Contents
Laurence-Moon syndrome
Laurence-Moon syndrome (LNMS or LMS), also called Laurence-Moon-Biedl syndrome (LMBBS) because of similarities with Bardet-Biedl syndrome (BBS), is a very rare genetic disorder that affects many parts of the body characterized by underactive pituitary gland (occurs when the pituitary gland doesn’t produce the right amount of hormones), ataxia (a neurological sign that causes problems coordinating how your muscles work, leading to awkward, unwieldy or clumsy movements), peripheral neuropathy (nerve outside of your brain and spinal cord also known as peripheral nerves are damaged causing them to stop working properly), spastic paraplegia (weakness and stiffness in your legs) and eye abnormalities called chorioretinal dystrophy primarily affecting the choroid (a layer of blood vessels between the retina and the sclera) and the retina (a light-sensitive layer of tissue at the back of your eyes that converts images into electrical signals for your brain) resulting in poor vision such as peripheral vision loss and vision loss at night, shortsightedness and abnormal eye movements with involuntary side-to-side movements of the eyes called nystagmus 1, 2, 3, 4, 5, 6, 7. People with Laurence-Moon-Biedl syndrome may have difficulties with functions of their brain, eyes, ears, stomach, kidneys, hands and feet. People with Laurence-Moon-Biedl syndrome often also demonstrate a tendency to short stature and obesity 7, 8.
In 1866, Laurence and Moon described four affected siblings with retinal dystrophy, obesity, and mental retardation 9. Three of them (males) also had small external genitalia and an abnormal gait 9. In 1920s, Bardet 10, 11 and Biedl 12, 13 separately reported a similar clinical features in individuals, who also had polydactyly (extra fingers and/or toes) and the condition was coined Laurence-Moon-Bardet-Biedl syndrome. The overlapping features of these cases suggested that the two disorders, Laurence-Moon-Bardet-Biedl syndrome and Bardet-Biedl syndrome (BBS) represented variable expression of a single condition 14, 15, 16 although this is controversial 17, 18. However, it is now generally considered that Bardet-Biedl syndrome (BBS) and Laurence-Moon syndrome (LMS) are distinct conditions 19.
Symptoms of Laurence-Moon-Biedl syndrome may start to appear in newborns. Laurence-Moon-Biedl syndrome is typically associated with syndactyly of the fingers (a condition where two or more fingers are fused together), polydactyly, intellectual impairment, obesity, undescended testicle (cryptorchidism), hypoplasia of the penis (also known as micropenis, is a rare condition that causes a penis to be smaller than normal), kidney impairment, sensorineural hearing loss, and a short height. Most patients with Laurence-Moon-Biedl syndrome will experience a gradual loss of vision due to retinitis pigmentosa (due to the loss of rod-cone photoreceptors), which begins with night blindness, worsens progressively with loss of color perception, and finally deteriorates into “tunnel vision”. Mild-to-moderate learning challenges attributed to weakened cognitive capacity are common in individuals with Laurence-Moon-Biedl syndrome. However, the suspected disabilities are not due to an underlying visual impairment. A smaller-than-average-size anterior pituitary gland has been observed in people with Laurence-Moon-Biedl syndrome, and as a result, these individuals are susceptible to various complications, including those related to the control of the body’s metabolism, emotional responses to stressors, physical development, low levels of thyroid-stimulating hormone (TSH), decreased levels of estrogen and testosterone, and underdeveloped reproductive organs 7.
Laurence-Moon syndrome or Laurence-Moon-Biedl syndrome is most commonly caused by changes (mutations) in the PNPLA6 (patatin like phospholipase domain containing 6) gene and is inherited in an autosomal recessive manner 5. Genes are specific sequences in DNA that provide instructions for the production of proteins. The PNPLA6 gene provides instructions for making a protein called neuropathy target esterase (NTE) 20, 7. The neuropathy target esterase (NTE) protein is an enzyme that is involved in the breakdown of certain fats (lipids). Specifically, neuropathy target esterase (NTE) breaks down a lipid called lysophosphatidylcholine, which is one of several compounds found in the outer membranes surrounding cells 20. The correct levels of lysophosphatidylcholine is critical to the stability of the cell membranes. The neuropathy target esterase (NTE) protein is an enzyme that is thought to drive the growth of nerve and non-nerve cells as they grow and mature. The PNPLA6 gene is notably associated not only with Laurence-Moon-Biedl syndrome but also Boucher-Neuhauser syndrome, Gordon-Holmes syndrome, Oliver-McFarlane syndrome, and spastic paraplegia type 39 20.
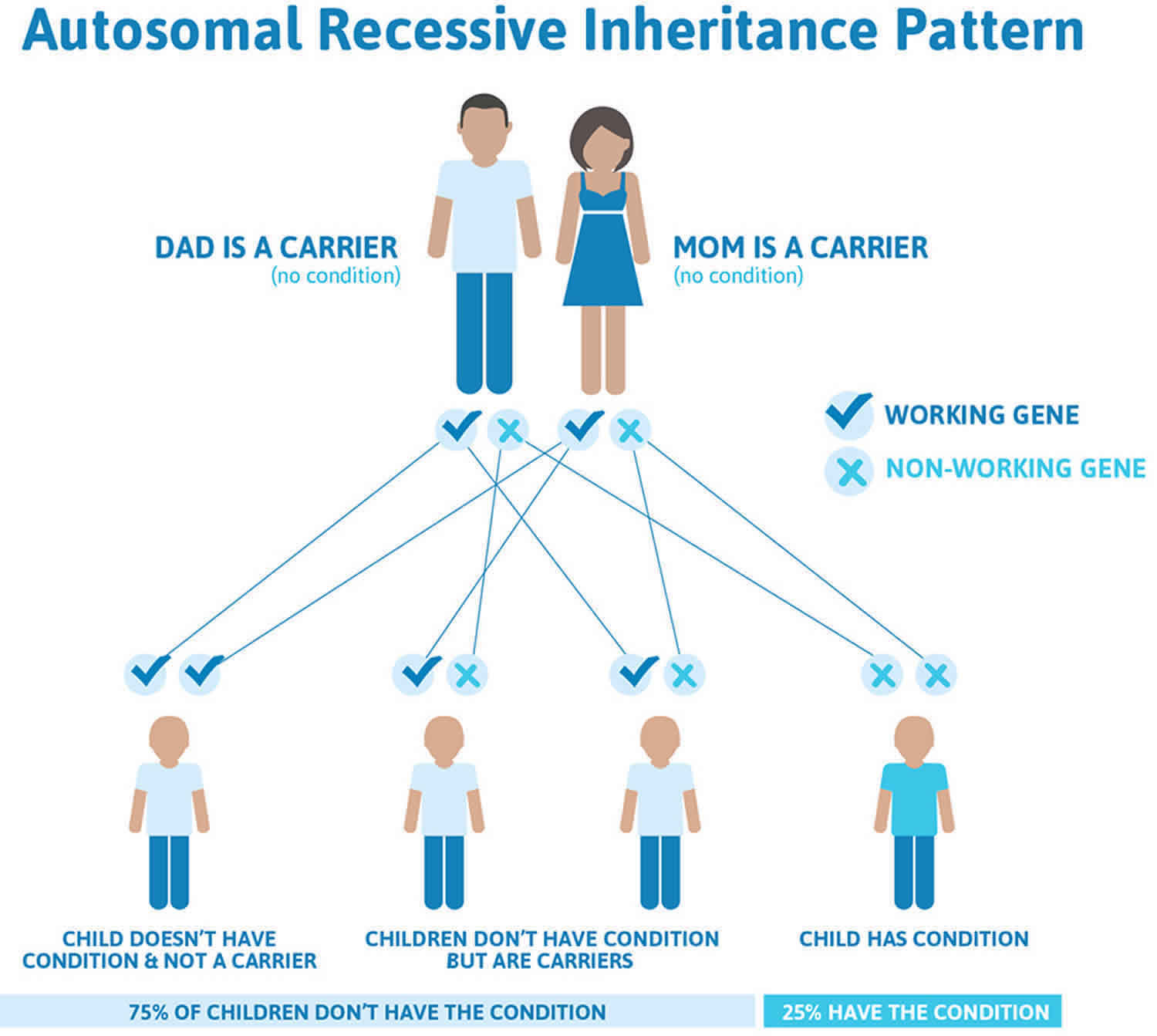
Laurence-Moon-Biedl syndrome follows an autosomal recessive pattern of inheritance (see Figure 4 below). Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% (1 in 4 chance) with each pregnancy. The risk to have a child who is a carrier like the parents is 50% (1 in 2 chance) with each pregnancy. The chance for a child to receive normal genes from both parents is 25% (1 in 4 chance). The risk is the same for males and females. As Laurence-Moon-Biedl syndrome is rare, a gene carrier is unlikely to have affected children unless their partner is also a carrier. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder 21.
In North America, it is estimated that 1 in 100,000 people are affected by Laurence-Moon-Biedl syndrome 7. In Europe, it is estimated that 1 in 125,000 to 1 in 160,000 people are affected by Laurence-Moon-Biedl syndrome 3. Kuwait and Newfoundland (off the east coast of Canada) are two places where the number of people affects with Laurence-Moon-Biedl syndrome are comparatively high affecting about 1:13,500 newborns and 1:17,500, respectively 22, 23. Consanguinity (union between two people who are related as second cousins or closer) is commonly practiced in various Middle Eastern countries, such as Kuwait, Saudi Arabia, Iran, and Pakistan 23. Consanguinity has been a chief contributing factor to Laurence-Moon-Biedl syndrome frequency. Families in these countries have approximately 0.1% homozygous genes of the total genome 23. In Pakistan, more than half of all marriages are consanguineous in nature, with 80% of them among first cousins, thereby increasing the possibility of homozygous mutations 23.
Due to the highly variable clinical presentation of Laurence-Moon-Biedl syndrome, no formal diagnostic criteria have been established for Laurence-Moon-Biedl syndrome or, for that matter, any PNPLA6-related disorders. Laurence-Moon-Biedl syndrome is diagnosed with molecular testing for mutations in the PNPLA6 gene 7, 24.
The management of Laurence-Moon-Bardet-Biedl syndrome involves a multidisciplinary approach and remains a challenge for clinicians. The treatments available for Laurence-Moon-Biedl syndrome are mainly toward managing the signs, symptoms and complications of the illness. Physical therapy aimed toward improving strength helps. Exercise can reduce the symptoms of spasticity. A dedicated regimen of nutritious, well-balanced meals and regular exercise is recommended, as there is an increased incidence of diabetes and abnormal cholesterol levels in patients with Laurence-Moon-Biedl syndrome 23. A low protein diet also slows the progression of kidney diseases in Laurence-Moon syndrome 25. The poor functional capacity of the anterior pituitary gland, resulting in slow metabolism, poor growth, and impaired fertility, can be managed with hormone replacement therapies. Levothyroxine can aid in increasing the body metabolism, resulting in reduced lethargy, hair loss, and obesity. Growth hormone supplementation reduces the psychosocial burden of short stature, whereas testosterone supplementation can be given in patients with markedly low levels to prevent underdeveloped genitalia. The extra digits are generally nonfunctional and can be removed for cosmetic purposes. Typically, retinal dystrophy is the first symptom that arises before the age of 10 years but affects almost all patients below the age of 20 years 26. Glasses can be used to treat this, and regular eye specialist (ophthalmologist) visits are recommended 27.
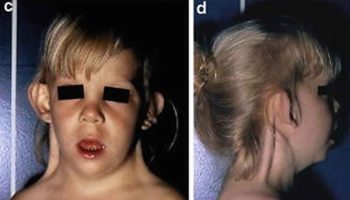
Figure 1. Laurence-Moon-Biedl syndrome face
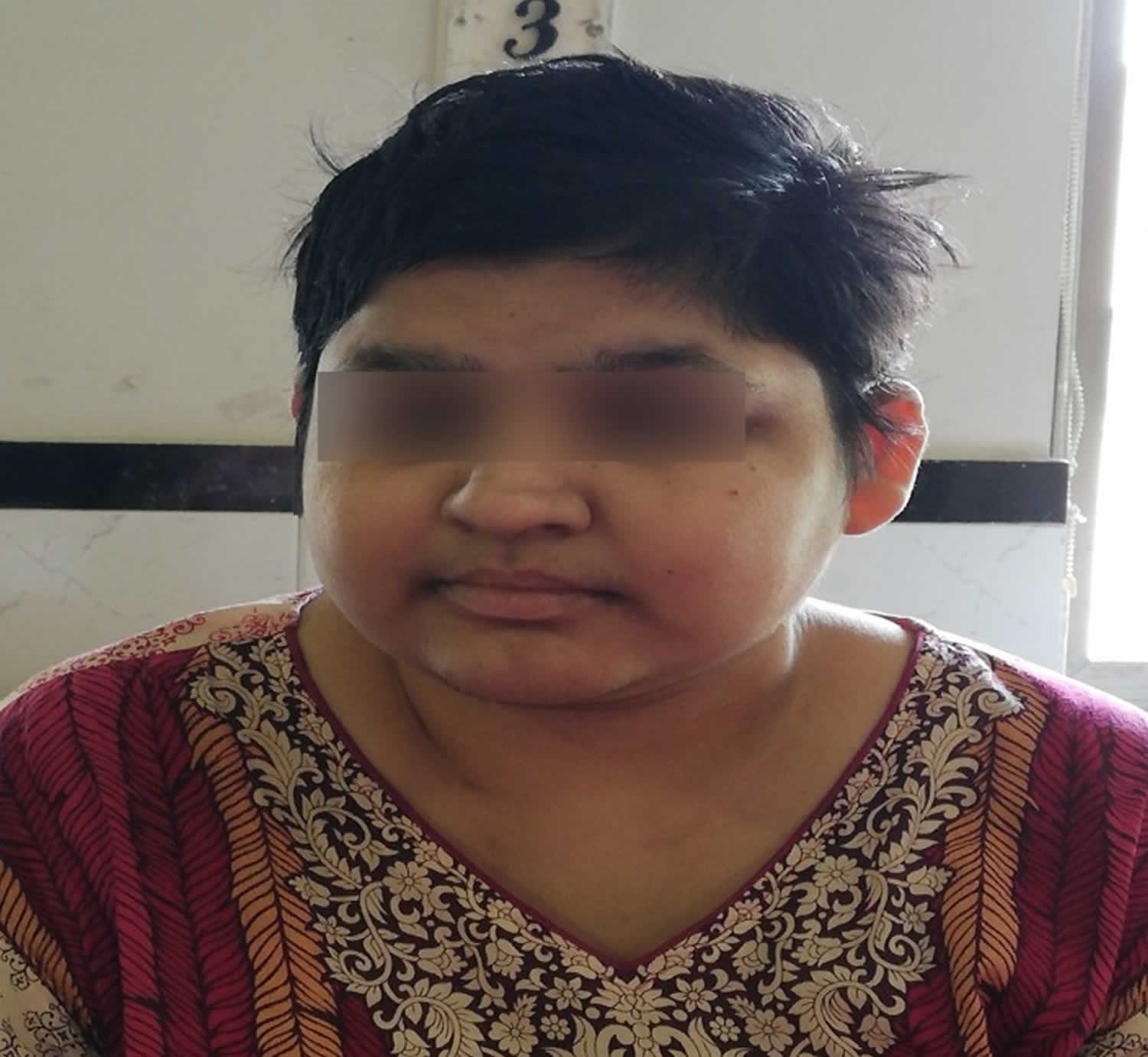
Figure 2. Laurence-Moon-Biedl syndrome hand (polydactyly)
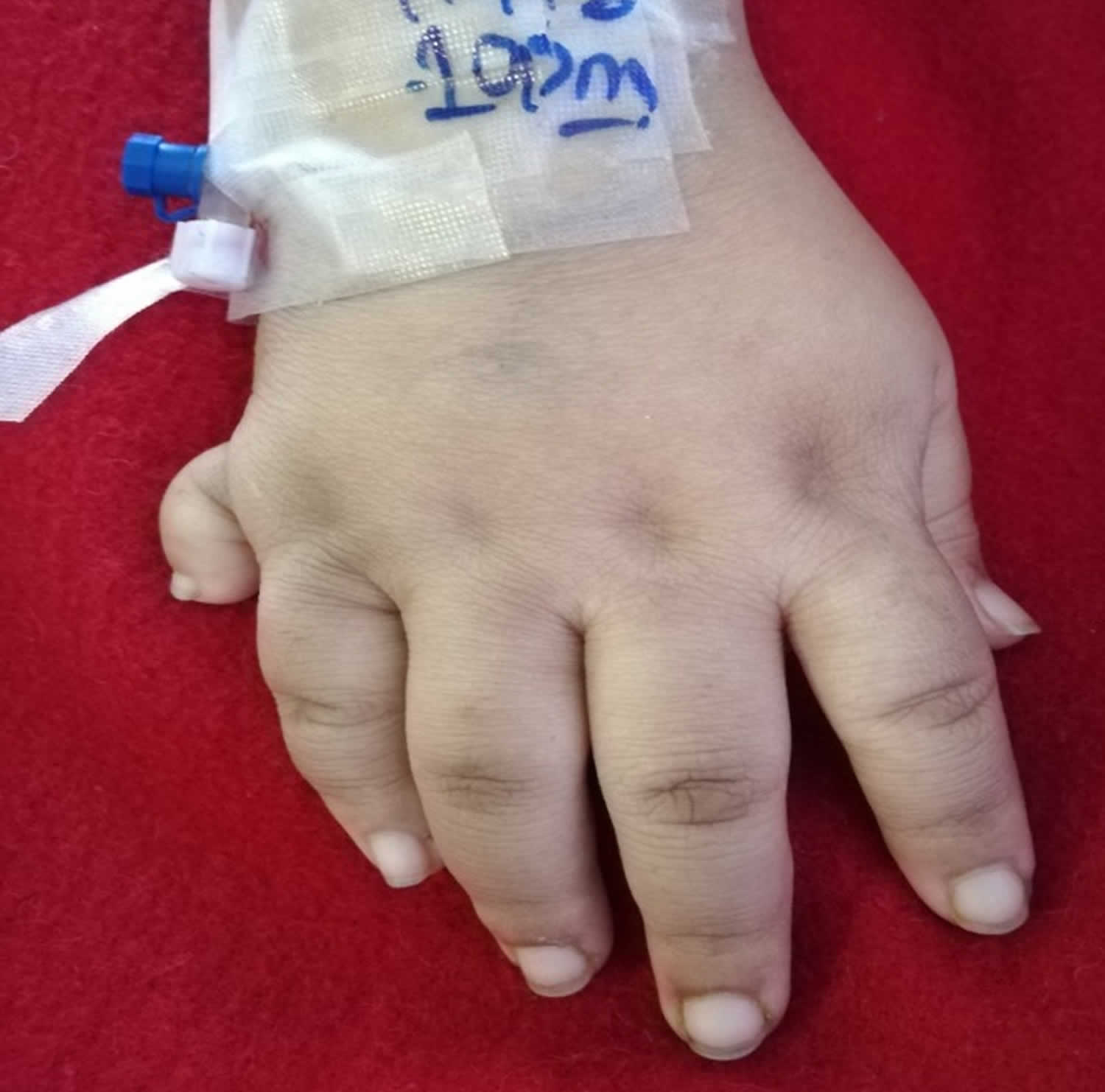
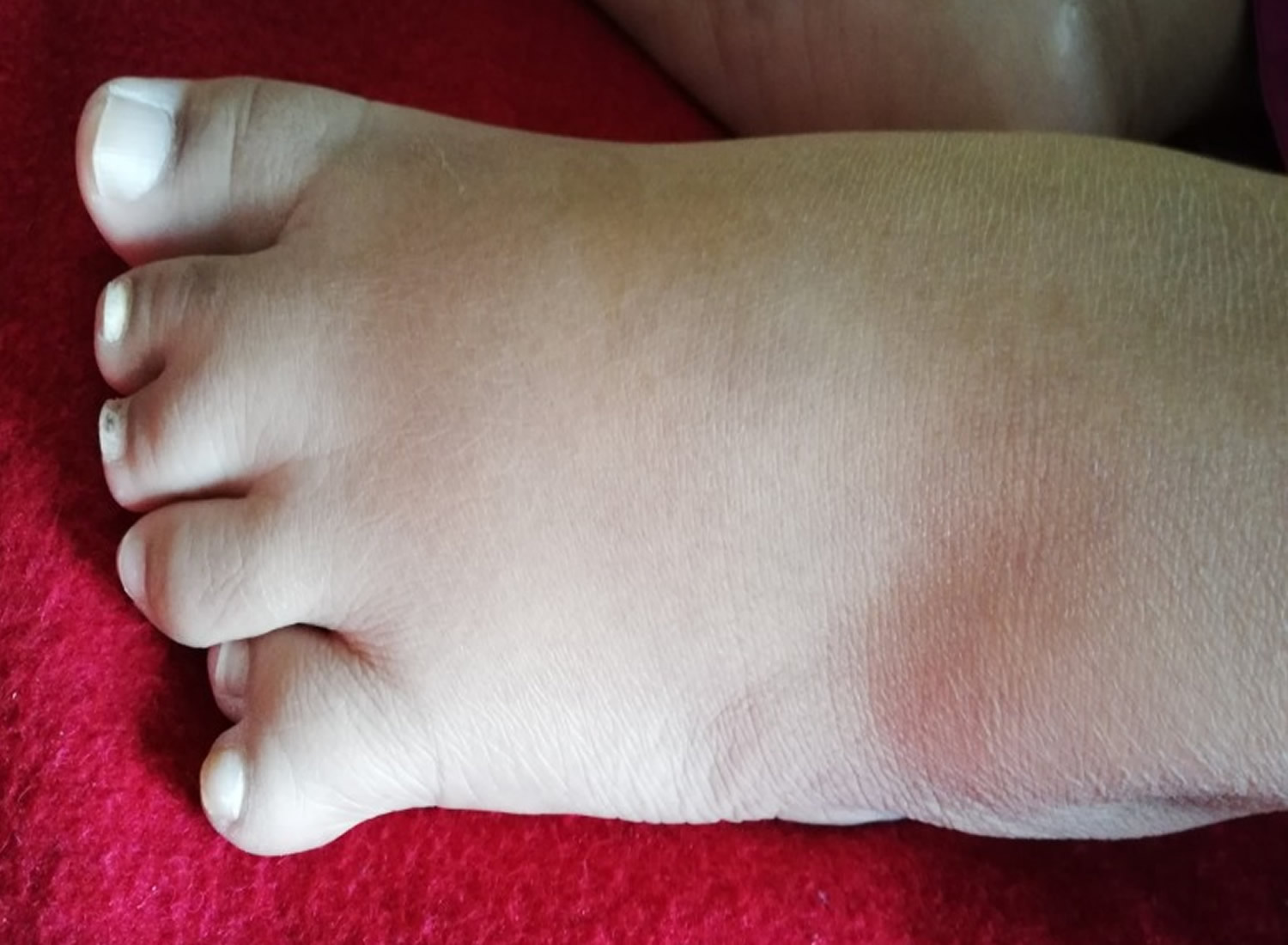
Figure 3. Laurence-Moon-Biedl syndrome foot (polydactyly)
Laurence-Moon-Biedl syndrome cause
Laurence-Moon syndrome or Laurence-Moon-Biedl syndrome is most commonly caused by changes (mutations) in the PNPLA6 (patatin like phospholipase domain containing 6) gene and is inherited in an autosomal recessive manner 5. Genes are specific sequences in DNA that provide instructions for the production of proteins. The PNPLA6 gene provides instructions for making a protein called neuropathy target esterase (NTE) 20, 7. The neuropathy target esterase (NTE) protein is an enzyme that is involved in the breakdown of certain fats (lipids). Specifically, neuropathy target esterase (NTE) breaks down a lipid called lysophosphatidylcholine, which is one of several compounds found in the outer membranes surrounding cells 20. The correct levels of lysophosphatidylcholine is critical to the stability of the cell membranes. The neuropathy target esterase (NTE) protein is an enzyme that is thought to drive the growth of nerve and non-nerve cells as they grow and mature. The PNPLA6 gene is notably associated not only with Laurence-Moon-Biedl syndrome but also Boucher-Neuhauser syndrome, Gordon-Holmes syndrome, Oliver-McFarlane syndrome, and spastic paraplegia type 39 20.
The neuropathy target esterase (NTE) protein is found most abundantly in your nervous system. Neuropathy target esterase (NTE) plays an important role in maintaining the stability of the membranes surrounding nerve cells (neurons) called myelin sheath. Myelin sheath is a protective, insulating layer made of protein and fatty substances that wraps around the axon of a nerve cell. Axons are long, thin nerve cell fibers (neurons) that transmit electrical signals from the cell body to other neurons or body tissues. Axons also transmit electrical impulses from muscle and gland cells to the brain. Most axons are covered by a protective myelin sheath that insulates the axon. The myelin sheath allows electrical nerve signals to travel faster, farther and efficiently along the nerve cell. Neurons communicate via electrical impulses that trigger the release of “chemical messengers” called neurotransmitters.
Neuropathy target esterase (NTE) may also play a role in the release of hormones from your pituitary gland, a process that requires particular changes in the cell membrane and appears to involve the lipids found there 20. The pituitary gland is located at the base of your brain and produces several hormones, including those that help direct sexual development and growth.
Researchers are unsure how mutations in the PNPLA6 gene lead to different combinations of features, resulting in the spectrum of PNPLA6-related disorders.
Laurence-Moon-Biedl syndrome inheritance pattern
Laurence-Moon-Biedl syndrome follows an autosomal recessive pattern of inheritance (Figure 4). Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% (1 in 4 chance) with each pregnancy. The risk to have a child who is a carrier like the parents is 50% (1 in 2 chance) with each pregnancy. The chance for a child to receive normal genes from both parents is 25% (1 in 4 chance). The risk is the same for males and females. As Laurence-Moon-Biedl syndrome is rare, a gene carrier is unlikely to have affected children unless their partner is also a carrier.
Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
If you are born to parents who both carry the same PNPLA6 gene, you have a 25% (1 in 4) chance of inheriting the PNPLA6 gene from both parents and developing Laurence-Moon-Biedl syndrome (see Figure 4). You have a 50% (1 in 2) chance of inheriting one variant PNPLA6 gene. This would make you PNPLA6 gene mutation carrier.
In other words, for a child born to a couple who both carry the PNPLA6 gene mutation (but do not have signs of disease), the expected outcome for each pregnancy is:
- A 25% chance that the child is born with two normal genes (healthy)
- A 50% chance that the child is born with one normal and one variant gene (carrier, without disease)
- A 25% chance that the child is born with two variant genes (at risk for the disease)
In North America, it is estimated that 1 in 100,000 people are affected by Laurence-Moon-Biedl syndrome 7. In Europe, it is estimated that 1 in 125,000 to 1 in 160,000 people are affected by Laurence-Moon-Biedl syndrome 3. Kuwait and Newfoundland (off the east coast of Canada) are two places where the number of people affects with Laurence-Moon-Biedl syndrome are comparatively high affecting about 1:13,500 newborns and 1:17,500, respectively 22, 23. Consanguinity (union between two people who are related as second cousins or closer) is commonly practiced in various Middle Eastern countries, such as Kuwait, Saudi Arabia, Iran, and Pakistan 23. Consanguinity has been a chief contributing factor to Laurence-Moon-Biedl syndrome frequency 21. Families in these countries have approximately 0.1% homozygous genes of the total genome 23. In Pakistan, more than half of all marriages are consanguineous in nature, with 80% of them among first cousins, thereby increasing the possibility of homozygous mutations 23.
Figure 4. Laurence-Moon-Biedl syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://findageneticcounselor.nsgc.org) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://www.acmg.net/ACMG/Directories.aspx) has a searchable database of medical genetics clinic services in the United States.
Laurence-Moon-Biedl syndrome symptoms
The primary features of Laurence-Moon-Biedl syndrome include chorioretinal dystrophy primarily affecting the choroid (a layer of blood vessels between the retina and the sclera) and the retina (a light-sensitive layer of tissue at the back of your eyes that converts images into electrical signals for your brain) resulting in poor vision such as peripheral vision loss and vision loss at night, shortsightedness and abnormal eye movements with involuntary side-to-side movements of the eyes called nystagmus, extra number of fingers or toes (polydactyly), truncal obesity, hypogonadism (a condition where the body’s sex glands [ovaries in women or testicles in men], produce little to no sex hormones), kidney abnormalities, and mental retardation or learning disabilities. Laurence-Moon-Biedl syndrome may also present with other secondary abnormalities, including speech disorders, developmental delay, ataxia, poor coordination/clumsiness, diabetes insipidus, polyuria/polydipsia, diabetes mellitus, left ventricular hypertrophy, liver fibrosis, spasticity, and hearing loss and crowding of teeth 28. People with Laurence-Moon-Biedl syndrome often also demonstrate a tendency to short stature, hypermobile or lax joints, and early osteoarthritis 29, 7, 30. Less often, fingers and toes may be abnormally short in length. This finding is called a “brachydactyly” and can be especially expected to affect the thumb 7. The thumb may additionally be placed subtly closer to the wrist than expected. Finally, the feet may also be overall short in length, of wide width, and carry a flat arch 7.
In regard to skeletal changes, patients with Laurence-Moon-Biedl syndrome may notice or be told that they have some slight changes in the basic shape of their teeth. Teeth are made up of two major segments, the body and roots. Patients with Laurence-Moon-Biedl syndrome may experience taurodontism, in which the development of tooth’s body is enlarged relative to the roots. Most commonly, it is the flat, molar teeth at the back of the mouth that are affected. Most commonly, taurodontism is noted at the time of first dental x-rays, where the teeth will appear more rectangular than expected.
People with Laurence-Moon-Biedl syndrome are also often burdened by problems with the coordination of their body’s movements. Many patients report a significant degree of clumsiness and often walk with legs in a wide-based stance. Walking heel-to-toe may be difficult. Such impairment is specifically the result of problems with the cerebellum, the sub-section of the brain responsible for coordination. The dysfunction of the cerebellum can lead to dysfunction of the spinal nerve conduction pathways that communicate signals between the brain and muscles. This results in a complex constellation of movement irregularities. The term ataxia is used to describe this loss of control over coordinated bodily movements, and can make it difficult to speak, eat, walk, and maintain balance. Ataxia is accompanied by spasticity, a continuous contraction of muscles in an involuntary manner. The term contracture is used to describe a hardening and shortening of muscles and surrounding connective tissue.
People with Laurence-Moon-Biedl syndrome also often experience other problems related to functions controlled by the brain.
Most patients with Laurence-Moon-Biedl syndrome will experience a gradual loss of vision. Laurence-Moon-Biedl syndrome patients typically arrive in their first or second decade of life with visual acuity impairment that progresses to blindness at a young age 30. The term “retinitis pigmentosa”, a rod-cone variant of progressive pigmentary retinal degeneration, is used to describe the particular, gradual-onset, vision loss that progresses according to a particular pattern 14. Retinitis pigmentosa begins with a night blindness that worsens with a loss of the ability to distinguish colors from one another, finally deteriorating into “tunnel vision”.
Mild-to-moderate learning difficulties are common in individuals with Laurence-Moon-Biedl syndrome. Often, learning disabilities are attributed to weakened cognitive capacity. Some individuals affected with Laurence-Moon-Biedl syndrome may have true learning disabilities due to dysfunction of brain development. However, it is important to be sure that suspected disabilities (such as delayed speech or reading skills) are not due to underlying visual impairment. If the learning disability is rooted in neurological impairments, they are often associated with symptoms of poor coordination, gross and fine motor skills, and social milestones in childhood such as inability to play complicated games with other children.
People living with Laurence-Moon-Biedl syndrome have been found to have smaller than average size anterior pituitary glands and can suffer from a series of different complications as a result. The anterior portion of the pituitary gland is responsible for regulating many functions including the body’s metabolism, emotional responses to stressors, physical bodily growth, and reproductive capacity. The body’s metabolism is controlled by thyroid stimulating hormone (TSH). With low levels of thyroid stimulating hormone (TSH), people will experience hypothyroidism (underactive thyroid) many underactive thyroid symptoms such as fatigue, sensitivity to cold, poor ability to concentrate, weight gain, constipation, shortness of breath. The body will potentially change its common behaviors. For example, skin will become dry and course, hair will fall out, reflexes will slow.
People with Laurence-Moon-Biedl syndrome often have decreased levels of the sex hormones estrogen and testosterone. This is also due to the small size of the pituitary gland, a small gland located in the brain that is responsible for producing the chemical signals that orchestrate the production of sex hormones. Hypogonadism (a condition where the body’s sex glands [ovaries in women or testicles in men], produce little to no sex hormones) is a noteworthy clinical feature that is more commonly diagnosed at an early age in males due to evident micropenis and small testes 1. As a result of a weak signal to produce estrogen and testosterone, the reproductive organs of both men and women living with Laurence-Moon-Biedl syndrome may be underdeveloped, resulting in reduced fertility or even infertility. In females, the uterus, fallopian tubes, and ovaries are often underdeveloped, along with menstrual abnormalities, contribute to the reproductive abnormalities of Laurence-Moon-Biedl syndrome 1. Menstruation cycles can be delayed from the average first age of onset and when they do begin, may follow an irregular cycle. Males with Laurence-Moon-Biedl syndrome may have an underdeveloped set of testicles that may be undescended. However, delayed puberty remains usual in both boys and girls 1.
Cognitive disorders and kidney abnormalities are also a frequent clinical manifestation. Laurence-Moon-Biedl syndrome patients are often confused, along with impeded memory, poor judgment skills, uncoordinated, and clumsy motor movements 1. While kidney impairment remains the major cause of mortality since the end-stage renal disease (ESRD) also known as kidney failure is a frequent complication in such patients 1.
Other less common features that have been reported in patients living with Laurence-Moon-Biedl syndrome include a skull shape shorter than average, termed “brachycephaly” and electrical abnormalities of the heart. People with Laurence-Moon-Biedl syndrome may also experience a loss of hearing, increased incidence of diabetes, and urinary and genital structural malformations.
Laurence-Moon-Biedl syndrome diagnosis
The presence of four primary features on their own or three primary coupled with two secondary features are the clinical grounds for making a Laurence-Moon-Biedl syndrome diagnosis 23, 31, 32.
Laurence-Moon-Biedl syndrome primary set of symptoms 23, 1:
- Cone-rod dystrophy (retinitis pigmentosa or photoreceptors disease),
- Polydactyly,
- Obesity,
- Learning disabilities,
- Hypogonadism,
- Kidney abnormalities
Laurence-Moon-Biedl syndrome secondary features 23, 33:
- Speech disorders,
- Brachydactyly,
- Developmental delay,
- Polyuria/polydipsia,
- Ataxia,
- Poor coordination/clumsiness,
- Diabetes mellitus,
- Left ventricular hypertrophy,
- Liver fibrosis,
- Spasticity,
- Hearing loss.
Apart from these features, short stature, crowding of teeth, hypermobile or lax joints, and early osteoarthritis are also reported 30.
Forsythe and Beales 34 stipulated that the existence of either at least 4 primary characteristics or 3 primary and 2 secondary features, is sufficient as having Laurence-Moon-Biedl syndrome. However, confusion still exists between Lawrence-Moon syndrome (LMS) and Bardet-Biedl syndrome (BBS) 35. Pigmentary retinal degeneration, mental retardation, and hypogonadism are common in both, whereas spastic paraplegia is predominant in Laurence-Moon-Biedl syndrome and polydactyly and obesity are seen in Bardet-Biedl syndrome (BBS). Because of some common features, some researchers believe Bardet-Biedl syndrome (BBS) to be a part of Lawrence-Moon syndrome (LMS) 14, 22, 36.
Due to the highly variable clinical presentation of Laurence-Moon-Biedl syndrome, no formal diagnostic criteria have been established for Laurence-Moon-Biedl syndrome or, for that matter, other PNPLA6-related disorders. Laurence-Moon-Biedl syndrome is typically identified during assessments of developmental delays. A definitive diagnosis of Laurence-Moon-Biedl syndrome is made by molecular testing for mutations in the PNPLA6 gene 7, 24.
Laurence-Moon-Biedl syndrome differential diagnosis
The following conditions have notably been considered in the differential diagnosis for patients with Laurence-Moon-Biedl syndrome given the similar presentation of symptoms 37, 38. Without genetic testing, it can be very difficult to clinically differentiate these conditions.
Bardet-Biedl Syndrome (BBS)
Bardet-Biedl syndrome (BBS) is a complex genetic disorder that affects many parts of the body including the retina 39, 40. Individuals with Bardet-Biedl syndrome (BBS) have a retinal degeneration similar to retinitis pigmentosa (RP). The retina is a light-sensitive layer of tissue at the back of your eyes that converts images into electrical signals for your brain. Rod and cone photoreceptors in the retina convert light into electrical signals that the brain interprets as vision. People with Bardet-Biedl syndrome (BBS) with retinal degeneration that is characterized by rod-cone dystrophy experience a gradual decline in their vision, because the photoreceptors (specialized cells in the retina that convert light into electrical signals that the brain can use for vision) degenerate. People with Bardet-Biedl syndrome (BBS) also have at least three additional non-eye features such as intellectual disability, obesity, polydactyly (a condition where a person has more than the normal number of fingers or toes), hypogonadism (a condition where the body’s sex glands [ovaries in women or testicles in men], produce little to no sex hormones), or kidney abnormalities as primary signs and symptoms 39, 40.
Vision loss is one of the major features of Bardet-Biedl syndrome 39, 40. Loss of vision occurs as the light-sensing tissue at the back of the eye (the retina) gradually deteriorates. Problems with night vision become apparent by mid-childhood, followed by blind spots that develop in the side (peripheral) vision. Over time, these blind spots enlarge and merge to produce tunnel vision. Most people with Bardet-Biedl syndrome also develop blurred central vision (poor visual acuity) and become legally blind by adolescence or early adulthood.
Obesity is another characteristic feature of Bardet-Biedl syndrome 39, 40. Abnormal weight gain typically begins in early childhood and continues to be an issue throughout life. Complications of obesity can include type 2 diabetes, high blood pressure (hypertension), and abnormally high cholesterol levels (hypercholesterolemia).
Other major signs and symptoms of Bardet-Biedl syndrome include the presence of extra fingers or toes (polydactyly), intellectual disability or learning problems, and abnormalities of the genitalia. Most affected males produce reduced amounts of sex hormones (hypogonadism), and they are usually unable to father biological children (infertile) 39, 40. Many people with Bardet-Biedl syndrome also have kidney abnormalities, which can be serious or life-threatening 39, 40.
In the absence of one of these 4 primary clinical features (i.e., vision loss, intellectual disability, obesity, polydactyly), the diagnosis of Bardet-Biedl syndrome (BBS) is made when at least two secondary features are observed, including hepatic fibrosis, diabetes mellitus, reproductive and developmental abnormalities, growth retardation, speech delays, or cardiovascular problems 39, 40.
Additional features of Bardet-Biedl syndrome can include impaired speech, delayed development of motor skills such as standing and walking, behavioral problems such as emotional immaturity and inappropriate outbursts, and clumsiness or poor coordination 39, 40. Distinctive facial features, dental abnormalities, unusually short or fused fingers or toes, and a partial or complete loss of the sense of smell (anosmia) have also been reported in some people with Bardet-Biedl syndrome 39, 40. Additionally, Bardet-Biedl syndrome can affect the heart, liver, and digestive system.
Bardet-Biedl syndrome can result from mutations in at least 22 different BBS genes 39, 41. The proteins produced from BBS genes are are known or suspected to play critical roles in the maintenance and function of cilia. Cilia are microscopic, finger-like projections that stick out from the surface of many types of cells. Cilia are involved in cell movement and many different chemical signaling pathways. Cilia are also necessary for the perception of sensory input such as sight, hearing, and smell.
Mutations in BBS genes lead to problems with the structure and function of cilia. Defects in these cell structures probably disrupt important chemical signaling pathways during development and lead to abnormalities of sensory perception. Researchers believe that defective cilia are responsible for most of the features of Bardet-Biedl syndrome.
About one-quarter of all cases of Bardet-Biedl syndrome result from mutations in the BBS1 gene. Another 20 percent of cases are caused by mutations in the BBS10 gene. The other BBS genes each account for only a small percentage of all cases of Bardet-Biedl syndrome. In about 25 percent of people with Bardet-Biedl syndrome, the cause of the disorder is unknown.
In individuals with Bardet-Biedl syndrome who have mutations in one of the BBS genes, mutations in additional genes may be involved in causing or modifying the course of the disorder. Studies suggest that these modifying genes may be known BBS genes or other genes. The additional genetic changes could help explain the variability in the signs and symptoms of Bardet-Biedl syndrome. However, this phenomenon appears to be uncommon, and it has not been found consistently in scientific studies.
Boucher-Neuhauser Syndrome (BNS)
Boucher-Neuhauser Syndrome (BNS) is a rare genetic condition that is characterized by 3 specific clinical features that include ataxia (cerebellar ataxia), chorioretinal dystrophy and hypogonadotropic hypogonadism (poor sexual development resulting from poor pituitary gland function) 42. Ataxia describes difficulty with coordination and balance. In Boucher-Neuhäuser syndrome, it arises from a loss of cells (atrophy) in the part of the brain involved in coordinating movements called the cerebellum 42. Affected individuals have an unsteady walking style (gait) and frequent falls. Another key feature of Boucher-Neuhäuser syndrome is hypogonadotropic hypogonadism, which is a condition affecting the production of hormones that direct sexual development 42. Affected individuals have a delay in development of the typical signs of puberty, such as the growth of facial hair and deepening of the voice in males, and the start of monthly periods (menstruation) and breast development in females. Other hormone abnormalities lead to short stature in some affected individuals. The third characteristic feature of Boucher-Neuhäuser syndrome is eye abnormalities, most commonly chorioretinal dystrophy 42. Chorioretinal dystrophy refers to problems with the light-sensitive tissue that lines the back of the eye (the retina) and a nearby tissue layer called the choroid 42. These eye abnormalities lead to impaired vision. People with Boucher-Neuhäuser syndrome (BNS) can also have abnormal eye movements, including involuntary side-to-side movements of the eyes called nystagmus. People with Boucher-Neuhäuser syndrome (BNS) can have additional medical problems, including muscle stiffness (spasticity); impaired speech (dysarthria); developmental delays and difficulty processing, learning, or remembering information (cognitive impairment).
The key features of Boucher-Neuhäuser syndrome can begin anytime from infancy to adulthood, although at least one feature usually occurs by adolescence. Ataxia is often the initial symptom of the disorder, but vision problems or delayed puberty can be the earliest finding. Vision and movement problems worsen slowly throughout life and can result in blindness or the need for a wheelchair for mobility in the most severely affected individuals.
Most cases of Boucher-Neuhäuser syndrome (BNS) are caused by mutations in the PNPLA6 gene and another gene called RNF216. RNF216 gene is responsible for the tagging of cell proteins destined for degradation. The protein produced from the RNF216 gene is involved in a cellular process, called ubiquitination, by which unneeded proteins are tagged with a molecule called ubiquitin. The ubiquitin tag signals for the protein to be broken down. One of several proteins tagged by RNF216 is a protein found in nerve cells (neurons) that plays a role in a process called synaptic plasticity. Synaptic plasticity is the ability of the connections between neurons (synapses) to change and adapt over time in response to experience. This process is critical for learning and memory.
Researchers speculate that as-yet-unidentified mutations in the PNPLA6 gene or changes in other genes are involved in the remainder of cases.
The PNPLA6 gene provides instructions for making a protein called neuropathy target esterase (NTE), which helps regulate the amount of certain fats (lipids) that make up the outer membrane surrounding cells. The correct levels of these lipids are critical to the stability and function of cell membranes. In particular, the NTE protein breaks down (metabolizes) a lipid called lysophosphatidylcholine, which in high amounts can damage cells. NTE is found most abundantly in the nervous system and is thought to help maintain the stability of membranes surrounding nerve cells (neurons). NTE is also thought to play a role in the release of hormones from the pituitary gland, a process that requires particular changes in the cell membrane. The pituitary gland is located at the base of the brain and produces several hormones, including those that help direct sexual development and growth.
PNPLA6 gene mutations are thought to impair NTE’s function. However, it is unclear how these mutations cause Boucher-Neuhäuser syndrome. Researchers speculate that impairment of lysophosphatidylcholine metabolism alters the balance of lipids in the cell membrane. This imbalance may damage neurons, leading to the movement and vision problems that characterize Boucher-Neuhäuser syndrome. The imbalance may also impair the release of hormones involved in sexual development, accounting for the delayed puberty in affected individuals.
Gordon-Holmes Syndrome (GHS)
Gordon-Holmes Syndrome (GHS) is a rare genetic condition characterized by ataxic movement abnormalities with hypogonadotropic hypogonadism 43. One of the key features of Gordon Holmes syndrome (GHS) is reduced production of hormones that direct sexual development (hypogonadotropic hypogonadism) 43. Many affected individuals have a delay in development of the typical signs of puberty, such as the growth of facial hair and deepening of the voice in males, and the start of monthly periods (menstruation) and breast development in females. Some never undergo puberty. While some people with Gordon Holmes syndrome seem to have normal puberty, they develop other problems with the reproductive system later in life 43.
In early adulthood, people with Gordon-Holmes syndrome (GHS) develop neurological problems usually beginning with speech difficulties (dysarthria) 43. A subset of patients with Gordon-Holmes syndrome (GHS) may demonstrate exaggerated, brisk reflexes. As the condition worsens, affected individuals have problems with balance and coordination (cerebellar ataxia), often leading to difficulties with activities of daily living and a need for wheelchair assistance. Some affected individuals also develop memory problems and a decline in intellectual function (dementia).
Gordon Holmes syndrome can be caused by mutations in the RNF216 or PNPLA6 gene 43. Some people with Gordon-Holmes syndrome (GHS) do not have mutations in these genes, indicating that mutations in other genes are likely involved in Gordon Holmes syndrome 43. The protein produced from the RNF216 gene is involved in a cellular process, called ubiquitination, by which unneeded proteins are tagged with a molecule called ubiquitin. The ubiquitin tag signals for the protein to be broken down. One of several proteins tagged by RNF216 is a protein found in nerve cells (neurons) that plays a role in a process called synaptic plasticity. Synaptic plasticity is the ability of the connections between neurons (synapses) to change and adapt over time in response to experience. This process is critical for learning and memory.
RNF216 gene mutations impair the ability of the RNF216 protein to tag unneeded proteins to be broken down. Impaired breakdown of the neuronal protein disrupts normal synaptic connections and plasticity, which likely contributes to dementia in people with Gordon Holmes syndrome. It is unclear how a lack of RNF216 protein function causes hypogonadotropic hypogonadism or cerebellar ataxia.
The PNPLA6 gene provides instructions for making a protein called neuropathy target esterase (NTE), which helps regulate the amount of certain fats (lipids) that make up the outer membrane surrounding cells. The correct levels of these lipids are critical to the stability and function of cell membranes. NTE is found most abundantly in the nervous system and is thought to help maintain the stability of membranes surrounding neurons. NTE is also thought to play a role in the release of hormones from the pituitary gland, a process that requires particular changes in the cell membrane. The pituitary gland is located at the base of the brain and produces several hormones, including those that help direct sexual development and growth.
PNPLA6 gene mutations are thought to impair NTE’s function. Researchers speculate that such an impairment alters the balance of lipids in the cell membrane. This imbalance may damage neurons in the brain, causing cerebellar ataxia, and impair the pituitary gland’s release of hormones involved in sexual development, leading to hypogonadotropic hypogonadism. Individuals with Gordon Holmes syndrome caused by PNPLA6 gene mutations do not appear to develop dementia.
Joubert syndrome
Joubert syndrome is a rare severe genetic disorder in which children do not live beyond three-years of age 44. The hallmark feature of Joubert syndrome is a combination of brain abnormalities that together are known as the molar tooth sign, which can be seen on brain imaging studies such as magnetic resonance imaging (MRI) 44. The molar tooth sign in the brain MRI scan results from the abnormal development of structures near the back of the brain, including the cerebellar vermis and the brainstem 44. The molar tooth sign got its name because the characteristic brain abnormalities resemble the cross-section of a molar tooth when seen on an MRI 44. Most babies with Joubert syndrome have low muscle tone (hypotonia) in infancy, which contributes to difficulty coordinating movements (ataxia) in early childhood. Other characteristic features of Joubert syndrome include episodes of unusually fast (hyperpnea) or slow (apnea) breathing in infancy, and abnormal eye movements (ocular motor apraxia) 44. Most affected individuals have delayed development and intellectual disability, which can range from mild to severe 44. Distinctive facial features can also occur in Joubert syndrome; these include a broad forehead, arched eyebrows, droopy eyelids (ptosis), widely spaced eyes (hypertelorism), low-set ears, and a triangle-shaped mouth 44.
Joubert syndrome can also include a broad range of additional signs and symptoms. Joubert syndrome is sometimes associated with other eye abnormalities such as retinal dystrophy, which can cause vision loss, and coloboma, which is a gap or split in a structure of the eye, kidney disease including polycystic kidney disease and nephronophthisis, liver disease, skeletal abnormalities such as the presence of extra fingers and toes, or hormone (endocrine) problems 44. A combination of the characteristic features of Joubert syndrome and one or more of these additional signs and symptoms once characterized several separate disorders. Together, those disorders were referred to as Joubert syndrome and related disorders (JSRD) 44. Now, however, any instances that involve the molar tooth sign on MRI scan, including those with these additional signs and symptoms, are usually considered Joubert syndrome 44.
Joubert syndrome can be caused by mutations in more than 30 genes 44. The proteins produced from these genes are known or suspected to play roles in cell structures called primary cilia. Primary cilia are microscopic, finger-like projections that stick out from the surface of cells and are involved in sensing the physical environment and in chemical signaling. Primary cilia are important for the structure and function of many types of cells, including brain cells (neurons) and certain cells in the kidneys and liver. Primary cilia are also necessary for the perception of sensory input, which is interpreted by the brain for sight, hearing, and smell.
Mutations in the genes associated with Joubert syndrome lead to problems with the structure and function of primary cilia. Defects in these cell structures can disrupt important chemical signaling pathways during development. Although researchers believe that defective primary cilia are responsible for most of the features of these disorders, it is not completely understood how they lead to specific developmental abnormalities.
Mutations in the genes known to be associated with Joubert syndrome account for about 60 to 90 percent of all cases of this condition. In the remaining cases, the genetic cause is unknown 44.
McKusick-Kaufman Syndrome (MKKS)
McKusick-Kaufman Syndrome (MKKS or MKS) also called hydrometrocolpos-polydactyly syndrome is a very rare, autosomal recessive developmental disorder presenting in the neonatal period that affects the development of the hands, feet, heart, and reproductive system. McKusick-Kaufman syndrome (MKKS) is characterized by a combination of 3 clinical features: extra fingers and/or toes (polydactyly), congenital heart defects and genital abnormalities 45, 46, 47, 48, 49, 50. The most common genital abnormality and the hallmark of McKusick-Kaufman Syndrome (MKKS) in female patients is hydrometrocolpos, an accumulation of fluid in the vagina and uterus as a result of the accumulation of cervical secretions from maternal estrogen stimulation, due to a blocked vaginal outlet 46. The blockage allows fluid to build up in the vagina and uterus, stretching these organs and leading to a fluid-filled mass. Hydrometrocolpos can be caused by a number of factors, including vaginal agenesis (failure of the distal third of the vagina to develop), vaginal atresia (a rare developmental defect where the lower part of the vagina doesn’t form properly), an imperforate hymen, persistent urogenital sinus, cloacal malformation, and transverse vaginal membrane 45. McKusick-Kaufman syndrome (MKKS) female patients may have an absent vagina with mucoid accumulations within an intact uterus within the abdomen 45, 46. Female with McKusick-Kaufman Syndrome (MKKS) may alternatively have a double vaginal or uterine structures. Hydrometrocolpos obstructs adjacent structures either in prenatal or postnatal life 48. In a fetus, urinary bladder obstruction may lead to proximal urinary tract dilation that results in oligohydramnios malformation sequence and diaphragm compression, which ultimately results in lung hypoplasia 48. In utero rectal compression, intestinal obstruction can rarely lead to perforation and peritonitis 51, 52. This usually presents as Hirschsprung’s disease (a rare birth defect that occurs when nerves in the intestine don’t develop properly) 53, 54. Inferior vena cava compression may result in hydrops fetalis and pedal edema 54. After birth, bladder displacement results in micturition defects which can cause recurrent pyelonephritis and chronic renal failure 52. Older children or teenagers present with menstrual pain, primary amenorrhea, constipation, lower back pain, or urinary retention 55.
McKusick-Kaufman syndrome (MKKS) in males can include undescended testes (cryptorchidism), abnormal opening of the urethra on the underside of the penis (hypospadias) and a downward-curving penis (chordee) 45, 46. Either sex may have fibrosis or degeneration of their ureters, the conduit for urine formed by the kidneys destined for storage in the bladder 45.
Polydactyly (extra fingers or toes) or brachydactyly (fingers and toes that are abnormally short in length) is present in 90% of McKusick-Kaufman syndrome (MKKS) cases 47. In people with McKusick-Kaufman syndrome, the extra fingers and toes are typically on the same side of the hand or foot as the pinky or little toe also known as postaxial polydactyly 45. The congenital heart defects in individuals with McKusick-Kaufman syndrome can include an atrial septal defect (ASD) or a ventricular septal defect (VSD), which are openings in the wall (septum) that separates the upper or lower chambers of the heart or a complex congenital heart malformation 45.
The signs and symptoms of McKusick-Kaufman syndrome (MKS) overlap significantly with those of another genetic disorder, Bardet-Biedl syndrome (BBS). However, Bardet-Biedl syndrome (BBS) has several features that are not typically seen in people with McKusick-Kaufman syndrome (MKKS). These include a gradual loss of vision, developmental disabilities, kidney abnormalities, and obesity 45. Because some of these features are not apparent at birth, the two conditions can be difficult to tell apart in infancy and early childhood.
Both McKusick-Kaufman syndrome and Bardet-Biedl syndrome (BBS) belong to a group of conditions called ciliopathies. Ciliopathies are inherited disorders that affect the structure or function of cilia, the microscopic, finger-like projections found on the surface of cells. Cilia are involved in signaling pathways that transmit information between cells.
Mutations in the MKKS gene also called the BBS6 gene have been found to cause McKusick-Kaufman syndrome 45. The MKKS gene (BBS6 gene) provides instructions for making a protein that plays an important role in early development, specifically in the formation of the limbs, heart, and reproductive system. The protein’s structure suggests that it may belong to a family of proteins called chaperonins. Proteins must be folded into the correct shape to function properly, and chaperonins help them do that.
The MKKS protein is thought to play a role in cell division and cell transport. Specifically, the MKKS protein is thought to transport molecules between the cytoplasm and the nucleus of the cell. The MKKS protein also combines with other proteins to form a complex within the cell. This complex is used to assemble the molecules involved in transporting materials that support the function of cilia.
Though it is not clear exactly how mutations in the MKKS gene (BBS6 gene) lead to the specific signs and symptoms of McKusick-Kaufman syndrome, two particular variants in the MKKS gene (BBS6 gene) appear to impair the protein’s ability to transport a molecule involved in regulating gene activity. This change likely affects the activity of certain genes that are critical during early development.
Once the diagnosis of McKusick-Kaufman syndrome (MKS) is confirmed, immediate decompression of the dilated uterus in female babies improves the prognosis for kidney function and lung capacities 47. Surgical intervention should be performed early to stop infections in the urinary tract. Molecular genetic analysis may be useful for treating and counseling females with hydrometrocolpos 56.
Meckel-Gruber syndrome
Meckel-Gruber syndrome also known as Meckel syndrome is rare genetic disorder that affects the structure and function of cilia (ciliopathy) that affect many parts of the body like Bardet-Biedl syndrome (BBS) 57. Meckel-Gruber syndrome signs and symptoms include retinal degeneration, extra fingers and toes (polydactyly), kidney cysts (enlarged kidneys with numerous fluid-filled cysts), occipital encephalocele which is a sac-like protrusion of the brain through an opening at the back of the skull and buildup of scar tissue (fibrosis) in the liver as well as poor lung function resulting from low amniotic fluid volumes within the placenta during the pregnancy 57.
Other signs and symptoms of Meckel-Gruber syndrome vary widely among affected individuals 57. Numerous abnormalities of the brain and spinal cord (central nervous system) have been reported in people with Meckel-Gruber syndrome, including a group of birth defects known as neural tube defects. These defects occur when a structure called the neural tube, a layer of cells that ultimately develops into the brain and spinal cord, fails to close completely during the first few weeks of embryonic development 57. Meckel-Gruber syndrome can also cause problems with development of the eyes and other facial features, heart, bones, urinary system, and genitalia 57.
Because of their serious health problems, most individuals with Meckel-Gruber syndrome die before or shortly after birth 57. Most often, affected infants die of respiratory problems or kidney failure.
Meckel syndrome can be caused by changes (mutations) in thirteen genes: B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRIP1L, TCTN2, TCTN3, TMEM67, TMEM107, TMEM216, TMEM231 and TMEM237 58. Mutations in these 13 genes account for 75 percent of all Meckel-Gruber syndrome cases; the remaining 25 percent have unknown genetic causes 58. Most of these genes are also responsible for a neurological disorder called Joubert syndrome, leading to the concept that Meckel syndrome is the extreme lethal form of Joubert syndrome 58. Mutations in several other genes have been identified in people with features similar to those of Meckel-Gruber syndrome, although it is unclear whether these individuals actually have Meckel-Gruber syndrome or a related disorder often described as a “Meckel-like phenotype” 57.
The proteins produced by these genes are known to influence cell structures or function called cilia. Cilia are microscopic, finger-like projections that stick out from the surface of cells and are involved in signaling pathways that transmit information between cells. Cilia are important for the structure and function of many types of cells, especially in the kidneys, liver, eyes and brain. Mutations in these gene cause problems in the function of the primary cilia, resulting in various defects dependent of the cell type. Early defective ciliary function can be responsible for developmental abnormalities, specifically in the kidneys, brain, limbs, heart.
Mutations in the genes associated with Meckel-Gruber syndrome lead to problems with the structure and function of cilia. Defects in these cell structures probably disrupt important chemical signaling pathways during early development. Although researchers believe that defective cilia are responsible for most of the features of Meckel syndrome, it remains unclear how they lead to specific developmental abnormalities of the brain, kidneys, and other parts of the body.
Oliver-McFarlane Syndrome (OMCS)
Oliver-McFarlane Syndrome (OMCS) is a rare genetic disorder that is characterized by congenital trichomegaly, pigmentary retinal degeneration (chorioretinal dystrophy), short stature, intellectual disability, and multiple pituitary hormone deficiencies (growth hormone (GH), gonadotrophins and thyroid stimulating hormone (TSH) deficiencies) 59, 60, 5, 61, 62, 9, 63, 64, 65, 66, 67. Thyroid and growth hormones deficiencies may be present at birth and, if untreated, result in intellectual impairment and profound short stature. Congenital hypogonadism occurs in half of patients, and nearly all have documented hypogonadotropic hypogonadism during puberty, with subsequent reproductive dysfunction. Chorioretinal atrophy is typically noted in the first five years of life. Half of reported cases have spinocerebellar involvement, including ataxia, spastic paraplegia and peripheral neuropathy.
Oliver-McFarlane syndrome (OMCS) also causes a number of physical features including a large frontal skull, prominent chin, mid-front scalp hair loss, bushy eyebrows, and long eyelashes.
Oliver-McFarlane syndrome (OMCS) signs and symptoms include 5:
- Hair abnormalities: Long eyelashes and eyebrows, sparse scalp hair
- Eye problems: Severe chorioretinal atrophy, pigmentary retinal degeneration, retinopathy
- Growth issues: Short stature, early growth retardation, bone age that is less than actual age
- Hormonal deficiencies: Pituitary hormone deficiencies, including growth hormone, gonadotropins, and thyroid-stimulating hormone
- Neurological issues: Ataxia, spastic paraplegia, peripheral neuropathy
- Intellectual disability: If left untreated, OMCS can lead to intellectual impairment
Oliver-McFarlane Syndrome (OMCS) is associated with mutations in the PNPLA6 gene. Fourteen cases have been reported to date, including two sibships, suggesting autosomal-recessive inheritance 68, 64.
Spastic Paraplegia Type 39 (SPG39)
Spastic Paraplegia Type 39 (SPG39) is a rare, progressive, inherited motor neuron disease that describes the association of upper motor neuron involvement and/or cerebellar ataxia, peripheral neuropathy, and/or reduced cognitive capacity 69, 70. Spastic Paraplegia Type 39 (SPG39) is associated with mutations in the PNPLA6 gene. Spastic paraplegia type 39 (SPG39) usually becomes apparent in childhood and early adulthood with muscle wasting (amyotrophy) in the upper and lower extremities, progressive muscle stiffness (spasticity), exaggerated reflexes (hyperreflexia), abnormal gait, axonal motor neuropathy, reduced cognitive functioning, and cerebellar ataxia. MRI imaging may show cerebellar and/or spinal cord atrophy. Treatments for spastic paraplegia include physical therapy, oral antispastic drugs, botulinum toxin therapy, and surgical baclofen pump implantation. Pure hereditary spastic paraplegia usually doesn’t affect life expectancy, and most people can live relatively independent and active lives.
Laurence-Moon-Biedl syndrome treatment
Since there is currently no recognized therapy for Laurence-Moon-Biedl syndrome, early intervention is essential to give children the best chance at a normal life. The treatments available for Laurence-Moon-Biedl syndrome are oriented towards managing the signs, symptoms and complications of the condition 7. People with Laurence-Moon-Biedl syndrome often experience lack of balance and coordination or a disturbance of gait (ataxia), muscle stiffness (spasticity), and contracture (a permanent of muscles, tendons, and other soft tissues that causes a joint to become stiff and limit normal movement) that compromise their ability to move comfortably. Physical therapy aimed towards improving strength and agility is key. Often walking can be assisted by tools such as ankle-foot orthotic braces, weight-bearing walkers, etc. Physical exercise can reduce the symptoms of ataxia, spasticity, and prevent contracture. A dedicated regimen of nutritious, well-balanced meals and regular exercise is recommended to avoid some of the less common but equally severe aspects that have been noted to affect patients with Laurence-Moon-Biedl syndrome. There is most notably, an increased incidence of diabetes and abnormal cholesterol levels in patients with Laurence-Moon-Biedl syndrome.
The underactive anterior pituitary gland that results in slowed metabolism, poor growth, and impaired fertility can be managed with hormone replacement therapies 71. Levothyroxine is a medication that mimics the functions of the thyroid hormone and can aid in speeding up the metabolism of the body, resulting in reduced symptoms of lethargy, hair loss, and obesity. Growth hormone supplementation can be offered to reduce the burden of short stature in patients identified as children.
It is important for patients with Laurence-Moon-Biedl syndrome to be under the care of an eye specialist (ophthalmologist). There is no cure for the vision problems that accompanies Laurence-Moon-Biedl syndrome, but ophthalmologists can help create corrective lenses against developing problems. Individuals with Laurence-Moon-Biedl syndrome should undergo regular eye examinations and keep up with their changing prescriptions. Since visual impairment is a major hurdle to learning in the classroom, special services might be organized on an individual basis between a child’s doctor and their school.
For patients living with Laurence-Moon-Biedl syndrome, it is important to recognize that the many difficulties described above are associated with the brain’s neurological function. It is therefore important to protect baseline function capacity of the brain. Inactivity and obesity exacerbate nerve damage (neuropathy). Alcohol and recreational drugs should be avoided.
Generally, very little information exists to guide women living with rare diseases who are interested in becoming pregnant. Pregnancy is well known to take many physical demands on a woman’s body and women who are living with Laurence-Moon-Biedl syndrome are generally warned that symptoms of ataxia may worsen or even develop for the first time during pregnancy. Pregnant women with Laurence-Moon-Biedl syndrome should be followed closely by obstetricians that are well trained in dealing with high-risk pregnancies.
Patients living with Laurence-Moon-Biedl syndrome experience alterations to the shape of their teeth. Taurodontism (a rare dental condition where the pulp chamber of a tooth enlarges, resulting in a tooth that resembles a bull’s tooth) is generally noted at the dental office at the time of first x-rays. Extra care may be needed in brushing and cleaning all aspects of the affected teeth, and dentists may need special tools to examine all aspects of the affected teeth.
Laurence-Moon-Biedl syndrome prognosis
Laurence-Moon-Biedl syndrome is a very rare genetic condition with multi-organ involvement, which can be serious or life-threatening and have poor prognosis 1. Laurence-Moon-Bardet-Biedl syndrome have a shorter life expectancy than the general population and kidney abnormalities are the most common cause of death particularly in the third and fourth decades of life 72, 73. Managing kidney issues and other complications can improve quality of life and life expectancy 72. Kidney problems are the primary cause of mortality, with life expectancy often being lower than that of the general population. Therefore, early kidney screenings and regular kidney function evaluations and urinalysis form part of the necessary follow-up protocol 74.
Patients with Laurence-Moon-Biedl syndrome frequently experience kidney issues in addition to the five primary symptoms of this illness. Blood pressure control and frequent testing need to be stressed since there is a high risk of progression to end-stage renal failure (kidney failure), especially in the third and fourth decades of life 72. In a recent study involving a cohort of 54 Bardet-Biedl syndrome (BBS) individuals, it was shown that urine-concentrating defects may predict the progression of kidney insufficiency 75.
- Kumar A, Husain A Sr, Saleem A, Khawaja UA, Virani S. Laurence-Moon-Bardet-Biedl Syndrome: A Rare Case With a Literature Review. Cureus. 2020 Nov 5;12(11):e11355. doi: 10.7759/cureus.11355[↩][↩][↩][↩][↩][↩][↩][↩]
- Pandya M, Daigavane S. A Rare Presentation of Laurence-Moon-Bardet-Biedl Syndrome: Atypical Retinitis Punctata Albescens and Non-alcoholic Fatty Liver Disease. Cureus. 2024 Feb 12;16(2):e54064. doi: 10.7759/cureus.54064[↩]
- Khan OA, Majeed R, Saad M, Khan A, Ghassan A. Rarity of Laurence Moon Bardet Biedl Syndrome and its Poor Management in the Pakistani Population. Cureus. 2019 Feb 21;11(2):e4114. doi: 10.7759/cureus.4114[↩][↩][↩][↩][↩][↩]
- Al Fareh N, Abbas M, Alosaimi FD. Psychosis in Laurence-Moon Syndrome: A Case Report. Cureus. 2024 Oct 21;16(10):e72003. doi: 10.7759/cureus.72003[↩]
- Hufnagel RB, Arno G, Hein ND, Hersheson J, Prasad M, Anderson Y, Krueger LA, Gregory LC, Stoetzel C, Jaworek TJ, Hull S, Li A, Plagnol V, Willen CM, Morgan TM, Prows CA, Hegde RS, Riazuddin S, Grabowski GA, Richardson RJ, Dieterich K, Huang T, Revesz T, Martinez-Barbera JP, Sisk RA, Jefferies C, Houlden H, Dattani MT, Fink JK, Dollfus H, Moore AT, Ahmed ZM. Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes. J Med Genet. 2015 Feb;52(2):85-94. doi: 10.1136/jmedgenet-2014-102856[↩][↩][↩][↩][↩]
- The endocrine and metabolic characteristics of a large Bardet-Biedl syndrome clinic population. Mujahid S, Hunt KF, Cheah YS, et al. J Clin Endocrinol Metab. 2018;103:1834–1841. doi: 10.1210/jc.2017-01459[↩]
- Laurence-Moon Syndrome. https://rarediseases.org/rare-diseases/laurence-moon-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Indraneel KS, VRajalakshmi, Dayanandan Y, Reddy NM. A Case of Laurence Moon Bardet Biedl Syndrome. J Assoc Physicians India. 2023 Jan;71(1):1.[↩]
- Laurence JZ, Moon RC. Four cases of “retinitis pigmentosa” occurring in the same family, and accompanied by general imperfections of development. 1866. Obes Res. 1995 Jul;3(4):400-3. doi: 10.1002/j.1550-8528.1995.tb00166.x[↩][↩][↩]
- Bardet G. Sur un syndrome d’obesite congenitale avec polydactylie et retinite pigmentaire (contribution a l’etude des formes cliniques de l’obesite hypophysaire) These de Paris (Le Grand) 1920;470:107.[↩]
- Bardet G. On congenital obesity syndrome with polydactyly and retinitis pigmentosa (a contribution to the study of clinical forms of hypophyseal obesity. Obes Res. 1995;3:387–399. doi: 10.1002/j.1550-8528.1995.tb00165.x[↩]
- Biedl A. Ein Geschwister mit adiposogenitaler Dystrophie. Dtsch Med Wochenschr. 1922;48:1630.[↩]
- Biedl A. A pair of siblings with adiposo-genital dystrophy. Obes Res. 1995;3:404. doi: 10.1002/j.1550-8528.1995.tb00167.x[↩]
- Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, Heath O, McManamon PJ, O’Leary E, Pryse-Phillips W. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989 Oct 12;321(15):1002-9. doi: 10.1056/NEJM198910123211503[↩][↩][↩]
- Solis-Cohen S, Weiss E. Dystrophia adiposogenitalis, with atypical retinitis pigmentosa and mental deficiency—The Laurence–Biedl syndrome. A report of four cases in one family. Am J Med Sci. 1925;169:489–505.[↩]
- Klein D, Amman F. The syndrome of Laurence-Moon-Bardet-Biedl and allied disease in Switzerland. Clinical, genetic and epidemiological studies. J Neurol Sci. 1969;9:479–513. doi: 10.1016/0022-510x(69)90091-4[↩]
- Schachat AP, Maumenee IH. Bardet–Biedl syndrome and related disorders. Arch Ophthalmol. 1982;100:285–288. doi: 10.1001/archopht.1982.01030030287011[↩]
- Laurence-Moon and Bardet-Biedl syndromes. Lancet. 1988 Nov 19;2(8621):1178. https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(88)90242-5/fulltext[↩]
- Bardet-Biedl Syndrome. https://rarediseases.org/rare-diseases/bardet-biedl-syndrome[↩]
- PNPLA6 gene. https://medlineplus.gov/genetics/gene/pnpla6[↩][↩][↩][↩][↩][↩][↩]
- Sahu JK, Jain V. Laurence-Moon-Bardet-Biedl syndrome. JNMA J Nepal Med Assoc. 2008 Oct-Dec;47(172):235-7.[↩][↩]
- Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, Stefanelli M, Murphy C, Cramer BC, Dean JC, Beales PL, Katsanis N, Bassett AS, Davidson WS, Parfrey PS. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet A. 2005 Feb 1;132A(4):352-60. doi: 10.1002/ajmg.a.30406[↩][↩][↩]
- Khan BA, Shahid A, Bin Nazir M, Khan KS, Punshi A. Laurence-Moon-Bardet-Biedl Syndrome: A Case Report. Cureus. 2019 Sep 10;11(9):e5618. doi: 10.7759/cureus.5618[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Synofzik M, Hufnagel RB, Züchner S. PNPLA6 Disorders. 2014 Oct 9 [Updated 2021 Jun 10]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK247161[↩][↩]
- Obesity control and low protein diet preserve or even improve renal functions in Bardet-Biedl syndrome: a report of two cases. Dervisoglu E, Isgoren S, Kasgari D, Demir H, Yilmaz A. Med Sci Monit. 2011;17:12–14. doi: 10.12659/MSM.881320[↩]
- Natural course of visual functions in the Bardet-Biedl syndrome. Fulton AB, Hansen RM, Glynn RJ. Arch Ophthalmol. 1993;111:1500–1506. doi: 10.1001/archopht.1993.01090110066026[↩]
- Bardet- Biedl syndrome: a rare case report from North India. Sumir K, Bharat BM, Jyotistema M. Indian J Dermatol Venereol Leprol. 2012;78:228. doi: 10.4103/0378-6323.93656[↩]
- Forsyth RL, Gunay-Aygun M. GeneReviews®. Seattle: University of Washington; 2003. Bardet-Biedl syndrome overview.[↩]
- Laurence-Moon-Bardet-Biedl syndrome: a case report. Khan BA, Shahid A, Bin Nazir M, Khan KS, Punshi A. Cureus. 2019;11:0. doi: 10.7759/cureus.5618[↩]
- Abbasi A, Butt N, Sultan B, Munir SM. Hypokalemic paralysis and megaloblastic anaemia in Laurence-Moon-Bardet-Biedl syndrome. J Coll Physicians Surg Pak. 2009 Mar;19(3):186-8.[↩][↩][↩]
- Ahmed SN, Shahin MA, Chowdhury R, Ahammad AM, Shazzad MN, Alam MR, et al. A 13-year-old female with Bardet-Biedl syndrome – a case report. Bangladesh J Med. 2015; 26: 31-34. https://www.researchgate.net/publication/283708876_A_13-year-old_Female_with_Bardet-_Biedl_Syndrome_-_A_Case_Report[↩]
- Qadar LT, Ahmed ZM, Munawar M, Hasan CA, Iqbal SU. Laurence-Moon-Bardet-Biedl Syndrome with Coexisting Abdominal Distension and Positive Fluid Thrill: A Rare Manifestation Reported in Karachi, Pakistan. Cureus. 2019 Jun 11;11(6):e4885. doi: 10.7759/cureus.4885[↩]
- Mahmood SH, Khan M, Qadar LT, Yousuf F, Hasan M. A Unique Manifestation of Bardet-Biedl Syndrome with Otolaryngologic Symptoms and Bronchopneumonia in a One-year-old Girl. Cureus. 2019 Sep 21;11(9):e5717. doi: 10.7759/cureus.5717[↩]
- Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013 Jan;21(1):8-13. doi: 10.1038/ejhg.2012.115[↩]
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999 Jun;36(6):437-46. https://pmc.ncbi.nlm.nih.gov/articles/instance/1734378/pdf/v036p00437.pdf[↩]
- Haque M, Alam M, Begum S, Rahman S. Bangabandhu Sheikh Mujib Med Univ J. Vol. 9. Bangabandhu Sheikh Mujib Medical University Journal; 2016. Bardet-Biedl syndrome; pp. 119–122.[↩]
- Kmoch S, Majewski J, Ramamurthy V, Cao S, Fahiminiya S, Ren H, MacDonald IM, Lopez I, Sun V, Keser V, Khan A, Stránecký V, Hartmannová H, Přistoupilová A, Hodaňová K, Piherová L, Kuchař L, Baxová A, Chen R, Barsottini OG, Pyle A, Griffin H, Splitt M, Sallum J, Tolmie JL, Sampson JR, Chinnery P; Care4Rare Canada; Banin E, Sharon D, Dutta S, Grebler R, Helfrich-Foerster C, Pedroso JL, Kretzschmar D, Cayouette M, Koenekoop RK. Mutations in PNPLA6 are linked to photoreceptor degeneration and various forms of childhood blindness. Nat Commun. 2015 Jan 9;6:5614. doi: 10.1038/ncomms6614[↩]
- Liu J, He Y, Lwin C, Han M, Guan B, Naik A, Bender C, Moore N, Huryn LA, Sergeev YV, Qian H, Zeng Y, Dong L, Liu P, Lei J, Haugen CJ, Prasov L, Shi R, Dollfus H, Aristodemou P, Laich Y, Németh AH, Taylor J, Downes S, Krawczynski MR, Meunier I, Strassberg M, Tenney J, Gao J, Shear MA, Moore AT, Duncan JL, Menendez B, Hull S, Vincent AL, Siskind CE, Traboulsi EI, Blackstone C, Sisk RA, Miraldi Utz V, Webster AR, Michaelides M, Arno G, Synofzik M, Hufnagel RB. Neuropathy target esterase activity defines phenotypes among PNPLA6 disorders. Brain. 2024 Jun 3;147(6):2085-2097. doi: 10.1093/brain/awae055[↩]
- Bardet-Biedl syndrome. https://medlineplus.gov/genetics/condition/bardet-biedl-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Bardet-Biedl syndrome. https://rarediseases.info.nih.gov/diseases/6866/x[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- What is Bardet-Biedl Syndrome? https://bbsuk.org.uk/what-is-bardet-biedl-syndrome[↩]
- Boucher-Neuhäuser syndrome. https://medlineplus.gov/genetics/condition/boucher-neuhauser-syndrome[↩][↩][↩][↩][↩]
- Gordon Holmes syndrome. https://medlineplus.gov/genetics/condition/gordon-holmes-syndrome[↩][↩][↩][↩][↩][↩]
- Joubert syndrome. https://medlineplus.gov/genetics/condition/joubert-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- McKusick-Kaufman syndrome. https://medlineplus.gov/genetics/condition/mckusick-kaufman-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Slavotinek AM. McKusick-Kaufman Syndrome. 2002 Sep 10 [Updated 2020 Dec 3]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1502[↩][↩][↩][↩]
- Yewalkar S.P., Yadav V.K., Khadse G.J. The McKusick-Kaufman hydrometrocolpos-polydactyly syndrome: a rare case report. Indian J. Radiol. Imag. 2013;23(2):183. doi: 10.4103/0971-3026.116573[↩][↩][↩]
- Ullah I, Rauf S, Ali S, Khan KS, Zahid T, Malik J, Afridi RU, Asghar MS. A case of McKusick-Kaufman syndrome with perinatal diagnosis: Case report and literature review. Ann Med Surg (Lond). 2022 Jun 6;79:103926. doi: 10.1016/j.amsu.2022.103926[↩][↩][↩]
- Traisrisilp K., Nunthapiwat S., Luewan S., Tongsong T. Fetal hydrometrocolpos with pre‐axial mirror polydactyly as a new variant of McKusick‐Kaufman syndrome. J. Clin. Ultrasound. 2021;49(1):62–65. doi: 10.1002/jcu.22882[↩]
- Thorat JV, Tambolkar S. Persistent Urogenital Sinus Leading to Hydrometrocolpos in a Female Child With Features of McKusick-Kaufman Syndrome. Cureus. 2024 Jun 8;16(6):e61957. doi: 10.7759/cureus.61957[↩]
- Halim A., Afzal T., Fatima S., Riaz S. A newborn with rare McKusick syndrome. J. Coll Physicians Surg. Pak. 2018;28(6):S140–S142. doi: 10.29271/jcpsp.2018.06.S140[↩]
- Rosenberg H.K., Chaudhry H. In: Diagnostic Ultrasound. fourth ed. Rumack C.M., editor. Elsevier Mosby Publishers; Philadelphia: 2011. Pediatric pelvic sonography; pp. 1936–1938.[↩][↩]
- Kanso, Khadija & Abou Merhi, Bassem & Zeidan, Marwan & Ibrahim, Soukaina & Ghandour, Fatima & Iskandarani, Fadi & el Houwayek, Eliane & Yassine, Fatima & Chokr, Imad. (2018). A Rare Case Report of Hydrometrocolpos in a Female Newborn. International Journal of Current Research and Review. Vol 10. 22-24. 10.7324/IJCRR.2018.1025[↩]
- Nagaraj B.R., Basavalingu D., Paramesh V.M., Nagendra P.D.K. Radiological diagnosis of neonatal hydrometrocolpos-a case report. J. Clin. Diagn. Res.: J. Clin. Diagn. Res. 2016;10(3):TD18. doi: 10.7860/JCDR/2016/18537.7510[↩][↩]
- Ayrim A.A., Gozdemir E., Turhan N., et al. Acute urinary retention associated with an imperforate hymen and haematocolpos. Gynecol. Obstet. Reprod. Med. 2016;15:105–107.[↩]
- Adam A., Hellig J., Mahomed N., Lambie L. Recurrent urinary tract infections in a female child with polydactyly and a pelvic mass: consider the McKusick-Kaufman syndrome. Urology. 2017;103:224–226. doi: 10.1016/j.urology.2017.01.024[↩]
- Meckel syndrome. https://medlineplus.gov/genetics/condition/meckel-syndrome[↩][↩][↩][↩][↩][↩][↩]
- Meckel Syndrome. https://rarediseases.org/rare-diseases/meckel-syndrome[↩][↩][↩]
- Oliver GL, McFarlane DC. Congenital trichomegaly: with associated pigmentary degeneration of the retina, dwarfism, and mental retardation. Arch Ophthalmol 1965;74:169–71. doi:10.1001/archopht.1965.00970040171008[↩]
- Chang TS, McFarlane DC, Oliver G, Willis NR. Congenital trichomegaly, pigmentary degeneration of the retina and growth retardation (Oliver-McFarlane syndrome): 28-year follow-up of the first reported case. Can J Ophthalmol. 1993 Jun;28(4):191-3.[↩]
- Corby DG, Lowe RS Jr, Haskins RC, Hebertson LM. Trichomegaly, pigmentary degeneration of the retina, and growth retardation. A new syndrome originating in utero. Am J Dis Child. 1971 Apr;121(4):344-5. doi: 10.1001/archpedi.1971.02100150118018[↩]
- Haritoglou C, Rudolph G, Kalpadakis P, Boergen KP. Congenital trichomegaly (Oliver-McFarlane syndrome): a case report with 9 years’ follow up. Br J Ophthalmol. 2003 Jan;87(1):119-20. doi: 10.1136/bjo.87.1.119[↩]
- Patton MA, Harding AE, Baraitser M. Congenital trichomegaly, pigmentary retinal degeneration, and short stature. Am J Ophthalmol. 1986 Apr 15;101(4):490-1. doi: 10.1016/0002-9394(86)90656-2[↩]
- Haimi M, Gershoni-Baruch R. Autosomal recessive Oliver-McFarlane syndrome: retinitis pigmentosa, short stature (GH deficiency), trichomegaly, and hair anomalies or CPD syndrome (chorioretinopathy-pituitary dysfunction). Am J Med Genet A. 2005 Oct 15;138A(3):268-71. doi: 10.1002/ajmg.a.30953[↩][↩]
- Zaun H, Stenger D, Zabransky S, Zankl M. Das Syndrom der langen Wimpern (“Trichomegaliesyndrom”, Oliver-McFarlane) [The long-eyelash syndrome (trichomegaly syndrome, Oliver-McFarlane)]. Hautarzt. 1984 Mar;35(3):162-5. German.[↩]
- Sonmez S, Forsyth RJ, Matthews DS, Clarke M, Splitt M. Oliver-McFarlane syndrome (chorioretinopathy-pituitary dysfunction) with prominent early pituitary dysfunction: differentiation from choroideremia-hypopituitarism. Clin Dysmorphol. 2008 Oct;17(4):265-7. doi: 10.1097/MCD.0b013e328306a374[↩]
- Sheng X, Zhang S, Peter Boergen K, Li H, Liu Y. Oliver-McFarlane syndrome in a chinese boy: retinitis pigmentosa, trichomegaly, hair anomalies and mental retardation. Ophthalmic Genet. 2015 Mar;36(1):70-4. doi: 10.3109/13816810.2013.824003[↩]
- Mathieu M, Goldfarb A, Berquin P, Boudailliez B, Labeille B, Piussan C. Trichomegaly, pigmentary degeneration of the retina and growth disturbances. A probable autosomal recessive disorder. Genet Couns. 1991;2(2):115-8.[↩]
- Rainier S, Bui M, Mark E, Thomas D, Tokarz D, Ming L, Delaney C, Richardson RJ, Albers JW, Matsunami N, Stevens J, Coon H, Leppert M, Fink JK. Neuropathy target esterase gene mutations cause motor neuron disease. Am J Hum Genet. 2008 Mar;82(3):780-5. doi: 10.1016/j.ajhg.2007.12.018[↩]
- Viertauer S, Kurth I, Eggermann K, Eggers C. Novel phenotype with prominent cerebellar oculomotor dysfunction in spastic paraplegia type 39. J Neurol. 2022 Dec;269(12):6476-6482. doi: 10.1007/s00415-022-11313-6[↩]
- Laurence-Moon-Biedl syndrome: its occurrence in a Negro child; treatment with gonadotropin and androgen. Scott RB, Johnson PT. Am J Dis Child. 1942;63:733–741.[↩]
- Bardet-Biedl syndrome with end-stage kidney disease in a four-year-old Romanian boy: a case report. Mihai CM, Marshall JD, Stoicescu RM. J Med Case Rep. 2011;5:378. doi: 10.1186/1752-1947-5-378[↩][↩][↩]
- Williams B, Jenkins D, Walls J. Chronic renal failure; an important feature of the Laurence-Moon-Biedl syndrome. Postgrad Med J. 1988 Jun;64(752):462-4. doi: 10.1136/pgmj.64.752.462[↩]
- Kaleem S, Srirangadhamu Gopu S, Ishfaq L, Afroze S, Parvez M, Mulaka GSR, Venugopal V. Laurence-Moon-Bardet Biedl Syndrome With Cholelithiasis. Cureus. 2023 Oct 19;15(10):e47316. doi: 10.7759/cureus.47316[↩]
- Urine concentrating defect as presenting sign of progressive renal failure in Bardet-Biedl syndrome patients. Zacchia M, Blanco FD, Torella A, et al. Clin Kidney J. 2021;14:1545–1551. doi: 10.1093/ckj/sfaa182[↩]




{kind=link}