features, dental abnormalities, unusually short or fused fingers or toes, and a partial or complete loss of the sense of smell (anosmia) have also been reported in some people with Bardet-Biedl syndrome 1, 2. They have a decreased ability to sense smells due to a change in the size to a brain center called the “olfactory bulb”. This is a relatively a mild problem but may impact safety if people are unable to sense for example, a gas leak from the stove 5.
In the absence of one of these 4 primary clinical features (i.e., vision loss, intellectual disability, obesity, polydactyly), the diagnosis of Bardet-Biedl syndrome (BBS) is made when at least two secondary features are observed, including hepatic fibrosis, diabetes mellitus, reproductive and developmental abnormalities, growth retardation, speech delays, or cardiovascular problems 1, 2.
Some individuals with Bardet-Biedl syndrome may also experience problems with their liver and digestive system. The liver is responsible for many body processes. Among them, it produces a green-brown digestive fluid that the body needs to break food down properly. Specifically, the liver conducts bile through thin ducts that can develop dilation or stricture and leak digestive fluid into the liver, where it causes damage in the form of scarring. More rarely, problems with digestive system may be due to Hirschsprung disease. Hirschsprung disease describes an absence of the nerves normally found in the colon that control the innate motion of the colon and move food along the tract.
A subgroup of affected individuals may exhibit some distinct facial features. These features including deep-set, widely-spaced eyes with downward-slanting lid folds, a flat nasal bridge with nostrils that flare forward, and a long groove (philtrum) in the center of the upper lip. Individuals may have a high-arched palate, with fewer teeth than expected The teeth may have short roots and lie crowded within the mouth.
Figure 1. Bardet-Biedl syndrome diagnostic features
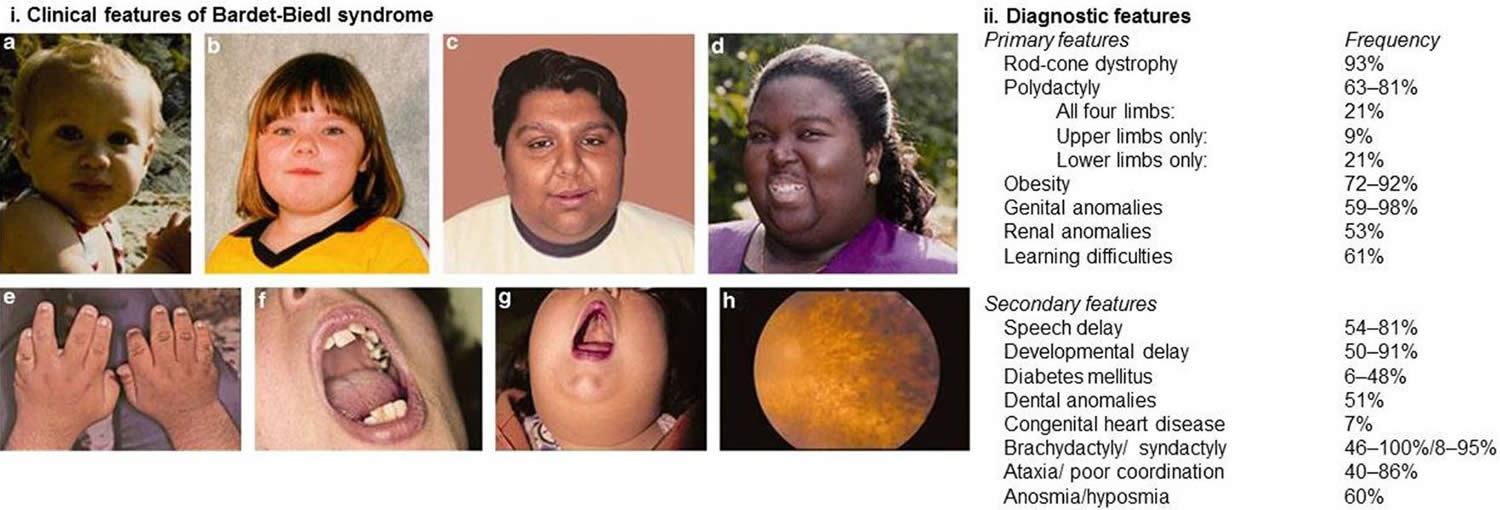
Footnotes: (i) Clinical and diagnostic features associated with Bardet–Biedl syndrome. (A–D) Typical facial features are often subtle and not always present. Typical facial features include malar hypoplasia, a depressed nasal bridge, deep set eyes, and retrognathia. (E) Brachydactyly. (F) Dental crowding. (G) High palate. (H) Rod-cone dystrophy. (ii) Diagnostic features of Bardet–Biedl syndrome. At least four major features or three major and two minor features are required to make a clinical diagnosis.
[Source 23 ]Figure 2. Bardet-Biedl syndrome facial features
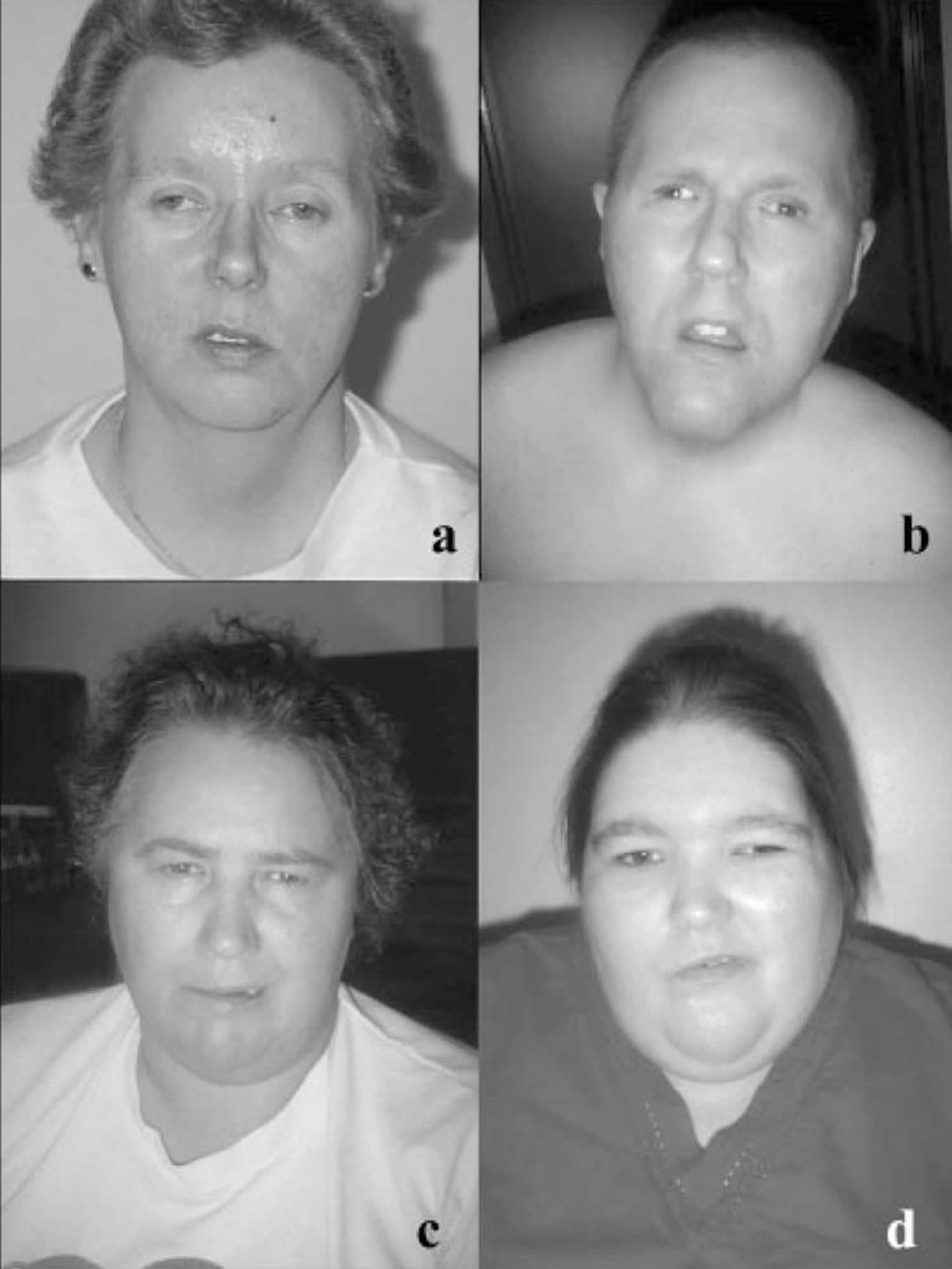
Footnotes: Facial features of four unrelated Bardet-Biedl syndrome Newfoundland patients showing asymmetric, expressionless facies, bitemporal narrowing, short narrow palpebral fissures, long shallow philtrum (b), small downturned mouth and thin upper lip (a, b, and d). Patient (a) has a mutation in the BBS1 gene, patients (b) and (c) have mutations in the MKKS/BBS6 gene, and patient (d) shows linkage to the BBS5 locus.
[Source 24 ]Figure 3. Bardet-Biedl syndrome moon face and polydactyly
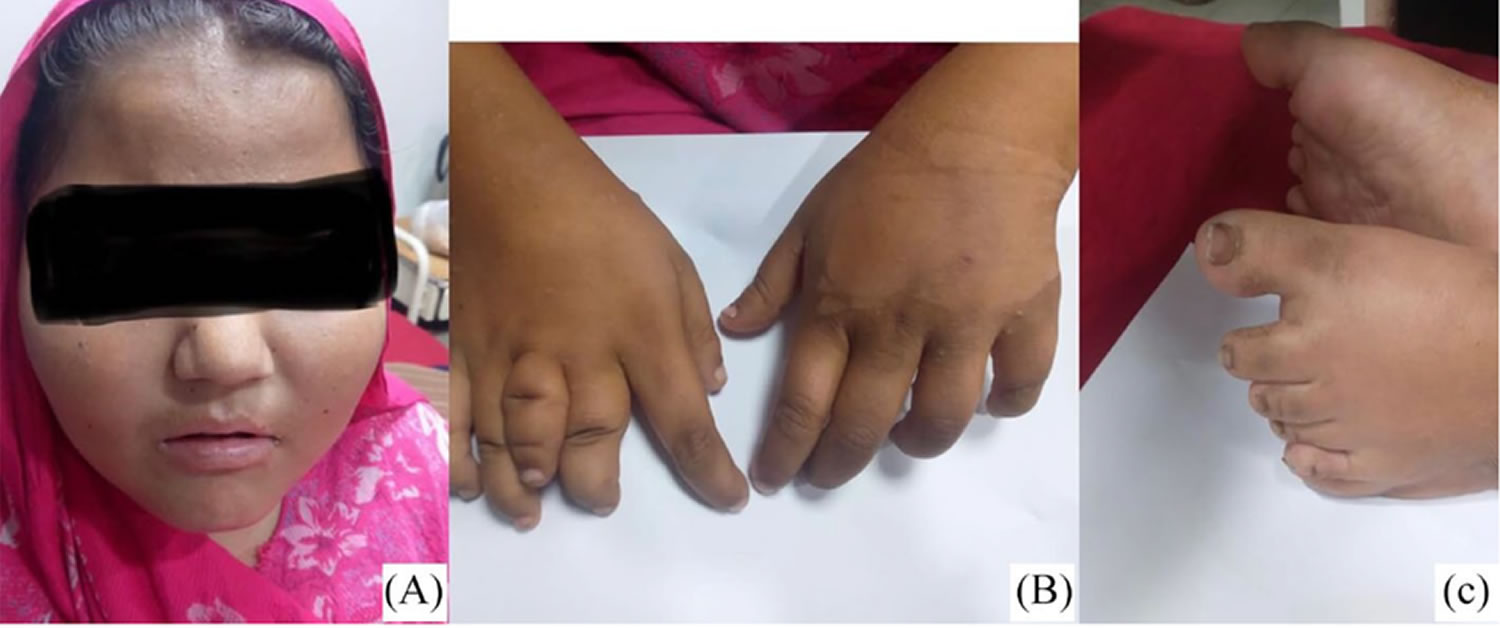
Footnote: Bardet-Biedl syndrome clinical features (A) moon face, (B) polydactyly of the right hand and left feet (C).
[Source 25 ]Figure 4. Bardet-Biedl syndrome retinal dystrophy
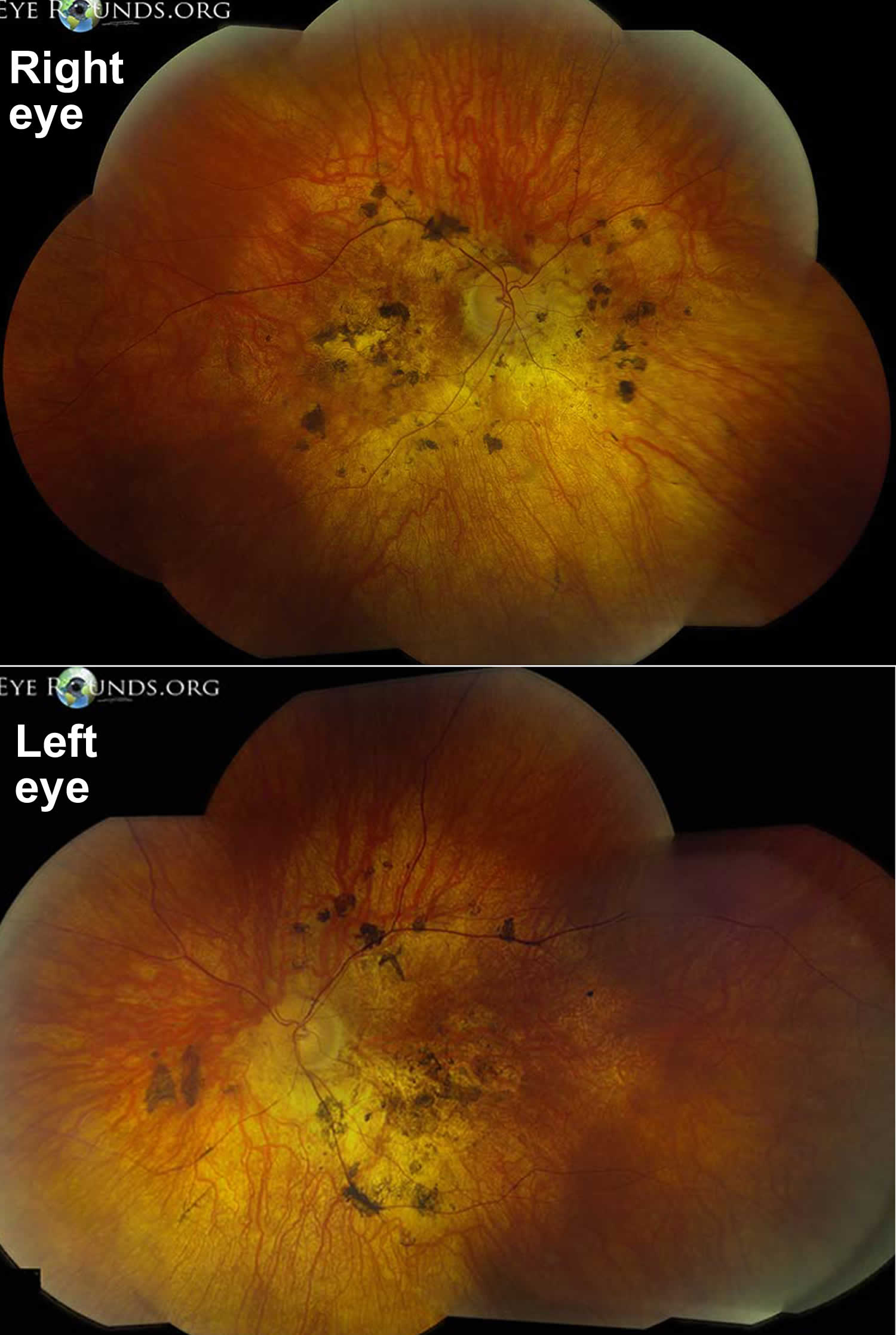
Bardet-Biedl syndrome cause
Bardet-Biedl syndrome can result from mutations in at least 26 different BBS genes 1, 3, 18. The proteins produced from BBS genes are are known or suspected to play critical roles in the maintenance and function of cilia 27, 28, 29, 30. Cilia are microscopic, finger-like projections that stick out from the surface of many types of cells. Cilia are involved in cell movement and many different chemical signaling pathways. Cilia are also necessary for the perception of sensory input such as sight, hearing, and smell.
Mutations in BBS genes lead to problems with the structure and function of cilia. Defects in these cell structures probably disrupt important chemical signaling pathways during development and lead to abnormalities of sensory perception. Researchers believe that defective cilia are responsible for most of the features of Bardet-Biedl syndrome.
About one-quarter of all cases of Bardet-Biedl syndrome result from mutations in the BBS1 gene. Another 20 percent of cases are caused by mutations in the BBS10 gene. The other BBS genes each account for only a small percentage of all cases of Bardet-Biedl syndrome. In about 25 percent of people with Bardet-Biedl syndrome, the cause of Bardet-Biedl syndrome is unknown. People with mutations in the BBS1 gene seem to have milder eye involvement. In comparison, people with mutations in the BBS2, BBS3 and BBS4 genes experience classic deterioration of their vision. People with mutations in the BBS10 gene generally have significantly increased tendency to obesity and insulin resistance.
Bardet-Biedl syndrome Genetics
In individuals with Bardet-Biedl syndrome who have mutations in one of the BBS genes, mutations in additional genes (CCDC28B, MKS1, MKS3 and C2ORF86 genes) may be involved in causing or modifying the course of the disorder 31. Studies suggest that these modifying genes (CCDC28B, MKS1, MKS3 and C2ORF86 genes) may be known BBS genes or other genes. The additional genetic changes could help explain the variability in the signs and symptoms of Bardet-Biedl syndrome. However, this phenomenon appears to be uncommon, and it has not been found consistently in scientific studies.
Variable expressivity is the hallmark of Bardet-Biedl syndrome 32; patients with the same genotype and even siblings frequently manifest symptoms differently. As a result, although genotype–phenotype correlations exist on a population basis, it is not possible to make individual predictions about clinical signs and symptoms of Bardet-Biedl syndrome 33. As a group, people with mutations in BBS1 gene are usually less severely affected than patients with mutations in other BBS genes 23. On average, people with mutations in BBS1 gene develop visual deterioration later in life 34, are less likely to develop kidney disease 33 and more likely to have a better endocrine biochemical profile 35 with a lower prevalence of metabolic syndrome 36, 37. It is not possible to outline if this is a consequence of the common missense mutation BBS1 p.M390R gene identified in 80% of Northern European patients 38 or if the milder phenotype is representative of an overall less severe phenotype associated with BBS1 gene mutations 37.
Further research added complexity to the genetics of Bardet-Biedl syndrome. There are occasional observations on biallelic BBS gene mutation carriers, who remain healthy by the time of the investigation; this suggests incomplete penetrance at least for some genes and/or types of mutations 39, 40, 41. At the same time, those patients who are affected by the disease and carry a homozygous mutation in one of the BBS genes often carry an additional heterozygous mutation in another BBS gene. These observations suggested that Bardet-Biedl syndrome would be a candidate for ‘triallelic inheritance’, whereby a third mutation is required to either manifest the condition or adding mutational load 42, 43, 44. In practice, the phenotypic variability observed in patients with the same genotype and within families is likely to reflect a complex interplay between multiple genetic factors and environmental influences 23.
In the future, it may be possible to identify phenotypic modifiers and further clarify the cause for variability through analysis of the “Omics” (genomics, epigenomics, transcriptomics, proteomics, and metabolomics) whereby the complex interplay of genes, transcription, protein expression, and metabolism is considered as part of the phenotypic analysis 45, 46.
Table 1. Bardet-Biedl syndrome Genes
| Gene1 (BBS Designation2) | % of all Bardet-Biedl syndrome 1 | Distinguishing Clinical Features / Comments | Allelic Disorder(s) 4 |
|---|---|---|---|
| BBIP1 (BBS18) | <1% | Reported in 2 unrelated persons w/multiple major features of Bardet-Biedl syndrome but w/o polydactyly 5 | None |
| BBS1 | 23.40% |
| |
| BBS2 | 9.60% |
| Nonsyndromic retinitis pigmentosa |
| BBS4 | 5.30% |
| None |
| BBS5 | 3.70% | Relatively more “syndromic” | |
| BBS7 | 4.20% | Relatively more “syndromic” w/↑ penetrance of renal anomalies | |
| BBS9 | 3.40% | ↑ penetrance of renal anomalies | |
| TTC8 (BBS8) | 2.00% | Relatively less “syndromic” w/↓ penetrance of renal anomalies | Nonsyndromic retinitis pigmentosa |
| ARL6 (BBS3) | 5.10% |
| |
| BBS10 | 14.50% |
| None |
| BBS12 | 6.40% | Significant adiposity | |
| MKKS (BBS6) | 6.30% | More likely to have congenital heart defect & genitourinary malformations | McKusick-Kaufman syndrome 10 |
| CFAP418 (formerly known as C8orf37) (BBS21) | 1.60% | ↑ penetrance of polydactyly | Nonsyndromic retinitis pigmentosa Cone-rod dystrophy w/polydactyly (OMIM 614500) |
| CEP164 | <1% | Reported in 1 person suspected of having primary ciliary dyskinesia due to unexplained cough & bronchiectasis, but reverse phenotyping revealed features of Bardet-Biedl syndrome 11 | Isolated nephronophthisis |
| CEP290 (BBS14) | 6.30% | Significant clinical overlap w/other ciliopathies | Joubert syndrome Leber congenital amaurosis Meckel syndrome (OMIM 611134) Senior-Løken syndrome (OMIM 610189) See footnote 10. |
| IFT27 (BBS19) | <1% |
| None |
| IFT74 (BBS20) 13 | <1% |
| |
| IFT172 (BBS20) 13 | 1.00% | Typical Bardet-Biedl syndrome features | Nonsyndromic retinitis pigmentosa Short-rib thoracic dysplasia w/ or w/o polydactyly (OMIM 615630) |
| LZTFL1 (BBS17) | <1% |
| None |
| MKS1 (BBS13) | 1.00% | Ophthalmology exam may show bone-spicule hyperpigmentation & attenuated arteries 16 | Joubert syndrome Meckel syndrome (OMIM 249000) |
| SCAPER | Unknown | Linkage & functional studies in 2 consanguineous families w/multiple persons w/features of Bardet-Biedl syndrome support causation 17 | intellectual disability disorder & retinitis pigmentosa (OMIM 618195) |
| SCLT1 | <1% | Reported in 4 persons from 2 families:
| Possible assoc between variation in SCLT1 & orofacial digital syndrome IX (OMIM 258865) |
| SDCCAG8 (BBS16) | 4.30% | Intronic variants reported 11 | Senior-Løken syndrome 10 (OMIM 613615) |
| TRIM32 (BBS11) | <1% | Identified in a consanguineous Bedouin family 19 | Limb-girdle muscular dystrophy (OMIM 254110) |
| WDPCP (BBS15) | <1% | Reported in 2 unrelated persons w/Bardet-Biedl syndrome (clinical data unavailable) 20 | congenital heart defects, hamartomas of tongue, & polysyndactyly (OMIM 217085) |
Abbreviations: AVCD = atrioventricular canal defect; BBS = Bardet-Biedl syndrome; CHD = congenital heart defect; ID = intellectual disability; PCD = primary ciliary dyskinesia
Footnotes:
1. Genes are listed in alphabetic order.
2. Included when BBS designation differs from gene
3. Determined by data of 923 individuals with BBS [899 from a meta-analysis of genotype-phenotype associations by Niederlova et al 47, 17 from a report by Shamseldin et al 48 not included in the meta-analysis, and 7 from additional case reports supporting causation of rare genes].
4. Link to OMIM gene description provided if no GeneReview available
5. Scheidecker et al 49, Shamseldin et al 48
6. Use of the word “syndromic” refers to syndromic score used by Niederlova et al 47 calculated as number of major features present in an individual divided by five (all major features excluding reproductive system anomalies [excluded due to differences in male and female physiology]).
7. Fan et al 50
8. Gouronc et al 51
9. Fieggen et al 52
10. These disorders have phenotypic overlap with BBS and should be considered in the differential diagnosis of BBS.
11. Shamseldin et al 48
12. Aldahmesh et al 53, Schaefer et al 54
13. In the literature, both IFT74 and IFT172 are associated with BBS20, so the recommendation is to discard the use of “BBS20.”
14. Lindstrand et al 55, Kleinendorst et al 56
15. Marion et al 57, Schaefer et al 58
16. Xing et al 59
17. Wormser et al 60
18. Morisada et al 61, Shamseldin et al 48
19. Chiang et al 62
20. Kim et al 63, Shamseldin et al 48
Bardet-Biedl syndrome inheritance pattern
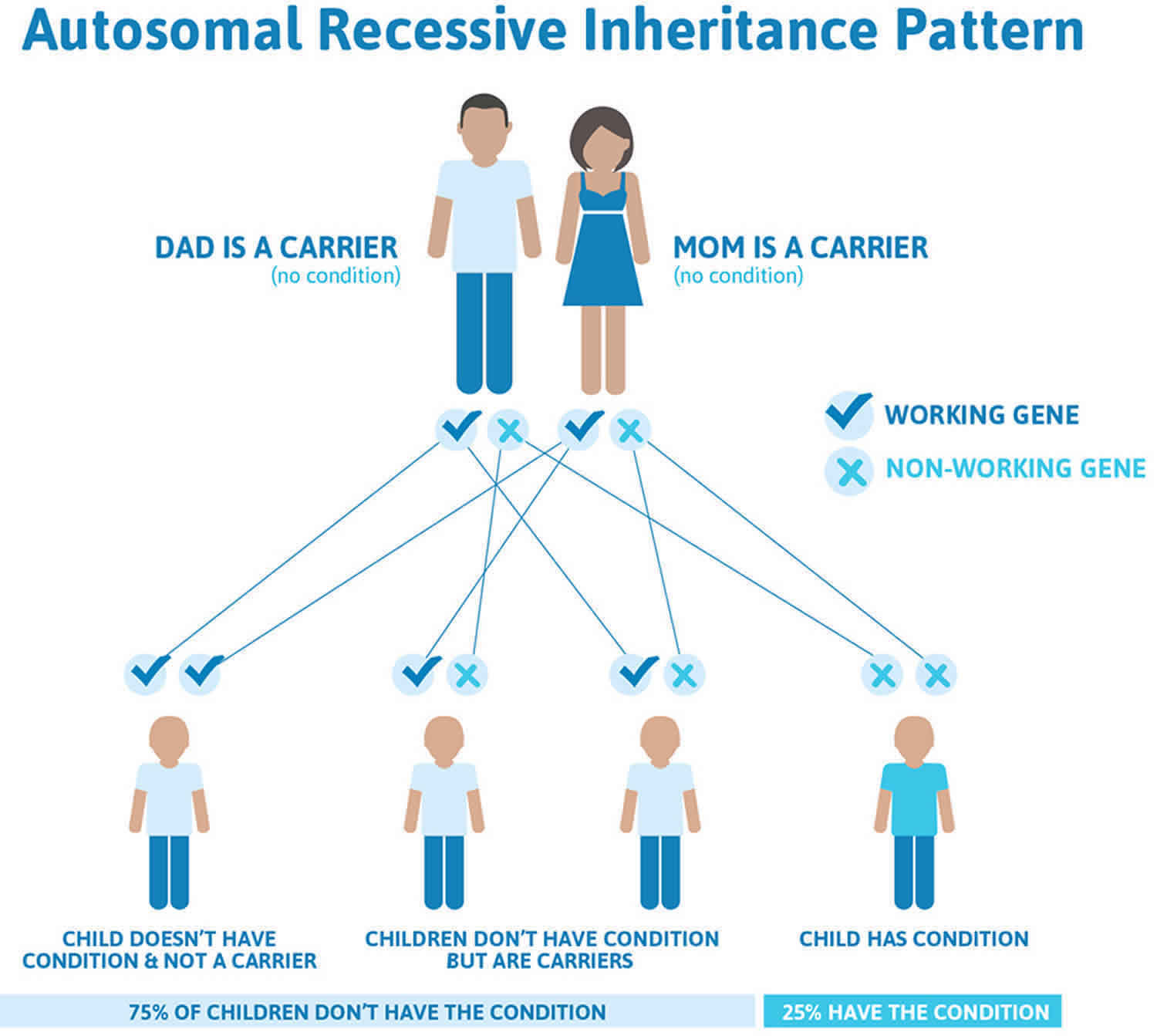
Bardet-Biedl syndrome is typically inherited in an autosomal recessive pattern, which means both copies of a Bardet-Biedl syndrome gene (BBS gene) in each cell have mutations. Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% (1 in 4 chance) with each pregnancy. The risk to have a child who is a carrier like the parents is 50% (1 in 2 chance) with each pregnancy. The chance for a child to receive normal genes from both parents is 25% (1 in 4 chance). The risk is the same for males and females. As Laurence-Moon-Biedl syndrome is rare, a gene carrier is unlikely to have affected children unless their partner is also a carrier.
Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
If you are born to parents who both carry the same BBS gene (Bardet-Biedl syndrome gene), you have a 25% (1 in 4) chance of inheriting the BBS gene (Bardet-Biedl syndrome gene) from both parents and developing Laurence-Moon-Biedl syndrome (see Figure 5). You have a 50% (1 in 2) chance of inheriting one variant BBS gene (Bardet-Biedl syndrome gene). This would make you BBS gene (Bardet-Biedl syndrome gene) mutation carrier.
In other words, for a child born to a couple who both carry the BBS gene (Bardet-Biedl syndrome gene) mutation (but do not have signs of disease), the expected outcome for each pregnancy is:
- A 25% chance that the child is born with two normal genes (healthy)
- A 50% chance that the child is born with one normal and one variant gene (carrier, without disease)
- A 25% chance that the child is born with two variant genes (at risk for the disease).
In most of North America and Europe, Bardet-Biedl syndrome has a prevalence of 1 in 125,000 to 1 in 160,000 newborns 15, 6, 1. Bardet-Biedl syndrome (BBS) also occurs more frequently, due to increased marriages among consanguineous, affecting about 1 in 13,500 newborns in the Bedouin population of Kuwait; 1:6900 in Jahra district, 1 in 17,000 in the island of Newfoundland (off the east coast of Canada) and 1:3700 in Faroe Islands 19, 20, 21, 22, 1.
Figure 5. Bardet-Biedl syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://findageneticcounselor.nsgc.org) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://www.acmg.net/ACMG/Directories.aspx) has a searchable database of medical genetics clinic services in the United States.
Bardet-Biedl syndrome signs and symptoms
Vision loss is one of the major features of Bardet-Biedl syndrome 1, 2. Most patients with Bardet-Biedl syndrome will experience the loss of light-sensing tissue at the back of the eye called the retina 5. The retina is part of the eye involved in detecting and decoding incoming images. Incoming light is focused onto the retina at the back of the eye. The retina is composed of cells called “rods and cones”. They translate incoming light into nerve impulses the brain can use. This gradual loss of the rod and cone cells on the retina is described as “retinal dystrophy”. Symptoms associated with cone-rod dystrophy may not become apparent until 7 or 8 years of age when children begin to complain of problems with night vision with an inability to see in dimly lit environments, such as a sidewalk lit only by streetlights 5. This “night blindness” may progress to variable degrees by blind spots that develop in the side (peripheral) vision. In most people, the vision becomes progressively weaker through the first and second decades of life 5. Over time, these blind spots enlarge and merge to produce tunnel vision. Affected individuals often first lose peripheral vision, and see only what is directly in front of their focus point. They see in what is termed ‘tunnel vision’ 5. Most people with Bardet-Biedl syndrome also eventually lose central vision (poor visual acuity) and become legally blind by mid-teens or early adulthood 5. In some people, the degeneration of the retina may follow a characteristic course, referred to as “retinitis pigmentosa” (RP). Retinitis pigmentosa (RP) begins with night blindness, followed by a loss of the ability to discriminate colors from one another, and finally to a progressive tunnel vision 5. Additional effects on the eye characteristic to individuals with Bardet-Biedl syndrome include: lazy eye (strabismus), clouding of the lens of the eyes (cataracts), and an increased pressure within the eyes that can result in damage to the optic nerve conducting signals to the brain (glaucoma) 5.
Truncal obesity is another characteristic feature of Bardet-Biedl syndrome 1, 2, 5. The term ‘truncal obesity’ refers to a condition where fat is disproportionately distributed onto the abdomen and chest rather than the arms and legs 5. Individuals can be described as having an apple-shape body type. Weight is usually normal at birth but weight gain is quickly evident through the first year of life in as many as 90% of people with Bardet-Biedl syndrome 5. Complications of obesity can include type 2 diabetes, high blood pressure (hypertension), and abnormally high cholesterol levels (hypercholesterolemia). Type 2 diabetes has been estimated to affect up to 45% of patients with Bardet-Biedl syndrome 5. High blood pressure (hypertension), and abnormally high cholesterol levels (hypercholesterolemia) may further complicate problems with the heart and blood vessels seen in patients with Bardet-Biedl syndrome. The heart functions as a pump for the blood, moving the blood through the vessels that bring it throughout the body. The heart relies on valves that keep the flow moving in the forward direction. With age, stiffening of the heart valves is completely normal. The stiffening is due to calcium laying down on the valves, and this process if described by the word “stenosis”. People with Bardet-Biedl syndrome may experience stenosis of their heart valves prematurely 5. People with Bardet-Biedl syndrome may also have defects of the heart’s muscular walls 5. The heart muscle is designed such that every motion is smoothly orchestrated. Defects in the heart muscle predispose people with Bardet-Biedl syndrome to heart beat abnormalities, referred to as “arrhythmias” 5.
Other major signs and symptoms of Bardet-Biedl syndrome include the presence of extra fingers or toes (polydactyly), intellectual disability or learning problems, and abnormalities of the genitalia. People with Bardet-Biedl syndrome may be born with an extra digit near the pinky or an extra toe near the fifth “little” toe (polydactyly). This finding occurs in approximately 70 percent of patients. Specifically, the presence of an extra toe is more common than that of an extra finger 5. In medical terminology, this is described as ‘postaxial polydactyly’. Fingers and toes may also show webbing, called “syndactyly”. Syndactyly is especially common between the second and third toes 5. Fingers and toes may occasionally be abnormally short in length. This characteristic is called “brachydactyly” 5. The feet may overall be short in length, of wide width and carry a flat arch.
Another major feature of Bardet-Biedl syndrome is a small size and poor function of the male gonads, termed “testicular hypogonadism”. This may manifest as a small penis, failure of the testes to descend into the scrotum termed “cryptorchidism” or a delay in the onset of puberty. Undescended testicles are a concern because they are associated with a greater risk for testicular cancer and should be surgically managed. An undescended testicle needs to be treated surgically with a procedure called orchiopexy before a child is 2 years old to increase his chance for fertility later in life. Affected females may also have complex genital and urinary tract abnormalities. Affected females may demonstrate an underdeveloped uterus, fallopian tubes, or ovaries. Menstruation cycles are often delayed from the average first age of onset and may also follow an irregular cycle. Pregnant women with Bardet-Biedl syndrome should be followed closely by obstetricians that are well trained in dealing with high-risk pregnancies.
Problems with fertility arise in both men and women. Most affected males produce reduced amounts of sex hormones (testosterone), and they are usually unable to father biological children (infertile) 1, 2.
Some individuals with Bardet-Biedl syndrome may also have abnormalities of the structure and function of the kidneys, which can be serious or life-threatening 1, 2. Kidney defects are highly variable but generally result in an accumulation of urine in the kidneys that results in inappropriate pressures within the kidneys, leading to stretching of important structures 5. The dilation resulting from this fluid accumulation is called “hydronephrosis” 5. It can be monitored by medical imaging such as ultrasound, abdominal x-ray, etc. One common risk that accompanies hydronephrosis includes bacterial infection of the kidneys. The inflammation associated with infection of the kidneys is called “pyelonephritis” 5. These complications to kidney functions can often predispose individuals with Bardet-Biedl syndrome to end-stage renal disease (ESRD) also known as kidney failure. Other clinical feature of Bardet-Biedl syndrome include the development of kidney cysts and damage to the microscopic filtration unit of the kidney 5. Kidneys are responsible for filtering the blood and damage to the filtration systems will result in urine that is dark red blood, possibly even foamy. In scenarios of kidney failure, patients may require dialysis and kidney transplantation. In the scenario that a patient requires kidney transplantation, the use of kidney-protective immunosuppressive medications have been associated with an extra increase in weight gain. This extra weight gain can further complicate any pre-existing diabetes and heart conditions.
Mild-to-moderate learning difficulties are common in individuals with Bardet-Biedl syndrome 5. Often, learning disabilities are attributed to weakened cognitive capacity. Some individuals affected with Bardet-Biedl syndrome may have true learning disabilities due to dysfunction of brain development 5. However, it is important to be sure that suspected disabilities (eg: delayed speech or reading skills) are not due to underlying vision impairment 5. Neurological impairments may manifest in poor coordination, gross and fine motor skills, and social milestones (eg: ability to play complicated games with other children). Many patients report a significant degree of clumsiness and often walk with legs in a wide-based stance. Walking heel-to-toe may be difficult 5.
Additional features of Bardet-Biedl syndrome can include impaired speech, delayed development of motor skills such as standing and walking, behavioral problems such as emotional immaturity and inappropriate outbursts, and clumsiness or poor coordination 1, 2. Distinctive facial features, dental abnormalities, unusually short or fused fingers or toes, and a partial or complete loss of the sense of smell (anosmia) have also been reported in some people with Bardet-Biedl syndrome 1, 2. They have a decreased ability to sense smells due to a change in the size to a brain center called the “olfactory bulb”. This is a relatively a mild problem but may impact safety if people are unable to sense for example, a gas leak from the stove 5.
In the absence of one of these 4 primary clinical features (i.e., vision loss, intellectual disability, obesity, polydactyly), the diagnosis of Bardet-Biedl syndrome (BBS) is made when at least two secondary features are observed, including hepatic fibrosis, diabetes mellitus, reproductive and developmental abnormalities, growth retardation, speech delays, or cardiovascular problems 1, 2.
Some individuals with Bardet-Biedl syndrome may also experience problems with their liver and digestive system. The liver is responsible for many body processes. Among them, it produces a green-brown digestive fluid that the body needs to break food down properly. Specifically, the liver conducts bile through thin ducts that can develop dilation or stricture and leak digestive fluid into the liver, where it causes damage in the form of scarring. More rarely, problems with digestive system may be due to Hirschsprung disease. Hirschsprung disease describes an absence of the nerves normally found in the colon that control the innate motion of the colon and move food along the tract.
A subgroup of affected individuals may exhibit some distinct facial features. These features including deep-set, widely-spaced eyes with downward-slanting lid folds, a flat nasal bridge with nostrils that flare forward, and a long groove (philtrum) in the center of the upper lip. Individuals may have a high-arched palate, with fewer teeth than expected The teeth may have short roots and lie crowded within the mouth.
Kidney Disease in Bardet-Biedl syndrome
Bardet–Biedl syndrome has classically been associated with polycystic kidney disease, a typical feature of ciliopathies with kidney manifestations 33, 36, 64, 65, 66, 67. The prevalence of kidney disease in Bardet-Biedl syndrome has been estimated at 53 to 82% 33, 36, 65. A recent study of 350 patients from the United Kingdom estimated that 50% of patients will develop functional kidney disease and demonstrated that cystic or dysplastic disease only accounts for 30% of patients with kidney disease, where the remainder have hydronephrosis, scarred or atrophic kidneys, loss of corticomedullary function, or developmental abnormalities 33. Around 8% of patients go on to develop end-stage renal disease (kidney failure) requiring dialysis or transplantation 33. The majority of patients who develop end-stage renal disease do so in early childhood (before the age of 5), and in most cases, deterioration is rapid with frequent requirement for dialysis within the first year of life 33. Some patients develop sudden kidney failure in adulthood for unknown reasons, and a further group of patients develop end-stage kidney disease as a result of comorbidities including type 2 diabetes and hypertension 33. The prevalence of these comorbidities is thought to be higher in Bardet-Biedl syndrome patients than the normal population and in 1 study of 69 patients were found in 22 and 35%, respectively 37. The risk of type 2 diabetes relates to obesity and is treated using standard protocols. Many patients with structural kidney abnormalities do not go on to develop functional kidney disease 37.
Bardet-Biedl syndrome diagnosis
Bardet-Biedl syndrome is generally diagnosed based on the presence of at least 4 major features (eg: visual problems due to retinal dystrophy, truncal obesity, post-axial polydactyly) or 3 major features and at least 2 minor features in accordance with the diagnostic criteria published by Beales et al. 68. Figure 1 above demonstrates the clinical features associated with Bardet-Biedl syndrome and highlights the relative frequencies at which these features are observed. However, since the signs and symptoms of Bardet-Biedl syndrome vary among affected individuals, even among members of the same family, some patients may not have a clear diagnosis for many years. Difficulties with Bardet-Biedl syndrome diagnosis arise when a child demonstrates learning disabilities and problems with obesity but who was not born with any congenital abnormality. For these children, diagnosis may remain uncertain until they begin to manifest vision loss symptoms. Diagnosis of retinal disease may require consultation with an eye specialist (ophthalmologist) and an examination including an electroretinogram (ERG). The electroretinogram (ERG) is a procedure that measures the electrical response of the retina to light stimulation.
The age at which people with Bardet-Biedl syndrome are diagnosed is extremely variable and is driven by the age of onset of symptomatic rod-cone dystrophy 23. While this may manifest in infancy, it is more usually seen between the ages of 5 and 10 years of age and typically presents with night blindness 68. Isolated polydactyly at birth or obesity, generally seen from infancy, do not usually prompt referral. Siblings of affected children are generally diagnosed earlier. Antenatal diagnosis is extremely rare in the absence of a family history, but BBS may be suspected from the identification of echogenic kidneys and polydactyly on ultrasound scanning 23. Children presenting with kidney anomalies or kidney failure may be diagnosed earlier than those without, but there are insufficient data to confirm this 23. A subset of individuals present with isolated rod-cone dystrophy with notable absence of other BBS-related features and are often diagnosed in adulthood 23. These individuals are now being picked up because of the introduction of panel-based genetic testing and major diagnostic studies such as the UK 100,000 genomes project 69 and the Deciphering Developmental Disorders (exome) study 70. They were previously overlooked as there are many causes of rod-cone dystrophy, and it was not understood that BBS genes could cause this feature in isolation.
Genetic testing may help confirm the diagnosis for some patients (e.g., individuals with certain gene mutations including the common BBS1 p.M390R, BBS2 p.Y24X, BBS2 p.R275X, and BBS10 c.91fsX5 mutations). The use of whole exome sequencing (WES) and whole genome sequencing (WGS) may increase coverage, aid in the discovery of novel genes, and allow for the identification of non-coding variants. However, along with increased expense, such gene testing may not be covered by insurance and are available only through research laboratories with a special interest in Bardet-Biedl syndrome 18. The disadvantage of the more advanced diagnostic genetic sequencing techniques is the identification of pathogenic variants in non-BBS genes and of variants of unknown significance (VUS) in BBS genes 71. This can result in a diagnostic conundrum in particular where patients manifest only one or two non-specific major diagnostic criteria such as obesity and/or learning difficulties.
Table 2. Bardet-Biedl syndrome clinical features
| Primary features | Secondary features |
|---|---|
|
|
Bardet-Biedl syndrome differential diagnosis
The conditions below are ones which may clinically resemble Bardet-Biedl syndrome. The list below can be useful in establishing a differential diagnosis. Without genetic testing, it can be very difficult to clinically differentiate these conditions.
Laurence-Moon syndrome
Laurence-Moon syndrome (LNMS or LMS), also called Laurence-Moon-Biedl syndrome (LMBBS) because of similarities with Bardet-Biedl syndrome (BBS), is a very rare genetic disorder that affects many parts of the body characterized by underactive pituitary gland (occurs when the pituitary gland doesn’t produce the right amount of hormones), ataxia (a neurological sign that causes problems coordinating how your muscles work, leading to awkward, unwieldy or clumsy movements), peripheral neuropathy (nerve outside of your brain and spinal cord also known as peripheral nerves are damaged causing them to stop working properly), spastic paraplegia (weakness and stiffness in your legs) and eye abnormalities called chorioretinal dystrophy primarily affecting the choroid (a layer of blood vessels between the retina and the sclera) and the retina (a light-sensitive layer of tissue at the back of your eyes that converts images into electrical signals for your brain) resulting in poor vision such as peripheral vision loss and vision loss at night, shortsightedness and abnormal eye movements with involuntary side-to-side movements of the eyes called nystagmus 73, 74, 75, 76, 77, 78, 79. People with Laurence-Moon-Biedl syndrome may have difficulties with functions of their brain, eyes, ears, stomach, kidneys, hands and feet. People with Laurence-Moon-Biedl syndrome often also demonstrate a tendency to short stature and obesity 79, 80.
Symptoms of Laurence-Moon-Biedl syndrome may start to appear in newborns. Laurence-Moon-Biedl syndrome is typically associated with syndactyly of the fingers (a condition where two or more fingers are fused together), polydactyly, intellectual impairment, obesity, undescended testicle (cryptorchidism), hypoplasia of the penis (also known as micropenis, is a rare condition that causes a penis to be smaller than normal), kidney impairment, sensorineural hearing loss, and a short height. Most patients with Laurence-Moon-Biedl syndrome will experience a gradual loss of vision due to retinitis pigmentosa (due to the loss of rod-cone photoreceptors), which begins with night blindness, worsens progressively with loss of color perception, and finally deteriorates into “tunnel vision”. Mild-to-moderate learning challenges attributed to weakened cognitive capacity are common in individuals with Laurence-Moon-Biedl syndrome. However, the suspected disabilities are not due to an underlying visual impairment. A smaller-than-average-size anterior pituitary gland has been observed in people with Laurence-Moon-Biedl syndrome, and as a result, these individuals are susceptible to various complications, including those related to the control of the body’s metabolism, emotional responses to stressors, physical development, low levels of thyroid-stimulating hormone (TSH), decreased levels of estrogen and testosterone, and underdeveloped reproductive organs 79.
Laurence-Moon syndrome or Laurence-Moon-Biedl syndrome is most commonly caused by changes (mutations) in the PNPLA6 (patatin like phospholipase domain containing 6) gene and is inherited in an autosomal recessive manner 77. Genes are specific sequences in DNA that provide instructions for the production of proteins. The PNPLA6 gene provides instructions for making a protein called neuropathy target esterase (NTE) 81, 79. The neuropathy target esterase (NTE) protein is an enzyme that is involved in the breakdown of certain fats (lipids). Specifically, neuropathy target esterase (NTE) breaks down a lipid called lysophosphatidylcholine, which is one of several compounds found in the outer membranes surrounding cells 81. The correct levels of lysophosphatidylcholine is critical to the stability of the cell membranes. The neuropathy target esterase (NTE) protein is an enzyme that is thought to drive the growth of nerve and non-nerve cells as they grow and mature. The PNPLA6 gene is notably associated not only with Laurence-Moon-Biedl syndrome but also Boucher-Neuhauser syndrome, Gordon-Holmes syndrome, Oliver-McFarlane syndrome, and spastic paraplegia type 39 81.
Laurence-Moon-Biedl syndrome follows an autosomal recessive pattern of inheritance (see Figure 5 below). Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% (1 in 4 chance) with each pregnancy. The risk to have a child who is a carrier like the parents is 50% (1 in 2 chance) with each pregnancy. The chance for a child to receive normal genes from both parents is 25% (1 in 4 chance). The risk is the same for males and females. As Laurence-Moon-Biedl syndrome is rare, a gene carrier is unlikely to have affected children unless their partner is also a carrier. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder 82.
In North America, it is estimated that 1 in 100,000 people are affected by Laurence-Moon-Biedl syndrome 79. In Europe, it is estimated that 1 in 125,000 to 1 in 160,000 people are affected by Laurence-Moon-Biedl syndrome 75. Kuwait and Newfoundland (off the east coast of Canada) are two places where the number of people affects with Laurence-Moon-Biedl syndrome are comparatively high affecting about 1:13,500 newborns and 1:17,500, respectively 24, 83. Consanguinity (union between two people who are related as second cousins or closer) is commonly practiced in various Middle Eastern countries, such as Kuwait, Saudi Arabia, Iran, and Pakistan 83. Consanguinity has been a chief contributing factor to Laurence-Moon-Biedl syndrome frequency. Families in these countries have approximately 0.1% homozygous genes of the total genome 83. In Pakistan, more than half of all marriages are consanguineous in nature, with 80% of them among first cousins, thereby increasing the possibility of homozygous mutations 83.
Due to the highly variable clinical presentation of Laurence-Moon-Biedl syndrome, no formal diagnostic criteria have been established for Laurence-Moon-Biedl syndrome or, for that matter, any PNPLA6-related disorders. Laurence-Moon-Biedl syndrome is diagnosed with molecular testing for mutations in the PNPLA6 gene 79, 84.
The management of Laurence-Moon-Bardet-Biedl syndrome involves a multidisciplinary approach and remains a challenge for clinicians. The treatments available for Laurence-Moon-Biedl syndrome are mainly toward managing the signs, symptoms and complications of the illness. Physical therapy aimed toward improving strength helps. Exercise can reduce the symptoms of spasticity. A dedicated regimen of nutritious, well-balanced meals and regular exercise is recommended, as there is an increased incidence of diabetes and abnormal cholesterol levels in patients with Laurence-Moon-Biedl syndrome 83. A low protein diet also slows the progression of kidney diseases in Laurence-Moon syndrome 85. The poor functional capacity of the anterior pituitary gland, resulting in slow metabolism, poor growth, and impaired fertility, can be managed with hormone replacement therapies. Levothyroxine can aid in increasing the body metabolism, resulting in reduced lethargy, hair loss, and obesity. Growth hormone supplementation reduces the psychosocial burden of short stature, whereas testosterone supplementation can be given in patients with markedly low levels to prevent underdeveloped genitalia. The extra digits are generally nonfunctional and can be removed for cosmetic purposes. Typically, retinal dystrophy is the first symptom that arises before the age of 10 years but affects almost all patients below the age of 20 years 86. Glasses can be used to treat this, and regular eye specialist (ophthalmologist) visits are recommended 87.
Alstrom syndrome
Alstrom syndrome (ALSS or ALMS) is a very rare autosomal recessive genetic disorder that is characterized by progressive loss of vision and hearing, childhood obesity, a form of heart disease that enlarges and weakens the heart muscle (dilated cardiomyopathy), type 2 diabetes, slowly progressive kidney and liver dysfunction and short stature 88, 89, 90. The signs and symptoms of Alström syndrome vary in severity, and not all affected individuals have all of the characteristic features of the disorder. Many of the signs and symptoms of Alstrom syndrome begin in infancy or early childhood, although some appear later in life.
Initial symptoms usually include an insensitivity to light and series of rapid, involuntary eye movements described under the term, “nystagmus”. Additional features sometimes associated with Alstrom syndrome include dysfunction of heart muscle and a skin condition called acanthosis nigricans, which causes the skin in body folds and creases to become thick, dark, and velvety 88, 89, 90. Intelligence is not affected. Alström syndrome can also cause serious or life-threatening medical problems involving the liver, kidneys, bladder, and lungs.
Mutations in the ALMS1 gene cause Alström syndrome. The ALMS1 gene provides instructions for making a protein whose function is unknown. Mutations in this gene probably lead to the production of an abnormally short, nonfunctional version of the ALMS1 protein. This protein is normally present at low levels in most tissues, so a loss of the protein’s normal function may help explain why the signs and symptoms of Alström syndrome affect many parts of the body.
There is no specific treatment for Alstrom syndrome. Treatment for symptoms may include:
- Diabetes medicine
- Hearing aids
- Heart medicine
- Thyroid hormone replacement
Meckel-Gruber syndrome
Meckel-Gruber syndrome also known as Meckel syndrome is rare genetic disorder that affects the structure and function of cilia (ciliopathy) that affect many parts of the body like Bardet-Biedl syndrome (BBS) 91. Meckel-Gruber syndrome signs and symptoms include retinal degeneration, extra fingers and toes (polydactyly), kidney cysts (enlarged kidneys with numerous fluid-filled cysts), occipital encephalocele which is a sac-like protrusion of the brain through an opening at the back of the skull and buildup of scar tissue (fibrosis) in the liver as well as poor lung function resulting from low amniotic fluid volumes within the placenta during the pregnancy 91.
Other signs and symptoms of Meckel-Gruber syndrome vary widely among affected individuals 91. Numerous abnormalities of the brain and spinal cord (central nervous system) have been reported in people with Meckel-Gruber syndrome, including a group of birth defects known as neural tube defects. These defects occur when a structure called the neural tube, a layer of cells that ultimately develops into the brain and spinal cord, fails to close completely during the first few weeks of embryonic development 91. Meckel-Gruber syndrome can also cause problems with development of the eyes and other facial features, heart, bones, urinary system, and genitalia 91.
Because of their serious health problems, most individuals with Meckel-Gruber syndrome die before or shortly after birth 91. Most often, affected infants die of respiratory problems or kidney failure.
Meckel syndrome can be caused by changes (mutations) in thirteen genes: B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRIP1L, TCTN2, TCTN3, TMEM67, TMEM107, TMEM216, TMEM231 and TMEM237 92. Mutations in these 13 genes account for 75 percent of all Meckel-Gruber syndrome cases; the remaining 25 percent have unknown genetic causes 92. Most of these genes are also responsible for a neurological disorder called Joubert syndrome, leading to the concept that Meckel syndrome is the extreme lethal form of Joubert syndrome 92. Mutations in several other genes have been identified in people with features similar to those of Meckel-Gruber syndrome, although it is unclear whether these individuals actually have Meckel-Gruber syndrome or a related disorder often described as a “Meckel-like phenotype” 91.
The proteins produced by these genes are known to influence cell structures or function called cilia. Cilia are microscopic, finger-like projections that stick out from the surface of cells and are involved in signaling pathways that transmit information between cells. Cilia are important for the structure and function of many types of cells, especially in the kidneys, liver, eyes and brain. Mutations in these gene cause problems in the function of the primary cilia, resulting in various defects dependent of the cell type. Early defective ciliary function can be responsible for developmental abnormalities, specifically in the kidneys, brain, limbs, heart.
Mutations in the genes associated with Meckel-Gruber syndrome lead to problems with the structure and function of cilia. Defects in these cell structures probably disrupt important chemical signaling pathways during early development. Although researchers believe that defective cilia are responsible for most of the features of Meckel syndrome, it remains unclear how they lead to specific developmental abnormalities of the brain, kidneys, and other parts of the body.
McKusick-Kaufman syndrome (MKKS)
McKusick-Kaufman Syndrome (MKKS or MKS) also called hydrometrocolpos-polydactyly syndrome is a very rare, autosomal recessive developmental disorder presenting in the neonatal period that affects the development of the hands, feet, heart, and reproductive system. McKusick-Kaufman syndrome (MKKS) is characterized by a combination of 3 clinical features: extra fingers and/or toes (polydactyly), congenital heart defects and genital abnormalities 93, 94, 95, 96, 97, 98. The most common genital abnormality and the hallmark of McKusick-Kaufman Syndrome (MKKS) in female patients is hydrometrocolpos, an accumulation of fluid in the vagina and uterus as a result of the accumulation of cervical secretions from maternal estrogen stimulation, due to a blocked vaginal outlet 94. The blockage allows fluid to build up in the vagina and uterus, stretching these organs and leading to a fluid-filled mass. Hydrometrocolpos can be caused by a number of factors, including vaginal agenesis (failure of the distal third of the vagina to develop), vaginal atresia (a rare developmental defect where the lower part of the vagina doesn’t form properly), an imperforate hymen, persistent urogenital sinus, cloacal malformation, and transverse vaginal membrane 93. McKusick-Kaufman syndrome (MKKS) female patients may have an absent vagina with mucoid accumulations within an intact uterus within the abdomen 93, 94. Female with McKusick-Kaufman Syndrome (MKKS) may alternatively have a double vaginal or uterine structures. Hydrometrocolpos obstructs adjacent structures either in prenatal or postnatal life 96. In a fetus, urinary bladder obstruction may lead to proximal urinary tract dilation that results in oligohydramnios malformation sequence and diaphragm compression, which ultimately results in lung hypoplasia 96. In utero rectal compression, intestinal obstruction can rarely lead to perforation and peritonitis 99, 100. This usually presents as Hirschsprung’s disease (a rare birth defect that occurs when nerves in the intestine don’t develop properly) 101, 102. Inferior vena cava compression may result in hydrops fetalis and pedal edema 102. After birth, bladder displacement results in micturition defects which can cause recurrent pyelonephritis and chronic renal failure 100. Older children or teenagers present with menstrual pain, primary amenorrhea, constipation, lower back pain, or urinary retention 103.
McKusick-Kaufman syndrome (MKKS) in males can include undescended testes (cryptorchidism), abnormal opening of the urethra on the underside of the penis (hypospadias) and a downward-curving penis (chordee) 93, 94. Either sex may have fibrosis or degeneration of their ureters, the conduit for urine formed by the kidneys destined for storage in the bladder 93.
Polydactyly (extra fingers or toes) or brachydactyly (fingers and toes that are abnormally short in length) is present in 90% of McKusick-Kaufman syndrome (MKKS) cases 95. In people with McKusick-Kaufman syndrome, the extra fingers and toes are typically on the same side of the hand or foot as the pinky or little toe also known as postaxial polydactyly 93. The congenital heart defects in individuals with McKusick-Kaufman syndrome can include an atrial septal defect (ASD) or a ventricular septal defect (VSD), which are openings in the wall (septum) that separates the upper or lower chambers of the heart or a complex congenital heart malformation 93.
The signs and symptoms of McKusick-Kaufman syndrome (MKS) overlap significantly with those of another genetic disorder, Bardet-Biedl syndrome (BBS). However, Bardet-Biedl syndrome (BBS) has several features that are not typically seen in people with McKusick-Kaufman syndrome (MKKS). These include a gradual loss of vision, developmental disabilities, kidney abnormalities, and obesity 93. Because some of these features are not apparent at birth, the two conditions can be difficult to tell apart in infancy and early childhood.
Both McKusick-Kaufman syndrome and Bardet-Biedl syndrome (BBS) belong to a group of conditions called ciliopathies. Ciliopathies are inherited disorders that affect the structure or function of cilia, the microscopic, finger-like projections found on the surface of cells. Cilia are involved in signaling pathways that transmit information between cells.
Mutations in the MKKS gene also called the BBS6 gene have been found to cause McKusick-Kaufman syndrome 93. The MKKS gene (BBS6 gene) provides instructions for making a protein that plays an important role in early development, specifically in the formation of the limbs, heart, and reproductive system. The protein’s structure suggests that it may belong to a family of proteins called chaperonins. Proteins must be folded into the correct shape to function properly, and chaperonins help them do that.
The MKKS protein is thought to play a role in cell division and cell transport. Specifically, the MKKS protein is thought to transport molecules between the cytoplasm and the nucleus of the cell. The MKKS protein also combines with other proteins to form a complex within the cell. This complex is used to assemble the molecules involved in transporting materials that support the function of cilia.
Though it is not clear exactly how mutations in the MKKS gene (BBS6 gene) lead to the specific signs and symptoms of McKusick-Kaufman syndrome, two particular variants in the MKKS gene (BBS6 gene) appear to impair the protein’s ability to transport a molecule involved in regulating gene activity. This change likely affects the activity of certain genes that are critical during early development.
Once the diagnosis of McKusick-Kaufman syndrome (MKS) is confirmed, immediate decompression of the dilated uterus in female babies improves the prognosis for kidney function and lung capacities 95. Surgical intervention should be performed early to stop infections in the urinary tract. Molecular genetic analysis may be useful for treating and counseling females with hydrometrocolpos 104.
Biemon 2 syndrome
Biemond 2 syndrome also called Biemond syndrome type 2 (BS2) is an extremely rare autosomal recessive inherited disorder that affects the development and nervous system. Biemond 2 syndrome (BS2) is characterized by absence of tissue from the colored portion of the eye (iris coloboma), intellectual disability, short stature, obesity, hypogonadism, genitourinary abnormalities, hydrocephalus, facial dysostosis and an extra finger near the pinky or an extra toe near the fifth toe (postaxial polydactyly). Biemond 2 syndrome is also known as hypogonadism-short stature-coloboma-preaxial polydactyly syndrome. Biemond 2 syndrome shares some features with Bardet-Biedl syndrome (BBS). Causal genes for Biemond 2 syndrome have not yet been identified. There have been no further descriptions in the literature since 1997 105.
Prader-Willi syndrome
Prader-Willi syndrome is a rare genetic disorder characterized by weak resting muscle strength (hypotonia), feeding difficulties, and failure to gain weight through infancy (failure to thrive). In later childhood, features of Prader-Willi syndrome include short stature, genital abnormalities, excessive appetite, obesity, and type 2 diabetes. Progressive obesity presents in most people with Prader-Willi syndrome because of a lack of feeling satisfied after completing a meal (satiety) that leads to overeating. All individuals with Prader-Willi syndrome have some degree of cognitive impairment that ranges from borderline normal with learning disabilities to severe intellectual disability. Behavior problems are common and can manifest as temper tantrums, obsessive-compulsive behavior, or skin picking. Prader-Willi syndrome occurs when the genes in a specific region of chromosome 15 do not function. The abnormal genes usually result from random errors in development, but are sometimes be inherited from a parent. There’s no cure for Prader-Willi syndrome, but early diagnosis and treatment can help improve quality of life.
Bardet-Biedl syndrome treatment
The primary treatment goal for patients with Bardet-Biedl syndrome involves treating the specific symptoms affecting each individual 5. Early intervention for anticipated problems can ensure that people with Bardet-Biedl syndrome reach their greatest potential. As many body systems are involved, care often requires the coordinated effort of a team of specialists 5.
Puberty is a stressful period in the life of any person. It is beneficial to individuals with Bardet-Biedl syndrome to seek the guidance of an experienced counselor. Patients with low hormone levels may be prescribed supplements under the guidance of an endocrinologist 5.
Some of the physical abnormalities associated with Bardet-Biedl syndrome can be corrected with surgery, including extra digits, and some genitourinary abnormalities and congenital heart defects 5. Kidney transplantation may be appropriate later in life if severe kidney disease develops. Surgery is a point of particular concern for patients with Bardet-Biedl syndrome. General anesthesia requires a series of highly coordinated steps that rely on the anatomy of the airways. Some patients with Bardet-Biedl syndrome may have significant anatomical anomalies in the airways and this might result in increased difficulty holding the airway open during general anesthesia 5. If this is the case, anesthetic medications may be introduced in the form of direct nerve blocks to a region of the body with while the patient is breathing for themselves 5.
As obesity is a common component to Bardet-Biedl syndrome, this is a particularly important factor to address 5. Obesity manifests typically by an age of two-three years 5. An active lifestyle incorporating physical activities can make a significant impact. Both diet and exercise programs are also highly recommended. Good diet management can prevent obesity that manifest in later life. Consulting with a primary care physician and a dietician can help in planning for adequate nutrition and prevention of excess weight gain 5. If problems with high cholesterol and diabetes exist, they are treated as in the general population. Weightloss surgery (bariatric surgery) with gastric banding or sleeve surgery has been attempted only in very few patients with Bardet-Biedl syndrome. In those patients, weight loss surgery was associated with a weight loss of 25% maintained at 12 months after procedure. Long-term follow-up of these patients is being conducted to determine the possible role for weight loss surgery in patients with Bardet-Biedl syndrome.
The eye problems are of central concern with Bardet-Biedl syndrome 5. The first symptom onset is usually that of night-blindness, typically seen around age 8-9 years of age 5. Vitamin A deficiency can worsen visual difficulties and age-appropriate vitamin and mineral supplements can help support best function 5. While there are currently no proven therapies available to cure the retinal dystrophy associated with Bardet-Biedl syndrome, care under the supervision of an ophthalmologist is critical. Ophthalmologists can help correct refractive errors (e.g., myopia/near-sightedness or hyperopia/far-sightedness) and low-visual acuity problems. Individuals with Bardet-Biedl syndrome should undergo regular eye examinations and keep up with their changing prescription lenses 5. As visual impairment is a major hurdle to learning in the classroom, special services might be organized between a child’s physician and their school.
In 2022, the U.S. Food and Drug Administration (FDA) approved setmelanotide (Imcivree) as a treatment option for chronic weight management in adults and children 6 years and older with obesity due to Bardet-Biedl syndrome 5.
Table 3. Recommended Surveillance in individuals with Bardet-Biedl Syndrome
| System/Concern | Initial Evaluation | Frequency of Surveillance 1 |
|---|---|---|
| Constitutional |
| At every health care visit |
| Eyes/Vision | Ophthalmologic consultation in:
| Annually or as directed by ophthalmologist |
| Oral/dental abnormalities | Routine dental care | Every 6 mos starting at age 1 yr |
| Cardiovascular & other thoraco- abdominal abnormalities |
|
|
| Respiratory | Monitor for:
| Annually |
| Gastrointestinal |
| |
| Liver |
| Annually if normal Persons w/liver disease should be monitored as directed by hepatologist. |
| Renal |
| Annually if normal Persons w/kidney disease should be monitored as directed by nephrologist. |
| Urologic | Ask about symptoms of neurogenic bladder & bladder outflow obstruction. | Annually |
| Metabolic syndrome |
| Annually starting at age 4 yrs if normal Those w/metabolic syndrome will require more frequent monitoring by experienced provider. |
| Hypothyroidism | Check thyroid gland function. | Annually |
| Hypogonadism |
| Annual lab assessment starting at age 13 yrs if indicated |
| Musculoskeletal | Skeletal survey | As needed if signs/symptoms of scoliosis, polydactyly, or joint disease |
| Development |
| Routine developmental assessments during early childhood School-aged persons should have annual individualized education program (IEP)/504 plans. |
| Psychiatric/ Behavioral | Neuropsychiatric eval if signs/symptoms of atypical behaviors or mood disorder | As needed |
| Genetic counseling | By genetics professionals 4 | To inform affected persons & their families re nature, mode of inheritance, & implications of BBS to facilitate medical & personal decision making |
| Family support & resources | Assess:
|
Abbreviations: BUN = blood urea nitrogen; CBC = complete blood cell count; FSH = follicle-stimulating hormone; HDL = high-density lipoproteins; HgbA1c = hemoglobin A1c; IBD = inflammatory bowel disease; IEP = individualized education program; LDL = low-density lipoproteins; LH = luteinizing hormone; MOI = mode of inheritance; PT = prothrombin time; PTT = partial thromboplastin time; SNHL = sensorineural hearing loss; US = ultrasound
Footnotes:
1. Recommended frequencies shown are for individuals who are stable and well-controlled. In many instances more frequent evaluations are needed. Individuals should be evaluated by a medical geneticist every one to two years, as they can help with coordination of care.
2. Prenatal ultrasonography may detect renal cysts but can be normal in 39% of individuals with renal abnormalities detected postnatally 106
3. MRI of the brain may show diffuse white matter loss predominantly in the occipital region, reduced grey matter in subcortical regions (caudate, putamen, thalamus), reduced hippocampal volume, and hippocampal dysgenesis 107, 108
4. Medical geneticist or certified genetic counselor.
Bardet-Biedl syndrome prognosis
Bardet-Biedl syndrome has poor prognosis with early onset of blindness, obesity, high blood pressure and diabetes mellitus 109. Kidney impairment is frequent and an important cause of death 109. Survival is substantially reduced. Bardet-Biedl syndrome have a shorter life expectancy than the general population with 25% of Bardet-Biedl syndrome patients had died by 44 years, whereas at that age 98% of unaffected siblings were still alive 109. In a prospective cohort study (1987 to 1993) to determine the natural history of Bardet-Biedl syndrome involving 21 families with Bardet-Biedl syndrome (BBS) 109, 38 patients with BBS and 58 unaffected siblings were identified. Age of onset of blindness, hypertension, diabetes, renal impairment, and death was determined. The prevalence of obesity, gonadal dysfunction, and renal structural abnormalities was assessed. All but 5 BBS patients (86%) were legally blind, 26% being blind by the age of 13 years and 50% by 18 years 109. Eighty-eight percent were above the 90th percentile for height and weight. Twenty-five (66%) patients had hypertension, 25% of BBS patients by age 26 years, and 50% by age 34 years, whereas in the unaffected group, 25% had hypertension by age 49 years 109. Twelve (32%) BBS patients developed type 2 diabetes, compared with none of the unaffected group. Only 2 patients were insulin dependent. Twenty-five percent of BBS patients had diabetes by the age of 35 years. In 12 women of reproductive age, 1 (8%) had primary gonadal failure. In 10 men, 4 had primary testicular failure. Nine (25%) patients developed kidney impairment, with 25% of the BBS group affected by the age of 48 years. Imaging procedures of the kidney were performed in 25 patients with normal kidney function. Whereas fetal lobulation and calyceal cysts/diverticula/clubbing were characteristic, occurring in 96% of patients, 20% (n = 5) had diffuse and 4% (n = 1) focal cortical loss 109. Eight patients (out of 38 patients) with Bardet-Biedl syndrome died, 3 with end-stage renal failure and 3 with chronic renal failure 109. On life-table analysis, 25% of BBS patients had died by 44 years, whereas at that age 98% of unaffected siblings were still alive 109.
Managing kidney issues and other complications can improve quality of life and life expectancy 110. Therefore, early kidney screenings and regular kidney function evaluations and urinalysis form part of the necessary follow-up protocol 111.
- Bardet-Biedl syndrome. https://medlineplus.gov/genetics/condition/bardet-biedl-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Bardet-Biedl syndrome. https://rarediseases.info.nih.gov/diseases/6866/x[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- What is Bardet-Biedl Syndrome? https://bbsuk.org.uk/what-is-bardet-biedl-syndrome[↩][↩][↩]
- Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, Heath O, McManamon PJ, O’Leary E, Pryse-Philips W. The cardinal manifestations of Bardet–Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Eng J Med. 1989;321:1002–1009. doi: 10.1056/NEJM198910123211503[↩]
- Bardet-Biedl Syndrome. https://rarediseases.org/rare-diseases/bardet-biedl-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Beales PL, Warner AM, Hitman GA, Thakker R, Flinter FA. Bardet–Biedl syndrome: A molecular and phenotypic study of 18 families. J Med Genet. 1997;34:92–98. doi: 10.1136/jmg.34.2.92[↩][↩][↩]
- Riise R, Andreasson S, Borgstrom M, Wright AF, Tommerup N, Rosenberg T, Tornqvist K. Intrafamilial variation of the phenotype in Bardet–Biedl syndrome. Br J Ophthalmol. 1997;81:378–385. doi: 10.1136/bjo.81.5.378[↩]
- Laurence JZ, Moon RC. Four cases of “retinitis pigmentosa” occurring in the same family, and accompanied by general imperfections of development. 1866. Obes Res. 1995 Jul;3(4):400-3. doi: 10.1002/j.1550-8528.1995.tb00166.x[↩][↩]
- Bardet G. Sur un syndrome d’obesite congenitale avec polydactylie et retinite pigmentaire (contribution a l’etude des formes cliniques de l’obesite hypophysaire) These de Paris (Le Grand) 1920;470:107.[↩]
- Bardet G. On congenital obesity syndrome with polydactyly and retinitis pigmentosa (a contribution to the study of clinical forms of hypophyseal obesity. Obes Res. 1995;3:387–399. doi: 10.1002/j.1550-8528.1995.tb00165.x[↩]
- Biedl A. Ein Geschwister mit adiposogenitaler Dystrophie. Dtsch Med Wochenschr. 1922;48:1630.[↩]
- Biedl A. A pair of siblings with adiposo-genital dystrophy. Obes Res. 1995;3:404. doi: 10.1002/j.1550-8528.1995.tb00167.x[↩]
- Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, Heath O, McManamon PJ, O’Leary E, Pryse-Phillips W. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989 Oct 12;321(15):1002-9. doi: 10.1056/NEJM198910123211503[↩]
- Solis-Cohen S, Weiss E. Dystrophia adiposogenitalis, with atypical retinitis pigmentosa and mental deficiency—The Laurence–Biedl syndrome. A report of four cases in one family. Am J Med Sci. 1925;169:489–505.[↩]
- Klein D, Amman F. The syndrome of Laurence-Moon-Bardet-Biedl and allied disease in Switzerland. Clinical, genetic and epidemiological studies. J Neurol Sci. 1969;9:479–513. doi: 10.1016/0022-510x(69)90091-4[↩][↩][↩]
- Schachat AP, Maumenee IH. Bardet–Biedl syndrome and related disorders. Arch Ophthalmol. 1982;100:285–288. doi: 10.1001/archopht.1982.01030030287011[↩]
- Laurence-Moon and Bardet-Biedl syndromes. Lancet. 1988 Nov 19;2(8621):1178. https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(88)90242-5/fulltext[↩]
- Forsyth RL, Gunay-Aygun M. Bardet-Biedl Syndrome Overview. 2003 Jul 14 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1363[↩][↩][↩][↩][↩]
- Farag TI, Teebi AS. High incidence of Bardet Biedl syndrome among the Bedouin. Clin Genet. 1989;36(6):463–464. doi: 10.1111/j.1399-0004.1989.tb03378.x[↩][↩]
- Moore SJ, Green JS, Fan Y, et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet A. 2005;132A(4):352–360. doi: 10.1002/ajmg.a.30406[↩][↩]
- Hjortshøj TD, Grønskov K, Brøndum-Nielsen K, Rosenberg T. A novel founder BBS1 mutation explains a unique high prevalence of Bardet-Biedl syndrome in the Faroe Islands. Br J Ophthalmol. 2009;93(3):409–413. doi: 10.1136/bjo.2007.131110[↩][↩]
- Farag TI, Teebi AS. Bardet–Biedl and Laurence–Moon syndromes in a mixed Arab population. Clin Genet. 1988;33:78–82. doi: 10.1111/j.1399-0004.1988.tb03414.x[↩][↩]
- Forsythe E, Kenny J, Bacchelli C, Beales PL. Managing Bardet-Biedl Syndrome-Now and in the Future. Front Pediatr. 2018 Feb 13;6:23. doi: 10.3389/fped.2018.00023[↩][↩][↩][↩][↩][↩][↩]
- Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, Stefanelli M, Murphy C, Cramer BC, Dean JC, Beales PL, Katsanis N, Bassett AS, Davidson WS, Parfrey PS. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet A. 2005 Feb 1;132A(4):352-60. doi: 10.1002/ajmg.a.30406[↩][↩]
- Hassan S, Khan QA, Saravanan P, Iram S, Rohail S, Belay NF, Afzal M, Hadi FA, Pande H. Megaloblastic Anemia in Bardet-Biedl Syndrome: A Rare Case Report. Clin Med Insights Case Rep. 2023 Aug 14;16:11795476231193896. doi: 10.1177/11795476231193896[↩]
- Bardet-Biedl Syndrome. http://www.eyerounds.org/atlas/pages/bardet-biedl.htm#gsc.tab=0[↩]
- Loktev AV, Zhang Q, Beck JS, Searby CC, Scheetz TE, Bazan JF, Slusarski DC, Sheffield VC, Jackson PK, Nachury MV. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev Cell. 2008;15:854–65. doi: 10.1016/j.devcel.2008.11.001[↩]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–13. doi: 10.1016/j.cell.2007.03.053[↩]
- Seo S, Zhang Q, Bugge K, Breslow DK, Searby CC, Nachury MV, Sheffield VC. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS Genet. 2011;7:e1002358. doi: 10.1371/journal.pgen.1002358[↩]
- Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell. 2010;141:1208–19. doi: 10.1016/j.cell.2010.05.015[↩]
- BARDET-BIEDL SYNDROME 1; BBS1. https://omim.org/entry/209900[↩]
- Mitchison HM, Valente EM. Motile and non-motile cilia in human pathology: from function to phenotypes. J Pathol (2017) 241(2):294–309. 10.1002/path.4843[↩]
- Forsythe E, Sparks K, Best S, Borrows S, Hoskins B, Sabir A, et al. Risk factors for severe renal disease in Bardet-Biedl syndrome. J Am Soc Nephrol (2017) 28(3):963–70. 10.1681/ASN.2015091029[↩][↩][↩][↩][↩][↩][↩][↩]
- Daniels AB, Sandberg MA, Chen J, Weigel-DiFranco C, Fielding Hejtmancic J, Berson EL. Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch Ophthalmol. 2012 Jul;130(7):901-7. doi: 10.1001/archophthalmol.2012.89[↩]
- Feuillan PP, Ng D, Han JC, Sapp JC, Wetsch K, Spaulding E, et al. Patients with Bardet-Biedl syndrome have hyperleptinemia suggestive of leptin resistance. J Clin Endocrinol Metab (2011) 96(3):E528–35. 10.1210/jc.2010-2290[↩]
- Imhoff O, Marion V, Stoetzel C, Durand M, Holder M, Sigaudy S, et al. Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin J Am Soc Nephrol (2011) 6(1):22–9. 10.2215/CJN.03320410[↩][↩][↩]
- Forsythe E, Sparks K, Hoskins BE, Bagkeris E, McGowan BM, Carroll PV, et al. Genetic predictors of cardiovascular morbidity in Bardet-Biedl syndrome. Clin Genet (2015) 87(4):343–9. 10.1111/cge.12373[↩][↩][↩][↩]
- Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet (2013) 21(1):8–13. 10.1038/ejhg.2012.115[↩]
- Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259. doi: 10.1126/science.1063525[↩]
- Beales PL, Badano JL, Ross AJ, Ansley SJ, Hoskins BE, et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am J Hum Genet. 2003;72:1187–1199. doi: 10.1086/375178[↩]
- Estrada-Cuzcano A, Koenekoop RK, Senechal A, De Baere EB, de Ravel T, et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch Ophthalmol. 2012;130:1425–1432. doi: 10.1001/archophthalmol.2012.2434[↩]
- Katsanis N, Eichers ER, Ansley SJ, Lewis RA, Kayserili H, Hoskins BE, et al. BBS4 is a minor contributor to Bardet-Biedl syndrome and may also participate in triallelic inheritance. Am J Hum Genet (2002) 71(1):22–9. 10.1086/341031[↩]
- Hichri H, Stoetzel C, Laurier V, Caron S, Sigaudy S, Sarda P, et al. Testing for triallelism: analysis of six BBS genes in a Bardet-Biedl syndrome family cohort. Eur J Hum Genet (2005) 13(5):607–16. 10.1038/sj.ejhg.5201372[↩]
- Abu-Safieh L, Al-Anazi S, Al-Abdi L, Hashem M, Alkuraya H, Alamr M, et al. In search of triallelism in Bardet-Biedl syndrome. Eur J Hum Genet (2012) 20(4):420–7. 10.1038/ejhg.2011.205[↩]
- Topol EJ. Individualized medicine from prewomb to tomb. Cell (2014) 157(1):241–53. 10.1016/j.cell.2014.02.012[↩]
- Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol (2017) 18(1):83. 10.1186/s13059-017-1215-1[↩]
- Niederlova V, Modrak M, Tsyklauri O, Huranova M, Stepanek O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum Mutat. 2019 Nov;40(11):2068-2087. doi: 10.1002/humu.23862[↩][↩]
- Shamseldin HE, Shaheen R, Ewida N, et al. The morbid genome of ciliopathies: an update. Genet Med. 2020 Jun;22(6):1051-1060. doi: 10.1038/s41436-020-0761-1. Epub 2020 Feb 14. Erratum in: Genet Med. 2022 Apr;24(4):966. doi: 10.1016/j.gim.2022.01.019[↩][↩][↩][↩][↩]
- Scheidecker S, Etard C, Pierce NW, Geoffroy V, Schaefer E, Muller J, Chennen K, Flori E, Pelletier V, Poch O, Marion V, Stoetzel C, Strähle U, Nachury MV, Dollfus H. Exome sequencing of Bardet-Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18). J Med Genet. 2014 Feb;51(2):132-6. doi: 10.1136/jmedgenet-2013-101785[↩]
- Fan Y, Esmail MA, Ansley SJ, Blacque OE, Boroevich K, Ross AJ, Moore SJ, Badano JL, May-Simera H, Compton DS, Green JS, Lewis RA, van Haelst MM, Parfrey PS, Baillie DL, Beales PL, Katsanis N, Davidson WS, Leroux MR. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat Genet. 2004 Sep;36(9):989-93. doi: 10.1038/ng1414[↩]
- Gouronc A, Zilliox V, Jacquemont ML, Darcel F, Leuvrey AS, Nourisson E, Antin M, Alessandri JL, Doray B, Gueguen P, Payet F, Randrianaivo H, Stoetzel C, Scheidecker S, Flodrops H, Dollfus H, Muller J. High prevalence of Bardet-Biedl syndrome in La Réunion Island is due to a founder variant in ARL6/BBS3. Clin Genet. 2020 Aug;98(2):166-171. doi: 10.1111/cge.13768[↩]
- Fieggen K, Milligan C, Henderson B, Esterhuizen AI. Bardet Biedl syndrome in South Africa: A single founder mutation. S Afr Med J. 2016 May 25;106(6 Suppl 1):S72-4. doi: 10.7196/SAMJ.2016.v106i6.11000[↩]
- Aldahmesh MA, Li Y, Alhashem A, Anazi S, Alkuraya H, Hashem M, Awaji AA, Sogaty S, Alkharashi A, Alzahrani S, Al Hazzaa SA, Xiong Y, Kong S, Sun Z, Alkuraya FS. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum Mol Genet. 2014 Jun 15;23(12):3307-15. doi: 10.1093/hmg/ddu044[↩]
- Schaefer E, Delvallée C, Mary L, Stoetzel C, Geoffroy V, Marks-Delesalle C, Holder-Espinasse M, Ghoumid J, Dollfus H, Muller J. Identification and Characterization of Known Biallelic Mutations in the IFT27 (BBS19) Gene in a Novel Family With Bardet-Biedl Syndrome. Front Genet. 2019 Jan 30;10:21. doi: 10.3389/fgene.2019.00021[↩]
- Lindstrand A, Frangakis S, Carvalho CM, Richardson EB, McFadden KA, Willer JR, Pehlivan D, Liu P, Pediaditakis IL, Sabo A, Lewis RA, Banin E, Lupski JR, Davis EE, Katsanis N. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am J Hum Genet. 2016 Aug 4;99(2):318-36. doi: 10.1016/j.ajhg.2015.04.023[↩]
- Kleinendorst L, Alsters SIM, Abawi O, Waisfisz Q, Boon EMJ, van den Akker ELT, van Haelst MM. Second case of Bardet-Biedl syndrome caused by biallelic variants in IFT74. Eur J Hum Genet. 2020 Jul;28(7):943-946. doi: 10.1038/s41431-020-0594-z[↩]
- Marion V, Stutzmann F, Gérard M, De Melo C, Schaefer E, Claussmann A, Hellé S, Delague V, Souied E, Barrey C, Verloes A, Stoetzel C, Dollfus H. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet–Biedl syndrome with situs inversus and insertional polydactyly. J Med Genet. 2012 May;49(5):317-21. doi: 10.1136/jmedgenet-2012-100737[↩]
- Schaefer E, Lauer J, Durand M, Pelletier V, Obringer C, Claussmann A, Braun JJ, Redin C, Mathis C, Muller J, Schmidt-Mutter C, Flori E, Marion V, Stoetzel C, Dollfus H. Mesoaxial polydactyly is a major feature in Bardet-Biedl syndrome patients with LZTFL1 (BBS17) mutations. Clin Genet. 2014 May;85(5):476-81. doi: 10.1111/cge.12198[↩]
- Xing DJ, Zhang HX, Huang N, Wu KC, Huang XF, Huang F, Tong Y, Pang CP, Qu J, Jin ZB. Comprehensive molecular diagnosis of Bardet-Biedl syndrome by high-throughput targeted exome sequencing. PLoS One. 2014 Mar 7;9(3):e90599. doi: 10.1371/journal.pone.0090599[↩]
- Wormser O, Gradstein L, Yogev Y, et al. SCAPER localizes to primary cilia and its mutation affects cilia length, causing Bardet-Biedl syndrome. Eur J Hum Genet. 2019 Jun;27(6):928-940. doi: 10.1038/s41431-019-0347-z[↩]
- Morisada N, Hamada R, Miura K, Ye MJ, Nozu K, Hattori M, Iijima K. Bardet-Biedl syndrome in two unrelated patients with identical compound heterozygous SCLT1 mutations. CEN Case Rep. 2020 Aug;9(3):260-265. doi: 10.1007/s13730-020-00472-y[↩]
- Chiang AP, Beck JS, Yen HJ, Tayeh MK, Scheetz TE, Swiderski RE, Nishimura DY, Braun TA, Kim KY, Huang J, Elbedour K, Carmi R, Slusarski DC, Casavant TL, Stone EM, Sheffield VC. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc Natl Acad Sci U S A. 2006 Apr 18;103(16):6287-92. doi: 10.1073/pnas.0600158103[↩]
- Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, Lewis RA, Johnson CA, Attie-Bittach T, Katsanis N, Wallingford JB. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science. 2010 Sep 10;329(5997):1337-40. doi: 10.1126/science.1191184[↩]
- Putoux A, Mougou-Zerelli S, Thomas S, Elkhartoufi N, Audollent S, Le MM, et al. BBS10 mutations are common in ‘Meckel’-type cystic kidneys. J Med Genet (2010) 47(12):848–52. 10.1136/jmg.2010.079392[↩]
- Harnett JD, Green JS, Cramer BC, Johnson G, Chafe L, McManamon P, et al. The spectrum of renal disease in Laurence-Moon-Biedl syndrome. N Engl J Med (1988) 319(10):615–8. 10.1056/NEJM198809083191005[↩][↩]
- Marion V, Schlicht D, Mockel A, Caillard S, Imhoff O, Stoetzel C, et al. Bardet-Biedl syndrome highlights the major role of the primary cilium in efficient water reabsorption. Kidney Int (2011) 79(9):1013–25. 10.1038/ki.2010.538[↩]
- Putoux A, Attie-Bitach T, Martinovic J, Gubler MC. Phenotypic variability of Bardet-Biedl syndrome: focusing on the kidney. Pediatr Nephrol (2012) 27(1):7–15. 10.1007/s00467-010-1751-3[↩]
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999 Jun;36(6):437-46. https://pmc.ncbi.nlm.nih.gov/articles/instance/1734378/pdf/v036p00437.pdf[↩][↩]
- Samuel GN, Farsides B. Public trust and ‘ethics review’ as a commodity: the case of Genomics England Limited and the UK’s 100,000 genomes project. Med Health Care Philos. 2018 Jun;21(2):159-168. doi: 10.1007/s11019-017-9810-1[↩]
- Wright CF, Fitzgerald TW, Jones WD, et al. DDD study. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet. 2015 Apr 4;385(9975):1305-14. doi: 10.1016/S0140-6736(14)61705-0[↩]
- Vears DF, Senecal K, Borry P. Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur J Med Genet (2017) 60(10):553–8. 10.1016/j.ejmg.2017.07.016[↩]
- Forsythe, E., Beales, P. Bardet–Biedl syndrome. Eur J Hum Genet 21, 8–13 (2013). https://doi.org/10.1038/ejhg.2012.115[↩]
- Kumar A, Husain A Sr, Saleem A, Khawaja UA, Virani S. Laurence-Moon-Bardet-Biedl Syndrome: A Rare Case With a Literature Review. Cureus. 2020 Nov 5;12(11):e11355. doi: 10.7759/cureus.11355[↩]
- Pandya M, Daigavane S. A Rare Presentation of Laurence-Moon-Bardet-Biedl Syndrome: Atypical Retinitis Punctata Albescens and Non-alcoholic Fatty Liver Disease. Cureus. 2024 Feb 12;16(2):e54064. doi: 10.7759/cureus.54064[↩]
- Khan OA, Majeed R, Saad M, Khan A, Ghassan A. Rarity of Laurence Moon Bardet Biedl Syndrome and its Poor Management in the Pakistani Population. Cureus. 2019 Feb 21;11(2):e4114. doi: 10.7759/cureus.4114[↩][↩]
- Al Fareh N, Abbas M, Alosaimi FD. Psychosis in Laurence-Moon Syndrome: A Case Report. Cureus. 2024 Oct 21;16(10):e72003. doi: 10.7759/cureus.72003[↩]
- Hufnagel RB, Arno G, Hein ND, Hersheson J, Prasad M, Anderson Y, Krueger LA, Gregory LC, Stoetzel C, Jaworek TJ, Hull S, Li A, Plagnol V, Willen CM, Morgan TM, Prows CA, Hegde RS, Riazuddin S, Grabowski GA, Richardson RJ, Dieterich K, Huang T, Revesz T, Martinez-Barbera JP, Sisk RA, Jefferies C, Houlden H, Dattani MT, Fink JK, Dollfus H, Moore AT, Ahmed ZM. Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes. J Med Genet. 2015 Feb;52(2):85-94. doi: 10.1136/jmedgenet-2014-102856[↩][↩]
- The endocrine and metabolic characteristics of a large Bardet-Biedl syndrome clinic population. Mujahid S, Hunt KF, Cheah YS, et al. J Clin Endocrinol Metab. 2018;103:1834–1841. doi: 10.1210/jc.2017-01459[↩]
- Laurence-Moon Syndrome. https://rarediseases.org/rare-diseases/laurence-moon-syndrome[↩][↩][↩][↩][↩][↩]
- Indraneel KS, VRajalakshmi, Dayanandan Y, Reddy NM. A Case of Laurence Moon Bardet Biedl Syndrome. J Assoc Physicians India. 2023 Jan;71(1):1.[↩]
- PNPLA6 gene. https://medlineplus.gov/genetics/gene/pnpla6[↩][↩][↩]
- Sahu JK, Jain V. Laurence-Moon-Bardet-Biedl syndrome. JNMA J Nepal Med Assoc. 2008 Oct-Dec;47(172):235-7.[↩]
- Khan BA, Shahid A, Bin Nazir M, Khan KS, Punshi A. Laurence-Moon-Bardet-Biedl Syndrome: A Case Report. Cureus. 2019 Sep 10;11(9):e5618. doi: 10.7759/cureus.5618[↩][↩][↩][↩][↩]
- Synofzik M, Hufnagel RB, Züchner S. PNPLA6 Disorders. 2014 Oct 9 [Updated 2021 Jun 10]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK247161[↩]
- Obesity control and low protein diet preserve or even improve renal functions in Bardet-Biedl syndrome: a report of two cases. Dervisoglu E, Isgoren S, Kasgari D, Demir H, Yilmaz A. Med Sci Monit. 2011;17:12–14. doi: 10.12659/MSM.881320[↩]
- Natural course of visual functions in the Bardet-Biedl syndrome. Fulton AB, Hansen RM, Glynn RJ. Arch Ophthalmol. 1993;111:1500–1506. doi: 10.1001/archopht.1993.01090110066026[↩]
- Bardet- Biedl syndrome: a rare case report from North India. Sumir K, Bharat BM, Jyotistema M. Indian J Dermatol Venereol Leprol. 2012;78:228. doi: 10.4103/0378-6323.93656[↩]
- Alstrom syndrome. https://medlineplus.gov/ency/article/001665.htm[↩][↩]
- Alstrom syndrome. https://medlineplus.gov/genetics/condition/alstrom-syndrome[↩][↩]
- Alstrom syndrome. https://rarediseases.org/mondo-disease/alstrom-syndrome[↩][↩]
- Meckel syndrome. https://medlineplus.gov/genetics/condition/meckel-syndrome[↩][↩][↩][↩][↩][↩][↩]
- Meckel Syndrome. https://rarediseases.org/rare-diseases/meckel-syndrome[↩][↩][↩]
- McKusick-Kaufman syndrome. https://medlineplus.gov/genetics/condition/mckusick-kaufman-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Slavotinek AM. McKusick-Kaufman Syndrome. 2002 Sep 10 [Updated 2020 Dec 3]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1502[↩][↩][↩][↩]
- Yewalkar S.P., Yadav V.K., Khadse G.J. The McKusick-Kaufman hydrometrocolpos-polydactyly syndrome: a rare case report. Indian J. Radiol. Imag. 2013;23(2):183. doi: 10.4103/0971-3026.116573[↩][↩][↩]
- Ullah I, Rauf S, Ali S, Khan KS, Zahid T, Malik J, Afridi RU, Asghar MS. A case of McKusick-Kaufman syndrome with perinatal diagnosis: Case report and literature review. Ann Med Surg (Lond). 2022 Jun 6;79:103926. doi: 10.1016/j.amsu.2022.103926[↩][↩][↩]
- Traisrisilp K., Nunthapiwat S., Luewan S., Tongsong T. Fetal hydrometrocolpos with pre‐axial mirror polydactyly as a new variant of McKusick‐Kaufman syndrome. J. Clin. Ultrasound. 2021;49(1):62–65. doi: 10.1002/jcu.22882[↩]
- Thorat JV, Tambolkar S. Persistent Urogenital Sinus Leading to Hydrometrocolpos in a Female Child With Features of McKusick-Kaufman Syndrome. Cureus. 2024 Jun 8;16(6):e61957. doi: 10.7759/cureus.61957[↩]
- Halim A., Afzal T., Fatima S., Riaz S. A newborn with rare McKusick syndrome. J. Coll Physicians Surg. Pak. 2018;28(6):S140–S142. doi: 10.29271/jcpsp.2018.06.S140[↩]
- Rosenberg H.K., Chaudhry H. In: Diagnostic Ultrasound. fourth ed. Rumack C.M., editor. Elsevier Mosby Publishers; Philadelphia: 2011. Pediatric pelvic sonography; pp. 1936–1938.[↩][↩]
- Kanso, Khadija & Abou Merhi, Bassem & Zeidan, Marwan & Ibrahim, Soukaina & Ghandour, Fatima & Iskandarani, Fadi & el Houwayek, Eliane & Yassine, Fatima & Chokr, Imad. (2018). A Rare Case Report of Hydrometrocolpos in a Female Newborn. International Journal of Current Research and Review. Vol 10. 22-24. 10.7324/IJCRR.2018.1025[↩]
- Nagaraj B.R., Basavalingu D., Paramesh V.M., Nagendra P.D.K. Radiological diagnosis of neonatal hydrometrocolpos-a case report. J. Clin. Diagn. Res.: J. Clin. Diagn. Res. 2016;10(3):TD18. doi: 10.7860/JCDR/2016/18537.7510[↩][↩]
- Ayrim A.A., Gozdemir E., Turhan N., et al. Acute urinary retention associated with an imperforate hymen and haematocolpos. Gynecol. Obstet. Reprod. Med. 2016;15:105–107.[↩]
- Adam A., Hellig J., Mahomed N., Lambie L. Recurrent urinary tract infections in a female child with polydactyly and a pelvic mass: consider the McKusick-Kaufman syndrome. Urology. 2017;103:224–226. doi: 10.1016/j.urology.2017.01.024[↩]
- Biemond syndrome type 2. https://rarediseases.org/mondo-disease/biemond-syndrome-type-2[↩]
- Mary L, Chennen K, Stoetzel C, et al. Bardet-Biedl syndrome: Antenatal presentation of forty-five fetuses with biallelic pathogenic variants in known Bardet-Biedl syndrome genes. Clin Genet. 2019 Mar;95(3):384-397. doi: 10.1111/cge.13500[↩]
- Baker K, Northam GB, Chong WK, Banks T, Beales P, Baldeweg T. Neocortical and hippocampal volume loss in a human ciliopathy: A quantitative MRI study in Bardet-Biedl syndrome. Am J Med Genet A. 2011 Jan;155A(1):1-8. doi: 10.1002/ajmg.a.33773[↩]
- Keppler-Noreuil KM, Blumhorst C, Sapp JC, Brinckman D, Johnston J, Nopoulos PC, Biesecker LG. Brain tissue- and region-specific abnormalities on volumetric MRI scans in 21 patients with Bardet-Biedl syndrome (BBS). BMC Med Genet. 2011 Jul 27;12:101. doi: 10.1186/1471-2350-12-101[↩]
- O’Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J. The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kidney Dis. 1996 Jun;27(6):776-83. doi: 10.1016/s0272-6386(96)90513-2[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Bardet-Biedl syndrome with end-stage kidney disease in a four-year-old Romanian boy: a case report. Mihai CM, Marshall JD, Stoicescu RM. J Med Case Rep. 2011;5:378. doi: 10.1186/1752-1947-5-378[↩]
- Kaleem S, Srirangadhamu Gopu S, Ishfaq L, Afroze S, Parvez M, Mulaka GSR, Venugopal V. Laurence-Moon-Bardet Biedl Syndrome With Cholelithiasis. Cureus. 2023 Oct 19;15(10):e47316. doi: 10.7759/cureus.47316[↩]





{kind=link}