Contents
What is homocysteine
Homocysteine (Hcy) is a sulfur containing amino acid, which is an intermediate product in the normal biosynthesis of the amino acids methionine and cysteine that is typically present in very small amounts in all cells of your body 1. Homocysteine is an amino acid produced via demethylation of dietary methionine, which is abundant in animal protein 2. Vitamin B6 (Pyridoxine), Folic acid (vitamin B9), Riboflavin (vitamin B2), and Vitamin B12 (Cyanocobalamin) are all required for methionine metabolism, and deficiency of each of these vitamins result in elevated plasma homocysteine. That is because your body normally converts homocysteine into other products quickly. Since vitamin B6 (pyridoxine), vitamin B12 (cyanocobalamin) and folate (vitamin B9) are necessary to metabolize (break down) homocysteine, increased levels of the homocysteine amino acid may be a sign of deficiency in those vitamins. Elevations in plasma homocysteine are commonly found as a result of vitamin deficiencies, polymorphisms of enzymes of methionine metabolism, and kidney disease. Although homocysteine is naturally present in our bodies, too much homocysteine in your blood (hyperhomocysteinemia) has been associated with an increased risk of heart and blood vessel disease (cardiovascular disease), including venous thrombosis (blood clots in veins), atherosclerosis (hardening of the arteries), high blood pressure, coronary heart disease (coronary artery disease), and stroke 3, 4, 5, 6. Most research indicates that a plasma homocysteine level less than 10 micromoles/L (<10 μmol/L) is associated with a lower risk of heart and blood vessel disease (cardiovascular disease) 7. The association between increased homocysteine (Hcy) levels and atherosclerosis-related conditions, including carotid plaque formation, coronary artery calcification and peripheral arterial disease, is well documented. A meta-analysis of 26 studies revealed that each 5 μmol/L rise in homocysteine (Hcy) levels results in a 20% increase in the risk of coronary heart disease (coronary artery disease), irrespective of common risk factors such as high blood pressure (hypertension), smoking, and diabetes 8. Notably, elevated homocysteine (Hcy) levels account for approximately 10% of coronary artery disease cases in the population, with a strong dose-dependent positive relationship between plasma homocysteine (Hcy) concentrations and cardiovascular disease risk 9. Further research has confirmed that individuals with homocysteine concentrations ≥ 10 μmol/L are at more than double the risk of developing peripheral arterial disease 5, 10. The Multi-Ethnic Study of Atherosclerosis (MESA) demonstrated that homocysteine levels exceeding 12 μmol/L were associated with over twice the risk of coronary artery calcification and descending thoracic aortic calcification, even after accounting for other cardiovascular risk factors 11. Several mechanisms have been proposed for how homocysteine leads to cardiovascular disease risk, including damaging blood vessel walls and supporting the formation of inappropriate blood clots, but direct links haven not been confirmed. Furthermore, there are also several studies that indicate no benefit of lowering elevated serum homocysteine and lipoprotein-A to reduce the risk of cardiovascular disease, prevent recurrent stroke, heart attack (myocardial infarction), and death with folic acid and B vitamin supplements 12, 13, 14.
At present, the use of homocysteine levels for risk assessment of cardiovascular disease, peripheral vascular disease, and stroke is uncertain, given that several trials investigating folic acid and vitamin B supplementation indicate no benefit or lowering of cardiovascular disease risk 12, 13, 14, 15, 16, 17. Additionally, a 2012 study of multiple datasets, including 50,000 people with coronary heart disease, called the potential for a cause-and-effect relationship between homocysteine levels and heart disease into question 15. A 2015 review of studies concluded that although there is a relationship between homocysteine and cardiovascular disease, there is other evidence that precludes homocysteine in being considered a biomarker for the disease 16. According to the American Heart Association (AHA), homocysteine levels do not predict cardiovascular disease development. Lowering homocysteine levels does not improve clinical outcomes, nor does it prevent future cardiovascular and thromboembolic diseases with treatment 18, 19, 20, 21. The American Heart Association (AHA) does acknowledge the relationship between homocysteine levels and heart attack/stroke survival rates but doesn’t consider elevated homocysteine an independent risk factor for cardiovascular disease. While the American Heart Association does not recommend the widespread use of folic acid and vitamin B to reduce risk of heart attack and stroke, it does promote a balanced, healthy diet. It advises health care practitioners to consider overall risk factors and diet in managing cardiovascular disease.
In the Vitamin Intervention for Stroke Prevention (VISP) trial, Toole et al. 12 aimed to assess whether high-dose vitamin supplementation (pyridoxine [B6], folic acid, and cobalamin [B12]) could reduce the risk of stroke recurrence, heart attack (myocardial infarction), or mortality in individuals who experienced a non-disabling ischemic stroke. Although the high-dose group achieved a 2-µmol/L greater reduction in homocysteine levels than the low-dose group, there was no significant difference in the rates of stroke, myocardial infarction, or death 12. The recurrence rates were 9.2% in the high-dose group and 8.8% in the low-dose group. The Vitamin Intervention for Stroke Prevention (VISP) trial concluded that B-vitamin supplementation for homocysteine reduction did not significantly reduce recurrent vascular events 12. However, the lack of a placebo group raises the possibility that the low-dose group achieved the maximum benefit, with no additional advantages from higher doses.
The Norwegian Vitamin (NORVIT) trial, led by Bønaa et al. 22, explored the effects of B-vitamin therapy on recurrent cardiovascular events in patients after acute heart attack (acute myocardial infarction). Although homocysteine levels were reduced by 27% after treatment, there was no significant reduction in recurrent myocardial infarction, strokes, or coronary heart disease-related deaths 22. In contrast to the placebo group, the treatment group exhibited a trend toward increased adverse cardiovascular events, with a relative risk of 1.22, suggesting potential harm from homocysteine-lowering therapy 22. However, the Norwegian Vitamin (NORVIT) trial had several confounding factors, including most events occurring within the first year post acute myocardial infarction—a period when even statins may not provide full protection. Additionally, the concurrent initiation of homocysteine-lowering therapy with other pleiotropic drugs might have obscured any minor benefits of such therapy. The study ultimately concluded against recommending B-vitamin therapy for secondary prevention after acute myocardial infarction because of potential adverse outcomes.
The Heart Outcomes Prevention Evaluation (HOPE)-2 study evaluated the impact of homocysteine-lowering therapy on major cardiovascular events in patients with vascular disease or diabetes 13. Despite a 2.4 µmol/L reduction in homocysteine levels over 5 years, no significant reduction was observed in the composite endpoints of cardiovascular death, myocardial infarction, or stroke 13. Although stroke incidence was reduced in the treatment group, there was an increase in hospitalizations for unstable angina 13. The Heart Outcomes Prevention Evaluation (HOPE)-2 trial concluded that homocysteine-lowering therapy with folic acid and B vitamins did not significantly reduce cardiovascular event risk and should not be recommended for this purpose. However, the study’s recruitment criteria did not ensure low baseline folate levels, and its statistical power was limited, particularly for subgroups in non-folate-fortified regions.
The Western Norway B Vitamin Intervention Trial (WENBIT) by Løland et al. 23, investigated the effects of homocysteine-lowering therapy on cardiovascular disease progression in patients after percutaneous coronary intervention (coronary angioplasty with stenting). Despite a 22% reduction in homocysteine levels, the treatment had no significant effect on cardiovascular disease progression. A post hoc analysis suggested that folic acid and vitamin B12 supplementation could increase the risk of rapid stenosis progression 23. The authors concluded that homocysteine-lowering therapy did not benefit coronary artery progression and might even promote adverse outcomes in some patients.
In the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) trial, Armitage et al 24 assessed the impact of homocysteine-lowering therapy on major vascular events in patients with myocardial infarction. Over a follow-up period of 6.7 years, the therapy reduced homocysteine levels by 3.8 µmol/L but did not reduce the incidence of major vascular events, including vascular mortality, stroke, and coronary events 24. The study concluded that homocysteine-lowering therapy with folic acid and vitamin B12 does not reduce cardiovascular risk in high-risk individuals 24. However, the short follow-up period and the potential variability in the homocysteine-lowering effects according to the stage of atherosclerosis were noted as limitations. The trial also did not determine whether individuals with severely elevated homocysteine levels could benefit from extended treatment. These studies, along with findings from Albert et al. 25 and Ebbing et al. 26, suggest that B-vitamin supplementation does not significantly lower cardiovascular disease risk or prevent adverse outcomes in high-risk populations.
Hyperhomocysteinemia (elevated homocysteine levels) is defined as fasting plasma homocysteine levels exceeding 15 μmol/L (>15 μmol/L), has emerged as a contributor to atherosclerosis 27, 28, 29. Homocysteine was first identified as an important biological compound in 1932 and linked with human disease in 1962 with the identification of homocystinuria, a rare inherited disorder of amino acid metabolism caused by homozygous cystathionine beta-synthase deficiency where the body can’t properly process methionine, leading to a buildup of homocysteine in the blood and urine, potentially causing various health problems. Homocystinuria is characterized by markedly elevated total homocysteine levels, manifests clinically as mental retardation, seizures, ectopia lentis (a condition where the eye’s natural lens is dislocated or moved out of its normal position, potentially causing vision problems), and childhood-onset strokes 30, 31. Homocysteinuria (homocystinuria), was later associated with premature occlusive cardiovascular disease, even in children. In homocystinuria, one of several different genes is altered, leading to a dysfunctional enzyme that does not allow the normal breakdown of methionine to homocysteine. Methionine is one of the eleven essential amino acids that must come from your diet because the body cannot produce it. Without the proper enzyme to break them down, homocysteine and methionine begin to build up in the body. Babies with homocystinuria will appear normal at birth but within a few years begin to develop signs such as a dislocated lens in the eye, a long slender build, long thin fingers, skeletal abnormalities, osteoporosis, and a greatly increased risk of thromboembolism and of atherosclerosis that can lead to premature cardiovascular disease. The buildup of homocysteine in the arteries may also cause intellectual disability, mental illness, slightly low IQ, behavioral disorders, and seizures. Some of those may be alleviated if the condition is detected early, which is why all states screen newborns for homocystinuria 32. There is no cure for homocystinuria. About half of people with homocystinuria respond to vitamin B6 (pyridoxine). Those who do respond will need to take vitamin B6 (pyridoxine), vitamin B9 (folate), and vitamin B12 supplements for the rest of their lives. Those who do not respond to vitamin B supplements will need to eat a low-methionine diet. Most will need to be treated with trimethylglycine, a medicine also known as betaine. Neither a low-methionine diet nor medicine will improve existing intellectual disability. Medicine and diet should be closely monitored by a doctor who has experience treating homocystinuria. Genetic counseling is recommended for people with a family history of homocystinuria who want to have children. Prenatal diagnosis of homocystinuria is available. This involves culturing amniotic cells or chorionic villi to test for cystathionine synthase (the enzyme that is missing in homocystinuria).
Homocysteine is an independent cardiovascular disease risk factor modifiable by nutrition and possibly exercise. Homocysteine-lowering strategies are also being investigated for their ability to reduce the risk of heart and blood vessel disease (cardiovascular disease). Nutrients involved in the metabolism of homocysteine include folate (vitamin B9), vitamin B12 (cyanocobalamin), vitamin B6 (pyridoxine), riboflavin (vitamin B2), and choline. Although supplementation with folate (vitamin B9), vitamin B12 (cyanocobalamin) and vitamin B6 (pyridoxine) successfully lowers homocysteine concentration in the blood, no significant effect on heart and blood vessel disease (cardiovascular disease) risk has been demonstrated. There is some evidence that riboflavin (vitamin B2) supplementation may lower homocysteine and blood pressure in individuals with a certain genetic predisposition.
Although homocysteine concentration in plasma is only about 10 micromolar (μM), even moderate hyperhomocysteinemia is associated with increased incidence of cardiovascular disease and Alzheimer’s disease. These observations led to research investigating the relationship of elevated homocysteine levels and cardiovascular disease in a wide variety of populations including middle age and elderly men and women with and without traditional risk factors for cardiovascular disease 33, 34. Moreover, homocysteine is found to be associated with cystathionine beta-synthase deficiency, cystathioninuria, methylenetetrahydrofolate reductase deficiency, and sulfite oxidase deficiency, which are inborn errors of metabolism.
Mutations in the gene responsible for the enzyme methylenetetrahydrofolate reductase (MTHFR) of C-to-T substitution at nucleotide 677, which is quite common in most populations with a homozygosity rate of 10 to 15%, is associated with moderate elevated homocysteine levels (hyperhomocysteinemia), especially in the context of marginal folate (vitamin B9) intake. Plasma homocysteine (Hcy) is inversely related to plasma creatinine in patients with kidney disease. This is due to an impairment in homocysteine (Hcy) removal in kidney disease. The role of these factors, and of modifiable lifestyle factors, in affecting methionine metabolism and in determining plasma homocysteine levels.
Experimental studies using cellular and animal models have elucidated multiple mechanisms by which hyperhomocysteinemia (elevated homocysteine levels) promotes atherogenesis (the process of forming plaques in the intima layer of arteries). These include the inhibition of endothelial cell proliferation, suppression of high-density lipoprotein (HDL or “good” cholesterol) synthesis, promotion of smooth muscle cell growth, and increased oxidation of low-density lipoprotein (LDL or “bad” cholesterol). Furthermore, hyperhomocysteinemia (elevated homocysteine levels) has been shown to enhance the production of chemokines and reactive oxygen species (ROS), which can destabilize atherosclerotic plaques, increasing their susceptibility to rupture and subsequent occlusion 7.
Homocysteine test that measures the amount of homocysteine (Hcy) in your blood and/or urine can help assess your heart disease or stroke risk, identify vitamin deficiencies, or monitor people with heart disease. While some research suggests that lowering homocysteine levels with B vitamins may not reduce the risk of heart disease, addressing vitamin deficiencies and managing other risk factors can be important for your overall health.
Note that homocysteine blood level is a more sensitive test for folate and vitamin B12 deficiency than folate and vitamin B12 levels. A normal homocysteine blood level excludes significant folate and vitamin B12 deficiency, though homocysteine blood level has limited specificity because elevations occur in other inherited and acquired disorders.
Homocysteine is present in plasma in four different forms: around 1% circulates as free thiol, 70–80% remains disulphide-bound to plasma proteins, mainly albumin and 20–30% combines with itself to form the dimer homocysteine or with other thiols 35. Homocysteine is a key determinant of the methylation cycle 36. It is methylated to methionine, which undergoes S-adenosylation and forms S-adenosylmethionine (SAM) 36. S-adenosylmethionine (SAM) is the principal methyl donor for all methylation reactions in cells 36. Condensation of methionine with ATP, leads to the formation of S-adenosylmethionine (SAM) via a reaction facilitated by SAM synthetase which is sometimes referred to as methionine adenosyltransferase (MAT) (see Figure 3 below) 37, 38, 39. The methyl group attached to the tertiary sulphur of S-adenosylmethionine (SAM) can be transferred and therefore can cause methylation of other substances. This methylation is accompanied by energy loss, so this reaction is irreversible. The demethyation reaction leads to the formation of S-adenosylhomocysteine (SAH) 37. S-adenosylhomocysteine (SAH) is a thioether (a sulfur bonded to two alkyl or aryl groups) analogous to methionine. The S-adenosylmethionine [SAM]/S-adenosylhomocysteine [SAH] ratio defines the methylation potential of a cell 36. Hydrolysis of S-adenosylhomocysteine (SAH) is catalyzed by S-adenosylhomocysteine hydrolase (SAHH) leads to the formation of homocysteine and adenosine (see Figure 3 below) 37. This homocysteine can be used in one of two ways:
- In case of methionine deficiency, homocysteine can be re-methylated to form methionine 37. The enzyme N5, N10-methylenetetrahydrofolate reductase converts homocysteine to methionine 1.
- In presence of sufficient methionine, homocysteine is instead used to produce cysteine 37. Cystathionine-β-synthase is an enzyme (with pyridoxine (vitamin B6) as an essential cofactor) that converts homocysteine to cysteine 1. Homocysteine is synthesized from the essential amino acid methionine, therefore cysteine is not an essential amino acid as long as sufficient methionine is available 37.
Figure 1. Homocysteine
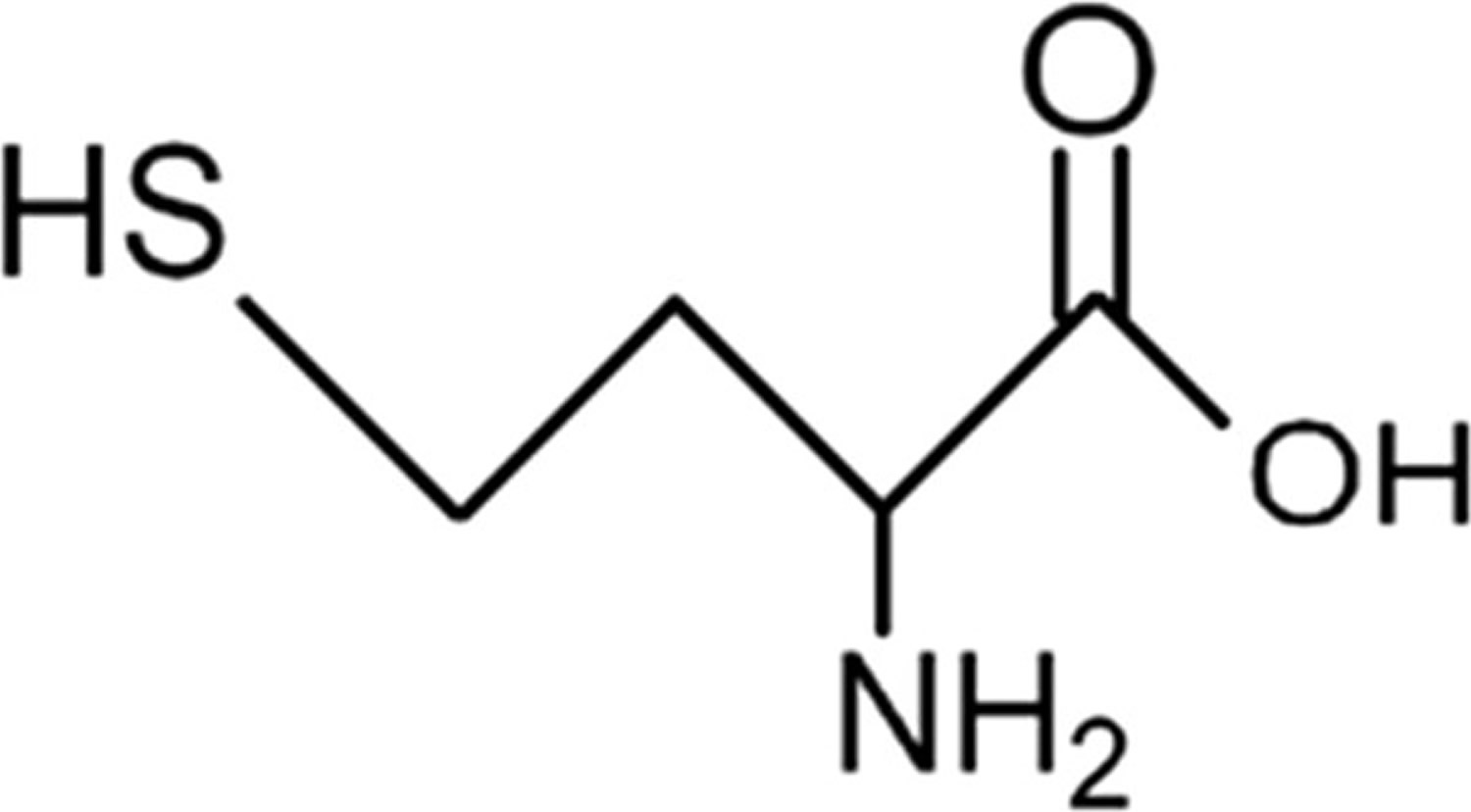
Figure 2. Homocysteine metabolism
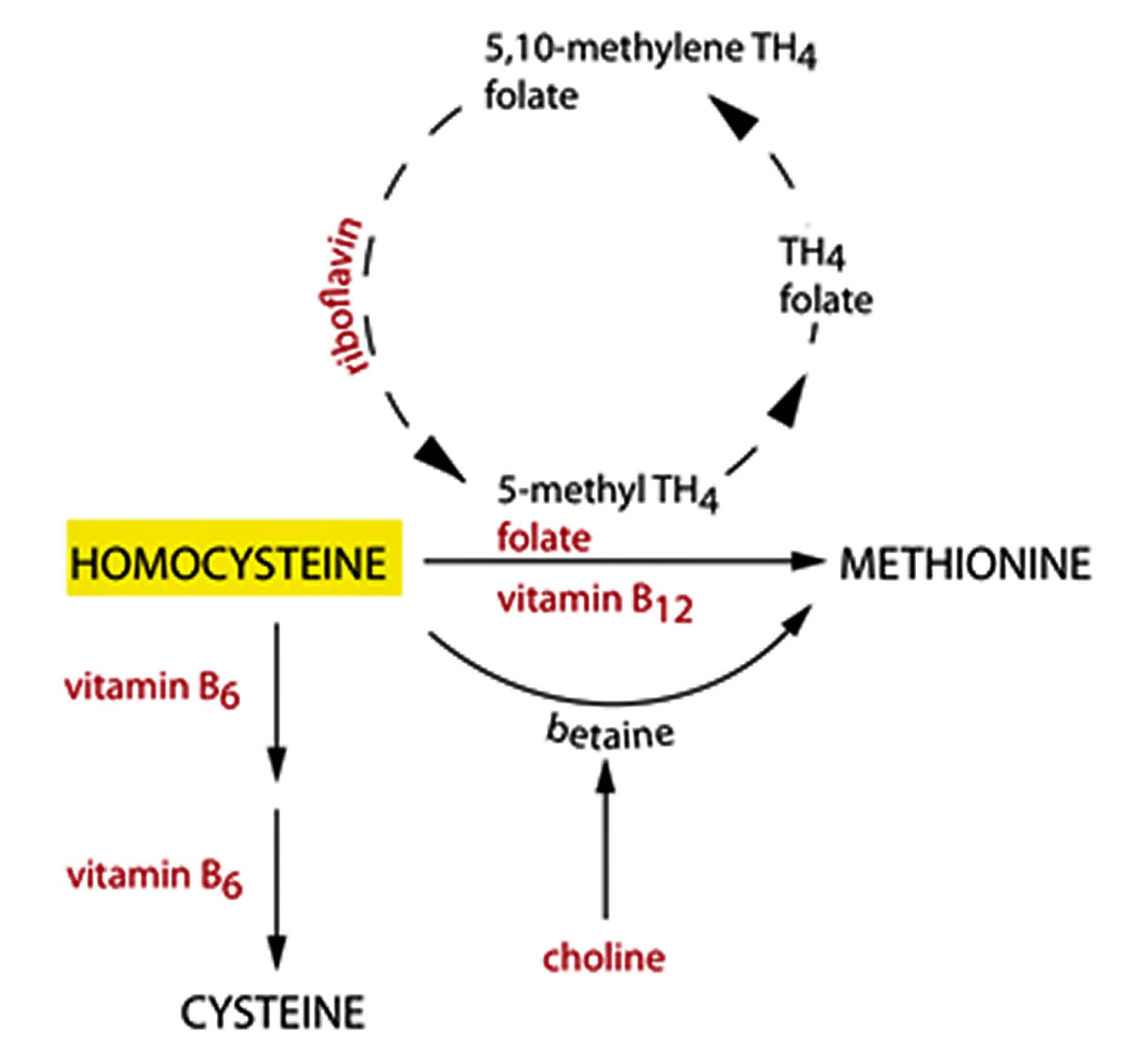
Figure 3. Homocysteine biosynthesis and metabolism
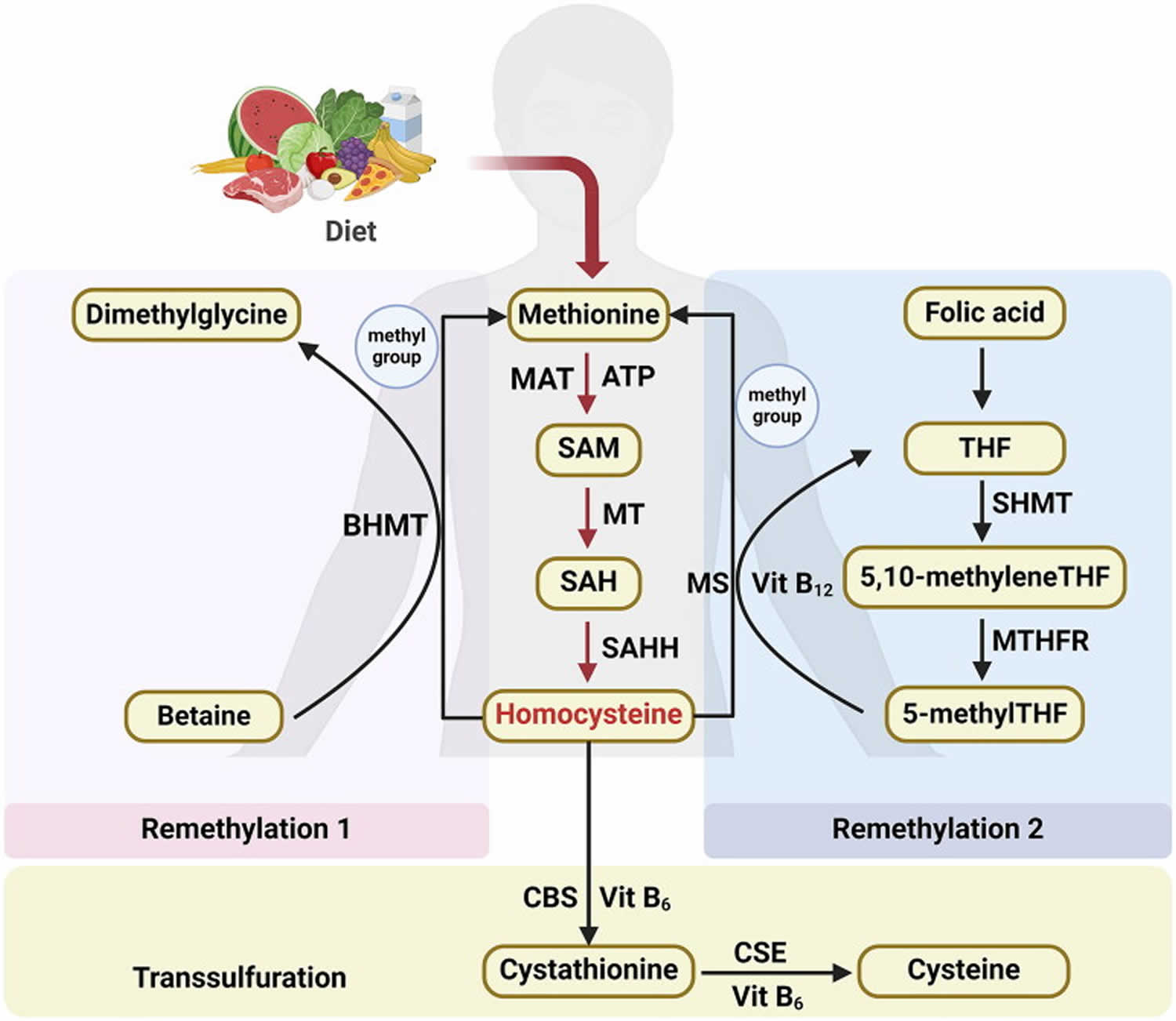
Footnote: Concise pathway for homocysteine biosynthesis and metabolism.
Abbreviations: BHMT = Betaine-homocysteine S-methyltransferase; MAT = Methionine adenosyltransferase; SAM = S-adenosylmethionine; MT = Methyltransferase; SAH = S-adenosylhomocysteine; SAHH = S-adenosylhomocysteine hydrolase; THF = Hydrolase tetrahydrofolate; SHMT = Serine hydroxymethyltransferase; MS = Methionine synthase; 5-methylTHF = N-5-methyltetrahydrofolate; MTHFR = 5:10-methylenetetrahydrofolate reductase; CBS = Cystathionine beta-synthase; CSE = Cystathionine gamma-lyase; vit = Vitamin.
[Source 7 ]Homocysteine biosynthesis
Humans can only synthesize homocysteine (Hcy) through the transmethylation of the essential amino acid methionine, which is obtained from the diet 7. Methionine is an essential amino acid found in meat, fish, and dairy products. Humans must obtain methionine through their diet. Methionine undergoes ATP-dependent activation and is subsequently converted into S-adenosylmethionine (SAM) via a reaction facilitated by SAM synthetase, which is sometimes referred to as methionine adenosyltransferase (MAT). Subsequently, S-adenosylmethionine (SAM) undergoes a particular methyltransferase (MT)-mediated process, leading to its conversion into S-adenosylhomocysteine (SAH), which then undergoes rapid hydrolysis via SAH hydrolase (SAHH), resulting in the formation of homocysteine and adenosine 38, 39.
Homocysteine metabolism
Homocysteine metabolism is governed by two distinct pathways: remethylation and transsulfuration.
Within the remethylation route, homocysteine (Hcy) is transformed into methionine by gaining a methyl group. This essential methyl group is derived from betaine in the liver and kidneys (denoted as Remethylation 1) or from N-5-methyltetrahydrofolate (5-methylTHF) across various tissues (referred to as Remethylation 2) (Figure 3) 7.
- Remethylation 1 is characterized by the action of betaine-homocysteine S-methyltransferase (BHMT) 2, a cytosolic enzyme that relies on zinc. Betaine-homocysteine S-methyltransferase (BHMT) mediates the donation of a methyl group from betaine to homocysteine, culminating in the production of dimethylglycine and methionine 40.
- Remethylation 2 intricately connects the metabolism of homocysteine to the internal dynamics of the folate cycle within cells 41. The produced tetrahydrofolate (THF) is further transformed into 5,10-methylene THF via a catalytic process facilitated by serine hydroxymethyltransferase (SHMT). Afterward, 5,10-methylene THF is further refined into 5-methylTHF via the action of 5,10-methylenetetrahydrofolate reductase (MTHFR) 42, 43. With vitamin B12 acting as a cofactor, methionine synthase (MS) catalyzes the transfer of a methyl group from 5-methylTHF to homocysteine, thereby producing methionine and regenerating tetrahydrofolate (THF) 44.
During the transsulfuration pathway, homocysteine (Hcy) is further metabolized to cysteine. This transformation begins with the enzyme cystathionine beta-synthase (CBS), which catalyzes the formation of cystathionine from homocysteine. Subsequently, cystathionine gamma-lyase (CSE) converts cystathionine into the amino acid cysteine. The entire biochemical pathway relies on the presence of vitamin B6.
Homocysteine and the nervous system
In the last decade, epidemiological observations have pointed towards a plausible association between hyperhomocysteinemia and CNS neurodegenerative disorders. Several studies demonstrated that homocysteine is capable of triggering neuronal damage via oxidative stress, DNA damage and activation of pro-apoptotic factors in cell cultures or animal models 45. In an experiment, SH-SY5Y neuroblastoma cells were modified to act as neuronal cells, by incubating them with retinoic acid, which induced their differentiation towards a neuronal-like phenotype 45. This was followed by incubation with/without D,L-homocysteine in a concentration range from 20 μM to 80 μM 45. The exposure to homocysteine induced a time and concentration dependent reduction of cell viability in comparison with controls. The highest cytotoxicity was portrayed by 80 μM homocysteine which produced 80% of cell death after 5 days of incubation 45. A significant reduction of cell viability to 35% was also observed after 5 days of incubation with 40 μM homocysteine. Cell exposure to homocysteine for a period of 3 days did not induce any significant change in Reactive Oxygen Species (ROS) levels, but incubation with homocysteine for 5 days resulted in a 4.4-fold increase in ROS production 45. Homocysteine notably triggered significant levels of genotoxic stress which was indicated by the assessment of DNA fragmentation by Comet assay. But the levels of genotoxic stress was significant only after a longer time of exposure, as shown by the number of Comet positive cells, which was significantly increased only after 5 days of incubation with homocysteine 45. Bax and Bcl-2 mRNA levels in cells showed an increase by two-fold and 14-fold, respectively, in the case of 5 days exposure to homocysteine 45. A time-dependent effect of homocysteine was also evident. The mRNA levels for the cyclins D1, E1, and A1 were increased by two-fold, six-fold, and five-fold, respectively, in cells exposed to homocysteine for 3 days, but the mRNA levels in case of cyclin B1 were not affected in the 3 day period 45. The mRNA levels of all cyclins returned to the basal levels after 5 days of incubation with homocysteine. A decrease in both mRNA and protein level, of p21, another key protein regulator of DNA damage induced cell death, was noted after 3 days of incubation with homocysteine, followed by a dramatic p21 up-regulation and protein synthesis at 5 days . Further down the timeline, a significant upregulation of p16 was observed, concomitantly with the reduction by 35% of phosphorylated pRB 45. These proteins are check-point regulators of G1-S phase progression through the inhibition of cyclin D-cdk4 complex and the direct binding and sequestration of the transcription factor E2F, respectively 45. Therefore, this indicates the arrest of cell cycle at G1 phase 45. The results suggest that prolonged exposure to mildly elevated homocysteine concentrations triggers oxidative and genotoxic stress in neuronal-like cells 45.
The effect of homocysteine on the brain
By adulthood, the folate related enzymes involved in purine and pyrimidine synthesis, decline almost tenfold. Hence, this leads us to believe that the provision of methyl groups for S-adenosylmethionine and methylation reactions coupled with recycling of homocysteine through methionine synthase may be dominant function of adult brain folate metabolism 46. The brain has a limited capacity for homocysteine metabolism. Folate plays an important role in the brain so a crucial mechanism is in play to protect the brain from folate deficiency. The level of 5 tetrahydrofolate in the cerebrospinal fluid is 3 times that of the plasma level and there exists an active process to maintain it 46. Methionine synthase is the only enzyme in the brain (neural tissue) that is capable of converting homocysteine to methionine. Cobalamin is a cofactor (hence essential) 46.
The brain tissue utilizes three mechanisms to maintain a low level of homocysteine 46:
- Efficient recycling through cobalamin dependent methionine synthase (given an adequate supply of cobalamine and folate),
- Catabolism through cystathione beta synthase to cystathione a non-noxious product,
- Export to external circulation 46.
In the brain and elsewhere disruption of homocysteine metabolism may result from nutritional imbalance, genetic defects or as a result of drug therapy 46.
The direct effect of homocysteine on the nervous system
The action of homocysteine as a neurotransmitter: homocysteine and its related compounds may have a role as an excitatory agonist on the NMDA subtype of glutamate receptors and recent evidence also points to the involvement of NMDA modulatory sites 46. It has also been shown that homocysteine, besides acting as a partial agonist at glutamate receptors also acts as a partial antagonist of glycine co-agonist site of the NMDA receptor 46. In the presence of normal glycine levels and normal physiological conditions homocysteine does not cause toxicity below millimolar concentrations. However in case of a head trauma or stroke, there is an elevation in glycine levels in which instance the neurotoxic effect of homocysteine as an agonist outweighs its neuroprotective antagonist effect. This may cause neuronal damage via calcium ion influx or free radical generation 46.
One evaluative experiment to discover the direct effect of homocysteine on the central nervous system involved local application of homocysteine by two different methods of drug delivery to the central nervous system of rats- pressure ejection and ionophoresis 47. Extracellular recordings were taken from neurons of cerebral cortex, cerebellum and midbrain. The recordings after either method of administration portrayed a dose-dependent increase in neuronal activity by D, L-homocysteine and L-glutamate in 67% of cells tested with both drugs. The similarity in the dose required of D,L-homocysteine and L-Glutamate, points out that D,L-homocysteine seems to be as potent as the latter. This data indicates that homocysteine seems to have an excitatory action on neurons, and this finding may account for neurological symptoms associated with disorders of amino acid metabolism 47. Some studies also suggest that elevated homocysteine levels may be associated with alterations in mental health such as cognitive impairment, dementia, depression, Alzheimer’s and Parkinson’s disease 46.
Homocysteine levels
Normal homocysteine levels: <5 μmol/L (less than 5 μmol/L)
Total concentration of homocysteine in plasma of healthy humans (fasting) is low and its level is between 5 and 15 μmol/L when assessed with the use of HPLC, or 5 to 12 μmol/L when immunoassay methods are used 48. Although there is no definitive cutoff value for normal plasma homocysteine levels, total homocysteine levels below 15 µmol/L are generally considered normal 49.
Elevated homocysteine levels
The definition of hyperhomocysteinemia differs between studies 1. Hyperhomocysteinemia is defined as a medical condition characterized by an abnormally high level (above 15 μmol/L) of homocysteine in the blood 50.
- 5-15 μmol/L (Mild elevation)
- 15-30 μmol/L (Moderate elevation) 35
- 30-100 μmol/L (Intermediate elevation) 35
- >100 μmol/L (Severe elevation) 35. Severe hyperhomocysteinemia are rare and usually result from genetic mutations affecting homocysteine metabolism, such as defects in enzymes such as cystathionine beta-synthase (classic homocystinuria), methionine synthase (MS), and 5,10-methylenetetrahydrofolate reductase (MTHFR) 51. For example, 5,10-methylenetetrahydrofolate reductase (MTHFR) genetic mutations are associated with mild to moderate hyperhomocysteinemia owing to reduced enzyme activity, whereas cystathionine beta-synthase (CBS) deficiencies can dramatically increase homocysteine levels in severe cases (see classic homocystinuria).
What does abnormal homocysteine test result mean?
In cases of suspected malnutrition or vitamin B12 or folate deficiency, homocysteine levels may be elevated. If an individual does not get enough B vitamins and/or folate through diet or supplements, then the body may not be able to convert homocysteine to forms that can be used by the body. In this case, the level of homocysteine in the blood can increase.
Studies from the mid- to late-1990s suggested that people who have elevated homocysteine levels have a much greater risk of heart attack or stroke than those with average levels. Investigating the link between high homocysteine levels and heart disease remains an active area of research. At present, however, the use of homocysteine levels for risk assessment of cardiovascular disease, peripheral vascular disease, and stroke is uncertain given that several trials investigating folic acid and B vitamin supplementation indicate no benefit or lowering of cardiovascular disease risk.
Additionally, a 2012 study of multiple datasets, including 50,000 people with coronary heart disease, called the potential for a cause-and-effect relationship between homocysteine levels and heart disease into question. The American Heart Association does acknowledge the relationship between homocysteine levels and heart attack/stroke survival rates but doesn’t consider elevated homocysteine a major risk factor for cardiovascular disease.
While the American Heart Association does not recommend widespread use of folic acid and B vitamins to reduce risk of heart attack and stroke, it does promote a balanced, healthy diet and advise health practitioners to consider overall risk factors and diet in managing cardiovascular disease.
In newborn testing, greatly increased concentrations of homocysteine in the urine and blood mean that it is likely that an infant has homocystinuria and indicates the need for further testing to confirm the cause of the increase.
When test results suggest homocystinuria, liver or skin biopsy samples are sometimes tested to determine whether the enzyme cystathionine beta synthase is present. The absence of this enzyme is the most common cause of homocystinuria. Genetic tests may be ordered to test for one or more of the most common gene mutations. If someone has a strong family history of early atherosclerosis or a family member has been diagnosed with homocystinuria, then that person should be tested for the gene mutation that was found in the family member.
Could any medications I may be taking have an effect on my homocysteine level?
Yes. There are numerous drugs that may either increase or decrease the amount of homocysteine in your body. You should always keep your healthcare provider and pharmacist aware of any medications, traditional or herbal, that you are taking since they may affect the test results. Azaribine, carbamazepine, methotrexate, nitrous oxide, and phenytoin can all cause increased levels of homocysteine. Oral contraceptives can also affect the metabolism of homocysteine.
Possible causes of elevated homocysteine levels
There are two types of hyperhomocysteinemia: (1) the rare but severe forms are due to major genetic mutations of the enzymes implicated in homocysteine metabolism; (2) the more common forms cause moderately elevated homocysteine levels related to a pathogenesis such as genetic and environmental factors 1.
- Genetic disorders
- Consider vitamin B6 (Pyridoxine), Folic acid (vitamin B9) and Vitamin B12 (Cyanocobalamin) deficiencies
- Kidney disease
- Smoking
Homocysteine levels can increase with age, when a person smokes, and with the use of drugs such as carbamazepine, methotrexate, and phenytoin. Homocysteine levels are lower in women than in men. Women’s concentrations increase after menopause, possibly due to decreased estrogen production.
Hyperhomocysteinemia may arise from genetic defects of enzymes involved in homocysteine metabolism. The enzymes involved can be 5, 10-methylene tetrahydrofolate reductase (MTFHR), methionine synthase (MS), and cystathionine-beta-synthase (CBS) 52. The most common one that is detected worldwide and has a high incidence in different populations, is single nucleotide polymorphisms of 5,10-methylene tetrahydrofolate reductase (MTFHR) which has been associated with mild (13–24 μmol/L) and moderate (25–60 μmol/L) hyperhomocysteinemia 52. Hankey et al. 53 stated that the most common enzyme defect associated with moderately raised total homocysteine is a point mutation (C-to-T substitution at nucleotide 677) in the coding region of the gene for MTHFR, which is associated with a thermo labile MTHFR variant that has about half-normal activity 53. The most common of the genetic causes of severe hyperhomocysteinemia and classic homocystinuria (congenital homocystinuria) is believed to be homozygous deficiency of CBS (cystathionine-beta-synthase) which results in an increase of up to 40-fold in fasting total homocysteine. Other rarer causes of severe hyperhomocysteinemia are considered to be homozygous deficiency of MTHFR, deficiency of methionine synthase, and impaired activity of methionine synthase due to genetic disorders of vitamin B12 metabolism 53.
Mild to moderate hyperhomocysteinemia can also arise from nutritional deficiencies of folate (folic acid), vitamin B6 (pyridoxine), and vitamin B12 52. Blood levels of folate, vitamin B12 and to a lesser extent, vitamin B6 are related inversely to total homocysteine; therefore a person with a nutritional deficiency that leads to low blood concentrations of the aforementioned is at increased risk of hyperhomocysteinemia 52. Folic acid (as 5-methylTHF) acts as a methyl donor in the conversion of homocysteine into methionine, a methionine synthase (MS)-catalyzed reaction that requires vitamin B12 as a co-factor 54. Vitamin B6 (pyridoxine) is essential for cystathionine-beta-synthase (CBS), as it converts homocysteine into cystathionine and subsequently cysteine. Deficiencies of these vitamins lead to elevated homocysteine levels.
Several diseases such as kidney failure, liver disease and underactive thyroid (hypothyroidism), cancer, psoriasis, and diabetes as well as various drugs, alcohol, tobacco, coffee, older age, male sex and menopause, are believed to be associated with moderately elevated homocysteine concentrations 55, 56, 57, 58. A rise in serum creatinine also leads to a rise in fasting total homocysteine 53. The major route of homocysteine clearance from plasma is the kidney, and the rise is due to defective metabolism of homocysteine by the kidney 53. Total homocysteine levels are found to be considerably higher in patients with chronic renal disease than the moderately raised concentrations commonly found in patients with atherothrombotic vascular disease, and this may be the probable cause that contributes to the high incidence of vascular complications in patients with chronic renal failure 53. Plasma homocysteine concentrations can be increased by various drugs (e.g. methotrexate, estrogen-based contraceptives) and diseases that interfere with folate, vitamin B6, and B12 metabolism, hence an abnormal homocysteine concentration may have a probable use as a diagnostic aid for some of these conditions 53.
Moderate hyperhomocysteinemia (15–30 µmol/L) is often caused by poor diet, mild folate, B12, or B6 deficiencies, heterozygosity for CBS (cystathionine-beta-synthase) defects, underactive thyroid (hypothyroidism), impaired kidney function, or intake of certain medications (e.g. methotrexate, estrogen-based contraceptives) 49. Intermediate hyperhomocysteinemia (30–100 µmol/L) is typically due to more severe B12 or folate deficiencies or kidney failure 49. Severe hyperhomocysteinemia (> 100 µmol/L) is usually caused by severe B12 deficiency or genetic conditions such as homocystinuria 59.
There has been an indication towards a significant correlation between hyperhomocysteinemia and cardiovascular disease and its complications such as heart attacks and strokes 60. It is believed that hyperhomocysteinemia leads to endothelial cell damage, reduction in the flexibility of vessels, and alters the process of haemostasis 60. Hyperhomocysteinemia may lead to an enhancement of the adverse effects of risk factors like hypertension, smoking, lipid and lipoprotein metabolism, as well as promotion of the development of inflammation 60. The prevalence of hyperhomocysteinemia may vary significantly between populations, and most likely depend on age, diet, and genetic background as well 55. Increasing age, male sex, smoking, coffee consumption, high blood pressure, unfavourable lipid profile, high creatinine and faulty diet are some of the factors associated with increased homocysteine levels 61. On the other hand, physical activity, moderate alcohol consumption, good folate and vitamin B12 status are associated with lower homocysteine levels. Vegetarians may be at a higher risk of hyperhomocysteinemia due to low plasma B12 levels but the difference is likely to be insignificant 61.
Management Options
- Consider vitamin supplements in those with genetic hyperhomocysteinemia
- Nutrients involved in the metabolism of homocysteine include folate, vitamin B12, vitamin B6, riboflavin, and choline. Although supplementation with folate, vitamin B6, and vitamin B12 successfully lowers homocysteine concentration in the blood, no significant effect on cardiovascular disease risk has been demonstrated. There is some evidence that riboflavin supplementation may lower homocysteine and blood pressure in individuals with a certain genetic predisposition. The consumption of folic acid supplements or cereals that are fortified with folic acid and to a lesser extent vitamin B6 and vitamin B-12, can lower blood homocysteine levels and may be beneficial in people with even mild genetic hyperhomocysteinemia.
- Manage other cardiovascular risk factors including obesity, exercise, diabetes, hypertension, cholesterol and cigarette smoking
Homocysteine test
The homocysteine test measures your blood homocysteine levels, a naturally occurring amino acid that serves in your body as an intermediate in the metabolism of methionine and cysteine. The homocysteine amino acid is typically broken down (metabolized) in the blood into other substances your body needs. So elevated homocysteine levels usually indicate a deficiency of vitamin B12, vitamin B6 (pyridoxine), or Folic acid (vitamin B9) because these vitamins help your body process homocysteine (Hcy).
The homocysteine test may be used a few different ways:
- A health practitioner may order a homocysteine test to determine if a person has a vitamin B12 or folate deficiency. The homocysteine concentration may be elevated before vitamin B12 and folate tests are abnormal. Some health practitioners may recommend homocysteine testing in malnourished individuals, the elderly, who often absorb less vitamin B12 from their diet, and individuals with poor nutrition, such as drug or alcohol addicts.
- Homocysteine may be ordered as part of a screen for people at high risk for heart attack or stroke. It may be useful in someone who has a family history of coronary artery disease but no other known risk factors, such as smoking, high blood pressure, or obesity. However, the exact role that homocysteine plays in the progression of cardiovascular disease has not been established, so the utility of the screening test continues to be questioned. Routine screening, such as that done for total cholesterol, has not been recommended.
- Tests for both a urine and blood homocysteine may be used to help diagnose homocystinuria if a health practitioner suspects that an infant or child may have this inherited disorder. In the U.S., all babies are routinely tested for excess methionine, a sign of homocystinuria, as part of their newborn screening. If a baby’s test is positive, then urine and blood homocysteine tests are often performed to confirm the findings.
When is homocysteine test ordered?
Homocysteine test may be ordered when a health practitioner suspects that a person may have a vitamin B12 deficiency and/or folate deficiency. Signs and symptoms are initially subtle and nonspecific. People with an early deficiency may be diagnosed before they experience any overt symptoms. Other affected people may experience a variety of mild to severe symptoms that can include:
- Diarrhea
- Dizziness
- Fatigue, weakness
- Loss of appetite
- Paleness
- Rapid heart rate
- Shortness of breath
- Sore tongue and mouth
- Tingling, numbness, and/or burning in the feet, hands, arms, and legs (with B12 deficiency)
Homocysteine testing may be ordered as part of assessing a person’s risk of cardiovascular disease, depending on the individual’s age and other risk factors. It may also be ordered following a heart attack or stroke to help guide treatment.
Homocysteine test is may be ordered when newborn screening detects an elevated level of methionine or if an infant or child has signs and symptoms of homocystinuria. Babies with this condition will appear normal at birth, but if not treated, they will, within a few years, begin to develop signs such as a dislocated lens in the eye, a long slender build, long thin fingers, and skeletal abnormalities.
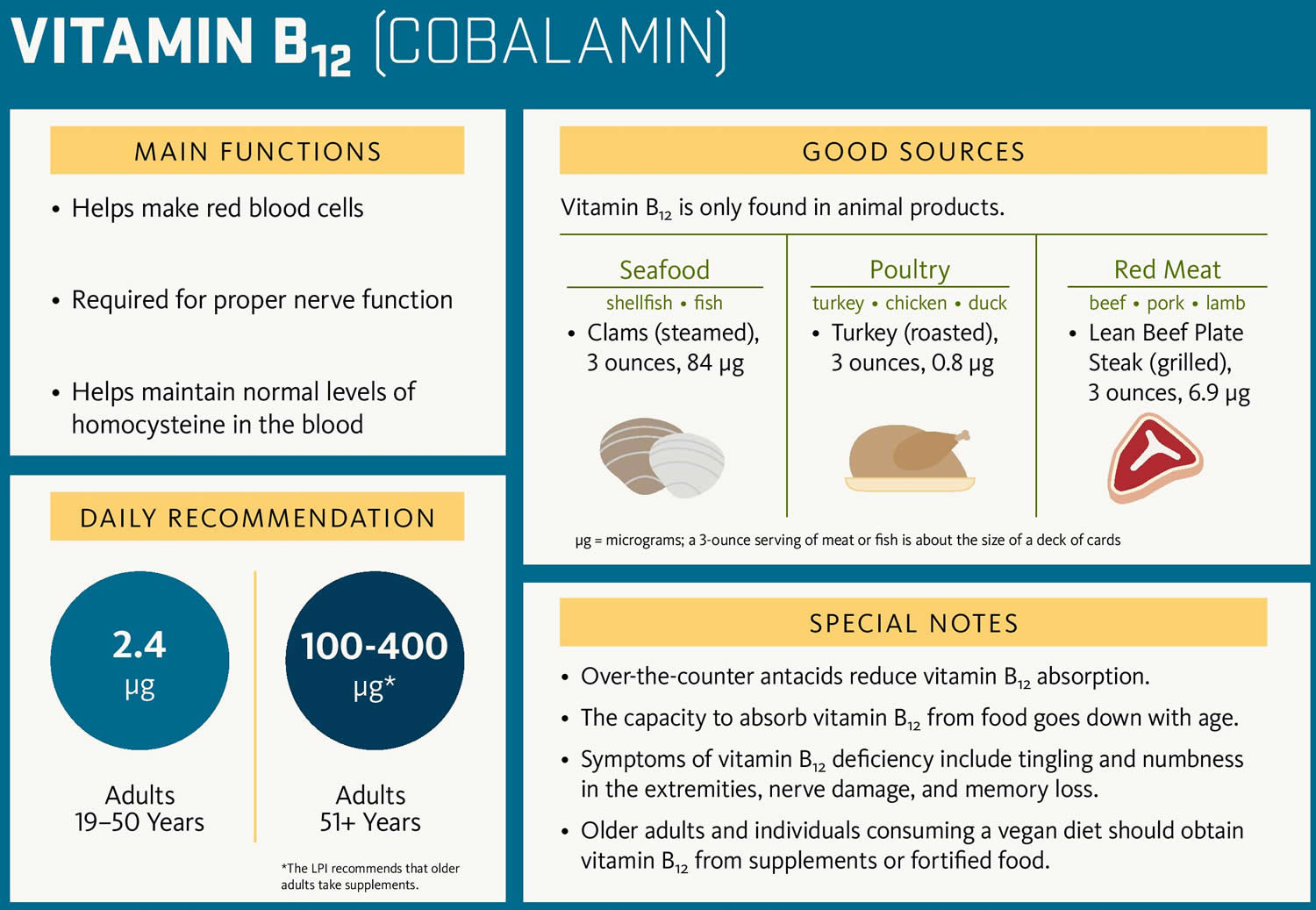
Vitamin B12 and folate deficiencies
Vitamin B12 and folate deficiencies are a lack of these two B complex vitamins that the body needs for several important functions. They are required to make normal red blood cells (RBCs), repair tissues and cells, synthesize DNA (the genetic material in cells). B12 is also important for normal nerve cell function. B12 and folate (also known as folic acid or vitamin B9) are nutrients that cannot be produced in the body and must be supplied by the diet. The body stores 3 to 6 years worth of B12 and about a 3 months’ supply of folate in the liver. So a B12 and/or folate deficiency reflects a chronic shortage of one or both of these vitamins.
In the U.S., B12 and folate deficiencies are not common in healthy adults because the body can store sufficient amounts and most adults eat enough foods or take supplements that contain these vitamins to meet their daily requirements. There are, however, people at risk of deficiency, such as the elderly, people with intestinal problems that prevent them from absorbing enough of the vitamins, heavy alcohol drinkers, and pregnant women, who need increased amounts of these vitamins.
B12 and folate deficiencies and their associated signs and symptoms can take months to years to manifest in adults. Infants and children will show signs of deficiency more rapidly because they have not yet had time to store sufficient amounts.
Over time, a deficiency in either B12 or folate can lead to macrocytic anemia, a condition in which red blood cells are enlarged. This production of fewer but larger red blood cells decreases the blood’s ability to carry oxygen. People with anemia may be weak, light-headed, and short of breath. Megaloblastic anemia, a type of macrocytic anemia, is characterized by the production of fewer but larger red blood cells in addition to some cellular changes in the bone marrow. Other laboratory findings associated with megaloblastic anemia include decreased white blood cell (WBC) count and platelet count.
A deficiency in B12 can also result in varying degrees of neuropathy or nerve damage that can cause tingling and numbness in the person’s hands and feet. In severe cases, mental changes that range from confusion and irritability to dementia may occur.
Pregnant women need increased folate for proper fetal development because of the added stress of rapidly growing fetal cells. A folate deficiency during pregnancy, especially in the early weeks when a woman might not know she is pregnant, may lead to premature birth and neural tube birth defects such as spina bifida in the child. To help prevent neural tube birth defects, the Food and Drug Administration mandated increased folate supplementation of grain products a number of years ago, which led to about a 50% decrease in neural tube defects in the U.S. Even so, it can be difficult sometimes to get enough folate from foods, so it is recommended that all women who may become pregnant take 400 micrograms of folate every day.
Figure 4. Folate-rich food sources

Figure 5. Vitamin B12 rich food sources
Vitamin B12 and folate deficiencies signs and symptoms
The initial signs and symptoms associated with B12 and folate deficiencies may be subtle and nonspecific. They may be related to the resulting megaloblastic anemia, nerve damage, and/or gastrointestinal changes. People with an early deficiency may be diagnosed before they experience any noticeable symptoms. Other affected people may experience a variety of mild to severe signs and symptoms that can include:
- Diarrhea
- Dizziness
- Fatigue, muscle weakness
- Loss of appetite
- Pale skin
- Rapid heart rate, irregular heartbeats
- Shortness of breath
- Sore tongue and mouth
- Tingling, numbness, and/or burning in the feet, hands, arms, and legs (with B12 deficiency)
- Confusion or forgetfulness
- Paranoia
Vitamin B12 and folate deficiencies causes
There are a variety of causes of B12 and/or folate deficiencies. They include:
Insufficient dietary intake
B12 is found in animal products such as red meat, fish, poultry, milk, and eggs. Folate, also called folic acid or vitamin B9, is found in leafy green vegetables, citrus fruits, dry beans, yeast, and fortified cereals.
The human body stores several years’ worth of B12 in the liver. Since a variety of foods consumed by Americans contain B12, a dietary deficiency of this vitamin is extremely rare in the U.S. It may be seen, for example, in people with generally poor nutrition or malnutrition, in vegans who do not consume any animal products, including milk and eggs, and in breastfed infants of vegans. In adults, dietary deficiencies do not usually cause symptoms until stores of the vitamins within the body have been depleted. Deficiencies in children and infants, however, show up fairly quickly since they have not had time to store as much of the vitamins as adults.
Folate deficiency used to be a common, but in 1997 the U.S. government mandated supplementation of cereals, breads, and other grain products with folic acid. Because folate is stored in tissue in smaller quantities than B12, folate must be consumed more regularly than B12.
Malabsorption
Both B12 and folate deficiencies may be seen in people who have conditions that interfere with absorption of the vitamins in the small intestine. Vitamin B12 absorption occurs in a series of steps. B12 is normally released from food by stomach acid and then, in the small intestine, is bound to intrinsic factor (IF), a protein made by parietal cells in the stomach. This B12-IF complex is then absorbed by the small intestine, bound by carrier proteins (transcobalamins), and enters the circulation. If a disease or condition interferes with any of these steps, then B12 absorption is impaired.
Some examples of these conditions include:
- Pernicious anemia is the most common cause of B12 deficiency. A protein called intrinsic factor, made by parietal cells that line the stomach, is needed for B12 absorption. In pernicious anemia, inflammation damages the parietal cells, leading to little or no intrinsic factor, thus preventing the intestines from absorbing B12. With insufficient B12, the body produces enlarged but fewer RBCs. Because of the larger than normal RBCs, this is often referred to as megaloblastic or macrocytic anemia.
- Celiac disease
- Inflammatory bowel disease, including Crohns disease and ulcerative colitis
- Bacterial overgrowth or the presence of parasites in the intestines
- Reduced stomach acid production; stomach acid is necessary to separate B12 from the protein in food. This is the most common cause of B12 deficiency in the elderly and in individuals on drugs that suppress gastric acid production.
- Surgery that removes part of the stomach (and the parietal cells) or the intestines may greatly reduce absorption of nutrients. This is a concern that is considered when gastric by-pass procedures are performed.
- Chronic pancreatitis
Increased need
All pregnant women need increased amounts of folate for proper fetal development. If a woman has a folate deficiency prior to pregnancy, it will be intensified during the pregnancy and may lead to premature birth and neural tube defects in the child. It is recommended that women with any chance of becoming pregnant take folic acid supplements because neural tube defects can develop in the first few weeks of pregnancy, before many women realize they are pregnant.
People with cancer that has spread (metastasized) or with a chronic hemolytic anemia such as due to sickle cell disease have an increased need for folate.
Other causes:
- Heavy alcohol drinking and chronic alcoholism can cause B12 and/or folate deficiency through a combination of poor nutrition and a decrease in the amount of B12 released from dietary proteins.
- Some drugs can cause B12 deficiency. For example, the diabetes drug metformin prevents B12 from being absorbed, while omeprazole (an acid reflux drug also known as Prilosec) reduces gastric acids and prevents B12 release from food.
- Anti-seizure medications such as phenytoin can decrease folate by blocking folate absorption.
- Methotrexate, an anti-cancer drug, affects body metabolism and use of folate.
Vitamin B12 and folate deficiencies diagnosis
Laboratory testing is used to detect a vitamin deficiency, determine its severity, establish it as the underlying cause of someone’s symptoms, and to monitor the effectiveness of treatment. The anemia and large red blood cells (RBCs) associated with a vitamin B12 or folate deficiency are often initially detected during a routine complete blood count (CBC). Additional laboratory testing is performed as follow up to identify the specific deficiency.
Laboratory Tests
To diagnose or monitor B12 and folate deficiencies:
- B12 blood level. If low, a deficiency is indicated, but it does not identify the cause. A low level of intrinsic factor may be a cause, for example. This test also may be ordered to monitor the effectiveness of treatment.
- Folate level. Either serum or RBC folate levels may be tested; if either is low, it indicates a deficiency. The tests may also be ordered to monitor the effectiveness of treatment. Pregnant women may be given this test at prenatal checkups.
- Complete blood count (CBC). This group of tests is ordered routinely to evaluate the health of blood cells. It determines the number of cell types and can give an indication of the physical characteristics of some of the cells. With both B12 and folate deficiencies, the amount of hemoglobin and the red blood cell count may be low and the RBCs are abnormally large (macrocytic or megaloblastic), resulting in an anemia. White blood cells and platelets also may be decreased.
- Methylmalonic acid (MMA). This test may be ordered to help detect mild or early B12 deficiency.
- Homocysteine. This test is seldom ordered but may be elevated in both B12 and folate deficiency.
To help determine the cause of a B12 deficiency:
- Intrinsic factor antibody. The antibody prevents intrinsic factor from carrying out its function, that is, to carry vitamin B12 and allow B12 to be absorbed at a specific segment of the small intestine.
- Parietal cell antibody. An antibody against the parietal cells that produce intrinsic factor. This antibody can disrupt the production of intrinsic factor and is present in a large percentage of those with pernicious anemia, but it may also be seen in other autoimmune disorders.
- Gastrin. A hormone that regulates the production of acid in the stomach during the digestive process. Increased gastrin is sometimes seen in pernicious anemia.
- Schilling test. Once frequently ordered to confirm a diagnosis of pernicious anemia, this test is generally no longer available.
Vitamin B12 and folate deficiencies treatment
Treatment for B12 and folate deficiencies frequently involves supplementation, which may be long-term or lifetime, depending on the underlying cause. People who lack intrinsic factor or have conditions causing general malabsorption require injections of B12. Folate is given as an oral supplement.
It is generally recommended that all women contemplating having a child or who may become pregnant take folic acid supplements to ensure that they have a sufficient store for normal fetal development and to prevent neural tube defects.
Individuals deficient in both B12 and folate will require replenishment of both. If someone with a B12 deficiency only takes folic acid supplements, the macrocytic anemia may be resolved but the underlying neuropathy caused by the B12 deficiency will persist. Appropriate treatment should resolve symptoms but may not reverse all of the nerve damage.
Homocystinuria
Homocystinuria is an inherited disorder in which the body is unable to process certain building blocks of proteins (amino acids) properly. There are multiple forms of homocystinuria, which are distinguished by their signs and symptoms and genetic cause. The most common form of homocystinuria also known as classic homocystinuria or cystathionine beta-synthase (CBS) deficiency is characterized by nearsightedness (myopia), dislocation of the lens at the front of the eye (ectopia lentis), an increased risk of abnormal blood clotting, and brittle bones that are prone to fracture (osteoporosis) or other skeletal abnormalities. Some affected individuals also have developmental delay and learning problems.
Less common forms of homocystinuria can cause intellectual disability, failure to grow and gain weight at the expected rate (failure to thrive), seizures, problems with movement, and a blood disorder called megaloblastic anemia. Megaloblastic anemia occurs when a person has a low number of red blood cells (anemia), and the remaining red blood cells are larger than normal (megaloblastic).
The signs and symptoms of homocystinuria typically develop within the first year of life, although some mildly affected people may not develop features until later in childhood or adulthood.
The most common form of homocystinuria affects at least 1 in 200,000 to 335,000 people worldwide. The disorder appears to be more common in some countries, such as Ireland (1 in 65,000), Germany (1 in 17,800), Norway (1 in 6,400), and Qatar (1 in 1,800). The rarer forms of homocystinuria each have a small number of cases reported in the scientific literature.
Homocystinuria causes
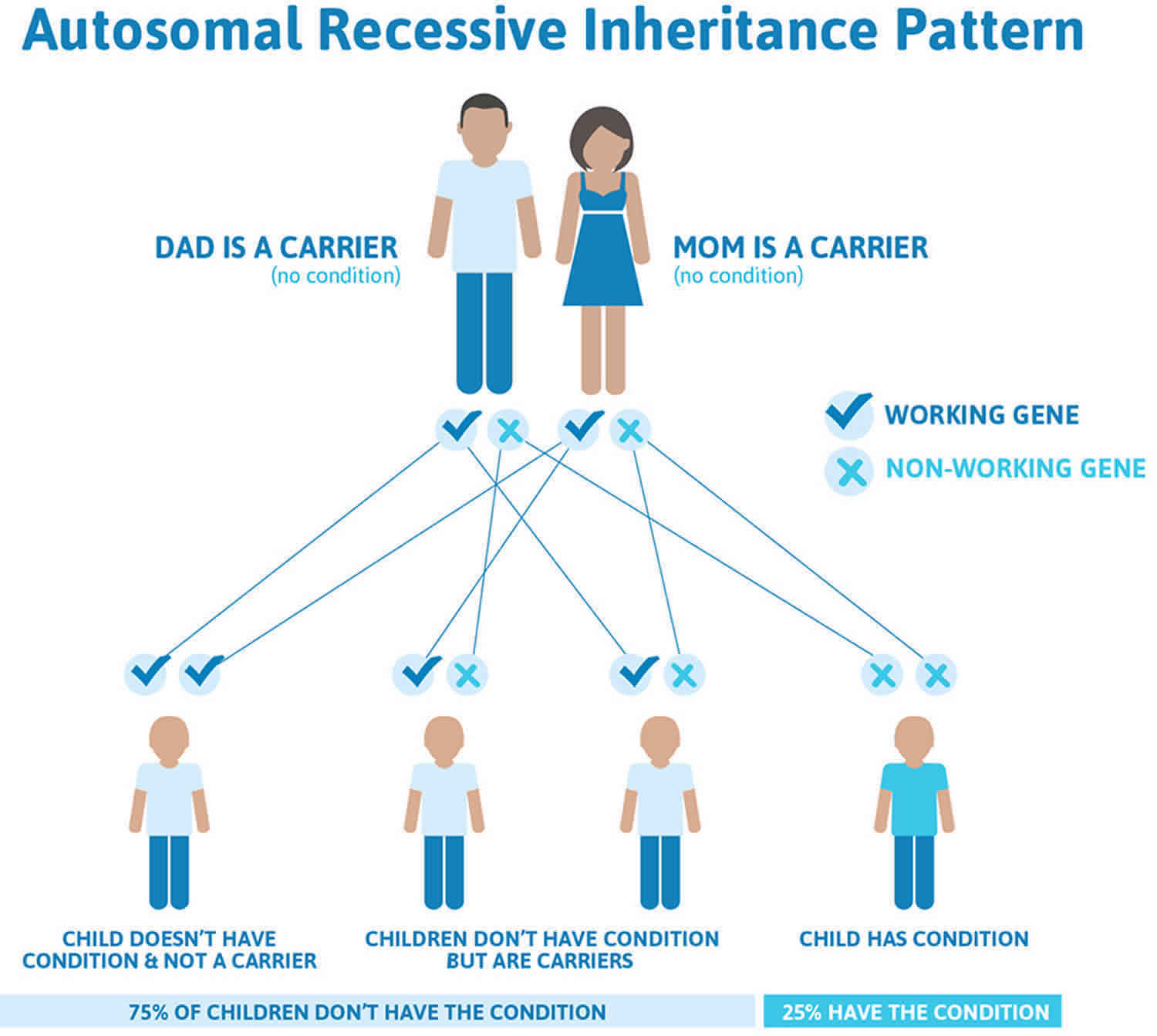
Homocystinuria is inherited in families as an autosomal recessive trait. This means that the child must inherit a non-working copy of the gene from each parent to be seriously affected. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition.
Although people who carry one mutated copy and one normal copy of the cystathionine beta-synthase (CBS) gene do not have homocystinuria, they are more likely than people without a CBS mutation to have shortages (deficiencies) of vitamin B12 and folic acid.
People with homocystinuria have several physical features in common with Marfan syndrome, including skeletal and eye changes.
Homocystinuria Genetic Changes
Mutations in the CBS, MTHFR, MTR, MTRR, and MMADHC genes cause homocystinuria. The CBS gene provides instructions for making an enzyme called cystathionine beta-synthase (CBS). This enzyme acts in a chemical pathway and is responsible for using vitamin B6 to convert building block of proteins (amino acid) called homocysteine and serine to a molecule called cytathionine. Another enzyme then converts cystathionine to the amino acid cysteine, which is used to build proteins or is broken down and excreted in urine. Additionally, other amino acids, including methionine, are produced in this pathway.
Mutations in the CBS gene cause the most common form of homocystinuria. The CBS gene provides instructions for producing an enzyme called cystathionine beta-synthase. This enzyme acts in a chemical pathway and is responsible for converting the amino acid homocysteine to a molecule called cystathionine. As a result of this pathway, other amino acids, including methionine, are produced. Mutations in the CBS gene disrupt the function of cystathionine beta-synthase, preventing homocysteine from being used properly. As a result, this amino acid and toxic byproducts substances build up in the blood. Some of the excess homocysteine is excreted in urine.
Rarely, homocystinuria can be caused by mutations in several other genes. The enzymes made by the MTHFR, MTR, MTRR, and MMADHC genes play roles in converting homocysteine to methionine. Mutations in any of these genes prevent the enzymes from functioning properly, which leads to a buildup of homocysteine in the body. Researchers have not determined how excess homocysteine and related compounds lead to the signs and symptoms of homocystinuria.
Homocystinuria is inherited in families as an autosomal recessive trait. This means that the child must inherit a non-working copy of the gene from each parent to be seriously affected. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Although people who carry one altered copy and one normal copy of the CBS gene do not have homocystinuria, they are more likely than people without a CBS variant to have shortages (deficiencies) of vitamin B12 and folic acid.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://findageneticcounselor.nsgc.org) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://www.acmg.net/ACMG/Directories.aspx) has a searchable database of medical genetics clinic services in the United States.
Figure 6. Homocystinuria autosomal recessive inheritance pattern
Homocystinuria Prevention
Genetic counseling is recommended for people with a family history of homocystinuria who want to have children. Prenatal diagnosis of homocystinuria is available. This involves culturing amniotic cells or chorionic villi to test for cystathionine synthase (the enzyme that is missing in homocystinuria).
If there are known gene defect in the parents or family, samples from chorionic villus sampling (CVS) or amniocentesis can be used to test for these defects.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://findageneticcounselor.nsgc.org) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://www.acmg.net/ACMG/Directories.aspx) has a searchable database of medical genetics clinic services in the United States.
Homocystinuria signs and symptoms
Newborn infants appear healthy. Early symptoms, if present, are not obvious.
Symptoms may occur as mildly delayed development or failure to thrive. Increasing visual problems may lead to diagnosis of homocystinuria.
The health care provider may notice that the child is tall and thin.
Other signs include:
- Curved spine (scoliosis)
- Deformity of the chest
- Dislocated lens of the eye
If there is poor or double vision, an eye doctor (ophthalmologist) will perform a dilated eye exam to look for dislocation of the lens or nearsightedness.
There may be a history of blood clots. Intellectual disability or mental illness is also possible.
Other symptoms include:
- Chest deformities (pectus carinatum, pectus excavatum)
- Flush across the cheeks
- High arches of the feet
- Intellectual disability
- Knock knees
- Long limbs
- Mental disorders
- Nearsightedness
- Spidery fingers (arachnodactyly)
- Tall, thin build
Infants who develop homocystinuria due to cystathionine beta-synthase (CBS) deficiency (classical homocystinuria) may fail to grow, or grow longer too fast, often have difficulty with gain weight at the expected rate (failure to thrive) and have developmental delays 62. By approximately age three, additional, more specific symptoms and findings may become apparent. These may include partial dislocation (subluxation) of the lens of the eyes (ectopia lentis), associated “quivering” (iridodonesis) of the colored region of the eyes (iris), severe nearsightedness (myopia), and other eye (ocular) abnormalities. Although intelligence may be normal in some cases, many children may be affected by progressive intellectual disability. In addition, some may develop psychiatric disturbances and/or episodes of uncontrolled electrical activity in the brain (seizures). Affected individuals also tend to be thin with unusually tall stature for their family; long, slender fingers and toes (arachnodactyly); and elongated arms and legs (“marfanoid” features). In addition, affected individuals may be at risk for the development of blood clots that can become lodged within certain large and small blood vessels (thromboembolisms), potentially leading to life-threatening complications.
Homocystinuria complications
Most serious complications result from blood clots. These episodes can be life threatening.
Dislocated lenses of the eyes can seriously damage vision. Lens replacement surgery may be needed.
Intellectual disability is a serious consequence of the disease. But, it can be reduced if diagnosed early.
Homocystinuria Diagnosis
Exams and Tests
Tests that may be ordered include any of the following:
- Amino acid screen of blood and urine
- Genetic testing
- Liver biopsy and enzyme assay
- Skeletal x-ray
- Skin biopsy with a fibroblast culture
- Standard ophthalmic exam
Homocystinuria Treatment
There is no cure for homocystinuria. About half of people with the disease respond to vitamin B6 (also known as pyridoxine). Those who do respond will need to take vitamin B6 (pyridoxine), B9 (folate), and B12 supplements for the rest of their lives. Those who do not respond will need to eat a low-methionine diet. Most will need to be treated with trimethylglycine a medicine also known as betaine.
Individuals on low-methionine diet require methionine-free supplemental metabolic foods usually in the form of a formula to provide them with other essential amino acids. A low protein, low methionine diet when started during infancy before any complications have developed has been effective in preventing or delaying the onset of symptoms.
A low protein, low methionine diet may be combined with cysteine supplementation. Cysteine is an amino acid that is often low in individuals with homocystinuria due to CBS deficiency. When methionine is broken down (metabolized) it produces cystine. Since individuals with homocystinuria cannot properly breakdown methionine, this may cause low levels of cysteine in some individuals, however good control of homocysteine levels and use of metabolic foods (i.e. formulas) often attenuates the need for supplementation.
Neither a low-methionine diet nor medicine will improve existing intellectual disability. Medicine and diet should be closely supervised by a doctor who has experience treating homocystinuria.
When individuals who are not responsive to vitamin B6 (pyridoxine) therapy are diagnosed later during childhood or adolescence, maintaining the dietary restrictions often proves difficult. The diet is usually not well-tolerated when it is begun in individuals diagnosed in childhood or adolescence.
Individuals with homocystinuria due to CBS deficiency, especially those who do not respond to vitamin B6 (pyridoxine) therapy may be treated with betaine (trimethylglycine), which can be used to lower the levels of homocysteine in the body. Betaine is often used an adjunct (add-on) to individuals on a low protein, low methionine treatment. Betaine for oral solution (Cystadane) is approved by the U.S. Food and Drug Administration (FDA) as a treatment for homocystinuria due to CBS deficiency.
Specific symptoms of homocystinuria due to CBS deficiency are treated as appropriate. For example, dislocation of the lenses of the eyes (ectopia lentis) or certain skeletal malformations may be treated surgically. However, affected individuals who undergo any surgery should receive particular care because homocystinuria due to CBS deficiency may increase the risk of post-surgical thromboembolic complications.
Figure 7. Pyridoxine vitamin B6 rich food sources

Homocystinuria Outlook (Prognosis)
Although no cure exists for homocystinuria, vitamin B therapy can help about half of people affected by homocystinuria.
If the diagnosis is made in childhood, starting a low-methionine diet quickly may prevent some intellectual disability and other complications of the disease. For this reason, some states screen for homocystinuria in all newborns.
People whose blood homocysteine levels continue to rise are at increased risk for blood clots. Clots can cause serious medical problems and shorten lifespan.
- Homocysteine as a risk factor for cardiovascular disease: should we (still) worry about? Faeh D, Chiolero A, Paccaud F. Swiss Med Wkly. 2006 Dec 2; 136(47-48):745-56. https://www.ncbi.nlm.nih.gov/pubmed/17225194/[↩][↩][↩][↩][↩]
- Venes D, Clarence WT. In: Taber’s Cyclopedic Medical Dictionary. 21. Venes D, editor. Philadelphia: F.A. Davis; 2005. p. 1089.[↩]
- Christen WG, Ajani UA, Glynn RJ, et al. Blood levels of homocysteine and increased risks of cardiovascular disease: causal or casual? Arch Intern Med. 2000;160(4):422–434. doi: 10.1001/archinte.160.4.422[↩]
- Habib SS, Al-Khlaiwi T, Almushawah A, et al. Homocysteine as a predictor and prognostic marker of atherosclerotic cardiovascular disease: a systematic review and meta-analysis. Eur Rev Med Pharmacol Sci. 2023;27(18):8598–8608. doi: 10.26355/eurrev_202309_33784[↩]
- Rong D, Liu J, Jia X, et al. Hyperhomocysteinaemia is an independent risk factor for peripheral arterial disease in a Chinese Han population. Atherosclerosis. 2017;263:205–210. doi: 10.1016/j.atherosclerosis.2017.05.006[↩][↩]
- Zhang T, Jiang Y, Zhang S, et al. The association between homocysteine and ischemic stroke subtypes in Chinese: a meta-analysis. Medicine. 2020;99(12):e19467. doi: 10.1097/MD.0000000000019467[↩]
- Tian W, Ju J, Guan B, Wang T, Zhang J, Song L, Xu H. Role of hyperhomocysteinemia in atherosclerosis: from bench to bedside. Ann Med. 2025 Dec;57(1):2457527. doi: 10.1080/07853890.2025.2457527[↩][↩][↩][↩][↩]
- Humphrey LL, Fu R, Rogers K, et al. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc. 2008;83(11):1203–1212. doi: 10.4065/83.11.1203[↩]
- Boushey CJ, Beresford SA, Omenn GS, et al. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA. 1995;274(13):1049–1057. doi: 10.1001/jama.1995.03530130055028[↩]
- Liu M, Fan F, Liu B, et al. Joint effects of plasma homocysteine concentration and traditional cardiovascular risk factors on the risk of new-onset peripheral arterial disease. Diabetes Metab Syndr Obes. 2020;13:3383–3393. doi: 10.2147/DMSO.S267122[↩]
- Karger AB, Steffen BT, Nomura SO, et al. Association between homocysteine and vascular calcification incidence, prevalence, and progression in the MESA cohort. J Am Heart Assoc. 2020;9(3):e013934. doi: 10.1161/JAHA.119.013934[↩]
- Toole JF, Malinow MR, Chambless LE, Spence JD, Pettigrew LC, Howard VJ, Sides EG, Wang CH, Stampfer M. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA. 2004 Feb 4;291(5):565-75. doi: 10.1001/jama.291.5.565[↩][↩][↩][↩][↩]
- Lonn E, Yusuf S, Arnold MJ, Sheridan P, Pogue J, Micks M, McQueen MJ, Probstfield J, Fodor G, Held C, Genest J Jr; Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med. 2006 Apr 13;354(15):1567-77. doi: 10.1056/NEJMoa060900 Erratum in: N Engl J Med. 2006 Aug 17;355(7):746.[↩][↩][↩][↩][↩]
- Smolders B, Lemmens R, Thijs V. Lipoprotein (a) and stroke: a meta-analysis of observational studies. Stroke. 2007 Jun;38(6):1959-66. doi: 10.1161/STROKEAHA.106.480657[↩][↩]
- Clarke R, Bennett DA, Parish S, et al. MTHFR Studies Collaborative Group. Homocysteine and coronary heart disease: meta-analysis of MTHFR case-control studies, avoiding publication bias. PLoS Med. 2012 Feb;9(2):e1001177. doi: 10.1371/journal.pmed.1001177[↩][↩]
- Ganguly P, Alam SF. Role of homocysteine in the development of cardiovascular disease. Nutr J. 2015 Jan 10;14:6. doi: 10.1186/1475-2891-14-6[↩][↩]
- Saposnik G, Ray JG, Sheridan P, McQueen M, Lonn E; Heart Outcomes Prevention Evaluation 2 Investigators. Homocysteine-lowering therapy and stroke risk, severity, and disability: additional findings from the HOPE 2 trial. Stroke. 2009 Apr;40(4):1365-72. doi: 10.1161/STROKEAHA.108.529503[↩]
- Lee M, Hong KS, Chang SC, Saver JL. Efficacy of homocysteine-lowering therapy with folic Acid in stroke prevention: a meta-analysis. Stroke. 2010 Jun;41(6):1205-12. doi: 10.1161/STROKEAHA.109.573410[↩]
- Li Y, Huang T, Zheng Y, Muka T, Troup J, Hu FB. Folic Acid Supplementation and the Risk of Cardiovascular Diseases: A Meta-Analysis of Randomized Controlled Trials. J Am Heart Assoc. 2016 Aug 15;5(8):e003768. doi: 10.1161/JAHA.116.003768[↩]
- Martí-Carvajal AJ, Solà I, Lathyris D, Dayer M. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev. 2017 Aug 17;8(8):CD006612. doi: 10.1002/14651858.CD006612.pub5[↩]
- Malinow MR, Bostom AG, Krauss RM. Homocyst(e)ine, diet, and cardiovascular diseases: a statement for healthcare professionals from the Nutrition Committee, American Heart Association. Circulation. 1999 Jan 5-12;99(1):178-82. doi: 10.1161/01.cir.99.1.178[↩]
- Bønaa KH, Njølstad I, Ueland PM, Schirmer H, Tverdal A, Steigen T, Wang H, Nordrehaug JE, Arnesen E, Rasmussen K; NORVIT Trial Investigators. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med. 2006 Apr 13;354(15):1578-88. doi: 10.1056/NEJMoa055227[↩][↩][↩]
- Løland KH, Bleie O, Blix AJ, Strand E, Ueland PM, Refsum H, Ebbing M, Nordrehaug JE, Nygård O. Effect of homocysteine-lowering B vitamin treatment on angiographic progression of coronary artery disease: a Western Norway B Vitamin Intervention Trial (WENBIT) substudy. Am J Cardiol. 2010 Jun 1;105(11):1577-84. doi: 10.1016/j.amjcard.2010.01.019[↩][↩]
- Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group; Armitage JM, Bowman L, Clarke RJ, Wallendszus K, Bulbulia R, Rahimi K, Haynes R, Parish S, Sleight P, Peto R, Collins R. Effects of homocysteine-lowering with folic acid plus vitamin B12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA. 2010 Jun 23;303(24):2486-94. doi: 10.1001/jama.2010.840[↩][↩][↩]
- Albert CM, Cook NR, Gaziano JM, et al. Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: a randomized trial. JAMA. 2008;299(17):2027–2036. doi: 10.1001/jama.299.17.2027[↩]
- Ebbing M, Bleie Ø, Ueland PM, et al. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA. 2008;300(7):795–804. doi: 10.1001/jama.300.7.795[↩]
- McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969 Jul;56(1):111-28. https://pmc.ncbi.nlm.nih.gov/articles/instance/2013581/pdf/amjpathol00422-0110.pdf[↩]
- Steed MM, Tyagi SC.. Mechanisms of cardiovascular remodeling in hyperhomocysteinemia. Antioxid Redox Signal. 2011;15(7):1927–1943. doi: 10.1089/ars.2010.3721[↩]
- Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA. 2002 Oct 23-30;288(16):2015-22. doi: 10.1001/jama.288.16.2015[↩]
- Carson NA, Neill DW.. Metabolic abnormalities detected in a survey of mentally backward individuals in Northern Ireland. Arch Dis Child. 1962;37(195):505–513. doi: 10.1136/adc.37.195.505[↩]
- Gerritsen T, Vaughn JG, Waisman HA.. The identification of homocystine in the urine. Biochem Biophys Res Commun. 1962;9(6):493–496. doi: 10.1016/0006-291x(62)90114-6[↩]
- CBS (homocystinemia/cystathionine beta-synthase deficiency). https://www.newbornscreening.info/cbs-homocystinemia-cystathionine-beta-synthase-deficiency/[↩]
- Exercise, nutrition, and homocysteine. Int J Sport Nutr Exerc Metab. 2006 Aug;16(4):341-61. https://www.ncbi.nlm.nih.gov/pubmed/17136938[↩]
- Homocysteine and cardiovascular disease: interactions between nutrition, genetics and lifestyle. Can J Appl Physiol. 2004 Dec;29(6):773-80. https://www.ncbi.nlm.nih.gov/pubmed/15630149[↩]
- Homocysteine and vascular disease. Hankey GJ, Eikelboom JW. Lancet. 1999 Jul 31; 354(9176):407-13. https://www.ncbi.nlm.nih.gov/pubmed/10437885/[↩][↩][↩][↩]
- Epigenetic modifications: basic mechanisms and role in cardiovascular disease (2013 Grover Conference series). Loscalzo J, Handy DE. Pulm Circ. 2014 Jun; 4(2):169-74. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4070783/[↩][↩][↩][↩]
- Harvey RA, Ferrier DR. In: Lippincott’s Illustrated Reviews, Biochemistry. 5. Rhyner S, editor. Philadelphia: Wolters Kluwer Health; 2011. pp. 264–5.[↩][↩][↩][↩][↩][↩]
- Jakubowski H. Homocysteine modification in protein structure/function and human disease. Physiol Rev. 2019;99(1):555–604. doi: 10.1152/physrev.00003.2018[↩][↩]
- Škovierová H, Vidomanová E, Mahmood S, et al. The molecular and cellular effect of homocysteine metabolism imbalance on human health. Int J Mol Sci. 2016;17(10):1733. doi: 10.3390/ijms17101733[↩][↩]
- Teng Y-W, Mehedint MG, Garrow TA, et al. Deletion of betaine-homocysteine S-methyltransferase in mice perturbs choline and 1-carbon metabolism, resulting in fatty liver and hepatocellular carcinomas. J Biol Chem. 2011;286(42):36258–36267. doi: 10.1074/jbc.M111.265348[↩]
- Hama Y, Hamano T, Shirafuji N, et al. Influences of Folate Supplementation on Homocysteine and Cognition in Patients with Folate Deficiency and Cognitive Impairment. Nutrients. 2020;12(10):3138. doi: 10.3390/nu12103138[↩]
- Moretti R, Caruso P.. The controversial role of homocysteine in neurology: from labs to clinical practice. Int J Mol Sci. 2019;20(1):231. doi: 10.3390/ijms20010231[↩]
- Blom HJ, Smulders Y.. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J Inherit Metab Dis. 2011;34(1):75–81. doi: 10.1007/s10545-010-9177-4[↩]
- Ueno A, Hamano T, Enomoto S, et al. Influences of vitamin B12 supplementation on cognition and homocysteine in patients with vitamin B12 deficiency and cognitive impairment. Nutrients. 2022;14(7):1494. doi: 10.3390/nu14071494[↩]
- Curro M, Gugliandolo A, Gangemi C, Risitano R, Ientile R, Caccamo D. Toxic effects of mildly elevated homocysteine concentrations in neuronal-like cells. Neurochem Res. 2014;39:1485–95. doi: 10.1007/s11064-014-1338-7. https://www.ncbi.nlm.nih.gov/pubmed/24867323[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Carmel R, Jacobsen DW. In: Homocysteine in health and disease. Carmel R, Jacobsen DW, editors. Cambridge: Cambridge UP; 2001. pp. 183–93.[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- The effect of local application of homocysteine on neuronal activity in the central nervous system of the rat. Wuerthele SE, Yasuda RP, Freed WJ, Hoffer BJ. Life Sci. 1982 Dec 13; 31(24):2683-91. https://www.ncbi.nlm.nih.gov/pubmed/6130457/[↩][↩]
- [Hyperhomocysteinemia in patients with cardiovascular disease]. Baszczuk A, Kopczyński Z. Postepy Hig Med Dosw (Online). 2014 Jan 2; 68():579-89. https://www.ncbi.nlm.nih.gov/pubmed/24864108/[↩]
- Refsum H, Smith AD, Ueland PM, et al. Facts and recommendations about total homocysteine determinations: an expert opinion. Clin Chem. 2004;50(1):3–32. doi: 10.1373/clinchem.2003.021634[↩][↩][↩]
- Influence of folic acid on plasma homocysteine levels & arterial endothelial function in patients with unstable angina. Guo H, Chi J, Xing Y, Wang P. Indian J Med Res. 2009 Mar; 129(3):279-84. https://www.ncbi.nlm.nih.gov/pubmed/19491420/[↩]
- Strauss E, Supinski W, Radziemski A, et al. Is hyperhomocysteinemia a causal factor for heart failure? The impact of the functional variants of MTHFR and PON1 on ischemic and non-ischemic etiology. Int J Cardiol. 2017;228:37–44. doi: 10.1016/j.ijcard.2016.11.213[↩]
- Curro M, Gugliandolo A, Gangemi C, Risitano R, Ientile R, Caccamo D. Toxic effects of mildly elevated homocysteine concentrations in neuronal-like cells. Neurochem Res. 2014;39:1485–95. doi: 10.1007/s11064-014-1338-7 https://www.ncbi.nlm.nih.gov/pubmed/24867323[↩][↩][↩][↩]
- Hankey GJ, Eikelboom JW. Homocysteine and vascular disease. Lancet. 1999;354:407–13. doi: 10.1016/S0140-6736(98)11058-9. https://www.ncbi.nlm.nih.gov/pubmed/10437885[↩][↩][↩][↩][↩][↩][↩]
- Miller JW, Nadeau MR, Smith D, et al. Vitamin B-6 deficiency vs. folate deficiency: comparison of responses to methionine loading in rats. Am J Clin Nutr. 1994;59(5):1033–1039. doi: 10.1093/ajcn/59.5.1033[↩]
- Faeh D, Chiolero A, Paccaud F. Homocysteine as a risk factor for cardiovascular disease: should we (still) worry about it? Swiss Med Wkly. 2006;136:745–56 https://www.ncbi.nlm.nih.gov/pubmed/17225194[↩][↩]
- Yuan S, Mason AM, Carter P, et al. Homocysteine, B vitamins, and cardiovascular disease: a Mendelian randomization study. BMC Med. 2021;19(1):97. doi: 10.1186/s12916-021-01977-8[↩]
- Ganguly P, Alam SF.. Role of homocysteine in the development of cardiovascular disease. Nutr J. 2015;14(1):6. doi: 10.1186/1475-2891-14-6[↩]
- Zhou C, Wu J, Fang S.. Meta-analysis of B vitamin supplementation on plasma homocysteine, cardiovascular and all-cause mortality. Clin Nutr. 2013;32(2):314. doi: 10.1016/j.clnu.2013.01.001[↩]
- Yap S. Classical homocystinuria: vascular risk and its prevention. J Inherit Metab Dis. 2003;26(2-3):259–265. doi: 10.1023/a:1024497419821[↩]
- Baszczuk A, Kopczynski Z. Hyperhomocysteinemia in patients with cardiovascular disease [Abstract] Postepy Hig Med Dosw. 2014;68:579. doi: 10.5604/17322693.1102340 https://www.ncbi.nlm.nih.gov/pubmed/24864108[↩][↩][↩]
- Shenov V, Mehendale V, Prabhu K, Shetty R, Rao P. Correlation of serum homocysteine levels with the severity of coronary artery disease. Ind J Clin Biochem. 2014;29(3):339–44. doi: 10.1007/s12291-013-0373-5 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4062675/[↩][↩]
- Classical Homocystinuria. https://rarediseases.org/videos/classical-homocystinuria/[↩]
{kind=link}