Contents
- Kidney Stone
- What is the urinary tract and how does it work?
- Types of kidney stones
- Kidney stone causes
- Risk factors for developing kidney stones
- Kidney Stone pathophysiology
- Kidney stone prevention
- Kidney stone signs and symptoms
- Kidney stone complications
- Kidney stone diagnosis
- Kidney stone treatment
- Kidney stones prognosis
- What is Renal Tubular Acidosis
- What is Fanconi’s syndrome
- What is Cystinuria
- What is Urinary Tract Infections
- Bacterial Urinary Tract Infections
- Bacterial Urinary Tract Infections pathophysiology
- Risk factors for Bacterial Urinary Tract Infections
- Bacterial Urinary Tract Infections causes
- Bacterial Urinary Tract Infections prevention
- Classification of Bacterial Urinary Tract Infections
- Bacterial Urinary Tract Infections signs and symptoms
- Bacterial Urinary Tract Infections diagnosis
- Bacterial Urinary Tract Infections treatment
- What are Cystic Kidney diseases
- What is Polycystic Kidney Disease?
- What is acquired cystic kidney disease?
- Acquired cystic kidney disease causes
- Acquired cystic kidney disease signs and symptoms
- Acquired cystic kidney disease complications
- Acquired cystic kidney disease diagnosis
- Acquired cystic kidney disease treatment
- Eating, Diet, and Nutrition for Acquired Cystic Kidney Disease
- Nutrition for Advanced Chronic Kidney Disease in Adults
- What is medical nutrition therapy (MNT)?
- Why is knowing about calories important for someone with advanced chronic kidney disease?
- Why is knowing about protein important for someone with advanced chronic kidney disease?
- What is the right meat portion size?
- Why is knowing about fat important for someone with advanced chronic kidney disease?
- Why is knowing about sodium important for someone with advanced chronic kidney disease?
- Why is knowing about potassium important for someone with advanced chronic kidney disease?
- Why is knowing about phosphorus important for someone with advanced chronic kidney disease?
- Why is regulating fluid intake important for someone with advanced chronic kidney disease?
- Is there kidney stone home remedy?
Kidney Stone
Kidney stones are hard, pebble-like pieces of material that form in one or both of your kidneys when high levels of certain minerals are in your urine 1. Kidney stones rarely cause permanent damage if treated by a health care professional 1. The scientific name for a kidney stone is renal calculus or nephrolith. You may hear health care professionals call this condition nephrolithiasis, urolithiasis, or urinary stones. Your urine has various wastes dissolved in it. When there is too much waste your urine combined with too little liquid, crystals begin to form. The crystals attract other elements and join together to form a solid that will get larger unless it is passed out of your body with the urine. Usually, these chemicals are eliminated in the urine by the body’s master chemist: the kidney. In most people, having enough liquid washes them out or other chemicals in urine stop a stone from forming. The stone-forming chemicals are calcium, oxalate, urate, cystine, xanthine, and phosphate.
There are five types of kidney stones: calcium oxalate, calcium phosphate (brushite), uric acid, struvite (magnesium ammonium phosphate), and cystine. The majority (65 to 75%) of kidney stones are composed of either pure or mostly of calcium salts, including those of calcium oxalate, mixed calcium oxalate with uric acid, and calcium phosphate (brushite) 2. Uric acid, cystine, and magnesium ammonium phosphate (struvite) compose the remainder of kidney stones 2.
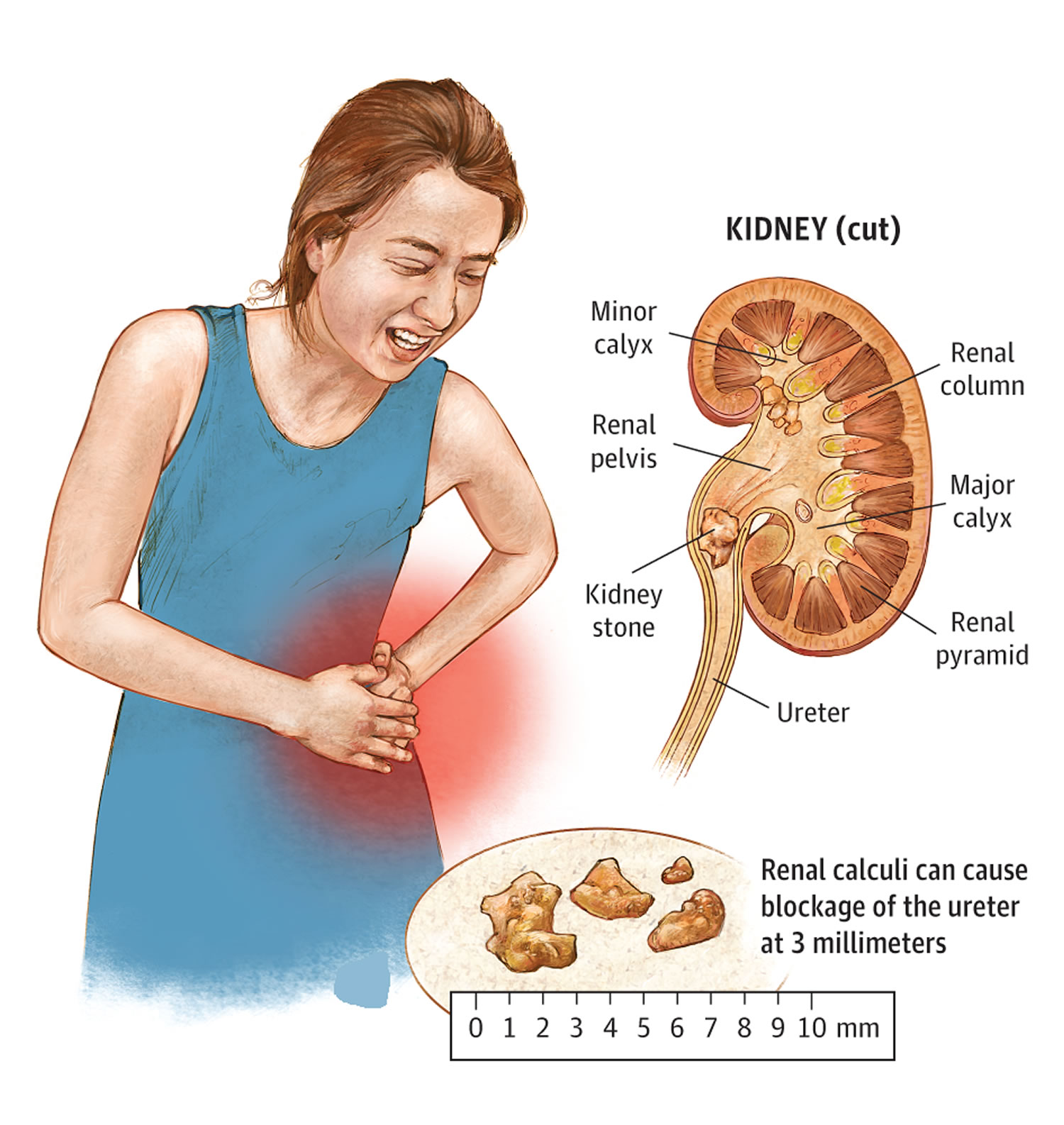
In the United States the lifetime risk for kidney stone formation is 12% in men and 5% in women. Recurrence rates of new kidney stone formation are high. If untreated, kidney stones will recur at the rate of 50% in 5 to 10 years 2. After it is formed, the stone may stay in your kidney or travel down the urinary tract into the ureter. Sometimes, tiny stones move out of the body in the urine without causing too much pain. But stones that don’t move may cause a back-up of urine in the kidney, ureter, the bladder, or the urethra. This can present with renal colic symptoms (kidney stone pain), and may cause urinary tract obstruction and/or infection. Common symptoms of kidney stones include severe pain in lower back, blood in your urine, nausea, vomiting, fever and chills, or urine that smells bad or looks cloudy.
Each year, more than half a million people go to emergency rooms for kidney stone problems. It is estimated that one in ten people will have a kidney stone at some time in their lives. In fact, acute passage of a kidney stone is the 9th most common cause for visits to an emergency room 3. An episode of renal colic (kidney stone pain) has a sudden onset, with fluctuation and intensification over 15 to 45 minutes. Kidney stones may obstruct the urinary tract and impair kidney function. There is increased risk of infection with chronic obstruction. Bleeding may be chronic and accompany obstruction. The size, number, and metabolic composition of new stones strongly influence the natural history and complication rates.
Kidney stones affect up to 5% of the population, with a lifetime risk of passing a kidney stone of about 8-10% 4, 5. About 1/1000 adults in the US is hospitalized annually because of kidney stones, which are also found in about 1% of all autopsies 6. Up to 12% of men and 8% of women will develop a urinary calculus by age 70 5, 6.
Increased incidence of kidney stones in the industrialized countries is associated with improved standards of living and is strongly associated with race or ethnicity and region of residence 7. The prevalence of kidney stones in the United States increased from 3.8% in the late 1970s to 8.8% in the late 2000s 8. The prevalence of kidney stones was 10% during 2013–2014 8. The risk of kidney stones is about 11% in men and 9% in women. Other diseases such as high blood pressure, diabetes, and obesity may increase your risk for kidney stones. A seasonal variation is also seen, with high urinary calcium oxalate saturation in men during summer and in women during early winter 9. Men are more likely to develop kidney stones than women and kidney stones form twice as often in men as women 10. The peak age in men is 30 years; women have a bimodal age distribution, with peaks at 35 and 55 years 10. Once a kidney stone forms, the probability that a second stone will form within five to seven years is approximately 50% 4. In postmenopausal women, the occurrence of kidney stones is associated with a history of hypertension and a low dietary intake of magnesium and calcium 11.
Kidney stones vary in size and shape. Kidney stones vary from small as a grain of sand or as large as a pea to kidney stones several centimeters in diameter. Rarely, some kidney stones are as big as golf balls. A large kidney stone, called a staghorn calculus, can fill an entire renal calyceal system.
Kidney stones may be smooth or jagged and are usually yellow or brown. Treatment for kidney stones usually depends on their size, location, and what they are made of 12. Most ureteral stones under 5 mm pass spontaneously 10. About 90% of ureteric stones smaller than 5 mm pass spontaneously, compared with about 50% of stones between 5 mm and 10 mm, so conservative management is preferred for ureteric stones 13. Depending on the size of the stone, the average time to pass the stone ranges between one week and three weeks, and the passage of the stone is most accurately assessed by a plain film (kidney-ureter-bladder view) every one to two weeks to monitor progression. An observation period of three to four weeks is reasonable unless urgent intervention is indicated for intractable symptoms, infection, or obstruction 10.
A small kidney stone may pass through your urinary tract on its own, causing little or no pain. A larger kidney stone may get stuck along the way. A kidney stone that gets stuck can block your flow of urine, causing severe pain or bleeding.
If you have symptoms of kidney stones, including severe pain or bleeding, seek care right away. A doctor, such as a urologist, can treat any pain and prevent further problems, such as a urinary tract infection (UTI).
Depending on your situation, you may need nothing more than to take pain medication and drink lots of water to pass a kidney stone. Drinking as much as 2 to 3 quarts (1.8 to 3.6 liters) of water a day will keep your urine dilute and may prevent stones from forming. Unless your doctor tells you otherwise, drink enough fluid — ideally mostly water — to produce clear or nearly clear urine. To relieve mild pain, your doctor may recommend pain relievers such as ibuprofen (Advil, Motrin IB, others) or naproxen sodium (Aleve). Your doctor may also give you a medication to help pass your kidney stone. This type of medication, known as an alpha blocker, relaxes the muscles in your ureter, helping you pass the kidney stone more quickly and with less pain. Examples of alpha blockers include tamsulosin (Flomax) and the drug combination dutasteride and tamsulosin (Jalyn).
In other instances — for example, if the stones become lodged in the urinary tract, are associated with a urinary infection or cause complications — surgery may be needed. Larger stones may need to be broken up or removed with surgery. Your type of surgery will depend on the size and location of your kidney stones.
The main types of surgery for removing kidney stones are:
- Shockwave lithotripsy (SWL) or using sound waves to break up stones.
- Extracorporeal shock wave lithotripsy (ESWL) uses sound waves to create strong vibrations (shock waves) that break the stones into tiny pieces that can be passed in your urine. The procedure lasts about 45 to 60 minutes and can cause moderate pain, so you may be under sedation or light anesthesia to make you comfortable.
- Extracorporeal shock wave lithotripsy (ESWL) can cause blood in your urine, bruising on the back or abdomen, bleeding around your kidney and other adjacent organs, and discomfort as the stone fragments pass through the urinary tract.
- Using a scope to remove stones (ureteroscopy).
- Ureteroscopy is carried out under general or local anesthesia. Ureteroscopy involves passing a long, thin telescope called a ureteroscope through the tube urine passes through on its way out of your body called the urethra and into your bladder. It’s then passed up into your ureter, which connects your bladder to your kidney. Once the stone is located, special tools can snare the stone or break it into pieces that will pass in your urine. Your surgeon may either try to gently remove the stone using another instrument, or they may use laser energy to break it up into small pieces so it can be passed naturally in your urine.
- Percutaneous nephrolithotomy (PCNL).
- Percutaneous nephrolithotomy (PCNL) is always carried out under general anaesthetic, where you’re asleep. Percutaneous nephrolithotomy (PCNL) involves using a thin telescopic instrument called a nephroscope. A small cut (incision) is made in your back and the nephroscope is passed through it and into your kidney. The stone is either pulled out or broken into smaller pieces using a laser or pneumatic energy. Your doctor may then place a small tube (stent) in the ureter to relieve swelling and promote healing. Your doctor may recommend percutaneous nephrolithotomy (PCNL) if extracorporeal shock wave lithotripsy (ESWL) is unsuccessful.
- Parathyroid gland surgery.
- Some calcium phosphate stones are caused by overactive parathyroid glands (hyperparathyroidism), which are located on the four corners of your thyroid gland, just below your Adam’s apple. When these glands produce too much parathyroid hormone (hyperparathyroidism), your calcium levels can become too high and kidney stones may form as a result. Hyperparathyroidism sometimes occurs when a small, benign tumor forms in one of your parathyroid glands or you develop another condition that leads these glands to produce more parathyroid hormone. Removing the growth from the gland stops the formation of kidney stones. Or your doctor may recommend treatment of the condition that’s causing your parathyroid gland to overproduce the hormone.
Your doctor may also recommend preventive treatment to reduce your risk of recurrent kidney stones if you’re at increased risk of developing them again. Prevention of kidney stones may include a combination of lifestyle changes and medications. Ask your doctor for a referral to a dietitian who can help you develop an eating plan that reduces your risk of kidney stones.
You may reduce your risk of kidney stones if you:
- Drink water throughout the day. For people with a history of kidney stones, doctors usually recommend drinking enough fluids to pass about 2.1 quarts (2 liters) of urine a day. Your doctor may ask that you measure your urine output to make sure that you’re drinking enough water. If you live in a hot, dry climate or you exercise frequently, you may need to drink even more water to produce enough urine. If your urine is light and clear, you’re likely drinking enough water.
- Eat fewer oxalate-rich foods. If you tend to form calcium oxalate stones, your doctor may recommend restricting foods rich in oxalates. These include rhubarb, beets, okra, spinach, Swiss chard, sweet potatoes, nuts, tea, chocolate, black pepper and soy products.
- Choose a diet low in salt and animal protein. Reduce the amount of salt you eat and choose nonanimal protein sources, such as legumes. Consider using a salt substitute, such as Mrs. Dash.
- Continue eating calcium-rich foods, but use caution with calcium supplements. Calcium in food doesn’t have an effect on your risk of kidney stones. Continue eating calcium-rich foods unless your doctor advises otherwise. Ask your doctor before taking calcium supplements, as these have been linked to increased risk of kidney stones. You may reduce the risk by taking supplements with meals. Diets low in calcium can increase kidney stone formation in some people.
Figure 1. Kidney with kidney stones
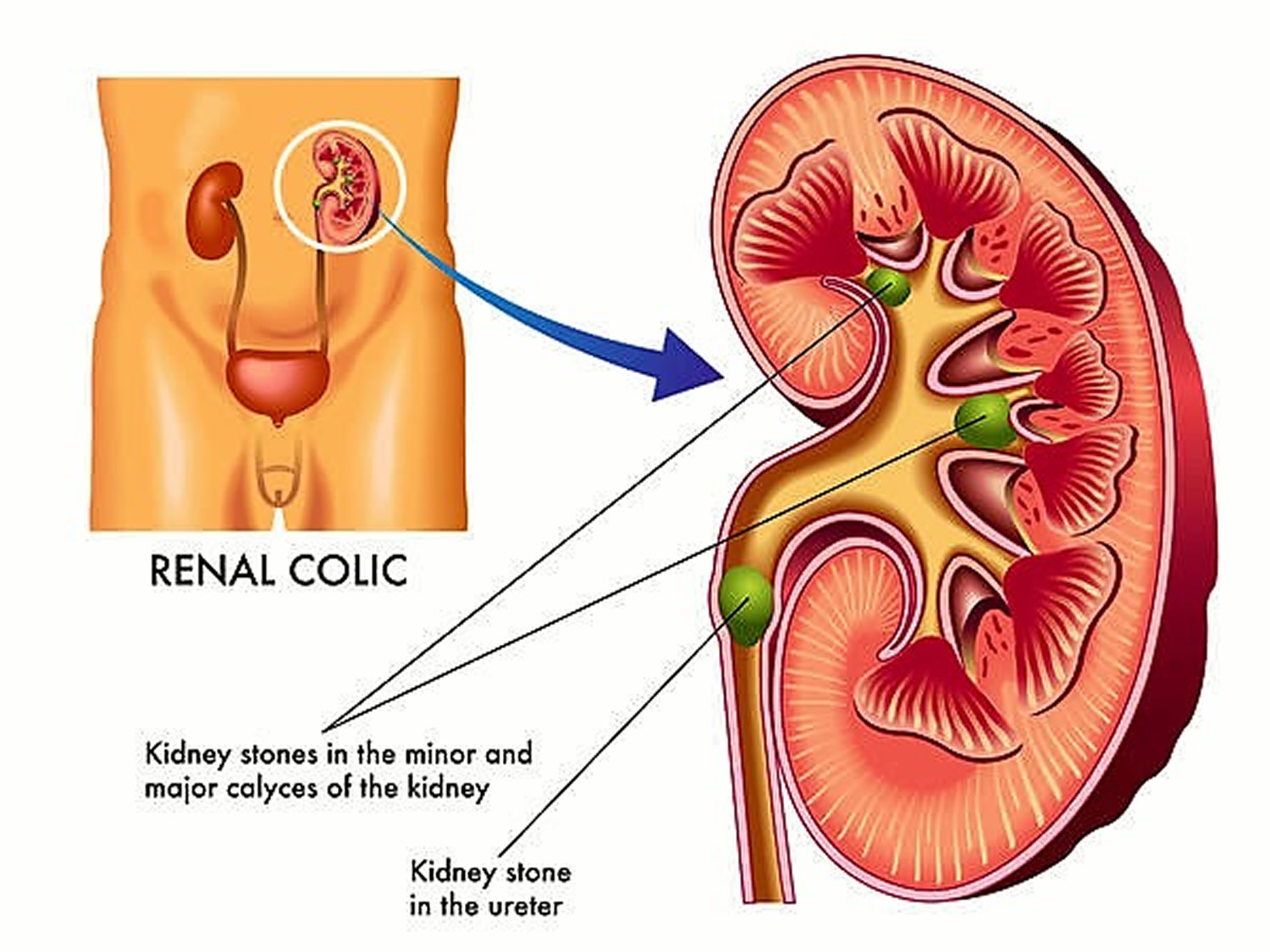
Make an appointment with your doctor if you have any signs and symptoms that worry you.
Seek immediate medical attention if you experience:
- Pain so severe that you can’t sit still or find a comfortable position
- Pain accompanied by nausea and vomiting
- Pain accompanied by fever and chills
- Blood in your urine
- Difficulty passing urine
How common are kidney stones?
Each year, more than half a million people go to emergency rooms for kidney stone problems 8. It is estimated that one in ten people will have a kidney stone at some time in their lives.
The prevalence of kidney stones in the United States increased from 3.8% in the late 1970s to 8.8% in the late 2000s. The prevalence of kidney stones was 10% during 2013–2014. The risk of kidney stones is about 11% in men and 9% in women 14. Other diseases such as high blood pressure, diabetes, and obesity may increase the risk for kidney stones.
Who is more likely to develop kidney stones?
Men are more likely to develop kidney stones than women. The risk of kidney stones is about 11% in men and 9% in women 14. If you have a family history of kidney stones, you are more likely to develop them. You are also more likely to develop kidney stones again if you’ve had them once. You may also be more likely to develop a kidney stone if you don’t drink enough liquids.
People with certain conditions
You are more likely to develop kidney stones if you have certain conditions, including:
- a blockage of the urinary tract
- chronic, or long-lasting, inflammation of the bowel
- cystic kidney diseases , which are disorders that cause fluid-filled sacs to form on the kidneys
- cystinuria
- digestive problems or a history of gastrointestinal tract surgery
- gout , a disorder that causes painful swelling of the joints
- hypercalciuria , a condition that runs in families in which urine contains unusually large amounts of calcium; this is the most common condition found in people who form calcium stones
- hyperoxaluria , a condition in which urine contains unusually large amounts of oxalate
- hyperparathyroidism, a condition in which the parathyroid glands release too much parathyroid hormone (PTH), causing extra calcium in the blood
- hyperuricosuria, a disorder in which too much uric acid is in the urine
- obesity
- repeated, or recurrent urinary tract infections (UTIs)
- renal tubular acidosis, a disease that occurs when the kidneys fail to remove acids into the urine, which causes a person’s blood to remain too acidic
People who take certain medicines
You are more likely to develop kidney stones if you are taking one or more of the following medicines over a long period of time:
- diuretics, often called water pills, which help rid your body of water
- calcium-based antacids
- indinavir, a protease inhibitor used to treat HIV infection
- topiramate, an anti-seizure medication
I think I have a kidney stone. What do I do?
See a doctor as soon as possible. You may be asked to drink extra fluid in an attempt to flush out the kidney stone out in the urine. If you strain your urine and can save a piece of the stone that has passed, bring it to your doctor. Or, the stone may need to be removed with surgery.
Why do doctors examine the contents of the kidney stone?
There are five types of kidney stones. Studying the stone can help understand why you have it and how to reduce the risk of further stones. The most common type of stone contains calcium. Calcium is a normal part of a healthy diet. The kidney usually removes extra calcium that the body doesn’t need. Often people with stones keep too much calcium. This calcium combines with waste products like oxalate to form a stone. The most common combination is called calcium oxalate.
Less common types of stones are: Infection-related stones, containing magnesium and ammonia called struvite stones and stones formed from monosodium urate crystals, called uric acid stones, which might be related to obesity and dietary factors. The rarest type of stone is a cvstine stone that tends to run in families.
Can children get kidney stones?
Yes. Kidney stones are found in children as young as 5 years. In fact, kidney stone is so common in children that some hospitals conduct ‘stone’ clinics for pediatric patients. The increase of kidney stones in children in the United States has been attributed to several factors, mostly related to food choices. The two most important reasons are not drinking enough fluids and eating foods that are high in salt. Kids should eat less salty potato chips and french fries. There are other salty foods: sandwich meats, canned soups, packaged meals, and even some sports drinks. Sodas and other sweetened beverages can also increase the risk of stones if they contain high fructose corn syrup.
Special Considerations
Children
More children are developing kidney stones, which is attributed to the corresponding rise in diabetes, obesity, and hypertension in this population 15, 16. Because increasing age is a risk factor for kidney stones, adolescents are more likely to form stones than younger children. The underlying causes and resulting treatments differ in children and adults. Children with kidney stones are more likely to have anatomic and metabolic abnormalities 16, increased urinary calcium excretion, decreased urinary oxalate and citrate excretion, and much higher urinary calcium oxalate saturations than children with no history of kidney stones 15. Children with cystinuria and other hereditary forms of kidney stones are at increased risk of decline in renal function compared with age-matched controls, although progression to end-stage renal disease is uncommon 15.
Pregnant women
Pregnant women are twice as likely to have calcium phosphate stones compared with age-matched nonpregnant women, and are two to three times more likely to have calcium phosphate stones than oxalate stones 17. The incidence of kidney stones during pregnancy increases in the second and third trimesters. Women have an increased glomerular filtration rate and higher urinary calcium excretion throughout pregnancy, with higher urine pH in the second and third trimesters, which may predispose them to calcium phosphate stones. Ultrasonography is considered the imaging modality of choice in pregnant women. Kidney stones during pregnancy increase the risk of urinary tract infections, and pregnant women with renal colic have nearly double the risk of pre-term delivery compared with women who do not have kidney stones 18.
What is the urinary tract and how does it work?
The urinary tract is the body’s drainage system for removing urine, which is composed of wastes and extra fluid. In order for normal urination to occur, all body parts in the urinary tract need to work together in the correct order 19.
- The urinary tract is the body’s drainage system for removing urine, which is composed of wastes and extra fluid.
- The urinary tract is important because it filters wastes and extra fluid from the bloodstream and removes them from the body.
- In order for normal urination to occur, all body parts in the urinary tract need to work together in the correct order.
- The kidneys are two bean-shaped organs, each about the size of a fist.
- Every day, the kidneys filter about 120 to 150 quarts (114 liters – 142 liters) of blood to produce about 1 to 2 quarts (950 ml – 1.9 liter) of urine.
- Ureters are the thin tubes of muscle—one on each side of the bladder—that carry urine from each of the kidneys to the bladder.
- The bladder, located in the pelvis between the pelvic bones, is a hollow, muscular, balloon-shaped organ that expands as it fills with urine.
- Bladder emptying is known as urination.
- During urination, the bladder empties through the urethra, located at the bottom of the bladder.
- The ureters, bladder, and urethra move urine from the kidneys and store it until releasing it from the body.
- The amount of urine a person produces depends on many factors, such as the amounts of liquid and food a person consumes and the amount of fluid lost through sweat and breathing.
Kidneys. The kidneys are two bean-shaped organs, each about the size of a fist. They are located just below the rib cage, one on each side of the spine. Every day, the kidneys filter about 120 to 150 quarts (114 liters- 142 liters) of blood to produce about 1 to 2 quarts (950 ml – 1.90 liter) of urine. The kidneys work around the clock; a person does not control what they do. The kidneys remove wastes and extra water from the blood and make urine. To keep the body working properly, the kidneys balance the salts and minerals—such as calcium, phosphorus, sodium, and potassium—that circulate in the blood. The kidneys also release hormones that help make red blood cells, regulate blood pressure, and keep bones strong.
Ureters. Ureters are the thin tubes of muscle—one on each side of the bladder—that carry urine from each of the kidneys to the bladder.
Bladder. The bladder, located in the pelvis between the pelvic bones, is a hollow, muscular, balloon-shaped organ that expands as it fills with urine. Although a person does not control kidney function, a person does control when the bladder empties. Bladder emptying is known as urination. The bladder stores urine until the person finds an appropriate time and place to urinate. A normal bladder acts like a reservoir and can hold 1.5 to 2 cups (325 ml – 470 ml) of urine. How often a person needs to urinate depends on how quickly the kidneys produce the urine that fills the bladder. The muscles of the bladder wall remain relaxed while the bladder fills with urine. As the bladder fills to capacity, signals sent to the brain tell a person to find a toilet soon. During urination, the bladder empties through the urethra, located at the bottom of the bladder.
Three sets of muscles work together like a dam, keeping urine in the bladder between trips to the bathroom.
The first set is the muscles of the urethra itself. The area where the urethra joins the bladder is the bladder neck. The bladder neck, composed of the second set of muscles known as the internal sphincter, helps urine stay in the bladder. The third set of muscles is the pelvic floor muscles, also referred to as the external sphincter, which surround and support the urethra.
To urinate, the brain signals the muscular bladder wall to tighten, squeezing urine out of the bladder. At the same time, the brain signals the sphincters to relax. As the sphincters relax, urine exits the bladder through the urethra.
Figure 2. The Urinary Tract
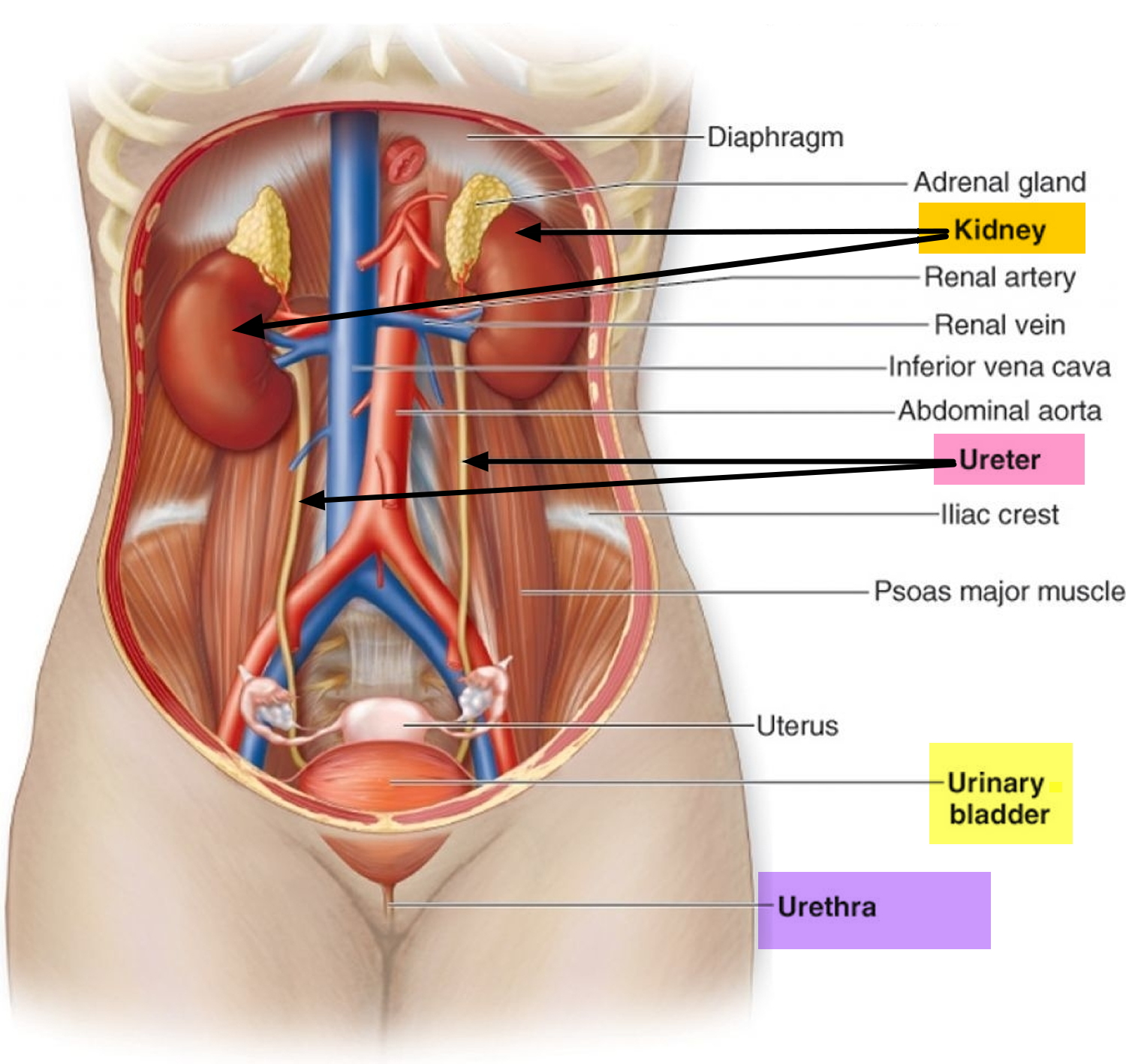
Figure 3. Normal Kidney Anatomy
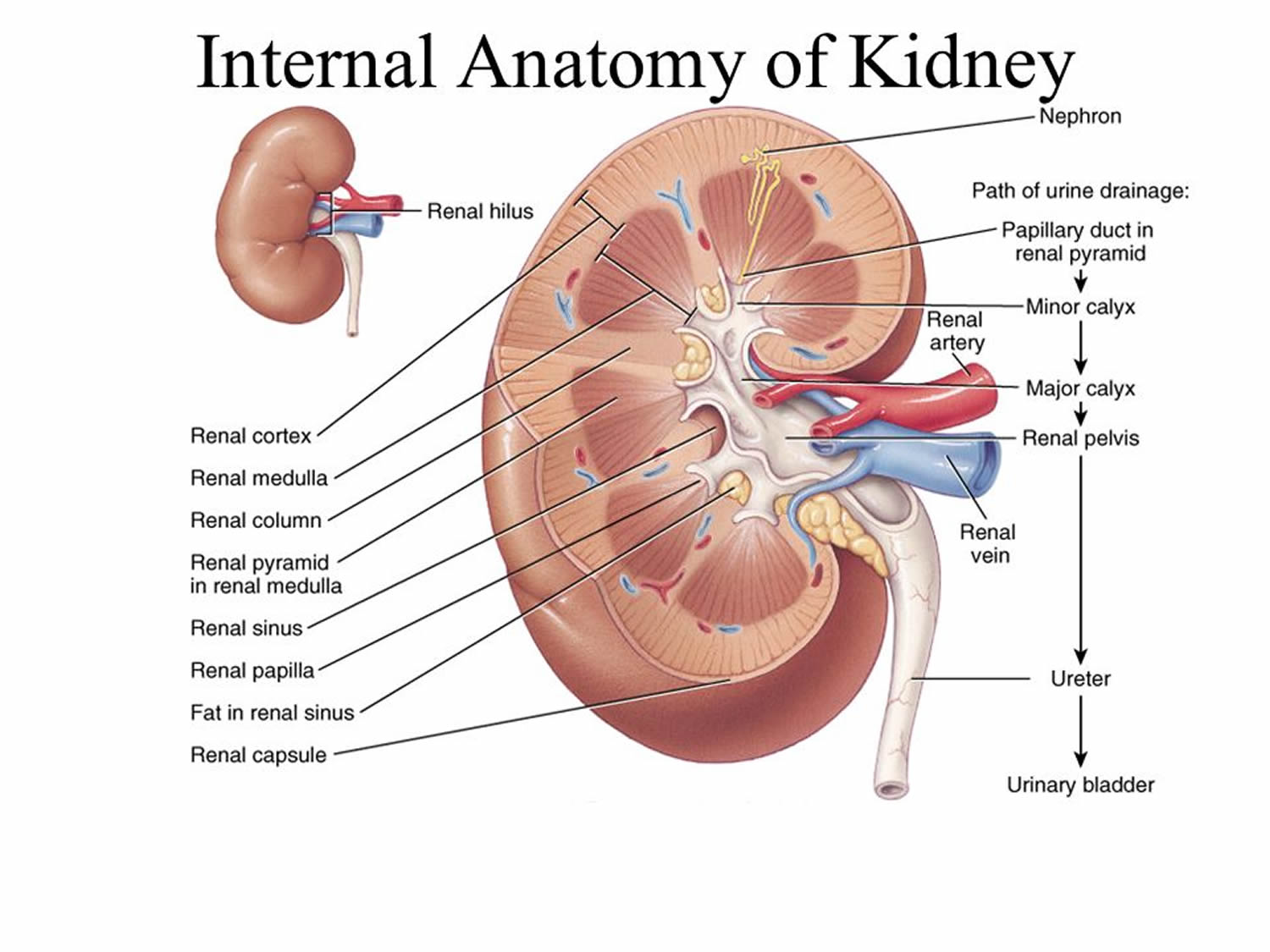
Why is the urinary tract important?
The urinary tract is important because it filters wastes and extra fluid from the bloodstream and removes them from the body. Normal, functioning kidneys
- prevent the buildup of wastes and extra fluid in the body
- keep levels of electrolytes, such as potassium and phosphate, stable
- make hormones that help regulate blood pressure
- make red blood cells
- keep bones strong.
The ureters, bladder, and urethra move urine from the kidneys and store it until releasing it from the body.
What affects the amount of urine a person produces?
The amount of urine a person produces depends on many factors, such as the amounts of liquid and food a person consumes and the amount of fluid lost through sweat and breathing. Certain medications, medical conditions, and types of food can also affect the amount of urine produced. Children produce less urine than adults; the amount produced depends on their age.
Types of kidney stones
Knowing the type of kidney stone you have helps determine its cause, and may give clues on how to reduce your risk of getting more kidney stones in the future. If possible, try to save your kidney stone if you pass one so that you can bring it to your doctor for analysis.
There are 5 main types of kidney stones. About 85% of kidney stones in the US are composed of calcium, mainly calcium oxalate (70%) (see Table 1: Composition of Kidney Stones); 15% are calcium phosphate; 10% are uric acid; 2% are cystine; most of the remainder are magnesium ammonium phosphate (struvite).
Treatment for kidney stones usually depends on their size, location, and what they are made of 12.
The prevalence of kidney stones (nephrolithiasis) is increasing in women and with increasing age. Table 2 includes rates of different types of kidney stones in children and adults 17, 20. Contributing risk factors for kidney stones are obesity, insulin resistance, gastrointestinal pathology, living in warmer climates, and certain dietary patterns and medications 15, 21.
Table 1. Composition of Kidney Stones
| Composition | Percentage of All Calculi | Common Causes |
|---|---|---|
| Calcium oxalate | 70% | Hypercalciuria Hyperparathyroidism Hypocitruria Renal tubular acidosis |
| Calcium phosphate | 15% | Hypercalciuria Hyperparathyroidism Hypocitruria Renal tubular acidosis |
| Cystine | 2% | Cystinuria |
| Magnesium ammonium phosphate (struvite) | 3% | Urinary Tract Infection (UTI) caused by urea-splitting bacteria |
| Uric acid | 10% | Urine pH < 5.5 Occasionally hyperuricosuria |
Table 2. Incidence of Kidney Stones in Children and Adults
| Type | Children (%) | Adult (%) |
|---|---|---|
| Calcium oxalate | 45 to 65 | 56 to 61 |
| Calcium phosphate | 24 to 30 | 8 to 18* |
| Cystine | 5 to 8 | 1 |
| Struvite (magnesium ammonium phosphate) | 7 to 13 | 2 to 4 |
| Uric acid | 2 to 4 | 9 to 17 |
| Other | 4 | 2 |
Footnote: *Incidence is as high as 75 percent in pregnant women
[Source 22 ]Calcium stones
Calcium stones, including calcium oxalate stones and calcium phosphate stones, are the most common types of kidney stones. Calcium oxalate stones are more common than calcium phosphate stones. Oxalate is a substance made daily by your liver or absorbed from your diet. Certain fruits and vegetables, as well as nuts and chocolate, have high oxalate content. Dietary factors, high doses of vitamin D, intestinal bypass surgery and several metabolic disorders can increase the concentration of calcium or oxalate in urine.
Calcium stones may also occur in the form of calcium phosphate. This type of stone is more common in metabolic conditions, such as renal tubular acidosis. It may also be associated with certain medications used to treat migraines or seizures, such as topiramate (Topamax, Trokendi XR, Qudexy XR).
Most patients (up to 80%) with calcium stones have one or more of the metabolic risk factors and about 25% of stones are idiopathic (cause is unknown) in origin.
Metabolic risk factors for calcium stones:
- Hypercalciuria (40-60%)
- Hyperuricosuria (25%)
- Hyperoxaluria
- Hypocitriuria
- Other (vitamin A deficiency, hot climates, immobilisation, urinary tract anomalies)
Calcium from food does not increase your chance of having calcium oxalate stones. Normally, extra calcium that isn’t used by your bones and muscles goes to your kidneys and is flushed out with urine. When this doesn’t happen, the calcium stays in the kidneys and joins with other waste products to form a kidney stone.
- For calcium stones, risk factors vary by population. The main risk factor in the US is hypercalciuria (condition of elevated calcium in the urine), a hereditary condition present in 50% of men and 75% of women with calcium calculi; thus, patients with a family history of calculi are at increased risk of recurrent calculi 6. These patients have normal serum calcium, but urinary calcium is elevated > 250 mg/day (> 6.2 mmol/day) in men and > 200 mg/day (> 5.0 mmol/day) in women 6.
- Hypocitruria (urinary citrate < 350 mg/day [1820 μmol/day]), present in about 40 to 50% of calcium calculi-formers, promotes calcium calculi formation because citrate normally binds urinary calcium and inhibits the crystallization of calcium salts 6. The activity of citrate is thought to be related to its concentration in urine, where it exhibits a dual action, opposing crystal formation by both thermodynamic and kinetic mechanisms. Citrate retards stone formation by inhibiting the calcium oxalate nucleation process and the growth of both calcium oxalate and calcium phosphate stones, largely by its ability to bind with urinary calcium and reduce the free calcium concentration, thereby reducing the supersaturation of urine. Citrate binds to the calcium oxalate crystal surface, inhibiting crystal growth and aggregation 23. There is also evidence that citrate blocks the adhesion of calcium oxalate monohydrate crystals to renal epithelial cells 24. Medical interventions to increase urinary citrate are a primary focus in the medical management of urolithiasis 25. The amount of diet-derived citrate that may escape in body conversion to bicarbonate is reportedly minor 26. Nonetheless, a prior study reported increased urinary citrate after 1 week on 4 ounces lemon juice per day, diluted in 2 L water, in stone formers with hypocitraturia 27. Two retrospective studies showed an effect in calcium stone formers of lemon juice and/or lemonade consumption on urinary citrate 28, but a recent clinical trial showed no influence of lemonade on urinary citrate 29.
- Hypocitraturia, if severe and/or persistent, usually requires pharmacologic therapy in the form of potassium citrate, which enhances urine pH and also citrate excretion. The identification and promotion of consumption of fluids that add to the crystal inhibitory potential of urine is appealing, not only to promote fluid intake but to enhance urinary citrate excretion. Citric acid is a naturally-occurring organic acid present in multiple fruits, such as lemon, lime, grapefruit, tangerine, and orange and their juices 30. Data on the citric acid content of fresh fruit juices and commercially-available fruit juice beverages may therefore prove useful in constructing nutrition therapy regimens for calcium stone formers.Lemon and lime juice, both from the fresh fruit and from juice concentrates, provide more citric acid per liter than ready-to-consume grapefruit juice, ready-to-consume orange juice, and orange juice squeezed from the fruit 30. Lemon and lime juices are rich sources of citric acid, containing 1.44 and 1.38 g/oz, respectively, comprising as much as 8% of the dry fruit weight 31. These data concur with those previously reported 32. As lemon and lime juice contain 38 and 35 mg potassium/oz, respectively, about the same as grapefruit juice and about 60% that of orange juice, ingestion of lemon or lime juice on a daily basis could provide dietary alkali that would decrease renal tubular reabsorption of citrate, resulting in enhanced urinary citrate excretion. The distribution of lemon or lime juice in ample water or other fluid, consumed throughout the day, would also add to the volume of fluids ingested, resulting in enhanced urine output 33 and reduced urine supersaturation.Further research should determine the bioavailability of dietary citric acid from various sources and characterize the response to dietary citric acid in kidney stone formers who are hypocitraturic, as well as those who are normocitraturic. The impact of diet-derived citrate on urinary concentrations among calcium stone formers consuming different diets (e.g., high fruit/vegetable intake versus low fruit/vegetable intake; high meat intake versus low meat intake) should be assessed, as dietary patterns are known to influence urinary citrate concentrations 34.
- About 5 to 8% of calculi are caused by renal tubular acidosis. About 1 to 2% of patients with calcium calculi have primary hyperparathyroidism 6. Rare causes of hypercalciuria are sarcoidosis, vitamin D intoxication, hyperthyroidism, multiple myeloma, metastatic cancer, and hyperoxaluria.
- Hyperoxaluria (urinary oxalate > 40 mg/day [> 440 μmol/day]) can be primary or caused by excess ingestion of oxalate-containing foods (eg, rhubarb, spinach, cocoa, nuts and nut products, peanuts [peanuts are legumes not nuts], wheat bran, pepper, tea) or by excess oxalate absorption due to various enteric diseases (eg, bacterial overgrowth syndromes, chronic pancreatic or biliary disease) or ileojejunal (eg, bariatric) surgery.
- Other risk factors include taking high doses of vitamin C (ie, > 2000 mg/day), a calcium-restricted diet (possibly because dietary calcium binds dietary oxalate), and mild hyperuricosuria. Mild hyperuricosuria, defined as urinary uric acid > 800 mg/day (> 5 mmol/day) in men or > 750 mg/day (> 4 mmol/day) in women, is almost always caused by excess intake of purine (in proteins, usually from meat, fish, and poultry); it may cause calcium oxalate calculus formation (hyperuricosuric calcium oxalate nephrolithiasis) 6.
Uric acid stones
Uric acid is the end product of purine metabolism and is either derived from exogenous (dietary) sources or produced endogenously during cell turnover. For example, eating a lot of fish, shellfish, and meat—especially organ meat—may increase uric acid in urine. Uric acid stones can form in people who lose too much fluid because of chronic diarrhea or malabsorption, those who eat a high-protein diet, and those with diabetes or metabolic syndrome. Certain genetic factors also may increase your risk of uric acid stones.
Chronic metabolic acidosis can result in protein metabolism and thus increased excretion of urate and formation of kidney stones 35. Pure uric acid stones are rare but recur frequently. Low urinary pH (pH < 5.5) – urine acidity – is the most common and important factor in uric acid kidney stone formation or rarely with severe hyperuricosuria (urinary uric acid > 1500 mg/day [> 9 mmol/day]), which crystallizes undissociated uric acid; in normouricosuric stone disease the primary defect seems to be in the renal excretion of ammonia and is linked to an insulin resistant state 36. Hyperuricosuria occurs in 10% of patients with calcium stones, where uric acid crystals form the nidus for deposition of calcium and oxalate. A history of gout doubles the risk of kidney stones in men 37. Uric acid crystals may comprise the entire calculus or, more commonly, provide a nidus on which calcium or mixed calcium and uric acid calculi can form.
Struvite stones
Struvite stones or magnesium ammonium phosphate calculi (infection stones) may form after you have a urinary tract infection (UTI) caused by urea-splitting bacteria (eg, Proteus sp, Klebsiella sp) 6. They can develop suddenly and become large quickly, sometimes with few symptoms or little warning. The struvite stones must be treated as infected foreign bodies and removed in their entirety. Unlike other types of calculi, magnesium ammonium phosphate calculi occur 3 times more frequently in women 6.
Cystine stones
Cystine stones result from a hereditary disorder called cystinuria that is passed down through families. Cystinuria causes the amino acid cystine to leak through your kidneys and into the urine. Less than 3% of urinary tract stones are cystine stones 38.
Kidney stone causes
Kidney stones often have no definite, single cause, although several factors may increase your risk. Kidney stones form when your urine contains more crystal-forming substances — such as calcium, oxalate and uric acid — than the fluid in your urine can dilute. At the same time, your urine may lack substances that prevent crystals from sticking together, creating an ideal environment for kidney stones to form.
If you have a family history of kidney stones, you are more likely to develop them. You are also more likely to develop kidney stones again if you’ve had them once.
You may also be more likely to develop a kidney stone if you don’t drink enough liquids.
General risk factors for developing kidney stone include disorders that increase urinary salt concentration, either by increased excretion of calcium or uric acid salts, or by decreased excretion of urinary citrate.
Vitamin D deficiency has been shown to be more prevalent in the stone-forming population 39. However, repletion of vitamin D with supplements in this population has been controversial. The Women’s Health Initiative study demonstrated a 17% increased risk of stone formation among 36 282 postmenopausal women randomized to calcium carbonate plus vitamin D (1 000 mg/day and 400 IU/day, respectively) versus placebo 40. However, a recent small randomized controlled trial comparing 6 weeks of low (1 000 IU/day) versus high (50 000 IU weekly) dose vitamin D supplementation among 21 vitamin D deficient stone formers found no significant change in urinary calcium after treatment in either group, although only the higher dose group showed a significant increase in serum vitamin D levels 41. If vitamin D supplementation is indicated, urine calcium should be monitored with 24 hour urine studies 42, 43.
You are more likely to develop kidney stones if you have these conditions, including:
- anatomical abnormalities that increase the risk of stone disease
- a blockage of the urinary tract
- chronic, or long-lasting, inflammation of the bowel
- cystic kidney diseases , which are disorders that cause fluid-filled sacs to form on the kidneys
- cystinuria
- digestive problems or a history of gastrointestinal tract surgery
- gout, a disorder that causes painful swelling of the joints
- hypercalciuria , a condition that runs in families in which urine contains unusually large amounts of calcium; this is the most common condition found in
- people who form calcium stones
- hyperoxaluria , a condition in which urine contains unusually large amounts of oxalate
- hyperparathyroidism, a condition in which the parathyroid glands release too much parathyroid hormone, causing extra calcium in the blood
- hyperuricosuria, a disorder in which too much uric acid is in the urine
- obesity
- repeated, or recurrent, urinary tract infections (UTIs)
- renal tubular acidosis, a disease that occurs when the kidneys fail to remove acids into the urine, which causes a person’s blood to remain too acidic.
Anatomical abnormalities that increase the risk of kidney stone disease
- Obstruction of the pelviureteral junction
- Hydronephrotic renal pelvis or calices
- Calyceal diverticulum
- Horseshoe kidney
- Ureterocele
- Vesicoureteral reflux
- Ureteral stricture
- Tubular ectasia (medullary sponge kidney)
Medications Associated with Kidney Stone Formation
Table 3. Medications Associated with Kidney Stone Formation
| Type of medication | Examples |
|---|---|
| Agents that decrease uric acid production | Allopurinol (Zyloprim) |
| Laxatives (specific to ammonium urate stones), especially if abused | Overuse of any laxative resulting in electrolyte losses |
| Antibiotics | Sulfonamides, ampicillin, amoxicillin, ceftriaxone (Rocephin), quinolones, furans, pyridines |
| Carbonic anhydrase inhibitors | Acetazolamide, topiramate (Topamax) |
| Ephedra alkaloids (banned in the United States) | Herbal products used as stimulants and appetite suppressants |
| Potassium channel blockers | Amiodarone, sotalol (Betapace), dalfampridine (Ampyra; multiple sclerosis therapy) |
| Potassium-sparing diuretics | Triamterene (Dyrenium) |
| Reverse transcriptase inhibitors and protease inhibitors | HAART (highly active antiretroviral therapy) |
| Sulfonylureas | Various therapies for type 2 diabetes mellitus |
Does Obesity Increase Risk and Will Weight Loss Reduce Your Risk for Kidney Stones?
Obesity contributes to risk of kidney stones more than dietary factors. The associated changes in body composition pose biophysical challenges associated with disturbed thermogenesis and dehydration. Because body fat is hydrophobic, the proportion of body water decreases with increasing obesity, which can lead to dehydration 44. Additionally, the decrease in surface area to body volume complicates heat exchange and metabolic rate 45. Obesity is a proinflammatory state associated with electrolyte imbalances and altered urine chemistry. Obese persons with kidney stones are predisposed to hyperuricemia, gout, hypocitraturia, hyperuricosuria, and uric acid stones 46. A recent retrospective analysis found that patients with diabetes and kidney stones excrete more oxalate and have lower urine pH, which is partly a result of higher sulfate excretion and less acid excreted as ammonium ions 47, 48.Patients with kidney disease who are obese or have diabetes may have a lesser genetic predisposition to kidney stones and greater responsiveness to environmental modification, such as a healthy diet and hydration.
Weight loss may improve or undermine management of kidney stones, depending on how it is achieved. Weight loss could be detrimental to prevention of kidney stones if associated with a high animal protein diet, laxative abuse, rapid loss of lean tissue, or poor hydration. High acid diets, such as the Atkins diet, increase the risk of uric acid stones 49. Therefore, diet advice should be based on the type of kidney stone.
Should people with a history of kidney stones reduce their fructose intake?
Increased dietary fructose has been associated with up to a 38 percent higher risk of kidney stones 50. Increased fructose intake increases urinary calcium excretion in persons with magnesium deficiency, and fructose is the only dietary carbohydrate known to raise uric acid levels. Additionally, sugar-sweetened beverages and orange juice have been linked to gout 51.
Hypercalciuria
Hypercalciuria is defined as excretion of urinary calcium exceeding 200 mg in a 24 hour collection or an excess of 4 mg calcium/kg/24 hour 10. Hypercalciuria has also been variously defined as 24 hour urinary calcium >300 mg/day in men, >250 mg/day in women or >200 mg/day in either sex while on a diet restricted in calcium, sodium, and animal protein 52, 53.
Hypercalciuria is the most common metabolic abnormality in patients with calcium oxalate stones and results from various mechanisms 10. Hypercalciuria may be a manifestation of systemic diseases such as primary hyperparathyroidism or sarcoidosis, but is considered idiopathic if no underlying cause can be identified 54. While idiopathic hypercalciuria has been shown to have a genetic predisposition in some cases, it can also be influenced by environmental factors such as diet 55. Although kidney stone formers have a higher risk of bone fracture than those in the general population56, it is not clear that hypercalciuria is a cause. In a retrospective study of 250 men and 182 post-menopausal women on or off estrogen therapy, no significant relationship was found between urine calcium levels and bone mineral density. Hypercalciuria may not cause low bone mineral density and increased fracture rate among stone formers 57.
A normal calcium intake (1 000–1 200 mg/day elemental calcium or approximately three servings of dairy daily) is recommended for patients with idiopathic hypercalciuria 58, 59. Both dairy and non-dairy dietary sources of calcium have been shown to have a protective effect against incident stone formation 60. On the other hand, severe calcium restriction should be avoided as it may accelerate bone loss and lead to hyperoxaluria due to the interaction between calcium and oxalate in the intestinal lumen by which calcium binds to oxalate and forms a calcium oxalate complex. In the setting of low calcium intake, excess uncomplexed oxalate is absorbed, ultimately leading to increased urinary oxalate excretion 61, 62, 63. Calcium in the form of food is preferred over calcium supplements as supplementation has been shown in epidemiologic studies to be associated with an increased risk of incident stone formation 64, 65, 66. If calcium supplements are indicated, they should be taken with meals, allowing the ingested calcium to complex with oxalate, thereby reducing intestinal oxalate absorption and counteracting the effect of increased urinary calcium.
- Absorptive hypercalciuria: Increased absorption of calcium from the gut results in increased circulating calcium, resulting in increased renal filtered load. The exact mechanism is unknown but seems to be inherited in an autosomal dominant fashion, and the jejunal mucosa is hyper-responsive to vitamin D. Absorptive hypercalciuria is very common, but most patients remain asymptomatic and do not experience stone formation.
- Renal hypercalciuria: Increased excretion of calcium in urine results from impaired renal tubular absorption of calcium. This occurs in about 2% of patients with recurrent stone formation.
- Resorptive hypercalciuria: Increased resorption of bone occurs as a result of primary hyperparathyroidism. This occurs in about 5% of patients with recurrent stone formation. The risk of renal stones is increased in primary hyperparathyroidism and returns to baseline about 10 years after parathyroidectomy. Patients who had stones before undergoing parathyroidectomy have a 27 times greater risk of stone formation after parathyroidectomy than do patients without hyperparathyroidism 67.
Hyperuricosuria
Uric acid is the end product of purine metabolism and is either derived from exogenous (dietary) sources (animal protein is a rich source of purines) or produced endogenously during cell turnover. Chronic metabolic acidosis can result in protein metabolism and thus increased excretion of urate and formation of kidney stones. Urinary uric acid has been shown to reduce the effectiveness of naturally occurring macromolecular inhibitors of calcium oxalate crystallization 68. In addition, protein derived from animal sources increases stone risk by increasing urinary calcium and oxalate and reducing pH and citrate. Animal protein provides an acid load through the high content of sulfur-containing amino acids, which leads to a state of mild chronic metabolic acidosis, low urine pH and hypocitraturia 69, 70. Since high protein diets are often additionally devoid of sufficient fruits and vegetables, hypocitraturia may also ensue from the lack of alkali 71. Interestingly, a study comparing idiopathic calcium stone formers with a control group found a higher mean renal acid load in the stone formers, even though animal protein intake was similar between the two groups 72. The authors attributed these findings to a lower intake of fruits and vegetables among stone formers 72. Furthermore, a small metabolic study simulating the three phases of the Atkins diet (a low carbohydrate, high protein diet) demonstrated increases in urinary uric acid and calcium and decreases in pH and citrate during both the stringent induction phase and the less stringent maintenance phases of the diet compared to baseline 73. While the acid load conferred by animal protein has been presumed to promote hypercalciuria by increasing bone resorption 74, Maalouf and colleagues 75 found that administration of potassium citrate failed to prevent protein-induced hypercalciuria, suggesting that the hypercalciuria may be attributable to a renal etiology.
Pure uric acid stones are rare but recur frequently. Low urinary pH (pH < 5.5) is the most common and important factor in uric acid nephrolithiasis; in normouricosuric stone disease the primary defect seems to be in the renal excretion of ammonia and is linked to an insulin resistant state. Hyperuricosuria occurs in 10% of patients with calcium stones, where uric acid crystals form the nidus for deposition of calcium and oxalate. A history of gout doubles the risk of kidney stones in men.
The recommended dietary allowance of protein is 0.8 g/kg/day 76, and animal protein restriction should include all forms of meat, including beef, poultry, and fish. A 3-phase randomized, crossover metabolic study in 25 normal subjects comparing three different animal protein sources revealed higher levels of urinary uric acid during the fish phase compared to the beef or poultry phase, although urinary saturation of calcium oxalate did not simply reflect urinary uric acid levels 77. Additionally, the question often arises among stone patients whether ingestion of whey protein, a dairy-derived protein supplement popular among athletes because it is thought to increase muscle mass and improve exercise performance, is also a risk factor for stone formation. A recent 2-phase metabolic study in which whey protein or albumin was given to 18 healthy volunteers on a controlled diet for 3 days showed no significant change from baseline in urinary stone risk factors with either supplement 78.
Hyperoxaluria
Hyperoxaluria occurs in approximately 10%–15% of calcium stone formers. Hyperoxaluria is defined as urinary excretion of oxalate in excess of 45 mg/day 10. Increased urinary oxalate can occur as a consequence of excessive dietary intake (oxalate gluttons), endogenous oxalate overproduction or intestinal oxalate overabsorption (enteric hyperoxaluria).
On the basis of the mechanism, hyperoxaluria is classified as follows:
- Enteric hyperoxaluria: This results from increased intestinal absorption due to ileal disease (Crohn’s disease, ileal bypass) or short bowel syndrome, low calcium intake, or gastrointestinal decolonisation of Oxalobacter formigenes. Oxalobacter is an intestinal bacterium that degrades dietary oxalate, and decolonisation of the gut results in increased absorption of oxalate. Oral administration of Oxalobacter has been shown to decrease urinary oxalate concentration in animals and humans 79, 80.
- Increased ingestion (oxalate gluttons): Dietary oxalate contributes to about half of the urinary oxalate and is inversely proportional to calcium intake in healthy people without gastrointestinal disease 81. Spinach, rhubarb, beets, chocolate, nuts, tea, wheat bran, strawberries, and soya foods are known to increase urinary oxalate concentrations 82. Vitamin C supplementation may increase urinary oxalate excretion and the risk of calcium oxalate crystallisation in patients who form calcium stones 83. Ingestion of grapefruit juice increases excretion of both oxalate and citrate in urine with no net change in its lithogenicity 84.
- Primary hyperoxaluria: This is an inborn error of metabolism (glycolic aciduria).
In normal individuals, approximately 10% of ingested oxalate is absorbed while the rest is eliminated through the stool 85, 86. For reasons not clearly understood, calcium oxalate stone formers absorb a slightly higher proportion of oxalate from the intestine than do normal subjects 85. Oxalate absorption depends not only on the amount of dietary oxalate, but also on dietary calcium intake. Higher calcium diets lead to reduced oxalate absorption, while calcium-restricted diets are associated with enhanced oxalate absorption and subsequently increased urinary oxalate excretion 86, 87. As such, a normal calcium diet in association with oxalate restriction is recommended in hyperoxaluric patients. High oxalate foods such as spinach, rhubarb, beets, nuts, chocolate, potatoes, bran, legumes and tea should be avoided 88. Some juices have been found to have a high oxalate content, including cranberry 89, grapefruit 90 and carambola juice (starfruit) 91. Spinach is frequently added to homemade fruit and vegetable juices, raising the oxalate content 92. Since the leaves are not cooked or processed to enhance removal or reduction of soluble oxalate, this practice may pose a threat to calcium oxalate stone formers. The addition of small amounts of calcium ions, especially in the form of calcium chloride, has been recommended by some to convert the oxalate into an insoluble form that is less likely to be absorbed in the digestive tract 92. However, any added calcium should be considered when calculating total dietary calcium intake.
Interestingly, no studies have directly shown a correlation between urinary oxalate and recurrent idiopathic calcium oxalate stone formation, and therefore recommendation of dietary oxalate restriction is empiric. Indeed, Noori and colleagues 93 randomized 57 patients with hyperoxaluria and recurrent calcium oxalate stones to a low oxalate diet or to the Dietary Approaches to Stop Hypertension (DASH) diet, which is a diet high in fruits, vegetables, nuts and legumes (high oxalate content) and low in sodium and red and processed meats. Although urinary oxalate increased in the group assigned to the DASH diet and decreased in those adhering to a low oxalate (4.8 mg/day vs. −4.2 mg/day, respectively), urinary saturation of calcium oxalate declined more on the DASH diet (−2.14) than on the low oxalate diet (−0.90), suggesting that other dietary measures had greater impact on reducing urinary stone risk than a low oxalate diet.
Enzymatic defects in the oxalate biosynthetic pathway lead to markedly high levels of urinary oxalate leading to aggressive calcium oxalate stone formation and oxalosis. Among the three forms of primary hyperoxaluria (I–III), renal failure is typically seen only with primary hyperoxaluria type 1. Strict dietary oxalate restriction is recommended in all forms of primary hyperoxaluria 94.
Patients with malabsorptive disorders from intestinal resection, roux-en-Y gastric bypass surgery, Crohn’s disease, celiac sprue, pancreatitis or use of fat-malabsorbing medications such as orlistat, are at risk of enteric hyperoxaluria because luminal calcium binds to poorly absorbed fatty acids, leading to higher levels of uncomplexed oxalate that is subsequently absorbed and excreted in the urine 95. In these patients, strict dietary oxalate restriction, along with a low fat diet and use of calcium supplements with meals to bind luminal oxalate is an effective strategy for stone prevention.
In experimental animals, testosterone promotes stone formation by suppressing osteopontin expression in the kidney and increasing urinary oxalate excretion. Estrogen seems to inhibit stone formation by increasing osteopontin expression in the kidney and decreasing urinary oxalate excretion 96.
Finally, O. formigenes is a Gram-negative anaerobic bacterium that resides in the intestine and uses oxalate as its sole source for energy and growth 97. Animal models have shown that absence of O. formigenes colonization can result in reduced degradation of oxalate in the intestinal lumen as well as reduced enteric oxalate secretion 98. Other animal models demonstrated that colonization with O. formigenes in addition to a low fat diet decreased urinary oxalate excretion 99. A case-control study in human subjects noted that despite a strong inverse correlation between colonization and risk of recurrent stone formation, no significant difference in median urinary oxalate levels was detected between patients who were or were not colonized with O. formigenes 100. Further work is needed to clarify the therapeutic role of this organism or its enzymes in preventing calcium oxalate stone formation.
Hypocitriuria
Hypocitraturia has been reported in 15%–63% of patients with kidney stones and is often seen in conjunction with other metabolic disorders 101. Hypocitriuria is defined as urinary citrate excretion of < 250 mg in 24 hours. Urinary citrate forms a soluble complex with calcium that inhibits the formation and propagation of crystals 10. It is a common correctable cause of recurrent pure calcium phosphate or brushite stones. Women excrete more citrate and have lower incidence of stone formation than men. Citrate is an important inhibitor of calcium stone formation because it directly inhibits nucleation, agglomeration and growth of calcium oxalate and/or calcium phosphate crystals and by complexing with calcium to reduce urinary saturation of calcium salts 102. Renal citrate excretion is modulated primarily by acid–base status; acidosis increases citrate reabsorption and alkalosis enhances citrate production and excretion in the renal proximal tubule.
Urinary citrate is mainly derived endogenously through the tricarboxylic acid cycle and is excreted by renal tubular cells. Intracellular acidosis, acidic diets (diets rich in animal proteins), and hypokalaemia decrease urinary citrate excretion. Fruits such as oranges and grapefruits are the main exogenous sources of urinary citrate. Hormonal replacement therapy in postmenopausal women results in higher urinary calcium excretion, but it also increases urinary excretion of citrate and leads to net inhibition of crystal precipitation, thereby decreasing the risk of calcium stones 103.
Fruits and vegetables increase urinary citrate because of their high alkali content, but not all fruits and juices have the same citraturic effect. Orange juice has shown the most consistent benefit because it has a high content of potassium citrate that confers an alkali load 104, 105, 106. Lemonade, which is high in citric acid, does not affect urine pH and has less citraturic effect. While fruit juices offer a more palatable and less costly therapy than potassium citrate medication, fruit juices can be high in calories and oxalate content and this may temper their use 90, 107. Fruits with a high malic acid (precursor to citrate) content, such as pears, may theoretically increase urinary citrate but few studies have examined this 108. Unfortunately, no citrus fruits or juices have been tested in a randomized trial to assess their benefit in reducing stone recurrence rates.
The Dietary Approaches to Stop Hypertension (DASH) diet, as an overall healthy diet, has been suggested to reduce the rate of stone formation 109. The high alkali content, among other factors, may contribute to improvement in urinary stone risk factors 110. Among three large cohorts of both men and women [Nurses’ Health Study I (NHS 1), Nurses’ Health Study II (NHS 2), and Health Professionals Follow-up Study (HPFS)], those subjects adhering most closely to the DASH diet had the lowest risk of incident stone formation on multivariate analysis 111. That is to say, when patients were given a score relating to how closely their diet resembled the DASH diet, those in the lowest quintile of DASH scores had the highest rate of incident stone formation in Health Professionals Follow-up Study (HPFS) (odds ratio (OR) = 1.53; Nurses’ Health Study 1 (OR = 1.47), and Nurses’ Health Study 2 (OR = 1.37).
Struvite (triple phosphate) and cystine stones
Various anatomical abnormalities promote urine stasis and increase the risk of stone formation by promoting precipitation of crystals. Urinary infection with urea splitting organisms (Proteus, Klebsiella, Serratia, and Mycoplasma) creates alkaline urine that promotes the formation of struvite stones. Urinary saturation with struvite occurs only when supranormal excretion of ammonia and alkaline urine occur together. Alkalaemia suppresses renal ammoniagenesis, but the hydrolysis of urea by bacteria liberates ammonia that alkalises urine.
Cystinuria (cystine stones) is an autosomal recessive trait, with an inborn error in the transport of dicarboxylic acids—cystine, ornithine, lysine, and arginine, commonly known as “COLA.” The low solubility of cystine results in its precipitation and stone formation.
Urinary glycoproteins
Various urinary glycoproteins (Tamm-Horsfall proteins, bikunin, nephrocalcin, urinary prothrombin fragment 1) are inhibitors of stone formation. Their deficiency may promote stone formation.
Drugs that may increase the risk of stone disease
- Decongestants: ephedrine, guaifenesin
- Diuretics: triamterene
- Protease inhibitors: indinavir
- Anticonvulsants: felbamate, topiramate, and zonisamide
Risk factors for developing kidney stones
Factors that increase your risk of developing kidney stones include:
- Family or personal history of kidney stone. If someone in your family has had kidney stones, you’re more likely to develop stones, too. If you’ve already had one or more kidney stones, you’re at increased risk of developing another.
- Dehydration. Not drinking enough water each day can increase your risk of kidney stones. People who live in warm, dry climates and those who sweat a lot may be at higher risk than others.
- Certain diets. Eating a diet that’s high in protein, sodium (salt) and sugar may increase your risk of some types of kidney stones. This is especially true with a high-sodium diet. Too much salt in your diet increases the amount of calcium your kidneys must filter and significantly increases your risk of kidney stones.
- Obesity. High body mass index (BMI), large waist size and weight gain have been linked to an increased risk of kidney stones.
- Digestive diseases and surgery. Gastric bypass surgery, inflammatory bowel disease or chronic diarrhea can cause changes in the digestive process that affect your absorption of calcium and water, increasing the amounts of stone-forming substances in your urine.
- Other medical conditions such as renal tubular acidosis, cystinuria, hyperparathyroidism and repeated urinary tract infections also can increase your risk of kidney stones.
- Certain supplements and medications, such as vitamin C, dietary supplements, laxatives (when used excessively), calcium-based antacids, and certain medications used to treat migraines or depression, can increase your risk of kidney stones.
Kidney Stone pathophysiology
Kidney stones may remain within the kidney or renal collecting system or be passed into the ureter and bladder. During passage, the stones may irritate the ureter and may become lodged, obstructing urine flow and causing hydroureter and sometimes hydronephrosis. Common areas of lodgment include the following:
- Ureteropelvic junction
- Distal ureter (at the level of the iliac vessels)
- Ureterovesical junction
Larger kidney stones are more likely to become lodged. Typically, a kidney stone must have a diameter > 5 mm to become lodged.
Kidney stones ≤ 5 mm are likely to pass spontaneously.
Even partial obstruction causes decreased glomerular filtration, which may persist briefly after the calculus has passed. With hydronephrosis and elevated glomerular pressure, renal blood flow declines, further worsening renal function. Generally, however, in the absence of infection, permanent renal dysfunction occurs only after about 28 days of complete obstruction.
Secondary infection can occur with long-standing obstruction, but most patients with calcium-containing calculi do not have infected urine.
Kidney stone prevention
In a patient who has passed a first calcium calculus, the likelihood of forming a 2nd calculus is about 15% at 1 yr, 40% at 5 yr, and 80% at 10 yr. Drinking large amounts of fluids— that will achieve a urine volume of at least 2.5 liters daily—is recommended for prevention of all stones 112. Recovery and analysis of the calculus, measurement of calculus-forming substances in the urine, and the clinical history are needed to plan other prophylactic measures.
In < 3% of patients, no metabolic abnormality is found. These patients seemingly cannot tolerate normal amounts of calculus-forming salts in their urine without crystallization. Thiazide diuretics, potassium citrate, and increased fluid intake may reduce their calculus production rate.
For hypercalciuria, patients may receive thiazide diuretics (eg, chlorthalidone 25 mg po once/day or indapamide 1.25 mg po once/day) to lower urine calcium excretion and thus prevent urinary supersaturation with calcium oxalate. Patients are encouraged to increase their fluid intake to ≥ 3 L/day. A diet that is low in sodium and high in potassium is recommended. Even with a high potassium intake, supplementation with potassium citrate is recommended to prevent hypokalemia. Restriction of dietary animal protein is also recommended.
For patients with hypocitruria, potassium citrate (20 mEq po bid) enhances citrate excretion. A normal calcium intake (eg, 1000 mg or about 2 to 3 dairy servings per day) is recommended, and calcium restriction is avoided. Oral orthophosphate has not been thoroughly studied.
Hyperoxaluria prevention varies. Patients with small-bowel disease can be treated with a combination of high fluid intake, calcium loading (usually in the form of calcium citrate 400 mg po bid with meals), cholestyramine, and a low-oxalate, low-fat diet. Hyperoxaluria may respond to pyridoxine 100 to 200 mg po once/day, possibly by increasing transaminase activity, because this activity is responsible for the conversion of glyoxylate, the immediate oxalate precursor, to glycine.
In hyperuricosuria, intake of animal protein should be reduced. If the diet cannot be changed, allopurinol 300 mg each morning lowers uric acid production. For uric acid calculi, the urine pH must be increased to between 6 and 6.5 by giving an oral alkalinizing drug that contains potassium (eg, potassium citrate 20 mEq bid) along with increased fluid intake.
Infection with urea-splitting bacteria requires culture-specific antibiotics and complete removal of all calculi. If eradication of infection is impossible, long-term suppressive therapy (eg, with nitrofurantoin) may be necessary. In addition, acetohydroxamic acid can be used to reduce the recurrence of struvite calculi.
To prevent recurrent cystine calculi, urinary cystine levels must be reduced to < 250 mg cystine/L of urine. Any combination of increasing urine volume along with reducing cystine excretion (eg, with alpha-mercaptopropionylglycine or penicillamine) should reduce the urinary cystine concentration.
Lifestyle changes
You may reduce your risk of kidney stones if you 113:
- Drink water throughout the day. For people with a history of kidney stones, doctors usually recommend drinking enough fluids to pass about 2.1 quarts (2 liters) of urine a day. Your doctor may ask that you measure your urine output to make sure that you’re drinking enough water. If you live in a hot, dry climate or you exercise frequently, you may need to drink even more water to produce enough urine. If your urine is light and clear, you’re likely drinking enough water.
- Eat fewer oxalate-rich foods. If you tend to form calcium oxalate stones, your doctor may recommend restricting foods rich in oxalates. These include rhubarb, beets, okra, spinach, Swiss chard, sweet potatoes, nuts, tea, chocolate, black pepper and soy products.
- Choose a diet low in salt and animal protein. Reduce the amount of salt you eat and choose nonanimal protein sources, such as legumes. Consider using a salt substitute, such as Mrs. Dash.
- Continue eating calcium-rich foods, but use caution with calcium supplements. Calcium in food doesn’t have an effect on your risk of kidney stones. Continue eating calcium-rich foods unless your doctor advises otherwise. Ask your doctor before taking calcium supplements, as these have been linked to increased risk of kidney stones. You may reduce the risk by taking supplements with meals. Diets low in calcium can increase kidney stone formation in some people.
Ask your doctor for a referral to a dietitian who can help you develop an eating plan that reduces your risk of kidney stones (https://www.eatright.org/find-a-nutrition-expert).
For prevention of calcium oxalate, cystine, and uric acid stones, urine should be alkalinized
For prevention of calcium oxalate, cystine, and uric acid stones, urine should be alkalinized [increase urine pH to 6.5 to 7] 114, 115. Western diets are characteristically high in acid-producing foods, such as grains, dairy products, legumes, and meat. Alkalinizing urine involves eating a diet high in fruits and vegetables, taking supplemental or prescription citrate, or drinking alkaline mineral waters 116.
How To Alkalinize Your Urine
Alkalinize urine (i.e., increase urine pH to 6.5 to 7) with dietary changes or oral supplementation, or until 24-hour urine citrate levels are in the normal range:
- Potassium citrate: 10 to 20 mEq orally with meals (prescription required)
- Calcium citrate: two 500-mg tablets per day with meals (each tablet contains 120 mg of calcium and 6 mEq of bicarbonate)
For prevention of calcium phosphate and struvite stones, urine should be acidified
For prevention of calcium phosphate and struvite stones, urine should be acidified [lower urine pH to 7 or less] 117. Cranberry juice or betaine can lower urine pH without the adverse effects associated with acid-producing foods. Although table salt (sodium chloride) also lowers urine pH, it can increase blood pressure, insulin excretion, and urine calcium excretion.
How to Acidify Your Urine
Acidify urine (i.e., lower urine pH to 7 or less) with dietary changes or oral supplementation:
- Cranberry juice: at least 16 oz per day
- Betaine: 650 mg orally three times per day with meals
Can Bacterial Infection Trigger Recurrence?
Bacteria exert both pathogenic and protective roles. Struvite stones are associated with recurrent infections because of high urinary pH levels from urease splitting bacteria and the body’s inability to rid the urinary tract of bacteria that become embedded in the stones 118.
Oxalobacter formigenes is an anaerobic bacterium that colonizes the intestinal tract, where it metabolizes oxalate to formate and carbon dioxide. Absence of O. formigenes colonization predisposes persons to oxalate stones 119. Preliminary studies of O. formigenes ingestion in healthy patients 119 and in patients with primary hyperoxaluria demonstrated up to a 90 percent decrease in urinary oxalate levels 120. Larger studies of this potential therapy are ongoing.
Specific treatments to prevent recurrent stones
Calcium stones
Normocalciuria
- Oral administration of potassium citrate—increases urine pH and citrate excretion in the urine.
Hypercalciuria
- Thiazide diuretics—decrease urinary calcium excretion by augmenting tubular reabsorption of calcium, but do not decrease intestinal absorption in absorptive hypercalciuria; the effect may be attenuated or lost after two or more years of treatment
- Addition of potassium citrate may help to control the diuretic induced hypokalaemia
- If magnesium loss is a concern because of chronic diuretic use, consider potassium magnesium citrate
- Potassium phosphate—may suppress calcitriol synthesis and thereby decrease calcium absorption
Hyperuricaemia or hyperuricosuria
- Allopurinol—to inhibit uric acid synthesis and decrease urinary uric acid excretion
- Potassium citrate should be given in addition to increase urine pH, as uric acid precipitates in acidic urine
Hyperoxaluria
- No specific drugs are available to reduce oxalate excretion in the urine
- Pyridoxine, a cofactor in the alanine-glycoxylate pathway, may reduce production of oxalate by inducing enzyme activity; in an observational study, high intake of vitamin B6 (> 40 mg/day) was inversely associated with risk of oxalate stone formation in women
- Calcium supplementation (250-1000 mg four times a day) to control enteric hyperoxaluria; urinary oxalate may decrease, but a concurrent rise in calcium may negate the beneficial effect
- Cholestyramine reduces intestinal absorption of oxalate, but no trials have shown its efficacy in preventing recurrent stones
- Probiotic treatment with Oxalobacter formigenes has recently been shown to significantly reduce oxalate excretion in both animals and humans; however, trials are pending to show its role in clinical practice 79, 121.
Hypocitriuria
- Potassium citrate—to increase citrate excretion
Struvite stones
- Treatment of infection is mandatory and may be needed in the long term
- Acetohydroxamic acid, a urease inhibitor, has been shown to reduce the urinary saturation of struvite but is associated with high frequency of side effects (deep vein thrombosis, haemolytic anaemia), which limits its use.
Cystine stones
- Treatment must include increasing urine output to about 3 l/day and adequate alkalinisation (urine pH > 7.0) with potassium citrate.
- In addition, specific agents such as α mercaptopropionylglycine or d-penicillamine that form soluble complexes with cystine are used.
Kidney stone signs and symptoms
Large kidney stones remaining in the kidney or renal collecting system are often asymptomatic unless they cause obstruction and/or infection or passes into one of the ureters 6. The ureters are the tubes that connect the kidneys and bladder. If a kidney stone becomes lodged in the ureters, it may block the flow of urine and cause the kidney to swell and the ureter to spasm, which can be very painful. Severe pain, often accompanied by nausea and vomiting, usually occurs when kidney stones pass into the ureter and cause acute obstruction. Sometimes gross hematuria also occurs.
Kidney stone signs and symptoms may include:
- Severe, sharp pain in the side and back, below the ribs
- Pain that radiates to the lower abdomen and groin
- Pain that comes in waves and fluctuates in intensity
- Pain or burning sensation while urinating
- A constant need to urinate, urinating more often than usual or urinating in small amounts
- Pink, red, or brown blood in your urine, also called hematuria
- Inability to urinate or can only urinate a small amount
- Cloudy or bad-smelling urine.
Pain (renal colic) is of variable intensity but is typically excruciating and intermittent, often occurs cyclically, and lasts 20 to 60 min. Nausea and vomiting are common. Pain caused by a kidney stone may change — for instance, shifting to a different location or increasing in intensity — as the stone moves through your urinary tract. Pain in the flank or kidney area that radiates across the abdomen suggests upper ureteral or renal pelvic obstruction. Pain that radiates along the course of the ureter into the genital region suggests lower ureteral obstruction. Suprapubic pain along with urinary urgency and frequency suggests a distal ureteral, ureterovesical, or bladder stone.
The person with kidney stone may be in obvious extreme discomfort, often ashen and diaphoretic (sweating heavily). The person with kidney stone with renal colic may be unable to lie still and may pace, writhe, or constantly shift position. The abdomen may be somewhat tender on the affected side as palpation increases pressure in the already-distended kidney (costovertebral angle tenderness), but peritoneal signs (guarding, rebound, rigidity) are lacking.
For some patients, the first symptom is hematuria or either gravel or a calculus in the urine. Other patients may have symptoms of a urinary tract infection, such as fever, dysuria (painful urination) or cloudy or foul-smelling urine.
See a health care professional right away if you have any of these symptoms. These symptoms may mean you have a kidney stone or a more serious condition.
Your pain may last for a short or long time or may come and go in waves. Along with pain, you may have:
- nausea
- vomiting
Other symptoms include:
- fever and chills if an infection is present
Kidney stone complications
Complications of kidney stones are rare if you seek treatment from a health care professional before problems occur.
Kidney stones increase the risk of developing chronic kidney disease. lf you have had one stone, you are at increased risk of having another stone. Those who have developed one stone are at approximately 50% risk for developing another within 5 to 7 years.
If kidney stones are not treated, they can cause:
- hematuria, or blood in the urine
- severe pain
- urinary tract infections (UTIs), including kidney infections (pyelonephritis)
- loss of kidney function
Kidney stone diagnosis
If your doctor suspects that you have a kidney stone, you may have diagnostic tests and procedures, such as:
- Blood testing. Blood tests may reveal too much calcium or uric acid in your blood. Blood test results help monitor the health of your kidneys and may lead your doctor to check for other medical conditions.
- Urine testing. The 24-hour urine collection test may show that you’re excreting too many stone-forming minerals or too few stone-preventing substances. For this test, your doctor may request that you perform two urine collections over two consecutive days.
- Imaging tests. Imaging tests may show kidney stones in your urinary tract. High-speed or dual energy computerized tomography (CT) may reveal even tiny stones. Simple abdominal X-rays are used less frequently because this kind of imaging test can miss small kidney stones. Ultrasound, a noninvasive test that is quick and easy to perform, is another imaging option to diagnose kidney stones.
- Analysis of passed stones. You may be asked to urinate through a strainer to catch stones that you pass. Lab analysis will reveal the makeup of your kidney stones. Your doctor uses this information to determine what’s causing your kidney stones and to form a plan to prevent more kidney stones.
Diagnosis of a kidney stone starts with a medical history, physical examination, and imaging tests. Your doctors will want to know the exact size and shape of the kidney stones. This can be done with a high resolution CT scan from the kidneys down to the bladder or an x-ray called a “KUB x-ray” (kidney-ureter-bladder x-ray) which will show the size of the stone and its position. The KUB x-ray is often obtained by the surgeons to determine if the stone is suitable for shock wave treatment. The KUB test may be used to monitor your stone before and after treatment, but the CT scan is usually preferred for diagnosis. In some people, doctors will also order an intravenous pyelogram or lVP, a special type of X- ray of the urinary system that is taken after injecting a dye.
Second, your doctors will decide how to treat your stone. The health of your kidneys will be evaluated by blood tests and urine tests. Your overall health, and the size and location of your stone will be considered.
Later, your doctor will want to find the cause of the stone. The stone will be analyzed after it comes out of your body, and your doctor will test your blood for calcium, phosphorus and uric acid. The doctor may also ask that you collect your urine for 24 hours to test for calcium and uric acid.
Lab tests
Urine tests can show whether your urine contains high levels of minerals that form kidney stones. Urine and blood tests can also help a health care professional find out what type of kidney stones you have.
Urinalysis. Urinalysis involves a health care professional testing your urine sample. You will collect a urine sample at a doctor’s office or at a lab, and a health care professional will test the sample. Urinalysis can show whether your urine has blood in it and minerals that can form kidney stones. White blood cells and bacteria in the urine mean you may have a urinary tract infection. Macroscopic or microscopic hematuria is common, but urine may be normal despite multiple kidney stones. Pyuria (pus in urine) with or without bacteria may be present. Pyuria suggests infection, particularly if combined with suggestive clinical findings, such as foul-smelling urine or a fever. A stone and various crystalline substances may be present in the sediment. If so, further testing is usually necessary because the composition of the calculus and crystals cannot be determined conclusively by microscopy. The only exception is when typical hexagonal crystals of cystine are found in a concentrated, acidified specimen, confirming cystinuria.
Blood tests. A health care professional may take a blood sample from you and send the sample to a lab to test. The blood test can show if you have high levels of certain minerals in your blood that can lead to kidney stones.
Imaging tests
Health care professionals use imaging tests to find kidney stones. The tests may also show problems that caused a kidney stone to form, such as a blockage in the urinary tract or a birth defect. You do not need anesthesia for these imaging tests.
Computed tomography (CT) scans. Noncontrast helical CT scan is the initial imaging study. This study can detect the location of a calculus as well as the degree of obstruction. Moreover, helical CT scan may also reveal another cause of the pain (eg, aortic aneurysm). For patients who have recurrent calculi, cumulative radiation exposure from multiple CT scans is a concern. However, the routine use of low-dose renal CT can meaningfully reduce cumulative radiation dose with little loss of sensitivity 122. For patients with typical symptoms, ultrasonography or plain abdominal x-rays can usually confirm presence of a calculus with minimal or no radiation exposure. MRI may not identify calculi. Although a CT scan without contrast medium is most commonly used to view your urinary tract, a health care professional may give you an injection of contrast medium. Contrast medium is a dye or other substance that makes structures inside your body easier to see during imaging tests. You’ll lie on a table that slides into a tunnel-shaped device that takes the x-rays. CT scans can show the size and location of a kidney stone, if the stone is blocking the urinary tract, and conditions that may have caused the kidney stone to form.
Abdominal x-ray. An abdominal x-ray is a picture of the abdomen that uses low levels of radiation and is recorded on film or on a computer. An x-ray technician takes an abdominal x-ray at a hospital or outpatient center, and a radiologist reads the images. Although most urinary calculi are demonstrable on plain x-ray, neither their presence nor their absence obviates the need for more definitive imaging, so this study can be avoided except in some patients with suspected recurrent calculi. Both renal ultrasonography and excretory urography (previously called intravenous urography) can identify calculi and hydronephrosis. However, ultrasonography is less sensitive for small or ureteral calculi in patients without hydronephrosis, and excretory urography is time consuming and exposes the patient to the risk of IV contrast agents. Abdominal x-rays can show the location of kidney stones in the urinary tract. Not all stones are visible on abdominal x-ray. These studies are generally used when helical CT is unavailable.
Kidney stone treatment
Treatment for kidney stones varies, depending on the type of stone, the location, and the cause. The American Urological Association produces a clincal guideline to provide doctors with a clinical framework for the diagnosis, prevention and follow-up of adult patients with kidney stones based on the best available published literature 123.
The treatment for kidney stones is similar in children and adults. Small kidney stones may pass through your urinary tract without treatment. You may be asked to drink a lot of water. Doctors try to let the stone pass without surgery. You may also get medication to help make your urine less acid. If you’re able to pass a kidney stone, a health care professional may ask you to catch the kidney stone in a special container. A health care professional will send the kidney stone to a lab to find out what type it is.
Larger kidney stones or kidney stones that block your urinary tract or cause great pain may need urgent treatment. If you are vomiting and dehydrated, you may need to go to the hospital and get fluids through an IV. The health care professional also may prescribe pain medicine.
Identifying the cause
The kidney stone is obtained by straining the urine (or, if necessary, during operative removal) and sent to the laboratory for stone analysis. Some calculi are brought in by patients. Urine specimens that show microscopic crystals are sent for crystallography.
In patients with a single calcium kidney stone and no additional risk factors for kidney stones, evaluation to exclude hyperparathyroidism is sufficient. Evaluation entails urinalysis and determination of plasma calcium concentration on 2 separate occasions. Predisposing factors, such as recurrent calculi, a diet high in animal protein, or use of vitamin C or D supplements, should be sought.
Patients with a strong family history of calculi, conditions that might predispose to calculi formation (eg, sarcoidosis, bone metastases, multiple myeloma), or conditions that would make it difficult to treat calculi (eg, solitary kidney, urinary tract anomalies) require evaluation for all possible causative disorders and risk factors. This evaluation should include serum electrolytes, uric acid, and calcium on 2 separate occasions. Follow-up determination of parathyroid hormone levels is done if necessary. Urine tests should include routine urinalysis and 2 separate 24-h urine collections to determine urine volume, pH, and excretion of calcium, uric acid, citrate, oxalate, sodium, and creatinine 124.
Acute Management of Kidney Stones in Adults
Oral hydration and pain management are part of the acute treatment of all stone types (Table 4) 125. A long-term randomized controlled trial (RCT) among recurrent idiopathic calcium oxalate stone formers demonstrated that a fluid intake of at least 2 L/day reduced the risk of stone recurrence by about 56% 126. For stones measuring 10 mm or less, antispasmodics such as calcium channel blockers and alpha blockers relax the smooth muscle of the ureters and have been shown to hasten stone passage by five to seven days 127. Co-administration of oral corticosteroids leads to little or no improvement in outcomes 128. People who are unable to take oral fluids or medications and people with low blood pressure or other signs of early hemodynamic instability should be treated intravenously. If signs of possible infection are present (e.g., fever, pyuria), initial management should include empiric antibiotics that cover gram-negative bacilli (e.g., Enterobacteriaceae species) and gram-positive cocci (e.g., staphylococci, enterococci) according to local susceptibility patterns. Referral to a urologist should be expedited if the patient also shows radiologic evidence of obstruction (hydronephrosis).
Table 4. Acute Management of Kidney Stones in Adults
| Management type | Therapy | Dosage | |
|---|---|---|---|
| Fluids | Oral intake of water, or intravenous normal saline if patient is unable to take oral fluids | At least 2 L of water per 24 hours | |
| 0.9% normal saline solution if blood pressure is low; consider decreasing sodium chloride in patients with calciuria (5% dextrose in water and 0.45% normal saline) | |||
| Antispasmodics to facilitate stone passage* | Alpha blockers | ||
| Doxazosin (Cardura) | 4 mg orally per day | ||
| Tamsulosin (Flomax) | 0.4 mg orally per day | ||
| Calcium channel blockers | |||
| Nifedipine (Procardia, sustained release) | 30 mg orally per day | ||
| Pain management† | Opioid narcotics | ||
| Codeine/acetaminophen | One or two tablets (5 to 10 mg codeine/325 to 500 mg acetaminophen) orally every four to six hours as needed | ||
| Hydrocodone/acetaminophen (Vicodin) | 5 to 10 mg orally every four to six hours as needed | ||
Footnote: People who are unable to take oral medications and people with low blood pressure or other signs of early hemodynamic instability should be treated intravenously.
* Often administered for up to four weeks before performing follow-up imaging studies to determine whether the stone has passed.
† Avoid using nonsteroidal anti-inflammatory drugs because they tend to lower kidney blood flow and glomerular filtration.
[Source 22 ]Medical expulsive therapy
Although increasing fluids (either oral or IV) has traditionally been recommended, increased fluid administration has not been proven to speed the passage of calculi. Patients with calculi < 1 cm in diameter who have no infection or obstruction, whose pain is controlled with analgesics, and who can tolerate liquids can be treated at home with analgesics and alpha-receptor blockers (eg, tamsulosin 0.4 mg po once/day) to facilitate calculus passage. Calculi that have not passed within 6 to 8 wk typically require removal. In patients with infection and obstruction, initial treatment is relief of obstruction with a ureteral stent and treatment of the infection followed by removal of calculi as soon as possible.
Kidney stone removal
The technique used for kidney stone removal depends on the location and size of the kidney stone. About 10-20% of all kidney stones need radiological or surgical intervention to remove the stone.
A urologist can remove the kidney stone or break it into small pieces with the following treatments:
- Shock wave lithotripsy also called extracorporeal shock wave lithotripsy (ESWL). The doctor can use extracorporeal shock wave lithotripsy (ESWL) to blast the kidney stone into small pieces. The smaller pieces of the kidney stone then pass through your urinary tract. A doctor can give you anesthesia during this outpatient procedure. Extracorporeal shock wave lithotripsy (ESWL) uses sound waves to create strong vibrations (shock waves) that break the stones into tiny pieces that can be passed in your urine. The procedure lasts about 45 to 60 minutes and can cause moderate pain, so you may be under sedation or light anesthesia to make you comfortable. ESWL can cause blood in the urine, bruising on the back or abdomen, bleeding around the kidney and other adjacent organs, and discomfort as the stone fragments pass through the urinary tract.
- Cystoscopy and ureteroscopy. During cystoscopy, the doctor uses a cystoscope to look inside the urethra and bladder to find a stone in your urethra or bladder. During ureteroscopy, the doctor uses a ureteroscope, which is longer and thinner than a cystoscope, to see detailed images of the lining of the ureters and kidneys. The doctor inserts the cystoscope or ureteroscope through the urethra to see the rest of the urinary tract. Once the stone is found, the doctor can remove it or break it into smaller pieces. The doctor performs these procedures in the hospital with anesthesia. You can typically go home the same day.
- Percutaneous nephrolithotomy (PCNL). Percutaneous nephrolithotomy (PCNL) involves using a thin telescopic instrument called a nephroscope to locate and remove the kidney stone. The doctor inserts the nephroscope directly into your kidney through a small cut (incision) made in your back. The stone is either pulled out or broken into smaller pieces using a laser or pneumatic energy. For larger kidney stones, the doctor also may use a laser to break the kidney stones into smaller pieces. Your doctor may then place a small tube (stent) in the ureter to help urine flow or a stone to pass and to relieve swelling and promote healing. Percutaneous nephrolithotomy (PCNL) is always carried out under general anaesthetic, where you’re asleep. You may have to stay in the hospital for several days after the procedure. Once the kidney stone is removed, your doctor sends the kidney stone or its pieces to a lab to find out what type it is. Your doctor may recommend percutaneous nephrolithotomy (PCNL) if extracorporeal shock wave lithotripsy (ESWL) is not successful.
Your doctor may also ask you to collect your urine for 24 hours after the kidney stone has passed or been removed. Your doctor can then measure how much urine you produce in a day, along with mineral levels in your urine. You are more likely to form stones if you don’t make enough urine each day or have a problem with high mineral levels.
For symptomatic calculi < 1 cm in diameter in the renal collecting system or proximal ureter, extracorporeal shock wave lithotripsy (ESWL) is a reasonable first option for therapy.
For larger calculi or if extracorporeal shock wave lithotripsy (ESWL) is unsuccessful, ureteroscopy (done in a retrograde fashion) with holmium laser lithotripsy is usually used. Sometimes removal is possible using an endoscope inserted anterograde through the kidney.
For renal stones > 2 cm, percutaneous nephrolithotomy (PCNL) , with insertion of a nephroscope directly into the kidney, is the treatment of choice.
For midureteral calculi, ureteroscopy with holmium laser lithotripsy is usually the treatment of choice. Shock wave lithotripsy is an alternative.
For distal ureteral calculi, endoscopic techniques (ureteroscopy), such as direct removal and use of intracorporeal lithotripsy (eg, pneumatic, electrohydraulic, laser), are considered by many to be the procedures of choice. Shock wave lithotripsy can also be used.
Kidney stone dissolution
Uric acid stones in the upper or lower urinary tract occasionally may be dissolved by prolonged alkalinization of the urine with potassium citrate 20 mEq oral twice to thre times daily, but chemical dissolution of calcium calculi is not possible and of cystine calculi is difficult.
Further Evaluation
Further evaluation identifies modifiable risk factors and guides individualized treatment and prevention. The medical history should identify conditions associated with increased risk of kidney stones (e.g., inflammatory bowel disease, bowel surgery, gout, diabetes mellitus, obesity or recent changes in weight, metabolic syndromes, hyperparathyroidism-associated conditions, frequent urinary tract infections, chronic kidney disease) 129. A family medical history should also be obtained.
A medication history establishes temporal associations; identifies medications recently discontinued, off-label use of medications, and use of herbal preparations and supplements; and screens for illicit drug use. Medications contribute to kidney stones (Table 3) through various mechanisms by forming urine crystals and altering urine characteristics (e.g., changing urine pH, reducing urine volume) 130, 131. For example, carbonic anhydrase inhibitors contribute to calcium phosphate stone formation by causing a mild systemic acidosis and paradoxically high urine pH, hypercalciuria, and low urine citrate 132, 133. Some antibiotics may increase urine oxalate by reducing the intestinal bacteria that break down oxalate.
Evaluation
- A clinician should perform a screening evaluation consisting of a detailed medical and dietary history, serum chemistries and urinalysis on a patient newly diagnosed with kidney or ureteral stones.
- Clinicians should obtain serum intact parathyroid hormone (PTH) level as part of the screening evaluation if primary hyperparathyroidism is suspected.
- When a stone is available, clinicians should obtain a stone analysis at least once.
- Clinicians should obtain or review available imaging studies to quantify stone burden.
- Clinicians should perform additional metabolic testing in high-risk or interested first-time stone formers and recurrent stone formers.
- Metabolic testing should consist of one or two 24-hour urine collections obtained on a random diet and analyzed at minimum for total volume, pH, calcium, oxalate, uric acid, citrate, sodium, potassium and creatinine.
- Clinicians should not routinely perform “fast and calcium load” testing to distinguish among types of hypercalciuria.
Kidney Stone Diet
- All kidney stone formers should drinking enough water to achieve a urine volume of at least 2.5 liters daily is the most important thing you can do to prevent kidney stones. Unless you have kidney failure, many health care professionals recommend that you drink six to eight, 8-ounce (237.2 ml) glasses a day. Talk with a health care professional about how much liquid you should drink.
- Studies have shown that being overweight increases your risk of kidney stones. A dietitian can help you plan meals to help you lose weight.
- Studies have shown that the Dietary Approaches to Stop Hypertension (DASH) diet can reduce the risk of kidney stones 109.
- If you have already had kidney stones, ask your health care professional which type of kidney stone you had. Based on the type of kidney stone you had, you may be able to prevent kidney stones by making changes in how much sodium, animal protein, calcium, or oxalate is in the food you eat. You may need to change what you eat and drink for these types of kidney stones:
- Calcium Oxalate Stones
- Calcium Phosphate Stones
- Uric Acid Stones
- Cystine Stones
- A dietitian who specializes in kidney stone prevention can help you plan meals to prevent kidney stones. Search for credentialed nutrition and dietetics practitioners by location, specialty, language or insurance and payment options here (https://www.eatright.org/find-a-nutrition-expert).
Calcium Oxalate Stones
- Reduce oxalate. If you’ve had calcium oxalate stones, you may want to avoid these foods to help reduce the amount of oxalate in your urine:
- nuts and nut products
- peanuts—which are legumes, not nuts, and are high in oxalate
- rhubarb
- spinach
- wheat bran
- Talk with a dietitian or health care professional about other food sources of oxalate and how much oxalate should be in what you eat.
- Reduce sodium. Your chance of developing kidney stones increases when you eat more sodium. Sodium is a part of salt. Sodium is in many canned, packaged, and fast foods. It is also in many condiments, seasonings, and meats. Most Americans consume too much sodium. Talk with a health care professional about how much sodium should be in what you eat. Adults should aim to consume less than 2,300 mg a day 134. One teaspoon of table salt has 2,325 milligrams (mg) of sodium. If you have had calcium oxalate or calcium phosphate stones, you should follow this guideline, even if you take medicine to prevent kidney stones.
- Check labels for ingredients and hidden sodium, such as:
- sodium bicarbonate, the chemical name for baking soda
- baking powder, which contains sodium bicarbonate and other chemicals
- disodium phosphate
- monosodium glutamate, or MSG
- sodium alginate
- sodium nitrate or nitrite
- Here are some tips to help you reduce your sodium intake:
- Check the Percent Daily Value (%DV) for sodium on the Nutrition Facts label found on many foods. Low in sodium is 5% or less, and high in sodium is 20% or more.
- Consider writing down how much sodium you consume each day.
- When eating out, ask about the sodium content in the food.
- Cook from scratch. Avoid processed and fast foods, canned soups and vegetables, and lunch meats.
- Look for foods labeled: sodium free, salt free, very low sodium, low sodium, reduced or less sodium, light in sodium, no salt added, unsalted, and lightly salted.
- Check labels for ingredients and hidden sodium, such as:
- Limit animal protein. Eating animal protein may increase your chances of developing kidney stones. Although you may need to limit how much animal protein you eat each day, you still need to make sure you get enough protein. Consider replacing some of the meat and animal protein you would typically eat with beans, dried peas, and lentils, which are plant-based foods that are high in protein and low in oxalate. Talk with a health care professional about how much total protein you should eat and how much should come from animal or plant-based foods.
- A health care professional may tell you to limit eating animal protein, including:
- beef, chicken, and pork, especially organ meats
- eggs
- fish and shellfish
- milk, cheese, and other dairy products
- A health care professional may tell you to limit eating animal protein, including:
- Get enough calcium from foods. Even though calcium sounds like it would be the cause of calcium stones, it’s not. In the right amounts, calcium can block other substances in your digestive tract that may cause stones. Talk with a health care professional about how much calcium you should eat to help prevent getting more calcium oxalate stones and to support strong bones. It may be best to get calcium from low-oxalate, plant-based foods such as calcium-fortified juices, cereals, breads, some kinds of vegetables, and some types of beans. Ask a dietitian or other health care professional which foods are the best sources of calcium for you.
Calcium Phosphate Stones
- Reduce sodium. Your chance of developing kidney stones increases when you eat more sodium. Sodium is a part of salt. Sodium is in many canned, packaged, and fast foods. It is also in many condiments, seasonings, and meats. Most Americans consume too much sodium. Talk with a health care professional about how much sodium should be in what you eat. Adults should aim to consume less than 2,300 mg a day 134. One teaspoon of table salt has 2,325 milligrams (mg) of sodium. If you have had calcium oxalate or calcium phosphate stones, you should follow this guideline, even if you take medicine to prevent kidney stones.
- Check labels for ingredients and hidden sodium, such as:
- sodium bicarbonate, the chemical name for baking soda
- baking powder, which contains sodium bicarbonate and other chemicals
- disodium phosphate
- monosodium glutamate, or MSG
- sodium alginate
- sodium nitrate or nitrite
- Here are some tips to help you reduce your sodium intake:
- Check the Percent Daily Value (%DV) for sodium on the Nutrition Facts label found on many foods. Low in sodium is 5% or less, and high in sodium is 20% or more.
- Consider writing down how much sodium you consume each day.
- When eating out, ask about the sodium content in the food.
- Cook from scratch. Avoid processed and fast foods, canned soups and vegetables, and lunch meats.
- Look for foods labeled: sodium free, salt free, very low sodium, low sodium, reduced or less sodium, light in sodium, no salt added, unsalted, and lightly salted.
- Check labels for ingredients and hidden sodium, such as:
- Limit animal protein. Eating animal protein may increase your chances of developing kidney stones. Although you may need to limit how much animal protein you eat each day, you still need to make sure you get enough protein.
- Consider replacing some of the meat and animal protein you would typically eat with these plant-based foods that are high in protein:
- legumes such as beans, dried peas, lentils, and peanuts
- soy foods, such as soy milk, soy nut butter, and tofu
- nuts and nut products, such as almonds and almond butter, cashews and cashew butter, walnuts, and pistachios
- sunflower seeds
- A health care professional may tell you to limit eating animal protein, including:
- beef, chicken, and pork, especially organ meats
- eggs
- fish and shellfish
- milk, cheese, and other dairy products
- Consider replacing some of the meat and animal protein you would typically eat with these plant-based foods that are high in protein:
- Get enough calcium from foods. Even though calcium sounds like it would be the cause of calcium stones, it’s not. In the right amounts, calcium can block other substances in the digestive tract that may lead to stones. Talk with a health care professional about how much calcium you should eat to help prevent getting more calcium phosphate stones and to support strong bones. It may be best to get calcium from plant-based foods such as calcium-fortified juices, cereals, breads, some kinds of vegetables, and some types of beans. Ask a dietitian or other health care professional which foods are the best sources of calcium for you.
Uric Acid Stones
- Limit animal protein. Eating animal protein may increase your chances of developing kidney stones. Although you may need to limit how much animal protein you eat each day, you still need to make sure you get enough protein. Talk with a health care professional about how much total protein you should eat and how much should come from animal or plant-based foods.
- Consider replacing some of the meat and animal protein you would typically eat with some of these plant-based foods that are high in protein:
- legumes such as beans, dried peas, lentils, and peanuts
- soy foods, such as soy milk, soy nut butter, and tofu
- nuts and nut products, such as almonds and almond butter, cashews and cashew butter, walnuts, and pistachios
- sunflower seeds
- A health care professional may tell you to limit eating animal protein, including:
- beef, chicken, and pork, especially organ meats
- eggs
- fish and shellfish
- milk, cheese, and other dairy products
- Consider replacing some of the meat and animal protein you would typically eat with some of these plant-based foods that are high in protein:
- Losing weight. Losing weight if you are overweight is especially important for people who have had uric acid stones.
Cystine Stones
Drinking enough liquid, mainly water, is the most important lifestyle change you can make to prevent cystine stones. Talk with a health care professional about how much liquid you should drink.
Medications to prevent future kidney stones
If you have had a kidney stone, your doctor also may prescribe medicines to prevent future kidney stones. Medications can control the amount of minerals and salts in your urine and may be helpful in people who form certain kinds of stones. The type of medication your doctor prescribes will depend on the kind of kidney stones you have and you may have to take the medicine for a few weeks, several months, or longer. For example, if you had struvite stones, you may have to take an oral antibiotic for 1 to 6 weeks, or possibly longer. If you had another type of stone, you may have to take a potassium citrate tablet 1 to 3 times daily. You may have to take potassium citrate for months or even longer until a health care professional says you are no longer at risk for kidney stones.
Here are some examples:
- Calcium stones. To help prevent calcium stones from forming, your doctor may prescribe a thiazide diuretic often called water pills to help rid your body of water, a phosphate-containing preparation or potassium citrate, which is used to raise the citrate and pH levels in urine.
- Uric acid stones. Your doctor may prescribe allopurinol (Zyloprim, Aloprim) to reduce uric acid levels in your blood and urine and a medicine to keep your urine alkaline. In some cases, allopurinol and an alkalizing agent such as potassium citrate may dissolve the uric acid stones.
- Struvite stones. To prevent struvite stones, your doctor may recommend strategies to keep your urine free of bacteria that cause infection, including drinking fluids to maintain good urine flow and frequent voiding. In rare cases long-term use of antibiotics in small or intermittent doses may help achieve this goal. Acetohydroxamic acid, a strong antibiotic, used with another long-term antibiotic medication to prevent infection. For instance, your doctor may recommend an antibiotic before and for a while after surgery to treat your kidney stones.
- Cystine stones. Along with suggesting a diet lower in salt and protein, your doctor may recommend that you drink more fluids so that you produce a lot more urine,. If that alone doesn’t help, your doctor may also prescribe a medication called mercaptopropionyl glycine (tiopronin) that increases the solubility of cystine in your urine or potassium citrate to raise the pH levels of your urine.
Talk with your doctor about your health history prior to taking kidney stone medicines. Some kidney stone medicines have minor to serious side effects. Side effects are more likely to occur the longer you take the medicine and the higher the dose. Tell your doctor about any side effects that occur when you take kidney stone medicine.
Table 5. Medications to prevent future kidney stones
| Medication | Rationale | Dose | Specifics/side effects | Monitoring |
|---|---|---|---|---|
| Calcium oxalate stones | ||||
| Thiazide | Hypercalciuria | Hydrochlorothiazide 25–50 mg BID, chlorthalidone 25–50 mg/day, indapamide 1.25–5 mg/day | Hypokalemia, hyperlipidemia, hyperuricemia, hyperglycemia, hypocitraturia, hyperuricosuria, fatigue, erectile dysfunction | basic metabolic profile, uric acid, lipid profile |
| Potassium citrate (oral) | Hypocitraturia, low urine pH | 10–30 mEq BID | gastrointestinal side effects | Serum creatinine & potassium |
| Potassium citrate (liquid) | Enteric hyperoxaluria, chronic diarrhea | 15–30 mEq TID–QID (titrate to reduce oxalate) | gastrointestinal side effects, take with two largest meals | Serum creatinine & potassium |
| Allopurinol | Hyperuricosuria | 100–300 mg/day | Hypertransaminasemia, Stevens–Johnson syndrome | Liver enzymes |
| Uric acid stones | ||||
| Potassium citrate (oral) a | Alkalinization | 10–30 mEq BID (titrate dose to pH 6–6.5) | gastrointestinal side effects | Serum creatinine & potassium |
| Sodium bicarbonate | Alkalinization | 650 mg BID–QID | Increased sodium load may increase risk of calcium stones | basic metabolic profile |
| Allopurinol | Hyperuricosuria 2nd line therapy when alkalinization not successful | 100–300 mg/day | Hypertransaminasemia, Stevens–Johnson syndrome | Liver enzymes |
| Cystine stones | ||||
| Tiopronin (α-mercaptopropionyl glycine) | Increase cystine solubility | Initial 400 mg/day titrate to effect | Hematologic effects, tachyphylaxis, proteinuria, nausea, diarrhea, vitamin B6 deficiency (long-term use) | complete blood count, basic metabolic profile, urine protein |
| Potassium citrate (oral) | Alkalinization | 10–30 mEq BID (titrate dose to pH 7–7.5) | gastrointestinal side effects | Serum creatinine & potassium |
| Struvite stones | ||||
| Acetohydroxamic acid | Urease-inhibitor | 250 mg BID–TID | Headache, anemia, thrombophlebitis, rash, tremulousness | complete blood count |
Footnote: a First-line therapy.
[Source 54 ]Calcium stones
Thiazides
Thiazide diuretics are recommended by both the American Urological Association and European Association of Urology guidelines for patients with recurrent calcium-based stones and hypercalciuria 135, 59. Thiazides enhance calcium reabsorption directly in the distal renal tubule and indirectly in the proximal renal tubule. A meta-analysis of six randomized controlled trials (RCTs) found that thiazides were associated with a 47% relative risk reduction in stone recurrence rates compared with placebo or no treatment 136. The benefit of thiazides may additionally extend to normocalciuric patients since the proportion of patients with documented hypercalciuria in these randomized controlled trials was variable, and indeed in some trials the metabolic background of patients was unknown.
Thiazide doses and regimens tested in randomized controlled trials include hydrochlorothiazide 25 mg twice daily, chlorthalidone 25 mg daily or indapamide 2.5 mg daily. Interestingly, one study noted that only 35% of patients treated with thiazides were prescribed a dose shown in randomized controlled trials to be beneficial for stone prevention 137. The most common side effect of thiazides is hypokalemia, which may lead to hypocitraturia due to intracellular acidosis. As such, potassium supplementation is recommended to prevent hypokalemia, to avoid increased cardiovascular risk and to minimize glucose intolerance. The choice of potassium citrate versus potassium chloride to prevent thiazide-induced hypokalemia should be based on initial or follow-up urine pH and citrate; if either is low, potassium citrate may be the preferred potassium supplement.
Because hypercalciuria has been shown to be a risk factor for osteoporosis, a multidisciplinary panel of stone experts authored a consensus paper in which they recommended that stone formers at increased risk of bone loss undergo measurement of bone mineral density by dual emission X-ray absorptiometry (DXA) 138. The fracture risk associated with stone disease varies by skeletal site and demonstrates a higher risk of incident wrist fracture, but not hip fracture, in women and men 139. The use of thiazides in hypercalciuric men was shown to reduce urinary calcium and improve bone mineral density in a short-term study 140. Likewise, Pak and colleagues 141 observed significant increases in bone mineral density at the lumbar spine, femoral neck and radial shaft in 28 recurrent idiopathic hypercalciuric stone formers treated with thiazides and potassium citrate for a mean of 3.7 years.
Potassium citrate
Potassium citrate is used to correct hypocitraturia, which occurs alone or in combination with other abnormalities in about 10%–60% of calcium stone formers 142. Barcelo and coworkers 143 showed a benefit of potassium citrate therapy among 57 recurrent, hypocitraturic calcium stone formers randomized to receive either 30–60 mEq of potassium citrate daily or placebo in a 3-year trial assessing stone remission rates. Patients in the potassium citrate group had a significantly higher remission rate compared to the placebo group (72% vs. 20%, respectively). Potassium citrate has also been shown to improve stone remission rates after shock wave lithotripsy. A recent meta-analysis evaluated the efficacy of potassium citrate in reducing stone recurrence rates 12 months after shock wave lithotripsy in four trials comprising 374 participants 144. A significantly lower stone recurrence rate was seen 12 months after shock wave lithotripsy in the potassium citrate group compared to the control group. Finally, patients with hypocitraturia due to distal renal tubular acidosis have also been shown to benefit from potassium citrate therapy, as the alkali load corrects the metabolic acidosis, increases urinary citrate excretion and reduces stone recurrence rates 145.
Potassium citrate may also reduce urinary calcium excretion as an additional benefit of treatment. Song and associates 146 observed a mean decrease in urine calcium of 30% among 22 hypocitraturic calcium oxalate stone formers receiving 30–60 mEq of potassium citrate daily for at least 3 months. They speculated that the reduction in urinary calcium may be accounted for by a decrease in bone turnover due to systemic alkalinization, by binding of calcium by citrate in the intestine and reducing calcium absorption or as a result of a direct effect on the distal renal tubule affecting calcium reabsorption 146.
The optimal dose and formulation of potassium citrate remains to be determined 102. However, potassium citrate is usually administered as a sustained-release wax matrix tablet in doses ranging from 10–30 mEq twice daily. However, for patients with chronic diarrhea, use of a liquid formulation is advised because the rapid intestinal transit in these patients precludes sufficient absorption of sustained-release potassium citrate 147.
The most common side effects of potassium citrate are gastrointestinal upset, abdominal pain, and diarrhea. Taking the medication with meals may prevent these symptoms in some patients. Periodic monitoring with serum electrolytes and creatinine is recommended. For patients at risk of hyperkalemia or those who are unable to tolerate potassium citrate, sodium alkali such as sodium bicarbonate or a combination of sodium citrate and citric acid may be considered, although the sodium load conferred by these medications may induce hypercalciuria.
Allopurinol
Hyperuricosuria is seen in approximately 20% of patients with calcium stones 148. Although dietary restriction of animal protein is recommended as first line therapy in patients with hyperuricosuria, allopurinol, a xanthine oxidase inhibitor that lowers serum and urinary uric acid, is indicated in patients who are unable to normalize urinary uric acid with diet alone or in those with a genetic predisposition to hyperuricosuria. Allopurinol may additionally have antioxidant effects that contribute to the prevention of calcium stone formation, independent of xanthine oxidase inhibition 149. Among four randomized controlled trials evaluating the effect of allopurinol in recurrent calcium oxalate stone formers, the only trial to show a significant benefit in reducing recurrence rates is one in which only hyperuricosuric, normocalciuric patients were enrolled 150.
Allopurinol is generally well tolerated but may, in rare cases, be associated with Stevens–Johnson syndrome. Any patient reporting a rash with allopurinol should be instructed to discontinue the medication immediately. Hypertransaminasemia is another side effect that is generally reversible with discontinuation of the medication. Liver enzymes should be monitored soon after starting allopurinol and periodically thereafter 151.
Pyridoxine (vitamin B6)
Nondietary urinary oxalate is derived from the conversion of glyoxylate to oxalate by the enzyme lactate dehydrogenase 152, 153. Pyridoxine, a component of vitamin B6, is a necessary cofactor for alanine:glyoxyalate aminotransferase (AGT) which is involved in the alternate metabolic conversion of glyoxylate to glycine. Patients with primary hyperoxaluria type 1 have a deficiency in AGT by which they are unable to divert glyoxylate metabolism to the alternate pathway. While the only curative treatment for this disease is liver transplantation, pyridoxine can be used to decrease urinary oxalate. A prospective, uncontrolled open label trial in which 12 patients with primary hyperoxaluria type I were given pyridoxine incrementally up to a dosage of 20 mg/kg per day showed a 25.5% mean relative reduction in urinary oxalate, with benefit seen in 50% of patients 154.
Pyridoxine (vitamin B6) has been proposed as a preventative treatment in patients with idiopathic hyperoxaluria as well. One retrospective study evaluating the effect of pyridoxine on urinary oxalate in 95 idiopathic hyperoxaluric stone formers found that 75% of patients treated with pyridoxine and dietary counseling versus only 52% of patients treated with dietary counseling alone showed a reduction in urinary oxalate 155. On the other hand, three large cohort studies (HPFS, NHS I, NHS II) found no association between vitamin B6 intake and risk of incident kidney stones 156. The American Urological Association Medical Management of Kidney Stones Guideline found insufficient evidence to recommend pyridoxine for the management of patients with idiopathic hyperoxaluria 135. However if it is used, pyridoxine should be started at a low dose and titrated in a stepwise fashion to no more than 200 mg/day 157. Doses excessing 500 mg/day should be avoided because of the risk of developing severe sensory neuropathy.
Uric acid stones
The prevention of uric acid stone formation involves the administration of alkali to increase urine pH. A target pH of 6–6.5 can effectively prevent and dissolve uric acid stones 158. Potassium citrate, titrated to achieve the target pH, is first-line therapy for uric acid stone formers, with sodium alkali (sodium bicarbonate) reserved for those unable to tolerate potassium citrate or for whom renal dysfunction or hyperkalemia precludes its use. Of note, even large amounts of uric acid are soluble if the urine pH is sufficiently high; in contrast, even small amounts of uric acid will precipitate out in acidic urine. As such, allopurinol should not constitute first line therapy for idiopathic uric acid stone formers and is reserved only for patients who continue to form uric acid stones despite adequate urinary alkalinization 135. In addition to pharmacologic treatment to prevent uric acid stones, lifestyle changes such as weight loss and exercise should be recommended by physicians for patients with metabolic syndrome.
Dissolution therapy for uric acid stones is also achievable with alkali therapy using a target urine pH of 6–6.5. However, the success of dissolution therapy relies on accurate diagnosis of pure uric acid stones. One group of investigators determined that computed tomography (CT) Hounsfield units ≤500, pH ≤ 5.5 and stone size > 4 mm on non-enhanced CT had an 86% sensitivity, 98% specificity, and 90% positive predictive value for diagnosing uric acid stones 159. In an in vivo study of 30 patients, dual energy CT was shown to have a 100% positive predictive value for detecting uric acid stones 160. Patients with 100% pure uric acid stones tend to be older (60 vs. 55 years), heavier (34.3 vs. 29 kg/m²), have higher serum uric acids (6.53 vs. 5.89 mg/dL) and have lower pH (5.62 vs. 5.89) than those with 10%–20% uric acid stone composition 161. Combining these demographics with stone size and Hounsfield units on CT scan may contribute to better diagnostic accuracy in identifying patients with pure uric acid stones who may be successfully treated with dissolution therapy. Complete dissolution may occur in as little as 6 weeks to 6 months or more 162, 163, likely due to the varying percentages of uric acid stone composition 161.
Cystine stones
The therapeutic goal in treating patients with cystinuria is to reduce cystine concentration or raise cystine solubility above the solubility limit of 250 mg/L. Reducing cystine concentration can be accomplished with increased fluid intake, typically enough fluid intake to produce a urine volume of at least 3 L/day, or by reducing cystine excretion. Restriction of sodium and animal protein have been shown in randomized trials to decrease urinary cystine excretion 164, 165, 166. Like all stone formers, cystinuric patients are advised to limit their sodium intake to less than 2300 mg/day (100 mEq/day) 135.
Cystine solubility is influenced by urine pH and the presence of urinary macromolecules. Because cystine solubility increases significantly only above pH 7.5 and most cystinuric patients have high urine pH at baseline, urinary alkalinization has a limited therapeutic role, but constitutes second line therapy after diet and fluids, with the goal of treatment to achieve a urine pH of 7.0–7.5. At pH > 7, the risk of calcium phosphate stone formation becomes more pronounced, and repeat stone analysis, if one is available during treatment, should be pursued 167, 168. Potassium alkali (potassium citrate) is preferred over sodium alkali for urinary alkalinization because sodium enhances urinary cystine excretion.
For more severe cystinuria, cystine-binding thiol drugs (CBTD) constitute third line therapy 169, 170. Agents most commonly used include α-mercaptopropionyl glycine (tiopronin) and d-penicillamine. Thiol compounds contain sulfhydryl groups that undergo a disulfide exchange reaction with cystine to produce two molecules of cysteine bound to the cystine-binding thiol drugs (CBTD), a complex that is 50 times more soluble than cystine. The effect of the drugs is dose-dependent.
Tiopronin, as a second generation cystine-binding thiol drugs (CBTD), is considered the first line drug, with dosing starting at 200 mg two or three times daily and titrated to achieve a urine cystine concentration of less than 250 mg/L. Adverse effects include fever, gastrointestinal upset, asthenia, rash, joint aches, loss of taste, thrombocytopenia, aplastic anemia, proteinuria, and changes in mental status 171.
d-penicillamine has a more extensive side effect profile than tiopronin and therefore is used much less commonly 172. Dosing typically starts at 250 mg two or three times daily and is titrated to effect. One study reported adverse effects in 65% of patients taking tiopronin compared with 84% of those taking d-penicillamine 173. Adverse reactions necessitating cessation of treatment were also less common with tiopronin (31% vs. 69%, respectively). Long-term therapy with d-penicillamine may lead to pyridoxine (vitamin B6) deficiency, which may require oral supplementation (50 mg/day).
Captopril is a third generation angiotensin-converting-enzyme inhibitor that also contains a free sulfhydryl group and has been shown in vitro to increase cystine solubility 174. However, the recommended doses of captopril in vivo are not thought to be sufficient to induce a therapeutic effect on cystine stone formation and the drug has not been tested in rigorous clinical trials.
Recently, a widely available nutritional supplement, alpha-lipoic acid, has been investigated as a treatment for cystinuria 175. Using the SLC3A1−/− mouse model which grows urinary bladder stones at a rate of 1 mm³/day, treatment with alpha-lipoic acid was found to not only attenuate growth of existing cystine stones but also prevent initiation of new stone formation. The protective effect is thought to be derived from increased urinary cystine solubility due to excretion of downstream α-lipoic acid metabolites into the urine. Clinical trials are necessary to prove its efficacy in humans.
Although traditionally the goal of treatment has been to reduce urinary cystine concentration below the solubility level 169, most assays are unable to distinguish free cystine from soluble thiol drug-cysteine complexes. A proprietary test, cystine capacity (Litholink Corporation, Chicago, IL, USA), has been shown to reliably estimate stone forming propensity even in the presence of cystine-binding thiol drugs (CBTD) and offers an alternative means of monitoring therapeutic efficacy 176, 177.
Struvite stones
Struvite stones form as a consequence of repeated infection with urease-producing organisms, leading to alkaline urine and precipitation of magnesium ammonium phosphate crystals. Because these stones incorporate bacteria inside them, aggressive surgical stone removal is essential to eradicate the bacteria and prevent recurrent infections and stone. Culture-specific antibiotic therapy is recommended both pre-operatively and for some time post-operatively to sterilize the urine and prevent re-infection. A recent multi-institutional study on patterns of infection and colonization of struvite stones revealed that the bacteriology has shifted away from traditional urea-splitting organisms such as Proteus to Enterococcus and Escherichia coli 178. The high prevalence of infection with Enterococcus (18%) suggests that 1st and 2nd generation cephalopshorins may be ineffective and antibiotic coverage should include agents appropriate for Enterococcus. Nonetheless, there is a lack of evidence on the role of antibiotics in preventing stone growth/recurrence and the optimal duration of therapy.
Acetohydroxamic acid (AHA), a potent urease inhibitor, decreases the risk of struvite stone formation by preventing bacterial-induced urease from altering the urinary milieu. Acetohydroxamic acid has been shown in a number of randomized controlled trials to reduce stone growth and prolong the time interval to stone growth 179, 180, 181. However, adverse events are common, leading to high attrition rates. Side effects include tremor, palpitations, edema, proteinuria, headache, rash, alopecia, anemia, gastrointestinal discomfort, and thromboembolic phenomena. In light of these limitations, acetohydroxamic acid, along with suppressive antibiotics, are reserved for patients at high risk of recurrent struvite stone formation and/or those unable to undergo surgical stone removal. Using a lower dose of 250 mg twice daily has been proposed to reduce the overall adverse events, although deep vein thrombosis/pulmonary embolus was still prevalent 182.
Follow-up
The success of a medical prophylactic program hinges on improvement in urinary stone risk factors and ultimately on reduction in stone recurrence rate. By monitoring 24 hour urine parameters, a change in stone risk might be anticipated and corrected before stones recur. The frequency of follow-up depends on the metabolic activity of the individual. Periodic imaging can detect failure of medical therapy and prompt early surgical treatment and modulation of the medical regimen. In addition, follow-up should include blood and urine testing for adverse effects of treatment, such as hypokalemia with thiazides or proteinuria with α-mercaptopropionyl glycine (tiopronin).
- Clinicians should obtain a single 24-hour urine specimen for stone risk factors within six months of the initiation of treatment to assess response to dietary and/or medical therapy.
- After the initial follow-up, clinicians should obtain a single 24-hour urine specimen annually or with greater frequency, depending on stone activity, to assess patient adherence and metabolic response.
- Clinicians should obtain periodic blood testing to assess for adverse effects in patients on pharmacological therapy.
- Clinicians should obtain a repeat stone analysis, when available, especially in patients not responding to treatment.
- Clinicians should monitor patients with struvite stones for reinfection with urease-producing organisms and utilize strategies to prevent such occurrences.
- Clinicians should periodically obtain follow-up imaging studies to assess for stone growth or new stone formation based on stone activity (plain abdominal imaging, renal ultrasonography or low dose computed tomography [CT scan]).
Kidney stones prognosis
Kidney stones are painful, but most of the time can be removed from the body without causing lasting damage. Kidney stones that do not pass can become obstructive and can subsequently cause acute renal failure, or it can also become a nidus for infection, which can eventually be lethal.
Kidney stones often come back. This occurs more often if the cause is not found and treated. You are at risk for:
- Urinary tract infection
- Kidney damage or scarring if treatment is delayed for too long
Kidney stones have been associated with an increased risk of chronic kidney diseases 183, end-stage renal failure 184, cardiovascular diseases such as heart attack 185, 186, diabetes, and hypertension 187. It has been suggested that kidney stone may be a systemic disorder linked to the metabolic syndrome. Kidney stone is responsible for 2 to 3% of end-stage renal cases if it is associated with nephrocalcinosis 188.
If the patient undergoes nephrostomy tube placement, then there is a chance of bleeding, renal collecting system injury, injury of visceral organs, pulmonary complications, thromboembolic complications, and extrarenal stone migration 189.
What is Renal Tubular Acidosis
Renal tubular acidosis (RTA) is a general term for when your kidneys cannot properly remove acid from your body. Renal tubular acidosis (RTA) occurs when your kidneys are unable to adequately reclaim filtered bicarbonate [HCO3–] (a base) or excrete sufficient hydrogen ions (H+) because of defects in tubular transport 190, 191. The acid level in your blood then becomes too high causing your blood pH to fall below 7.35, a condition called acidosis. Some acid in the blood is normal, but too much acid can disturb many bodily functions. There are three main types of renal tubular acidosis that are characterized by: 1) a normal anion gap metabolic acidosis; 2) abnormalities in renal bicarbonate (HCO3-) absorption or new renal bicarbonate (HCO3-) generation; 3) changes in renal ammonium (NH4+), calcium (Ca2+), potassium (K+) and water (H2O) homeostasis; and 4) extrarenal manifestations that provide etiologic diagnostic clues 192, 193, 194.
The kidneys contain nephrons, which are hair-sized structures that are the basic filtering units of the kidneys. Each nephron consists of a glomerulus and a renal tubule. The renal tubule reabsorbs electrolytes such as sodium, chloride and potassium back into the blood so that not too much electrolyte is lost through the urine. The kidneys, through the tubules, reclaim bicarbonate, an electrolyte that helps to maintain the acid-base balance in the body, and then excrete acid through the urine. Acid is produced as a byproduct from a normal diet.
There are 3 main types of renal tubular acidosis.
- Type 1 renal tubular acidosis or distal renal tubular acidosis, occurs when there is a problem at the end or distal part of the tubules in the kidneys not being able to rid the body of the daily acid load. This results in an inability to lower urine pH regardless of the degree of acidosis or acidemia (acid level in the blood). Distal refers to being “distant” from the point of origin. In the nephron, it means the defect occurs away from the point where fluid enters the tubule. Distal renal tubular acidosis occurs because the kidneys fail to secrete acids into the urine.
- Type 2 renal tubular acidosis or proximal renal tubular acidosis, occurs when there is a problem in the beginning or proximal part of the tubules in the kidneys.
- Type 3 renal tubular acidosis is rarely used as a classification now because it is thought to be a combination of type 1 and type 2 renal tubular acidosis with features of both distal and proximal renal tubular acidosis.
- Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis, occurs when the tubules are unable to remove enough potassium, which also interferes with the kidney’s ability to remove acid from the blood.
Table 6. Summary of renal tubular acidosis classification, diagnostic characteristics, and treatment options
| Type 1 RTA (Distal renal tubular acidosis) | Type 2 RTA (Proximal renal tubular acidosis) | Type 4 RTA (Hyperkalemic renal tubular acidosis) | |
|---|---|---|---|
| Primary defect | Decreased distal acid excretion or increased H+ membrane permeability | Decreased proximal reabsorption of bicarbonate (HCO3–) | Reduced excretion of acid and K+ in the collecting duct |
| Symptoms | Polydipsia, polyuria, muscle weakness, nephrolithiasis, nephrocalcinosis, growth retardation or failure to thrive, rickets | Muscle weakness or paralysis (if severely hypokalemic), growth retardation in early childhood | Often asymptomatic, occasional muscle weakness of cardiac arrhythmia |
| Urine pH | > 5.3 | < 5.5 | < 5.5 |
| Serum bicarbonate (HCO3-) | 10–20 mmol/L | 16–20 mmol/L | 16–22 mmol/L |
| Serum K+ | Low (< 3.5 mmol/L) | Low (< 3.5 mmol/L) | High (5.5–6.5 mmol/L) |
| Serum anion gap | Normal | Normal | Normal |
| Diagnostic tests | Positive urinary anion gap after ammonium (NH4+) loading test | Fractional excretion of bicarbonate (HCO3–) > 15% or urine pH > 7.5 after bicarbonate (HCO3–) loading test Glycosuria, hypophosphatemia, and hypouricemia indicates Fanconi syndrome | Urinary K+ < 40 mmol/L or fractional K+ excretion < 20%, abnormal serum aldosterone, with near-normal renal function |
| Treatment | |||
| Diet and lifestyle modifications | Increased citrus fruit and fluid intake, restricted intake of Na+, oxalate, fructose, and animal protein, normal Ca2+ intake | Limit acid-based foods (animal source protein), increase alkali-based foods (fruits and vegetables) | Dietary K+ restriction, increase alkali-based foods, limit acid-based foods |
| Pharmacotherapy | Sodium bicarbonate (NaHCO3) or potassium bicarbonate (KHCO3) (1–2 mmol/kg/day), potassium chloride (KCl) or potassium citrate (K-citrate) (in patients with severe hypokalemia) | Alkali therapy (usually K-citrate 10–15 mmol/kg/day), fluids, electrolytes, vitamin D, phosphate, hydrochlorothiazide | Low-dose fludrocortisone, loop diuretics (if fludrocortisone not tolerated), oral NaHCO3 if serum bicarbonate (HCO3–) < 22 mmol/L, K+ binders (patiromer or sodium zirconium cyclosilicate) |
Untreated renal tubular acidosis can affect a child’s growth, cause kidney stones, and other problems like bone or kidney disease. Fortunately, treatment is very helpful at preventing these things from happening. So it’s important to start treatment as soon as renal tubular acidosis (RTA) is diagnosed.
The pH is a number that shows how acidic or alkaline a substance is. A pH of less than 7 is acidic, and greater than 7 is alkaline. The pH of blood is about 7.4. A pH below 7.35 is called acidosis or acidemia. This is very unlikely to occur, as your body has multiple mechanisms for ensuring a very stable blood pH. Acidosis only becomes acidemia, where your blood pH is less than 7.35, when your body’s acid-base compensatory measures or buffering systems become overwhelmed. Your blood pH 7.4 is tightly regulated by your kidneys and respiratory system 195. The primary pH buffering system in the human body is the bicarbonate (HCO3–) and carbon dioxide (CO2). Bicarbonate (HCO3–) functions as an alkalotic substance. Carbon dioxide (CO2) functions as an acidic substance. Therefore, a decrease in serum bicarbonate (HCO3–) or an increase in CO2 (carbon dioxide) will make blood more acidic. The opposite is also true where an increase in bicarbonate (HCO3–) or a decrease in carbon dioxide (CO2) will make blood more alkaline. The carbon dioxide (CO2) levels are physiologically regulated by the pulmonary system through respiration, whereas the serum bicarbonate (HCO3–) levels are regulated through your kidneys by two mechanisms: bicarbonate [HCO3–] (a base) reclamation mainly in the proximal tubule and bicarbonate [HCO3–] (a base) generation predominantly in the distal nephron. Any excess acid is excreted in the urine. Your blood pH is not altered by your dietary intake.
For all types of RTA, drinking a solution of sodium bicarbonate or sodium citrate will lower the acid level in your blood. This alkali therapy can prevent kidney stones from forming and make your kidneys work more normally so kidney failure does not get worse.
Infants with type 1 renal tubular acidosis may need potassium supplements, but older children and adults rarely do because alkali therapy prevents the kidneys from excreting potassium into the urine.
Children with type 2 renal tubular acidosis will also drink an alkali solution (sodium bicarbonate or potassium citrate) to lower the acid level in their blood, prevent bone disorders and kidney stones, and grow normally. Some adults with type 2 RTA may need to take vitamin D supplements to help prevent bone problems.
People with type 4 renal tubular acidosis (hyperkalemic renal tubular acidosis) may need other medicines to lower the potassium levels in their blood.
If your RTA is caused by another condition, your health care professional will try to identify and treat it.
Who is more likely to have renal tubular acidosis?
You are more likely to have type 1 renal tubular acidosis (distal renal tubular acidosis) if you inherit specific genes from your parents or if you have certain autoimmune diseases such as Sjögren’s syndrome or lupus.
If you have Fanconi syndrome or are taking medicines to treat HIV or viral hepatitis, you are more likely to have type 2 RTA (proximal renal tubular acidosis). People who inherit genes for type 2 RTA from their parents may also have it. In adults, type 2 RTA (proximal renal tubular acidosis) can be a complication or side effect of multiple myeloma, exposure to toxins, or certain medications. In rare cases, type 2 RTA occurs in people who experience chronic rejection of a transplanted kidney.
If you have low levels of the hormone aldosterone, cannot urinate freely because of an obstruction, or had a kidney transplant, you are more likely to develop type 4 RTA ( (hyperkalemic renal tubular acidosis)). One in five people develop type 4 RTA if they experience rejection of a transplanted kidney or are taking immunosuppressive medications.
Renal tubular acidosis types
There are three main types of renal tubular acidosis that are characterized by: 1) a normal anion gap metabolic acidosis; 2) abnormalities in renal bicarbonate (HCO3-) absorption or new renal bicarbonate (HCO3-) generation; 3) changes in renal ammonium (NH4+), calcium (Ca2+), potassium (K+) and water (H2O) homeostasis; and 4) extrarenal manifestations that provide etiologic diagnostic clues 192, 193, 194.
- Type 1 renal tubular acidosis or distal renal tubular acidosis, occurs when there is a problem at the end or distal part of the tubules.
- Type 2 renal tubular acidosis or proximal renal tubular acidosis, occurs when there is a problem in the beginning or proximal part of the tubules.
- Type 3 renal tubular acidosis is rarely used as a classification now because it is thought to be a combination of type 1 and type 2 renal tubular acidosis with features of both distal and proximal renal tubular acidosis.
- Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis, occurs when the tubules are unable to remove enough potassium, which also interferes with the kidney’s ability to remove acid from the blood.
Type 1 renal tubular acidosis
Type 1 renal tubular acidosis or distal renal tubular acidosis occurs when there is a problem at the distal part of the tubules. Distal renal tubular acidosis occurs because the kidneys fail to secrete acids into the urine and evidence of kidney K+ wasting with either normal or minimally reduced GFR and persistently alkaline urine pH > 5.3 196. Distal type 1 RTA or classic renal tubular acidosis usually referred to as distal renal tubular acidosis is characterized by a buildup of acids in the blood as a consequence of the distal tubules in the kidneys not being able to rid the body of the daily acid load. This results in an inability to lower urine pH regardless of the degree of acidemia (acid level in the blood). Distal refers to being “distant” from the point of origin. In the nephron, it means the defect occurs away from the point where fluid enters the tubule.
Untreated type 1 renal tubular acidosis (distal renal tubular acidosis) causes children to grow more slowly and adults to develop progressive kidney disease and bone diseases. Adults and children with untreated type 1 RTA may develop kidney stones because of abnormal calcium deposits that build up in the kidneys. These deposits prevent the kidneys from working properly.
Type 1 renal tubular acidosis causes
There are different forms of type 1 renal tubular acidosis (distal renal tubular acidosis). On the basis of the underlying defect, type 1 renal tubular acidosis (distal renal tubular acidosis) may be classified as hereditary (primary) or acquired (secondary) 190.
Type 1 RTA may be inherited, which is also known as primary distal renal tubular acidosis. Researchers have identified at least three different genes that may cause the inherited form of type 1 RTA 197. The inherited type 1 renal tubular acidosis (primary distal renal tubular acidosis) are caused by a variation (mutation) in one of at least three different genes; the SLC4A1 gene, the ATP6V0A4 gene, and the ATP6V1B1 gene. A variation in the SLC4A1 gene is usually inherited in an autosomal dominant pattern, and less often in an autosomal recessive pattern. Variations in the ATP6V0A4 and ATP6V1B1 genes are usually inherited in an autosomal recessive pattern 197. In some affected individuals (~20% cases), no variation in these three genes can be identified suggesting that other, as-yet-unidentified genes can play a role in primary distal renal tubular acidosis 198, 199.
The SLC4A1 gene contains instructions for producing (encoding) a protein called anion exchanger 1 or AE1. This protein helps negatively-charged atoms cross cell membranes; specifically, it helps exchange chlorine ions for bicarbonate ions. Bicarbonate is an electrolyte that helps maintain the acid-base balance in the body and is filtered by the kidneys; but, most of the bicarbonate is still retained in the blood and the urine contains very small amounts. Theanion exchanger 1 (AE1) protein is found in the membranes of kidney cells and red blood cells. The kidneys reclaim filtered bicarbonate and then release acid into the urine to be excreted from the body. Researchers have speculated that a variation in the SLC4A1 gene prevents enough functional anion exchanger 1 (AE1) protein from reaching the cell membranes of kidney and red blood cells. Ultimately, this prevents the kidneys from releasing acid into the urine. Acid then builds up in the blood and tissues of the body (metabolic acidosis). The reason why some people develop metabolic acidosis and others do not is not fully understood. In red blood cells, anion exchanger 1 (AE1) protein cannot reach the red cell membrane resulting in red blood cells that break down prematurely. Some altered AE1 protein can still reach the membranes of red blood cells because it is helped by another protein called glycophorin A. This is most likely why many people with a disease-causing variation in the SLC4A1 gene do not develop hemolytic anemia.
Variations in the SLCA41 gene are usually inherited in an autosomal dominant pattern, and, less often, in an autosomal recessive pattern. Disease-causing variations in the SLC4A1 gene can be inherited from a parent or it can occur as a new (sporadic or de novo) mutation, which means that the gene variation has occurred at the time of the formation of the egg or sperm for that child only, and no other family member will be affected. Affected individuals can then pass on the altered gene in an autosomal dominant pattern.
The ATP6V0A4 and the ATP6V1B1 genes encode specific proteins that are part of a protein complex called vacuolar H+-ATPase (V-ATPase). This protein complex acts as a proton pump that helps to move positively-charged atoms (protons) across cell membranes, and helps to regulate acid levels of cells and their surrounding areas. These proteins are commonly found in cells of the inner ear and within the nephron, which is the basic filtering unit of the kidneys. These proteins have a role in regulating the amount of acid removed from the blood to the urine, and in maintaining the proper acid balance within the ear. Variations in the ATP6V0A4 and the ATP6V1B1 genes are inherited in an autosomal recessive pattern.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child.
Disorders inherited in a recessive pattern occur when an individual inherits two variants in a gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Inherited type 1 RTA (primary distal renal tubular acidosis) affects females and males in equal numbers. The exact number of people who have this disorder is unknown. Rare disorders like primary distal renal tubular acidosis often go misdiagnosed or undiagnosed, making it difficult to determine their true frequency in the general population.
Acquired (secondary) forms of type 1 RTA are caused by autoimmune disorders including Sjögren’s syndrome or systemic lupus erythematosus. Autoimmune disorders are ones in which the body’s immune system mistakenly attacks healthy tissue. Sometimes, in certain autoimmune disorders the immune system attacks the distal portion of the renal tubules. Distal renal tubular acidosis can also occur in people with sickle cell anemia, chronic obstructive uropathy, Ehlers-Danlos syndrome, hypogammaglobulinemia, chronic liver disease, and following a kidney transplant. Acquired forms of type 1 renal tubular acidosis can also be caused by certain medications (e.g., lithium, amphotericin B, NSAIDs, lead, antivirals), including some used for pain and bipolar disorder, conditions causing high calcium in the urine, blocked urinary tract, or rejection of a transplanted kidney.
Diseases and conditions related to type 1 renal tubular acidosis:
- a hereditary form of deafness
- renal medullary cystic disease
- sickle cell disease
- sickle cell anemia
- Ehlers-Danlos syndrome
- urinary tract infections
- amyloidosis, a buildup of abnormal protein, called amyloid, in the tissues and organs
- Fabry disease, an abnormal buildup in the body of a certain type of fatty substance
- High level of calcium in the blood (hypercalcemia)
- Sjögren syndrome, an autoimmune disorder in which the glands that produce tears and saliva are destroyed
- Systemic lupus erythematosus, an autoimmune disease in which the body’s immune system mistakenly attacks healthy tissue
- Wilson disease, an inherited disorder in which there is too much copper in the body’s tissues
- Use of certain medicines, such as amphotericin B, lithium, and nonsteroidal anti-inflammatory drugs (NSAIDs)
Type 1 renal tubular acidosis signs and symptoms
Inherited type 1 RTA (primary distal renal tubular acidosis) is a highly variable disorder; this means that the disorder affects people differently. Some individuals may only have slightly elevated acid levels and no accompanying symptoms (asymptomatic). Some individuals living with type 1 renal tubular acidosis (primary distal renal tubular acidosis) may experience kidney stones and others may not. Generally, people with an autosomal dominant pattern of inheritance have milder symptoms and a later age of onset of symptoms than people with an autosomal recessive pattern of inheritance. However, this is not always true and sometimes more severe complications such as growth failure or rickets (bowing of the bones) can affect individuals with dominantly-inherited primary distal renal tubular acidosis.
Symptoms of type 1 renal tubular acidosis (distal renal tubular acidosis) include any of the following:
- Confusion or decreased alertness
- Fatigue
- Impaired growth in children
- Increased breathing rate
- Kidney stones
- Nephrocalcinosis (too much calcium deposited in the kidneys)
- Osteomalacia (softening of the bones)
- Muscle weakness
Other symptoms may include:
- Bone pain
- Decreased urine output
- Increased heart rate or irregular heartbeat
- Muscle cramps
- Pain in the back, flank, or abdomen
- Skeletal abnormalities
Inherited type 1 RTA (primary distal renal tubular acidosis) can cause severe complications in infants, especially if unrecognized and untreated. Affected infants can experience vomiting, dehydration, and poor growth that can result in being short for their age and gender (short stature). Additional symptoms can include excessive thirst (polydipsia), urinating frequently (polyuria), constipation, muscle weakness, and fatigue. Sometimes, affected individuals may have diminished reflexes. Many of these symptoms are related to metabolic acidosis, a serious and often life-threatening condition. Parents should seek prompt medical attention if a baby shows signs of metabolic acidosis. Some children develop rickets, which is a condition characterized by improper hardening (calcification) of the bones leading to softening and distortion/bowing of the bones and bone pain. If unrecognized and untreated, primary distal renal tubular acidosis usually causes too much calcium to build up in the kidneys (nephrocalcinosis), and the formation of kidney stones (nephrolithiasis). If untreated, nephrocalcinosis can progress to cause damage to the kidneys resulting in chronic kidney disease (CKD) and reduced kidney function.
In severe instances, if untreated, extreme muscle weakness (muscle paralysis), abnormal heartbeats (cardiac arrhythmia), and episodes of having difficulty breathing or stopping breathing (respiratory arrest) can develop. These symptoms are related to low levels of potassium in the blood (hypokalemia). Potassium is an important electrolyte for the health of nerve and muscles. The kidneys excrete excess potassium through the urine. However, in primary distal renal tubular acidosis the kidneys sometimes excrete too much potassium. Hypokalemia may also contribute to excessive urination (polyuria).
A subset of individuals with the autosomal recessive forms develop sensorineural hearing loss. Sensorineural hearing loss occurs when the nerves within the ear cannot properly send sensory input (sound) to the brain, and is not caused by problems with the ear itself. The degree and progression of sensorineural hearing loss can vary from one child to another, but often affects both ears (bilateral) and is usually severe.
Affected individuals with an autosomal dominant type 1 renal tubular acidosis (primary distal renal tubular acidosis) usually experience a milder form of the disorder with onset of symptoms in adolescence or adulthood. Affected adults may develop reduced bone mass (osteopenia) and abnormal softening of the bones (osteomalacia) and bone pain. Weakened bones may be prone to fracture. Some individuals may develop an abnormal increase in red blood cell mass (erythrocytosis). They may develop kidney stones or kidney issues as adolescents or adults if the disorder is unrecognized and untreated.
Occasionally, individuals with an inherited variation in the SLC4A1 gene have experienced the premature breakdown of red blood cells, which leads to low levels of circulating red blood cells (hemolytic anemia). The main function of red blood cells is to deliver oxygen throughout the body. People with hemolytic anemia can experience shortness of breath (dyspnea), lightheadedness, fatigue, weakness, pale skin color, and headaches.
Type 1 renal tubular acidosis treatment
The treatment of inherited type 1 RTA (primary distal renal tubular acidosis) may require the coordinated efforts of a team of specialists. A kidney doctor (nephrologist) who specialize in diagnosing and treating kidney disorders may be a critical member of the care team. A pediatric nephrologist specializes in kidney disorders in children. Physicians who specialize in diagnosing and treating skeletal disorders (orthopedists), an audiologist to monitor hearing and other healthcare professionals may need to systematically and comprehensively help guide treatment.
The goal is to restore normal acid level and electrolyte balance in the body. This will help correct bone disorders and reduce calcium buildup in the kidneys (nephrocalcinosis) and kidney stones.
The underlying cause of distal renal tubular acidosis should be corrected if it can be identified.
Individuals with inherited type 1 RTA (primary distal renal tubular acidosis) are treated with alkali therapy. Alkali are chemical compounds that neutralize acids. Medicines that may be prescribed include potassium citrate, sodium bicarbonate, and thiazide diuretics. A mixture of sodium and potassium salts in the form of sodium citrate or potassium citrate liquid solutions is usually recommended. These are alkaline medicines that help correct the acidic condition of the body. Sodium bicarbonate and potassium supplements may correct the loss of potassium and calcium. Liquid preparations, however, have poor palatability and acceptance among patients. In this case, sodium bicarbonate tablets are used instead or in addition to the drinking solution.
Alkali therapy usually leads to normal growth in children, and can improve other symptoms including lowering the tendency to develop calcium build up in the kidneys and calcium stones and reverse bone disease. Alkali therapy usually consists of drinking a solution of sodium bicarbonate (baking soda) or sodium citrate every day to counteract the acids produced from eating each day. The dose and specific type of alkali therapy depends upon the bicarbonate and potassium concentrations in the blood serum. Children generally require larger doses; these doses are adjusted as a child ages. Most individuals do not experience symptoms (asymptomatic) when properly treated, except for irreversible kidney or skeletal damage that has occurred before treatment was begun.
If low potassium levels persist (hypokalemia), then affected individuals may require treatment with alkylating potassium salt like potassium citrate. Potassium citrate (versus sodium citrate) may also be recommended when calcium stones are present because sodium can increase calcium stone formation. Citrate salts like potassium citrate correct low levels of citrate (hypocitraturia) and prevent calcium stone formation.
The focus of treatment in individuals with severe hypokalemia that causes paralysis or breathing problems (respiratory compromise) should be to correct the low potassium levels with an intravenous potassium chloride.
Children with autosomal recessive primary distal renal tubular acidosis should receive routine hearing assessments through childhood to detect hearing loss. Hearing loss does not usually respond to alkali therapy. Other treatments can include supplementation with vitamin D or oral calcium supplements to help reduce skeletal abnormalities such as rickets or osteomalacia.
Type 2 renal tubular acidosis
Type 2 renal tubular acidosis or proximal renal tubular acidosis occurs when there is a problem in the beginning or proximal part of the tubules. Isolated proximal type 2 RTA is characterized by defects in the reabsorption of filtered bicarbonate (HCO3–) in the proximal tubule without defects in the transport of other solutes (Table 1) 200, 201. The threshold serum concentration for bicarbonate (HCO3–) reabsorption (normally approximately 25 mmol/L) is reduced, leading to delivery of larger quantities of filtered bicarbonate (HCO3–) to the distal nephron (which has a low capacity for HCO3– reabsorption) and urinary bicarbonate (HCO3–) wastage 200, 201. Reductions in serum bicarbonate (HCO3–) cause acidosis; however, the urine pH remains alkaline because of the presence of urinary bicarbonate (HCO3–) 201. When serum bicarbonate (HCO3–) concentrations decrease below the lower threshold (16–20 mmol/L), a new steady state is reached, whereby all filtered bicarbonate (HCO3–) is reabsorbed. At this point, the urine contains no bicarbonate (HCO3–) and is maximally acidic 200.
A diagnosis of proximal renal tubular acidosis (type 2 renal tubular acidosis) may be suspected in patients who present with hypokalemia, normal anion gap metabolic acidosis, and acidic urine (pH < 5.5) 202. Other signs of proximal tubular dysfunction, including hypophosphatemia, hypouricemia, euglycemic glycosuria, and proteinuria, are also consistent with a proximal renal tubular acidosis (type 2 renal tubular acidosis) diagnosis and are reflective of generalized proximal tubular dysfunction 202. Fractional urinary bicarbonate (HCO3–) excretion, which is normally less than 5% of filtered bicarbonate (HCO3–), is usually approximately 15% in patients with proximal proximal renal tubular acidosis (type 2 renal tubular acidosis) 203. Isolated proximal RTA, that is decreased bicarbonate (HCO3–) reabsorption without abnormalities in the transport of other solutes, is rare 201.
The cause of isolated type 2 renal tubular acidosis or proximal renal tubular acidosis may be inherited (autosomal recessive mutations in SLC4A4, the gene encoding electrogenic NBCe1) or acquired (e.g., due to carbonic anhydrase inhibitors) 204, 201. Patients with isolated proximal renal tubular acidosis (type 2 RTA) typically present with growth retardation in early childhood 205.
In patients with proximal renal tubular acidosis (type 2 RTA), hypokalemia develops as a result of the loss of proximal bicarbonate (HCO3–) reabsorption 202. Increased urinary excretion of bicarbonate (HCO3–) causes a decrease in intravascular volume, which leads to renin-angiotensin-aldosterone system (RAAS) stimulation. Impaired proximal bicarbonate (HCO3–) reabsorption also causes increased distal Na+ delivery. The increase in renin-angiotensin-aldosterone system (RAAS) activity results in elevated aldosterone levels that, combined with elevated distal Na+ concentrations, cause an increase in urinary K+ excretion (i.e., K+ wasting) that leads to hypokalemia 202.
Proximal renal tubular acidosis (type 2 RTA) may occur as an isolated defect in bicarbonate (HCO3–) reabsorption, but more typically occurs in association with Fanconi syndrome, characterized by a widespread proximal tubular dysfunction resulting in the loss of phosphate, glucose, uric acid, amino acids, and low molecular weight proteins, as well as bicarbonate (HCO3–) 200, 201. Although proximal RTA is not associated with nephrolithiasis or nephrocalcinosis, patients with proximal renal tubular acidosis (type 2 RTA) and Fanconi syndrome may develop skeletal abnormalities, such as osteomalacia 206, 207, 208. Skeletal abnormalities result from impaired phosphate reabsorption, which causes chronic hypophosphatemia due to renal phosphate wasting 202, 206, 209. Active vitamin D deficiency may be present as a result of impaired conversion of 25(OH) vitamin D3 to 1,25 (OH)2 vitamin D3 in the proximal tubule 207. Osteopenia may also be evident as a result of acidosis-induced bone demineralization 210.
Proximal renal tubular acidosis (type 2 RTA) in association with Fanconi syndrome can also occur following exposure to some medications, including tenofovir 201, ifosfamide 211, sodium valproate 212 and topiramate 213. Topiramate is a carbonic anhydrase inhibitor that can cause simultaneous defects in both proximal and distal acidification mechanisms, presenting as type 3 RTA 214. Proximal renal tubular acidosis (type 2 RTA) can occur secondary to metabolic diseases, such as hereditary fructose intolerance and glycogen storage disease 215, 216.
The most common cause of acquired Fanconi syndrome in adults is multiple myeloma 217. The clinical manifestations of anemia, hypercalcemia, and bone pain suggest a diagnosis of multiple myeloma. The patient’s condition is also complicated by proximal renal tubular acidosis (type 2 RTA) , characterized by impaired proximal bicarbonate (HCO3–) reabsorption, hypokalemia, and variable urine pH. As the most common cause of proximal RTA in adults is multiple myeloma, this diagnosis should be excluded in all adults with proximal RTA unless another cause is found.
Untreated type 2 renal tubular acidosis may cause children to grow more slowly. In addition, type 2 RTA may cause rickets, a bone disease and dental disease in both children and adults 218, 219. A very low potassium level (hypokalemia) can develop during treatment of type 2 RTA with alkali 220.
Type 4 renal tubular acidosis
Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis, is typically caused by selective aldosterone deficiency or intrinsic defects in the cortical collecting duct that lead to aldosterone resistance, which causes impaired distal H+ and K+ secretion 221, 202. Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis occurs when the tubules are unable to remove enough potassium, which also interferes with the kidney’s ability to remove acid from the blood. Hyperkalemic renal tubular acidosis (type 4 RTA) commonly develops in patients with diabetes or interstitial nephritis and is characterized by a disturbance in distal nephron function, leading to a reduction in the excretion of H+ and K+ in the cortical collecting duct that results in hyperkalemic, hyperchloremic, normal anion gap acidosis (Table 1) 222, 223.
In type 4 renal tubular acidosis, the hypoaldosteronism causes reduced principal cell Na+ reabsorption and a decrease in transepithelial voltage in the cortical collecting duct, which results in diminished excretion of H+ and K+. Increased serum K+ concentrations inhibit ammonium (NH4) synthesis in the proximal tubule, further reducing the kidney’s capacity to excrete acid. Ammonium (NH4) availability is critical for normal distal H+ secretion. Lack of adequate ammonium (NH4) buffer results in a drop in urine pH creating a steep pH gradient, which impedes distal H+ secretion. Aldosterone deficiency can cause Na+ wasting, leading to decreased plasma volume that stimulates proximal Na+ reabsorption. The reduction in distal Na+ delivery secondarily inhibits secretion of K+ and H+ in the distal nephron 221, 202. Hyperkalemia reduces ammonium (NH4) production in the proximal tubule and inhibits ammonium (NH4) transport in the thick ascending limb as the high luminal K+ concentrations compete with NH4+ for Na+/K+/2Cl– cotransporters and apical K+ channels 224. Reductions in urine ammonium (NH4) can be detected by a positive urine anion gap and a failure to increase the urine osmolal gap.
Patients with hyperkalemic renal tubular acidosis or type 4 RTA are often asymptomatic and are typically diagnosed during routine laboratory analyses. When symptomatic, high levels of potassium in the blood (hyperkalemia) can lead to muscle weakness or heart problems, such as slow or irregular heartbeats and cardiac arrest 225, 221, 202. Hyperkalemic renal tubular acidosis may be diagnosed by the presence of hyperkalemia, normal anion gap metabolic acidosis, and abnormal serum aldosterone levels, although the glomerular filtration rate (GFR) may be near-normal or only moderately reduced (45 to less than 60 mL/min/1.73 m²) 202, 226.
Patients with hyperkalemic renal tubular acidosis or type 4 RTA usually have diabetes with mild-to-moderate decreases in glomerular filtration rate (GFR), a serum bicarbonate [HCO3–] concentration of 18–22 mmol/L, and a serum K+ concentration of 5.5–6.5 mmol/L 202. Other causes of hyperkalemia and normal anion gap acidosis include selective aldosterone deficiency, or defects in K+ or H+ secretion resulting from aldosterone resistance in the kidney (sickle cell nephropathy) 202, 226. Urinary obstruction can give rise to type 4 RTA [52, 53]. In patients with obstructive uropathy and hyperkalemic metabolic acidosis, those who are unable to acidify their urine pH to less than 5.5 are thought to have a voltage-dependent defect in Na+ transport in the distal nephron (i.e., voltage-dependent distal RTA) 221, 227.
Patients with chronic kidney disease (CKD) are also at risk of developing hyperkalemic renal tubular acidosis or type 4 RTA because of the progressive loss of functional kidney mass 202. Initially, there is an adaptive increase in NH4+ production and acid secretion [56]; however, as kidney impairment progresses (GFR 30–40 mL/min/1.73 m²), this adaptive increase is unable to maintain sufficient net H+ excretion to keep pace with endogenous acid production. Patients develop a hyperchloremic normal gap acidosis, often referred to as RTA of kidney insufficiency 202. In patients with more advanced chronic kidney disease (CKD) (GFR < 15–20 mL/min/1.73 m²), the ability to excrete phosphate and other anions is reduced and a high anion gap metabolic acidosis develops; the acidosis at this stage is termed uremic acidosis. During this transition, patients frequently manifest features of both a normal and increased anion gap metabolic acidosis 202. Furthermore, patients with stage 3 to 5 chronic kidney disease (CKD) and hyperkalemia commonly develop metabolic acidosis as progressive kidney impairment leads to compromised maintenance of electrolyte and acid–base balance 228.
Pseudohypoaldosteronism is a genetic condition associated with hyperkalemic, hyperchloremic metabolic acidosis with normal kidney function and either normal or high aldosterone levels 221. Type 1 pseudohypoaldosteronism may be caused by mutations in genes encoding the mineralocorticoid receptor or epithelial Na+ channels (ENaC) 221, whereas type 2 pseudohypoaldosteronism is caused by mutations in genes encoding the with-no-lysine (WNK) family of kinases 229. Mutations in epithelial Na+ channels (ENaC) cause Na+ wasting and are characterized by increased aldosterone levels, while WNK mutations give rise to Na+ retention and either normal or low circulating aldosterone levels 229, 221.
Hyperkalemic type 4 RTA may also be caused by medications, including K+-sparing diuretics (e.g., spironolactone, eplerenone, and amiloride), antibiotics (e.g., trimethoprim and pentamidine), nonsteroidal anti-inflammatory drugs (NSAIDs), including cyclooxygenase-2 (COX-2) inhibitors, ACE inhibitors, and heparin or low molecular weight heparin 190, 230. These agents cause hyperkalemic type 4 RTA by reducing aldosterone synthesis (ACE inhibitors), release (NSAIDs and heparin), or receptor binding (spironolactone, eplerenone), or through inhibition of ENaC (amiloride, trimethoprim, and pentamidine) 190. Hyperkalemic renal tubular acidosis or type 4 RTA may also be caused by immunosuppressant therapy with calcineurin inhibitors (e.g., tacrolimus and ciclosporin) 231, 232, 233. Calcineurin inhibitors block K+ and H+ secretion from the collecting duct through inhibition of basolateral Na+/K+-ATPase and Na+/K+/2Cl– cotransporter activity 233, 234. Calcineurin inhibitors also suppress expression of mineralocorticoid receptors, resulting in aldosterone resistance 235.
Renal tubular acidosis causes
Type 1 renal tubular acidosis causes
Type 1 renal tubular acidosis or distal renal tubular acidosis, occurs when there is a problem at the end or distal part of the tubules in the kidneys not being able to rid the body of the daily acid load. This results in an inability to lower urine pH regardless of the degree of acidosis or acidemia (acid level in the blood). Distal refers to being “distant” from the point of origin. In the nephron, it means the defect occurs away from the point where fluid enters the tubule. Distal renal tubular acidosis occurs because the kidneys fail to secrete acids into the urine. On the basis of the underlying defect, type 1 renal tubular acidosis (distal renal tubular acidosis) may be classified as hereditary (primary) or acquired (secondary) 190.
Type 1 RTA may be inherited, which is also known as primary distal renal tubular acidosis. Researchers have identified at least three different genes that may cause the inherited form of type 1 RTA 197. The inherited type 1 renal tubular acidosis (primary distal renal tubular acidosis) are caused by a variation (mutation) in one of at least three different genes; the SLC4A1 gene, the ATP6V0A4 gene, and the ATP6V1B1 gene. A variation in the SLC4A1 gene is usually inherited in an autosomal dominant pattern, and less often in an autosomal recessive pattern. Variations in the ATP6V0A4 and ATP6V1B1 genes are usually inherited in an autosomal recessive pattern 197. In some affected individuals (~20% cases), no variation in these three genes can be identified suggesting that other, as-yet-unidentified genes can play a role in primary distal renal tubular acidosis 198, 199.
The SLC4A1 gene contains instructions for producing (encoding) a protein called anion exchanger 1 or AE1. This protein helps negatively-charged atoms cross cell membranes; specifically, it helps exchange chlorine ions for bicarbonate ions. Bicarbonate is an electrolyte that helps maintain the acid-base balance in the body and is filtered by the kidneys; but, most of the bicarbonate is still retained in the blood and the urine contains very small amounts. Theanion exchanger 1 (AE1) protein is found in the membranes of kidney cells and red blood cells. The kidneys reclaim filtered bicarbonate and then release acid into the urine to be excreted from the body. Researchers have speculated that a variation in the SLC4A1 gene prevents enough functional anion exchanger 1 (AE1) protein from reaching the cell membranes of kidney and red blood cells. Ultimately, this prevents the kidneys from releasing acid into the urine. Acid then builds up in the blood and tissues of the body (metabolic acidosis). The reason why some people develop metabolic acidosis and others do not is not fully understood. In red blood cells, anion exchanger 1 (AE1) protein cannot reach the red cell membrane resulting in red blood cells that break down prematurely. Some altered AE1 protein can still reach the membranes of red blood cells because it is helped by another protein called glycophorin A. This is most likely why many people with a disease-causing variation in the SLC4A1 gene do not develop hemolytic anemia.
Variations in the SLCA41 gene are usually inherited in an autosomal dominant pattern, and, less often, in an autosomal recessive pattern. Disease-causing variations in the SLC4A1 gene can be inherited from a parent or it can occur as a new (sporadic or de novo) mutation, which means that the gene variation has occurred at the time of the formation of the egg or sperm for that child only, and no other family member will be affected. Affected individuals can then pass on the altered gene in an autosomal dominant pattern.
The ATP6V0A4 and the ATP6V1B1 genes encode specific proteins that are part of a protein complex called vacuolar H+-ATPase (V-ATPase). This protein complex acts as a proton pump that helps to move positively-charged atoms (protons) across cell membranes, and helps to regulate acid levels of cells and their surrounding areas. These proteins are commonly found in cells of the inner ear and within the nephron, which is the basic filtering unit of the kidneys. These proteins have a role in regulating the amount of acid removed from the blood to the urine, and in maintaining the proper acid balance within the ear. Variations in the ATP6V0A4 and the ATP6V1B1 genes are inherited in an autosomal recessive pattern.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child.
Disorders inherited in a recessive pattern occur when an individual inherits two variants in a gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Inherited type 1 RTA (primary distal renal tubular acidosis) affects females and males in equal numbers. The exact number of people who have this disorder is unknown. Rare disorders like primary distal renal tubular acidosis often go misdiagnosed or undiagnosed, making it difficult to determine their true frequency in the general population.
Acquired (secondary) forms of type 1 RTA are caused by autoimmune disorders including Sjögren’s syndrome or systemic lupus erythematosus. Autoimmune disorders are ones in which the body’s immune system mistakenly attacks healthy tissue. Sometimes, in certain autoimmune disorders the immune system attacks the distal portion of the renal tubules. Distal renal tubular acidosis can also occur in people with sickle cell anemia, chronic obstructive uropathy, Ehlers-Danlos syndrome, hypogammaglobulinemia, chronic liver disease, and following a kidney transplant. Acquired forms of type 1 renal tubular acidosis can also be caused by certain medications (e.g., lithium, amphotericin B, NSAIDs, lead, antivirals), including some used for pain and bipolar disorder, conditions causing high calcium in the urine, blocked urinary tract, or rejection of a transplanted kidney.
Diseases and conditions related to type 1 renal tubular acidosis:
- a hereditary form of deafness
- renal medullary cystic disease
- sickle cell disease
- sickle cell anemia
- Ehlers-Danlos syndrome
- urinary tract infections
- amyloidosis, a buildup of abnormal protein, called amyloid, in the tissues and organs
- Fabry disease, an abnormal buildup in the body of a certain type of fatty substance
- High level of calcium in the blood (hypercalcemia)
- Sjögren syndrome, an autoimmune disorder in which the glands that produce tears and saliva are destroyed
- Systemic lupus erythematosus, an autoimmune disease in which the body’s immune system mistakenly attacks healthy tissue
- Wilson disease, an inherited disorder in which there is too much copper in the body’s tissues
- Use of certain medicines, such as amphotericin B, lithium, and nonsteroidal anti-inflammatory drugs (NSAIDs)
Type 2 renal tubular acidosis causes
Type 2 renal tubular acidosis or proximal renal tubular acidosis occurs when there is a problem in the beginning or proximal part of the tubules. Isolated proximal type 2 RTA is characterized by defects in the reabsorption of filtered bicarbonate (HCO3–) in the proximal tubule without defects in the transport of other solutes (Table 1) 200, 201.
Type 2 renal tubular acidosis (proximal renal tubular acidosis) may be inherited or caused by other inherited conditions such as:
- cystinosis, a rare disease in which cystine crystals are deposited in bones and other tissues
- hereditary fructose intolerance
- Wilson’s disease
Type 2 RTA can also be caused by acute lead poisoning or chronic exposure to cadmium. It can also occur in people treated with certain medications used in chemotherapy and to treat HIV, viral hepatitis, glaucoma, migraines, and seizures.
Type 2 renal tubular acidosis almost always occurs as part of Fanconi syndrome. The main features of Fanconi syndrome include:
- abnormal excretion of glucose, amino acids, citrate, bicarbonate, and phosphate into the urine
- low blood potassium levels
- low levels of vitamin D
The most common cause of acquired Fanconi syndrome in adults is multiple myeloma 217. The clinical manifestations of anemia, hypercalcemia, and bone pain suggest a diagnosis of multiple myeloma. As the most common cause of proximal RTA in adults is multiple myeloma, this diagnosis should be excluded in all adults with proximal RTA unless another cause is found.
Type 4 renal tubular acidosis causes
Type 4 renal tubular acidosis or hyperkalemic renal tubular acidosis, is typically caused by selective aldosterone deficiency or when the kidneys do not respond to the aldosterone hormone (aldosterone resistance). Aldosterone directs the kidneys to regulate the level of sodium, which also affects the levels of chloride and potassium, in the blood. Hyperkalemic renal tubular acidosis (type 4 RTA) commonly develops in patients with diabetes or interstitial nephritis and is characterized by a disturbance in distal nephron function, leading to a reduction in the excretion of H+ and K+ in the cortical collecting duct that results in hyperkalemic, hyperchloremic, normal anion gap acidosis (Table 1) 222, 223. Hypoaldosteronism causes reduced principal cell Na+ reabsorption and a decrease in transepithelial voltage in the cortical collecting duct, which results in diminished excretion of H+ and K+. Increased serum K+ concentrations inhibit ammonium (NH4) synthesis in the proximal tubule, further reducing the kidney’s capacity to excrete acid. Ammonium (NH4) availability is critical for normal distal H+ secretion. Lack of adequate ammonium (NH4) buffer results in a drop in urine pH creating a steep pH gradient, which impedes distal H+ secretion. Aldosterone deficiency can cause Na+ wasting, leading to decreased plasma volume that stimulates proximal Na+ reabsorption. The reduction in distal Na+ delivery secondarily inhibits secretion of K+ and H+ in the distal nephron 221, 202. Hyperkalemia reduces ammonium (NH4) production in the proximal tubule and inhibits ammonium (NH4) transport in the thick ascending limb as the high luminal K+ concentrations compete with NH4+ for Na+/K+/2Cl– cotransporters and apical K+ channels 224. Reductions in urine ammonium (NH4) can be detected by a positive urine anion gap and a failure to increase the urine osmolal gap.
Certain medicines that interfere with the kidney’s task of moving electrolytes between your blood and urine may also cause hyperkalemic renal tubular acidosis or type 4 RTA. Some of these include:
- blood pressure medicines called angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs)
- certain diuretics used to treat congestive heart failure that do not decrease potassium in the blood
- certain medicines to prevent blood from clotting
- some immunosuppressive medicines that prevent the rejection of transplanted organs
- painkillers called nonsteroidal anti-inflammatory drugs (NSAIDs)
- antibiotics used to treat pneumonia, urinary tract infections, and traveler’s diarrhea
Type 4 RTA can also occur when diseases or an inherited disorder affect how the kidneys work, such as:
- Addison’s disease, due to disease or removal of the adrenal glands
- congenital adrenal insufficiency
- aldosterone synthase deficiency
- Gordon syndrome
- amyloidosis
- diabetic kidney disease
- HIV/AIDS
- kidney transplant rejection
- lupus
- sickle cell disease
- urinary tract obstruction
Renal tubular acidosis symptoms
The major signs of type 1 renal tubular acidosis (distal renal tubular acidosis) and type 2 renal tubular acidosis (proximal renal tubular acidosis) are low levels of potassium (K+) and bicarbonate (HCO3–), a waste product produced by your body in the blood. The potassium level drops if your kidneys send too much potassium into your urine instead of returning it to the blood.
Because potassium (K+) helps regulate your nerve and muscle health and heart rate, low potassium levels (hypokalemia) can cause:
- extreme weakness
- irregular heartbeat
- paralysis
- death
The major signs of type 4 renal tubular acidosis (hyperkalemic renal tubular acidosis) are high potassium and low bicarbonate levels in the blood. Symptoms of type 4 RTA include 236:
- abdominal pain
- fatigue that does not go away
- weak muscles
- not feeling hungry
- weight change
Renal tubular acidosis diagnosis
A diagnosis of distal renal tubular acidosis is based upon identification of characteristic symptoms, a detailed patient and family history, a thorough clinical evaluation and a variety of specialized tests. The disorder may be suspected in individuals with unexplained metabolic acidosis and an elevated plasma chloride (so called normal anion gap metabolic acidosis). In all cases of metabolic acidosis, plasma or serum anion gap should be the first laboratory assessment; hyperchloremic metabolic acidosis with a normal anion gap is present in all types of renal tubular acidosis (Table 1) 237, 226.
If your blood is more acidic than it should be and your urine is less acidic than it should be, RTA may be the reason, but a health care professional will need to rule out other causes.
Tests that may be ordered include:
- Arterial blood gas
- Basic metabolic panel, (a group of blood tests that measure your sodium, potassium, and chloride levels, kidney function, and other chemicals and functions)
- A complete blood count (CBC) to evaluate for an infectious cause with elevated white blood count and fluid body status with hemoglobin and hematocrit values is useful.
- Urine pH
- Urine ketones or blood ketones
- Acid-load test
- Bicarbonate infusion test
- Urinalysis
- Genetic testing. Molecular genetic testing can confirm a diagnosis. Genetic counseling may be of benefit for affected individuals and their families. Molecular genetic testing can detect disease-causing variants in the specific genes known to cause primary distal renal tubular acidosis, but is available only as a diagnostic service at specialized laboratories. Psychosocial support for the entire family can be essential as well.
Confirmatory testing of distal versus proximal renal tubular acidosis involves assessment of the markers of urinary acid and bicarbonate (HCO3–) secretion 226. An ammonium (NH4+) loading test is used to confirm distal renal tubular acidosis in patients with hypokalemic, hyperchloremic metabolic acidosis and urine pH > 5.5; at 6 hour after oral ingestion of NH4Cl 100 mg/kg, patients with distal renal tubular acidosis will develop a positive urine anion gap (Urine anion gap [UAG] = Urine Na + Urine K – Urine Cl) 226. As the NH4Cl test is associated with gastrointestinal adverse effects (e.g., nausea and vomiting), the furosemide–fludrocortisone test may be used as an alternative method of diagnosing distal renal tubular acidosis 238, 239.
In patients with distal renal tubular acidosis, measurement of the urine-to-blood (U-B) pCO2 gradient during an NaHCO3 infusion can be used to diagnose H+-ATPase secretory defect 240. Patients with an H+-ATPase defect have an abnormally low urine-to-blood pCO2 gradient (30 mmHg or lower), indicating impaired distal H+ secretion 240. In patients with a gradient defect, as occurs with amphotericin B-induced distal renal tubular acidosis 241, the urine-to-blood pCO2 gradient is normal (greater than 30 mmHg), suggesting an intact H+ secretory mechanism 242. In these patients, secreted H+ diffuses back into the cell as a result of a luminal permeability defect 241.
A diagnosis of proximal renal tubular acidosis may be confirmed using a NaHCO3 loading test; during an intravenous infusion of NaHCO3, the increase in serum bicarbonate (HCO3–) above the reabsorption threshold will lead to fractional excretion of bicarbonate (HCO3–) greater than 15% or urine pH > 7.5 in patients with proximal renal tubular acidosis 226. Patients with proximal RTA should also be evaluated for Fanconi syndrome by assessment of serum and urine samples for glycosuria, hypophosphatemia, and hypouricemia.
The differential diagnosis of type 4 hyperkalemic renal tubular acidosis may be divided into conditions associated with hypoaldosteronism or those caused by cortical collecting duct defects. A urine pH of less than 5.5 indicates defects caused by impaired aldosterone activity and a more severe reduction in NH3 availability than impairment of H+ secretion 202. In contrast, when the primary defect is caused by structural damage to the cortical collecting duct, the urine pH is more alkaline 202. Selective aldosterone deficiency may be confirmed after other causes of hyperkalemia are excluded, including transcellular shifts in K+ or the use of KCl, K+-sparing diuretics, or RAAS inhibitors [52]. After correction of serum K+, persistently low aldosterone levels indicate aldosterone deficiency; in most cases plasma renin levels are low. In patients with hyperkalemia, urinary K+ below 40 mmol/L or fractional K+ excretion below 20% are indicative of a defect in kidney K+ secretion 221. In patients with hypoaldosteronism, but normal renin levels, possible causes include adrenal gland damage, Addison disease, critical illness (i.e., direct renal suppression), RAAS inhibitor therapy, or heparin-induced suppression of aldosterone synthesis 222.
Extrarenal conditions may mimic RTA by causing normal gap metabolic acidosis through increased production of endogenous H+ and accelerated loss of extrarenal bicarbonate (HCO3–) 202. Extrarenal metabolic acidosis is associated with elevated levels of H+ and urinary NH4+ excretion 202. Severe or chronic diarrhea commonly causes hyperchloremic metabolic acidosis through the loss of large amounts of gastrointestinal bicarbonate (HCO3–), particularly as bicarbonate (HCO3–) concentrations are usually higher in diarrheal fluid than in plasma 243, 226. The reduction in plasma volume triggers an increase in kidney NaCl reabsorption which, in combination with bicarbonate (HCO3–) losses, leads to normal anion gap metabolic acidosis 202. Low serum pH, as well as hypokalemia caused by gastrointestinal losses, promotes ammoniagenesis in the proximal tubule; the increased NH3 concentrations allow for increased H+ excretion by the distal nephron. This can result in an increased urine pH in patients with chronic diarrhea because of increases in kidney NH3 metabolism202. Chronic laxative abuse has been reported to mimic distal RTA through intestinal losses of bicarbonate (HCO3–) and K+ 244. The normal increase in urinary NH3 excretion in these situations is reflected by a negative urinary anion gap and an increased urine osmolal gap.
Calcium deposits in the kidneys and kidney stones may be seen on:
- X-rays
- Ultrasound
- CT scan
Plain x-rays (radiographs) or specialized imaging techniques such as computerized tomography (CT) scanning or ultrasonography can help to further confirm a diagnosis, or help determine the extent of disease. These tests can show the accumulation or deposition of calcium in the kidneys and help to rule out other conditions. During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of certain tissue structures. During ultrasonography, reflected sound waves are used to create an image of structures within the body including the kidneys.
Figure 4. Renal tubular acidosis diagnostic algorithm
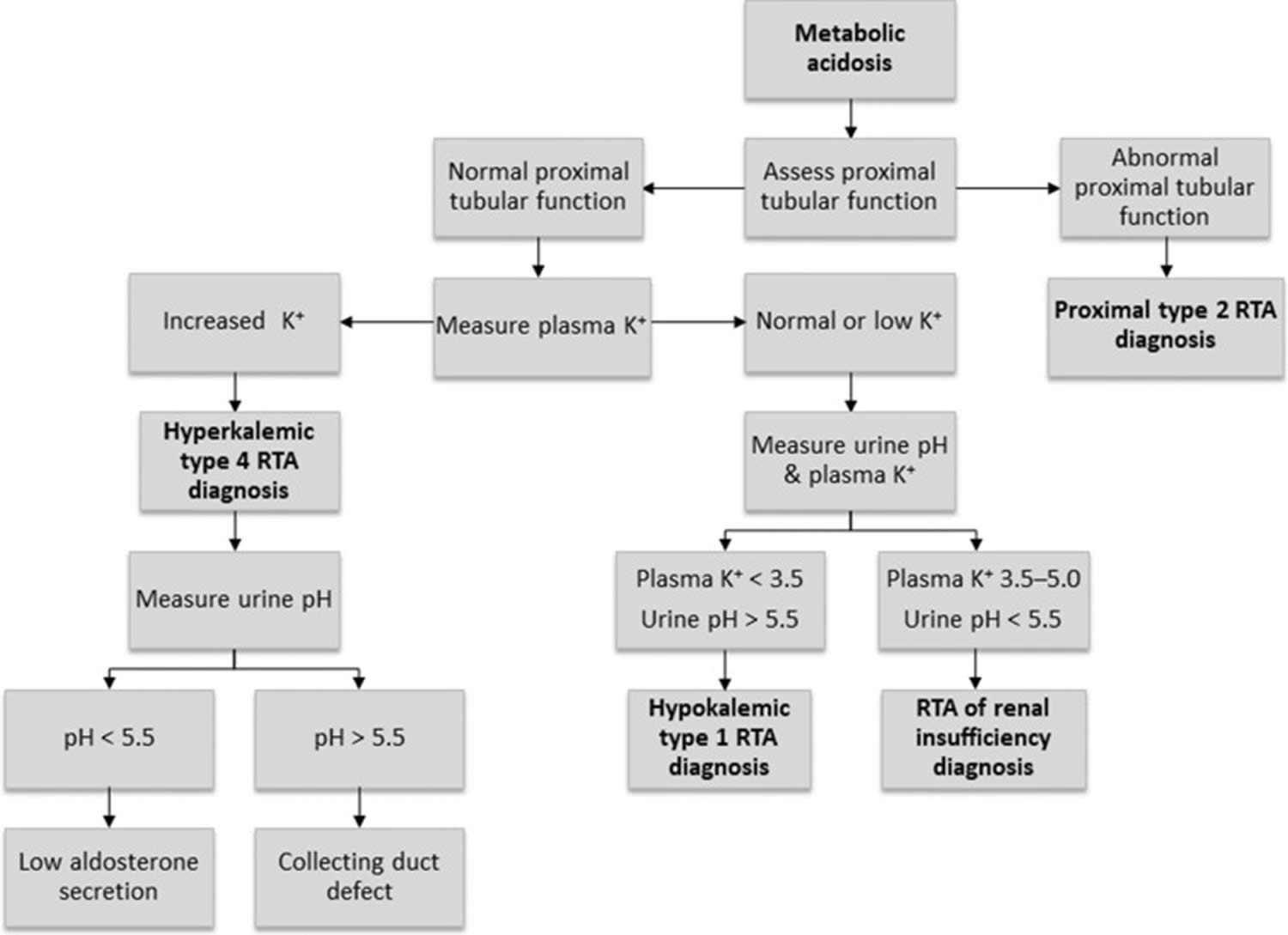
Arterial blood gas (ABG) analysis
Arterial blood gas (ABG) sampling, is a test often performed in an inpatient setting to assess the acid-base status of a patient. A needle is used to draw blood from an artery, often the radial artery, and the blood is analyzed to determine parameters such as the pH, arterial partial pressure of carbon dioxide (PaCO2), arterial partial pressure of oxygen (PaO2), bicarbonate (HCO3–), oxygen saturation (O2 Sat) and more. This allows the physician to understand the status of the patient better. ABGs are especially important in the critically ill. They are the main tool utilized in adjusting to the needs of a patient on a ventilator.
- Arterial partial pressure of carbon dioxide (PaCO2) as carbon dioxide tension, this measures the level of carbon dioxide in your blood.
- Arterial partial pressure of oxygen (PaO2) also known as oxygen tension, this measures how well oxygen is being transferred into your blood.
- Oxygen saturation (O2 Sat) is an assessment of the amount of oxygen in your blood that is based on measuring levels of hemoglobin. Hemoglobin is a protein found inside red blood cells that is responsible for carrying oxygen throughout the body.
- Bicarbonate (HCO3–) concentration: Bicarbonate (HCO3–) is an electrolyte, which is a type of mineral involved in managing your body’s acid-base balance. Most of the carbon dioxide (CO2) in your blood is stored in the form of bicarbonate, so this measurement helps reflect carbon dioxide (CO2) levels.
- Although not universal, some arterial blood gases tests include measurements of hemoglobin as well as altered forms of the hemoglobin protein. Examples of these potential additional measurements include:
- Methemoglobin: Methemoglobin is a form of hemoglobin that has been oxidized, changing its heme iron configuration from the ferrous (Fe2+) to the ferric (Fe3+) state. Unlike normal hemoglobin, methemoglobin does not bind oxygen and as a result cannot deliver oxygen to the tissues.
- Carboxyhemoglobin: Carboxyhemoglobin is a stable complex of carbon monoxide and hemoglobin that forms in red blood cells upon contact with carbon monoxide. This abnormal form of hemoglobin attaches to carbon monoxide and can interfere with oxygen’s ability to travel in the blood.
- Oxyhemoglobin: Oxyhemoglobin represents the fraction of oxygenated hemoglobin in relation to the total hemoglobin present, including non-oxygen-binding hemoglobins. In healthy individuals, oxyhemoglobin and oxygen saturation are approximately equal.
- Deoxyhemoglobin: This is the form of hemoglobin without oxygen in the blood.
The following are the most important Normal Values on an ABG:
- pH = 7.35 to 7.45
- Arterial partial pressure of carbon dioxide (PaCO2) = 35 to 45 mmHg
- Arterial partial pressure of oxygen (PaO2) = 75 to 100 mmHg
- Bicarbonate (HCO3–) = 22 to 26 mEq/L
- O2 Sat = greater than 95%
The ability to quickly and efficiently read an ABG is paramount to quality patient care.
- Look at the pH. Decide whether it is acidotic, alkalotic, or within the physiological range
- Arterial partial pressure of carbon dioxide (PaCO2) level determines respiratory contribution; a high level means the respiratory system is lowering the pH and vice versa.
- Bicarbonate (HCO3–) level denotes metabolic/kidney effect. An elevated bicarbonate (HCO3–) is raising the pH and vice versa.
- If the pH is acidotic, look for the number that corresponds with a lower pH. If it is a respiratory acidosis, the carbon dioxide (CO2) should be high. If the patient is compensating metabolically, the bicarbonate (HCO3–) should be high as well. A metabolic acidosis will be depicted with an bicarbonate (HCO3–) that is low.
- If the pH is alkalotic, again, determine which value is causing this. A respiratory alkalosis will mean the carbon dioxide (CO2) is low; a metabolic alkalosis should lend an bicarbonate (HCO3–) that is high. Compensation with either system will be reflected oppositely; for a respiratory alkalosis the metabolic response should be a low bicarbonate (HCO3–) and for metabolic alkalosis, the respiratory response should be a high carbon dioxide (CO2).
- If the pH level is in the physiological range but the arterial partial pressure of carbon dioxide (PaCO2) and/or bicarbonate (HCO3–) are not within normal limits, there is likely a mixed disorder. Also, compensation does not always occur; this is when clinical information becomes paramount.
- Sometimes it is difficult to ascertain whether a patient has a mixed disorder.
Other tests that are important to perform when analyzing the acid-base status of a patient include those that measure electrolyte levels and renal function. This helps the clinician gather information that can be used to determine the exact mechanism of the acid-base imbalance as well as the factors contributing to the disorders 245, 246.
Urinalysis
Urine pH is normally acidic, at less than 5.0. In acidemia, the urine normally becomes more acidic. If the urine pH is above 5.5 in the face of acidemia, this finding is consistent with a type 1 renal tubular acidosis (RTA). Alkaline urine is typical in salicylate toxicity.
Patients with ethylene glycol toxicity may present with calcium oxalate crystals, which appear needle shaped, in the urine.
Serum Anion Gap
The formula for anion gap is:
- Serum anion gap = (Na+) – [(HCO3– + Cl–)]
Where Na+ is plasma sodium concentration, HCO3– is plasma bicarbonate concentration, and Cl– is plasma chloride concentration. The anions are negatively charged ions like chloride [Cl–] and bicarbonate [HCO3–]. The anion gap is the difference between measured cations (positively charged ions like sodium [Na+] and potassium [K+]) and measured anions (negatively charged ions like chloride [Cl–] and bicarbonate [HCO3–]) 247. The most common application of the anion gap is classifying cases of metabolic acidosis, states of lower than normal blood pH. Specifically, classifying into either those that do and those that do not have unmeasured anions in the plasma. The human body is electrically neutral; therefore, in reality, does not have a true anion gap 247. A normal serum anion gap is measured to be 5 to 16 mEq/L, with autoanalyzers using an ion-selective electrode. However, the anion gap value is dependent on the type of instrument used to measure its components 243. Therefore, you should know the reference range of the analyzer used and, if known, the patient’s baseline anion gap, too.
The most common application of the anion gap is classifying cases of metabolic acidosis, states of lower than normal blood pH. Specifically, classifying into either those that do and those that do not have unmeasured anions in the plasma. The human body is electrically neutral; therefore, in reality, does not have a true anion gap 247. A normal serum anion gap is measured to be 5 to 16 mEq/L, with autoanalyzers using an ion-selective electrode. However, the anion gap value is dependent on the type of instrument used to measure its components 243. Therefore, you should know the reference range of the analyzer used and, if known, the patient’s baseline anion gap, too.
The anion gap is a calculation to determine the quantity of ionically active components within your blood that are not routinely measured. Serum anion gap is affected by the concentrations of all anions and cations which are not included in its calculations: i.e., albumin, globulin, potassium, calcium, magnesium, and organic and inorganic acids (see Figure 1). Because of the narrow extracellular concentration, most ions are omitted from the anion gap calculation. Since there are always components not directly measured, we expect this value to not equal 0. Most of this number is due to albumin (Alb); this anion is not accounted for in the anion gap formula, which is a large reason why the anion gap is not closer to zero. According to James Gamble 248, electrical neutrality in solution demands that the sum of the cations is equal to the sum of the anions (Figure 1). Sodium, chloride, bicarbonate, and albumin are quantitatively the major ions in the extracellular fluid compartment and are therefore used to calculate the anion gap 243. A true “ion gap,” however, does not exist in vivo which makes the anion gap a fundamental tool to evaluate acid-base disorders 249. Albumin is normally 4 mg/dL. Because of the large effect of albumin on anion gap, if a patient’s albumin level is abnormal, their expected anion gap will not be accurate 250. This can be corrected using simple math. The correction factor for albumin is 2.3–2.5 × [albumin], in g/dL 243. Therefore, each g/dL albumin decline will decrease the anion gap with about 2.5 mEq/L. To appreciate these facts, the anion gap formula should be: [Na+] − [Cl−] − [HCO3−] − 2.5 [albumin, in g/dL]. This equation is about zero in health, to stress the balance of ions, and also shows the relevance of albumin as a negative ion 243. As opposed to high anion gap acidosis which involves increased organic acid production, normal anion gap acidosis involves either increased production of chloride (hyperchloremic acidosis) or increased excretion of bicarbonate (HCO3–).
If a patient has an anion gap over 12, these mnemonics are helpful to remember the possible causes of the disorder 251, 252. The mnemonic MUDPILES has classically been used by clinicians to summarize the causes of high anion gap metabolic acidosis.
MUDPILES stands for:
- Methanol,
- Uremia,
- Diabetic ketoacidosis,
- Paraldehyde,
- Infection,
- Lactic acidosis,
- Ethylene glycol, and
- Salicylates.
A new mnemonic, GOLDMARK, has been suggested to be an improvement 250.
GOLDMARK is an anagram for:
- Glycols (ethylene and propylene),
- Oxoproline,
- Lactate,
- Methanol,
- Aspirin,
- Renal failure, or chronic kidney disease (CKD) and
- Ketones.
Urine Anion Gap
Calculating the urine anion gap is helpful in evaluating some cases of non-anion gap metabolic acidosis. The major measured urinary cations are Na+ and K+, and the major measured urinary anion is Cl-:
- Urine anion gap = Urine Na + Urine K – Urine Cl
In the face of metabolic acidosis, the kidneys increase the amount of NH3 synthesized to buffer the excess H+ and NH4 Cl excretion increases. The increased unmeasured ammonium (NH4+) thus increases the measured anion Cl- in the urine, and the net effect is a negative anion gap, representing a normal response to systemic acidification. The finding of a positive urine anion gap in the face of non-anion gap metabolic acidosis points toward a renal acidification defect (eg, renal tubular acidosis) 253.
Salicylate levels and Iron levels
Therapeutic salicylate levels range up to 20-35 mg/dL. Plasma levels exceeding 40-50 mg/dL are in the toxic range.
Plasma levels provide some information as to the severity of intoxication: 40-60 mg/dL is considered mild; 60-100 mg/dL is moderate; and greater than 100 mg/dL is considered severe.
Iron toxicity is associated with lactic acidosis. Iron levels greater than 300 mg/dL are considered toxic.
Special tests
Measuring the transtubular potassium gradient (TTKG) is useful in determining the cause of hyperkalemia or hypokalemia associated with metabolic acidosis.
- Transtubular Potassium Gradient (TTKG) = urine K+ × serum osmolality/serum K+ × urine osmolality
A transtubular potassium gradient (TTKG) of greater than 8 indicates that aldosterone is present and that the collecting duct is responsive to it. A transtubular potassium gradient (TTKG) of less than 5 in the presence of hyperkalemia indicates aldosterone deficiency or resistance. For the test to be interpretable, the urine Na+ level should be greater than 10 mEq/L and the urine osmolality should be greater than or equal to serum osmolality.
Plasma renin activity and plasma aldosterone levels are useful in determining the cause of the hyperkalemia and hypokalemia that accompany metabolic acidosis.
Calculation of fractional excretion of bicarbonate (FEHCO3–) is useful in the diagnosis of proximal renal tubular acidosis (RTA).
The ammonium chloride (NH4Cl) loading test is useful in patients with nephrocalcinosis and/or nephrolithiasis, who may have an incomplete form of distal renal tubular acidosis. These patients may not have a pH less than 7.35 or a drop in serum bicarbonate (HCO3–); metabolic acidosis can be induced by administration of NH4Cl (0.1 g/kg for 3 days). Under these circumstances of induced acidemia, a urine pH greater than 5.3 indicates distal renal tubular acidosis (RTA).
An alternative to the ammonium chloride (NH4Cl) loading test involves the simultaneous oral administration of furosemide to increase distal Na+ delivery and fludrocortisone to increase collecting duct Na+ absorption and proton secretion 254. Under these circumstances, a urine pH greater than 5.3 indicates distal renal tubular acidosis (RTA).
Measuring the urine-blood arterial partial pressure of carbon dioxide (PaCO2) gradient following an bicarbonate (HCO3–) load is useful in some patients with classic distal renal tubular acidosis to differentiate a permeability defect from other defects. This test is useful in patients with nephrocalcinosis in whom distal renal tubular acidosis (RTA) is suspected but urine is acidified appropriately in the face of metabolic acidosis. Some of these patients have a rate-dependent defect in proton secretion, revealed by a low urine-blood PaCO2 gradient following bicarbonate (HCO3–) loading.
Abdominal radiographs (eg, kidneys, ureters, bladder), CT scans, and/or renal ultrasound images may show renal stones or nephrocalcinosis in patients with distal renal tubular acidosis.
Renal tubular acidosis treatment
For all types of RTA, drinking a solution of sodium bicarbonate (NaHCO3) or sodium citrate will lower the acid level in your blood. This alkali therapy can prevent kidney stones from forming and make your kidneys work more normally so kidney failure does not get worse.
Infants with type 1 renal tubular acidosis may need potassium supplements, but older children and adults rarely do because alkali therapy prevents the kidneys from excreting potassium into the urine.
Children with type 2 renal tubular acidosis will also drink an alkali solution (sodium bicarbonate or potassium citrate) to lower the acid level in their blood, prevent bone disorders and kidney stones, and grow normally. Some adults with type 2 RTA may need to take vitamin D supplements to help prevent bone problems.
People with type 4 renal tubular acidosis (hyperkalemic renal tubular acidosis) may need other medicines to lower the potassium levels in their blood.
If your RTA is caused by another condition, your health care professional will try to identify and treat it.
Medications to treat renal tubular acidosis
Alkali therapy may be used to correct acidosis in patients with distal or proximal RTA 202. In patients with distal renal tubular acidosis (type 1 RTA), alkali therapy also corrects for hypokalemia. Alkali therapy with 1–2 mmol/kg/day sodium bicarbonate (NaHCO3) or potassium bicarbonate (KHCO3) is normally sufficient to equal daily acid production 190; however, in patients with kidney stones or nephrocalcinosis, the elevated Na+ load with sodium bicarbonate (NaHCO3) therapy may cause increased urine calcium excretion, which can precipitate kidney stone formation. In these patients, potassium citrate (K-citrate) administration is preferable; this will also increase urine citrate excretion and prevent recurrence of kidney stones 101. Patients with severe hypokalemia should also receive K+ replacement (i.e., with KCl or K-citrate) to prevent further lowering of serum K+ concentrations and symptomatic hypokalemia 202. Long-term treatment of distal renal tubular acidosis (type 1 RTA) generally requires a combination of sodium bicarbonate (NaHCO3) and potassium bicarbonate (KHCO3) 202, 255. Children with distal renal tubular acidosis (type 1 RTA) require sufficient sodium bicarbonate (NaHCO3) or potassium bicarbonate (KHCO3) (usually 4–8 mmol/kg/day) to maintain normal serum bicarbonate (HCO3–) concentrations and prevent growth retardation 190.
In contrast, treatment of proximal renal tubular acidosis (type 2 RTA) is often challenging and patients require larger quantities of alkali therapy (10–15 mmol/kg/day), which are usually administered as a K+ salt (e.g., K-citrate) to avoid worsening hypokalemia 190, 202. However, as exogenous alkali is rapidly excreted in the urine, correction of acidosis is often impossible despite administration of large amounts of alkali therapy 202. In addition to alkali therapy, patients with Fanconi syndrome are treated with fluid and electrolyte replacement to prevent volume depletion, as well as supplementation with vitamin D and phosphate (1–3 g/day) to prevent bone disease 256. Impaired proximal bicarbonate (HCO3–) reabsorption leads to most of the administered bicarbonate (HCO3–) being lost in the urine with minimal increases in serum concentrations 257, while increased distal Na+ delivery and elevated aldosterone levels cause increased renal K+ wasting 202. Hydrochlorothiazide may be beneficial in increasing bicarbonate (HCO3–) reabsorption capacity, preventing volume expansion, and increasing the effectiveness of alkali therapy; however, supplemental K+ or K+-sparing diuretics are needed to prevent hypokalemia 256. After initiation of therapy, patients should be closely monitored for severe electrolyte abnormalities. Children with proximal renal tubular acidosis (type 2 RTA) often require aggressive alkali therapy (5–15 mmol/kg/day) to mitigate bone disease and growth retardation 258, whereas alkali therapy is typically administered in adults with serum bicarbonate (HCO3–) concentrations of less than 18 mmol/L to prevent severe acidosis 202.
Sodium bicarbonate (NaHCO3) therapy is often associated with gastrointestinal adverse effects, most commonly bloating and belching, and should be taken on an empty stomach 259. If these adverse effects limit patient adherence, Na-citrate or enteric-coated sodium bicarbonate (NaHCO3) (where available) may be considered 259.
In patients with hyperkalemic type 4 renal tubular acidosis, lowering of serum K+ concentrations often leads to correction of metabolic acidosis 202. The reduction in serum K+ leads to increased NH3 production in the proximal tubule and medullary transfer, thereby increasing the availability of buffer supply for distal acidification 202. Any non-essential medications affecting renal K+ excretion or aldosterone synthesis or activity should be discontinued 202. Discontinuation of the COX-2 inhibitor celecoxib would be beneficial in reducing serum K+ concentrations. ACE inhibitors and ARBs reduce urinary K+ excretion through inhibition of aldosterone secretion in the adrenal gland and may cause hyperkalemia in patients with pre-existing conditions that cause impaired K+ excretion 260. For this reason, RAAS inhibitor therapy is avoided; however, ACE inhibitors and ARBs are normally continued in patients with CKD because of their cardiovascular and renoprotective benefits 202. Low-dose fludrocortisone therapy may also be effective in managing hyperkalemia and hyponatremia in patients with type 4 RTA who do not have heart failure or hypertension 221, 261. Since thiazide diuretics are largely ineffective in patients with an estimated GFR of less than 30 mL/min/1.73 m², loop diuretics and NaHCO3 therapy may be beneficial in patients with type 4 RTA, particularly when fludrocortisone is not tolerated. Loop diuretics may reduce serum K+ and help control volume overload by increasing Na+ delivery and flow rates to the cortical collecting duct while lowering blood pressure 221. The Kidney Disease-Improving Global Outcomes guidelines suggest administration of oral HCO3– therapy to maintain serum bicarbonate (HCO3–) in the normal range in patients with CKD and serum bicarbonate (HCO3–) less than 22 mmol/L 262. Sodium bicarbonate (NaHCO3) administration will correct metabolic acidosis and further minimize the risk of hyperkalemia, and the addition of Na+ would not be problematic in the setting of effective diuretic therapy. However, patients on sodium bicarbonate (NaHCO3) therapy should be closely monitored for volume overload and hypertension 202.
There are newer K+-binding agents available (i.e., patiromer and sodium zirconium cyclosilicate [sodium zirconium cyclosilicate]) for patients with hyperkalemic type 4 RTA, which can be used to treat hyperkalemia and improve acidosis. Patiromer is a polymeric cation exchange resin that binds K+ ions in exchange for calcium ions in the colon 263, whereas sodium zirconium cyclosilicate is non-polymeric, selective K+-binder, which entraps K+ and NH4+ ions in exchange for H+ and Na+ ions throughout the gastrointestinal tract 264. Both patiromer and sodium zirconium cyclosilicate are effective in patients receiving RAAS inhibitor therapy, which allows continuation of these therapeutic agents in the presence of hyperkalemia 265. sodium zirconium cyclosilicate has also been associated with significant increases in serum bicarbonate (HCO3–) concentrations by approximately 2 mmol/L versus placebo during the initial 48-h treatment period and 2–3 mmol/L during maintenance therapy for 1 month, regardless of chronic kidney disease (CKD) stage 265. This increase in serum bicarbonate (HCO3–) with sodium zirconium cyclosilicate is most likely as a result of gastrointestinal NH4+ binding ions and associated decreases in serum urea concentrations 266. Reducing serum K+ concentrations with secondary increases in NH3 synthesis may be an additional mechanism by which acidosis is improved with sodium zirconium cyclosilicate. Whether patiromer produces similar effects on serum bicarbonate (HCO3–) remains to be determined; however, treatment with sodium zirconium cyclosilicate may provide therapeutic benefits in patients with hyperkalemic renal tubular acidosis (type 4 RTA). Studies are needed to confirm the efficacy and safety of sodium zirconium cyclosilicate or patiromer in hyperkalemic renal tubular acidosis (type 4 RTA).
Renal tubular acidosis diet
In general, plant-based diets contain a lower net acid load and less bioavailable phosphate than animal-based diets 267. In patients with metabolic acidosis, the dietary acid load can be decreased by limiting acid-producing foods (e.g., animal protein) and increasing alkali-producing foods (e.g., fruits and vegetables) (Table 1) 268, 259.. Reduction in the consumption of dietary protein obtained from animal sources also results in increases in serum total carbon dioxide (CO2) concentrations 259.
Changes in dietary consumption of citrus fruit and juices, as well as restricted Na+, oxalate, fructose, and animal protein intake, in combination with normal calcium intake may also benefit patients with distal renal tubular acidosis and nephrolithiasis 101. Increased fruit and vegetable intake is associated with increased urinary citrate excretion in patients with hypocitraturia and nephrolithiasis 269, 101. In patients with hyperkalemic renal tubular acidosis, dietary restriction of K+ has previously been the standard of care 190; however, new data suggest that increased intake of alkali-producing fruits and vegetables (which are often high in K+) and limiting intake of acid-producing foods may correct acidosis 270.
What is Fanconi’s syndrome
Fanconi syndrome consists of multiple defects in kidney proximal tubular reabsorption, causing glucosuria (excretion of glucose into the urine), phosphaturia (excessive discharge of phosphates in the urine), generalized aminoaciduria (presence of amino acids in the urine) and bicarbonate wasting 271. It may be hereditary or acquired. Symptoms in children are failure to thrive, growth retardation, and rickets. Symptoms in adults are osteomalacia (softening of the bones) and muscle weakness. Diagnosis is by showing glucosuria, phosphaturia, and aminoaciduria 271. Treatment is sometimes bicarbonate and potassium replacement, removal of offending nephrotoxins (toxic agents or substances that inhibit, damage or destroy the cells and/or tissues of the kidneys), and measures directed at renal failure.
Fanconi syndrome causes
Fanconi syndrome can be:
- Hereditary
- Acquired
Hereditary Fanconi syndrome
This disorder usually accompanies another genetic disorder, particularly cystinosis 271. Cystinosis is an inherited (autosomal recessive) metabolic disorder in which cystine accumulates within cells and tissues (and is not excreted to excess in the urine as occurs in cystinuria). Besides renal tubular dysfunction, other complications of cystinosis include eye disorders, hepatomegaly, hypothyroidism, and other manifestations.
Fanconi syndrome may also accompany Wilson disease, hereditary fructose intolerance, galactosemia, oculocerebrorenal syndrome (Lowe syndrome), mitochondrial cytopathies, and tyrosinemia. Inheritance patterns vary with the associated disorder.
Acquired Fanconi syndrome
This disorder may be caused by various drugs, including certain cancer chemotherapy drugs (eg, ifosfamide, streptozocin), antiretrovirals (eg, didanosine, cidofovir), and outdated tetracycline 271. All of these drugs are nephrotoxic. Acquired Fanconi syndrome also may occur after renal transplantation and in patients with multiple myeloma, amyloidosis, intoxication with heavy metals or other chemicals, or vitamin D deficiency.
Fanconi syndrome pathophysioloy
Various defects of kidney proximal tubular transport function occur, including impaired resorption of glucose, phosphate, amino acids, bicarbonate, uric acid, water, potassium, and sodium. The aminoaciduria is generalized, and, unlike that in cystinuria, increased cystine excretion is a minor component. The basic pathophysiologic abnormality is unknown but may involve a mitochondrial disturbance. Low levels of serum phosphate cause rickets, which is worsened by decreased proximal tubular conversion of vitamin D to its active form.
Fanconi syndrome signs and symptoms
In hereditary Fanconi syndrome, the chief clinical features—proximal tubular acidosis, hypophosphatemic rickets, hypokalemia, polyuria, and polydipsia—usually appear in infancy.
When Fanconi syndrome occurs because of cystinosis, failure to thrive and growth retardation are common. The retinas show patchy depigmentation. Interstitial nephritis develops, leading to progressive renal failure that may be fatal before adolescence.
In acquired Fanconi syndrome, adults present with the laboratory abnormalities of renal tubular acidosis (proximal type 2—see Table 2. Features of Different Types of Renal Tubular Acidosis), hypophosphatemia, and hypokalemia. They may present with symptoms of bone disease (osteomalacia) and muscle weakness.
Fanconi syndrome diagnosis
- Urine testing for glucose, phosphates, and amino acids.
Diagnosis is made by showing the abnormalities of renal function, particularly glucosuria (in the presence of normal serum glucose), phosphaturia, and aminoaciduria. In cystinosis, slit-lamp examination may show cystine crystals in the cornea.
Fanconi syndrome treatment
- Sometimes sodium bicarbonate or potassium bicarbonate or sodium citrate or potassium citrate 271.
- Sometimes potassium supplementation 271.
Other than removing the offending nephrotoxin, there is no specific treatment.
Acidosis may be lessened by giving tablets or solutions of sodium bicarbonate or potassium bicarbonate or sodium citrate or potassium citrate, eg, Shohl’s solution (sodium citrate and citric acid; 1 mL is equivalent to 1 mmol of bicarbonate) given 1 mEq/kg bid to tid or 5 to 15 mL after meals and at bedtime 271.
Potassium depletion may require replacement therapy with a potassium-containing salt.
Hypophosphatemic rickets can be treated.
Kidney transplantation has been successful in treating renal failure. However, when cystinosis is the underlying disease, progressive damage may continue in other organs and eventually result in death 271.
What is Cystinuria
Cystinuria is a condition characterized by the buildup of the amino acid cystine, a building block of most proteins, in the kidneys and bladder. As the kidneys filter blood to create urine, cystine is normally absorbed back into the bloodstream. People with cystinuria cannot properly reabsorb cystine into their bloodstream, so the amino acid accumulates in their urine 272.
As urine becomes more concentrated in the kidneys, the excess cystine forms crystals. Larger crystals become stones that may lodge in the kidneys or in the bladder. Sometimes cystine crystals combine with calcium molecules in the kidneys to form large stones. These crystals and stones can create blockages in the urinary tract and reduce the ability of the kidneys to eliminate waste through urine. The stones also provide sites where bacteria may cause infections. Less than 3% of urinary tract stones are cystine stones 38.
Mutations in the SLC3A1 (type A) or SLC7A9 (type B) gene cause cystinuria 272. The SLC3A1 and SLC7A9 genes provide instructions for making the two parts (subunits) of a protein complex that is primarily found in the kidneys. Normally this protein complex controls the reabsorption of certain amino acids, including cystine, into the blood from the filtered fluid that will become urine. Mutations in either the SLC3A1 gene or SLC7A9 gene disrupt the ability of the protein complex to reabsorb amino acids, which causes the amino acids to become concentrated in the urine 272.
The primary defect results in diminished renal proximal tubular resorption of cystine and as the levels of cystine in the urine increase, the crystals typical of cystinuria form. Cystine is poorly soluble in acidic urine, so when its urinary concentration exceeds its solubility, crystals precipitate and cystine kidney stones form. The other amino acids that are reabsorbed by the protein complex do not create crystals when they accumulate in the urine 273.
Resorption of other dibasic amino acids (lysine, ornithine, arginine) is also impaired but causes no problems because these amino acids have an alternative transport system separate from that shared with cystine 273. Furthermore, they are more soluble than cystine in urine, and their increased excretion does not result in crystal or stone formation. Their absorption (and that of cystine) is also decreased in the small bowel 273.
Cystinuria affects approximately 1 in 10,000 people. Cystine stones are most common in young adults under age 40 38.
Cystinuria is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition 272.
Cystinuria causes
Cystinuria is caused by changes (mutations) in the SLC3A1 and SLC7A9 genes 274, 275. The SLC3A1 and SLC7A9 genes provide instructions for making the two parts (subunits) of a protein complex that is primarily found in the kidneys. Normally this protein complex controls the reabsorption of certain amino acids, including cystine, into the blood from the filtered fluid that will become urine. Mutations in either the SLC3A1 gene or SLC7A9 gene disrupt the ability of the protein complex to reabsorb amino acids, which causes the amino acids to become concentrated in the urine. As the levels of cystine in the urine increase, the crystals typical of cystinuria form. The other amino acids that are reabsorbed by the protein complex do not create crystals when they accumulate in the urine.
Cystinuria is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition 274. Recessive genetic disorders occur when an individual inherits two copies of an altered gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. People who are carriers of these genes typically have slightly elevated levels of cystine and lysine in the urine. The full implications of being a carrier are not known and require further investigation. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
Cystinuria prevention
There are medicines that can be taken so cystine does not form a stone. Ask your doctor about these medicines and their side effects. Any person with a known history of stones in the urinary tract should drink plenty of fluids to regularly produce a high amount of urine. This allows stones and crystals to leave the body before they become large enough to cause symptoms. Decreasing your intake of salt or sodium will help as well.
When high fluid intake and alkalinization do not reduce stone formation, other drugs may be tried.
- Penicillamine (7.5 mg/kg po qid in young children and 125 mg to 0.5 g po qid in older children) improves cystine solubility, but toxicity limits its usefulness. About half of all patients develop some toxic manifestation, such as fever, rash, arthralgias, or, less commonly, nephrotic syndrome, pancytopenia, or SLE-like reaction.
- Pyridoxine supplements (50 mg po once/day) should be given with penicillamine.
- Tiopronin (100 mg to 300 mg po qid) can be used instead of penicillamine to treat some children because it has a lower frequency of adverse effects.
- Captopril (0.3 mg/kg po tid) is not as effective as penicillamine but is less toxic. Close monitoring of response to therapy is very important.
Cystinuria symptoms
People with cystinuria excrete abnormally high levels of cystine in the urine. The level of cystine is so high that it remains undissolved in the urine. The amino acids lysine, arginine, and ornithine are also excreted in massive amounts by people with this disorder, but they dissolve more readily in the urine and are not associated with any particular symptoms.
The initial symptom of cystinuria is usually sharp pain in the lower back or side of the abdomen (renal colic). Other symptoms may include blood in the urine (hematuria), obstruction of the urinary tract, and/or infections of the urinary tract. Frequent recurrences ultimately may lead to kidney damage.
People with cystinuria typically produce jagged stones that are small, though some form very large stones. Stones may be accompanied by urinary “gravel,” which consists of yellowish-brown hexagonal crystals. All patients with urinary stones should be screened for cystinuria.
Cystinuria symptoms include:
- Blood in the urine
- Renal colic: flank pain or pain in the side or back. Pain is most often on one side; it is rarely felt on both sides. Pain is often severe. It may get worse over days. You may also feel pain in the pelvis, groin, genitals, or between the upper abdomen and back.
- Most commonly renal colic, may occur in infants but usually appear between ages 10 and 30.
- UTI and renal failure due to urinary tract obstruction may develop.
Cystinuria diagnosis
The disorder is most often diagnosed after an episode of kidney stones. Testing the stones shows that they are made of cystine.
Unlike calcium-containing stones, cystine stones do not show up well on plain x-rays.
Tests that may be done to detect these stones and diagnose the condition include:
- 24-hour urine collection
- Abdominal CT scan, MRI, or ultrasound
- Intravenous pyelogram (IVP)
- Urinalysis
- Analysis of collected kidney stones
- Measurement of urinary cystine excretion
Cystinuria treatment
The goal of treatment is to relieve symptoms and prevent more stones from forming. A person with severe symptoms may need to go into the hospital.
Treatment involves:
- drinking plenty of fluids, especially water, to produce large amounts of urine. You should drink at least 6 to 8 glasses per day.
In some cases, fluids may need to be given through a vein (by IV).
End-stage renal disease may develop. Decreasing urinary cystine excretion decreases renal toxicity and is accomplished by increasing urine volume with fluid intake sufficient to provide a urine flow rate of 3 to 4 L/day.
- Hydration is particularly important at night when urinary pH drops.
- Alkalinization of the urine to pH > 7.0 with potassium citrate or potassium bicarbonate 1 mEq/kg po tid to qid and in some cases acetazolamide 5 mg/kg (up to 250 mg) po at bedtime increases the solubility of cystine significantly.
- Mild restrictions of dietary sodium (100 mEq/day) and protein (0.8 to 1.0 g/kg/day) may help reduce cystine excretion.
Medicines may be prescribed to help dissolve the cystine crystals. Eating less salt can also decrease cystine release and stone formation.
You may need pain relievers to control pain in the kidney or bladder area when you pass stones. Smaller stones most often pass through the urine on their own. Larger stones may need extra treatments.
Some large stones may need to be removed with surgery:
- Extracorporeal shock wave lithotripsy (ESWL): Sound waves are passed through the body and are focused on the stones to break them into small, passable fragments. ESWL may not work well for cystine stones because they are very hard as compared with other types of stones.
- Percutaneous nephrostolithotomy or nephrolithotomy: A small tube is placed through the flank directly into the kidney. A telescope is then passed through the tube to fragment the stone under direct vision.
- Ureteroscopy for stones in the lower urinary tract.
Cystinuria prognosis
Cystinuria is a chronic, lifelong condition. Stones commonly return. However, the condition rarely results in kidney failure. It does not affect other organs.
Cystinuria complications
Cystinuria complications may include:
- Bladder injury from stone
- Kidney injury from stone
- Kidney infection (pyelonephritis)
- Ureteral obstruction
- Urinary tract infection (UTI)
What is Urinary Tract Infections
Urinary tract infections (UTIs) can be divided into upper tract infections, which involve the kidneys (pyelonephritis), and lower tract infections, which involve the bladder (cystitis), urethra (urethritis), and prostate (prostatitis) 276. However, in practice, and particularly in children, differentiating between the sites may be difficult or impossible. Moreover, infection often spreads from one area to the other. Although urethritis and prostatitis are infections that involve the urinary tract, the term urinary tract infection (UTI) usually refers to pyelonephritis (inflammation of the kidney as a result of bacterial infection) and cystitis (inflammation of the urinary bladder, often caused by infection and is usually accompanied by frequent painful urination).
Most cystitis and pyelonephritis are caused by bacteria. The most common nonbacterial pathogens are fungi (usually candidal species), and, less commonly, mycobacteria, viruses, and parasites. Nonbacterial pathogens usually affect patients who are immunocompromised; have diabetes, obstruction, or structural urinary tract abnormalities; or have had recent urinary tract instrumentation.
Other than adenoviruses (implicated in hemorrhagic cystitis), viruses have no major contribution to urinary tract infection (UTI) in immunocompetent patients.
The predominant parasitic causes of urinary tract infections are filariasis, trichomoniasis, leishmaniasis, malaria, and schistosomiasis. Of the parasitic diseases, only trichomoniasis is common in the US, usually as a sexually transmitted disease (STD) 276.
Urethritis is usually caused by an STD 276. Prostatitis is usually caused by a bacterium and is sometimes caused by an STD 276.
Bacterial Urinary Tract Infections
Bacterial UTIs can involve the urethra, prostate, bladder, or kidneys. Symptoms may be absent or include urinary frequency, urgency, dysuria (painful or difficult urination), lower abdominal pain, and flank (the side of a person’s body between the ribs and the hip) pain. Systemic symptoms and even sepsis (chemicals released into the bloodstream to fight the infection trigger inflammatory responses throughout the body) may occur with kidney infection. Diagnosis is based on analysis and culture of urine. Treatment is with antibiotics and removal of any urinary tract catheters and obstructions.
Among adults aged 20 to 50 yr, UTIs are about 50-fold more common in women. In women in this age group, most UTIs are cystitis or pyelonephritis. In men of the same age, most UTIs are urethritis or prostatitis. The incidence of UTI increases in patients > 50 yr, but the female:male ratio decreases because of the increasing frequency of prostate enlargement and instrumentation in men.
Bacterial Urinary Tract Infections pathophysiology
The urinary tract, from the kidneys to the urethral meatus, is normally sterile and resistant to bacterial colonization despite frequent contamination of the distal urethra with colonic bacteria. The major defense against UTI is complete emptying of the bladder during urination. Other mechanisms that maintain the tract’s sterility include urine acidity, vesicoureteral valve, and various immunologic and mucosal barriers.
About 95% of UTIs occur when bacteria ascend the urethra to the bladder and, in the case of pyelonephritis, ascend the ureter to the kidney. The remainder of UTIs are hematogenous. Systemic infection can result from UTI, particularly in the elderly. About 6.5% of cases of hospital-acquired bacteremia are attributable to UTI.
Uncomplicated UTI is usually considered to be cystitis or pyelonephritis that occurs in premenopausal adult women with no structural or functional abnormality of the urinary tract and who are not pregnant and have no significant comorbidity that could lead to more serious outcomes. Also, some experts consider UTIs to be uncomplicated even if they affect postmenopausal women or patients with well-controlled diabetes. In males, most UTIs occur in children or elderly patients, are due to anatomic abnormalities or instrumentation, and are considered complicated.
The rare UTIs that occur in men aged 15 to 50 yr are usually in men who have unprotected anal intercourse or in those who have an uncircumcised penis, and they are generally considered uncomplicated. UTIs in men this age who do not have unprotected anal intercourse or an uncircumcised penis are very rare and, although also considered uncomplicated, warrant evaluation for urologic abnormalities.
Complicated UTI can involve either sex at any age. It is usually considered to be pyelonephritis or cystitis that does not fulfill criteria to be considered uncomplicated. A UTI is considered complicated if the patient is a child, is pregnant, or has any of the following:
- A structural or functional urinary tract abnormality and obstruction of urine flow
- A comorbidity that increases risk of acquiring infection or resistance to treatment, such as poorly controlled diabetes, chronic kidney disease, or immunocompromise.
- Recent instrumentation or surgery of the urinary tract
Risk factors for Bacterial Urinary Tract Infections
Risk factors for development of UTI in women include the following:
- Sexual intercourse
- Diaphragm and spermicide use
- Antibiotic use
- New sex partner within the past year
- History of UTIs in 1st-degree female relatives
- History of recurrent UTIs
- First UTI at early age
Even use of condoms that are spermicide-coated increases risk of UTI in women. The increased risk of UTI in women using antibiotics or spermicides probably occurs because of alterations in vaginal flora that allow overgrowth of Escherichia coli (E.coli). In elderly women, soiling of the perineum due to fecal incontinence increases risk.
Anatomic, structural, and functional abnormalities are risk factors for UTI. A common consequence of anatomic abnormality is vesicoureteral reflux (VUR), which is present in 30 to 45% of young children with symptomatic UTI. VUR is usually caused by a congenital defect that results in incompetence of the ureterovesical valve. VUR can also be acquired in patients with a flaccid bladder due to spinal cord injury or after urinary tract surgery. Other anatomic abnormalities predisposing to UTI include urethral valves (a congenital obstructive abnormality), delayed bladder neck maturation, bladder diverticulum, and urethral duplications.
Structural and functional urinary tract abnormalities that predispose to UTI usually involve obstruction of urine flow and poor bladder emptying. Urine flow can be compromised by calculi and tumors. Bladder emptying can be impaired by neurogenic dysfunction (see Neurogenic Bladder), pregnancy, uterine prolapse, cystocele, and prostatic enlargement. UTI caused by congenital factors manifests most commonly during childhood. Most other risk factors are more common in the elderly.
Other risk factors for UTI include instrumentation (eg, bladder catheterization, stent placement, cystoscopy) and recent surgery.
Bacterial Urinary Tract Infections causes
The bacteria that most often cause cystitis and pyelonephritis are the following:
- Enteric (relating to the intestines), usually gram-negative aerobic bacteria (most often)
- Gram-positive bacteria (less often)
In normal genitourinary tracts, strains of Escherichia coli with specific attachment factors for transitional epithelium of the bladder and ureters account for 75 to 95% of cases. The remaining gram-negative urinary pathogens are usually other enterobacteria, typically Klebsiella or Proteus mirabilis, and occasionally Pseudomonas aeruginosa. Among gram-positive bacteria, Staphylococcus saprophyticus is isolated in 5 to 10% of bacterial UTIs. Less common gram-positive bacterial isolates are Enterococcus faecalis (group D streptococci) and Streptococcus agalactiae (group B streptococci), which may be contaminants, particularly if they were isolated from patients with uncomplicated cystitis.
In hospitalized patients, E. coli accounts for about 50% of cases. The gram-negative species Klebsiella, Proteus, Enterobacter, Pseudomonas, and Serratia account for about 40%, and the gram-positive bacterial cocci, E. faecalis, S. saprophyticus, and Staphylococcus aureus account for the remainder.
Bacterial Urinary Tract Infections prevention
In women who experience ≥ 3 UTIs/yr, behavioral measures are recommended, including increasing fluid intake, avoiding spermicides and diaphragm use, not delaying urination, wiping front to back after defecation, avoiding douching, and urinating immediately after sexual intercourse. Although some evidence shows that cranberry products prevent UTI in women, others do not; the optimal dose is unknown; and they can have high amounts of oxalates (possibly increasing risk of oxalate stones). Thus, most experts do not recommend use of cranberry products for prevention of symptomatic UTI in women.
If these techniques are unsuccessful, antibiotic prophylaxis should be considered. Common options are continuous and postcoital prophylaxis.
Continuous prophylaxis commonly begins with a 6 mo trial. If UTI recurs after 6 mo of prophylactic therapy, prophylaxis may be reinstituted for 2 or 3 yr. Choice of antibiotic depends on susceptibility patterns of prior infections. Common options are trimethoprim/sulfamethoxazole 40/200 mg orally once/day or 3 times per week, nitrofurantoin 50 or 100 mg orally once/day, cephalexin 125 to 250 mg orally once/day, and fosfomycin 3 g po q 10 days. Fluoroquinolones are effective but are not usually recommended because resistance is increasing. Also, fluoroquinolones are contraindicated in pregnant women and children. Nitrofurantoin is contraindicated if creatinine clearance is < 60 mL/min. Long-term use can rarely cause damage to the lungs, liver, and nervous system.
Postcoital prophylaxis in women may be more effective if UTIs are temporally related to sexual intercourse. Usually, a single dose of one of the drugs used for continuous prophylaxis (other than fosfomycin) is effective.
Contraception is recommended for women using a fluoroquinolone because these drugs can potentially injure a fetus. Although concern exists that antibiotics may decrease the effectiveness of oral contraceptives, pharmacokinetic studies have not shown a significant or consistent effect. Nonetheless, some experts still recommend that women who use oral contraceptives use barrier contraceptives while they are taking antibiotics.
In pregnant women, effective prophylaxis of UTI is similar to that in nonpregnant women, including use of postcoital prophylaxis. Appropriate patients include those with acute pyelonephritis during a pregnancy, patients with > 1 episode (despite treatment) of UTI or bacteriuria during pregnancy, and patients who required prophylaxis for recurrent UTI before pregnancy.
In postmenopausal women, antibiotic prophylaxis is similar to that described previously. Additionally, topical estrogen therapy markedly reduces the incidence of recurrent UTI in women with atrophic vaginitis or atrophic urethritis.
Classification of Bacterial Urinary Tract Infections
Urethritis
Infection of the urethra with bacteria (or with protozoa, viruses, or fungi) occurs when organisms that gain access to it acutely or chronically colonize the numerous periurethral glands in the bulbous and pendulous portions of the male urethra and in the entire female urethra. The sexually transmitted pathogens Chlamydia trachomatis (Chlamydial, Mycoplasmal, and Ureaplasmal Mucosal Infections), Neisseria gonorrhoeae (Gonorrhea infection), Trichomonas vaginalis (Trichomoniasis), and herpes simplex virus are common causes in both sexes.
Cystitis
Cystitis is infection of the bladder. It is common in women, in whom cases of uncomplicated cystitis are usually preceded by sexual intercourse (honeymoon cystitis). In men, bacterial infection of the bladder is usually complicated and usually results from ascending infection from the urethra or prostate or is secondary to urethral instrumentation. The most common cause of recurrent cystitis in men is chronic bacterial prostatitis.
Acute urethral syndrome
Acute urethral syndrome, which occurs in women, is a syndrome involving dysuria, frequency, and pyuria (dysuria-pyuria syndrome), which thus resembles cystitis. However, in acute urethral syndrome (unlike in cystitis), routine urine cultures are either negative or show colony counts that are lower than the traditional criteria for diagnosis of bacterial cystitis. Urethritis is a possible cause because causative organisms include Chlamydia trachomatis and Ureaplasma urealyticum, which are not detected on routine urine culture.
Noninfectious causes have been proposed, but supporting evidence is not conclusive, and most noninfectious causes usually cause little or no pyuria. Possible noninfectious causes include anatomic abnormalities (eg, urethral stenosis), physiologic abnormalities (eg, pelvic floor muscle dysfunction), hormonal imbalances (eg, atrophic urethritis), localized trauma, gastrointestinal system symptoms, and inflammation.
Asymptomatic bacteriuria
Asymptomatic bacteriuria is absence of UTI signs or symptoms in a patient whose urine culture satisfies criteria for UTI. Pyuria may or may not be present. Because it is asymptomatic, such bacteriuria is found mainly when high-risk patients are screened or when urine culture is done for other reasons.
Screening patients for asymptomatic bacteriuria is indicated for those at risk of complications if the bacteriuria is untreated. Such patients include:
- Pregnant women at 12 to 16 wks’ gestation or at the first prenatal visit, if later (because of the risk of symptomatic UTI, including pyelonephritis, during pregnancy; and adverse pregnancy outcomes, including low-birth-weight neonate and premature delivery).
- Patients who have had a kidney transplant within the previous 6 months.
- Young children with gross vesicoureteral reflux (the backward flow of urine from the bladder into the kidneys).
- Before certain invasive genitourinary procedures that can cause mucosal bleeding (eg, transurethral resection of the prostate).
Certain patients (eg, postmenopausal women; patients with controlled diabetes; patients with ongoing use of urinary tract foreign objects such as stents, nephrostomy tubes, and indwelling catheters) often have persistent asymptomatic bacteriuria and sometimes pyuria. However, such patients should not be screened because they are at low risk of complicated UTI due to the bacteriuria and thus do not require treatment. Also, in patients with indwelling catheters, treatment often fails to clear the bacteriuria and only leads to development of highly antibiotic-resistant organisms.
Acute pyelonephritis
Pyelonephritis is bacterial infection of the kidney parenchyma. The term should not be used to describe tubulointerstitial nephropathy unless infection is documented. In women, about 20% of community-acquired bacteremias are due to pyelonephritis. Pyelonephritis is uncommon in men with a normal urinary tract.
In 95% of cases of pyelonephritis, the cause is ascension of bacteria through the urinary tract. Although obstruction (eg, strictures, calculi, tumors, neurogenic bladder, VUR) predisposes to pyelonephritis, most women with pyelonephritis have no demonstrable functional or anatomic defects. In men, pyelonephritis is always due to some functional or anatomic defect. Cystitis alone or anatomic defects may cause reflux. The risk of bacterial ascension is greatly enhanced when ureteral peristalsis is inhibited (eg, during pregnancy, by obstruction, by endotoxins of gram-negative bacteria). Pyelonephritis is common in young girls and in pregnant women after bladder catheterization.
Pyelonephritis not caused by bacterial ascension is caused by hematogenous spread, which is particularly characteristic of virulent organisms such as S. aureus, P. aeruginosa, Salmonella species, and Candida species.
The affected kidney is usually enlarged because of inflammatory PMNs and edema. Infection is focal and patchy, beginning in the pelvis and medulla and extending into the cortex as an enlarging wedge. Cells mediating chronic inflammation appear within a few days, and medullary and subcortical abscesses may develop. Normal parenchymal tissue between foci of infection is common.
Papillary necrosis may be evident in acute pyelonephritis associated with diabetes, obstruction, sickle cell disease, pyelonephritis in renal transplants, pyelonephritis due to candidiasis, or analgesic nephropathy.
Although acute pyelonephritis is frequently associated with renal scarring in children, similar scarring in adults is not detectable in the absence of reflux or obstruction.
Bacterial Urinary Tract Infections signs and symptoms
Elderly patients and patients with a neurogenic bladder or an indwelling catheter may present with sepsis and delirium but without symptoms referable to the urinary tract.
When symptoms are present, they may not correlate with the location of the infection within the urinary tract because there is considerable overlap; however, some generalizations are useful.
In urethritis, the main symptoms are dysuria and, primarily in men, urethral discharge. Discharge can be purulent, whitish, or mucoid. Characteristics of the discharge, such as the amount of purulence, do not reliably differentiate gonococcal from nongonococcal urethritis.
Cystitis onset is usually sudden, typically with frequency, urgency, and burning or painful voiding of small volumes of urine. Nocturia, with suprapubic pain and often low back pain, is common. The urine is often turbid, and microscopic (or rarely gross) hematuria can occur. A low-grade fever may develop. Pneumaturia (passage of air in the urine) can occur when infection results from a vesicoenteric or vesicovaginal fistula or from emphysematous cystitis.
In acute pyelonephritis, symptoms may be the same as those of cystitis. One third of patients have frequency and dysuria. However, with pyelonephritis, symptoms typically include chills, fever, flank pain, colicky abdominal pain, nausea, and vomiting. If abdominal rigidity is absent or slight, a tender, enlarged kidney is sometimes palpable. Costovertebral angle percussion tenderness is generally present on the infected side. In urinary tract infection in children, symptoms often are meager and less characteristic.
Bacterial Urinary Tract Infections diagnosis
- Urinalysis
- Sometimes urine culture
Diagnosis by culture is not always necessary. If done, diagnosis by culture requires demonstration of significant bacteriuria in properly collected urine.
Urine collection
If a sexually transmitted disease (STD) is suspected, a urethral swab for STD testing is obtained prior to voiding. Urine collection is then by clean-catch or catheterization.
To obtain a clean-catch, midstream specimen, the urethral opening is washed with a mild, nonfoaming disinfectant and air dried. Contact of the urinary stream with the mucosa should be minimized by spreading the labia in women and by pulling back the foreskin in uncircumcised men. The first 5 mL of urine is not captured; the next 5 to 10 mL is collected in a sterile container.
A specimen obtained by catheterization is preferable in older women (who typically have difficulty obtaining a clean-catch specimen) and in women with vaginal bleeding or discharge. Many clinicians also use catheterization to obtain a specimen if evaluation includes a pelvic examination. Diagnosis in patients with indwelling catheters is discussed elsewhere (see Catheter-Associated Urinary Tract Infections (CAUTIs) : Diagnosis).
Testing, particularly culturing, should be done within 2 h of specimen collection; if not, the sample should be refrigerated.
Urine testing
Microscopic examination of urine is useful but not definitive. Pyuria is defined as ≥ 8 WBCs/μL of uncentrifuged urine, which corresponds to 2 to 5 WBCs/high-power field in spun sediment. Most truly infected patients have > 10 WBCs/μL. The presence of bacteria in the absence of pyuria, especially when several strains are found, is usually due to contamination during sampling. Microscopic hematuria occurs in up to 50% of patients, but gross hematuria is uncommon. WBC casts, which may require special stains to differentiate from renal tubular casts, indicate only an inflammatory reaction; they can be present in pyelonephritis, glomerulonephritis, and noninfective tubulointerstitial nephritis.
Pyuria in the absence of bacteriuria and of UTI is possible, for example, if patients have nephrolithiasis, a uroepithelial tumor, appendicitis, or inflammatory bowel disease or if the sample is contaminated by vaginal WBCs. Women who have dysuria and pyuria but without significant bacteriuria have the urethral syndrome or dysuria-pyuria syndrome.
Dipstick tests also are commonly used. A positive nitrite test on a freshly voided specimen (bacterial replication in the container renders results unreliable if the specimen is not tested rapidly) is highly specific for UTI, but the test is not very sensitive. The leukocyte esterase test is very specific for the presence of > 10 WBCs/μL and is fairly sensitive. In adult women with uncomplicated UTI with typical symptoms, most clinicians consider positive microscopic and dipstick tests sufficient; in these cases, given the likely pathogens, cultures are unlikely to change treatment but add significant expense.
Cultures are recommended in patients whose characteristics and symptoms suggest complicated UTI or an indication for treatment of bacteriuria. Common examples include the following:
- Pregnant women
- Postmenopausal women
- Men
- Prepubertal children
- Patients with urinary tract abnormalities or recent instrumentation
- Patients with immunosuppression or significant comorbidities
- Patients whose symptoms suggest pyelonephritis or sepsis
- Patients with recurrent UTIs (≥ 3/yr)
Samples containing large numbers of epithelial cells are contaminated and unlikely to be helpful. An uncontaminated specimen must be obtained for culture. Culture of a morning specimen is most likely to detect UTI. Samples left at room temperature for > 2 h can give falsely high colony counts due to continuing bacterial proliferation. Criteria for culture positivity include isolation of a single bacterial species from a midstream, clean catch, or catheterized urine specimen.
For asymptomatic bacteriuria, criteria for culture positivity based on the guidelines of the Infectious Diseases Society of America are:
- Two consecutive clean-catch, voided specimens (for men, one specimen) from which the same bacterial strain is isolated in colony counts of >105/mL
- Among women or men, in a catheter-obtained specimen, a single bacterial species is isolated in colony counts of > 102/mL
For symptomatic patients, culture criteria are:
- Uncomplicated cystitis in women: > 103/mL
- Uncomplicated cystitis in women: > 102/mL (This quantification may be considered to improve sensitivity to E. coli.)
- Acute, uncomplicated pyelonephritis in women: > 104/mL
- Complicated UTI: > 105/mL in women; or > 104/mL in men or from a catheter-derived specimen in women
- Acute urethral syndrome: > 102/mL of a single bacterial species
Any positive culture result, regardless of colony count, in a sample obtained via suprapubic bladder puncture should be considered a true positive.
In midstream urine, E. coliin mixed flora may be a true pathogen 277.
Occasionally, UTI is present despite lower colony counts, possibly because of prior antibiotic therapy, very dilute urine (specific gravity < 1.003), or obstruction to the flow of grossly infected urine. Repeating the culture improves the diagnostic accuracy of a positive result, ie, may differentiate between a contaminant and a true positive result.
Infection localization
Clinical differentiation between upper and lower UTI is impossible in many patients, and testing is not usually advisable. When the patient has high fever, costovertebral angle tenderness, and gross pyuria with casts, pyelonephritis is highly likely. The best noninvasive technique for differentiating bladder from kidney infection appears to be the response to a short course of antibiotic therapy. If the urine has not cleared after 3 days of treatment, pyelonephritis should be sought.
Symptoms similar to those of cystitis and urethritis can occur in patients with vaginitis, which may cause dysuria due to the passage of urine across inflamed labia. Vaginitis can often be distinguished by the presence of vaginal discharge, vaginal odor, and dyspareunia.
Other testing
Seriously ill patients require evaluation for sepsis, typically with complete blood count, electrolytes, lactate, blood urea nitrogen (BUN – test measures the amount of nitrogen in your blood that comes from the waste product urea), creatinine, and blood cultures. Patients with abdominal pain or tenderness are evaluated for other causes of an acute abdomen.
Patients who have dysuria/pyuria but no bacteriuria should have testing for an STD, typically using nucleic acid-based testing of swabs from the urethra and cervix.
Most adults do not require assessment for structural abnormalities unless the following occur:
- The patient has ≥ 2 episodes of pyelonephritis.
- Infections are complicated.
- Nephrolithiasis is suspected.
- There is painless gross hematuria or new renal insufficiency.
- Fever persists for ≥ 72 h.
Urinary tract imaging choices include ultrasonography, CT, and IVU. Occasionally, voiding cystourethrography, retrograde urethrography, or cystoscopy is warranted. Urologic investigation is not routinely needed in women with symptomatic cystitis or asymptomatic recurrent cystitis, because findings do not influence therapy. Children with UTI often require imaging.
Bacterial Urinary Tract Infections treatment
Antibiotics
Occasionally surgery (eg, to drain abscesses, correct underlying structural abnormalities, or relieve obstruction)
All forms of symptomatic bacterial UTI require antibiotics. For patients with troublesome dysuria, phenazopyridine may help control symptoms until the antibiotics do (usually within 48 h).
Choice of antibiotic should be based on the patient’s allergy and adherence history, local resistance patterns (if known), antibiotic availability and cost, and patient and provider tolerance for risk of treatment failure. Propensity for inducing antibiotic resistance should also be considered. When urine culture is done, choice of antibiotic should be modified when culture and sensitivity results are available to the most narrow-spectrum drug effective against the identified pathogen.
Surgical correction is usually required for obstructive uropathy, anatomic abnormalities, and neuropathic urinary tract lesions such as compression of the spinal cord. Catheter drainage of an obstructed urinary tract aids in prompt control of UTI. Occasionally, a renal cortical abscess or perinephric abscess requires surgical drainage. Instrumentation of the lower urinary tract in the presence of infected urine should be deferred if possible. Sterilization of the urine before instrumentation and antibiotic therapy for 3 to 7 days after instrumentation can prevent life-threatening urosepsis.
Urethritis
Sexually active patients with symptoms are usually treated presumptively for STDs pending test results. A typical regimen is ceftriaxone 250 mg IM plus either azithromycin 1 g orally once or doxycycline 100 mg po bid for 7 days. All sex partners within 60 days should be evaluated. Men diagnosed with urethritis should be tested for HIV and syphilis in accordance with the Centers for Disease Control and Prevention’s 2015 Sexually Transmitted Diseases Treatment Guidelines.
Cystitis
First-line treatment of uncomplicated cystitis is nitrofurantoin 100 mg po bid for 5 days (it is contraindicated if creatinine clearance is < 60 mL/min), trimethoprim/sulfamethoxazole 160/800 mg orally twice daily for 3 days, or fosfomycin 3 g orally once. Less desirable choices include a fluoroquinolone or a beta-lactam antibiotic. If cystitis recurs within a week or two, a broader spectrum antibiotic (eg, a fluoroquinolone) can be used and the urine should be cultured.
Complicated cystitis should be treated with empiric broad-spectrum antibiotics chosen based on local pathogens and resistance patterns and adjusted based on culture results. Urinary tract abnormalities must also be managed.
Acute urethral syndrome
Treatment depends on clinical findings and urine culture results:
- Women with dysuria, pyuria, and colony growth of > 102/mL of a single bacterial species on urine culture can be treated as for uncomplicated cystitis.
- Women who have dysuria and pyuria with no bacteriuria should be evaluated for an STD (including for N. gonorrhoeae and C. trachomatis).
- Women who have dysuria but neither pyuria nor bacteriuria do not have the true urethral syndrome. They should be evaluated for noninfectious causes of dysuria. Evaluation may include therapeutic trials, for example, of behavioral treatments (eg, biofeedback and pelvic musculature relaxation), surgery (for urethral stenosis), and drugs (eg, hormone replacement for suspected atrophic urethritis, anesthetics, antispasmodics).
Asymptomatic bacteriuria
Typically, asymptomatic bacteriuria in patients with diabetes, elderly patients, or patients with chronically indwelling bladder catheters should not be treated. However, patients at risk of complications from asymptomatic bacteriuria (see Asymptomatic bacteriuria) should have any treatable causes addressed and be given antibiotics as for cystitis. In pregnant women, only a few antibiotics can be safely used. Oral beta-lactams, sulfonamides, and nitrofurantoin are considered safe in early pregnancy, but trimethoprim should be avoided during the 1st trimester, and sulfamethoxazole should be avoided during the 3rd trimester, particularly near parturition. Patients with untreatable obstructive problems (eg, calculi, reflux) may require long-term suppressive therapy.
Acute pyelonephritis
Antibiotics are required. Outpatient treatment with oral antibiotics is possible if all of the following criteria are satisfied:
- Patients are expected to be adherent
- Patients are immunocompetent
- Patients have no nausea or vomiting or evidence of volume depletion or septicemia
- Patients have no factors suggesting complicated UTI
Ciprofloxacin 500 mg po bid for 7 days and levofloxacin 750 mg po once/day for 5 days are 1st-line antibiotics if < 10% of the uropathogens in the community are resistant. A 2nd option is usually trimethoprim/sulfamethoxazole 160/800 mg orally twice per day for 14 days. However, local sensitivity patterns should be considered because in some parts of the US, > 20% of E. coli are resistant to sulfa.
Patients not eligible for outpatient treatment should be hospitalized and given parenteral therapy selected on the basis of local sensitivity patterns. First-line antibiotics are usually renally excreted fluoroquinolones, such as ciprofloxacin and levofloxacin. Other choices, such as ampicillin plus gentamicin, broad-spectrum cephalosporins (eg, ceftriaxone, cefotaxime, cefepime), aztreonam, beta-lactam/beta-lactam inhibitor combinations (ampicillin/sulbactam, ticarcillin/clavulanate, piperacillin/tazobactam), and imipenem/cilastatin, are usually reserved for patients with more complicated pyelonephritis (eg, with obstruction, calculi, resistant bacteria, or a hospital-acquired infection) or recent urinary tract instrumentation.
Parenteral therapy is continued until defervescence and other signs of clinical improvement occur. In > 80% of patients, improvement occurs within 72 h. Oral therapy can then begin, and the patient can be discharged for the remainder of a 7- to14-day treatment course. Complicated cases require longer courses of IV antibiotics with total duration of 2 to 3 wk and urologic correction of anatomic defects.
Outpatient management can be considered in pregnant women with pyelonephritis, but only if symptoms are mild, close follow-up is available, and (preferably) pregnancy is < 24 wk gestation. Outpatient treatment is with cephalosporins (eg, ceftriaxone 1 to 2 g IV or IM, then cephalexin 500 mg po qid for 10 days). Otherwise, 1st-line IV antibiotics include cephalosporins, aztreonam, or ampicillin plus gentamicin. If pyelonephritis is severe, possibilities include piperacillin/tazobactam or meropenem. Fluoroquinolones and trimethoprim/sulfamethoxazole should be avoided. Because recurrence is common, some authorities recommend prophylaxis after the acute infection resolves with nitrofurantoin 100 mg orally or cephalexin 250 mg orally every night during the remainder of the pregnancy and for 4 to 6 wk after pregnancy.
What are Cystic Kidney diseases
A cyst is a fluid-filled sac. You may get simple kidney cysts as you age; they are usually harmless. There are also some diseases which cause kidney cysts. One type is polycystic kidney disease (PKD). It runs in families. In polycystic kidney disease, many cysts grow in the kidneys. This can enlarge the kidneys and make them work poorly. About half of people with the most common type of polycystic kidney disease end up with kidney failure. Polycystic kidney disease also causes cysts in other parts of the body, such as the liver.
Often, there are no symptoms at first. Later, symptoms include
- Pain in the back and lower sides
- Headaches
- Blood in the urine
Doctors diagnose polycystic kidney disease with imaging tests and family history. There is no cure. Treatments can help with symptoms and complications. They include medicines and lifestyle changes, and if there is kidney failure, dialysis or kidney transplants.
Figure 4. Polycystic Kidney Disease
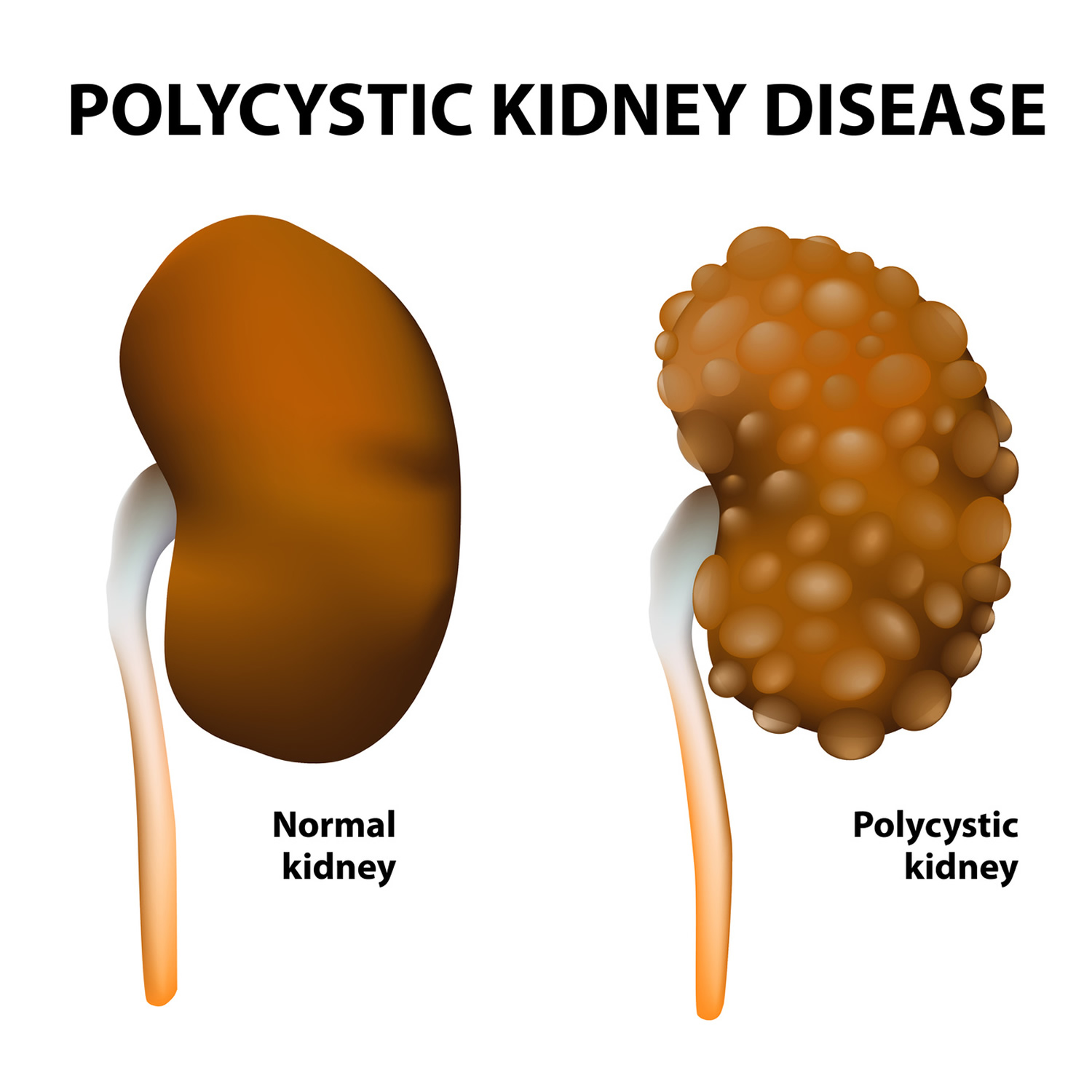
What are the differences between acquired cystic kidney disease and polycystic kidney disease ?
Acquired cystic kidney disease differs from polycystic kidney disease in several ways. Unlike acquired cystic kidney disease, polycystic kidney disease is a genetic, or inherited, disorder that can cause complications such as high blood pressure and problems with blood vessels in the brain and heart.
The following lists the differences:
People with Polycystic Kidney Disease
- are born with a gene that causes the disease
- have enlarged kidneys
- develop cysts in the liver and other parts of the body.
People with Acquired Cystic Kidney Disease
- do not have a disease-causing gene
- have kidneys that are normal-sized or smaller
- do not form cysts in other parts of the body.
In addition, for people with polycystic kidney disease, the presence of cysts marks the onset of their disease, while people with acquired cystic kidney disease already have chronic kidney disease when they develop cysts.
What is Polycystic Kidney Disease?
Polycystic kidney disease is is a genetic disorder that causes many fluid-filled cysts to grow in your kidneys and other organs. Clusters of fluid-filled sacs, called cysts, develop in the kidneys and interfere with their ability to filter waste products from the blood. Unlike the usually harmless simple kidney cysts that can form in the kidneys later in life, polycystic kidney disease cysts can change the shape of your kidneys, including making them much larger 278. The growth of cysts causes the kidneys to become enlarged and can lead to kidney failure. Cysts may also develop in other organs, particularly the liver.
Polycystic kidney disease is a fairly common genetic disorder. It affects about 500,000 people in the United States 279. The autosomal dominant form of the disease is much more common than the autosomal recessive form. Autosomal dominant polycystic kidney disease affects 1 in 500 to 1,000 people, while the autosomal recessive type occurs in an estimated 1 in 20,000 to 40,000 people 279.
Polycystic kidney disease also can cause other complications, or problems, such as dangerously high blood pressure, pain in the back or sides, blood in the urine (hematuria), recurrent urinary tract infections, kidney stones, and heart valve abnormalities, cysts in the liver and problems with blood vessels in your brain and heart 278. Additionally, people with polycystic kidney disease have an increased risk of an abnormal bulging (an aneurysm) in a large blood vessel called the aorta or in blood vessels at the base of the brain. Aneurysms can be life-threatening if they tear or rupture 279.
Polycystic kidney disease is associated with the following conditions:
- Aortic aneurysms
- Brain aneurysms
- Cysts in the liver, pancreas, and testes
- Diverticula of the colon
As many as half of people with polycystic kidney disease have cysts in the liver 280.
Polycystic kidney disease is a form of chronic kidney disease that reduces kidney function and may lead to kidney failure.
The two major forms of polycystic kidney disease are distinguished by the usual age of onset and the pattern in which it is passed through families. The autosomal dominant form (sometimes called Autosomal Dominant Polycystic kidney disease or ADPKD) has signs and symptoms that typically begin in adulthood, although cysts in the kidney are often present from birth or childhood. Autosomal dominant polycystic kidney disease can be further divided into type 1 and type 2, depending on the genetic cause. The autosomal recessive form of polycystic kidney disease (sometimes called Autosomal Recessive Polycystic kidney disease or ARPKD) is much rarer and is often lethal early in life. The signs and symptoms of this condition are usually apparent at birth or in early infancy.
How common is Polycystic kidney disease?
Polycystic kidney disease is one of the most common genetic disorders. Polycystic kidney disease affects about 500,000 people in the United States.
ADPKD affects 1 in every 400 to 1,000 people in the world, and Autosomal Recessive Polycystic kidney disease (ARPKD) affects 1 in 20,000 children 281, 282.
Who is more likely to have Polycystic kidney disease ?
Polycystic kidney disease affects people of all ages, races, and ethnicities worldwide. The disorder occurs equally in women and men.
What can you do to slow down polycystic kidney disease?
The sooner you know you or your child has polycystic kidney disease, the sooner you can keep the condition from getting worse. Getting tested if you or your child are at risk for polycystic kidney disease can help you take early action.
You also can take steps to help delay or prevent kidney failure. Healthy lifestyle practices such as being active, reducing stress, and quitting smoking can help.
Make lifestyle changes To Slow Down Polycystic Kidney Disease
- Be active for 30 minutes or more on most days. Regular physical activity can help you reduce stress, manage your weight, and control your blood pressure. If you are not active now, ask your health care provider about how much and what type of physical activity is right for you.
If you play contact sports, such as football or hockey, a health care provider should do a magnetic resonance imaging (MRI) test to see whether these sports are safe for you. Trauma to your body, especially to your back and sides, may cause kidney cysts to burst.
- Lose weight. Being overweight makes your kidneys work harder. Losing weight helps protect your kidneys.
- Aim for 7 to 8 hours of sleep each night. Getting enough sleep is important to your overall physical and mental health and can help you manage your blood pressure and blood glucose, or blood sugar.
- Reduce stress. Long-term stress can raise your blood pressure and even lead to depression. Some of the steps you take to manage your polycystic kidney disease are also healthy ways to cope with stress. For example, getting enough physical activity and sleep helps reduce stress.
- Quit smoking. Cigarette smoking can raise your blood pressure, making your kidney damage worse. Quitting smoking may help you meet your blood pressure goals, which is good for your kidneys and can lower your chances of having a heart attack or stroke. Quitting smoking is even more important for people with polycystic kidney disease who have aneurysms. An aneurysm is a bulge in the wall of a blood vessel.
- Change what you eat and drink. You may need to change what you eat and drink to help control your blood pressure and protect your kidneys. People with any kind of kidney disease, including polycystic kidney disease, should talk with a dietitian about which foods and drinks to include in their healthy eating plan and which may be harmful. Staying hydrated by drinking the right amount of fluid may help slow polycystic kidney disease’s progress toward kidney failure. Read more about what to eat or drink if you have polycystic kidney disease or are at risk for polycystic kidney disease.
Polycystic kidney disease causes
A gene mutation, or defect, causes polycystic kidney disease. In most polycystic kidney disease cases, a child got the gene mutation from a parent. In a small number of polycystic kidney disease cases, the gene mutation developed on its own, without either parent carrying a copy of the mutated gene. This type of mutation is called “spontaneous.”
The two main types of polycystic kidney disease are:
- Autosomal Dominant Polycystic kidney disease (ADPKD), which is usually diagnosed in adulthood
- Autosomal Recessive Polycystic kidney disease (ARPKD), which can be diagnosed in the womb or shortly after a baby is born.
Inheritance Pattern and Genetic Changes of Polycystic kidney disease
Mutations in the PKD1, PKD2, and PKHD1 genes cause polycystic kidney disease 279.
Mutations in either the PKD1 or PKD2 gene can cause autosomal dominant polycystic kidney disease; PKD1 gene mutations cause Autosomal Dominant Polycystic kidney disease (ADPKD) type 1, and PKD2 gene mutations cause Autosomal Dominant Polycystic kidney disease (ADPKD) type 2 279. These genes provide instructions for making proteins whose functions are not fully understood. Researchers believe that they are involved in transmitting chemical signals from outside the cell to the cell’s nucleus. The two proteins work together to promote normal kidney development, organization, and function. Mutations in the PKD1 or PKD2 gene lead to the formation of thousands of cysts, which disrupt the normal functions of the kidneys and other organs 279. People with mutations in the PKD2 gene, particularly women, typically have a less severe form of the disease than people with PKD1 mutations 279. The signs and symptoms, including a decline in kidney function, tend to appear later in adulthood in people with a PKD2 mutation.
Mutations in the PKHD1 gene cause autosomal recessive polycystic kidney disease 279. This gene provides instructions for making a protein whose exact function is unknown; however, the protein likely transmits chemical signals from outside the cell to the cell nucleus. Researchers have not determined how mutations in the PKHD1 gene lead to the formation of numerous cysts characteristic of polycystic kidney disease.
Although polycystic kidney disease is usually a genetic disorder, a small percentage of cases are not caused by gene mutations 279. These cases are called acquired polycystic kidney disease. This form of the disorder occurs most often in people with other types of kidney disease who have been treated for several years with hemodialysis (a procedure that filters waste products from the blood).
Most cases of polycystic kidney disease have an autosomal dominant pattern of inheritance 279. People with this condition are born with one mutated copy of the PKD1 or PKD2 gene in each cell. In about 90 percent of these cases, an affected person inherits the mutation from one affected parent. The other 10 percent of cases result from a new mutation in one of the genes and occur in people with no history of the disorder in their family.
Although one altered copy of a gene in each cell is sufficient to cause the disorder, an additional mutation in the second copy of the PKD1 or PKD2 gene may make cysts grow faster and increase the severity of the disease 279. The rate at which cysts enlarge and cause a loss of kidney function varies widely, and may be influenced by mutations in other genes that have not been identified.
Polycystic kidney disease also can be inherited in an autosomal recessive pattern. People with this form of the condition have two altered copies of the PKHD1 gene in each cell. The parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene.
Polycystic kidney disease prevention
Researchers have not yet found a way to prevent polycystic kidney disease. However, you may be able to slow polycystic kidney disease problems caused by high blood pressure, such as kidney damage. Aim for a blood pressure goal of less than 120/80. Work with a health care team to help manage your or your child’s polycystic kidney disease. The health care team will probably include a general practitioner and a nephrologist, a health care provider specializing in kidney health.
Polycystic kidney disease signs and symptoms
The signs and symptoms of Autosomal Dominant Polycystic kidney disease (ADPKD), such as pain, high blood pressure, and kidney failure, are also polycystic kidney disease complications. In many cases, Autosomal Dominant Polycystic kidney disease (ADPKD) does not cause signs or symptoms until your kidney cysts are a half inch or larger in size.
Early signs of Autosomal Recessive Polycystic kidney disease (ARPKD) in the womb are larger-than-normal kidneys and a smaller-than-average size baby, a condition called growth failure. The early signs of Autosomal Recessive Polycystic kidney disease (ARPKD) are also complications. However, some people with Autosomal Recessive Polycystic kidney disease (ARPKD) do not develop signs or symptoms until later in childhood or even adulthood.
Symptoms of polycystic kidney disease may include any of the following:
- Abdominal pain or tenderness
- Blood in the urine
- Excessive urination at night
- Flank pain on one or both sides
- Drowsiness
- Joint pain
- Nail abnormalities
Polycystic kidney disease complications
Health problems that may result from polycystic kidney disease include:
- Anemia
- Bleeding or rupture of cysts
- Chronic kidney disease
- End-stage kidney disease
- High blood pressure
- Infection of liver cysts
- Kidney stones
- Liver failure (mild to severe)
- Repeated urinary tract infections
Polycystic kidney disease diagnosis
An examination may show:
- Abdominal tenderness over the liver
- Enlarged liver
- Heart murmurs or other signs of aortic insufficiency or mitral insufficiency
- High blood pressure
- Growths in the kidneys or abdomen
Tests that may be done include:
- Cerebral angiography
- Complete blood count (CBC) to check for anemia
- Liver tests (blood)
- Urinalysis
People with a personal or family history of polycystic kidney disease who have headaches should be tested to determine if cerebral aneurysms are the cause.
Polycystic kidney disease and cysts on the liver or other organs may be found using the following tests:
- Abdominal CT scan
- Abdominal MRI scan
- Abdominal ultrasound
- Intravenous pyelogram (IVP)
If several members of your family have polycystic kidney disease, genetic tests can be done to determine whether you carry the polycystic kidney disease gene.
Polycystic kidney disease treatment
Currently, no treatment can prevent the cysts from forming or enlarging 280.
The goal of treatment is to control symptoms and prevent complications. Treatment may include:
- Blood pressure medicines. Two types of blood pressure medicines, angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs), may slow kidney disease and delay kidney failure.
- Diuretics (water pills)
- Low-salt diet
Any urinary tract infection should be treated quickly with antibiotics.
Cysts that are painful, infected, bleeding, or causing a blockage may need to be drained. There are usually too many cysts to make it practical to remove each cyst.
Surgery to remove 1 or both kidneys may be needed. Treatments for end-stage kidney disease may include dialysis or a kidney transplant.
Polycystic kidney disease prognosis
The disease gets worse slowly. Eventually, it may lead to end-stage kidney failure. It is also associated with liver disease, including infection of liver cysts.
Treatment may relieve symptoms for many years.
People with polycystic kidney disease who don’t have other diseases may be good candidates for a kidney transplant.
What is acquired cystic kidney disease?
Acquired cystic kidney disease (ACKD) happens in people who have chronic kidney disease, especially if they are on dialysis. Unlike polycystic kidney disease, the kidneys are normal sized, and cysts do not form in other parts of the body. Acquired cystic kidney disease often has no symptoms. Usually, the cysts are harmless and do not need treatment. If they do cause complications, treatments include medicines, draining the cysts, or surgery.
Acquired cystic kidney disease occurs in children and adults who have:
- Chronic kidney disease (CKD)—a condition that develops over many years and may lead to end-stage kidney disease. The kidneys of people with CKD gradually lose their ability to filter wastes, extra salt, and fluid from the blood properly.
- End-stage kidney disease (ESRD) is the last stage (stage five) of chronic kidney disease (CKD). When CKD, polycystic kidney disease (PKD) or other kidney diseases develop into end-stage kidney disease, blood-filtering treatments called dialysis or a kidney transplant is necessary to live.
The cysts are more likely to develop in people who are on kidney dialysis. The chance of developing acquired cystic kidney disease increases with the number of years a person is on dialysis. However, the cysts are caused by chronic kidney disease or kidney failure, not dialysis treatments.
How common is acquired cystic kidney disease?
Acquired cystic kidney disease becomes more common the longer a person has chronic kidney disease.
- About 7 to 22 percent of people with chronic kidney disease already have acquired cystic kidney disease before starting dialysis treatments.
- Almost 60 percent of people on dialysis for 2 to 4 years develop acquired cystic kidney disease 283.
- About 90 percent of people on dialysis for 8 years develop acquired cystic kidney disease 283.
Acquired cystic kidney disease causes
Researchers do not fully understand what causes cysts to grow in the kidneys of people with chronic kidney disease. The fact that these cysts occur only in the kidneys and not in other parts of the body, as in polycystic kidney disease, indicates that the processes that lead to cyst formation take place primarily inside the kidneys 284.
Acquired cystic kidney disease signs and symptoms
A person with acquired cystic kidney disease often has no symptoms. However, the complications of acquired cystic kidney disease can have signs and symptoms.
Acquired cystic kidney disease complications
People with acquired cystic kidney disease may develop the following complications:
- an infected cyst, which can cause fever and back pain.
- blood in the urine, which can signal that a cyst in the kidney is bleeding.
- tumors in the kidneys. People with acquired cystic kidney disease are more likely than people in the general population to have cancerous kidney tumors. However, the chance of cancer spreading is lower in people with acquired cystic kidney disease than that of other kidney cancers not associated with acquired cystic kidney disease, and the long-term outlook is better 283.
Acquired cystic kidney disease diagnosis
A doctor may diagnose a person with acquired cystic kidney disease based on
- medical history
- imaging tests
Medical History
Taking a medical history may help a health care provider diagnose acquired cystic kidney disease. A health care provider may suspect acquired cystic kidney disease if a person who has been on dialysis for several years develops symptoms such as fever, back pain, or blood in the urine.
Imaging Tests
To confirm the diagnosis, the health care provider may order one or more imaging tests. A radiologist—a doctor who specializes in medical imaging—interprets the images from these tests, and the patient does not need anesthesia.
- Ultrasound uses a device, called a transducer, that bounces safe, painless sound waves off organs to create an image of their structure. A specially trained technician performs the procedure in a health care provider’s office, an outpatient center, or a hospital. The images can show cysts in the kidneys as well as the kidneys’ size and shape.
- Computerized tomography (CT) scans use a combination of x-rays and computer technology to create images. For a CT scan, a nurse or technician may give the patient a solution to drink and an injection of a special dye, called contrast medium. CT scans require the patient to lie on a table that slides into a tunnel-shaped device where an x-ray technician takes the x-rays. An x-ray technician performs the procedure in an outpatient center or a hospital. CT scans can show cysts and tumors in the kidneys.
- Magnetic resonance imaging (MRI) is a test that takes pictures of the body’s internal organs and soft tissues without using x-rays. A specially trained technician performs the procedure in an outpatient center or a hospital. Although the patient does not need anesthesia, a health care provider may give people with a fear of confined spaces light sedation, taken by mouth. An MRI may include the injection of contrast medium. With most MRI machines, the patient will lie on a table that slides into a tunnel-shaped device that may be open-ended or closed at one end. Some machines allow the patient to lie in a more open space. During an MRI, the patient, although usually awake, must remain perfectly still while the technician takes the images, which usually takes only a few minutes. The technician will take a sequence of images from different angles to create a detailed picture of the kidneys. During the test, the patient will hear loud mechanical knocking and humming noises from the machine.
Sometimes a doctor may discover acquired cystic kidney disease during an imaging exam for another condition. Images of the kidneys may help the doctor distinguish acquired cystic kidney disease from polycystic kidney disease.
Acquired cystic kidney disease treatment
If acquired cystic kidney disease is not causing complications, a person does not need treatment. A health care provider will treat infections with antibiotics—medications that kill bacteria. If large cysts are causing pain, a health care provider may drain the cyst using a long needle inserted into the cyst through the skin.
When a surgeon transplants a new kidney into a patient’s body to treat kidney failure, acquired cystic kidney disease in the damaged kidneys, which usually remain in place after a transplant, often disappears.
A surgeon may perform an operation to remove tumors or suspected tumors. In rare cases, a surgeon performs an operation to stop cysts from bleeding.
Eating, Diet, and Nutrition for Acquired Cystic Kidney Disease
No specific diet will prevent or delay acquired cystic kidney disease. In general, a diet designed for people on hemodialysis or peritoneal dialysis reduces the amount of wastes that accumulate in the body between dialysis sessions.
- Eating & Nutrition for Hemodialysis
Hemodialysis removes extra fluid from your body. However, hemodialysis can remove only so much fluid at a time safely. If you come to your hemodialysis with too much fluid in your body, your treatment may make you feel ill. You may get muscle cramps or have a sudden drop in blood pressure that causes you to feel dizzy or sick to your stomach.
Your choices about what to eat and drink while on hemodialysis can make a difference in how you feel and can make your treatments work better.
Between dialysis treatment sessions, wastes can build up in your blood and make you sick. You can reduce waste buildup by controlling what you eat and drink.
You can match what you eat and drink with what your kidney treatments remove.
Some foods cause wastes to build up quickly between your dialysis sessions. If your blood contains too much waste, your kidney treatment session may not remove them all.
What is my dry weight?
Your dry weight is your weight after a hemodialysis session has removed all extra fluid from your body. Controlling your liquid intake helps you stay at your proper dry weight. If you let too much fluid build up between sessions, it is harder to achieve your dry weight. Your health care provider can help you figure out what dry weight is right for you.
My dry weight goal: _____________
What you should eat and drink while on hemodialysis
You will need to carefully plan your meals and keep track of the amount of liquids you eat and drink. It helps to limit or avoid foods and beverages that have lots of:
- potassium
- phosphorus
- sodium—for example, vegetable juice and sports drinks.
Your dialysis center has a renal dietitian to help you plan your meals. A renal dietitian has special training in caring for the food and nutrition needs of people with kidney disease.
Use this information to help you learn how to eat right to feel right on hemodialysis. Read one section at a time.
Keep a copy of this information handy to remind yourself of foods you can eat and foods to avoid.
What you need to know about potassium
Healthy kidneys keep the right amount of potassium in your blood to keep your heart beating at a steady pace. Potassium levels can rise between hemodialysis sessions and affect your heartbeat. Eating too much potassium can be dangerous to your heart and may even cause death.
To control potassium levels, limit potassium-rich foods such as avocados, bananas, kiwis, and dried fruit. Choose fruits and vegetables that are lower in potassium. Have very small portions of foods that are higher in potassium, such as one or two cherry tomatoes on a salad or a few raisins in your oatmeal.
You can remove some of the potassium from potatoes by dicing or shredding them and then boiling them in a full pot of water.
To remove some of the potassium from potatoes:
- Dice potatoes into small pieces.
- Or, grate potatoes into shreds.
- And then boil potatoes in a full pot of water.
Your renal dietitian will give you more specific information about the potassium content of foods.
What you need to know about phosphorus
Too much phosphorus in your blood pulls calcium from your bones. Losing calcium may make your bones weak and likely to break. Also, too much phosphorus may make your skin itch. Limiting phosphorus can be hard because foods that contain phosphorus, such as meat and milk, also contain the protein you need. You should be careful to eat enough protein; however, not so much that you get too much phosphorus. Processed and packaged foods contain especially high levels of phosphorus. You can also find phosphorus naturally in foods such as poultry, fish, nuts, peanut butter, beans, cola, tea, and dairy products. Usually, people on hemodialysis should only have a 1/2 cup of milk per day.
Your renal dietitian will give you more specific information about phosphorus.
You may need to take a phosphate binder such as sevelamer (Renvela), calcium acetate (PhosLo), lanthanum carbonate (Fosrenol), or calcium carbonate to control the phosphorus in your blood between hemodialysis sessions. These medicines, the phosphorus binder “seals” the phosphorus from food and moves it out through stool so the phosphorous does not enter the bloodstream.
What you need to know about protein
Renal dietitians encourage most people on hemodialysis to eat high-quality protein because it produces less waste for removal during dialysis. High-quality protein comes from meat, poultry, fish, and eggs. Avoid processed meats such as hot dogs and canned chili, which have high amounts of sodium and phosphorus.
What you need to know about sodium
Sodium is a part of salt. Sodium is found in many canned, packaged, frozen, and fast foods. Sodium is also found in many condiments, seasonings, and meats. Too much sodium makes you thirsty, which makes you drink more liquid.
Try to eat fresh, naturally low-sodium foods. Look for products labeled “low sodium,” especially in canned and frozen foods.
Do not use salt substitutes because they contain potassium. Talk with your renal dietitian about spices you can use to flavor your food. Your renal dietitian can help you find spice blends without sodium or potassium.
What you need to know about calories
All foods contain calories, and you need calories for energy. Many people on hemodialysis do not have a good appetite and do not get enough calories. If you find you do not feel like eating, talk with your renal dietitian to find healthy ways to add calories to your diet. Vegetable oils—such as olive oil, canola oil, and safflower oil—are good sources of calories and are the healthiest way to add fat to your diet if you need to gain weight. Use them generously on breads, rice, and noodles only if your renal dietitian tells you to add calories to your diet.
Butter and margarines are rich in calories; however, they are mainly saturated fat. Saturated fats and trans fats can clog your arteries. Use them less often. Soft margarine that comes in a tub is better than stick margarine. Choose a soft margarine with less saturated and trans fats.
Talk with your renal dietitian about the types and amounts of fat you need in your diet. Everyone will have different needs that a renal dietitian can help address.
Hard candy, sugar, honey, jam, and jelly provide calories and energy without fat or adding other things that your body does not need. If you have diabetes, be careful about eating sweets and talk with your renal dietitian before adding sweets to your food plan.
Should you take vitamin and mineral supplements?
Do not take nutritional supplements you can buy over the counter. These supplements may contain vitamins or minerals that are harmful to you. For safety reasons, talk with your health care provider before using probiotics, dietary supplements, or any other medicine together with or in place of the treatment your health care provider prescribes.
Why is it important to keep track of how much liquid you eat or drink?
You may feel better if you keep track of and limit how much liquid you eat and drink. Excess fluid can build up in your body and may cause
- swelling and weight gain between dialysis sessions
- changes in your blood pressure
- your heart to work harder, which can lead to serious heart trouble
- a buildup of fluid in your lungs, making it hard for you to breathe
Your health care provider can help you figure out how much liquid is right for you.
One way to limit how much liquid you have is to limit the salt in the foods you eat. Salt makes you thirsty, so you drink more. Avoid salty foods such as chips and pretzels.
Your renal dietitian will give you other tips to help you limit how much liquid you consume while making sure you don’t feel too thirsty.
What foods count as liquid and why?
Foods that are liquid at room temperature, such as soup, contain water. Gelatin, pudding, ice cream, and other foods that include a lot of liquid in the recipe also count. Most fruits and vegetables contain water, such as melons, grapes, apples, oranges, tomatoes, lettuce, and celery. When you count up how much liquid you have in a day, be sure to count these foods.
Nutrition for Advanced Chronic Kidney Disease in Adults
A person may prevent or delay some health problems from chronic kidney disease by eating the right foods and avoiding foods high in sodium, potassium, and phosphorus 285. Learning about calories, fats, proteins, and fluids is important for a person with advanced chronic kidney disease. Protein foods such as meat and dairy products break down into waste products that healthy kidneys remove from the blood.
As chronic kidney disease progresses, nutritional needs change. A health care provider may recommend that a patient with reduced kidney function choose foods carefully.
Chronic kidney disease usually takes a long time to develop and does not go away. In chronic kidney disease, the kidneys continue to work—just not as well as they should. Wastes may build up so gradually that the body becomes used to having those wastes in the blood. Salts containing phosphorus and potassium may rise to unsafe levels, causing heart and bone problems. Anemia—low red blood cell count—can result from chronic kidney disease because the kidneys stop making enough erythropoietin, a hormone that causes bone marrow to make red blood cells. After months or years, chronic kidney disease may progress to permanent kidney failure, which requires a person to have a kidney transplant or regular blood filtering treatments called dialysis.
What is medical nutrition therapy (MNT)?
Medical nutrition therapy is the use of nutrition counseling by a registered dietitian to help promote a medical or health goal. A health care provider may refer a patient to a registered dietitian to help with the patient’s food plan. Many insurance policies cover medical nutrition therapy when recommended by a health care provider. Anyone who qualifies for Medicare can receive a benefit for medical nutrition therapy from a registered dietitian or nutrition professional when a health care provider provides a referral indicating that the person has diabetes or kidney disease.
One way to locate a qualified dietitian is to contact the Academy of Nutrition and Dietetics at www.eatright.org and click on “Find a Registered Dietitian.” Users can enter their address or ZIP code for a list of dietitians in their area. A person looking for dietary advice to prevent kidney damage should click on “Renal (Kidney) Nutrition” in the specialty field. Dietitians who specialize in helping people with chronic kidney disease are called renal dietitians.
Why is knowing about calories important for someone with advanced chronic kidney disease?
As chronic kidney disease progresses, people often lose their appetites because they find that foods do not taste the same. As a result, they consume fewer calories—important units of energy in food—and may lose too much weight. Renal dietitians can help people with advanced chronic kidney disease find healthy ways to add calories to their diet if they are losing too much weight.
Why is knowing about protein important for someone with advanced chronic kidney disease?
Protein is an essential part of any diet. Proteins help build and maintain muscle, bone, skin, connective tissue, internal organs, and blood. They help fight disease and heal wounds. But proteins also break down into waste products that must be removed from the blood by the kidneys. Eating more protein than the body needs may put an extra burden on the kidneys and cause kidney function to decline faster.
Health care providers recommend that people with chronic kidney disease eat moderate or reduced amounts of protein. However, restricting protein could lead to malnutrition, so people with chronic kidney disease need to be careful. The typical American diet contains more than enough protein. Learning about portion sizes can help people limit protein intake without endangering their health.
What is the right meat portion size?
Most people—with or without chronic kidney disease—can get the daily protein they need by eating two 3-ounce servings of meat or meat substitute. A 3-ounce serving of meat is about the size of a deck of cards or the palm of a person’s hand.
A renal dietitian can help people learn about the amount and sources of protein in their diet. Animal protein in egg whites, cheese, chicken, fish, and red meats contain more of the essential nutrients a body needs. With careful meal planning, a well-balanced vegetarian diet can also provide these nutrients. A renal dietitian can help people with advanced chronic kidney disease make small adjustments in their eating habits that can result in significant protein reduction. For example, people can lower their protein intake by making sandwiches using thinner slices of meat and adding lettuce, cucumber slices, apple slices, and other garnishes. The following table lists some higher-protein foods and suggestions for lower-protein alternatives that are better choices for people with chronic kidney disease trying to limit their protein intake.
When kidney function declines to the point where dialysis becomes necessary, patients should include more protein in their diet because dialysis removes large amounts of protein from the blood.
Why is knowing about fat important for someone with advanced chronic kidney disease?
Everyone should know about fat sources because eating the wrong kinds of fat and too much fat increases the risk of clogged blood vessels and heart problems. Fat provides energy, helps produce hormonelike substances that regulate blood pressure and other heart functions, and carries fat-soluble vitamins. Everyone needs dietary fat, but some fats are healthier than others. People with chronic kidney disease are at higher risk of having a heart attack or stroke. Therefore, people with chronic kidney disease should be especially careful about how dietary fat affects their heart health.
People with advanced chronic kidney disease should talk with a dietitian about healthy and unhealthy sources of fat. Saturated fats and trans-fatty acids can raise blood cholesterol levels and clog blood vessels. Saturated fats are found in animal products such as red meat, poultry, whole milk, and butter. These fats are usually solid at room temperature. Trans-fatty acids are often found in commercially baked goods such as cookies and cakes and in fried foods like doughnuts and french fries.
A dietitian can suggest healthy ways to include fat in the diet, especially if more calories are needed. Vegetable oils such as corn or safflower oil are healthier than animal fats such as butter or lard. Hydrogenated vegetable oils should be avoided because they are high in trans-fatty acids. Monounsaturated fats—olive, peanut, and canola oils—are healthy alternatives to animal fats. The table below shows the sources of fats, broken down into three types of fats that should be eaten less often and good fats that can be eaten more often.
Why is knowing about sodium important for someone with advanced chronic kidney disease?
Too much sodium in a person’s diet can be harmful because it causes blood to hold fluid. People with chronic kidney disease need to be careful not to let too much fluid build up in their bodies. The extra fluid raises blood pressure and puts a strain on the heart and kidneys. A dietitian can help people find ways to reduce the amount of sodium in their diet. Nutrition labels provide information about the sodium content in food. The U.S. Food and Drug Administration advises that healthy people should limit their daily sodium intake to no more than 2,300 milligrams (mg), the amount found in 1 teaspoon of table salt. People who are at risk for a heart attack or stroke because of a condition such as high blood pressure or kidney disease should limit their daily sodium intake to no more than 1,500 mg. Choosing sodium-free or low-sodium food products will help them reach that goal.
Sodium is found in ordinary table salt and many salty seasonings such as soy sauce and teriyaki sauce. Canned foods, some frozen foods, and most processed meats have large amounts of salt. Snack foods such as chips and crackers are also high in salt.
Alternative seasonings such as lemon juice, salt-free seasoning mixes, and hot pepper sauce can help people reduce their salt intake. People with advanced chronic kidney disease should avoid salt substitutes that use potassium, such as AlsoSalt or Nu-Salt, because chronic kidney disease limits the body’s ability to eliminate potassium from the blood.
Why is knowing about potassium important for someone with advanced chronic kidney disease?
Keeping the proper level of potassium in the blood is essential. Potassium keeps the heart beating regularly and muscles working right. Problems can occur when blood potassium levels are either too low or too high. Damaged kidneys allow potassium to build up in the blood, causing serious heart problems. Potassium is found in many fruits and vegetables, such as bananas, potatoes, avocados, and melons. People with advanced chronic kidney disease may need to avoid some fruits and vegetables. Blood tests can indicate when potassium levels have climbed above normal range. A renal dietitian can help people with advanced chronic kidney disease find ways to limit the amount of potassium they eat. The potassium content of potatoes and other vegetables can be reduced by boiling them in water. The following table gives examples of some high-potassium foods and suggestions for low-potassium alternatives for people with advanced chronic kidney disease.
Why is knowing about phosphorus important for someone with advanced chronic kidney disease?
Damaged kidneys allow phosphorus, a mineral found in many foods, to build up in the blood. Too much phosphorus in the blood pulls calcium from the bones, making the bones weak and likely to break. Too much phosphorus may also make skin itch. Foods such as milk and cheese, dried beans, peas, colas, canned iced teas and lemonade, nuts, and peanut butter are high in phosphorus. A renal dietitian can help people with advanced chronic kidney disease learn how to limit phosphorus in their diet.
As chronic kidney disease progresses, a person may need to take a phosphate binder such as sevelamer hydrochloride (Renagel), lanthanum carbonate (Fosrenol), calcium acetate (PhosLo), or calcium carbonate (Tums) to control the phosphorus in the blood. These medications act like sponges to soak up, or bind, phosphorus while it is in the stomach. Because it is bound, the phosphorus does not get into the blood. Instead, it is removed from the body in the stool.
Why is regulating fluid intake important for someone with advanced chronic kidney disease?
People with advanced chronic kidney disease may need to limit how much they drink because damaged kidneys can’t remove extra fluid. The fluid builds up in the body and strains the heart. Patients should tell their health care provider about any swelling around the eyes or in the legs, arms, or abdomen.
Is there kidney stone home remedy?
Due to the complex and multifactoral causes of kidney stones that involve many diseases that may include urinary tract infections, sexually transmitted disease, hypercalciuria, hyperparathyroidism, cystinuria, hypocitruria, sarcoidosis, bone metastases, multiple myeloma, solitary kidney, urinary tract anomalies, renal tubular acidosis, hyperuricosuria, etc., there is no home remedy available to get rid of kidney stone safely and prevent long term kidney stone complications. Recovery and analysis of the kidney stone, measurement of kidney stone-forming substances in the urine, and the clinical tests (e.g. blood tests, kidney function tests, 24 hour urine test, CT scan, etc.) are needed to plan proper kidney stone diagnosis, management, prevention and treatment. Complications of kidney stones like infections, chronic kidney disease and end-stage kidney disease where you’ll need a kidney transplant are rare if you seek treatment from a health care professional before these problems occur. Therefore we do not recommend any home remedy.
However there are general measures you can do to prevent recurrent kidney stone formation:
- Low urine volume is the most common abnormality and the single most important preventable factor to correct so as to avoid kidney stone recurrences. The only home remedy would be to drink large amounts of water— to achieve a urine volume of at least 2.5 liters daily to prevent kidney stone from forming. People who have had cystine stones may need to drink even more. The amount of fluid each person needs to drink depends on the weather and the person’s activity level—people who work or exercise in hot weather need more fluid to replace the fluid they lose through sweat. A 24-hour urine collection may be used to determine the volume of urine produced during a day. If the volume of urine produced is too low, the person can be advised to increase fluid intake. Drinking enough fluid is the most important thing a person can do to prevent kidney stones.
- Risk of a recurrent stone is about 50% within five to seven years 10.
- Diets low in salt (< 1500 mg/day) and animal proteins (< 52 g/day) are helpful in decreasing the frequency of recurrent calcium oxalate stones 286.
- Decrease intake of animal protein (≤ 52 g/day): Reduces production of metabolic acids, resulting in a lower level of acid induced calcium excretion; increases excretion of citrate that forms a soluble complex with calcium; and reduces supersaturation with respect to calcium oxalate and limits the excretion of uric acid 286. Meats and other animal protein—such as eggs and fish—contain purines, which break down into uric acid in the urine. Foods especially rich in purines include organ meats, such as liver. People who form uric acid stones should limit their meat consumption to 6 ounces each day. Animal protein may also raise the risk of calcium stones by increasing the excretion of calcium and reducing the excretion of citrate into the urine. Citrate prevents kidney stones, but the acid in animal protein reduces the citrate in urine.
- Restrict salt intake (< 1500 mg/day of sodium chloride): Dietary and urinary sodium is directly correlated with urinary calcium excretion, and lower urinary excretion of sodium reduces urinary calcium excretion 286. Sodium, often from salt, causes the kidneys to excrete more calcium into the urine. High concentrations of calcium in the urine combine with oxalate and phosphorus to form stones. Reducing sodium intake is preferred to reducing calcium intake. The risk of kidney stones increases with increased daily sodium consumption. People who form calcium oxalate or calcium phosphate stones should limit their intake to the American Heart Association level, even if they take medications to prevent kidney stones.
- Normal calcium intake (≥ 800 mg/day): Low calcium diets increase urinary oxalate excretion, which may result in more stone formation and possibly a negative calcium balance 286. Calcium from food does not increase the risk of calcium oxalate stones. Calcium in the digestive tract binds to oxalate from food and keeps it from entering the blood, and then the urinary tract, where it can form stones. People who form calcium oxalate stones should include 800 mg of calcium in their diet every day, not only for kidney stone prevention but also to maintain bone density. A cup of low-fat milk contains 300 mg of calcium. Other dairy products such as yogurt are also high in calcium. For people who have lactose intolerance and must avoid dairy products, orange juice fortified with calcium or dairy with reduced lactose content may be alternatives. Calcium supplements may increase the risk of calcium oxalate stones if they are not taken with food. Some studies suggest citrus drinks like lemonade and orange juice protect against kidney stones because they contain citrate, which stops crystals from growing into stones. But watch for the added sugar and extra calories from the citrus drinks which can lead to weight gain and obesity. Studies have shown that being overweight increases the risk of kidney stones, particularly uric acid stones.
- Decrease dietary oxalate: Reduce the intake of foods rich in oxalate—spinach, rhubarb, chocolate, nuts and wheat bran 82. Some of the oxalate in urine is made by the body. However, eating certain foods with high levels of oxalate can increase the amount of oxalate in the urine, where it combines with calcium to form calcium oxalate stones. Foods that have been shown to increase the amount of oxalate in urine include — spinach, rhubarb, nuts, chocolate and wheat bran. Avoiding these foods may help reduce the amount of oxalate in the urine.
- Cranberry juice: Decreases oxalate and phosphate excretion and increases citrate excretion 287. Cranberry juice has antilithogenic properties and, as such, deserves consideration as a conservative therapeutic protocol in managing calcium oxalate urolithiasis.
- Studies have shown the Dietary Approaches to Stop Hypertension (DASH) diet can reduce the risk of kidney stones. The DASH diet is high in fruits and vegetables, moderate in low-fat dairy products, and low in animal protein. More information about the DASH diet can be found here What is the DASH Diet ?
Does the type of kidney stone you have affect food choices you should make?
Yes. If you have already had kidney stones, ask your health care professional which type of kidney stone you had. Based on the type of kidney stone you had, you may be able to prevent kidney stones by making changes in how much sodium, animal protein, calcium, or oxalate is in the food you eat.
You may need to change what you eat and drink for these types of kidney stones:
- Calcium Oxalate Stones
- Calcium Phosphate Stones
- Uric Acid Stones
- Cystine Stones
A dietitian who specializes in kidney stone prevention can help you plan meals to prevent kidney stones. Find a dietitian who can help you.
Calcium Oxalate Stones
Reduce oxalate
If you’ve had calcium oxalate stones, you may want to avoid these foods to help reduce the amount of oxalate in your urine:
- nuts and nut products
- peanuts—which are legumes, not nuts, and are high in oxalate
- rhubarb
- spinach
- wheat bran
Talk with a health care professional about other food sources of oxalate and how much oxalate should be in what you eat.
Reduce sodium
Your chance of developing kidney stones increases when you eat more sodium. Sodium is a part of salt. Sodium is in many canned, packaged, and fast foods. It is also in many condiments, seasonings, and meats.
Talk with a health care professional about how much sodium should be in what you eat.
Tips to Reduce Your Sodium Intake
Most Americans consume too much sodium. Adults should aim to consume less than 2,300 mg a day. One teaspoon of table salt has 2,325 milligrams (mg) of sodium. If you have had calcium oxalate or calcium phosphate stones, you should follow this guideline, even if you take medicine to prevent kidney stones.
Here are some tips to help you reduce your sodium intake:
- Check the Percent Daily Value (%DV) for sodium on the Nutrition Facts label found on many foods. Low in sodium is 5% or less, and high in sodium is 20% or more.
- Consider writing down how much sodium you consume each day.
- When eating out, ask about the sodium content in the food.
- Cook from scratch. Avoid processed and fast foods, canned soups and vegetables, and lunch meats.
- Look for foods labeled: sodium free, salt free, very low sodium, low sodium, reduced or less sodium, light in sodium, no salt added, unsalted, and lightly salted.
Check labels for ingredients and hidden sodium, such as:
- sodium bicarbonate, the chemical name for baking soda
- baking powder, which contains sodium bicarbonate and other chemicals
- disodium phosphate
- monosodium glutamate, or MSG
- sodium alginate
- sodium nitrate or nitrite
Limit animal protein
Eating animal protein may increase your chances of developing kidney stones.
A health care professional may tell you to limit eating animal protein, including:
- beef, chicken, and pork, especially organ meats
- eggs
- fish and shellfish
- milk, cheese, and other dairy products
Although you may need to limit how much animal protein you eat each day, you still need to make sure you get enough protein. Consider replacing some of the meat and animal protein you would typically eat with beans, dried peas, and lentils, which are plant-based foods that are high in protein and low in oxalate.
Talk with a health care professional about how much total protein you should eat and how much should come from animal or plant-based foods.
Get enough calcium from foods
Even though calcium sounds like it would be the cause of calcium stones, it’s not. In the right amounts, calcium can block other substances in the digestive tract that may cause stones. Talk with a health care professional about how much calcium you should eat to help prevent getting more calcium oxalate stones and to support strong bones. It may be best to get calcium from low-oxalate, plant-based foods such as calcium-fortified juices, cereals, breads, some kinds of vegetables, and some types of beans. Ask a dietitian or other health care professional which foods are the best sources of calcium for you.
Calcium Phosphate Stones
Reduce sodium
Your chance of developing kidney stones increases when you eat more sodium. Sodium is a part of salt. Sodium is in many canned, packaged, and fast foods. It is also in many condiments, seasonings, and meats.
Talk with a health care professional about how much sodium should be in what you eat.
Limit animal protein
Eating animal protein may increase your chances of developing kidney stones.
A health care professional may tell you to limit eating animal protein, including:
- beef, chicken, and pork, especially organ meats
- eggs
- fish and shellfish
- milk, cheese, and other dairy products
Although you may need to limit how much animal protein you have each day, you still need to make sure you get enough protein. Consider replacing some of the meat and animal protein you would typically eat with some of these plant-based foods that are high in protein:
- legumes such as beans, dried peas, lentils, and peanuts
- soy foods, such as soy milk, soy nut butter, and tofu
- nuts and nut products, such as almonds and almond butter, cashews and cashew butter, walnuts, and pistachios
- sunflower seeds
Talk with a health care professional about how much total protein you should eat and how much should come from animal or plant-based foods.
Get enough calcium from foods
Even though calcium sounds like it would be the cause of calcium stones, it’s not. In the right amounts, calcium can block other substances in the digestive tract that may lead to stones. Talk with a health care professional about how much calcium you should eat to help prevent getting more calcium phosphate stones and to support strong bones. It may be best to get calcium from plant-based foods such as calcium-fortified juices, cereals, breads, some kinds of vegetables, and some types of beans. Ask a dietitian or other health care professional which foods are the best sources of calcium for you.
Uric Acid Stones
Limit animal protein
Eating animal protein may increase your chances of developing kidney stones.
A health care professional may tell you to limit eating animal protein, including
- beef, chicken, and pork, especially organ meats
- eggs
- fish and shellfish
- milk, cheese, and other dairy products
Although you may need to limit how much animal protein you have each day, you still need to make sure you get enough protein. Consider replacing some of the meat and animal protein you would typically eat with some of these plant-based foods that are high in protein:
- legumes such as beans, dried peas, lentils, and peanuts
- soy foods, such as soy milk, soy nut butter, and tofu
- nuts and nut products, such as almonds and almond butter, cashews and cashew butter, walnuts, and pistachios
- sunflower seeds
Talk with a health care professional about how much total protein you should eat and how much should come from animal or plant-based foods.
Losing weight if you are overweight is especially important for people who have had uric acid stones.
Cystine Stones
Drinking enough liquid, mainly water, is the most important lifestyle change you can make to prevent cystine stones. Talk with a health care professional about how much liquid you should drink.
Kidney stones summary
Kidney stones formation now appear to be related to chronic conditions that require long-term medical management, dietary and lifestyle changes and sometimes drug therapy and/or surgery. Kidney stones are a risk factor for chronic kidney disease and progression to end-stage renal disease 288. Persons with kidney stones are more likely to have traditional risk factors for chronic kidney disease (e.g., hypertension, preexisting kidney disease, diabetes, proteinuria, albuminuria), as well as non-traditional factors (e.g., interstitial nephritis, chronic pyelonephritis, female sex) 289.
A family history of kidney stones (increases your risk by three times), insulin resistant states, a history of hypertension, primary hyperparathyroidism, a history of gout, chronic metabolic acidosis, and surgical menopause are all associated with increased risk of kidney stones 10. Incidence of stones is higher in patients with an anatomical abnormality of the urinary tract that may result in urinary stasis. Most patients (up to 80%) with calcium stones have one or more of the metabolic risk factors and about 25% of stones are idiopathic in origin. Various drugs can also increase your risk of kidney stone disease.
Your kidneys remove waste from your blood by filtering the blood and making urine. Kidney stones formation are complex and involves many diseases that may include urinary tract infections (eg, hypercalciuria, urinary tract infections, hyperparathyroidism, cystinuria, hypocitruria, sarcoidosis, bone metastases, multiple myeloma, solitary kidney, urinary tract anomalies, renal tubular acidosis, hyperuricosuria, etc.) that will require medical diagnosis, treatment and management. Incidence of kidney stones is higher in patients with an anatomical abnormality of the urinary tract that may result in urinary stasis. Complications of kidney stones are rare if you seek treatment from a health care professional before problems occur.
- Kidney stones can form when substances in the urine—such as calcium, oxalate, and phosphorus— become highly concentrated. Diet is one of several factors that can promote or inhibit kidney stone formation.
- Four major types of kidney stones can form: calcium stones, uric acid stones, struvite stones, and cystine stones.
- Drinking enough fluids — that will achieve a urine volume of at least 2.5 liters daily— is the most important thing a person can do to prevent all kidney stones 112).
- Sodium, often from salt, causes the kidneys to excrete more calcium into the urine. High concentrations of calcium in the urine combine with oxalate and phosphorus to form stones. Reducing sodium intake (to less than 1500 mg per day) is preferred to reducing calcium intake.
- Meats and other animal protein—such as eggs and fish—contain purines, which break down into uric acid in the urine.
- Calcium from food does not increase the risk of calcium oxalate stones. Calcium in the digestive tract binds to oxalate from food and keeps it from entering the blood, and then the urinary tract, where it can form stones.
Kidney stones may remain within the kidney or renal collecting system or be passed into the ureter and bladder. During passage, the stones may irritate the ureter and may become lodged, obstructing urine flow and causing hydroureter and sometimes hydronephrosis.
Even partial obstruction causes decreased glomerular filtration (renal function), which may persist briefly after the calculus has passed. With hydronephrosis and elevated glomerular pressure, renal blood flow declines, further worsening renal function. Generally, however, in the absence of infection, permanent renal dysfunction occurs only after about 28 days of complete obstruction.
Secondary infection can occur with long-standing obstruction, but most patients with calcium-containing calculi do not have infected urine.
Therefore, if you suspect that you may have kidney stone or have symptoms of urinary tract stones, see your health care provider ASAP to prevent permanent kidney damage. Recovery and analysis of the kidney stone, measurement of kidney stone-forming substances in the urine, and the clinical history are needed to plan proper kidney stone diagnosis, management, prevention and treatment.
- Kidney stone. https://www.niddk.nih.gov/health-information/urologic-diseases/kidney-stones/definition-facts[↩][↩]
- Favus M. Nephrolithiasis. [Updated 2016 Dec 11]. In: De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279069[↩][↩][↩]
- Song L, Maalouf NM. Nephrolithiasis. [Updated 2020 Mar 9]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279069[↩]
- Asplin JR, Favus MJ, Coe FL. Nephrolithiasis. In: Brenner BM, ed. Brenner and Rector’s the kidney. 5th ed. Philadelphia: Saunders, 1996: 1893-935.[↩][↩]
- Favus MJ, Feingold KR. Kidney Stone Emergencies. [Updated 2018 Sep 13]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK278956[↩][↩]
- Merck Sharp & Dohme Corp., Merck Manual. Urinary Calculi. https://www.merckmanuals.com/professional/genitourinary-disorders/urinary-calculi/urinary-calculi[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Time trends in reported prevalence of kidney stones in the United States: 1976-1994. Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC. Kidney Int. 2003 May; 63(5):1817-23. https://www.ncbi.nlm.nih.gov/pubmed/12675858/[↩]
- Kidney Stones. https://www.kidney.org/atoz/content/kidneystones[↩][↩][↩]
- Gender differences in seasonal variation of urine stone risk factors. Parks JH, Barsky R, Coe FL. J Urol. 2003 Aug; 170(2 Pt 1):384-8. https://www.ncbi.nlm.nih.gov/pubmed/12853781/[↩]
- Parmar MS. Kidney stones. BMJ : British Medical Journal. 2004;328(7453):1420-1424. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC421787/[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Hall WD, Pettinger M, Oberman A, Watts NB, Johnson KC, Paskett ED, et al. Risk factors for kidney stones in older women in Southern United States. Am J Med Sci 2001;322: 12-8. https://www.ncbi.nlm.nih.gov/pubmed/11465241[↩]
- U.S. Department of Health and Human Services. The National Institute of Diabetes and Digestive and Kidney Diseases Health Information Center. Definition & Facts for Kidney Stones. https://www.niddk.nih.gov/health-information/urologic-diseases/kidney-stones/definition-facts[↩][↩]
- Segura JW, Preminger GM, Assimos DG, Dretler SP, Kahn RL, Lingeman JE, et al. Ureteral stones clinical guidelines panel summary report on the management of ureteral calculi. J Urol 1997;158: 1915-21. https://www.ncbi.nlm.nih.gov/pubmed/9334635[↩]
- U.S. Department of Health and Human Services. Chapter 9: Urinary tract stones. In: Litwin MS, Saigal CS, eds. Urologic diseases in America. https://www.niddk.nih.gov/-/media/Files/Strategic-Plans/urologic/Trackstar-Urologic-Diseases-Chap-09.pdf[↩][↩]
- Acar B, Inci Arikan F, Emeksiz S, Dallar Y. Risk factors for nephrolithiasis in children. World J Urol. 2008;26(6):627–630.[↩][↩][↩][↩]
- Sas DJ, Hulsey TC, Shatat IF, Orak JK. Increasing incidence of kidney stones in children evaluated in the emergency department. J Pediatr. 2010;157(1):132–137.[↩][↩]
- Ross AE, Handa S, Lingeman JE, Matlaga BR. Kidney stones during pregnancy: an investigation into stone composition. Urol Res. 2008;36(2):99–102.[↩][↩]
- Swartz MA, Lydon-Rochelle MT, Simon D, Wright JL, Porter MP. Admission for nephrolithiasis in pregnancy and risk of adverse birth outcomes. Obstet Gynecol. 2007;109(5):1099–1104.[↩]
- U.S. Department of Health and Human Services. The National Institute of Diabetes and Digestive and Kidney Diseases. Health Information Center. The Urinary Tract & How It Works. https://www.niddk.nih.gov/health-information/urologic-diseases/urinary-tract-how-it-works[↩]
- Costa-Bauza A, Ramis M, Montesinos V, et al. Type of renal calculi: variation with age and sex. World J Urol. 2007;25(4):415–21.[↩]
- Pietrow PK, Karellas ME. Medical management of common urinary calculi. Am Fam Physician. 2006;74(1):86–94.[↩]
- Treatment and Prevention of Kidney Stones: An Update. Am Fam Physician. 2011 Dec 1;84(11):1234-1242. http://www.aafp.org/afp/2011/1201/p1234.html[↩][↩][↩]
- Ryall RL. Urinary inhibitors of calcium oxalate crystallization and their potential role in stone formation. World J Urol. 1997;15:155 https://www.ncbi.nlm.nih.gov/pubmed/9228722[↩]
- Sheng X, Jung T, Wesson JA, et al. Adhesion at calcium oxalate crystal surfaces and the effect of urinary constituents. Proc Natl Acad Sci USA. 2005;102:267. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC544292/[↩]
- Heilberg IP. Update on dietary recommendations and medical treatment of renal stone disease. Nephrol Dial Transplant. 2000;15:117 https://www.ncbi.nlm.nih.gov/pubmed/10607782[↩]
- Meschi T, Maggiore U, Fiaccadori E, et al. The effect of fruits and vegetables on urinary stone risk factors. Kidney Int. 2004;66:2402. https://www.ncbi.nlm.nih.gov/pubmed/15569332[↩]
- Seltzer MA, Low RK, McDonald M, et al. Dietary manipulation with lemonade to treat hypocitraturic calcium nephrolithiasis. J Urol. 1996;156:907. https://www.ncbi.nlm.nih.gov/pubmed/8709360[↩]
- Kang D, Haleblian GE, Sur RL, et al. Long-term lemonade based dietary manipulation in patients with hypocitraturic nephrolithia-sis. J Urol. 2007;177:1358 https://www.ncbi.nlm.nih.gov/pubmed/17382731[↩]
- Odvina CV. Comparative value of orange juice versus lemonade in reducing stone-forming risk. Clin J Am Soc Nephrol. 2006;1:1269 http://cjasn.asnjournals.org/content/1/6/1269.long[↩]
- PENNISTON KL, NAKADA SY, HOLMES RP, ASSIMOS DG. Quantitative Assessment of Citric Acid in Lemon Juice, Lime Juice, and Commercially-Available Fruit Juice Products. Journal of endourology / Endourological Society. 2008;22(3):567-570. doi:10.1089/end.2007.0304. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2637791/[↩][↩]
- Penniston KL, Nakada SY, Holmes RP, et al. : Quantitative assessment of citric acid in lemon juice, lime juice, and commercially-available fruit juice products. J Endourol 2008;22:567–570 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2637791/[↩]
- Dietary manipulation with lemonade to treat hypocitraturic calcium nephrolithiasis. Seltzer MA, Low RK, McDonald M, Shami GS, Stoller ML. J Urol. 1996 Sep; 156(3):907-9. https://www.ncbi.nlm.nih.gov/pubmed/8709360/[↩]
- Lemonade therapy increases urinary citrate and urine volumes in patients with recurrent calcium oxalate stone formation. Penniston KL, Steele TH, Nakada SY. Urology. 2007 Nov; 70(5):856-60. https://www.ncbi.nlm.nih.gov/pubmed/17919696/[↩]
- Meschi T, Maggiore U, Fiaccadori E, et al. The effect of fruits and vegetables on urinary stone risk factors. Kidney Int. 2004;66:2402 https://www.kidney-international.org/article/S0085-2538(15)50347-5/fulltext[↩]
- Carbohydrate intolerance and kidney stones in children in the Goldfields. Baldwin DN, Spencer JL, Jeffries-Stokes CA. J Paediatr Child Health. 2003 Jul; 39(5):381-5. https://www.ncbi.nlm.nih.gov/pubmed/12887672/[↩]
- Pathophysiologic basis for normouricosuric uric acid nephrolithiasis. Sakhaee K, Adams-Huet B, Moe OW, Pak CY. Kidney Int. 2002 Sep; 62(3):971-9. https://www.ncbi.nlm.nih.gov/pubmed/12164880/[↩]
- The association between gout and nephrolithiasis in men: The Health Professionals’ Follow-Up Study. Kramer HJ, Choi HK, Atkinson K, Stampfer M, Curhan GC. Kidney Int. 2003 Sep; 64(3):1022-6. https://www.ncbi.nlm.nih.gov/pubmed/12911552/[↩]
- U.S. National Library of Medicine. Cystinuria. https://medlineplus.gov/ency/article/000346.htm[↩][↩][↩]
- Ticinesi A, Nouvenne A, Ferraro PM, Folesani G, Lauretani F, Allegri F, Guerra A, Cerundolo N, Aloe R, Lippi G, Maggio M, Gambaro G, Borghi L, Meschi T. Idiopathic Calcium Nephrolithiasis and Hypovitaminosis D: A Case-control Study. Urology. 2016 Jan;87:40-5. doi: 10.1016/j.urology.2015.10.009[↩]
- Jackson RD, LaCroix AZ, Gass M, Wallace RB, Robbins J, Lewis CE, Bassford T, Beresford SA, Black HR, Blanchette P, Bonds DE, Brunner RL, Brzyski RG, Caan B, Cauley JA, Chlebowski RT, Cummings SR, Granek I, Hays J, Heiss G, Hendrix SL, Howard BV, Hsia J, Hubbell FA, Johnson KC, Judd H, Kotchen JM, Kuller LH, Langer RD, Lasser NL, Limacher MC, Ludlam S, Manson JE, Margolis KL, McGowan J, Ockene JK, O’Sullivan MJ, Phillips L, Prentice RL, Sarto GE, Stefanick ML, Van Horn L, Wactawski-Wende J, Whitlock E, Anderson GL, Assaf AR, Barad D; Women’s Health Initiative Investigators. Calcium plus vitamin D supplementation and the risk of fractures. N Engl J Med. 2006 Feb 16;354(7):669-83. doi: 10.1056/NEJMoa055218. Erratum in: N Engl J Med. 2006 Mar 9;354(10):1102.[↩]
- Ferroni MC, Rycyna KJ, Averch TD, Semins MJ. Vitamin D Repletion in Kidney Stone Formers: A Randomized Controlled Trial. J Urol. 2017 Apr;197(4):1079-1083. doi: 10.1016/j.juro.2016.10.057[↩]
- Eisner BH, Thavaseelan S, Sheth S, Haleblian G, Pareek G. Relationship between serum vitamin D and 24-hour urine calcium in patients with nephrolithiasis. Urology. 2012 Nov;80(5):1007-10. doi: 10.1016/j.urology.2012.04.041[↩]
- Johri N, Jaeger P, Ferraro PM, Shavit L, Nair D, Robertson WG, Gambaro G, Unwin RJ. Vitamin D deficiency is prevalent among idiopathic stone formers, but does correction pose any risk? Urolithiasis. 2017 Dec;45(6):535-543. doi: 10.1007/s00240-016-0954-x[↩]
- Batmanghelidj F, Kohlstadt I. Water: a driving force in the musculoskeletal system. In: Scientific Evidence for Musculoskeletal, Bariatric and Sports Nutrition. Boca Raton, Fla.: Taylor & Francis; 2006:127–135.[↩]
- Livingston EH, Kohlstadt I. Simplified resting metabolic rate-predicting formulas for normal-sized and obese individuals. Obes Res. 2005;13(7):1255–1262.[↩]
- Ekeruo WO, Tan YH, Young MD, et al. Metabolic risk factors and the impact of medical therapy on the management of nephrolithiasis in obese patients. J Urol. 2004;172(1):159–163.[↩]
- Eisner BH, Porten SP, Bechis SK, Stoller ML. Diabetic kidney stone formers excrete more oxalate and have lower urine pH than nondiabetic stone formers. J Urol. 2010;183(6):2244–2248.[↩]
- Maalouf NM, Cameron MA, Moe OW, Sakhaee K. Metabolic basis for low urine pH in type 2 diabetes. Clin J Am Soc Nephrol. 2010;5(7):1277–1281.[↩]
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab. 1988;66(1):140–146.[↩]
- Taylor EN, Curhan GC. Fructose consumption and the risk of kidney stones. Kidney Int. 2008;73(2):207–212.[↩]
- Choi HK, Willett W, Curhan G. Fructose-rich beverages and risk of gout in women. JAMA. 2010;304(20):2270–2278.[↩]
- Worcester EM, Coe FL. New insights into the pathogenesis of idiopathic hypercalciuria. Semin Nephrol. 2008 Mar;28(2):120-32. doi: 10.1016/j.semnephrol.2008.01.005[↩]
- Pak CY, Sakhaee K, Moe OW, Poindexter J, Adams-Huet B, Pearle MS, Zerwekh JE, Preminger GM, Wills MR, Breslau NA, Bartter FC, Brater DC, Heller HJ, Odvina CV, Wabner CL, Fordtran JS, Oh M, Garg A, Harvey JA, Alpern RJ, Snyder WH, Peters PC. Defining hypercalciuria in nephrolithiasis. Kidney Int. 2011 Oct;80(7):777-82. doi: 10.1038/ki.2011.227[↩]
- Sorokin I, Pearle MS. Medical therapy for nephrolithiasis: State of the art. Asian J Urol. 2018 Oct;5(4):243-255. doi: 10.1016/j.ajur.2018.08.005[↩][↩]
- Coe FL, Parks JH, Moore ES. Familial idiopathic hypercalciuria. N Engl J Med. 1979 Feb 15;300(7):337-40. doi: 10.1056/NEJM197902153000703[↩]
- Melton LJ 3rd, Crowson CS, Khosla S, Wilson DM, O’Fallon WM. Fracture risk among patients with urolithiasis: a population-based cohort study. Kidney Int. 1998 Feb;53(2):459-64. doi: 10.1046/j.1523-1755.1998.00779.x[↩]
- Sakhaee K, Maalouf NM, Poindexter J, Adams-Huet B, Moe OW. Relationship between Urinary Calcium and Bone Mineral Density in Patients with Calcium Nephrolithiasis. J Urol. 2017 Jun;197(6):1472-1477. doi: 10.1016/j.juro.2017.01.002[↩]
- Pearle MS, Goldfarb DS, Assimos DG, Curhan G, Denu-Ciocca CJ, Matlaga BR, Monga M, Penniston KL, Preminger GM, Turk TM, White JR; American Urological Assocation. Medical management of kidney stones: AUA guideline. J Urol. 2014 Aug;192(2):316-24. https://www.auajournals.org/doi/10.1016/j.juro.2014.05.006[↩]
- Skolarikos A, Straub M, Knoll T, Sarica K, Seitz C, Petřík A, Türk C. Metabolic evaluation and recurrence prevention for urinary stone patients: EAU guidelines. Eur Urol. 2015 Apr;67(4):750-63. doi: 10.1016/j.eururo.2014.10.029[↩][↩]
- Taylor EN, Curhan GC. Dietary calcium from dairy and nondairy sources, and risk of symptomatic kidney stones. J Urol. 2013 Oct;190(4):1255-9. doi: 10.1016/j.juro.2013.03.074[↩]
- Sakhaee K, Maalouf NM, Sinnott B. Clinical review. Kidney stones 2012: pathogenesis, diagnosis, and management. J Clin Endocrinol Metab. 2012 Jun;97(6):1847-60. doi: 10.1210/jc.2011-3492[↩]
- Hess B, Jost C, Zipperle L, Takkinen R, Jaeger P. High-calcium intake abolishes hyperoxaluria and reduces urinary crystallization during a 20-fold normal oxalate load in humans. Nephrol Dial Transplant. 1998 Sep;13(9):2241-7. doi: 10.1093/ndt/13.9.2241[↩]
- Domrongkitchaiporn S, Sopassathit W, Stitchantrakul W, Prapaipanich S, Ingsathit A, Rajatanavin R. Schedule of taking calcium supplement and the risk of nephrolithiasis. Kidney Int. 2004 May;65(5):1835-41. doi: 10.1111/j.1523-1755.2004.00587.x[↩]
- Curhan GC, Willett WC, Speizer FE, Spiegelman D, Stampfer MJ. Comparison of dietary calcium with supplemental calcium and other nutrients as factors affecting the risk for kidney stones in women. Ann Intern Med. 1997 Apr 1;126(7):497-504. doi: 10.7326/0003-4819-126-7-199704010-00001[↩]
- Curhan GC, Willett WC, Knight EL, Stampfer MJ. Dietary factors and the risk of incident kidney stones in younger women: Nurses’ Health Study II. Arch Intern Med. 2004 Apr 26;164(8):885-91. doi: 10.1001/archinte.164.8.885[↩]
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med. 1993 Mar 25;328(12):833-8. doi: 10.1056/NEJM199303253281203[↩]
- Risk of renal stone events in primary hyperparathyroidism before and after parathyroid surgery: controlled retrospective follow up study. Mollerup CL, Vestergaard P, Frøkjaer VG, Mosekilde L, Christiansen P, Blichert-Toft M. BMJ. 2002 Oct 12; 325(7368):807. https://www.ncbi.nlm.nih.gov/pubmed/12376441/[↩]
- Zerwekh JE, Holt K, Pak CY. Natural urinary macromolecular inhibitors: attenuation of inhibitory activity by urate salts. Kidney Int. 1983 Jun;23(6):838-41. doi: 10.1038/ki.1983.103[↩]
- Remer T, Manz F. Potential renal acid load of foods and its influence on urine pH. J Am Diet Assoc. 1995 Jul;95(7):791-7. doi: 10.1016/S0002-8223(95)00219-7[↩]
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab. 1988 Jan;66(1):140-6. doi: 10.1210/jcem-66-1-140[↩]
- Nouvenne A, Ticinesi A, Morelli I, Guida L, Borghi L, Meschi T. Fad diets and their effect on urinary stone formation. Transl Androl Urol. 2014 Sep;3(3):303-12. doi: 10.3978/j.issn.2223-4683.2014.06.01[↩]
- Vezzoli G, Dogliotti E, Terranegra A, Arcidiacono T, Macrina L, Tavecchia M, Pivari F, Mingione A, Brasacchio C, Nouvenne A, Meschi T, Cusi D, Spotti D, Montanari E, Soldati L. Dietary style and acid load in an Italian population of calcium kidney stone formers. Nutr Metab Cardiovasc Dis. 2015 Jun;25(6):588-93. doi: 10.1016/j.numecd.2015.03.005[↩][↩]
- Reddy ST, Wang CY, Sakhaee K, Brinkley L, Pak CY. Effect of low-carbohydrate high-protein diets on acid-base balance, stone-forming propensity, and calcium metabolism. Am J Kidney Dis. 2002 Aug;40(2):265-74. doi: 10.1053/ajkd.2002.34504[↩]
- Buclin T, Cosma M, Appenzeller M, Jacquet AF, Décosterd LA, Biollaz J, Burckhardt P. Diet acids and alkalis influence calcium retention in bone. Osteoporos Int. 2001;12(6):493-9. doi: 10.1007/s001980170095[↩]
- Maalouf NM, Moe OW, Adams-Huet B, Sakhaee K. Hypercalciuria associated with high dietary protein intake is not due to acid load. J Clin Endocrinol Metab. 2011 Dec;96(12):3733-40. doi: 10.1210/jc.2011-1531[↩]
- Eisenstein J, Roberts SB, Dallal G, Saltzman E. High-protein weight-loss diets: are they safe and do they work? A review of the experimental and epidemiologic data. Nutr Rev. 2002 Jul;60(7 Pt 1):189-200. doi: 10.1301/00296640260184264[↩]
- Tracy CR, Best S, Bagrodia A, Poindexter JR, Adams-Huet B, Sakhaee K, Maalouf N, Pak CY, Pearle MS. Animal protein and the risk of kidney stones: a comparative metabolic study of animal protein sources. J Urol. 2014 Jul;192(1):137-41. doi: 10.1016/j.juro.2014.01.093[↩]
- Hattori CM, Tiselius HG, Heilberg IP. Whey protein and albumin effects upon urinary risk factors for stone formation. Urolithiasis. 2017 Oct;45(5):421-428. doi: 10.1007/s00240-017-0975-0[↩]
- Rapid reversal of hyperoxaluria in a rat model after probiotic administration of Oxalobacter formigenes. Sidhu H, Allison MJ, Chow JM, Clark A, Peck AB. J Urol. 2001 Oct; 166(4):1487-91. https://www.ncbi.nlm.nih.gov/pubmed/11547118/[↩][↩]
- Oxalobacter formigenes and its potential role in human health. Duncan SH, Richardson AJ, Kaul P, Holmes RP, Allison MJ, Stewart CS. Appl Environ Microbiol. 2002 Aug; 68(8):3841-7. https://www.ncbi.nlm.nih.gov/pubmed/12147479/[↩]
- Contribution of dietary oxalate to urinary oxalate excretion. Holmes RP, Goodman HO, Assimos DG. Kidney Int. 2001 Jan; 59(1):270-6. https://www.ncbi.nlm.nih.gov/pubmed/11135080/[↩]
- Grentz L, Massey LK. Contribution of dietary oxalate to urinary oxalate in health and disease. Topics in Clinical Nutrition 2002;17: 60-70.[↩][↩]
- Effect of vitamin C supplements on urinary oxalate and pH in calcium stone-forming patients. Baxmann AC, De O G Mendonça C, Heilberg IP. Kidney Int. 2003 Mar; 63(3):1066-71. https://www.ncbi.nlm.nih.gov/pubmed/12631089/[↩]
- Effect of grapefruit juice on urinary lithogenicity. Goldfarb DS, Asplin JR. J Urol. 2001 Jul; 166(1):263-7. https://www.ncbi.nlm.nih.gov/pubmed/11435883/[↩]
- Hesse A, Schneeberger W, Engfeld S, Von Unruh GE, Sauerbruch T. Intestinal hyperabsorption of oxalate in calcium oxalate stone formers: application of a new test with [13C2]oxalate. J Am Soc Nephrol. 1999 Nov;10 Suppl 14:S329-33.[↩][↩]
- Holmes RP, Goodman HO, Assimos DG. Contribution of dietary oxalate to urinary oxalate excretion. Kidney Int. 2001 Jan;59(1):270-6. doi: 10.1046/j.1523-1755.2001.00488.x[↩][↩]
- von Unruh GE, Voss S, Sauerbruch T, Hesse A. Dependence of oxalate absorption on the daily calcium intake. J Am Soc Nephrol. 2004 Jun;15(6):1567-73. doi: 10.1097/01.asn.0000127864.26968.7f[↩]
- Massey LK. Dietary influences on urinary oxalate and risk of kidney stones. Front Biosci. 2003 May 1;8:s584-94. doi: 10.2741/1082[↩]
- Gettman MT, Ogan K, Brinkley LJ, Adams-Huet B, Pak CY, Pearle MS. Effect of cranberry juice consumption on urinary stone risk factors. J Urol. 2005 Aug;174(2):590-4; quiz 801. doi: 10.1097/01.ju.0000165168.68054.f8[↩]
- Goldfarb DS, Asplin JR. Effect of grapefruit juice on urinary lithogenicity. J Urol. 2001 Jul;166(1):263-7.[↩][↩]
- Huynh NK, Nguyen HVH. Effects of Juice Processing on Oxalate Contents in Carambola Juice Products. Plant Foods Hum Nutr. 2017 Sep;72(3):236-242. doi: 10.1007/s11130-017-0615-4[↩]
- Bong WC, Vanhanen LP, Savage GP. Addition of calcium compounds to reduce soluble oxalate in a high oxalate food system. Food Chem. 2017 Apr 15;221:54-57. doi: 10.1016/j.foodchem.2016.10.031[↩][↩]
- Noori N, Honarkar E, Goldfarb DS, Kalantar-Zadeh K, Taheri M, Shakhssalim N, Parvin M, Basiri A. Urinary lithogenic risk profile in recurrent stone formers with hyperoxaluria: a randomized controlled trial comparing DASH (Dietary Approaches to Stop Hypertension)-style and low-oxalate diets. Am J Kidney Dis. 2014 Mar;63(3):456-63. doi: 10.1053/j.ajkd.2013.11.022[↩]
- Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012 Jun 12;8(8):467-75. doi: 10.1038/nrneph.2012.113[↩]
- Solomon LR, Nixon AC, Ogden L, Nair B. Orlistat-induced oxalate nephropathy: an under-recognised cause of chronic kidney disease. BMJ Case Rep. 2017 Nov 12;2017:bcr2016218623. doi: 10.1136/bcr-2016-218623[↩]
- The influence of sex hormones on renal osteopontin expression and urinary constituents in experimental urolithiasis. Yagisawa T, Ito F, Osaka Y, Amano H, Kobayashi C, Toma H. J Urol. 2001 Sep; 166(3):1078-82. https://www.ncbi.nlm.nih.gov/pubmed/11490302/[↩]
- Allison MJ, Dawson KA, Mayberry WR, Foss JG. Oxalobacter formigenes gen. nov., sp. nov.: oxalate-degrading anaerobes that inhabit the gastrointestinal tract. Arch Microbiol. 1985 Feb;141(1):1-7. doi: 10.1007/BF00446731[↩]
- Hatch M, Cornelius J, Allison M, Sidhu H, Peck A, Freel RW. Oxalobacter sp. reduces urinary oxalate excretion by promoting enteric oxalate secretion. Kidney Int. 2006 Feb;69(4):691-8. doi: 10.1038/sj.ki.5000162[↩]
- Canales BK, Hatch M. Oxalobacter formigenes colonization normalizes oxalate excretion in a gastric bypass model of hyperoxaluria. Surg Obes Relat Dis. 2017 Jul;13(7):1152-1157. doi: 10.1016/j.soard.2017.03.014[↩]
- Kaufman DW, Kelly JP, Curhan GC, Anderson TE, Dretler SP, Preminger GM, Cave DR. Oxalobacter formigenes may reduce the risk of calcium oxalate kidney stones. J Am Soc Nephrol. 2008 Jun;19(6):1197-203. doi: 10.1681/ASN.2007101058[↩]
- Zuckerman JM, Assimos DG. Hypocitraturia: pathophysiology and medical management. Rev Urol. 2009 Summer;11(3):134-44. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2777061[↩][↩][↩][↩]
- Phillips R, Hanchanale VS, Myatt A, Somani B, Nabi G, Biyani CS. Citrate salts for preventing and treating calcium containing kidney stones in adults. Cochrane Database Syst Rev. 2015 Oct 6;2015(10):CD010057. doi: 10.1002/14651858.CD010057.pub2[↩][↩]
- Dey J, Creighton A, Lindberg JS, Fuselier HA, Kok DJ, Cole FE, et al. Estrogen replacement increased the citrate and calcium excretion in rates in postmenopausal women with recurrent urolithiasis. J Urol 2002;167: 169-71. https://www.ncbi.nlm.nih.gov/pubmed/11743298[↩]
- Odvina CV. Comparative value of orange juice versus lemonade in reducing stone-forming risk. Clin J Am Soc Nephrol. 2006 Nov;1(6):1269-74. doi: 10.2215/CJN.00800306[↩]
- Hönow R, Laube N, Schneider A, Kessler T, Hesse A. Influence of grapefruit-, orange- and apple-juice consumption on urinary variables and risk of crystallization. Br J Nutr. 2003 Aug;90(2):295-300. doi: 10.1079/bjn2003897[↩]
- Wabner CL, Pak CY. Effect of orange juice consumption on urinary stone risk factors. J Urol. 1993 Jun;149(6):1405-8. doi: 10.1016/s0022-5347(17)36401-7[↩]
- Tracy CR, Pearle MS. Update on the medical management of stone disease. Curr Opin Urol. 2009 Mar;19(2):200-4. doi: 10.1097/MOU.0b013e328323a81d[↩]
- Manfredini R, De Giorgi A, Storari A, Fabbian F. Pears and renal stones: possible weapon for prevention? A comprehensive narrative review. Eur Rev Med Pharmacol Sci. 2016;20(3):414-25. https://www.europeanreview.org/wp/wp-content/uploads/414-425.pdf[↩]
- Taylor EN, Fung TT, Curhan GC. DASH-style diet associates with reduced risk for kidney stones. J Am Soc Nephrol. 2009 Oct;20(10):2253-9. doi: 10.1681/ASN.2009030276[↩][↩]
- Taylor EN, Stampfer MJ, Mount DB, Curhan GC. DASH-style diet and 24-hour urine composition. Clin J Am Soc Nephrol. 2010 Dec;5(12):2315-22. doi: 10.2215/CJN.04420510[↩]
- Ferraro PM, Taylor EN, Gambaro G, Curhan GC. Dietary and Lifestyle Risk Factors Associated with Incident Kidney Stones in Men and Women. J Urol. 2017 Oct;198(4):858-863. doi: 10.1016/j.juro.2017.03.124[↩]
- The American Urological Association. Guidelines for managing kidney stones. Medical Management of Kidney Stones 2014. http://www.auanet.org/guidelines/medical-management-of-kidney-stones-(2014[↩][↩]
- Kidney stones. https://www.mayoclinic.org/diseases-conditions/kidney-stones/diagnosis-treatment/drc-20355759[↩]
- Trinchieri A, Esposito N, Castelnuovo C. Dissolution of radiolucent renal stones by oral alkalinization with potassium citrate/potassium bicarbonate. Arch Ital Urol Androl. 2009;81(3):188–191.[↩]
- Sakhaee K, Nicar M, Hill K, Pak CY. Contrasting effects of potassium citrate and sodium citrate therapies on urinary chemistries and crystallization of stone-forming salts. Kidney Int. 1983;24(3):348–352.[↩]
- Long LO, Park S. Update on nephrolithiasis management. Minerva Urol Nefrol. 2007;59(3):317–325.[↩]
- Pizzarelli F, Peacock M. Effect of chronic administration of ammonium sulfate on phosphatic stone recurrence. Nephron. 1987;46(3):247–252.[↩]
- Jarrar K, Boedeker RH, Weidner W. Struvite stones: long term follow up under metaphylaxis. Ann Urol (Paris). 1996;30(3):112–117.[↩]
- Siva S, Barrack ER, Reddy GP, et al. A critical analysis of the role of gut Oxalobacter formigenes in oxalate stone disease. BJU Int. 2009;103(1):18–21.[↩][↩]
- Hoppe B, Beck B, Gatter N, et al. Oxalobacter formigenes: a potential tool for the treatment of primary hyperoxaluria type 1. Kidney Int. 2006;70(7):1305–1311.[↩]
- Oxalobacter formigenes and its potential role in human health. Duncan SH, Richardson AJ, Kaul P, Holmes RP, Allison MJ, Stewart CS. Appl Environ Microbiol. 2002 Aug; 68(8):3841-7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC124017/[↩]
- Zilberman DE, Tsivian M, Lipkin ME, et al. Low dose computerized tomography for detection of urolithiasis—its effectiveness in the setting of the urology clinic. J Urol 185(3):910-914, 2011. https://www.ncbi.nlm.nih.gov/pubmed/21239024[↩]
- The American Urological Association. Guidelines for managing kidney stones. Medical Management of Kidney Stones 2014. https://www.auanet.org/documents/Guidelines/PDF/Kidney-Stones-Medica-Management-Guideline-JU.pdf[↩]
- Pearle MS, Goldfarb DS, Assimos DG, et al. Medical management of kidney stones—AUA guideline. J Urology192(2):316-24, 2014.https://www.ncbi.nlm.nih.gov/pubmed/24857648[↩]
- Lotan Y, Buendia Jiménez I, Lenoir-Wijnkoop I, Daudon M, Molinier L, Tack I, Nuijten MJ. Increased water intake as a prevention strategy for recurrent urolithiasis: major impact of compliance on cost-effectiveness. J Urol. 2013 Mar;189(3):935-9. doi: 10.1016/j.juro.2012.08.254[↩]
- Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol. 1996 Mar;155(3):839-43.[↩]
- Preminger GM, Tiselius HG, Assimos DG, et al.; EAU/AUA Nephrolithiasis Guideline Panel. 2007 guideline for the management of ureteral calculi. J Urol. 2007;178(6):2418–2434.[↩]
- Dellabella M, Milanese G, Muzzonigro G. Medical-expulsive therapy for distal ureterolithiasis: randomized prospective study on role of corticosteroids used in combination with tamsulosinsimplifited treatment regimen and health-related quality of life. Urology. 2005;66(4):712–715.[↩]
- Paige NM, Nagami GT. The top 10 things nephrologists wish every primary care physician knew. Mayo Clin Proc. 2009;84(2):180–186.[↩]
- Saltel E, Angel JB, Futter NG, Walsh WG, O’Rourke K, Mahoney JE. Increased prevalence and analysis of risk factors for indinavir nephrolithiasis. J Urol. 2000;164(6):1895–1897.[↩]
- Dick WH, Lingeman JE, Preminger GM, Smith LH, Wilson DM, Shirrell WL. Laxative abuse as a cause for ammonium urate renal calculi. J Urol. 1990;143(2):244–247.[↩]
- Sterrett SP, Penniston KL, Wolf JS Jr, Nakada SY. Acetazolamide is an effective adjunct for urinary alkalization in patients with uric acid and cystine stone formation recalcitrant to potassium citrate. Urology. 2008;72(2):278–281.[↩]
- Welch BJ, Graybeal D, Moe OW, Maalouf NM, Sakhaee K. Biochemical and stone-risk profiles with topiramate treatment. Am J Kidney Dis. 2006;48(4):555–563.[↩]
- Dietary Guidelines for Americans. https://health.gov/our-work/nutrition-physical-activity/dietary-guidelines[↩][↩]
- Pearle MS, Goldfarb DS, Assimos DG, Curhan G, Denu-Ciocca CJ, Matlaga BR, Monga M, Penniston KL, Preminger GM, Turk TM, White JR; American Urological Assocation. Medical management of kidney stones: AUA guideline. J Urol. 2014 Aug;192(2):316-24. doi: 10.1016/j.juro.2014.05.006[↩][↩][↩][↩]
- Fink HA, Wilt TJ, Eidman KE, Garimella PS, MacDonald R, Rutks IR, Brasure M, Kane RL, Ouellette J, Monga M. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med. 2013 Apr 2;158(7):535-43. doi: 10.7326/0003-4819-158-7-201304020-00005. Erratum in: Ann Intern Med. 2013 Aug 6;159(3):230-2.[↩]
- Vigen R, Weideman RA, Reilly RF. Thiazides diuretics in the treatment of nephrolithiasis: are we using them in an evidence-based fashion? Int Urol Nephrol. 2011 Sep;43(3):813-9. doi: 10.1007/s11255-010-9824-6[↩]
- Gambaro G, Croppi E, Coe F, Lingeman J, Moe O, Worcester E, Buchholz N, Bushinsky D, Curhan GC, Ferraro PM, Fuster D, Goldfarb DS, Heilberg IP, Hess B, Lieske J, Marangella M, Milliner D, Preminger GM, Reis Santos JM, Sakhaee K, Sarica K, Siener R, Strazzullo P, Williams JC; Consensus Conference Group. Metabolic diagnosis and medical prevention of calcium nephrolithiasis and its systemic manifestations: a consensus statement. J Nephrol. 2016 Dec;29(6):715-734. doi: 10.1007/s40620-016-0329-y[↩]
- Taylor EN, Feskanich D, Paik JM, Curhan GC. Nephrolithiasis and Risk of Incident Bone Fracture. J Urol. 2016 May;195(5):1482-1486. doi: 10.1016/j.juro.2015.12.069[↩]
- Adams JS, Song CF, Kantorovich V. Rapid recovery of bone mass in hypercalciuric, osteoporotic men treated with hydrochlorothiazide. Ann Intern Med. 1999 Apr 20;130(8):658-60. doi: 10.7326/0003-4819-130-8-199904200-00012[↩]
- Pak CY, Heller HJ, Pearle MS, Odvina CV, Poindexter JR, Peterson RD. Prevention of stone formation and bone loss in absorptive hypercalciuria by combined dietary and pharmacological interventions. J Urol. 2003 Feb;169(2):465-9. doi: 10.1097/01.ju.0000047341.55340.19[↩]
- Levy FL, Adams-Huet B, Pak CY. Ambulatory evaluation of nephrolithiasis: an update of a 1980 protocol. Am J Med. 1995 Jan;98(1):50-9. doi: 10.1016/S0002-9343(99)80080-1[↩]
- Barcelo P, Wuhl O, Servitge E, Rousaud A, Pak CY. Randomized double-blind study of potassium citrate in idiopathic hypocitraturic calcium nephrolithiasis. J Urol. 1993 Dec;150(6):1761-4. doi: 10.1016/s0022-5347(17)35888-3[↩]
- Carvalho M, Erbano BO, Kuwaki EY, Pontes HP, Liu JWTW, Boros LH, Asinelli MO, Baena CP. Effect of potassium citrate supplement on stone recurrence before or after lithotripsy: systematic review and meta-analysis. Urolithiasis. 2017 Oct;45(5):449-455. doi: 10.1007/s00240-016-0950-1[↩]
- Preminger GM, Sakhaee K, Skurla C, Pak CY. Prevention of recurrent calcium stone formation with potassium citrate therapy in patients with distal renal tubular acidosis. J Urol. 1985 Jul;134(1):20-3. doi: 10.1016/s0022-5347(17)46963-1[↩]
- Song Y, Hernandez N, Shoag J, Goldfarb DS, Eisner BH. Potassium citrate decreases urine calcium excretion in patients with hypocitraturic calcium oxalate nephrolithiasis. Urolithiasis. 2016 Apr;44(2):145-8. doi: 10.1007/s00240-015-0819-8[↩][↩]
- Sakhaee K, Pak C. Superior calcium bioavailability of effervescent potassium calcium citrate over tablet formulation of calcium citrate after Roux-en-Y gastric bypass. Surg Obes Relat Dis. 2013 Sep-Oct;9(5):743-8. doi: 10.1016/j.soard.2011.11.011[↩]
- Coe FL. Treated and untreated recurrent calcium nephrolithiasis in patients with idiopathic hypercalciuria, hyperuricosuria, or no metabolic disorder. Ann Intern Med. 1977 Oct;87(4):404-10. doi: 10.7326/0003-4819-87-4-404[↩]
- Arowojolu O, Goldfarb DS. Treatment of calcium nephrolithiasis in the patient with hyperuricosuria. J Nephrol. 2014 Dec;27(6):601-5. doi: 10.1007/s40620-014-0084-x[↩]
- Pearle MS, Roehrborn CG, Pak CY. Meta-analysis of randomized trials for medical prevention of calcium oxalate nephrolithiasis. J Endourol. 1999 Nov;13(9):679-85. doi: 10.1089/end.1999.13.679[↩]
- Morgan MS, Pearle MS. Medical management of renal stones. BMJ. 2016 Mar 14;352:i52. doi: 10.1136/bmj.i52[↩]
- Holmes RP, Assimos DG. Glyoxylate synthesis, and its modulation and influence on oxalate synthesis. J Urol. 1998 Nov;160(5):1617-24.[↩]
- Williams HE, Wandzilak TR. Oxalate synthesis, transport and the hyperoxaluric syndromes. J Urol. 1989 Mar;141(3 Pt 2):742-9. doi: 10.1016/s0022-5347(17)40999-2[↩]
- Hoyer-Kuhn H, Kohbrok S, Volland R, Franklin J, Hero B, Beck BB, Hoppe B. Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin J Am Soc Nephrol. 2014 Mar;9(3):468-77. doi: 10.2215/CJN.06820613[↩]
- Ortiz-Alvarado O, Miyaoka R, Kriedberg C, Moeding A, Stessman M, Monga M. Pyridoxine and dietary counseling for the management of idiopathic hyperoxaluria in stone-forming patients. Urology. 2011 May;77(5):1054-8. doi: 10.1016/j.urology.2010.08.002[↩]
- Ferraro PM, Taylor EN, Gambaro G, Curhan GC. Vitamin B6 intake and the risk of incident kidney stones. Urolithiasis. 2018 Jun;46(3):265-270. doi: 10.1007/s00240-017-0999-5[↩]
- Jaeger P, Portmann L, Jacquet AF, Burckhardt P. La pyridoxine peut normaliser l’oxalurie dans la lithiase rénale idiopathique [Pyridoxine can normalize oxaluria in idiopathic renal lithiasis]. Schweiz Med Wochenschr. 1986 Dec 13;116(50):1783-6. French.[↩]
- Maalouf NM, Cameron MA, Moe OW, Sakhaee K. Novel insights into the pathogenesis of uric acid nephrolithiasis. Curr Opin Nephrol Hypertens. 2004 Mar;13(2):181-9. doi: 10.1097/00041552-200403000-00006[↩]
- Spettel S, Shah P, Sekhar K, Herr A, White MD. Using Hounsfield unit measurement and urine parameters to predict uric acid stones. Urology. 2013 Jul;82(1):22-6. doi: 10.1016/j.urology.2013.01.015[↩]
- Bonatti M, Lombardo F, Zamboni GA, Pernter P, Pycha A, Mucelli RP, Bonatti G. Renal stones composition in vivo determination: comparison between 100/Sn140 kV dual-energy CT and 120 kV single-energy CT. Urolithiasis. 2017 Jun;45(3):255-261. doi: 10.1007/s00240-016-0905-6[↩]
- Reichard C, Gill BC, Sarkissian C, De S, Monga M. 100% uric acid stone formers: what makes them different? Urology. 2015 Feb;85(2):296-8. doi: 10.1016/j.urology.2014.10.029[↩][↩]
- Trinchieri A, Esposito N, Castelnuovo C. Dissolution of radiolucent renal stones by oral alkalinization with potassium citrate/potassium bicarbonate. Arch Ital Urol Androl. 2009 Sep;81(3):188-91.[↩]
- Sinha M, Prabhu K, Venkatesh P, Krishnamoorthy V. Results of urinary dissolution therapy for radiolucent calculi. Int Braz J Urol. 2013 Jan-Feb;39(1):103-7. doi: 10.1590/S1677-5538.IBJU.2013.01.13[↩]
- Jaeger P, Portmann L, Saunders A, Rosenberg LE, Thier SO. Anticystinuric effects of glutamine and of dietary sodium restriction. N Engl J Med. 1986 Oct 30;315(18):1120-3. doi: 10.1056/NEJM198610303151803[↩]
- Lindell A, Denneberg T, Edholm E, Jeppsson JO. The effect of sodium intake on cystinuria with and without tiopronin treatment. Nephron. 1995;71(4):407-15. doi: 10.1159/000188760[↩]
- Rodman JS, Blackburn P, Williams JJ, Brown A, Pospischil MA, Peterson CM. The effect of dietary protein on cystine excretion in patients with cystinuria. Clin Nephrol. 1984 Dec;22(6):273-8.[↩]
- Sakhaee K, Poindexter JR, Pak CY. The spectrum of metabolic abnormalities in patients with cystine nephrolithiasis. J Urol. 1989 Apr;141(4):819-21. doi: 10.1016/s0022-5347(17)41019-6[↩]
- Parks JH, Coe FL, Evan AP, Worcester EM. Urine pH in renal calcium stone formers who do and do not increase stone phosphate content with time. Nephrol Dial Transplant. 2009 Jan;24(1):130-6. doi: 10.1093/ndt/gfn420[↩]
- Barbey F, Joly D, Rieu P, Méjean A, Daudon M, Jungers P. Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol. 2000 May;163(5):1419-23. doi: 10.1016/s0022-5347(05)67633-1[↩][↩]
- Saravakos P, Kokkinou V, Giannatos E. Cystinuria: current diagnosis and management. Urology. 2014 Apr;83(4):693-9. doi: 10.1016/j.urology.2013.10.013[↩]
- Tiopronin. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=211843[↩]
- DeBerardinis RJ, Coughlin CR 2nd, Kaplan P. Penicillamine therapy for pediatric cystinuria: experience from a cohort of American children. J Urol. 2008 Dec;180(6):2620-3. doi: 10.1016/j.juro.2008.08.057[↩]
- Pak CY, Fuller C, Sakhaee K, Zerwekh JE, Adams BV. Management of cystine nephrolithiasis with alpha-mercaptopropionylglycine. J Urol. 1986 Nov;136(5):1003-8. doi: 10.1016/s0022-5347(17)45188-3[↩]
- Sloand JA, Izzo JL. Captopril Reduces Urinary Cystine Excretion in Cystinuria. Arch Intern Med. 1987;147(8):1409–1412. doi:10.1001/archinte.1987.00370080045011[↩]
- Zee T, Bose N, Zee J, Beck JN, Yang S, Parihar J, Yang M, Damodar S, Hall D, O’Leary MN, Ramanathan A, Gerona RR, Killilea DW, Chi T, Tischfield J, Sahota A, Kahn A, Stoller ML, Kapahi P. α-Lipoic acid treatment prevents cystine urolithiasis in a mouse model of cystinuria. Nat Med. 2017 Mar;23(3):288-290. doi: 10.1038/nm.4280[↩]
- Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int. 2006 Mar;69(6):1041-7. doi: 10.1038/sj.ki.5000104[↩]
- Dolin DJ, Asplin JR, Flagel L, Grasso M, Goldfarb DS. Effect of cystine-binding thiol drugs on urinary cystine capacity in patients with cystinuria. J Endourol. 2005 Apr;19(3):429-32. doi: 10.1089/end.2005.19.429[↩]
- Parkhomenko E, De Fazio A, Tran T, Thai J, Blum K, Gupta M. A Multi-Institutional Study of Struvite Stones: Patterns of Infection and Colonization. J Endourol. 2017 May;31(5):533-537. doi: 10.1089/end.2016.0885[↩]
- Griffith DP, Gleeson MJ, Lee H, Longuet R, Deman E, Earle N. Randomized, double-blind trial of Lithostat (acetohydroxamic acid) in the palliative treatment of infection-induced urinary calculi. Eur Urol. 1991;20(3):243-7. doi: 10.1159/000471707[↩]
- Williams JJ, Rodman JS, Peterson CM. A randomized double-blind study of acetohydroxamic acid in struvite nephrolithiasis. N Engl J Med. 1984 Sep 20;311(12):760-4. doi: 10.1056/NEJM198409203111203[↩]
- Griffith DP, Khonsari F, Skurnick JH, James KE. A randomized trial of acetohydroxamic acid for the treatment and prevention of infection-induced urinary stones in spinal cord injury patients. J Urol. 1988 Aug;140(2):318-24. doi: 10.1016/s0022-5347(17)41592-8[↩]
- Iqbal MW, Youssef RF, Neisius A, Kuntz N, Hanna J, Ferrandino MN, Preminger GM, Lipkin ME. Contemporary Management of Struvite Stones Using Combined Endourologic and Medical Treatment: Predictors of Unfavorable Clinical Outcome. J Endourol. 2016 Jul;30(7):771-7. doi: 10.1089/end.2013.0257[↩]
- Sigurjonsdottir V. K., L.Runolfsdottir H., Indridason O. S., et al. Impact of nephrolithiasis on kidney function. BMC Nephrology. 2015;16(1):p. 149. doi: 10.1186/s12882-015-0126-1[↩]
- El-Zoghby Z. M., Lieske J. C., Foley R. N., et al. Urolithiasis and the risk of ESRD. Clinical Journal of the American Society of Nephrology. 2012;7(9):1409–1415. doi: 10.2215/cjn.03210312[↩]
- Rule A. D., Roger V. L., Melton L. J., et al. Kidney stones associate with increased risk for myocardial infarction. Journal of the American Society of Nephrology. 2010;21(10):1641–1644. doi: 10.1681/asn.2010030253[↩]
- Worcester E. M., Coe F. L. Nephrolithiasis. Primary Care. 2008;35(2):369–391. doi: 10.1016/j.pop.2008.01.005[↩]
- Taylor E. N., Stampfer M. J., Curhan G. C. Obesity, weight gain and the risk of kidney stones. Journal of the American Medical Association. 2005;293(4):455–462. doi: 10.1001/jama.293.4.455[↩]
- Courbebaisse M., Prot-Bertoye C., Bertocchio J., et al. Nephrolithiasis of adult: from mechanisms to preventive medical treatment. Revue Medicale Internationale. 2017;38(1):44–52. doi: 10.1016/j.revmed.2016.05.013[↩]
- Expert Panel on Interventional Radiology; Scheidt MJ, Hohenwalter EJ, Pinchot JW, Ahmed O, Bjurlin MA, Braun AR, Kim CY, Knavel Koepsel EM, Schramm K, Sella DM, Weiss CR, Lorenz JM. ACR Appropriateness Criteria® Radiologic Management of Urinary Tract Obstruction. J Am Coll Radiol. 2020 May;17(5S):S281-S292. doi: 10.1016/j.jacr.2020.01.039[↩]
- Soleimani M, Rastegar A. Pathophysiology of renal tubular acidosis: core curriculum 2016. Am J Kidney Dis. 2016;68:488–498. doi: 10.1053/j.ajkd.2016.03.422[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Palmer BF, Kelepouris E, Clegg DJ. Renal Tubular Acidosis and Management Strategies: A Narrative Review. Adv Ther. 2021 Feb;38(2):949-968. doi: 10.1007/s12325-020-01587-5[↩][↩]
- Kurtz I. Renal Tubular Acidosis: H+/Base and Ammonia Transport Abnormalities and Clinical Syndromes. Adv Chronic Kidney Dis. 2018 Jul;25(4):334-350. doi: 10.1053/j.ackd.2018.05.005[↩][↩]
- Renal Tubular Acidosis. https://www.niddk.nih.gov/health-information/kidney-disease/renal-tubular-acidosis[↩][↩]
- Roth KS, Chan JC. Renal tubular acidosis: a new look at an old problem. Clin Pediatr (Phila). 2001 Oct;40(10):533-43. doi: 10.1177/000992280104001001[↩][↩]
- Hamm LL, Nakhoul N, Hering-Smith KS. Acid-Base Homeostasis. Clin J Am Soc Nephrol. 2015 Dec 7;10(12):2232-42. doi: 10.2215/CJN.07400715[↩]
- Trepiccione F, Prosperi F, de la Motte LR, et al. New findings on the pathogenesis of distal renal tubular acidosis. Kidney Dis (Basel) 2017;3:98–105. doi: 10.1159/000478781[↩]
- Primary Distal Renal Tubular Acidosis. https://rarediseases.org/rare-diseases/primary-distal-renal-tubular-acidosis[↩][↩][↩][↩]
- Mohebbi N, Wagner CA. Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis. J Nephrol. 2018 Aug;31(4):511-522. doi: 10.1007/s40620-017-0447-1[↩][↩]
- Gil-Peña H, Mejía N, Santos F. Renal tubular acidosis. J Pediatr. 2014 Apr;164(4):691-698.e1. doi: 10.1016/j.jpeds.2013.10.085[↩][↩]
- Kashoor I, Batlle D. Proximal renal tubular acidosis with and without Fanconi syndrome. Kidney Res Clin Pract. 2019;38:267–281. doi: 10.23876/j.krcp.19.056[↩][↩][↩][↩][↩]
- Haque SK, Ariceta G, Batlle D. Proximal renal tubular acidosis: a not so rare disorder of multiple etiologies. Nephrol Dial Transplant. 2012;27:4273–4287. doi: 10.1093/ndt/gfs493[↩][↩][↩][↩][↩][↩][↩][↩]
- Palmer BF, Clegg DJ. Hyperchloremic normal gap metabolic acidosis. Minerva Endocrinol. 2019 Dec;44(4):363-377. doi: 10.23736/S0391-1977.19.03059-1[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Unwin RJ, Capasso G. The renal tubular acidoses. J R Soc Med. 2001;94:221–225. doi: 10.1177/014107680109400506[↩]
- Igarashi T, Inatomi J, Sekine T, et al. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet. 1999;23:264–266. doi: 10.1038/15440[↩]
- Igarashi T, Sekine T, Inatomi J, Seki G. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol. 2002;13:2171–2177. doi: 10.1097/01.ASN.0000025281.70901.30[↩]
- Gutierrez JO, Zurita MF, Zurita LA. Sjögren’s Syndrome Associated with Fanconi’s Syndrome and Osteomalacia. Am J Case Rep. 2018 Apr 3;19:392-396. doi: 10.12659/ajcr.907503[↩][↩]
- Yamaguchi S, Maruyama T, Wakino S, Tokuyama H, Hashiguchi A, Tada S, Homma K, Monkawa T, Thomas J, Miyashita K, Kurihara I, Yoshida T, Konishi K, Hayashi K, Hayashi M, Itoh H. A case of severe osteomalacia caused by Tubulointerstitial nephritis with Fanconi syndrome in asymptomotic primary biliary cirrhosis. BMC Nephrol. 2015 Nov 11;16:187. doi: 10.1186/s12882-015-0184-4[↩][↩]
- Lemann J Jr, Adams ND, Wilz DR, Brenes LG. Acid and mineral balances and bone in familial proximal renal tubular acidosis. Kidney Int. 2000 Sep;58(3):1267-77. doi: 10.1046/j.1523-1755.2000.00282.x[↩]
- Brenner RJ, Spring DB, Sebastian A, et al. Incidence of radiographically evident bone disease, nephrocalcinosis, and nephrolithiasis in various types of renal tubular acidosis. N Engl J Med. 1982;307:217–221. doi: 10.1056/NEJM198207223070403[↩]
- Lee YS, Kim BK, Lee HJ, Dan J. Pathologic femoral neck fracture due to Fanconi syndrome induced by adefovir dipivoxil therapy for hepatitis B. Clin Orthop Surg. 2016;8:232–236. doi: 10.4055/cios.2016.8.2.232[↩]
- Negro A, Regolisti G, Perazzoli F, Davoli S, Sani C, Rossi E. Ifosfamide-induced renal Fanconi syndrome with associated nephrogenic diabetes insipidus in an adult patient. Nephrol Dial Transplant. 1998;13:1547–1549. doi: 10.1093/ndt/13.6.1547[↩]
- Knights M, Thekkekkara T, Morris A, Finlay E. Sodium valproate-induced Fanconi type proximal renal tubular acidosis. BMJ Case Rep. 2016;2016:bcr2015213418[↩]
- Izzedine H, Launay-Vacher V, Deray G. Topiramate-induced renal tubular acidosis. Am J Med. 2004;116:281–282. doi: 10.1016/j.amjmed.2003.08.021[↩]
- Sacré A, Jouret F, Manicourt D, Devuyst O. Topiramate induces type 3 renal tubular acidosis by inhibiting renal carbonic anhydrase. Nephrol Dial Transplant. 2006;21:2995–2996. doi: 10.1093/ndt/gfl251[↩]
- Nagai T, Matsuo N, Tsuchiya Y, Cho H, Hasegawa Y, Igarashi Y. Proximal renal tubular acidosis associated with glycogen storage disease, type 9. Acta Paediatr Scand. 1988;77:460–463. doi: 10.1111/j.1651-2227.1988.tb10681.x[↩]
- Richardson RM, Little JA, Patten RL, Goldstein MB, Halperin ML. Pathogenesis of acidosis in hereditary fructose intolerance. Metabolism. 1979;28:1133–1138. doi: 10.1016/0026-0495(79)90152-5[↩]
- Mathur M, Chacko B, Vankalakunti M, Patil C. Fanconi syndrome due to light chain proximal tubulopathy in a patient with multiple myeloma. Saudi J Kidney Dis Transpl. 2016;27:805–807. doi: 10.4103/1319-2442.185268[↩][↩]
- Lacruz RS, Habelitz S, Wright JT, Paine ML. DENTAL ENAMEL FORMATION AND IMPLICATIONS FOR ORAL HEALTH AND DISEASE. Physiol Rev. 2017 Jul 1;97(3):939-993. doi: 10.1152/physrev.00030.2016[↩]
- Chanchlani R, Nemer P, Sinha R, Nemer L, Krishnappa V, Sochett E, Safadi F, Raina R. An Overview of Rickets in Children. Kidney Int Rep. 2020 Apr 11;5(7):980-990. doi: 10.1016/j.ekir.2020.03.025[↩]
- Menegussi J, Tatagiba LS, Vianna JGP, Seguro AC, Luchi WM. A physiology-based approach to a patient with hyperkalemic renal tubular acidosis. J Bras Nefrol. 2018 Oct-Dec;40(4):410-417. doi: 10.1590/2175-8239-JBN-3821[↩]
- Batlle D, Arruda J. Hyperkalemic forms of renal tubular acidosis: clinical and pathophysiological aspects. Adv Chronic Kidney Dis. 2018;25:321–333. doi: 10.1053/j.ackd.2018.05.004[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Karet FE. Mechanisms in hyperkalemic renal tubular acidosis. J Am Soc Nephrol. 2009;20:251–254. doi: 10.1681/ASN.2008020166[↩][↩][↩]
- Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med. 2015;373:548–559. doi: 10.1056/NEJMra1503102[↩][↩]
- Mount DB. Thick ascending limb of the loop of Henle. Clin J Am Soc Nephrol. 2014;9:1974–1986. doi: 10.2215/CJN.04480413[↩][↩]
- Montford JR, Linas S. How Dangerous Is Hyperkalemia? J Am Soc Nephrol. 2017 Nov;28(11):3155-3165. doi: 10.1681/ASN.2016121344[↩]
- Yaxley J, Pirrone C. Review of the Diagnostic Evaluation of Renal Tubular Acidosis. Ochsner J. 2016 Winter;16(4):525-530. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5158160[↩][↩][↩][↩][↩][↩][↩]
- Batlle DC, Arruda JA, Kurtzman NA. Hyperkalemic distal renal tubular acidosis associated with obstructive uropathy. N Engl J Med. 1981;304:373–380. doi: 10.1056/NEJM198102123040701[↩]
- Cook E, Davis J, Israni R, et al. Prevalence of metabolic acidosis among patients with CKD and hyperkalemia [abstract 89] Am J Kidney Dis. 2020;75:561–562.[↩]
- Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844[↩][↩]
- Harris RC, Breyer MD. Update on cyclooxygenase-2 inhibitors. Clin J Am Soc Nephrol. 2006;1:236–245. doi: 10.2215/CJN.00890805[↩]
- Lin W, Mou L, Tu H, et al. Clinical analysis of hyperkalemic renal tubular acidosis caused by calcineurin inhibitors in solid organ transplant recipients. J Clin Pharm Ther. 2017;42:122–124. doi: 10.1111/jcpt.12485[↩]
- Riveiro-Barciela M, Campos-Varela I, Tovar JL, et al. Hyperkalemic distal renal tubular acidosis caused by immunosuppressant treatment with tacrolimus in a liver transplant patient: case report. Transplant Proc. 2011;43:4016–4018. doi: 10.1016/j.transproceed.2011.09.064[↩]
- Schmoyer C, Mishra S, Fulco F. Tacrolimus-induced type IV renal tubular acidosis following liver transplantation. Case Rep Hepatol. 2017;2017:9312481[↩][↩]
- Tumlin JA, Sands JM. Nephron segment-specific inhibition of Na+/K+-ATPase activity by cyclosporin A. Kidney Int. 1993;43:246–251. doi: 10.1038/ki.1993.38[↩]
- Heering PJ, Kurschat C, Vo DT, Klein-Vehne N, Fehsel K, Ivens K. Aldosterone resistance in kidney transplantation is in part induced by a down-regulation of mineralocorticoid receptor expression. Clin Transplant. 2004;18:186–192. doi: 10.1046/j.1399-0012.2003.00154.x[↩]
- Adrenal Insufficiency & Addison’s Disease. https://www.niddk.nih.gov/health-information/endocrine-diseases/adrenal-insufficiency-addisons-disease[↩]
- Santos F, Ordonez FA, Claramunt-Taberner D, Gil-Pena H. Clinical and laboratory approaches in the diagnosis of renal tubular acidosis. Pediatr Nephrol. 2015;30:2099–2107. doi: 10.1007/s00467-015-3083-9[↩]
- Kyono Y, Nozu K, Nakagawa T, et al. Combination of furosemide and fludrocortisone as a loading test for diagnosis of distal renal tubular acidosis in a pediatric case. CEN Case Rep. 2020;9:81–86. doi: 10.1007/s13730-019-00432-1[↩]
- Walsh SB, Shirley DG, Wrong OM, Unwin RJ. Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment: an alternative to ammonium chloride. Kidney Int. 2007;71:1310–1316. doi: 10.1038/sj.ki.5002220[↩]
- Kim S, Lee JW, Park J, et al. The urine-blood PCO2 gradient as a diagnostic index of H+-ATPase defect distal renal tubular acidosis. Kidney Int. 2004;66:761–767. doi: 10.1111/j.1523-1755.2004.00801.x[↩][↩]
- Overview and pathophysiology of renal tubular acidosis and the effect on potassium balance. https://www.uptodate.com/contents/overview-and-pathophysiology-of-renal-tubular-acidosis-and-the-effect-on-potassium-balance[↩][↩]
- Stinebaugh BJ, Schloeder FX, Tam SC, Goldstein MB, Halperin ML. Pathogenesis of distal renal tubular acidosis. Kidney Int. 1981;19:1–7. doi: 10.1038/ki.1981.1[↩]
- Berend K. Review of the Diagnostic Evaluation of Normal Anion Gap Metabolic Acidosis. Kidney Dis (Basel). 2017 Dec;3(4):149-159. doi: 10.1159/000479279[↩][↩][↩][↩][↩][↩]
- Sidler M, Mohebbi N, Hoorn EJ, Wagner CA. Gut it out: laxative abuse mimicking distal renal tubular acidosis. Kidney Blood Press Res. 2019;44:1294–1299. doi: 10.1159/000501855[↩]
- Patel S, Sharma S. Respiratory Acidosis. [Updated 2022 Jun 21]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482430[↩]
- Rajkumar P, Pluznick JL. Acid-base regulation in the renal proximal tubules: using novel pH sensors to maintain homeostasis. Am J Physiol Renal Physiol. 2018 Nov 1;315(5):F1187-F1190. doi: 10.1152/ajprenal.00185.2018[↩]
- Pandey DG, Sharma S. Biochemistry, Anion Gap. [Updated 2019 Apr 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539757[↩][↩][↩]
- Gamble JL. Extracellular fluid and its maintenance. N Engl J Med. 1936;250:1150–1152.[↩]
- Berend K, de Vries AP, Gans RO. Physiological approach to assessment of acid-base disturbances. N Engl J Med. 2015 Jan 8;372(2):195. doi: 10.1056/NEJMc1413880[↩]
- Hopkins E, Sanvictores T, Sharma S. Physiology, Acid Base Balance. [Updated 2022 Sep 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507807[↩][↩]
- Burger MK, Schaller DJ. Metabolic Acidosis. [Updated 2022 Jul 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482146[↩]
- Marano M. Sobre el uso de la fórmula de Winters en la acidosis metabólica crónica [On the use of Winters’ formula in chronic metabolic acidosis]. Rev Psiquiatr Salud Ment. 2015 Jan-Mar;8(1):45-6. Spanish. doi: 10.1016/j.rpsm.2014.07.006[↩]
- Pereira PC, Miranda DM, Oliveira EA, Silva AC. Molecular pathophysiology of renal tubular acidosis. Curr Genomics. 2009 Mar;10(1):51-9. doi: 10.2174/138920209787581262[↩]
- Walsh SB, Shirley DG, Wrong OM, Unwin RJ. Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment: an alternative to ammonium chloride. Kidney Int. 2007 Jun;71(12):1310-6. doi: 10.1038/sj.ki.5002220[↩]
- Mohebbi N, Wagner CA. Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis. J Nephrol. 2018;31:511–522. doi: 10.1007/s40620-017-0447-1[↩]
- Karatzas A, Paridis D, Kozyrakis D, Tzortzis V, Samarinas M, Dailiana Z, Karachalios T. Fanconi syndrome in the adulthood. The role of early diagnosis and treatment. J Musculoskelet Neuronal Interact. 2017 Dec 1;17(4):303-306. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5749037[↩][↩]
- Sebastian A, McSherry E, Morris RC., Jr On the mechanism of renal potassium wasting in renal tubular acidosis associated with the Fanconi syndrome (type 2 RTA) J Clin Invest. 1971;50:231–243. doi: 10.1172/JCI106479[↩]
- Nash MA, Torrado AD, Greifer I, Spitzer A, Edelmann CM Jr. Renal tubular acidosis in infants and children. Clinical course, response to treatment, and prognosis. J Pediatr. 1972 May;80(5):738-48. doi: 10.1016/s0022-3476(72)80124-0[↩]
- Raphael KL. Metabolic acidosis in CKD: core curriculum 2019. Am J Kidney Dis. 2019;74:263–275. doi: 10.1053/j.ajkd.2019.01.036[↩][↩][↩][↩]
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004;351:585–592. doi: 10.1056/NEJMra035279[↩]
- Dobbin SJH, Petrie JR, Lean MEJ, McKay GA. Fludrocortisone therapy for persistent hyperkalaemia. Diabet Med. 2017;34:1005–1008. doi: 10.1111/dme.13359[↩]
- Kidney Disease Improving Global Outcomes KDIGO clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3:1–150. doi: 10.1038/kisup.2012.73[↩]
- Pitt B, Anker SD, Bushinsky DA, Kitzman DW, Zannad F, Huang IZ. Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double-blind, placebo-controlled study in patients with chronic heart failure (the PEARL-HF) trial. Eur Heart J. 2011;32:820–828. doi: 10.1093/eurheartj/ehq502[↩]
- Stavros F, Yang A, Leon A, Nuttall M, Rasmussen HS. Characterization of structure and function of ZS-9, a K+ selective ion trap. PLoS One. 2014;9:e114686. doi: 10.1371/journal.pone.0114686[↩]
- Roger SD, Lavin PT, Lerma EV, et al. Long-term safety and efficacy of sodium zirconium cyclosilicate for hyperkalaemia in patients with mild/moderate versus severe/end-stage chronic kidney disease: comparative results from an open-label, phase 3 study. Nephrol Dial Transplant. 2020 doi: 10.1093/ndt/gfz285[↩][↩]
- Roger SD, Spinowitz BS, Lerma EV, et al. Sodium zirconium cyclosilicate increases serum bicarbonate concentrations among patients with hyperkalaemia: exploratory analyses from three randomized, multi-dose, placebo-controlled trials. Nephrol Dial Transplant. 2020 doi: 10.1093/ndt/gfaa158[↩]
- Carrero JJ, Gonzalez-Ortiz A, Avesani CM, et al. Plant-based diets to manage the risks and complications of chronic kidney disease. Nat Rev Nephrol. 2020;16:525–542. doi: 10.1038/s41581-020-0297-2[↩]
- Navaneethan SD, Shao J, Buysse J, Bushinsky DA. Effects of treatment of metabolic acidosis in CKD: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2019;14:1011–1020. doi: 10.2215/CJN.13091118[↩]
- Meschi T, Maggiore U, Fiaccadori E, et al. The effect of fruits and vegetables on urinary stone risk factors. Kidney Int. 2004;66:2402–2410. doi: 10.1111/j.1523-1755.2004.66029.x[↩]
- Goraya N, Wesson DE. Management of the metabolic acidosis of chronic kidney disease. Adv Chronic Kidney Dis. 2017;24:298–304. doi: 10.1053/j.ackd.2017.06.006[↩]
- Merck Sharp & Dohme Corp., Merck Manual. Fanconi Syndrome. https://www.merckmanuals.com/professional/genitourinary-disorders/renal-transport-abnormalities/fanconi-syndrome#v1057806[↩][↩][↩][↩][↩][↩][↩][↩]
- U.S. Department of Health & Human Services. Genetic Home Reference. Cystinuria. https://ghr.nlm.nih.gov/condition/cystinuria[↩][↩][↩][↩]
- Merck Sharp & Dohme Corp., Merck Manual. Cystinuria. https://www.merckmanuals.com/professional/pediatrics/congenital-renal-transport-abnormalities/cystinuria[↩][↩][↩]
- Cystinuria. https://medlineplus.gov/genetics/condition/cystinuria[↩][↩]
- Cystinuria. https://rarediseases.org/rare-diseases/cystinuria[↩]
- Merck Sharp & Dohme Corp., Merck Manual. Introduction to Urinary Tract Infections (UTIs). https://www.merckmanuals.com/professional/genitourinary-disorders/urinary-tract-infections-utis/introduction-to-urinary-tract-infections-utis[↩][↩][↩][↩]
- Hooton TM, Roberts PL, Cox ME, Stapleton AE. Voided midstream urine culture and acute cystitis in premenopausal women. N Engl J Med 369(20):1883-1891, 2013. https://www.ncbi.nlm.nih.gov/pubmed/24224622[↩]
- U.S. Department of Health and Human Services. The National Institute of Diabetes and Digestive and Kidney Diseases Health Information Center. What Is Polycystic Kidney Disease ? https://www.niddk.nih.gov/health-information/kidney-disease/polycystic-kidney-disease/what-is-pkd[↩][↩]
- National Library of Medicine. Genetics Home Reference. Polycystic kidney disease. https://ghr.nlm.nih.gov/condition/polycystic-kidney-disease[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- U.S. National Library of Medicine. MedlinePlus. Polycystic kidney disease. https://medlineplus.gov/ency/article/000502.htm[↩][↩]
- Vicente E Torres MD, William M Bennett MD. Diagnosis of and screening for autosomal dominant polycystic kidney disease. UpToDate website. Updated: September 2, 2015.[↩]
- Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003;111(5 Pt 1):1072–1080.[↩]
- Torres VE, Grantham JJ. Cystic diseases of the kidney. In: Taal MW, ed. Brenner & Rector’s The Kidney. 9th ed. Philadelphia: W.B. Saunders; 2011: 1626–1667.[↩][↩][↩]
- Chapman AB, Rahbari-Ouskoui FF, Bennett WM. Acquired cystic disease of the kidneys in adults. https://www.uptodate.com/contents/acquired-cystic-disease-of-the-kidney-in-adults?source=search_result&search=acquired+cystic+disease+of+the+kidney+inadults&selectedTitle=1~150[↩]
- U.S. Department of Health and Human Services. The National Institute of Diabetes and Digestive and Kidney Diseases Health Information Center. Nutrition for Advanced Chronic Kidney Disease in Adults. https://www.niddk.nih.gov/health-information/kidney-disease/chronic-kidney-disease-ckd/eating-nutrition/nutrition-advanced-chronic-kidney-disease-adults[↩]
- Borghi L, Schianchi T, Meschi T, Guerra A, Allegri F, Maggiore U, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002;346: 77-84. http://www.nejm.org/doi/full/10.1056/NEJMoa010369[↩][↩][↩][↩]
- McHarg T, Rodgers A, Charlton K. Influence of cranberry juice on the urinary risk factors for calcium oxalate stone formation. BJU Int 2003;92: 765-8. https://www.ncbi.nlm.nih.gov/pubmed/14616463[↩]
- Rule AD, Bergstralh EJ, Melton LJ III, Li X, Weaver AL, Lieske JC. Kidney stones and the risk for chronic kidney disease. Clin J Am Soc Nephrol. 2009;4(4):804–811.[↩]
- Gambaro G, Favaro S, D’Angelo A. Risk for renal failure in nephrolithiasis. Am J Kidney Dis. 2001;37(2):233–243.[↩]





{kind=link}